DOI:
10.1039/D1TB02493K
(Paper)
J. Mater. Chem. B, 2022,
10, 2497-2503
Zwitterionic choline phosphate conjugated folate-poly (ethylene glycol): a general decoration of erythrocyte membrane-coated nanoparticles for enhanced tumor-targeting drug delivery†
Received
15th November 2021
, Accepted 18th December 2021
First published on 20th December 2021
Abstract
Erythrocyte membrane nanosystems have become one of the important research directions of disease treatment, especially for tumor treatment, and can enhance the long circulation time of anti-cancer drugs in vivo, and penetrate and accumulate in the tumor site effectively. However, erythrocyte membranes lack targeting properties and it is necessary to provide tumor-targeting function by modifying erythrocyte membranes. In this study, we report on a novel modification method of an erythrocyte membrane nanosystem to target tumors. Specifically, the tumor-targeting molecule folate-poly (ethylene glycol) (FA-PEG) was modified with a zwitterionic 2-(methyl acryloyoxy) ethyl choline phosphate (MCP) by the Michael addition reaction to obtain MCP-modified FA-PEG (MCP-PEG-FA). Based on the strong “N–P” tetravalent electrostatic interaction between MCP and phosphatidyl choline on the erythrocyte membranes, MCP-PEG-FA can be modified on the erythrocyte membrane encapsulated doxorubicin (DOX) loaded poly(lactic-co-glycolic acid) (PLGA) nanosystem to form a tumor-targeting erythrocyte membrane nanosystem (FA-RBC@PLGA-DOX). The results show that MCP-PEG-FA was synthesized and successfully bonded to the erythrocyte membrane nanosystem, and the FA-RBC@PLGA-DOX nanosystem had a better tumor-targeting function and tumor killing effect compared with those of the nanosystems without FA ligand modification. The universal modification method of erythrocyte membranes is successfully provided and can be applied to the treatment of various diseases.
1. Introduction
Chemotherapy is still one of the important means of tumor treatment.1 Nowadays, the commonly used anticancer drugs on the market include doxorubicin (DOX), paclitaxel, and cisplatin.2–4 DOX, an anthracycline antibiotic, is one of the most widely used anticancer drugs in the clinical treatment of a wide range of solid tumors.5,6 Nevertheless, the poor tissue selectivity of most anticancer drugs can lead to the easy removal of anticancer drugs, which cannot be concentrated in the tumor site effectively.7–9 In recent years, researchers have developed various nano-drug delivery systems with high drug delivery efficiency, such as liposomes, nanoparticles (NPs), polymer micelles, etc.10–12 They can effectively infiltrate and accumulate in tumor tissues, improve the efficiency of drug therapy and reduce drug side effects because of their enhanced permeability and retention (EPR) effects.13–16 However, there are still some challenges to be addressed: how to further prevent drug clearance, enhance the long circulation of drugs in vivo, and actively target tumors.
In recent years, the treatment of diseases based on erythrocyte (also known as red blood cells, RBC) membranes and their derivatives has developed rapidly.17 Benefitting from the natural superiority of erythrocyte membranes, such as good biocompatibility and long circulation time, RBC-coated NPs have been successfully applied in tumor treatment.18–21 In addition, erythrocyte membranes are widely studied because of their advantages such as a wide range of sources, easy extraction and easy preservation. Compared with PEG-coated nanoparticles which are commonly used to avoid the removal of reticuloendothelial systems (RESs), erythrocyte membrane nanosystems can not only prolong the drug circulation time in vivo, but can also solve immune rejection through homology recognition without generating antibodies.22–24 However, erythrocyte membranes are not intelligent enough because they do not have the ability to target tumors actively.25
In order to improve the tumor targeting ability of RBC-coated NPs, erythrocyte membranes can be combined with the targeted molecule by physical coupling or chemical coupling, which are the most common methods.18,26–29 For example, Zhu et al.30 decorated the surface of erythrocytes with a photoactivatable molecular trigger, 2-(1-hexyloxyethyl)-2-devinyl pyropheophorbide-α, via covalent modification. It not only makes use of the natural carrier properties of erythrocytes, but also can rapidly release loaded hemoglobin and tilapazamine at fixed points under short and mild laser irradiation, becoming a “Photoactivatable Bomb”. Physical coupling depends on the properties of the conjugate, and needs to have a positive charge or hydrophobicity because they are often coupled by electrostatic interactions or hydrophobic interactions, and erythrocyte membranes are hydrophobic and their surfaces are electronegative. Chemical coupling will inevitably change the chemical composition of erythrocyte membranes, so it cannot be a universal modification method.18 Currently, the universal method of membrane modification is through phospholipid–polymer insertion into erythrocyte membranes.18,31,32 It has a phosphate choline (PC) zwitterionic structure that can be inserted into the phosphatidylcholine head group of erythrocyte membranes, while attaching a functional structure at the other end to give erythrocyte membranes a special function. For instance, Chai et al.27 inserted streptavidin-PEG-DSPE into erythrocyte membranes to make them modifiable first, then modified the tumor-targeting peptide c(RGDyK) on the surface of erythrocyte membranes by using biotin–avidin interactions to obtain a promising erythrocyte membrane delivery system.
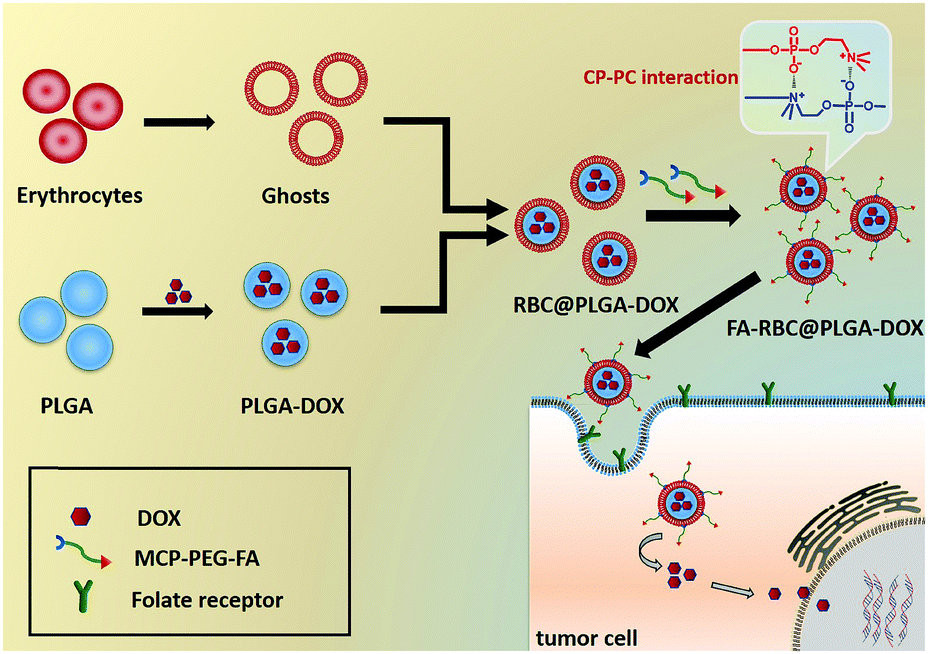
The choline phosphate (CP) zwitterionic structure, a “PC-inverse” structure inspired by cell membranes, has attracted the attention of researchers too. The charge distribution of CP and PC is the opposite, which results in a strong “N–P” tetravalent electrostatic interaction, so the CP can interact with PC liposomes and PC-based cell membranes.33–35 Yu et al.34 made use of the tetravalent electrostatic interaction between the CP and PC structures to make hyperbranched CP polymers a good bio-adhesive, which could bond erythrocytes at room temperature. They also used CP liposomes to deliver the drug, achieving better tumor targeting ability.36,37 In our previous work, CP was grafted onto the surface of the material and its zwitterionic properties were utilized to resist protein adsorption. And, its universal interaction with the PC-head group of cell membranes promotes cell adhesion, providing a new method for obtaining cell adhesion surfaces with abiotic contamination “background”.38–42 In this article, a universal modification method for erythrocyte membranes is proposed by using the interaction of PC–CP. In brief, we first synthesized zwitterionic choline phosphate conjugated folate-poly (ethylene glycol) (MCP-PEG-FA), a zwitterionic compound with a “CP” structure. Then, DOX was loaded into poly(lactic-co-glycolic acid) (PLGA) NPs as a model drug, which was co-extruded with erythrocyte membranes to obtain RBC@PLGA-DOX. Finally, the CP-PC interaction was formed between the MCP-PEG-FA and PC head group of erythrocyte membranes to obtain a tumor-targeted drug delivery system (FA-RBC@PLGA-DOX) inspired by cell membranes. This method is based on the interaction of CP–PC and does not destroy the structure of erythrocyte membranes, providing a universal method to modify erythrocyte membranes, and also creating a new idea for targeting drug delivery systems inspired by erythrocyte membranes.
2. Materials and methods
2.1 Materials
FA-PEG1000-NH2 (95%) was purchased from Pengshuo Biological Technology Co., Ltd, Shanghai, China. 2-Chloro-1,3,2-dioxaphospholane-2-oxide (COP, 95%) was purchased from TCI, Tokyo, Japan. 2-(Dimethyl-amino) ethyl methacrylate (DMAEMA, 99%) was obtained from Shanghai Aladdin Biochemical Technology Co., Ltd., China. Folic acid (FA, 95%) and EDTA spray coated tubes (10 mL) were purchased from Pengshida Laboratory Products Co., Ltd, Chengdu, China. The cell counting kit-8 (CCK-8) assay kit was obtained from Baoxin Biotechnology Co., LTD, Chengdu, China. DOX was purchased from Aasenbo Pharma Co., Ltd, Beijing, China. PLGA (MW.90000, 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50) was purchased from Meilun Biotechnology Co., LTD, Dalian, China. The radioimmunoprecipitation (RIPA) lysis buffer was purchased from Beyotime Biotechnology Co., LTD, Shanghai, China. All other solvents and chemicals were purchased from commercial sources and used as received, all solvents were of analytical grade or higher, unless otherwise noted. Ultrapure water with a resistivity of 18.2 MΩcm was used throughout.
:50) was purchased from Meilun Biotechnology Co., LTD, Dalian, China. The radioimmunoprecipitation (RIPA) lysis buffer was purchased from Beyotime Biotechnology Co., LTD, Shanghai, China. All other solvents and chemicals were purchased from commercial sources and used as received, all solvents were of analytical grade or higher, unless otherwise noted. Ultrapure water with a resistivity of 18.2 MΩcm was used throughout.
2.2 Synthesis of MCP-PEG-FA
As before, we synthesized MCP in two steps.41 And, MCP-PEG-FA was synthesized by the Michael addition reaction.43 Simply, 100 mg of FA-PEG-NH2 was dissolved in 8 mL of DMSO under nitrogen protection, 50 μL of triethylamine (TEA) was added and heated to 50 °C, under stirring to make the reactants mix evenly. About 100 mg of MCP was then added to the reaction system drop-by-drop through a syringe. After 48 h, the reaction was stopped and the product was dialyzed in ultrapure water for 3 days. Finally, the dialysis solution was freeze-dried to obtain the choline phosphate modified folate-poly (ethylene glycol), named MCP-PEG-FA. The chemical structure of MCP-PEG-FA was determined using 1H NMR and FT-IR.
2.3 Preparation of erythrocyte ghosts
Erythrocyte ghosts were prepared following a previously reported method with minor modifications.44 Animal experiments were approved by the Animal Research Committee of the Sichuan University. Whole blood from New Zealand female rabbits (purchased from Chengdu Dossy Experimental Animal Co., LTD., China) was obtained by cardiac puncture using an EDTA spray coated tube. To obtain erythrocyte ghosts, whole blood was centrifuged at 4000 rpm for 5 min to discard the serum and buffy coat. The packed erythrocytes were washed with cold PBS twice and then treated with 0.2 × PBS hypotonic solution at −20 °C for 30 min. The released hemoglobin was removed by centrifuging the suspension at 10000 rpm for 5 min and washed several times with ultrapure water, until the solution was colorless.
2.4 Preparation of PLGA NPs and PLGA-DOX NPs
10 mg of PLGA was dissolved in 2.5 mL of DMSO, and 400 μL of PLGA solution was dropped into 5 mL of ultrapure water in a slow stirring process to obtain PLGA NPs. For preparing the NP-loaded DOX, DOX and PLGA were mixed in DMSO, DOX:PLGA = 1:1 (W/W). PLGA NPs were prepared in the presence of DOX to form PLGA-DOX NPs loaded with drugs.
2.5 Preparation of FA-RBC@PLGA and FA-RBC@PLGA-DOX
PLGA NPs and erythrocyte ghosts were mixed at a weight ratio of about 1:1, and mixed evenly under ultrasonic treatment at 100 w for 2 min. Then, RBC@PLGA NPs were obtained through 400 nm and 200 nm polycarbonate films successively. Finally, 1 mL of RBC@PLGA NPs and MCP-PEG-FA (0.25 mg, 0.5 mg, or 1.0 mg) were mixed and incubated overnight in a 4 °C refrigerator to obtain FA-RBC@PLGA. And, FA-RBC@PLGA-DOX was prepared in a similar way. The drug-loading capacity of DOX was determined by combining with a corresponding calibration curve. The formulas for calculating the encapsulation efficiency and loading efficiency of the drug are listed as follows:
| % Encapsulation efficiency = [Amount of the drug loaded/Drug added] × 100 |
| % Loading efficiency = [Amount of the drug loaded/Amount of the NPs] × 100 |
The amount of drugs loaded into the NPs was calculated by recording the UV-visible spectra of DOX (λabs: 481 nm).
2.6 Characterization of FA-RBC@PLGA
2.6.1 Morphology.
The morphology of PLGA and FA-RBC@PLGA NPs was examined using transmission electron microscopy (TEM) (Tecnai G2 F20 S-TWIN, FEI, USA). FA-RBC@PLGA NPs were stained with 1% uranyl acetate.
2.6.2 Particle size and zeta potential.
The dynamic light scattering (DLS) and surface zeta potential of NPs were obtained using a Zetasizer (the Malvern Nano series, Malvern, UK). The test samples were diluted with ultrapure water (1:10) and analyzed in triplicate at 25 °C.
2.6.3 Protein content.
To investigate the type and content of membrane protein in erythrocyte ghosts and FA-RBC@PLGA NPs, the RIPA lysis buffer was used to extract the total proteins of the erythrocyte membrane. The proteins were analyzed by polyacrylamide gel electrophoresis at 150 V for 1 h and stained with Kaumas blue for 30 min.
2.7 Isothermal titration calorimetry (ITC)
MCP-PEG-FA and erythrocyte membranes were dissolved in 5% DMSO aqueous solution, the erythrocyte membrane concentration was about 1 μg mL−1, and that of MCP-PEG-FA was 1 mM. Typically, 19 consecutive injections of MCP-PEG-FA (2 μL each) were made into the chamber (300 μL) filled with erythrocyte membrane solution. The enthalpy change was obtained using an isothermal titration microcalorimeter (PEAQ-ITC, Malvern, USA).
2.8 Stability test
The FA-RBC@PLGA NP suspension was placed in a refrigerator at 4 °C and taken out at 0, 5, 7, 12, 21 and 28 d, respectively, to evaluate the long-term stability of the nanodrug delivery system by measuring its particle size distribution.
2.9 Cytotoxicity assay
The CCK-8 assay was performed to identify the cytotoxic effects of FA-RBC@PLGA NPs. HeLa cells were seeded at a density of 5 × 103 cells per well in a 96-well plate with RPMI 1640 medium (10% serum and 1% penicillin–streptomycin solution were added) at 37 °C in 5% CO2. After 24 h, the growth media were replaced with an equal volume of test samples (RBC@PLGA-DOX and FA-RBC@PLGA-DOX NPs) at various DOX concentrations in RPMI 1640 medium (1 μg mL−1, 2 μg mL−1, 4 μg mL−1, and 6 μg mL−1) for 24 h of incubation. Then, the culture medium was removed, 100μL of CCK-8 and RPMI 1640 medium were mixed (CCK-8:RPMI 1640 = 1:9) for another 2 h, and the optical density at 450 nm was determined with a microplate reader. And, considering the importance of biosafety of nanomaterials, the toxicity of PLGA, RBC@PLGA and FA-RBC@PLGA NPs to L929 cells was investigated by CCK-8 too.
2.10 Cellular uptake studies
HeLa cells, a model tumor cell line overexpressing the folate receptor, were used to confirm the successful functionalization of the FA-modified nanoparticles. HeLa cells were seeded at 5 × 103 cells in a 6-well plate. Then, RBC@PLGA-DOX and FA-RBC@PLGA-DOX (DOX final concentration = 2 μg mL−1) were added, respectively. To verify the targeting ability of FA, a group of cell surface folate receptors was saturated with 1 mM FA for 24 h prior to the addition of FA-RBC@PLGA-DOX. After the cells were washed with PBS and trypsinized, they were collected by centrifugation and resuspended in PBS for analysis by flow cytometry.
At the same time, cellular localization was visualized with a fluorescence microscope. The HeLa cells treated 2 h or 24 h with FA-RBC@PLGA-DOX or RBC@PLGA-DOX (DOX final concentration = 2 μg mL−1) were handled for observation. Before observation, the cells were washed with PBS. Cell nuclei were stained using a 300 nM 4′,6-diamidino-2-phenylindole (DAPI) solution for 10 min.
2.11 Statistical analysis
All experimental data in this study were shown as mean ± standard deviation (SD). Statistical comparisons were calculated by using Student's t-test, and p < 0.05 was considered to be statistically significant.
3. Results and discussion
3.1 Synthesis and characterization of MCP-PEG-FA
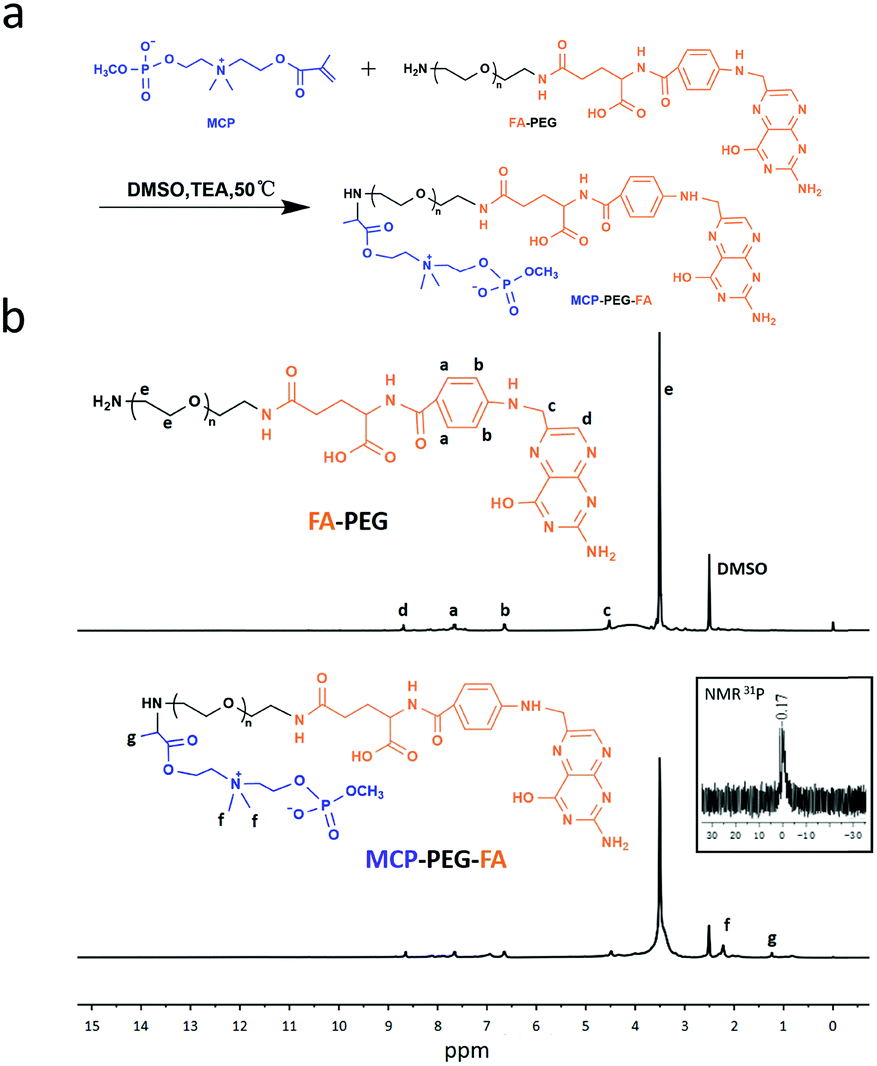
The preparation process of FA-RBC@PLGA NPs and the proposed tumor targeting mechanism of the strategy are schematically depicted in Fig. 1. According to our previous two-step synthesis,41 MCP was successfully synthesized and characterized by 1H NMR and 31P NMR (Fig. S1 and S2, ESI†). The targeting ligand MCP-PEG-FA was synthesized by the Michael addition reaction, and the structure was confirmed by 1H NMR (Fig. 2). The 1H NMR results (Fig. 2b) showed proton peaks at 3.6 ppm (ascribed to –CH2CH2O– in PEG), 6.7–8.6 ppm (chemical shifts of aromatic proton signals of FA), 2.22 ppm (methyl peak of N+(CH3)2 of MCP), and 1.24 ppm (methyl peak of –CH–CH3 of MCP). Moreover, 31P NMR spectra are shown in Fig. 2b as inset panels. The 31P NMR signal at σ 0.17 ppm is assigned to MCP. The FT-IR spectra of FA-PEG and MCP-PEG-FA are shown in Fig. S3 (ESI†). The peak at 1390 cm−1 is due to the P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretch which is blue shifted because of the electron-withdrawing effect of the P–O–C groups, and the peak of P–O–C stretching is at 990 cm−1. The above results indicate that MCP-PEG-FA has been synthesized successfully.
O stretch which is blue shifted because of the electron-withdrawing effect of the P–O–C groups, and the peak of P–O–C stretching is at 990 cm−1. The above results indicate that MCP-PEG-FA has been synthesized successfully.
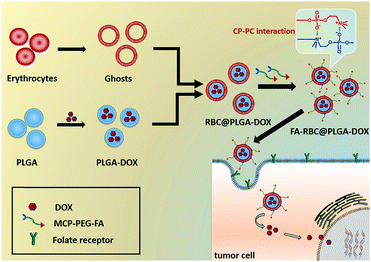 |
| | Fig. 1 Schematic illustration of FA-RBC@PLGA-DOX preparation and tumor targeting. | |
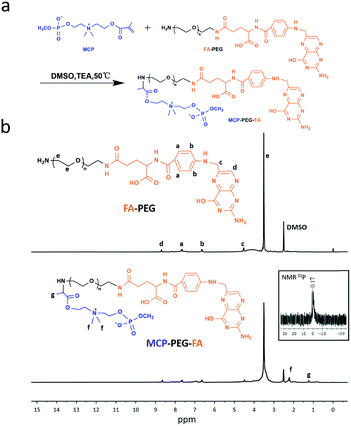 |
| | Fig. 2 (a) Synthetic route of MCP-PEG-FA. (b) 1H NMR spectrum of FA-PEG, 1H NMR spectrum and 31P NMR spectrum of MCP-PEG-FA. | |
3.2 Characterization of FA-RBC@PLGA
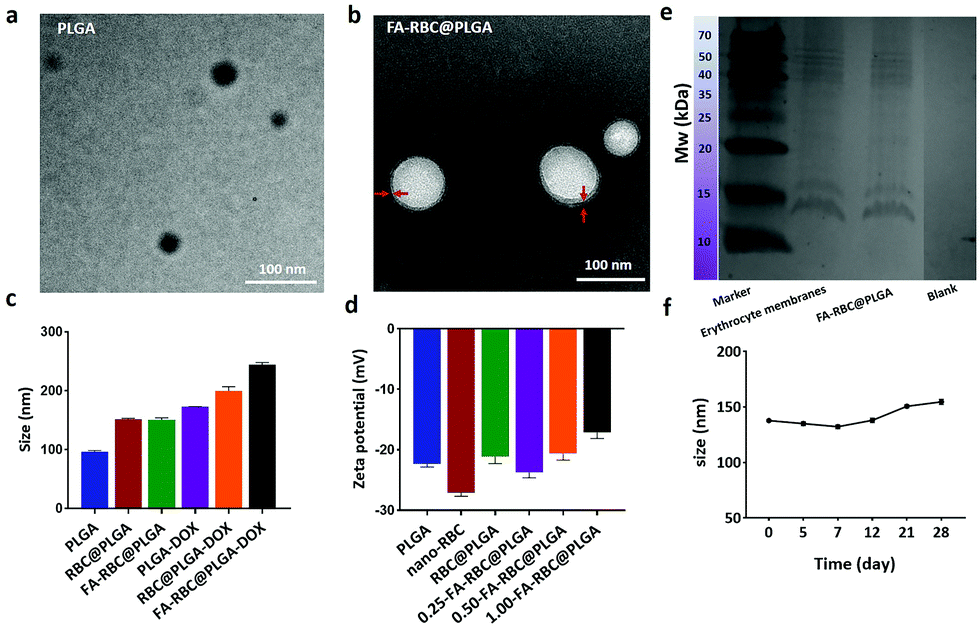
Several characterization studies of PLGA, RBC@PLGA and FA-RBC@PLGA were performed (Fig. 3). The TEM images showed the morphology of the PLGA NPs and the FA-RBC@PLGA (Fig. 3a and b). After erythrocyte membrane camouflage, there was an obvious core–shell structure. As can be seen, the particle size of PLGA was about 100 nm, and that of RBC@PLGA and FA-RBC@PLGA was about 150nm. After DOX loading, NP particle sizes were further increased (Fig. 3c). Furthermore, by combining with a corresponding calibration curve (Fig. S4, ESI†), the drug loading capacity of DOX was about 13.8%. The NP particle size from the TEM results was smaller than that from DLS analysis, which may be because of the shrinkage of NPs. Fig. 3d indicates the potentials of PLGA, nano-RBC and RBC@PLGA NPs of −22.3, −27.1, and −21.2 mV, respectively. Moreover, it can be seen that the potential also changed significantly in the RBC@PLGA NP solution with the increase of the mass of MCP-PEG-FA. Compositions of membrane proteins were analyzed via SDS-PAGE, as shown in Fig. 3e, the FA-RBC@PLGA formulation contained all major protein fractions of erythrocyte membranes. The stability of the FA-RBC@PLGA was also verified at 4 °C within 28 days by the change of particle size (Fig. 3f). During the test period, the average particle size of FA-RBC@PLGA increased slightly from 137.8 nm to 154.6 nm because of the swelling of the particles, demonstrating the high storage stability of FA-RBC@PLGA.
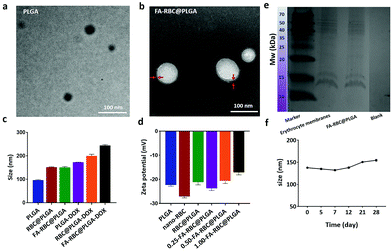 |
| | Fig. 3 Characterization of FA-RBC@PLGA. (a and b) The TEM images of PLGA and FA-RBC@PLGA (bar = 100 nm). (c) DLS analysis of PLGA, RBC@PLGA, FA-RBC@PLGA, PLGA-DOX, RBC@PLGA-DOX and FA-RBC@PLGA-DOX NPs. (d) Zeta potentials of PLGA, nano-RBC, RBC@PLGA, 0.25-FA-RBC@PLGA, 0.5-FA-RBC@PLGA and 1.0-FA-RBC@PLGA NPs (0.25-FA-RBC@PLGA means 1 mL RBC@PLGA NPs and 0.25 mg MCP-PEG-FA were mixed and incubated, and so on). (e) Membrane protein analysis of erythrocyte membranes and FA-RBC@PLGA. (f) The average particle size of FA-RBC@PLGA over 28 days. | |
3.3 Interaction force assay
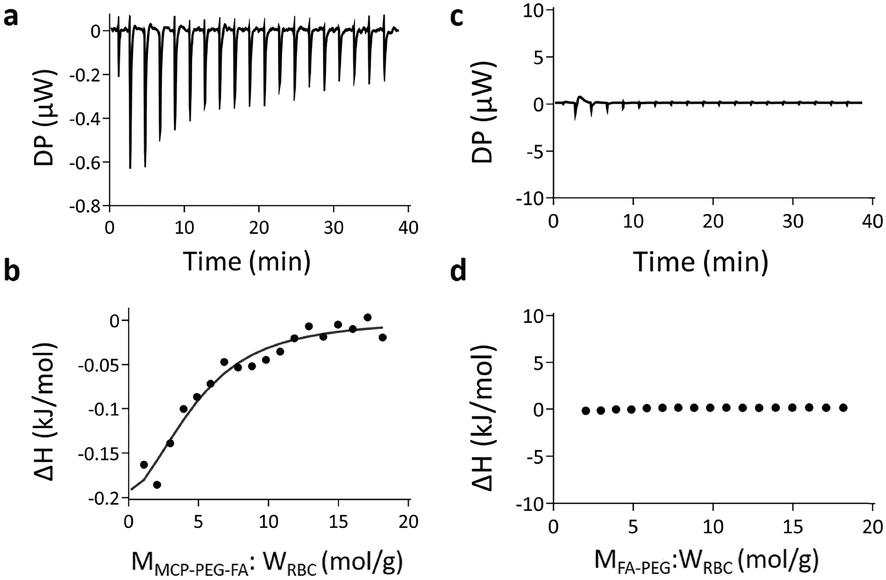
The interaction between erythrocyte membranes and MCP-PEG-FA was examined by ITC. It can be seen in Fig. 4a that when MCP-PEG-FA is continuously dropped into the solution of erythrocyte membranes, there is an obvious thermal change by ITC. This is also confirmed by the enthalpy simulation curve (Fig. 4b). However, when the same molarity of FA-PEG was dropped, the simulation curve is almost horizontal, meaning that there is no significant interaction between them (Fig. 4c and d). Therefore, it can be concluded that the tetravalent CP–PC electrostatic interaction between MCP-PEG-FA and erythrocyte membranes exists.
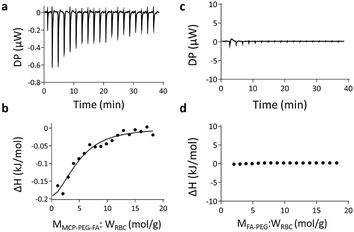 |
| | Fig. 4 (a) ITC of different amounts of MCP-PEG-FA added to erythrocyte membranes. (b) Enthalpy change simulation curve of MCP-PEG-FA added to erythrocyte membranes. (c) ITC of different amounts of FA-PEG added to erythrocyte membranes. (d) Enthalpy change simulation curve of FA-PEG added to erythrocyte membranes. They were all dissolved in 5% DMSO aqueous solution, the erythrocyte membrane concentration was about 1 μg mL−1, and the concentration of MCP-PEG-FA and FA-PEG was 1 mM. | |
3.4 Biosafety and tumor cell-killing in vitro
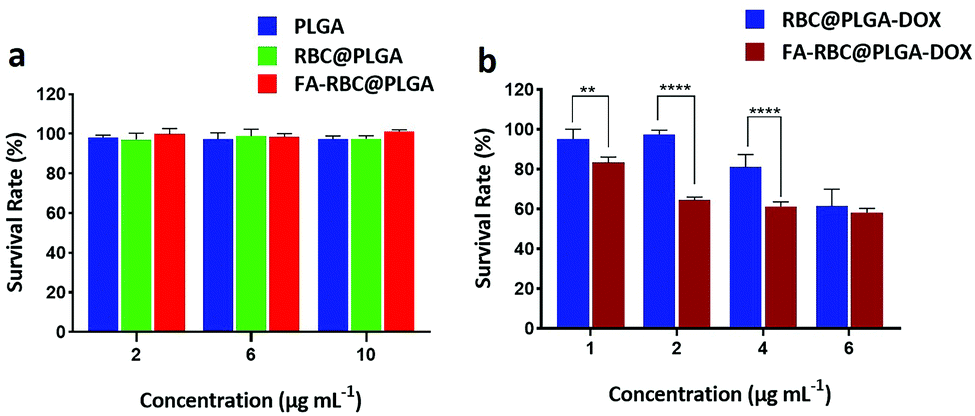
The cytotoxicity of FA-RBC@PLGA was detected by a standard CCK-8 assay (Fig. 5a). The concentration of PLGA, RBC@PLGA and FA-RBC@PLGA NPs was set at 2 μg mL−1, 6 μg mL−1 and 10 μg mL−1 of PLGA, then the survival viability of L929 cells was measured. By comparison, the survival rate of cells in the experimental group reached 95%, which means that these NPs have excellent biocompatibility.
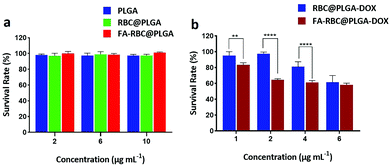 |
| | Fig. 5 (a) Cytotoxicity of PLGA, RBC@PLGA and FA-RBC@PLGA in L929 cells treated for 24 h. (b) Cytotoxicity of RBC@PLGA-DOX and FA-RBC@PLGA-DOX in HeLa cells treated for 24 h. Data are shown as mean ± SD, n = 6, **P < 0.01, ***P < 0.001 and ****P < 0.0001. | |
The cytotoxicity of FA-RBC@PLGA-DOX in HeLa cells was also detected via a standard CCK-8 assay after co-culture for 24 h (Fig. 5b). The results showed that at low concentrations of DOX (1 μg mL−1, 2 μg mL−1), RBC@PLGA-DOX basically had no killing effect on HeLa cells. However, when HeLa cells were treated with FA-RBC@PLGA-DOX (DOX = 2 μg mL−1), the cell survival rate was 64.4%, which was similar to that of RBC@PLGA-DOX (DOX = 6 μg mL−1), indicating that FA-RBC@PLGA-DOX can efficiently increase the cellular uptake and therefore upgrade the antitumor efficacy. Also, this result proves the success of the MCP-PEG-FA modified erythrocyte membrane strategy, and the uptake of DOX by HeLa cells was enhanced because of FA modification.
3.5 Folic acid targeting assay
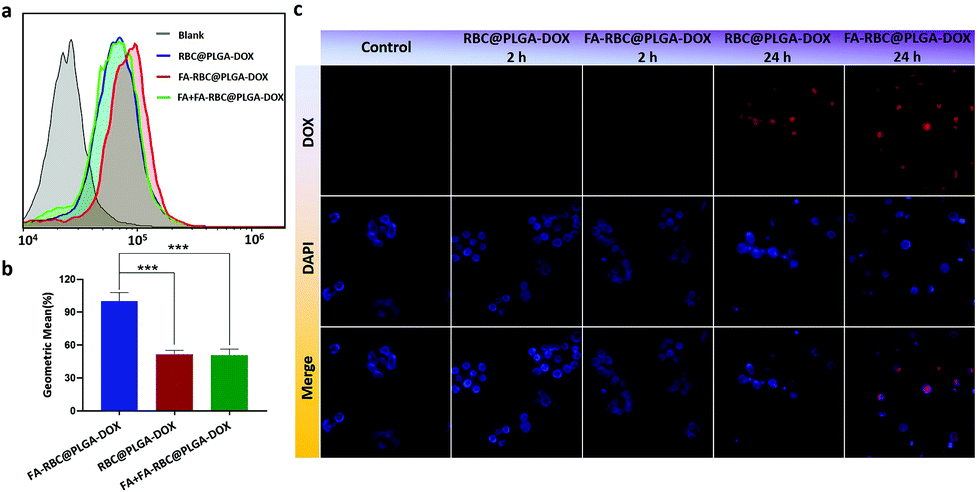
To further comfirm that the MCP-PEG-FA modified NPs can target HeLa cells which are highly expressed folate receptors, flow cytometry was used to measure the mean fluorescence intensity of DOX uptake in HeLa cells. RBC@PLGA-DOX is the blue curve in Fig. 6a, and there was a significant right shift compared with the blank on account of the fluorescence absorption of DOX. Compared with RBC@PLGA-DOX, the uptake intensity of FA-RBC@PLGA-DOX was higher, as indicated by the apparent rightward shift of the curve in the flow cytometry results (red curve in Fig. 6a). In contrast, when the folate receptors on the membrane surface were saturated with free FA (FA + FA-RBC@PLGA-DOX), this specific binding was inhibited by competition, and the uptake efficiency decreased, as indicated by a leftward shift on the green curve. In order to quantitatively show the difference of fluorescence intensity between the experimental group and the control group, we calculated the right shift distance of each curve compared to the blank group. With the fluorescence intensity of FA-RBC@PLGA-DOX defined as 100%, those of RBC@PLGA-DOX and FA + FA-RBC@PLGA-DOX were both only 50%, and the results reflect the success of the MCP-PEG-FA modification strategy (Fig. 6b). The location of DOX in HeLa cells transported by RBC@PLGA-DOX and FA-RBC@PLGA-DOX was observed by fluorescence microscopy (Fig. 6c). During the first 2 hours, the uptake of DOX was observed in the FA-RBC@PLGA-DOX, while almost no uptake was observed in the RBC@PLGA-DOX. And within 24 hours, the red fluorescence of FA-RBC@PLGA-DOX was enhanced with the extension of time, indicating that the uptake of DOX was increased, while the uptake was still not significant in RBC@PLGA-DOX. These results suggested that the uptake of nanoparticles by HeLa cells was significantly increased after modification with the target molecule MCP-PEG-FA on RBC@PLGA-DOX.
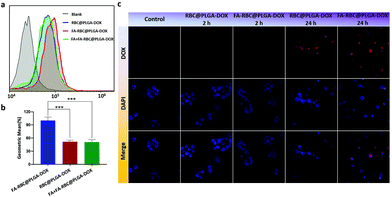 |
| | Fig. 6 (a and b) Flow cytometry analysis and relative fluorescence intensity analysis for RBC@PLGA-DOX and FA-RBC@PLGA-DOX (pretreated with or without FA) in HeLa cells. (c) Fluorescence images (400×) of RBC@PLGA-DOX and FA-RBC@PLGA-DOX in HeLa cells. Data are shown as mean ± SD, n = 3, ***P < 0.001. | |
4. Conclusions
In summary, we successfully developed a novel modification method for an erythrocyte membrane nanosystem using MCP-PEG-FA through the interaction of “CP–PC” to improve the tumor targeting ability of the erythrocyte membrane nanosystem. This strategy significantly enhanced the uptake capacity of DOX by HeLa cells and successfully improved the killing ability of DOX towards tumors. Based on this study, we believe that using MCP with a “PC-inverse” structure to modify erythrocyte membrane nanosystems through tetravalent electrostatic binding between CP and PC is a very promising scheme, which can also be used in the modification of other targeted drug delivery systems in the future.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 51925304 and 52173140), the Sichuan Province Science and Technology (2021YJ192), and the Opening Project of State Key Laboratory of Polymer Materials Engineering (Sichuan University) (Grant No. sklpme2020-4-13).
References
- E. Perez-Herrero and A. Fernandez-Medarde, Eur. J. Pharm. Biopharm., 2015, 93, 52–79 CrossRef CAS PubMed.
- H. Chen, Q. Luo, J. Wang, H. He, W. Luo, L. Zhang, Q. Xiao, T. Chen, X. Xu, W. Niu, Y. Ke and Y. Wang, ACS Appl. Mater. Interfaces, 2020, 12, 22673–22686 CrossRef CAS PubMed.
- Y. Li, N. Chen, M. Palmisano and S. Zhou, Mol. Pharmaceutics, 2015, 12, 1308–1317 CrossRef CAS PubMed.
- T. M. Harned, O. Kalous, A. Neuwelt, J. Loera, L. Ji, P. Iovine, R. Sposto, E. A. Neuwelt and C. P. Reynolds, Clin. Cancer Res., 2008, 14, 533–540 CrossRef CAS PubMed.
- J. Woodley-Cook, L. Y. Y. Shin, L. Swystun, S. Caruso, S. Beaudin and P. C. Liaw, Mol. Cancer Ther., 2006, 5, 3303–3311 CrossRef CAS PubMed.
- L. Gandhi, M. W. Harding, M. Neubauer, C. J. Langer, M. Moore, H. J. Ross, B. E. Johnson and T. J. Lynch, Cancer, 2007, 109, 924–932 CrossRef CAS PubMed.
- M. Li, K. Shi, X. Tang, J. Wei, X. Cun, Y. Long, Z. Zhang and Q. He, Nanomedicine, 2018, 14, 1833–1843 CrossRef CAS PubMed.
- W. Sun, J. Fan, S. Wang, Y. Kang, J. Du and X. Peng, ACS Appl. Mater. Interfaces, 2018, 10, 7832–7840 CrossRef CAS PubMed.
- X. Wang, J. Li, C. Xu, Y. Li, T. Gong, X. Sun, Y. Fu, Q. He and Z. Zhang, Bioconjugate Chem., 2014, 25, 2046–2054 CrossRef CAS PubMed.
- S. Jin, J. Wan, L. Meng, X. Huang, J. Guo, L. Liu and C. Wang, ACS Appl. Mater. Interfaces, 2015, 7, 19843–19852 CrossRef CAS PubMed.
- D. Zhu, W. Tao, H. Zhang, G. Liu, T. Wang, L. Zhang, X. Zeng and L. Mei, Acta Biomater., 2016, 30, 144–154 CrossRef CAS PubMed.
- F. Danhier, E. Ansorena, J. M. Silva, R. Coco, A. Le Breton and V. Preat, J. Controlled Release, 2012, 161, 505–522 CrossRef CAS PubMed.
- J. Huang, Y. Li, A. Orza, Q. Lu, P. Guo, L. Wang, L. Yang and H. Mao, Adv. Funct. Mater., 2016, 26, 3818–3836 CrossRef CAS PubMed.
- M. S. Chan, L. S. Liu, H. M. Leung and P. K. Lo, ACS Appl. Mater. Interfaces, 2017, 9, 11780–11789 CrossRef CAS PubMed.
- Y. J. Li, M. Dong, F. M. Kong and J. P. Zhou, Int. J. Pharm., 2015, 489, 83–90 CrossRef CAS PubMed.
- N. Dixit, K. Vaibhav, R. S. Pandey, U. K. Jain, O. P. Katare, A. Katyal and J. Madan, Biomed. Pharmacother., 2015, 69, 1–10 CrossRef CAS PubMed.
- Y. Zhao, R. Xie, N. Yodsanit, M. Ye, Y. Wang and S. Gong, Nano Today, 2020, 35, 100986 CrossRef CAS PubMed.
- G.
F. Luo, W. H. Chen, X. Zeng and X. Z. Zhang, Chem. Soc. Rev., 2021, 50, 945–985 RSC.
- Z. Wu, T. Li, J. Li, W. Gao, T. Xu, C. Christianson, W. Gao, M. Galarnyk, Q. He, L. Zhang and J. Wang, ACS Nano, 2014, 8, 12041–12048 CrossRef CAS PubMed.
- J. Zhang, C. Fu, S. Song, H. Du, D. Zhao, H. Huang, L. Zhang, J. Guan, Y. Zhang, X. Zhao, C. Ma, C.-L. Jia and D. Tománek, Nano Lett., 2020, 20, 1280–1285 CrossRef CAS PubMed.
- F. Pierige, S. Serafini, L. Rossi and M. Magnani, Adv. Drug Delivery Rev., 2008, 60, 286–295 CrossRef CAS PubMed.
- X. Wang, X. Gu, H. Wang, Y. Sun, H. Wu and S. Mao, Eur. J. Pharm. Sci., 2017, 96, 255–262 CrossRef CAS PubMed.
- X. Han, C. Wang and Z. Liu, Bioconjugate Chem., 2018, 29, 852–860 CrossRef CAS PubMed.
- P.-A. Oldenborg, A. Zheleznyak, Y.-F. Fang, C. F. Lagenaur, H. D. Gresham and F. P. Lindberg, Science, 2000, 288, 2051–2054 CrossRef CAS PubMed.
- W. L. Liu, T. Liu, M. Z. Zou, W. Y. Yu, C. X. Li, Z. Y. He, M. K. Zhang, M. D. Liu, Z. H. Li, J. Feng and X. Z. Zhang, Adv. Mater., 2018, 30, 1802006 CrossRef PubMed.
- Z. Chen, W. Wang, Y. Li, C. Wei, P. Zhong, D. He, H. Liu, P. Wang, Z. Huang, W. Zhu, Y. Zhou and L. Qin, Mol. Pharmaceutics, 2021, 18, 386–402 CrossRef CAS PubMed.
- Z. Chai, D. Ran, L. Lu, C. Zhan, H. Ruan, X. Hu, C. Xie, K. Jiang, J. Li, J. Zhou, J. Wang, Y. Zhang, R. H. Fang, L. Zhang and W. Lu, ACS Nano, 2019, 13, 5591–5601 CrossRef CAS PubMed.
- L. Luo, F. Zeng, J. Xie, J. Fan, S. Xiao, Z. Wang, H. Xie and B. Liu, J. Mater. Chem. B, 2020, 8, 4080–4092 RSC.
- L. Rao, Q. F. Meng, L. L. Bu, B. Cai, Q. Huang, Z. J. Sun, W. F. Zhang, A. Li, S. S. Guo, W. Liu, T. H. Wang and X. Z. Zhao, ACS Appl. Mater. Interfaces, 2017, 9, 2159–2168 CrossRef CAS PubMed.
- Y. X. Zhu, H. R. Jia, Y. Guo, X. Liu, N. Zhou, P. Liu and F. G. Wu, Small, 2021, 17, 2100753 CrossRef CAS PubMed.
- L. Yang, D.-F. K. Toh, M. S. Krishna, Z. Zhong, Y. Liu, S. Wang, Y. Gong and G. Chen, Biochemistry, 2020, 59, 4429–4438 CrossRef CAS PubMed.
- Y. Wang, X. Chen, D. He, Y. Zhou and L. Qin, Mol. Pharmaceutics, 2018, 15, 5728–5740 CrossRef CAS PubMed.
- X. Yu, Z. Liu, J. Janzen, I. Chafeeva, S. Horte, W. Chen, R. K. Kainthan, J. N. Kizhakkedathu and D. E. Brooks, Nat. Mater., 2012, 11, 468–476 CrossRef CAS PubMed.
- X. Yu, X. Yang, S. Horte, J. N. Kizhakkedathu and D. E. Brooks, Chem. Commun., 2013, 49, 6831–6833 RSC.
- X. Chen and J. Li, Mater. Chem. Front., 2020, 4, 750–774 RSC.
- W. Wang, S. Jiang, S. Li, X. Yan, S. Liu, X. Mao and X. Yu, Chem. Mater., 2021, 33, 774–781 CrossRef CAS.
- S. Li, W. Mei, X. Wang, S. Jiang, X. Yan, S. Liu and X. Yu, Chem. Commun., 2021, 57, 1372–1375 RSC.
- X. Chen, T. Chen, Z. Lin, X. E. Li, W. Wu and J. Li, Chem. Commun., 2015, 51, 487–490 RSC.
- X. Chen, H. Shang, S. Cao, H. Tan and J. Li, RSC Adv., 2015, 5, 76216–76220 RSC.
- X. Chen, M. Yang, B. Liu, Z. Li, H. Tan and J. Li, Langmuir, 2017, 33, 8295–8301 CrossRef CAS PubMed.
- X. Chen, Z. Lin, Y. Feng, H. Tan, X. Xu, J. Luo and J. Li, Small, 2019, 15, 1903784 CrossRef CAS PubMed.
- Q. Xin, T. Sun, M. Lu, Z. Wang, K. Song, D. Hu, Z. Li, J. Luo, X. Chen, J. Li and J. Weng, Colloids Surf., B, 2020, 185, 110630 CrossRef CAS PubMed.
- M. Lu, S. Yu, Z. Wang, Q. Xin, T. Sun, X. Chen, Z. Liu, X. Chen, J. Weng and J. Li, Eur. Polym. J., 2020, 134, 109821 CrossRef CAS.
- Y. Wang, K. Zhang, X. Qin, T. Li, J. Qiu, T. Yin, J. Huang, S. McGinty, G. Pontrelli, J. Ren, Q. Wang, W. Wu and G. Wang, Adv. Sci., 2019, 6, 1900172 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1tb02493k |
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 a,
Jun
Luo
a,
Jun
Luo