
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Thionated PDI supramolecular polymers: controlling aggregation mechanisms, morphology and function†
Henry E.
Symons
 ,
Maximilian J. L.
Hagemann
,
Robert L.
Harniman
and
Charl F. J.
Faul
*
,
Maximilian J. L.
Hagemann
,
Robert L.
Harniman
and
Charl F. J.
Faul
*
School of Chemistry, University of Bristol, Bristol, BS8 1TS, UK. E-mail: charl.faul@bristol.ac.uk
First published on 24th January 2022
Abstract
Controlling the self-assembly of functional organic molecules provides opportunities to optimise the interlinked properties and structures of these materials for specific applications. Perylene diimide (PDI) derivative, PDI1, was used as a model monomer to investigate the effects of heteroatom substitution in supramolecular polymer systems. In addition to the previously reported optoelectronic effects (including shifted absorbance and quenched fluorescence), thionation was found to have a significant impact on the self-assembly behaviour of PDI1. The thionated analogues of this species displayed a diverse range of self-assembled morphologies, rationalised through changes in intermolecular interactions. Furthermore, thionation results in a transition from highly cooperative to isodesmic assembly mechanism. Such mechanistic control should also be applicable to many other supramolecular systems based on either hydrogen-bonding or π-stacking interactions, and presents an important route for the regulation of dynamic polymer properties. Finally, thionation was found to result in a substantial increase in conductivity recorded within analogous supramolecular polymer structures.
Introduction
In the field of supramolecular polymers (SMPs), i.e., polymers connected by non-covalent, directional interactions between their constituent monomers,1 significant research efforts are focused on gaining control over the assembly behavior. Several promising routes are being explored to obtain this desired control, including chemical modification2–4 or altering the external environment, such as solvent or temperature.3,5–7 One of the most attractive building blocks for such assemblies are perylene diimides (PDIs), also called perylene bisimides (PBIs). PDIs possess a range of useful optoelectronic properties, including high fluorescence quantum yields, high chemical and thermal stability, and N-type semiconductivity. The change and control of the assembly behaviour can have a significant influence on the properties of the resulting SMPs, especially the optoelectronic properties, depending on whether the monomers undergo H- or J-aggregation.3The formation of one-dimensional self-assembled structures from PDI derivatives has been the subject of much research, owing largely to their fascinating array of potential applications.8 The unique properties of PDIs are well-suited to utilisation in the fields of electronics, photocatalysis, bioimaging and in thermoelectric devices, amongst others, as shown by Zhu et al. and others in the field.9–13 More recently, the ability of PDIs to readily form anionic species has been demonstrated to offer excellent material properties, for example as thermoelectric materials and as photosensitizers for hydrogen evolution reactions (HERs).14,15 The alignment of chromophores achieved in one-dimensional assemblies further enhances the desirable functionalities displayed by these materials, for example, through π-stacking-mediated exciton diffusion.8
In order to tune the properties, self-assembly and therefore applications, the versatility of the PDI motif has been exploited by selective functionalisation in a number of positions, for example by the laboratories of Würthner and Zhu.3,4,14–25 Imide substitution is the most facile route for functionalisation, and may be used to alter the aggregation behaviour and solubility of PDI derivatives. Such changes have minimal effect on the PDI optoelectronic properties, owing to nodes in the frontier molecular orbitals at the imide-nitrogen position.10,26 Conversely, substitution in the bay positions has strong substituent-dependent effects on the optoelectronic properties of the PDI chromophore.20 Targeted changes at the bay positions allow tailoring of these properties for particular applications. The resultant core distortion from this substitution pattern, however, has a significant impact on the self-assembly behaviour, in many cases inhibiting the formation of well-defined nanostructures.27
One area that has received comparatively little attention is functionalization within the core PDI moiety. Heteroatom substitution has been applied to a number of organic semiconductors, and has proven to be a versatile strategy to optimize their properties, including solubility, film morphology, and charge carrier mobility.28–31 This approach has only recently been utilised for both PDI and related naphthalene diimide (NDI) systems.32
Oxygen-to-sulphur exchange has a number of reported effects on the optoelectronic properties of these chromophores. Firstly, absorbance is bathochromically shifted significantly, resulting in a distinctive colour change. PDIs typically fluoresce very strongly with quantum yields close to unity.33–35 Thionation has been shown to result in complete quenching of this fluorescence, instead forming longer-lived triplet excited states leading to phosphoresence.36 These species may be desirable for their increased rate of charge separation in organic photovoltaic (OPV) devices via singlet fission, or for generating singlet oxygen for therapeutic applications.37–39
Despite the promising properties displayed by these thionated materials, a very limited number of studies have been conducted on their ability to form self-assembled structures. Differences in film-forming behaviour and solid-state absorbance spectra, however, suggest thionation has a number of effects and is worthy of further investigation. We herein present a comprehensive study on the effects of thionation on the self-assembly behaviour of a previously reported PDI derivative,5,40PDI1, shown in Scheme 1. We highlight the opportunities that thionation provides for fine control over the self-assembly behaviour of this known PDI derivative.
 | ||
| Scheme 1 General conditions used for the thionation of PDI-1 with Lawesson's reagent (LR). Chemical structures of products isolated from thionation with between one and four oxygen-to-sulphur substitutions. | ||
Results and discussion
Synthesis and characterisation
Thionation of PDI1 was achieved by reaction of the parent PDI with Lawesson's reagent (LR) in anhydrous toluene, as described in Scheme 1.36 This reaction generated a mixture of thionated products, with between one and six substitutions found in crude reaction mixtures. As demonstrated in Table S1 (ESI†), alterations to either the molar ratio of LR to PDI1 or the reaction length could be used to favour products with differing levels of thionation.Despite the more complex side chains employed in this study (when compared with previous studies on PDI thionation), separation of each product was successfully achieved with conventional chromatography using a combination of preparative TLC and silica gel column chromatography. By this method, products containing between one and four sulphur atoms were isolated, as shown in Scheme 1, and termed PDI1-S1 to PDI1-S4, respectively. Interestingly, when four substitutions occur, two distinct regioisomers are observed, denoted cisPDI1-S4 and transPDI1-S4, depending on the relative positions of the two core sulphur atoms. An additional product substituted with five sulphur atoms was also isolated, however yield was insufficient to allow extensive characterisation. Notably, two products were not obtained after purification: the third geminal S4-substituted isomer, and the fully-thionated S6-substituted product. Findings in previous studies suggest the former is energetically unfavoured, and likely only present in trace amounts unless favoured by steric blocking of one imide group.36,41,42 Peaks consistent with trace amounts of an S6-substituted product were observed in mass spectra of the crude reaction mixtures upon thionation of PDI1; however, despite extensive efforts, significant quantities of this product could not be isolated. All thionated products obtained in significant amounts were fully characterised by 1H and 13C NMR spectroscopy (Fig. S1–S10, ESI†) and MALDI-ToF mass spectrometry (Fig. S12–S16, ESI†). To confirm the identities of each regioisomer of PDI1-S4, additional 1H–1H ROESY NMR analysis was conducted on these two compounds and is shown in Fig. S11 (ESI†). In agreement with previous reports on thionated PDI systems,36,43 the trans isomer displayed additional through-space coupling interactions between the bridgehead protons, confirming their close proximity in this compound. Comparable peaks were not observed for cisPDI1-S4.
The complete absence of products exhibiting thionation of only the imide oxygen atoms indicates a remarkable preference for thionation of the side chain amide group compared with the imide positions.
Optoelectronic properties
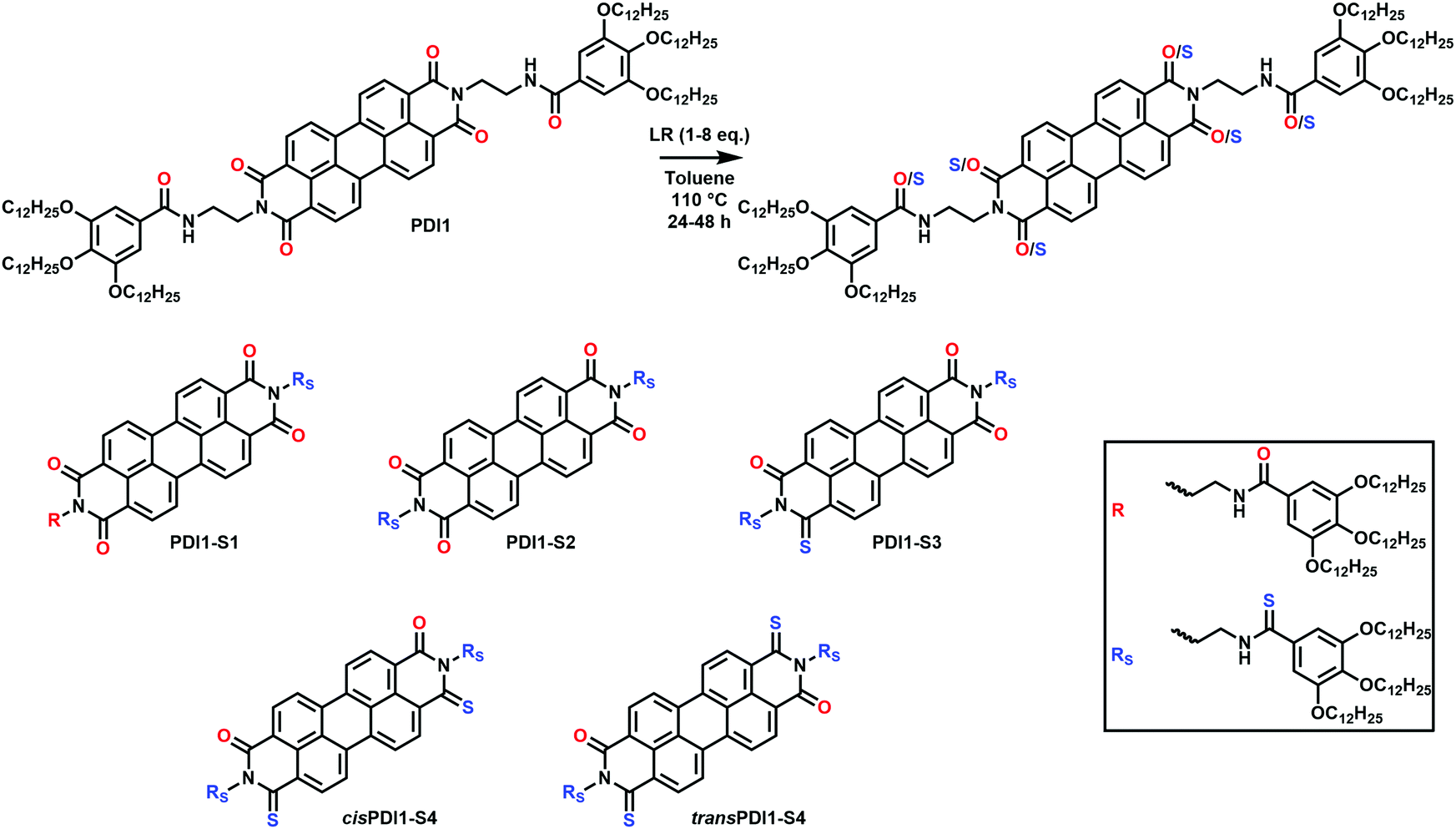
UV/Vis and fluorescence spectroscopy were used to investigate the optoelectronic properties of PDI1 and its thionated analogues. Spectra, shown in Fig. 1a, were recorded in chloroform (at a concentration of 1 × 10−5 M) to minimise aggregation.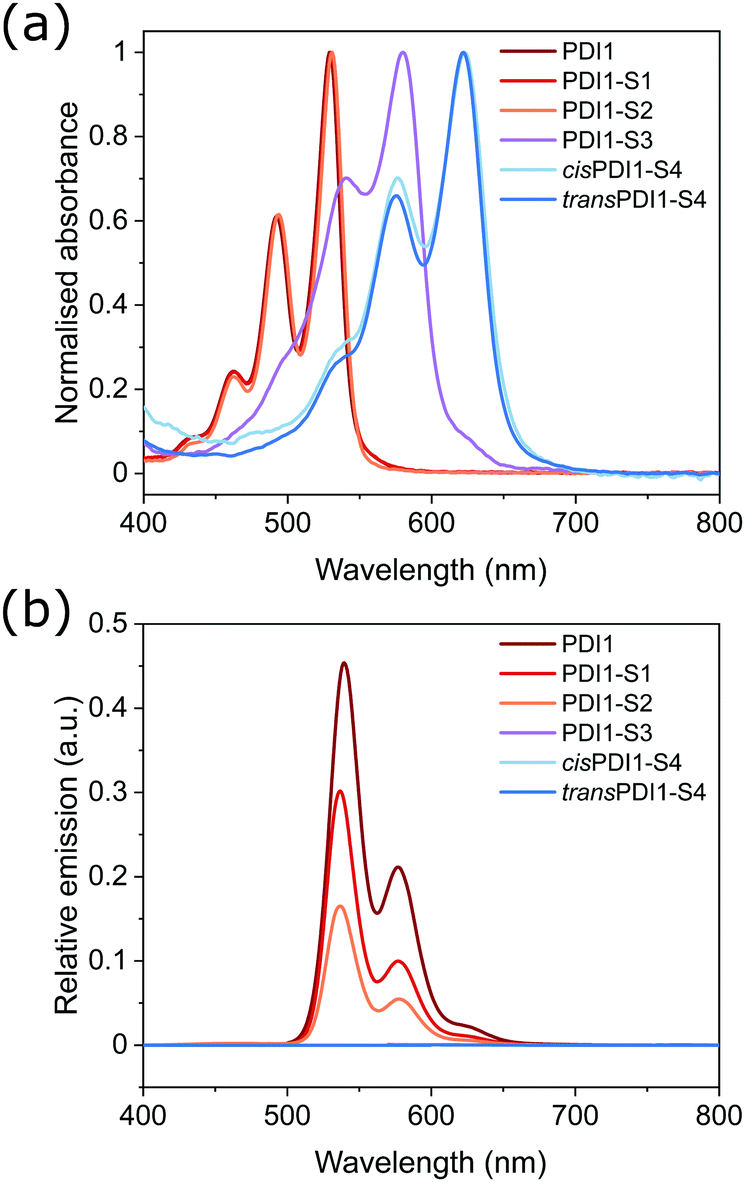 | ||
| Fig. 1 UV/Vis absorbance spectra (a), and fluorescence spectra (b), of PDI1, PDI1-S1, PDI1-S2, PDI1-S3, cisPDI1-S4, transPDI1-S4, and PDI1-S5. All spectra recorded at a concentration of 10−5 M in chloroform. Excitation wavelengths were chosen based on the maxima of the corresponding absorbance; 530 nm for PDI1, PDI1-S1, and PDI1-S2, 580 nm for PDI1-S3, 622 nm for cisPDI1-S4 and transPDI1-S4, and 670 nm for PDI1-S5. | ||
The absorbance spectrum of PDI1 displays a vibronic progression typical of PDI materials, with a maximum absorbance at 529 nm, and progressively weaker peaks at shorter wavelengths. PDI1-S1 and PDI1-S2 exhibit spectra almost identical to the parent PDI, with absorbance maxima at 530 and 531 nm, respectively. The absence of significant changes is consistent with substitution occurring only within the side chains, which does not affect the optoelectronic properties of the core PDI chromophore. Conversely, products containing 3 or more sulphur atoms displayed successive bathochromic shifts of their absorbance spectra, dependent on the number of thionated imide positions: PDI1-S3 displayed a maximum absorbance at 580 nm, whereas both S4-substituted PDIs exhibited identical maxima at 622 nm. This behaviour is consistent with previous reports on NDI and PDI thionation and is attributed to a concurrent increase in energy of the HOMO and decrease in that of the LUMO, and an increase in the electron affinity associated with increasing degree of thionation.32,36,44
The typically strong fluorescence of PDI materials is completely quenched in all derivatives exhibiting any degree of core thionation, as shown in Fig. 1b. This remarkably strong quenching effect has been attributed to a reordering of electronic states upon thionation, such that inter-system crossing to a triplet excited state is facilitated.36 Conversely, thionation of only the side chain amide groups resulted in a significant, though not complete, quenching as shown in the spectra of PDI1-S1 and PDI1-S2. This more gradual quenching, evidenced by a successive decrease in fluorescence intensity upon thionation of neither, one or both amide groups, is likely to occur due to more conventional mechanisms, such as the heavy atom effect.45,46 This effect is likely to be further exacerbated by the well-documented intramolecular hydrogen-bonding interactions between amide and imide groups within PDI1, resulting in a closer proximity of the heavier sulphur atoms to the PDI chromophore.5
Self-assembled morphology
To explore differences in behaviour between PDI1 and its thionated analogues, the solution self-assembly of all obtained derivatives was investigated in methylcyclohexane (MCH). This solvent has previously been demonstrated to strongly induce aggregation in PDI1 and therefore serves as an ideal medium to highlight morphological differences.5 TEM images of the typical morphologies shown by each derivative in this solvent are shown in Fig. 2. | ||
| Fig. 2 TEM images of the morphologies formed by the self-assembly of PDI1 (a), PDI1-S1 (b), PDI1-S2 (c), PDI1-S3 (d), cisPDI1-S4 (e), and transPDI1-S4 (f), in MCH at a concentration of 5 × 10−5 M. | ||
As reported elsewhere, at the dilute concentrations studied here (5 × 10−5 M), PDI1 (Fig. 2a) forms networks of interlinked nanofibers, likely driven predominantly by a combination of hydrogen-bonding and π-stacking interactions.5,40 Individual nanofibers were highly elongated, often multiple micrometres in length. Widths ranged from unimolecular strands of approximately 3 nm to ribbon-like structures more than 500 nm in width. The significant proportion of narrow supramolecular strands suggests they are relatively stable, with aggregation into larger bundles occurring only due to the high density of nanofibers present.
The assembly of PDI1-S1 was studied by a similar procedure, with typical morphologies observed shown in Fig. 2b. This compound also forms fibrous structures but with clear differences to those formed by the parent PDI1. Most notably, nanofiber lengths are significantly shorter, with typical lengths between 100 and 300 nm, and lengths greater than 500 nm uncommon.
Further increases to the degree of thionation have large effects on the self-assembly behaviour of PDI1 derivatives. As shown in Fig. 2c, PDI1-S2 displays no ordered nanostructures up to concentrations of 5 × 10−5 M in MCH. Instead, regions of increased electron density are visible in the TEM images of these samples. Despite the absence of ordered structures, the visibility of these regions by TEM without staining does not rule out self-assembly. PDI1-S3 displays similar behaviour, with amorphous films, shown in Fig. 2d, present across the majority of the substrate. In contrast to PDI1-S2, however, these morphologies exhibit higher electron densities, and possess occasional regions of greater anisotropy.
Unlike lesser thionated derivatives, both S4-substituted isomers formed well-defined SMPs similar to those displayed by the parent PDI1 (shown in Fig. 2e and f). Interestingly, significant differences between the morphologies formed by each regioisomer were evident. cisPDI1-S4 assembles into individual, well-dispersed supramolecular fibers. Nanofiber lengths ranged from hundreds of nanometres to multiple micrometres. The width of these fibers was predominantly between 2 and 20 nm, with a mean of 8.5 nm (width distribution shown in Fig. S17, ESI†). Conversely, nanofibers formed by the self-assembly of transPDI1-S4, although similar in length, possessed significantly greater widths. Typical widths of these ribbon-like structures were between 30 and 200 nm, though a limited number of thinner fibers and sections were also present, with a mean value of 70.4 nm (width distribution shown in Fig. S18, ESI†). Also notable are differences in the subsequent aggregation of the assembled nanofibers. Fibers formed by transPDI1-S4 showed a strong tendency to aggregate and were almost exclusively present in large clusters, or in extended fibrous networks. This behaviour is in stark contrast to that of cisPDI1-S4 nanofibers, which were dispersed (as single fibers) across the surface of the grid.
These morphological differences can be rationalised by considering changes in the relative strengths of the non-covalent interactions that lead to SMP formation. As discussed previously, the self-assembly of PDI1 is driven by aromatic stacking interactions of the PDI core complemented by hydrogen-bonding interactions between adjacent amide groups in both side chains. Computational studies on model thioamide systems have previously revealed a decrease in hydrogen bond acceptor ability and an increase in hydrogen bond donor ability compared with the corresponding oxoamide.47 For an amide group within a supramolecular polymer, acting as both donor and acceptor of hydrogen bonds, an overall decrease in interaction energy of approximately 1.25 kcal mol−1 upon thionation can be expected. Similar studies were carried out by Tilley et al. to assess the effect of imide thionation on the strength of stacking interactions between a pair of model PDI derivatives.41 Thionation was found to increase the interaction energy, with each successive oxygen–sulphur substitution leading to a comparable average increase of 1.38 kcal mol−1.
Based on these effects, a progressive decrease in interaction energy is expected upon the initial thionation of one or both side chain amides (in PDI1-S1 and PDI1-S2, respectively) compared with PDI1. The shorter nanofibers and absence of anisotropic structures shown by these two derivatives are in good agreement with this decrease in interaction strength. Subsequent core thionation leads to a successive increase in interaction strength between adjacent unimers. Notably, both S4-substituted PDI derivatives are expected to display similar interaction energies to the parent PDI1. Morphological studies corroborate this behaviour, with all three systems displaying similar nanofibrous structures. The expected changes in interaction energy upon thionation are summarised graphically in Fig. S19 (ESI†).
The apparent similarities in the morphologies formed by PDI1, cisPDI1-S4 and transPDI1-S4 prompted further investigation of these species by AFM, with typical structures observed shown in Fig. 3. On carbon-coated mica substrates, AFM revealed comparable nanofiber structures to those observed on the carbon-coated TEM grids used. Apparent increases in widths compared with TEM images are likely to be a result of either slight differences in sample preparation methodology, or peak broadening artefacts. All three PDI derivatives studied formed nanofibers with similar minimum heights of between 2.5–3.5 nm, likely indicative of supramolecular strands of unimolecular thickness.
 | ||
| Fig. 3 AFM height images of the morphologies formed by the self-assembly of PDI1 (a) cisPDI1-S4 (c), and transPDI1-S4 (e) in MCH at a concentration of 5 × 10−5 M. Height profiles along each corresponding coloured line, (b, d and f). Scale bars = 1 μm. | ||
In agreement with previous reports, PDI1 nanofibers were also found to display regions of greater width and height which likely arise due to lateral aggregation of individual supramolecular strands.5,40 Nanofibers of transPDI1-S4 showed similar behaviour with distinct heights of 2.5–3.5, 6–7 or 10–11 nm observed across the majority of structures. This incremental increase in height likely indicates overlapping of multiple fibers of unimolecular thickness. In contrast to the other two species, cisPDI1-S4 nanofibers showed exclusively unimolecular heights of 2.5–3.5 nm, regardless of width or length.
The unexpectedly dissimilar self-assembly behaviour of regioisomers cisPDI1-S4 and transPDI1-S4 may be attributed to the effects of heteroatom substitution on the aforementioned lateral aggregation of nanofibers. Relatively strong interactions between divalent sulphur atoms and aromatic systems are well-documented, most commonly in the context of proteins.48–51 Non-covalent interactions between Group 16 elements, often referred to as chalcogen bonding, are increasingly utilised in a range of supramolecular systems and may display strengths comparable with conventional hydrogen bonds.52,53 The strength of such interactions increases considerably from oxygen to sulphur and other larger chalcogen atoms.54
We postulate that both aforementioned interactions contribute to the aggregation of individual co-facially-stacked supramolecular strands into larger bundled structures. Additional S–S and S–π interactions should encourage this type of lateral aggregation on the sulphur-substituted edges of thionated PDI1 derivatives. In transPDI1-S4, sulphur atoms are positioned on opposite edges of the PDI motif and are therefore always exposed, leading to the formation of wider structures with a tendency to aggregate further into fibrous networks. For cisPDI1-S4, both sulphur atoms are positioned on the same edge of the PDI core. Upon lateral aggregation of individual strands, both sulphur atoms may be positioned internally, inhibiting further bundling of these structures. Such behaviour supports the remarkable preference of this derivative for nanofibers less than 10 nm in width, compared to those with widths almost an order of magnitude greater formed by transPDI1-S4.
Mechanism of self-assembly
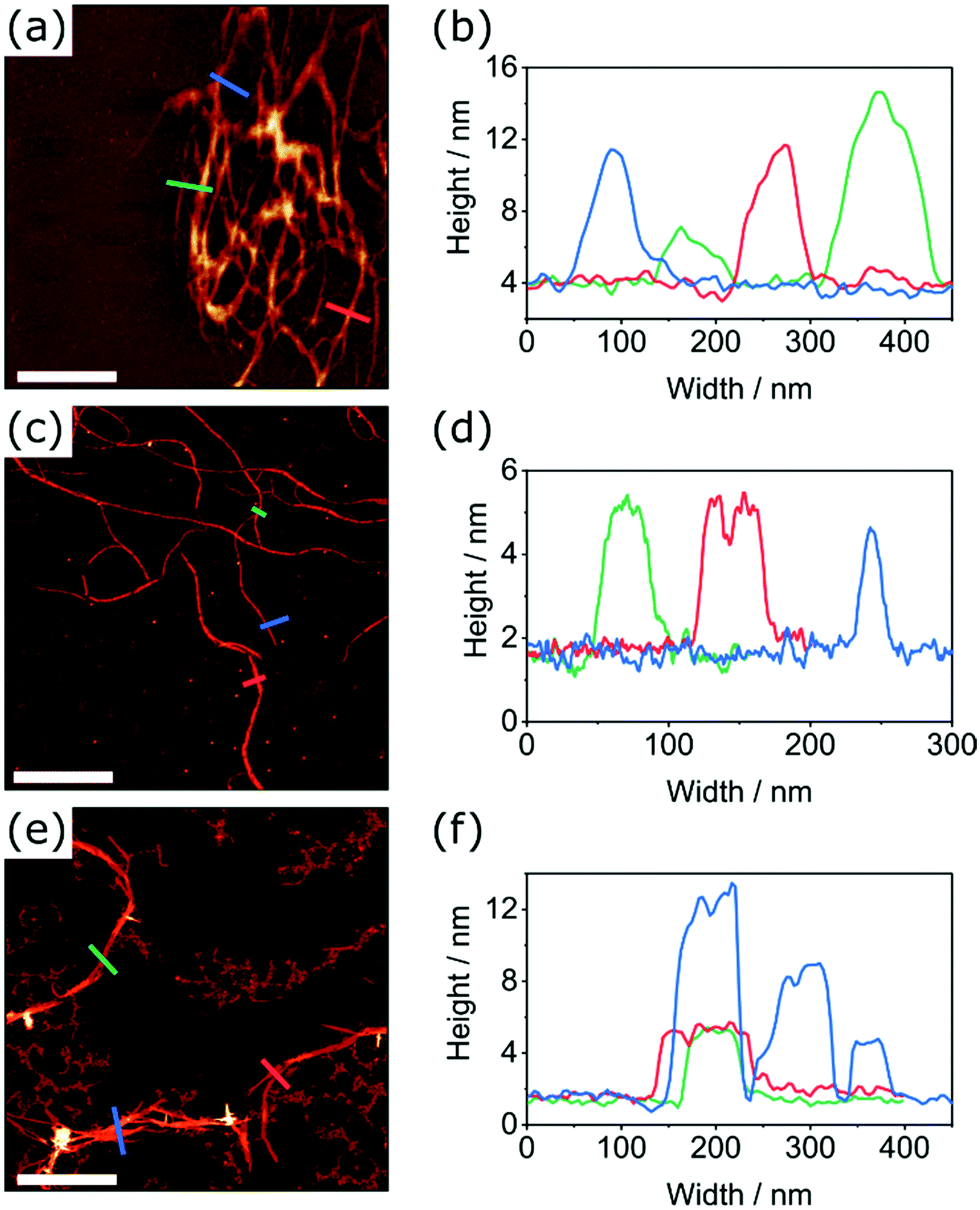
Unlike their covalent counterparts, the formation mechanisms of SMPs have significant effects on the properties of the resultant materials.55 Two predominant mechanisms exist; isodesmic, where the binding constant is independent of polymer length, and cooperative, where two distinct phases are present, characterised by different binding constants.56 Changes in the degree of aggregation as a function of temperature allow for the most reliable identification of polymerisation mechanism.57 In this study, we therefore employed variable-temperature UV-Vis spectroscopy measurements to investigate the effects of thionation on the mechanism of self-assembly.Changes in the absorbance profiles of PDI1 and transPDI1-S4 in MCH upon heating from 293 to 373 K are shown in Fig. 4. A concentration of 5 × 10−6 M was selected, such that near complete aggregation was observed at the lowest temperature, and a completely unimeric sample obtained at the highest temperature. Changes in absorbance for the remaining thionated derivatives are shown in Fig. S20 (ESI†). Prior to heating, all investigated materials displayed similar differences to the corresponding unimeric spectra shown in Fig. 1; λmax for each was shifted hypsochromically (by approximately 30–70 nm with respect to the corresponding unimer), and a new bathochromically shifted shoulder was apparent (at a wavelength 20–30 nm longer than the λmax of the corresponding unimer). These changes are consistent with H-aggregation, with the partially allowed J-band resulting from rotational displacement of PDI cores.58
 | ||
| Fig. 4 Changes in the UV/Vis absorbance spectra of PDI1 (a), and transPDI1-S4 (b), upon heating from 293 K (red profiles) to 373 K (black profiles) in 5 K intervals. Spectra recorded at a concentration of 5 × 10−6 M in MCH. | ||
Upon heating, spectral changes associated with disaggregation were observed for all samples. The change in absorbance at the λmax of the corresponding unimeric spectra was used to determine the degree of aggregation at each temperature for each sample using eqn (S1) (ESI†). Degree of aggregation was plotted as a function of temperature for each sample in Fig. 5. Data for each compound were fitted to either the cooperative model proposed by Smulders et al.,57,59 or to a sigmoidal curve (indicative of an isodesmic mechanism60) depending on which gave the lowest R2 parameter. Samples best modelled by a cooperative mechanism were initially fitted by eqn (S2) (ESI†) at temperatures below nucleation, and eqn (S3) (ESI†) at higher temperatures.
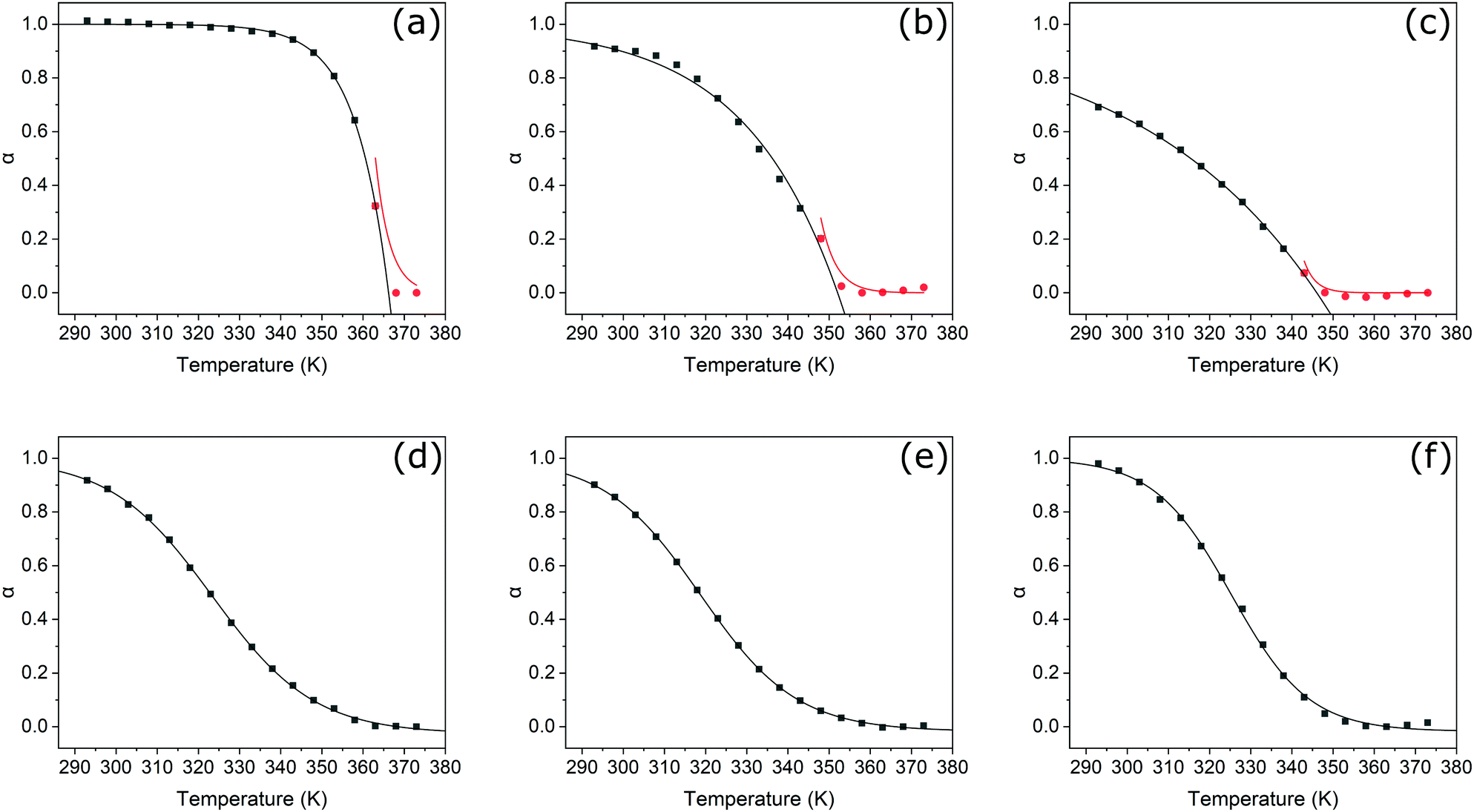 | ||
| Fig. 5 Changes in degree of aggregation, α, as a function of temperature in K, and fitting with either cooperative or isodesmic models, for PDI1 (a), PDI1-S1 (b), PDI1-S2 (c), PDI1-S3 (d), cisPDI1-S4 (e), and transPDI1-S4 (f). For species best fitted with a cooperative mechanism, data are separated into the nucleation phase (red circles), and elongation phase (black squares). Fitting curves for each regime are shown in the corresponding colour. | ||
In agreement with previous reports,5PDI1 displays a highly cooperative mechanism, characterised by a sharp transition between nucleation and elongation. Specifically, a very steep elongation curve is observed, with near complete aggregation persisting at a relatively high temperature of approximately 340 K. For the initial two sulphur substitutions (i.e., PDI1-S1 and PDI1-S2), the cooperative assembly mechanism of PDI1 is maintained. A clear transition between nucleation and elongation is visible, depicted by a change of symbol (black squares to red circles) in Fig. 5b and c. Successive thionation for these two derivatives, however, resulted in a significant reduction in the steepness of the elongation curve. Both derivatives therefore show incomplete aggregation, even at the lowest temperature studied.
Thermodynamic parameters extracted directly from the fitting function reveal a decrease in the magnitude of ΔHe, the enthalpic change during elongation, from −137.8 kJ mol−1 for PDI1 to −45.1 and −22.6 kJ mol−1 for PDI1-S1 and PDI1-S2, respectively. Eqn (S4) and (S5) (ESI†) were used to estimate the degree of polymerisation. At 293 K, a degree of polymerisation >10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 was determined for PDI1, alongside values of 390 for PDI1-S1 and 290 for PDI1-S2. Full thermodynamic parameters for cooperatively fitted systems are given in Table S2 (ESI†).
000 was determined for PDI1, alongside values of 390 for PDI1-S1 and 290 for PDI1-S2. Full thermodynamic parameters for cooperatively fitted systems are given in Table S2 (ESI†).
For systems best modelled by an isodesmic mechanism, the equilibrium constant, Ke, was estimated using eqn (S6) and (S7) (ESI†) at each temperature value. Thermodynamic parameters for polymerisation were then determined via Van’t Hoff plots, as shown in Fig. S21 (ESI†), and described by eqn (S8) and (S9) (ESI†). Compared with PDI1-S2, the magnitude of ΔH increased considerably for PDI1-S3 to a value of −78.7 kJ mol−1. Further increases in magnitude were observed for cisPDI1-S4 and transPDI1-S4, with values of −91.8 and −103.5 kJ mol−1, respectively. The degree of polymerisation at 293 K for each sample was estimated using eqn (S6) (ESI†). PDI1-S3 displayed a degree of polymerisation of 3.5, cisPDI1-S4 a value of 3.2, and transPDI1-S4 a value of 7.0. Full thermodynamic parameters derived for compounds that polymerised via isodesmic mechanisms are given in Table S3 (ESI†).
The overall trends in the enthalpic changes determined here are in good agreement with those predicted based on differences in intermolecular interactions upon thionation. The enthalpy loss for the polymerisation of PDI1 decreases significantly upon the initial thionation of side chain amide groups, indicative of a decrease in the strength of hydrogen–bonding interactions. Further thionation of the core imide groups then leads to enhancement of stacking interactions, reflected by a successive increase in the magnitude of ΔH from S2- to S4-substituted PDI1 analogues. The more negative value of ΔH displayed by the trans S4-substituted isomer compared to the cis isomer further supports the formation of additional energetically favourable S–S or S–π interactions, leading to lateral aggregation as described above. Changes in the estimated degree of polymerisation followed a similar pattern for PDI1, PDI1-S1, and PDI1-S2; however, the remaining three compounds investigated all displayed low values between of 3 and 7. Such behaviour is typical of isodesmic systems, where a significant proportion of the material may remain in a unimeric or oligomeric state.
Overall, a clear transition from a highly cooperative polymerisation process to one that proceeds isodesmically is evident upon successive oxygen-to-sulphur substitutions. This behaviour may also be rationalised by changes in the relative strengths of noncovalent interactions. A common source of cooperativity in SMP formation is the electronic influence in hydrogen-bonding interactions along a polymer chain.56 Conversely, supramolecular polymers formed predominantly by π-stacking interactions typically display isodesmic formation mechanisms.56 As previously discussed, amide thionation reduces overall hydrogen–bonding ability, whereas thionation of PDI imide positions increases their tendency to π-stack. Thionation in either position would therefore be expected to cause a shift from a binding mode with a large hydrogen–bonding contribution to one based largely on aromatic stacking. This change is consistent with the gradual transition of observed mechanism from cooperative to isodesmic upon thionation.
Electrostatic and conductivity measurements
The semiconductivity displayed by PDI-based materials is important in a range of applications. To assess if changes in conductivity were exhibited within individual SMPs upon thionation, we performed both electrostatic force microscopy (EFM) and PeakForce tunnelling AFM (PF-TUNA) on self-assembled samples of PDI1 and cisPDI1-S4. Comparison of electronic properties of these two systems (i.e., control PDI1 and thionated cisPDI1-S4) is possible as the fiber-like morphology of these compounds upon self-assembly is very similar and the non-contact lift mode and <1 nN force regime of EFM and PF-TUNA, respectively, ensure minimal disruption to the molecular organisation within the self-assembled architectures.When a conductive cantilever moves through an electrostatic field a force is induced dependent on the magnitude of the field. If oscillating at its resonance frequency, the induced force will alter the resonance creating a phase shift in the cantilever's motion. The concurrent measurements of the topography and electrostatic interaction (detailed methodology given in ESI†) for PDI1 and cisPDI1-S4 are shown in Fig. 6a and b, respectively, as 3-dimensional surfaces overlaid with the induced phase shifts as blue colour maps. Data are shown for short sections of nanofibers with comparable heights of 3.5–4.5 nm for each compound. Measured with the same conductive cantilever, qualitative comparison of the magnitude of the electrostatic field is possible when the induced phase shifts are normalised to the carbon background. Phase profiles, extracted orthogonally to nanofiber long-axes are presented in Fig. 6c, demonstrating a greater contrast in phase between nanofiber and substrate, and so greater electrostatic field, for thionated cisPDI1-S4 compared with the parent PDI1.
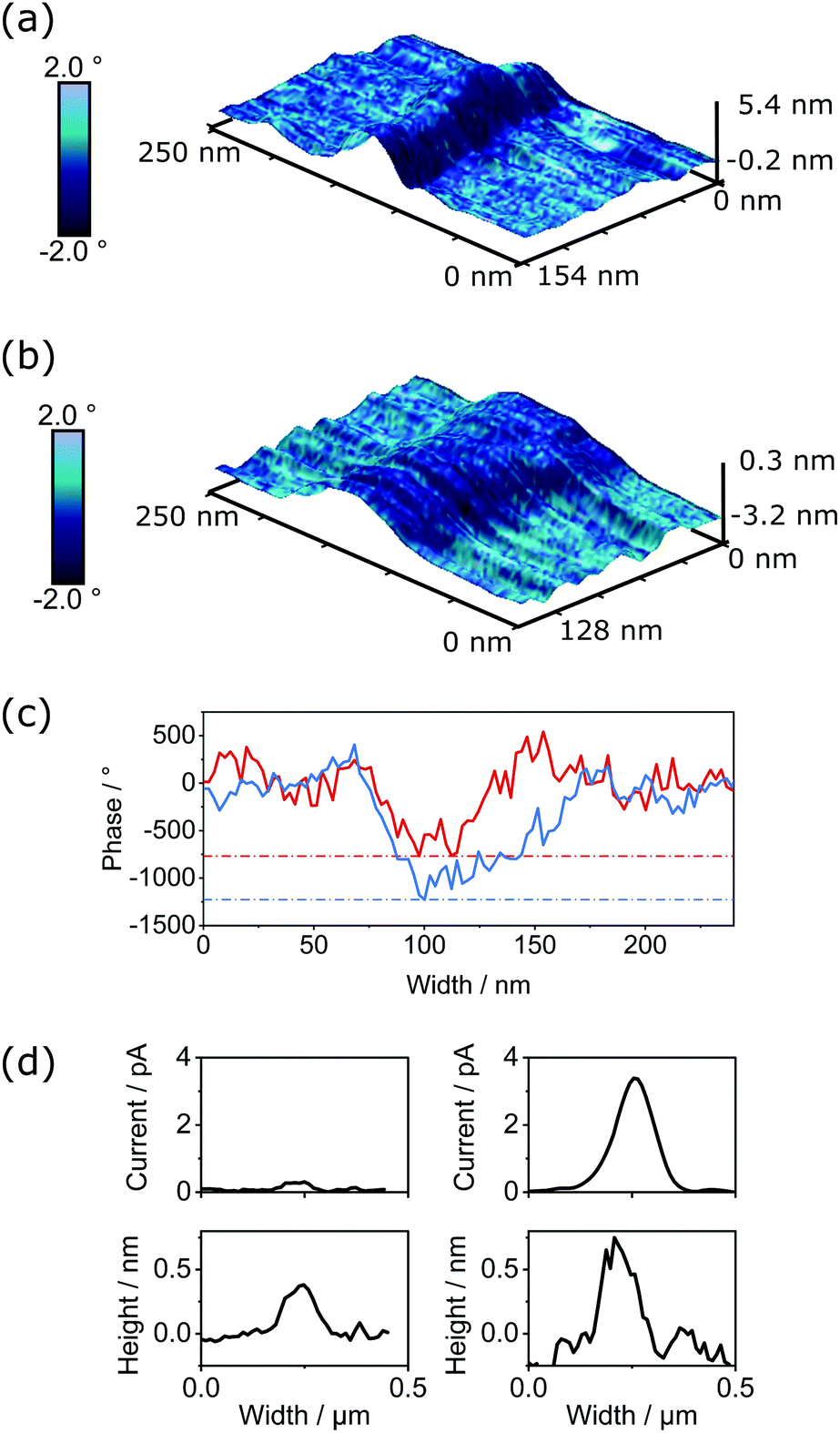 | ||
| Fig. 6 3D micrographs showing combined topological (vertical axis) and phase data (colourmap) obtained by EFM measurements on self-assembled nanofibers of PDI1 (a), and cisPDI1-S4 (b). (c) Corresponding phase profiles extracted from EFM data for PDI1 (solid red line) and cisPDI1-S4 (solid blue line). Data are normalised such that the mean background phase is 0° for both samples. Dashed lines of the corresponding colour indicate the greatest phase contrast compared with the substrate for each sample. (d) Height and current profiles extracted from PF-TUNA measurements for samples of PDI1 (left) and cisPDI1-S4 (right) nanofibers. | ||
The currents measured through thionated cisPDI1-S4 and the parent PDI1 by PF-TUNA under 500 mV potential are presented in Fig. 6d. While the potential was kept low to prevent possible damage to the delicate assemblies in the confined geometry beneath the cantilever tip it can clearly be observed that for a comparable height of fiber cisPDI1-S4 exhibits a greater degree of conductivity than that of parent PDI1. In light of the difference in electrostatic potential displayed by the two comparable supramolecular structures, it appears reasonable to infer that the greater level of conductivity observed in cisPDI1-S4 by PF-TUNA occurs upon the thionation processes described here.
PF-TUNA measurements were also carried out on a sample of transPDI1-S4 nanofibers. Peak current values observed for this compound, and the two other species measured by this method are compared in Fig. S22 (ESI†). For transPDI1-S4, we measured peak current values between those observed for PDI1 and cisPDI1-S4. It should, however, be noted that the current values obtained are dependent on many factors including the morphology of the sample. We have chosen to focus our comparison on PDI1 and cisPDI1-S4 as these compounds display similar self-assembled morphologies, meaning conductivity differences are likely a direct result of the oxygen-to-sulphur substitution. The notably different morphology displayed by transPDI1-S4, described above, could have a substantial influence on the peak current recorded for this compound.
Conclusions
In this work, we present a comprehensive investigation into the effects of thionation on PDI-based SMPs. Thionation provides a facile method by which to tune the optoelectronic properties of these materials by simple stoichiometric adjustments. Substitution within side-chain amide groups largely retains the optoelectronic properties of the parent PDI1, whereas core substitution results in significant changes. Namely, fluorescence is quenched, and absorbance is bathochromically shifted by approximately 50 nm for each successive substitution. Such readily adjustable optoelectronic properties could facilitate the tuning of supramolecular constructs for specific applications, for example to better match the solar spectrum in photovoltaic devices.61 Moreover, the formation of the longer-lived triplet excited states responsible for the fluorescence quenching may allow enhancement of exciton transport distances within self-assembled structures.36Profound changes in the self-assembly behaviour of these materials were also observed. Thionation has significant effects on the morphology of the self-assembled structures and the ability of each compound to form linear SMPs. Differences in behaviour are rationalised by considering changes in binding energy upon thionation, and the introduction of additional S–S and S–π interactions. Mechanistic investigations reveal that thionation evokes a transition from a highly cooperative to an isodesmic assembly model. Such mechanistic control should also be applicable to many other supramolecular systems based on either hydrogen–bonding or π-stacking interactions, and presents an important tool for the future design and regulation of dynamic polymer properties.62
Overall, the approach described provides a facile route to simultaneously exert control over the mechanism, morphology and functionality of self-assembled PDI materials. Thionated PDIs already show great promise in OFET devices,41 and further optimisation of their self-assembly, such as the work described here, should facilitate their application in the diverse range of fields for which one-dimensional PDI-based materials are well suited.8
Experimental
Synthesis
General procedure for thionation of PDI136
PDI1 was dried into a flask with Lawesson's reagent (1–8 eq.) and placed under a nitrogen atmosphere. Toluene (anhydrous, 2 mL) was added and the mixture heated to reflux for 24–48 hours. The mixture was cooled to room temperature, the solvent removed under reduced pressure, and the product purified by preparative TLC (silica, chloroform:toluene), followed by silica gel microcolumn chromatography (chloroform:hexane) to afford the product. Please see ESI† for detailed methodologies, conditions (p4–6) and associated spectra (Fig. S1–S16, ESI†).Self-assembly of PDI1 and thionated analogues
PDI1 and its thionated analogues were each dispersed in degassed MCH at concentrations of 5 × 10−5 or 5 × 10−6 M under nitrogen atmosphere. Samples were heated briefly to 90 °C until dissolved, allowed to cool to room temperature, then aged for at least 24 hours prior to microscopy studies or spectroscopic analysis. Please see ESI† for detailed sample preparation and analysis methodologies (p2–3, p23–24).All relevant underlying data are provided as ESI† accompanying this paper.
Author contributions
Henry E. Symons: conceptualization, formal analysis, investigation, methodology, validation, visualization, writing – original draft; Maximilian J. L. Hagemann: investigation, validation, writing – review and editing; Robert L. Harniman: investigation, methodology, resources, writing – review and editing; Charl F. J. Faul: conceptualization, project administration, resources, supervision, writing – review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
H. E. S. acknowledges the EPSRC for support (EP/N509619/1). M. J. L. H. acknowledges the EPSRC for support (EP/R513179/1). Mass spectrometric measurements were performed using instrumentation bought through the Core Capability for Chemistry Research—Strategic Investment in Mass Spectrometry EPSRC grant (EP/K03927X/1). AFM, EFM and PF-TUNA measurements were conducted in the Chemical Imaging Facility, School of Chemistry, University of Bristol with equipment funded by EPSRC under “Atoms to Applications” Grant EP/K035746/1. All authors thank the School of Chemistry for research support.Notes and references
- J. H. K. K. Hirschberg, L. Brunsveld, A. Ramzi, J. A. J. M. Vekemans, R. P. Sijbesma and E. W. Meijer, Nature, 2000, 407, 167–170 CrossRef CAS PubMed.
- L. Herkert, J. Droste, K. K. Kartha, P. A. Korevaar, T. F. A. de Greef, M. R. Hansen and G. Fernández, Angew. Chem., Int. Ed., 2019, 58, 11344–11349 CrossRef CAS PubMed.
- S. Ghosh, X. Q. Li, V. Stepanenko and F. Würthner, Chem. – Eur. J., 2008, 14, 11343–11357 CrossRef CAS PubMed.
- K. Balakrishnan, A. Datar, T. Naddo, J. Huang, R. Oitker, M. Yen, J. Zhao and L. Zang, J. Am. Chem. Soc., 2006, 128, 7390–7398 CrossRef CAS PubMed.
- S. Ogi, V. Stepanenko, K. Sugiyasu, M. Takeuchi and F. Würthner, J. Am. Chem. Soc., 2015, 137, 3300–3307 CrossRef CAS PubMed.
- A. Maciejewski, M. Szymanski and R. P. Steer, J. Phys. Chem., 1986, 90, 6314–6318 CrossRef CAS.
- S. Ahmed, B. Pramanik, K. N. A. Sankar, A. Srivastava, N. Singha, P. Dowari, A. Srivastava, K. Mohanta, A. Debnath and D. Das, Sci. Rep., 2017, 7, 9485 CrossRef PubMed.
- S. Chen, P. Slattum, C. Wang and L. Zang, Chem. Rev., 2015, 115, 11967–11998 CrossRef CAS PubMed.
- E. Rostami-Tapeh-Esmail, M. Golshan, M. Salami-Kalajahi and H. Roghani-Mamaqani, Dyes Pigm., 2020, 180, 108488 CrossRef CAS.
- C. Li and H. Wonneberger, Adv. Mater., 2012, 24, 613–636 CrossRef CAS PubMed.
- N. Wang, Y. Li, X. He, H. Gan, Y. Li, C. Huang, X. Xu, J. Xiao, S. Wang, H. Liu and D. Zhu, Tetrahedron, 2006, 62, 1216–1222 CrossRef CAS.
- Y. Li, H. Zheng, Y. Li, S. Wang, Z. Wu, P. Liu, Z. Gao, H. Liu and D. Zhu, J. Org. Chem., 2007, 72, 2878–2885 CrossRef CAS PubMed.
- C. Huang, F. Lu, Y. Li, H. Gan, T. Jiu, J. Xiao, X. Xu, S. Cui, H. Liu and D. Zhu, J. Nanosci. Nanotechnol., 2007, 7, 1472–1478 CrossRef CAS PubMed.
- Q. Jiang, H. Sun, D. Zhao, F. Zhang, D. Hu, F. Jiao, L. Qin, V. Linseis, S. Fabiano, X. Crispin, Y. Ma and Y. Cao, Adv. Mater., 2020, 32, 2002752 CrossRef CAS PubMed.
- Y. Xu, J. Zheng, J. O. Lindner, X. Wen, N. Jiang, Z. Hu, L. Liu, F. Huang, F. Würthner and Z. Xie, Angew. Chem., Int. Ed., 2020, 59, 10363–10367 CrossRef CAS PubMed.
- C. Huang, S. Barlow and S. R. Marder, J. Org. Chem., 2011, 76, 2386–2407 CrossRef CAS PubMed.
- S. L. Suraru and F. Würthner, Angew. Chem., Int. Ed., 2014, 53, 7428–7448 CrossRef CAS PubMed.
- J. P. Sun, A. D. Hendsbee, A. J. Dobson, G. C. Welch and I. G. Hill, Org. Electron., 2016, 35, 151–157 CrossRef CAS.
- C. Kaufmann, W. Kim, A. Nowak-Król, Y. Hong, D. Kim and F. Würthner, J. Am. Chem. Soc., 2018, 140, 4253–4258 CrossRef CAS PubMed.
- A. Nowak-Król and F. Würthner, Org. Chem. Front., 2019, 6, 1272–1318 RSC.
- Y. Liu, N. Wang, Y. Li, H. Liu, Y. Li, J. Xiao, X. Xu, C. Huang, S. Cui and D. Zhu, Macromolecules, 2005, 38, 4880–4887 CrossRef CAS.
- Y. Li, N. Wang, H. Gan, H. Liu, H. Li, Y. Li, X. He, C. Huang, S. Cui, S. Wang and D. Zhu, J. Org. Chem., 2005, 70, 9686–9692 CrossRef CAS PubMed.
- H. Qian, Z. Wang, W. Yue and D. Zhu, J. Am. Chem. Soc., 2007, 129, 10664–10665 CrossRef CAS PubMed.
- H. Qian, W. Yue, Y. Zhen, S. Di Motta, E. Di Donato, F. Negri, J. Qu, W. Xu, D. Zhu and Z. Wang, J. Org. Chem., 2009, 74, 6275–6282 CrossRef CAS PubMed.
- C. Jarrett-Wilkins, X. He, H. E. Symons, R. L. Harniman, C. F. J. Faul and I. Manners, Chem. – Eur. J., 2018, 24, 15556–15565 CrossRef CAS PubMed.
- C. Wang, J. Wang, N. Wu, M. Xu, X. Yang, Y. Lu and L. Zang, RSC Adv., 2017, 7, 2382–2387 RSC.
- F. Zhang, Y. Ma, Y. Chi, H. Yu, Y. Li, T. Jiang, X. Wei and J. Shi, Sci. Rep., 2018, 8, 8208 CrossRef PubMed.
- W. Zhang, Y. Liu and G. Yu, Adv. Mater., 2014, 26, 6898–6904 CrossRef CAS PubMed.
- Z. Zhao, Z. Yin, H. Chen, L. Zheng, C. Zhu, L. Zhang, S. Tan, H. Wang, Y. Guo, Q. Tang and Y. Liu, Adv. Mater., 2017, 29, 1602410 CrossRef PubMed.
- M. Winkler and K. N. Houk, J. Am. Chem. Soc., 2007, 129, 1805–1815 CrossRef CAS PubMed.
- J. Liu, B. Walker, A. Tamayo, Y. Zhang and T. Q. Nguyen, Adv. Funct. Mater., 2013, 23, 47–56 CrossRef CAS.
- P. Pahlavanlu and D. S. Seferos, Phosphorus, Sulfur Silicon Relat. Elem., 2019, 194, 664–668 CrossRef CAS.
- G. Seybold and G. Wagenblast, Dyes Pigm., 1989, 11, 303–317 CrossRef.
- F. Würthner, Chem. Commun., 2004, 1564–1579 RSC.
- H. Langhals, Heterocycles, 1995, 40, 477–500 CrossRef CAS.
- A. J. Tilley, R. D. Pensack, T. S. Lee, B. Djukic, G. D. Scholes and D. S. Seferos, J. Phys. Chem. C, 2014, 118, 9996–10004 CrossRef CAS.
- A. K. Pandey, Sci. Rep., 2015, 5, 7787 CrossRef CAS PubMed.
- B. T. Luppi, D. Majak, M. Gupta, E. Rivard and K. Shankar, J. Mater. Chem. A, 2019, 7, 2445–2463 RSC.
- R. Gao, X. Mei, D. Yan, R. Liang and M. Wei, Nat. Commun., 2018, 9, 2798 CrossRef PubMed.
- X. Q. Li, V. Stepanenko, Z. Chen, P. Prins, L. D. A. Siebbeles and F. Würthner, Chem. Commun., 2006, 3871–3873 RSC.
- A. J. Tilley, C. Guo, M. B. Miltenburg, T. B. Schon, H. Yan, Y. Li and D. S. Seferos, Adv. Funct. Mater., 2015, 25, 3321–3329 CrossRef CAS.
- N. Pearce, E. S. Davies, W. Lewis and N. R. Champness, ACS Omega, 2018, 3, 14236–14244 CrossRef CAS PubMed.
- Z. Liu, Y. Gao, X. Jin, Q. Deng, Z. Yin, S. Tong, W. Qing and Y. Huang, J. Mater. Chem. B, 2020, 8, 5535–5544 RSC.
- P. Pahlavanlu, A. J. Tilley, B. T. McAllister and D. S. Seferos, J. Org. Chem., 2017, 82, 12337–12345 CrossRef CAS PubMed.
- M. Zander, Z. Naturforsch., A: Phys. Sci., 1989, 44, 1116–1118 CrossRef CAS.
- A. Rodriguez-Serrano, V. Rai-Constapel, M. C. Daza, M. Doerr and C. M. Marian, Phys. Chem. Chem. Phys., 2015, 17, 11350–11358 RSC.
- H. J. Lee, Y. S. Choi, K. B. Lee, J. Park and C. J. Yoon, J. Phys. Chem. A, 2002, 106, 7010–7017 CrossRef CAS.
- R. J. Zauhar, C. L. Colbert, R. S. Morgan and W. J. Welsh, Biopolymers, 2000, 53, 233–248 CrossRef CAS PubMed.
- K. N. M. Daeffler, H. A. Lester and D. A. Dougherty, J. Am. Chem. Soc., 2012, 134, 14890–14896 CrossRef CAS PubMed.
- A. L. Ringer, A. Senenko and C. D. Sherrill, Protein Sci., 2007, 16, 2216–2223 CrossRef CAS PubMed.
- K. S. C. Reid, P. F. Lindley and J. M. Thornton, FEBS Lett., 1985, 190, 209–213 CrossRef CAS.
- C. B. Aakeroy, D. L. Bryce, G. R. Desiraju, A. Frontera, A. C. Legon, F. Nicotra, K. Rissanen, S. Scheiner, G. Terraneo, P. Metrangolo and G. Resnati, Pure Appl. Chem., 2019, 91, 1889–1892 CrossRef CAS.
- D. J. Pascoe, K. B. Ling and S. L. Cockroft, J. Am. Chem. Soc., 2017, 139, 15160–15167 CrossRef CAS PubMed.
- K. T. Mahmudov, M. N. Kopylovich, M. F. C. Guedes Da Silva and A. J. L. Pombeiro, Dalton Trans., 2017, 46, 10121–10138 RSC.
- D. B. Korlepara, W. R. Henderson, R. K. Castellano and S. Balasubramanian, Chem. Commun., 2019, 55, 3773–3776 RSC.
- T. F. A. De Greef, M. M. J. Smulders, M. Wolffs, A. P. H. J. Schenning, R. P. Sijbesma and E. W. Meijer, Chem. Rev., 2009, 109, 5687–5754 CrossRef CAS PubMed.
- M. M. J. Smulders, M. M. L. Nieuwenhuizen, T. F. A. De Greef, P. Van Der Schoot, A. P. H. J. Schenning and E. W. Meijer, Chem. – Eur. J., 2010, 16, 362–367 CrossRef CAS PubMed.
- F. Würthner, C. R. Saha-Möller, B. Fimmel, S. Ogi, P. Leowanawat and D. Schmidt, Chem. Rev., 2016, 116, 962–1052 CrossRef PubMed.
- M. M. J. Smulders, A. P. H. J. Schenning and E. W. Meijer, J. Am. Chem. Soc., 2008, 130, 606–611 CrossRef CAS PubMed.
- M. Hartlieb, E. D. H. Mansfield and S. Perrier, Polym. Chem., 2020, 11, 1083–1110 RSC.
- Y. Cui, H. Yao, J. Zhang, T. Zhang, Y. Wang, L. Hong, K. Xian, B. Xu, S. Zhang, J. Peng, Z. Wei, F. Gao and J. Hou, Nat. Commun., 2019, 10, 2515 CrossRef PubMed.
- C. Kulkarni, E. W. Meijer and A. R. A. Palmans, Acc. Chem. Res., 2017, 50, 1928–1936 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1tc04518k |
| This journal is © The Royal Society of Chemistry 2022 |