
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Development of polymer-based multifunctional composite particles of protease and peroxidase activities†
Szilárd
Sáringer
a,
Tamás
Valtner
a,
Árpád
Varga
 b,
József
Maléth
b and
István
Szilágyi
*a
b,
József
Maléth
b and
István
Szilágyi
*a
aMTA-SZTE Lendület Biocolloids Research Group, Interdisciplinary Excellence Center, Department of Physical Chemistry and Materials Science, University of Szeged, H-6720 Szeged, Hungary. E-mail: szistvan@chem.u-szeged.hu
bHAS-USZ Momentum Epithelial Cell Signaling and Secretion Research Group and HCEMM-SZTE Molecular Gastroenterology Research Group, Department of Medicine, University of Szeged, H-6720 Szeged, Hungary
First published on 27th October 2021
Abstract
A hybrid material (SL–PPN–HEP–HRP) of dual enzyme function was prepared by co-immobilization of papain (PPN) and horseradish peroxidase (HRP) on sulphate latex (SL) microspheres using heparin (HEP) polyelectrolyte as a building block in the sequential adsorption method. The doses of PPN, HEP and HRP were optimized in each step of the preparation process to achieve high functional and colloidal stability. The enzymes and the polyelectrolyte strongly adsorbed on the oppositely charged surfaces via electrostatic forces, and enzyme leakage was not observed from the hybrid material, as confirmed by colorimetric protein tests and microscopy measurements. It was found that the polyelectrolyte acted as a separator between PPN and HRP to prevent hydrolytic attack on the latter enzyme, which otherwise prevents the joint use of these important biocatalysts. Excellent colloidal stability was obtained for the SL–PPN–HEP–HRP composite and the embedded PPN and HRP showed remarkable protease and peroxidase activities, respectively, at least until five days after preparation. The present results offer a promising approach to develop biocatalytic systems of dual function, which are often required in manufacturing processes in the food industry, where the colloidal stability of such multifunctional materials is a key parameter to achieve remarkable efficiency.
Introduction
Enzymes are highly efficient biocatalysts applied for industrial-scale catalysis because of several distinct advantages.1 As such, their lower energy requirements, mitigation of waste generation, and simplified production routes have been partially realized in the pharmaceutical and food industries.2,3 In nature, almost all of the intracellular reactions are catalysed by the cooperation of various enzymes.4 The application of biocatalysts of multi-enzymatic functions is an important technology to produce many valuable chemicals in the industry, which integrates several biocatalytic transformations and thus, bridging the gap between single-enzyme catalysis and whole-cell catalysis.3By the inspiration from multi-enzymatic reactions in nature, researchers have attempted to construct multifunctional biocatalytic systems to produce desired chemicals or to decompose hazardous contents.5–7 In industrial and biomedical applications, it is necessary to immobilize the enzymes to prolong their catalytic activities and to provide enhanced stability and resistance against severe conditions, including extreme pH, temperature and presence of enzyme inhibitors, and to protect them against proteolysis and to achieve reduced susceptibility to contamination.8–10 Besides, immobilization of enzymes also offers several technical advantages, such as easier product recovery and purification.11 As a solid support for enzymes, nano or colloidal particles provide ideal characteristics for maintaining efficient biocatalytic activity for the enzymes leading to remarkable advantages over conventional supports. As summarized in recent reviews,5,12–14 various materials, such as graphene, carbon nanotubes, metal–organic frameworks, DNA nanostructures, polymers and metal oxides, have been applied for multi-enzyme immobilization. Different preparation strategies have been reported, which include fusion of enzymes, enzyme–scaffold complexes and parallel attachment of enzymes to solid supports.8,10 Among such systems, biocatalysts of hydrolytic and antioxidant properties have attracted considerable attention due to their widespread use in various industrial processes.
Accordingly, plant proteases, such as papain (PPN),15 are widely applied in the food industry for various purposes including brewing, tenderization of meat, coagulation of milk and as a digestive aid.16 Due to their hydrolytic attack on the peptide bonds and subsequent decomposition of proteins, proteases are also used to improve the flavour, nutritional value, solubility and digestibility of food proteins as well as to modify functional properties including coagulation and emulsification. Recently, several nanomaterials have been used as a matrix for PPN immobilization including magnetic nanoparticles,17 carbon nanotubes18 as well as polymeric micro- and nanoparticles.19,20
Besides, horseradish peroxidase (HRP)21 is one of the most extensively studied peroxidase enzymes because of its widespread applications. It catalyses the oxidation of various substrates using hydrogen peroxide or other peroxide derivatives. HRP has been applied for the removal of phenols from wastewater,22 organic compound syntheses23 and development of biosensors.24 Moreover, it is also being used in the food industry for improving nutritional quality of food. Removal of residual amount of hydrogen peroxide after cold sterilization of beer, rinsing and dairy treatment processes is carried out by HRP.2 Its advantages include high activity, simple detection of products, relatively good stability and ease of immobilization.24 The immobilization of HRP was performed with various solid supports such as silica nanoparticles,25 polysaccharide derivatives,26 chitosan,27 graphene28 or iron oxide.29
The above-mentioned examples clearly show that the combined use of PPN and HRP enzymes is desirable in certain industrial areas. On the one hand, the successful application requires separation of the enzymes to protect HRP from the proteolytic activity of PPN. This can be achieved by co-immobilization of the two enzymes. On the other hand, colloidal stability of these systems is a critical issue in the above applications. Indeed, aggregation of the carrier particles would lead to unsuccessful enzyme delivery processes due to the formation of irregularly shaped clusters giving rise to significant loss in enzymatic activity.30 The joint immobilization of HRP and PPN with the sequential adsorption method31 is a self-evident way to separate the enzymes in space on solid supports and to ensure sufficiently high colloidal stability of the composite suspension, as demonstrated earlier for immobilization of dual enzyme systems or enzyme mimicking compounds.32,33 Moreover, an intermediate polyelectrolyte layer may provide enhanced protection for HRP against PPN.
Therefore, the aim of the present study is to design stable dispersions of a latex particle (SL)-supported multi-enzymatic system involving heparin (HEP) polyelectrolyte in the sequential adsorption process (Scheme 1). Scattering techniques were used to optimize the charging and aggregation features as well as to gain information on the structure of the SL–PPN–HEP–HRP composites, while the proteolytic and peroxidase activities were assessed in biochemical test reactions.
 | ||
| Scheme 1 Schematic illustration of the synthesis and enzymatic activity of the SL–PPN–HEP–HRP composite. | ||
Experimental section
Materials
Spherical polystyrene latex (SL) particles functionalized with negatively charged sulphate groups and fluorophore conjugated secondary antibody (Donkey anti-mouse Alexa 647) were purchased from Thermo Fischer Scientific. The radius of the SL particles was (215 ± 4) nm, as determined by the manufacturer using transmission electron microscopy in the dried stage. The surface charge density of the particles was reported to be −12 mC m−2. Papain (PPN) from Carica papaya (3 U mg−1), heparin (HEP) sodium salt from porcine intestinal mucosa, Folin & Ciocalteu reagent, anti-HRP primary antibody, glucose oxidase, catalase, phosphate buffered saline (PBS), glucose and cysteamine hydrochloride were purchased from Sigma-Aldrich. Horseradish peroxidase (HRP), Coomassie brilliant blue, hydrogen peroxide, NaCl, NaH2PO4 (anhydrous), Na2HPO4 (anhydrous), orthophosphoric acid (85 w/w%), ethanol, trichloroacetic acid (TCA) and sodium carbonate were bought from WVR, while tyrosine and guaiacol were from Acros Organics. Paraformaldehyde (PFA) was acquired from Alfa Aesar and bovine serum albumin (BSA) was from PanBiotech. These chemicals were of analytical grade and were used as received. All solutions were diluted using ultrapure water obtained from a VWR Purity TU+ machine. The water and the salt solutions were filtered with a 0.1 μm syringe filter (Millex).Composite preparation
The SL–PPN–HEP–HRP composite material was built up based on the sequential adsorption method (Scheme 1). Accordingly, SL particles were added to a solution containing a calculated amount of PPN at pH 4. The sample was stirred for 1.5 h on a rotational stirrer and HEP solution of appropriate concentration was added to the dispersion. After 1.5 h additional stirring, HRP was added to the samples followed by continuous stirring for 1.5 h. The doses of PPN, HEP and HRP were 400, 200 and 10 mg g−1 SL, respectively, in the final composite.Electrophoretic mobility
Electrophoretic light scattering was used to determine the electrophoretic mobility of the particles with a Litesizer 500 instrument (Anton Paar) equipped with a 40 mW semiconductor laser operating at 658 nm wavelength. For all the measurements 1.5 mL samples were prepared, during which 0.15 mL of 100 mg L−1 particle stock dispersion was added to a 1.35 mL solution composed of a calculated amount of polyelectrolyte and/or enzyme as well as NaCl to set the ionic strength to 1 mM. The samples were allowed to rest for 2 h at room temperature before measuring the electrophoretic mobility after a 1 min equilibrating time in the device. The reported values were the average of 5 individual measurements, which were performed in omega shaped plastic cuvettes (Anton Paar). The standard deviation of the data was always within 5 mV.Dynamic light scattering
The hydrodynamic radius (rh) of the particles was determined by dynamic light scattering (DLS) with an ALV-NIBS High Performance Particle Sizer instrument. The cumulant fit was used to fit the correlation function, which was collected for 20 s at 173.5° scattering angle.34 To determine the aggregation rate coefficient (k) of the particle dimer formation, time-resolved DLS measurements were performed and the calculations were carried out as follows:35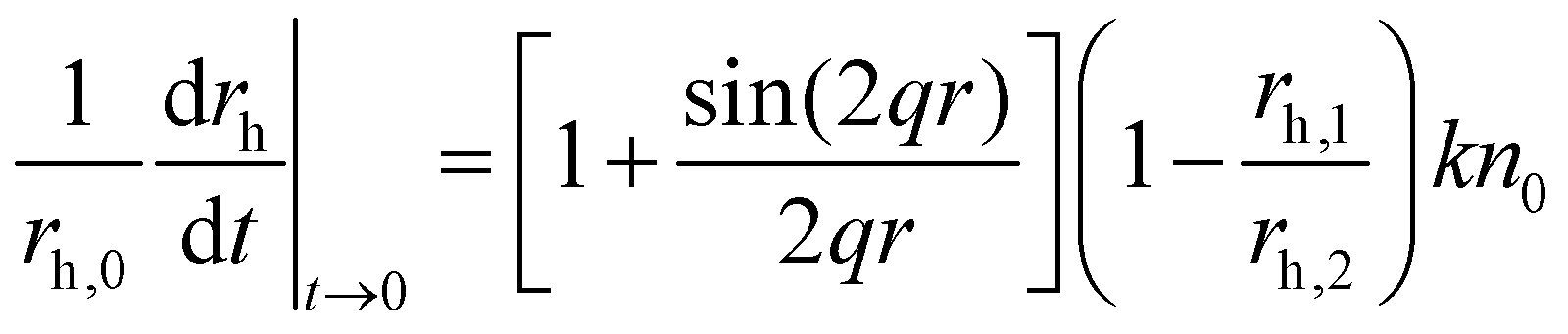 | (1) |
 | (2) |
The critical coagulation concentration (CCC) values were calculated with the formula described as39
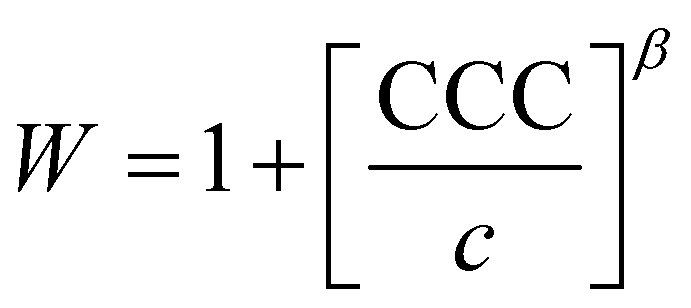 | (3) |
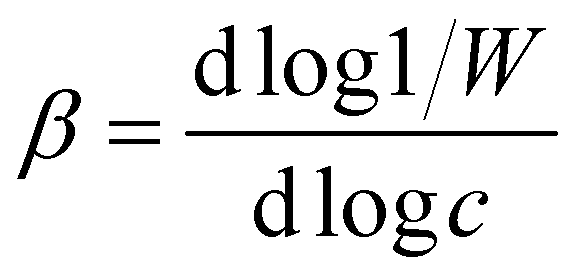 | (4) |
Determination of enzyme content
The amount of PPN enzyme in solution was determined by the Bradford protein test.40 Coomassie Brilliant Blue dye stock solution was prepared as follows. 100 mg of the dye was dissolved in the mixture of 50 mL of 95% ethanol and 100 mL of 85% phosphoric acid. The solution was then made up to 1000 mL with ultrapure water. Thereafter, standard solutions of PPN in the 1–100 mg L−1 range and SL–PPN at 1000 mg L−1 particle concentration were prepared. After mixing 0.4 mL of the test solution with 1.6 mL of the dye solution, the UV-Vis spectra of the samples were recorded after a 5 min equilibration time on a Thermo Fischer Genesys 10s spectrophotometer. The changes in the absorbance at 464 nm and 594 nm wavelength were monitored. The amount of PPN in the samples was calculated from the ratio of these absorbance values.Immunofluorescent labelling and dSTORM imaging
The immobilization of HRP on SL–PPN–HEP was confirmed by direct stochastic optical reconstruction microscopy (dSTORM). The particle suspensions were placed on cover glass (VWR). The samples were incubated for 20 min to promote adhesion. After 5 min long fixation with 4% PFA in PBS, specific binding sites were blocked by applying 10% BSA in PBS for 2 h at 37 °C. Anti-HRP primary antibody was used during a 2 h incubation step at room temperature. The sample was washed three times for 10 min with PBS. Fluorophore conjugated (Alexa 647) secondary antibody was applied prior to another washing step (three times for 10 min). For dSTORM imaging, cover glasses were placed on cavity slides filled with blinking buffer and sealed with a two component adhesive. The blinking buffer contained 100 U glucose oxidase, 2000 U catalase, 55.6 mM glucose and 100 mM cysteamine hydrochloride in 1 mL final volume completed with sterile PBS. dSTORM images were captured using Nanoimager S (Oxford Nanoimaging ONI Ltd).Hydrolytic activity measurements
The universal protease activity assay protocol based on the Lowry method41 with some modifications was used for determination of the protease-like function. Accordingly, a calibration curve was prepared by adding different amounts of 1.1 mM standard tyrosine solution to several samples and completed to a final volume of 2 mL with ultrapure water. The final tyrosine concentrations in the samples were between 0.01 and 1 μM. Note that tyrosine is the product of the hydrolysis in the test reaction. After 30 min incubation at 37 °C, the colour change in the samples was measured with a spectrophotometer at 660 nm. The absorbance recorded at this wavelength was used to determine the tyrosine concentration in the samples. For the estimation of protease-like activity, the concentration of the bare and immobilized PPN was 2500 mg L−1 in the stock solutions. For the measurements, 0.4 mL was added to 3.6 mL of 10 mM phosphate and 5 mM acetate buffer at pH 7. Thus, the PPN concentration was 250 mg L−1 in the buffered solution. During the measurements, 0.65 w/v% casein, as a substrate, was diluted with 50 mM phosphate buffer solution first. Thereafter, 0.1 mL of free or immobilized enzymes was added to 0.5 mL casein solution. The reaction was terminated after 20 min by adding 0.5 mL of 110 mM TCA solution and then, an appropriate volume of PPN solution or SL–PPN–HEP–HRP dispersion was added to the blank sample to reach the 1 mL final volume. After 30 min incubation at 37 °C, the samples were centrifuged at 10000 rpm for 10 min. Then, 0.75 mL of 500 mM sodium carbonate and 0.1 mL of Folin & Ciocalteus reagent were added to 3 mL of the supernatant. After 30 min incubation at 37 °C, the absorbance of the samples was recorded in the 400–800 nm range. The protease activity was determined from the absorbance values measured at 660 nm. One unit of protease activity was equivalent to the amount of enzyme that was required to release 1 μg of tyrosine per mL per min under the standard assay conditions described above. For the time dependent measurements, the stock solutions of 2500 mg L−1 concentration were stored under room temperature and the activities of the samples were measured each day for a week applying the above-mentioned steps.Peroxidase activity measurements
The guaiacol assay was used to determine the HRP activity of the samples.42 In brief, 0.125 mL of 25 mg L−1 stock solution containing immobilized or bare enzymes was added to the mixture of 0.125 mL of 100 mM phosphate buffer and an appropriate volume of 100 mM guaiacol solution. The samples were completed to a volume of 0.85 mL with water. Finally, 0.35 mL of 9 mM hydrogen peroxide was added into the cuvette and introduced into the UV-Vis spectrophotometer to follow the formation of the guaiacol degradation products at 470 nm wavelength. The reaction rate (v) was determined by linear regression of absorbance versus reaction time data. The Michaelis–Menten constant (Km) and the maximum reaction rate (vmax) were calculated with the Lineweaver–Burk equations as follows:42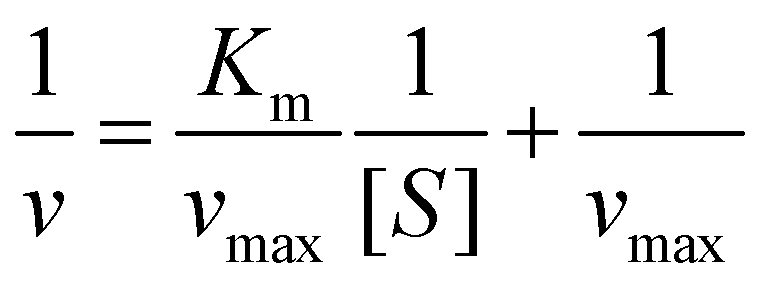 | (5) |
Results and discussion
PPN adsorption on SL
As the first step in the preparation of the SL–PPN–HEP–HRP composite, the adsorption of PPN on SL particles was studied in an earlier work of our group.43 It was found that PPN strongly adsorbed on the oppositely charged SL surface through electrostatic forces at pH 4, which is below the pI of the PPN enzyme.44 The charging and aggregation properties of the SL particles were explored by electrophoresis and time-resolved DLS measurements. The electrophoretic mobility of the latex particles changed from negative to positive on increasing the amount of enzyme added (Fig. S1, ESI†) indicating strong affinity to the oppositely charged particle surface. The stability ratio values were high at low and elevated enzyme doses referring to stable dispersions under these conditions. A U-shaped curve was formed in the intermediate regime with a minimum corresponding to the isoelectric point (IEP) determined in the electrophoretic mobility measurements. At this minimum, the stability ratios were unity within the experimental error indicating unstable dispersions, i.e., rapid particle aggregation. The theory developed by Derjaguin, Landau, Verwey and Overbeek (DLVO)45,46 may qualitatively explain this behaviour of the particles since they possessed sufficient charge at high and low PPN doses, i.e., strong repulsive forces were present due to the overlap of the electrical double layers. At the IEP, however, such a repulsive electrical double layer force was absent, and the suspensions were destabilized by the attractive van der Waals forces.It is important to note that the SL surface saturated with the adsorbed enzyme around 400 mg g−1 dose, as indicated by a plateau in the mobility above this PPN concentration. Moreover, the SL–PPN particles obtained at this dose formed a stable dispersion; therefore, these particles were chosen for the further steps in the sequential adsorption method to obtain the SL–PPN–HEP–HRP composite.
Our previous work43 on PPN adsorption was extended by quantifying this process with the Bradford method, an excellent tool to determine the PPN concentration in solutions.40,47 Accordingly, the SL–PPN dispersion (at 400 mg g−1 PPN dose) was filtered to remove the particles and the filtrate was analysed with the Bradford test. The obtained results including the ones from the reference measurements with different amounts of native PPN are shown in Fig. S2a (ESI†) and the absorbance ratios are presented in Fig S2b (ESI†). The absorbance decreased at 465 nm and increased at 595 nm on increasing PPN concentration. These changes in the absorption maxima are based on the binding of the applied dye to the PPN molecules. The enzyme concentration in the filtrate was found to be 2.1 mg L−1, which is 0.5% of the total amount of PPN. This value is within the experimental error of the test and clearly shows that more than 99% of the enzyme adsorbed on the SL and that no significant PPN partitioning between the bulk and the surface took place.
Considering the above results, the surface coverage can be estimated. Given the diameter of SL (430 nm) and PPN (7.2 nm),48 calculations yielded 12.45 mg of enzyme adsorbed on 1 g of particles in the case of monolayer formation. Therefore, the 400 mg g−1 dose at saturation indicates the development of PPN multilayers on the surface, which has been already assumed in our previous study43 to explain loss in the enzymatic activity. Note that, however, this calculation was based on a smooth latex surface and possible conformational changes of PPN were not considered upon adsorption on SL.
Functionalization with HEP
The net positive surface charge of SL–PPN allows the adsorption of the negatively charged HEP polyelectrolyte through electrostatic interactions.49 High colloidal stability was the major requirement to be achieved after the bilayer formation. Although polyelectrolyte multilayer formation was frequently reported on particles and planar surfaces as well,33,50–52 very limited information is available on sequential adsorption processes involving saturated enzyme layers on colloidal particles.32,53The electrophoretic mobility was first determined to probe the influence of the HEP concentration on the charging behaviour of the SL–PPN. Considering the change in the magnitude of the electrophoretic mobility on increasing the HEP dose, similar tendencies were observed as in the system containing SL and PPN. At low HEP doses, the SL–PPN possesses a positive net charge, while on increasing the HEP dose, the polyelectrolyte adsorption was clearly indicated by the decrease of mobility (Fig. 1).
 | ||
| Fig. 1 Electrophoretic mobility (red circles) and stability ratio (blue squares) data of the SL–PPN particles as a function of the HEP dose. The measurements were carried out at pH 4 and 1 mM ionic strength adjusted by NaCl. The mg g−1 unit refers to mg of HEP per 1 g of the particles. The solid lines serve to guide the eyes. | ||
Such an adsorption process led to charge neutralization at the IEP, which is located at 20 mg g−1 dose. Above the IEP, charge reversal occurred at higher doses. The adsorption continued until the mobility reached a plateau at 200 mg g−1 and further added polyelectrolytes remained dissolved in the bulk solution.
Stability ratio measurements revealed that the particles were stable at low and high polyelectrolyte doses. Like the dispersions containing SL and PPN (Fig. S1, ESI†), the stability ratios went through a minimum near the IEP. This behaviour is again in line with the DLVO theory, i.e., the acting interparticle forces are of electrostatic origin, and similar to findings reported earlier with other polyelectrolyte-particle systems.38,49,52,54,55 The minimum value of the stability ratios was around unity, which is indeed in agreement with the prediction of DLVO theory. On the other hand, there is a remarkable difference in the slopes of the stability ratios before and after the minimum value. At low polyelectrolyte doses, a smaller slope was observed, which indicates the occurrence of the non-DLVO destabilizing effect, which may originate from the adsorbed PPN–HEP layers. It is assumed that the patch–charge effect56,57 is responsible for this phenomenon as follows. At low polyelectrolyte doses, HEP adsorbed on the SL–PPN particles in islands or in so-called patches, while empty positively charged places (charges) were still available on the SL–PPN surface. The patches are electrostatically attracted by the opposite charges giving rise to the evolution of an attractive non-DLVO interaction in addition to the already existing van der Waals forces. The patch–charge interaction leads to faster aggregation of the particles and thus to lower stability ratios.
Overall, the above mobility and stability ratio data ensure that at 200 mg g−1 polyelectrolyte dose the SL–PPN–HEP particles are negatively charged and form a highly stable colloid.
Immobilization of HRP
The HRP enzyme is positively charged at pH 4 since its pI value is 8.8.21 Therefore, HRP is expected to adsorb on the negatively charged SL–PPN–HRP surface at least by electrostatic attraction. However, hydrophobic interactions and hydrogen bonding may also play a role in the adsorption process. One should note that extensive HRP adsorption may affect the charging and aggregation properties of the SL–PPN–HEP particles. It is important to avoid a significant decrease in the magnitude of surface charge upon enzyme immobilization, because it may lead to a weakening of the double layer repulsion and to particle aggregation.To explore the charging behaviour, the influence of HRP adsorption on the electrophoretic mobility of SL–PPN–HEP was probed by measuring the mobility at different HRP concentrations (Fig. S3, ESI†). One can observe that increasing the HRP dose did not lead to significant changes in the mobility, unlike PPN and HEP adsorption in the previous steps, due to its low charge density. The results revealed that for an enzyme dose lower than 100 mg g−1, the mobility values of SL–PPN–HEP were not affected by the added amount of HRP indicating a colloidal stability similar to that of the SL–PPN–HEP particles. Above this loading, the mobility slightly increased, which indicates weaker electrical double layer repulsion and consequently, an undesired decrease in colloidal stability. Considering these results, 10 mg g−1 dose was selected (SL–PPN–HEP–HRP). Note also that this dose was applied in our previous immobilization studies55,58 and this condition gave rise to remarkable peroxidase activities of the obtained composites.
The immobilization of HRP on the surface of the oppositely charged SL–PPN–HEP hybrid was confirmed by the dSTORM technique. The transmitted light image (Fig. 2a) showed several well-concentrated foci in the distant red range of light indicating the presence of HRP in the SL–PPN–HEP–HRP particles. After data acquisition (Fig. 2b), dSTORM revealed the spatial extent of the blinking events in Fig. 2c and the blinking frequency of the fluorophores along the white dashed line drawn in Fig. 2b as a function of distance (Fig. 2d). The dimension of the flashes shown in Fig. 2c and d is about 400 nm, which is in good agreement with the diameter of the SL nanoparticle used. These results provide unambiguous evidence for the adsorption of HRP on the SL–PPN–HEP composite and that the enzyme is strongly attached to the particulate support.
 | ||
| Fig. 2 Transmitted light image of the particles on a glass surface (a) and dSTORM picture of the marked focal point (b). Blinking events of the fluorophores were plotted along the X and Y axes (c) and the frequency of blinking events was plotted as a function of the distance (d) from the white dashed line shown in (b). | ||
An overview of the electrophoretic mobility, stability ratio and hydrodynamic radius data during the sequential adsorption process used for SL–PPN–HEP–HRP is shown in Fig. 3.
 | ||
| Fig. 3 Schematic illustration of the charging and aggregation tendencies during preparation of the SL–PPN–HEP–HRP composite by the sequential adsorption method. The evolution of the electrophoretic mobility (top) and stability ratios (bottom) as a function of the enzymes and polyelectrolytes is shown. The constant hydrodynamic radii of the SL–PPN–HEP–HRP composite confirm the absence of particle aggregation (bottom right corner). The particle concentration was 10 mg L−1 and the measurements were carried out at pH 4 and 1 mM ionic strength. | ||
The mobility of the SL particles at low PPN doses was around (−3.81 ± 0.06) × 10−8 m2 V−1 s−1. This negative charge resulted in stable colloids. The formation of a saturated PPN layer at 400 mg g−1 provided again stable dispersions and the magnitude of the mobility was (2.44 ± 0.03) × 10−8 m2 V−1 s−1. After functionalization with HEP at 200 mg g−1, the mobility was (−3.46 ± 0.04) × 10−8 m2 V−1 s−1 due to the high charge density of the polyelectrolyte. This saturated HEP layer provided high colloidal stability for the particles. Finally, the immobilization of HRP slightly decreased the mobility values to (−2.77 ± 0.08) × 10−8 m2 V−1 s−1. The hydrodynamic radius of the SL–PPN–HEP–HRP composite was (280 ± 6) nm. As shown by the time-resolved data, no aggregation was detected for the final hybrid particles.
Resistance against salt-induced aggregation
Ionic strength is an important factor in the preparation, stability and applications of biocatalytic systems.30,59,60 Therefore, the charging and aggregation features of the obtained particles were tested in a wide range of salt concentration. The ionic strength was systematically changed in the dispersion of SL, SL–PPN and SL–PPN–HEP–HRP and the electrophoretic mobility values as well as stability ratios were determined, and the tendencies were compared (Fig. 4). | ||
| Fig. 4 (a) Electrophoretic mobility and (b) stability ratio of SL, SL–PPN, and SL–PPN–HEP–HRP particles as a function of the ionic strength adjusted with NaCl. The measurements were carried out at pH 4 and the particle concentration was 10 mg L−1. | ||
On increasing the ionic strength, the magnitude of the mobility decreased for all the systems owing to the screening effect of the dissolved salt constituents on the surface charges (Fig. 4a). The mobility was close to zero at high salt concentrations, but no charge reversal was observed in the ionic strength regime investigated.
Time-resolved DLS measurements were carried out to investigate the aggregation behaviour at different electrolyte levels. The obtained stability ratios are shown in Fig. 4b. The tendency in the data followed the prediction of the DLVO theory.46 Accordingly, high stability ratios indicated stable dispersions at low salt concentrations, while they decreased by increasing the ionic strength. Rapid particle aggregation occurred after the critical coagulation concentration (CCC) values. The relatively high CCC data shed light on the remarkable colloidal stability of the particle systems. Similar results were reported earlier on polyelectrolyte-coated particle dispersions.49,52,54,58,61
The aggregation rates determined above the CCC values were 3.1 × 10−18 m3 s−1, 3.3 × 10−18 m3 s−1 and 3.4 × 10−18 m3 s−1 for SL, SL–PPN and SL–PPN–HEP–HRP, respectively. This reasonable agreement between the data indicates that the nature of the attractive forces was very similar for all particles and such forces originate most probably from van der Waals attractions. Nevertheless, for the SL–PPN–HEP–HRP system, stability ratio values lower than unity were observed in the fast aggregation regime after the CCC and the slope in the slow aggregation regime was also different from the other particle systems. This difference may originate from additional non-DLVO forces. First, such an acceleration in the aggregation is the result of the above discussed patch–charge interactions, which were frequently reported for highly charged polyelectrolytes adsorbing on oppositely charged surfaces.54,56,57 Second, steric hindrance between adsorbed polyelectrolyte chains may evolve upon approach of two particles, which leads to a non-electrostatic repulsion.38,52,54 This effect is more pronounced at lower ionic strength and hence, should be responsible for the different slope in the slow aggregation regime of the SL–PPN–HEP–HRP composite.
Protease activity
The hydrolytic activity of the immobilized and free PPN was determined on the basis of the hydrolysis of the casein substrate.41 The assay was carried out daily during five days for SL–PPN–HEP–HRP to probe its functional stability over time. The quantitative analysis was based on the characteristic absorbance values of the tyrosine product appearing at 660 nm wavelength (Fig. S4, ESI†), from which the normalized activities were calculated (see the Experimental section for details). The determined activity on the first day was (0.62 ± 0.01) U mL−1 for the composite and (0.71 ± 0.03) U mL−1 for the native PPN. Thus, the immobilized PPN possessed 85% activity compared to the free PPN on the first day, while this value was only 67% for the SL–PPN system.The decrease in the activity upon immobilization can be explained as follows. On the one hand, it is caused by the conformational changes of PPN, which occurred once attachment to the surface of the latex took place. Such a change may result in decreased activity due to the hindered interaction between the active centre and the substrate. On the other hand, a relatively high amount of PPN is adsorbed on SL, probably in more than one layer, and the blocking effect of the outer layers, i.e., the limited accessibility of substrate molecules to the inner enzymes, decreased the efficiency.
However, it was observed in several studies62–64 that PPN exhibited a high binding affinity to HEP and this binding is mediated mainly by electrostatic interactions. The presence of HEP may significantly change the PPN structure by increasing the α-helix content of the enzyme. This binding can result in an increase in the affinity of the enzyme for the substrate. It was also shown that the interaction between HEP and PPN is specific, since other sulphated glycosaminoglycans, namely dermatan sulphate and chondroitin sulphate, were able to neither increase the affinity of PPN to the substrate, nor change the α-helix content in PPN.65 To explore the PPN–HEP interaction and its effect on the protease activity, PPN assays were carried out in the presence of HEP, i.e., with PPN–HEP solution. The hydrolytic activity of the PPN–HEP system was about the same as the one determined for the native PPN enzyme. This result clearly indicates that possible interactions between PPN and HEP do not affect the protease function significantly.
Fig. 5 shows the change in the relative enzyme activities determined at different days. Although the enzymatic activity of the immobilized PPN decreases as a function of time, the hybrid material maintained significant protease-like activity even after 5 days. The decrease in the activity can be explained by the partial denaturation of the enzyme, the presence of PPN aggregates on the particles or by the accumulation of the products on the SL–PPN–HEP–HRP surface, which sterically hinders the interaction between the enzyme and the substrate. However, we do not have direct experimental proof for the exact reason of this loss in protease activity. Some decrease was also observed for the free PPN, but it was not significant, and the enzyme retained 95% of its activity even after 5 days.
 | ||
| Fig. 5 Relative protease-like activity of the PPN enzyme and the SL–PPN–HEP–HRP composite as a function of time. The applied PPN concentration was 22.7 mg L−1 in all measurements. | ||
The hydrolytic activities of the native and immobilized PPN were also assessed at 50 °C and 70 °C after a 1 hour incubation time (Fig. S5, ESI†). At the former temperature, the activities did not change significantly compared to the value measured without heat treatment, while at 70 °C, the hydrolytic efficiency decreased by 45% and 60% for PPN and SL–PPN, respectively.
Peroxidase activity
The peroxidase-like activity of the SL–PPN–HEP–HRP composite was tested in the guaiacol assay.42 This biochemical test is based on the detection of the degradation products of guaiacol under the combined effect of HRP and hydrogen peroxide. The formation of the tetra-guaiacol product was monitored by following the colour change of the solution over time. As mentioned in the Experimental section, the Lineweaver–Burk plot was used to analyse the result and from the double reciprocal reaction rate versus substrate concentration plot, the Michaelis–Menten constant (Km) and the maximum reaction rate (vmax) were calculated (Fig. S6, ESI†). The Km value corresponds to the affinity of the enzyme towards the substrate. For example, a decrease in the Km value refers to a higher affinity of the enzyme to guaiacol. vmax is the maximum reaction rate that can be achieved by the system once the active sites of the enzymes are completely saturated with the substrate.The determined vmax values for the PPN–HRP mixed solution and for the SL–PPN–HEP–HRP composite were (0.125 ± 0.070) mM s−1 and (0.069 ± 0.020) mM s−1, respectively, on the first day with freshly prepared samples. For these systems, the Km values were (3.52 ± 0.26) mM and (29.16 ± 9.71) mM, respectively. The lower activity of the immobilized HRP might originate from the interaction with HEP on the surface.
In the time dependent measurements, the activity of the immobilized enzymes decreased (Fig. 6); however, significant peroxidase-like activity was still detected after 5 days. In contrast, the PPN–HRP solution (i.e., mixing HRP and PPN without SL and HEP) lost its activity after 3 days due to the hydrolysis of HRP by the PPN enzyme.
 | ||
| Fig. 6 Relative maximum reaction rate (red bars, left axis) and the Michaelis–Menten constant (Km, blue bars, right axis) values of the SL–PPN–HEP–HRP composite as a function of time. The applied HRP concentration was 2.6 mg L−1. | ||
To further support our conclusions on the decrease of enzymatic activities of both PPN and HRP with time and upon immobilization, attempts were made to follow potential structural changes of the enzymes in the experiments. Nevertheless, neither scattering (SAXS) nor spectroscopy (FT-IR) techniques were useful due to the relatively large size of the carrier particles and their strong light scattering signal as well as their intense vibrational peaks in the fingerprint region.
Besides, the above results clearly indicate that the polyelectrolyte layer in the SL–PPN–HEP–HRP composite can protect HRP from proteolytic degradation by the PPN enzyme. Accordingly, the immobilized HRP exhibited good resistance to proteolysis by PPN. Such process usually takes place in the PPN–HRP mixed homogeneous solution. Similarly, it was reported earlier that the dissolved peroxidases were more easily inactivated in the presence of protease enzymes compared to their immobilized counterparts.66
The findings obtained with the enzymatic tests unambiguously pointed out that the SL–PPN–HEP–HRP composite containing the co-immobilized enzymes possesses significant protease and peroxidase activities, which are maintained at least for 5 days. This is a longer time than the one usually applied for PPN67 and HRP68 in industrial applications. Moreover, such a dual enzyme activity is accompanied with excellent colloidal stability making the developed hybrid a promising candidate for application in heterogeneous systems.
Conclusions
In conclusion, PPN and HRP enzymes were successfully co-immobilized on SL microspheres by the sequential adsorption method using HEP polyelectrolyte as a building block and a separator between the two enzymes. The polyelectrolyte and the enzyme doses were optimized to achieve high colloidal stability of the SL–PPN–HEP–HRP composite, as confirmed by the remarkable resistance achieved against salt-induced aggregation. The successful immobilization of the enzymes was proved by protein tests and by super-resolution microscopy and no enzyme leakage was detected. The protease and peroxidase activities of the immobilized enzymes were tested in biochemical assays. It was found that the enzymes lost some of their activities upon immobilization; however, the obtained SL–PPN–HEP–HRP composite was still very active, i.e., it possessed significant PPN and HRP activities. Time-dependent peroxidase activity measurements revealed that such a dual activity is maintained at least for 5 days, while in the mixed solution of the native enzymes, HRP lost its activity after 3 days due to hydrolysis by the PPN enzyme. These results indicate that the applied HEP layer successfully protected HRP from the protease-like action of PPN. Therefore, the obtained SL–PPN–HEP–HRP hybrid material acts as an immobilized multi-enzymatic system with protease and peroxidase activities and possesses the advantages of a heterogeneous catalyst such as easier separation from the reaction mixture. The appreciable colloidal and functional stability, which is a crucial requirement for biocatalysts to be applied in industrial manufacturing processes, also promotes the application of the developed composite.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are thankful for the financial assistance of the Eötvös Lóránd Research Network (96130 and 95026), the Hungarian Academy of Sciences (LP2017–18/2017) and the National Excellence Programme (20391-3/2018/FEKUSTRAT, TUDFO/47138-1/2019/ITM and TKP2020). The project has received funding from the EU's Horizon 2020 research and innovation program under grant agreement No. 739593. The support from the University of Szeged Open Access Fund (5541) is gratefully acknowledged.References
- S. J. Benkovic and S. Hammes-Schiffer, Science, 2003, 301, 1196–1202 CrossRef CAS PubMed.
- S. Raveendran, B. Parameswaran, S. B. Ummalyma, A. Abraham, A. K. Mathew, A. Madhavan, S. Rebello and A. Pandey, Food Technol. Biotechnol., 2018, 56, 16–30 CAS.
- J. Britton, S. Majumdar and G. A. Weiss, Chem. Soc. Rev., 2018, 47, 5891–5918 RSC.
- E. Ricca, B. Brucher and J. H. Schrittwieser, Adv. Synth. Catal., 2011, 353, 2239–2262 CrossRef CAS.
- K. L. Xu, X. X. Chen, R. C. Zheng and Y. G. Zheng, Front. Bioeng. Biotechnol., 2020, 8, 660 CrossRef PubMed.
- M. Vazquez-Gonzalez, C. Wang and I. Willner, Nat. Catal., 2020, 3, 256–273 CrossRef CAS.
- A. Grotzky, E. Atamura, J. Adamcik, P. Carrara, P. Stano, F. Mavelli, T. Nauser, R. Mezzenga, A. D. Schluter and P. Walde, Langmuir, 2013, 29, 10831–10840 CrossRef CAS.
- R. A. Sheldon and S. van Pelt, Chem. Soc. Rev., 2013, 42, 6223–6235 RSC.
- X. L. Wang, J. F. Shi, S. H. Zhang, H. Wu, Z. Y. Jiang, C. Yang, Y. X. Wang, L. Tang and A. F. Yan, J. Mater. Chem. B, 2015, 3, 6587–6598 RSC.
- E. T. Hwang and S. Lee, ACS Catal., 2019, 9, 4402–4425 CrossRef CAS.
- Y. Kulshrestha and Q. Husain, Enzyme Microb. Technol., 2006, 38, 470–477 CrossRef CAS.
- M. Bilal, M. Asgher, H. R. Cheng, Y. J. Yan and H. M. N. Iqbal, Crit. Rev. Biotechnol., 2019, 39, 202–219 CrossRef CAS PubMed.
- M. Massaro, G. Lazzara, S. Milioto, R. Noto and S. Riela, J. Mater. Chem. B, 2017, 5, 2867–2882 RSC.
- F. Kazenwadel, M. Franzreb and B. E. Rapp, Anal. Methods, 2015, 7, 4030–4037 RSC.
- J. Drenth, J. N. Jansonius, R. Koekoek, H. M. Swen and B. G. Wolthers, Nature, 1968, 218, 929–932 CrossRef CAS PubMed.
- J. Fernandez-Lucas, D. Castaneda and D. Hormigo, Trends Food Sci. Technol., 2017, 68, 91–101 CrossRef CAS.
- T. G. Hu, J. H. Cheng, B. B. Zhang, W. Y. Lou and M. H. Zong, Ind. Eng. Chem. Res., 2015, 54, 4689–4698 CrossRef CAS.
- Q. Wang, L. Y. Zhou, Y. J. Jiang and J. Gao, Enzyme Microb. Technol., 2011, 49, 11–16 CrossRef CAS.
- F. C. Vasconcellos, G. A. S. Goulart and M. M. Beppu, Powder Technol., 2011, 205, 65–70 CrossRef CAS.
- C. Chankhampan, J. Manosroi, H. Yamamoto, K. Tahara, W. Manosroi, Y. Kawashima and A. Manosroi, J. Exp. Nanosci., 2014, 9, 138–151 CrossRef CAS.
- M. Gajhede, D. J. Schuller, A. Henriksen, A. T. Smith and T. L. Poulos, Nat. Struct. Biol., 1997, 4, 1032–1038 CrossRef CAS.
- J. Cheng, S. M. Yu and P. Zuo, Water Res., 2006, 40, 283–290 CrossRef CAS PubMed.
- M. J. H. Van Haandel, F. C. E. Saraber, M. G. Boersma, C. Laane, Y. Fleming, H. Weenen and I. Rietjens, J. Agric. Food Chem., 2000, 48, 1949–1954 CrossRef CAS PubMed.
- V. Vojinovic, R. H. Carvalho, F. Lemos, J. M. S. Cabral, L. P. Fonseca and B. S. Ferreira, Biochem. Eng. J., 2007, 35, 126–135 CrossRef CAS.
- M. S. Sadjadi, N. Farhadyar and K. Zare, J. Nanosci. Nanotechnol., 2011, 11, 9304–9309 CrossRef CAS PubMed.
- T. Mohan, R. Rathner, D. Reishofer, M. Koller, T. Elschner, S. Spirk, T. Heinze, K. Stana-Kleinschek and R. Kargl, Biomacromolecules, 2015, 16, 2403–2411 CrossRef CAS.
- M. Monier, D. M. Ayad, Y. Wei and A. A. Sarhan, Int. J. Biol. Macromol., 2010, 46, 324–330 CrossRef CAS PubMed.
- M. B. Vineh, A. A. Saboury, A. A. Poostchi, A. M. Rashidi and K. Parivar, Int. J. Biol. Macromol., 2018, 106, 1314–1322 CrossRef PubMed.
- S. A. Mohamed, M. H. Al-Harbi, Y. Q. Almulaiky, I. H. Ibrahim and R. M. El-Shishtawy, Electron. J. Biotechnol., 2017, 27, 84–90 CrossRef CAS.
- T. L. Moore, L. Rodriguez-Lorenzo, V. Hirsch, S. Balog, D. Urban, C. Jud, B. Rothen-Rutishauser, M. Lattuada and A. Petri-Fink, Chem. Soc. Rev., 2015, 44, 6287–6305 RSC.
- G. Decher, Science, 1997, 277, 1232–1237 CrossRef CAS.
- S. Saringer, P. Rouster and I. Szilagyi, J. Colloid Interface Sci., 2021, 590, 28–37 CrossRef CAS PubMed.
- Z. Somosi, N. V. May, D. Sebok, I. Palinko and I. Szilagyi, Dalton Trans., 2021, 50, 2426–2435 RSC.
- H. Holthoff, S. U. Egelhaaf, M. Borkovec, P. Schurtenberger and H. Sticher, Langmuir, 1996, 12, 5541–5549 CrossRef CAS.
- G. Trefalt, I. Szilagyi, T. Oncsik, A. Sadeghpour and M. Borkovec, Chimia, 2013, 67, 772–776 CrossRef CAS.
- L. Ehrl, Z. Jia, H. Wu, M. Lattuada, M. Soos and M. Morbidelli, Langmuir, 2009, 25, 2696–2702 CrossRef CAS PubMed.
- M. Kobayashi, S. Yuki and Y. Adachi, Colloids Surf., A, 2016, 510, 190–197 CrossRef CAS.
- F. Iselau, T. P. Xuan, G. Trefalt, A. Matic, K. Holmberg and R. Bordes, Soft Matter, 2016, 12, 9509–9519 RSC.
- D. Grolimund, M. Elimelech and M. Borkovec, Colloids Surf., A, 2001, 191, 179–188 CrossRef CAS.
- M. M. Bradford, Anal. Biochem., 1976, 72, 248–254 CrossRef CAS.
- C. Cupp-Enyard, J. Visualized Exp., 2008, 19, e899 Search PubMed.
- D. R. Doerge, R. L. Divi and M. I. Churchwell, Anal. Biochem., 1997, 250, 10–17 CrossRef CAS PubMed.
- S. Saringer, R. A. Akula, A. Szerlauth and I. Szilagyi, J. Phys. Chem. B, 2019, 123, 9984–9991 CrossRef CAS PubMed.
- P. Alpay and D. A. Uygun, J. Mol. Catal. B: Enzym., 2015, 111, 56–63 CrossRef CAS.
- S. H. Behrens, D. I. Christl, R. Emmerzael, P. Schurtenberger and M. Borkovec, Langmuir, 2000, 16, 2566–2575 CrossRef CAS.
- B. Derjaguin and L. D. Landau, Acta Phys. Chim., 1941, 14, 633–662 Search PubMed.
- T. Zor and Z. Seliger, Anal. Biochem., 1996, 236, 302–308 CrossRef CAS PubMed.
- N. Zou and J. Plank, J. Phys. Chem. Solids, 2012, 73, 1127–1130 CrossRef CAS.
- M. Pavlovic, P. Rouster and I. Szilagyi, Nanoscale, 2017, 9, 369–379 RSC.
- R. F. de Oliveira, M. L. de Moraes, O. N. Oliveira and M. Ferreira, J. Phys. Chem. C, 2011, 115, 19136–19140 CrossRef.
- D. G. Ramirez-Wong, C. Bonhomme, S. Demoustier-Champagne and A. M. Jonas, J. Colloid Interface Sci., 2018, 514, 592–598 CrossRef CAS.
- Z. Somosi, M. Pavlovic, I. Palinko and I. Szilagyi, Nanomaterials, 2018, 8, 986 CrossRef.
- M. Pavlovic, S. Murath, X. Katona, N. B. Alsharif, P. Rouster, J. Maleth and I. Szilagyi, Sci. Rep., 2021, 11, 4321 CrossRef CAS.
- S. Saringer, P. Rouster and I. Szilagyi, Langmuir, 2019, 35, 4986–4994 CrossRef CAS.
- P. Rouster, M. Pavlovic, S. Saringer and I. Szilagyi, J. Phys. Chem. C, 2018, 122, 11455–11463 CrossRef CAS.
- Y. K. Leong, Colloid Polym. Sci., 1999, 277, 299–305 CrossRef CAS.
- I. Popa, G. Papastavrou and M. Borkovec, Phys. Chem. Chem. Phys., 2010, 12, 4863–4871 RSC.
- M. Pavlovic, P. Rouster, Z. Somosi and I. Szilagyi, J. Colloid Interface Sci., 2018, 524, 114–121 CrossRef CAS PubMed.
- A. vander Straeten, A. Bratek-Skicki, A. M. Jonas, C. A. Fustin and C. Dupont-Gillain, ACS Nano, 2018, 12, 8372–8381 CrossRef CAS PubMed.
- H. J. Kim, Y. Suma, S. H. Lee, J. A. Kim and H. S. Kim, J. Mol. Catal. B: Enzym., 2012, 83, 8–15 CrossRef CAS.
- J. Hierrezuelo, A. Sadeghpour, I. Szilagyi, A. Vaccaro and M. Borkovec, Langmuir, 2010, 26, 15109–15111 CrossRef CAS PubMed.
- P. C. Almeida, I. L. Nantes, C. C. A. Rizzi, W. A. S. Judice, J. R. Chagas, L. Juliano, H. B. Nader and I. L. S. Tersariol, J. Biol. Chem., 1999, 274, 30433–30438 CrossRef CAS PubMed.
- X. X. Zhang, X. Y. Liu, G. W. Su, M. M. Li, J. Liu, C. Y. Wang and D. Xu, Commun. Biol., 2021, 4, 198 CrossRef CAS PubMed.
- P. C. Almeida, I. L. Nantes, J. R. Chagas, C. C. A. Rizzi, A. Faljoni-Alario, E. Carmona, L. Juliano, H. B. Nader and I. L. S. Tersariol, J. Biol. Chem., 2001, 276, 944–951 CrossRef CAS PubMed.
- J. L. S. Tersariol, D. C. Pimenta, J. R. Chagas and P. C. Almeida, Braz. J. Med. Biol. Res., 2002, 35, 135–144 CrossRef PubMed.
- S. Akhtar, A. A. Khan and Q. Husain, J. Chem. Technol. Biotechnol., 2005, 80, 198–205 CrossRef CAS.
- K. Liburdi, C. Boselli, G. Giangolini, S. Amatiste and M. Esti, Foods, 2019, 8, 600 CrossRef CAS PubMed.
- S. E. Gilliland, J. Diary Sci., 1969, 52, 321–324 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Electrophoretic mobilities, stability ratios, data of Bradford, protease and peroxidase tests. See DOI: 10.1039/d1tb01861b |
| This journal is © The Royal Society of Chemistry 2022 |