DOI:
10.1039/D1TA11063B
(Paper)
J. Mater. Chem. A, 2022,
10, 7999-8014
Unlocking the potential of a novel hierarchical hybrid (Ni–Co)Se2@NiMoO4@rGO–NF core–shell electrode for high-performance hybrid supercapacitors†
Received
31st December 2021
, Accepted 22nd February 2022
First published on 24th February 2022
Abstract
Nano-hybridization of a core–shell structure integrating a transition metal selenide with transition metal oxides on the surface of a carbonaceous material embellished 3D substrate results in the formation of high-capacity electrode materials for use in energy storage devices thanks to the ample electroactive sites and relatively high electronic conductivity of the resultant materials compared to their individual components. Herein, oxalic acid templated and selenization processed porous Ni–Co–Se (NCSe) nanorods are vertically grown on the surface of an rGO–nickel foam (NF) substrate, followed by the embellishing of the surface of the NCSe nanorods with hydrothermally-prepared NiMoO4 (NMO) nanosheets to prepare a hierarchical NCSe@NMO@rGO–NF hybrid core–shell structure. The as-prepared NCSe@NMO@rGO–NF hybrid electrode exhibits tremendous electrochemical performance in terms of a high specific capacity (396.1 mA h g−1) at a current density of 1 A g−1, along with excellent capacity retention (87.6%) after 8000 consecutive charge–discharge (GCD) cycles. A hybrid supercapacitor (HSC) device was also assembled by sandwiching the NCSe@NMO@rGO–NF electrode with oxalic acid and metal–organic framework derived porous carbon/CNT coated NF (OA-MOF-PC-CNTs@NF) to unveil its practical applicability. The as-fabricated HSC shows a high energy density (63.2 W h kg−1) and power density (7983.5 W kg−1) alongside 89.4% capacitance retention after 8000 GCD cycles. Owing to their unique structural features and superlative electrochemical properties, the proposed method and selected materials can mitigate the issues related to electrochemical capacitors.
1. Introduction
The ever-mushrooming pursuit of portable and hybrid electronic devices has imperatively spurred the exploration of electrochemical supercapacitors (SCs) owing to their intrinsic features including swift charge–discharge rate, long-term cycling life-span, enhanced power density, high safety and long-term durability.1,2 Generally, the charge storage mechanism in SCs occurs via surface-controlled reactions that take place on the surface of electrode materials. Consequently, the charge transfer is hindered on or nearby the surface of the electrode, which causes SCs to bank minimal energy density, thereby impeding its potential applications for modern electronic gadgets and hybrid electric vehicles.3–5 Based on the equation, E = 1/2CSCV,2 for SCs the energy density (E) is precisely interrelated to the specific capacitance (CSC) and the operating voltage window (V).6 Accordingly, the construction of advanced SCs featuring high capacitance along with elongated working potential is a good approach for substantial optimizing the energy density of SCs. Thus, an efficient strategy to achieve SCs with optimal energy density without attenuation of their power output performance is the sandwiching of well-matched hybrid supercapacitors (HSCs), integrating pseudocapacitive (PC) or battery-type materials as a source of energy and electrochemical double-layer capacitor (EDLC) induced electrode materials as a source of power.5,7–12 In recent years, carbon-based materials and a diverse range of metal oxides/hydroxides/chalcogenides have been universally diagnosed as capacitive and battery-type materials, respectively.13–21 EDLC materials have long-term cyclic durability, but suffer from low electrochemical performance in HSC devices.1 In contrast, battery-type materials exhibit swift redox kinetics and outstanding theoretical capacity, nonetheless they always have disappointing rate performance and ultra-long cycling stability.22 Accordingly, it is imperative to construct advanced hierarchical nanostructured featured electrode materials that feature synergistic interactions to ameliorate the overall electrochemical performance, which is immensely desirable in the design of HSCs.
To date, transition metal selenides (TMSs) have garnered encyclopedic attention in diverse sectors, including water splitting, batteries, photocatalysis and SCs.23–26 Additionally, owing to the lower electronegativity (EN) value of selenium (Se) than oxygen (O) and sulfur (S), it has been divulged that TMSs have superior electronic conductivity and enhanced electrochemical activity compared to their correspondent oxide/sulfide counterparts.14,27–29 Additionally, compared to monometallic selenides, binary TMSs have attracted widespread attention owing to them having the highest electronic conductivity, synergistic effects on faradaic reactions, improved electrochemical activity, as well as ample structural deficiencies, which make them auspicious contenders in the construction of electrode materials for high-energy SCs.30–32 Despite these excellent electrochemical properties, the semiconductor nature of Ni–Co–Se however shields the charge transfer kinetics from active sites to the current collector resulting in minimal rate performance and specific energy density for energy storage and conversion systems. Designing the conductive current collector framework is assumed to be highly beneficial for the swift transport of electrons between the electrode and active materials, thereby boosting the electrochemical reaction.27,33 Moreover, owing to the eloquent volume transfiguration in the redox reaction, battery-type electrode materials experience tremendous structural deformation, and greatly experience swift capacity fading.34 Accordingly, the garnishing of chemically reduced graphene oxide (rGO) onto the 3D current collector not only serves as a conductive network tendering electronic “superhighways”, but rather it also confines the heterostructure growth of active materials and supports the structure, thereby suppressing possible structural agglomeration.35 However, the construction of active materials on carbon materials-decorated 3D substrates is not sufficient to procure the essences of practical implementation towards energy technologies. In this context, additional strategies to triumph over the shortcomings of TMSs have been considered, such as the meticulous architecture of a hierarchical heterostructure core–shell electrode, incorporating binary transition metal oxides (BTMOs) as a two dimensional (2D) protective shell layer with a one dimensional (1D) core-material onto a carbonaceous material embellished 3D substrate, thereby elevating the overall electrochemical performance of SCs in-terms of paramount specific capacity, ultra-fast ion-electron transfer and remarkable structural endurance capability.35,36
Recently, metal molybdates (notably, NiMoO4) have attracted a high amount of research interest as a protective shell layer owing to their immense electronic conductivity, low cost, prominent electrochemical features and environmental friendliness.37,38 Furthermore, the usage of NiMoO4 as a protective shell material during the construction of a core–shell nano-framework can be attributed to the coupling effect of nickel (Ni) and molybdenum (Mo) atoms. Meanwhile, throughout the reversible faradaic reactions in alkaline surroundings, Mo atoms furnish ample electronic conductivity, while at the same time Ni atoms tender enhanced capacitive behavior.37,39 Moreover, the ample electroactive surface area due to the octahedral and tetrahedral localities of Ni and Mo atoms along with their prominent nanoscale features could amplify the reversible redox reaction (pseudocapacitive nature) kinetics of Ni–Mo-based electrode materials.40–42 However, to the best of our knowledge, the design of a hybrid heterostructure core–shell nanoarchitecture using oxalic acid assisted hollow Ni–Co–Se nanotubes as a core and NiMoO4 nanosheets as a protective shell material on a carbonaceous materials-garnished 3D current collector has not been reported elsewhere. Moreover, the scrupulous construction of a core–shell nanostructure on an rGO embellished 3D current collector could be highly beneficial to produce a heterostructure core–shell electrode with intensified electrochemical performance for SCs. The decoration of rGO onto the current collector gives it multifarious advantages owing to its remarkable electronic conductivity and commodious surface area, as well as splendid surface functionalities features. Accordingly, it can be affirmed that the scrupulous construction of a Ni–Co–Se based nanorod–nanosheet hierarchical hybrid core–shell (Ni–Co–Se@NiMoO4@rGO–NF) electrode could be a beneficial approach to ameliorate the overall electrochemical features of hybrid supercapacitors (HSCs).
To this aim, we have designed the cost effective selenization of Ni–Co-oxalate coupled with the garnishing of 2D NiMoO4 nanosheets via a hydrothermal technique to develop a novel hybrid core–shell nanoarchitecture of Ni–Co–Se@NiMoO4 on an rGO–Ni foam substrate. There are ample research studies in which binary metal oxides/sulfides have been extensively used for the construction of core–shell electrodes for SCs. However, the construction of a meticulous architecture of a binary metal selenide (Ni–Co–Se) and TMO (NiMoO4) based core–shell electrode for SCs has not been reported previously. Imperatively, this is the first approach in which bimetallic oxalate (Ni–Co-oxalate) precursors have been scrupulously converted into hollow and porous Ni–Co–Se nanorods, which act as a core material and ultimately, NiMoO4 nanosheets (shell-layer) are decorated over it to achieve a hierarchical hybrid core–shell nanoarchitecture of Ni–Co–Se@NiMoO4 on a rGO–NF substrate. The novel Ni–Co–Se hollow nanotubes (core-network) possess high electrical conductivity and offer a plentiful electroactive surface area for the spontaneous insertion of electrolyte ions, and rapid faradaic reactions channels. Furthermore, the well-interconnected 2D NiMoO4 nanosheets (shell-framework) furnish a commodious electroactive surface area that provides curtailed ion-electron subways, enabling deep and feasible penetration of electrolyte ions during swift reversible redox reactions. The as-constructed hierarchical hybrid core–shell when employed as an additive free electrode delivers a remarkable specific capacity with splendid rate performance and ultra-long cycling durability. Furthermore, a HSC device was assembled using Ni–Co–Se@NiMoO4@rGO–NF as a cathode (positive electrode) and oxalic acid derived porous carbon (OADPC) in alkaline medium. The as-sandwiched Ni–Co–Se@NiMoO4@rGO–NF//OA-MOF-PC-CNT HSC device exhibits a high energy density of 63.1 W h kg−1 at a power density of 799.8 W kg−1 and ultra-long cycling performance (89.4% capacitance retention after 8000 cycles).
2. Experimental section
Materials and reagents
Nickel(II) nitrate hexahydrate (Ni(NO3)2·6H2O), cobalt(II) nitrate hexahydrate (Co(NO3)2·6H2O), oxalic acid dihydrate (C2H2O4·2H2O), graphite powder, poly(vinylidene fluoride) (PVDF), N-methyl-2-pyrrolidone (NMP), 2-methylimidazole (C4H6N2), sodium molybdate dihydrate (Na2MoO4·2H2O) and selenium (Se) powder were purchased from Sigma-Aldrich. Ethanol (C2H5OH), potassium hydroxide (KOH), carbon black and hydrochloric acid (HCl) were ordered from Daejung Chemical Co. Ltd., South Korea. All of the chemicals were of analytical grade and used without further purification.
Synthesis of reduced graphene oxide on Ni foam substrate (rGO–NF)
Prior to garnishing rGO, the Ni foam (NF) substrate was carefully cleaned with 2 M HCl via ultrasonication, followed by successive cleaning with de-ionized (DI) water and ethanol and dried at 60 °C for 12 h. Afterwards, the deposition of rGO on Ni foam was carried out according to our previously reported technique.43
Synthesis of Ni–Co–Se onto rGO–Ni foam (NCSe@rGO–NF) hollow and porous nanotubes
Firstly, a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 molar ratio of Ni(NO3)2·6H2O and Co(NO3)2·6H2O were dissolved in 50 ml of DI water and the obtained solution was labelled as solution A. In the meantime, solution B was prepared by dissolving 6.35 g of oxalic acid in 50 ml of DI water. Then, the rGO–NF substrate was kept inside solution A followed by subsequent dropwise addition of solution B and left under continuous magnetic stirring for 40 min. After the completion of the reaction, the as-obtained product was washed with water and ethanol repeatedly to remove partly attached species and dried at 60 °C overnight. The color of the NF substrate was white in appearance, manifesting the formation of vertically aligned Ni–Co-oxalate on rGO–NF (NCOx@rGO–NF). Secondly, for the selenization treatment, 2 mM Se powder, 4 mM NaBH4, 3 ml of DI water and 50 ml of absolute ethanol were mixed together and stirred for 30 min. After that, the as-prepared NCOx@rGO–NF sample was placed vertically into the growth solution and heated at 180 °C for 12 h. After the completion of the reaction, the sample was carefully washed and dried at 60 °C overnight in an oven. Finally, the as-obtained product was annealed under an Ar atmosphere to produce porous NCSe@rGO–NF nanorods.
:2 molar ratio of Ni(NO3)2·6H2O and Co(NO3)2·6H2O were dissolved in 50 ml of DI water and the obtained solution was labelled as solution A. In the meantime, solution B was prepared by dissolving 6.35 g of oxalic acid in 50 ml of DI water. Then, the rGO–NF substrate was kept inside solution A followed by subsequent dropwise addition of solution B and left under continuous magnetic stirring for 40 min. After the completion of the reaction, the as-obtained product was washed with water and ethanol repeatedly to remove partly attached species and dried at 60 °C overnight. The color of the NF substrate was white in appearance, manifesting the formation of vertically aligned Ni–Co-oxalate on rGO–NF (NCOx@rGO–NF). Secondly, for the selenization treatment, 2 mM Se powder, 4 mM NaBH4, 3 ml of DI water and 50 ml of absolute ethanol were mixed together and stirred for 30 min. After that, the as-prepared NCOx@rGO–NF sample was placed vertically into the growth solution and heated at 180 °C for 12 h. After the completion of the reaction, the sample was carefully washed and dried at 60 °C overnight in an oven. Finally, the as-obtained product was annealed under an Ar atmosphere to produce porous NCSe@rGO–NF nanorods.
Synthesis of Ni–Co–Se@NiMOO4 on a rGO–Ni foam (NCSe@NMO@rGO–NF) hybrid core–shell nanoarchitecture
A secondary hydrothermal reaction was carried out to embellish NiMoO4 nanosheets arrays (NSAs) on a rGO–NF substrate covered with hollow Ni–Co–Se nanotubes. In detail, 0.5 mmol of Ni(NO3)2·6H2O and 0.5 mmol of Na2MoO4·2H2O was dissolved in 50 ml of DI under stirring for 40 min. Then, the as-prepared solution was transferred into a Teflon-lined autoclave containing NCSe@rGO–NF, and the reaction was continued at 140 °C for 4 h. After the completion of the reaction, the as-obtained sample was repeatedly washed with water and ethanol to remove loosely adhered species followed by drying to produce a Ni–Co–Se@NiMoO4@rGO–NF hybrid heterostructure core–shell nanoarchitecture, indicated as NCSe@NMO@rGO–NF. For comparison, pure NiMoO4 (NMO) was also prepared on a rGO–NF substrate using a similar route in the absence of NCSe@rGO–NF, and nominated as NMO@rGO–NF. Mass loadings of NCSe@NMO@rGO–NF, NMO@rGO–NF and NCSe@NMO@rGO–NF of 1.8, 1.4 and 2.3 mg cm−2 were achieved, respectively.
Preparation of OA and MOF-derived porous carbon and carbon nanotube network (OA-MOF-PC-CNTs)
In order to synthesize OA-MOF-PC-CNTs, firstly, 1 mmol of Co(NO3)2·6H2O, and 1 mmol of Zn(NO3)2·6H2O were dissolved in a solution containing a mixture of methanol and water and designated as solution A. Next, solution B was prepared by dissolving 16 mmol of 2-methylimidazole in 40 ml of CH3OH. Similarly, solution C was obtained by dissolving 5 mmol of OA acid in 30 ml of DI water. Subsequently, solutions A, B and C were mixed together and continuously stirred for 30 min followed by ageing for 12 h. The as-obtained product was filtered, washed and dried under vacuum overnight. The as-obtained product was carbonized at 800 °C in a tube furnace at a heating ramp of 2 °C min−1 followed by etching with HCl, washing and drying to produce an OA-MOF porous carbon–CNT network (OA-MOF-PC-CNTS).
Physical characterization tools
Field-emission scanning electron microscopy (FE-SEM) was employed to analyze the surface morphologies of the as-prepared samples. The crystalline features coupled with the phase purities of the samples were scrutinized via X-ray diffraction (XRD) analysis. The inherent structural information of the samples was determined by transmission electron microscopy (TEM) analysis. Elemental distribution to elucidate the pertinent morphology was investigated by energy dispersive X-ray (EDX) analysis. X-ray photoelectron spectroscopy (XPS) analysis was adopted to explore the surface elemental composition and oxidation state of the each of the elements present in the samples.
Electrochemical characterization
A VersaStat 4 electrochemical workstation was employed to scrutinize the electrochemical properties of all of the electrodes for SC applications. Basic but imperative measurements like cyclic voltammetry (CV), galvanostatic charge–discharge (GCD), and electrochemical impedance spectroscopy (EIS) data were carried out in order to inspect the performance of the electrode materials. Ag/AgCl and Pt wire were used as reference and counter electrodes, respectively, coupled with the working electrode (active material garnished NF substrate) in a conventional three-electrode configuration. NCSe@NMO@rGO–NF and OADPC were used as positive and negative electrodes, respectively, in the assembly of a HSC, which was tested for its practical applicability. Cellulose membrane (CHMLAB GROUP, weight: 85 g m−2, thickness of 180 μm, ASH content < 0.06%) and 2 M KOH were used as a separator and electrolyte, respectively. The as-fabricated positive electrode was directly employed as a binder-free working electrode, while a negative electrode was prepared using a homogeneous slurry comprising active material (OADPC), carbon black, and organic binder (PVDF) in a ratio of 80:10:10 mixed and milled in the presence of NMP solvent on the NF substrate (immersed area: 1 × 1 cm2), followed by drying in a vacuum oven. The specific capacitance, specific capacity, energy density and power density were calculated employing the various equations given below.| | | E = 0.5 × CSP × ΔV2/3.6 | (3) |
where CSP is the specific capacitance (F g−1), QSC represents the specific capacity (mA h g−1), E signifies the energy density (W h kg−1), P indicates the power density (W kg−1), I refers the current density (A g−1), m is the mass of the active material (m), Δt is the discharge time (s), and ΔV is the working potential (V).
3. Results and discussion
The schematic diagram in Fig. 1 elucidates the hybrid core–shell structure of the additive-free Ni–Co–Se@NiMoO4@rGO-Ni foam (NCSe@NMO@rGO–NF) material by conversion of OA-assisted Ni–Co-oxalate precursors via selenization and subsequent hydrothermal treatment. The NF substrate was used as a current collector scaffold for the preparation of Ni–Co–Se@NiMoO4 owing to its appealing structural features, including an open porous framework, 3D intertwined stems and good electronic conductivity. Imperatively, the 3D interconnected stems in the NF promote swift electron transportation. Similarly, the 3D open porous framework allows facile insertion (or penetration) of electrolyte ions in alkaline growth solution. Furthermore, the electrical conductivity and effectual 3D porous framework for elevated electrochemical performance can be ameliorated by garnishing the 3D NF substrate with rGO. Primarily, owing to its excellent electronic conductivity and ample surface area, the decoration of rGO not only enhances the conductive nature of the NF substrate and hybrid core–shell heterostructure but also promotes electronic superhighways. Also, the decoration of rGO sheets reinforces the homogeneous growth of active material thereby curtailing the probable agglomeration and diminishing of the inaugurated structural deformation of the electrode materials after long-term durability testing. In the first step, Fig. 1a(i–iii) represents the chemical reduction of the graphene oxide (GO) using ascorbic acid (AA), which is considered to be a cost-effective and green reducing agent. The reduction process is initiated with an SN2 nucleophilic attack and subsequent water bath treatment, which restore the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C unsaturated framework by eliminating the saturated epoxide and hydroxyl groups from the basal planes of the GO sheets, suggesting the conversion of GO into rGO with the liberation of dehydroascorbic acid, which ultimately splits into guluronic acid and oxalic acid.44 Also, during this process, the intermediate product (dehydroascorbic acid) forms strong hydrogen bonds (H-bonding) with residual oxygen species, including carboxylic (–COOH) groups on the surrounding GO sheets, which inhibits the π–π stacking between the rGO sheets, thereby precluding possible aggregation and the forming of an immensely stable rGO–AA suspension.44,45Fig. 1b(i–vi) shows an in-depth scheme with possible formation mechanism of the NCSe@NMO@rGO–NF core–shell heterostructure. During the growth process, firstly in situ formed Ni2+ and Co2+ cations were adsorbed onto the rGO–NF surface. In the meantime, the oxalate (C2O42−) anions tendered by an aqueous solution of OA react with Ni2+ and Co2+ cations leading to the formation of a Ni–Co-oxalate hydrated complex compound, as proved by the formation of a white precipitate. After careful washing and drying, the as-formed product was converted into the respective Ni–Co–Se nanostructure via selenization. Herein, the reaction proceeds with the dissolution of Ni–Co-oxalate into Ni and Co ions. At the same time, Se2− ions derived via the reduction of sodium selenite (Na2Se2O3) by hydrazine hydrate (N2H4) react with Ni2+ and Co2+ ions followed by the annealing the sample under an inert (Ar) atmosphere to obtain a hierarchical porous Ni–Co–Se nanorod structure. The strong coordination of Ni and Co ions with Se ions is crucial to the formation of Ni–Co–Se, and the heating treatment under an Ar atmosphere was greatly responsible for forming the porous nanoarchitecture. In the following step, a second hydrothermal treatment was employed in which Ni2+ and MoO42− self assemble together forming NiMoO4 as a protective shell layer on the surface of porous Ni–Co–Se nanorods to form a hybrid NCSe@NMO@rGO–NF core–shell nanoarchitecture.
C unsaturated framework by eliminating the saturated epoxide and hydroxyl groups from the basal planes of the GO sheets, suggesting the conversion of GO into rGO with the liberation of dehydroascorbic acid, which ultimately splits into guluronic acid and oxalic acid.44 Also, during this process, the intermediate product (dehydroascorbic acid) forms strong hydrogen bonds (H-bonding) with residual oxygen species, including carboxylic (–COOH) groups on the surrounding GO sheets, which inhibits the π–π stacking between the rGO sheets, thereby precluding possible aggregation and the forming of an immensely stable rGO–AA suspension.44,45Fig. 1b(i–vi) shows an in-depth scheme with possible formation mechanism of the NCSe@NMO@rGO–NF core–shell heterostructure. During the growth process, firstly in situ formed Ni2+ and Co2+ cations were adsorbed onto the rGO–NF surface. In the meantime, the oxalate (C2O42−) anions tendered by an aqueous solution of OA react with Ni2+ and Co2+ cations leading to the formation of a Ni–Co-oxalate hydrated complex compound, as proved by the formation of a white precipitate. After careful washing and drying, the as-formed product was converted into the respective Ni–Co–Se nanostructure via selenization. Herein, the reaction proceeds with the dissolution of Ni–Co-oxalate into Ni and Co ions. At the same time, Se2− ions derived via the reduction of sodium selenite (Na2Se2O3) by hydrazine hydrate (N2H4) react with Ni2+ and Co2+ ions followed by the annealing the sample under an inert (Ar) atmosphere to obtain a hierarchical porous Ni–Co–Se nanorod structure. The strong coordination of Ni and Co ions with Se ions is crucial to the formation of Ni–Co–Se, and the heating treatment under an Ar atmosphere was greatly responsible for forming the porous nanoarchitecture. In the following step, a second hydrothermal treatment was employed in which Ni2+ and MoO42− self assemble together forming NiMoO4 as a protective shell layer on the surface of porous Ni–Co–Se nanorods to form a hybrid NCSe@NMO@rGO–NF core–shell nanoarchitecture.
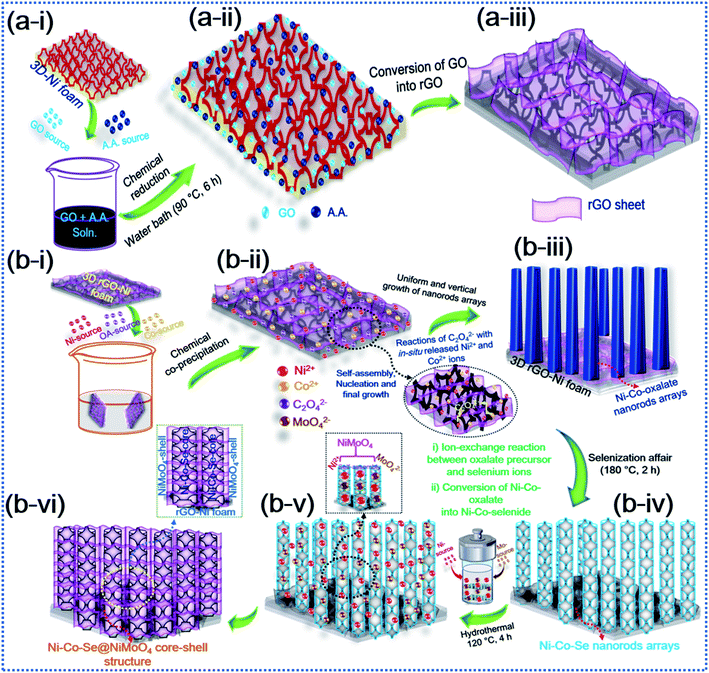 |
| | Fig. 1 Schematic illustration of the synthesis of the hierarchical hybrid NCSe@NMO@rGO–NF core–shell structure. | |
The surface morphology of the as-prepared various samples was scrutinized by employing FESEM analysis. Fig. S1a(i–iii)† depict the FESEM images of the bare NF surface, comprising irregularly aligned canes which satisfactorily promote the penetration of electrolyte ions and serve as subways for charge carrier transportation. Fig. S1b(i–iii)† show the surface morphology of rGO–NF, displaying the uniform decoration of 2D rGO-sheets on the surface of the NF. The garnishing of the 3D NF substrate with rGO sheets not only enhances the electrical conductivity but also creates a superhighway for swift electron transfer and stimulates uniform growth of active material that eliminates possible agglomeration. The digital image of the NF substrate before and after the garnishment of rGO is shown in Fig. S2c(i and ii),† manifesting the successful deposition of rGO on the NF substrate. Similarly, the surface morphologies of NCOx@rGO–NF, NCO@rGO–NF, NCSe@rGO–NF, NMO@rGO–NF and NCSe@NMO-rGO–NF are shown in Fig. 2. Fig. 2a(i–iii) display the vertically and uniformly grown NCOX nanorods, which serve as a template for the fabrication of the core–shell nanoarchitecture. Fig. 2b(i–iii) depict the surface morphology of the NCSe nanorods on rGO–NF. As elucidated from low and high magnification images, porous NCSe nanorods are vertically grown on rGO–NF. The porosity of the NCSe nanorods could be associated with the ion-exchange based selenization and annealing treatment under inert atmosphere. The low magnification FESEM images (Fig. 2c(i and ii)) display the uniform decoration of NMO nanosheets over the entire rGO–NF surface. The high magnification FESEM image (Fig. 2d(iii)) clearly shows the well-aligned porous nanosheets, which exhibit an open space and high electroactive surface area to boost the capacitive features of the electrode materials. Additionally, the FESEM images of the hybrid NCSe@NMO@rGO–NF core–shell structure are shown in Fig. 2d(i–iii). As expected, the low-magnification FESEM images (Fig. 2d(i and ii)) manifest the uniform embellishment of NMO nanosheets over the porous NCSe nanorods, forming a core–shell structure over the entire surface of the rGO–NF. The explicit view in Fig. 2d(iii) further suggests the garnishment of porous NMO nanosheets over the hollow NCSe nanorods, creating a continuous nanosheet to porous nanorod network, manifesting the successful formation of a hierarchical hybrid core–shell heterostructure. Importantly, the connectivity of porous NCSe nanorods with NMO nanosheets (1D–2D heterostructure hybrid system) leads to the formation of electroactive sites for redox reaction, open spaces for rapid ion-electron transportation and effectual electrolyte penetration, thereby uplifting the energy storage capacity of the electrode materials.
 |
| | Fig. 2 Low and high magnification FESEM images of (a) NCOX@rGO–NF (i–iii), (b) NCSe@rGO–NF (i–iii), (c) NMO@rGO–NF (i–iii) and (d) NCSe@NMO@rGO–NF (i–iii). | |
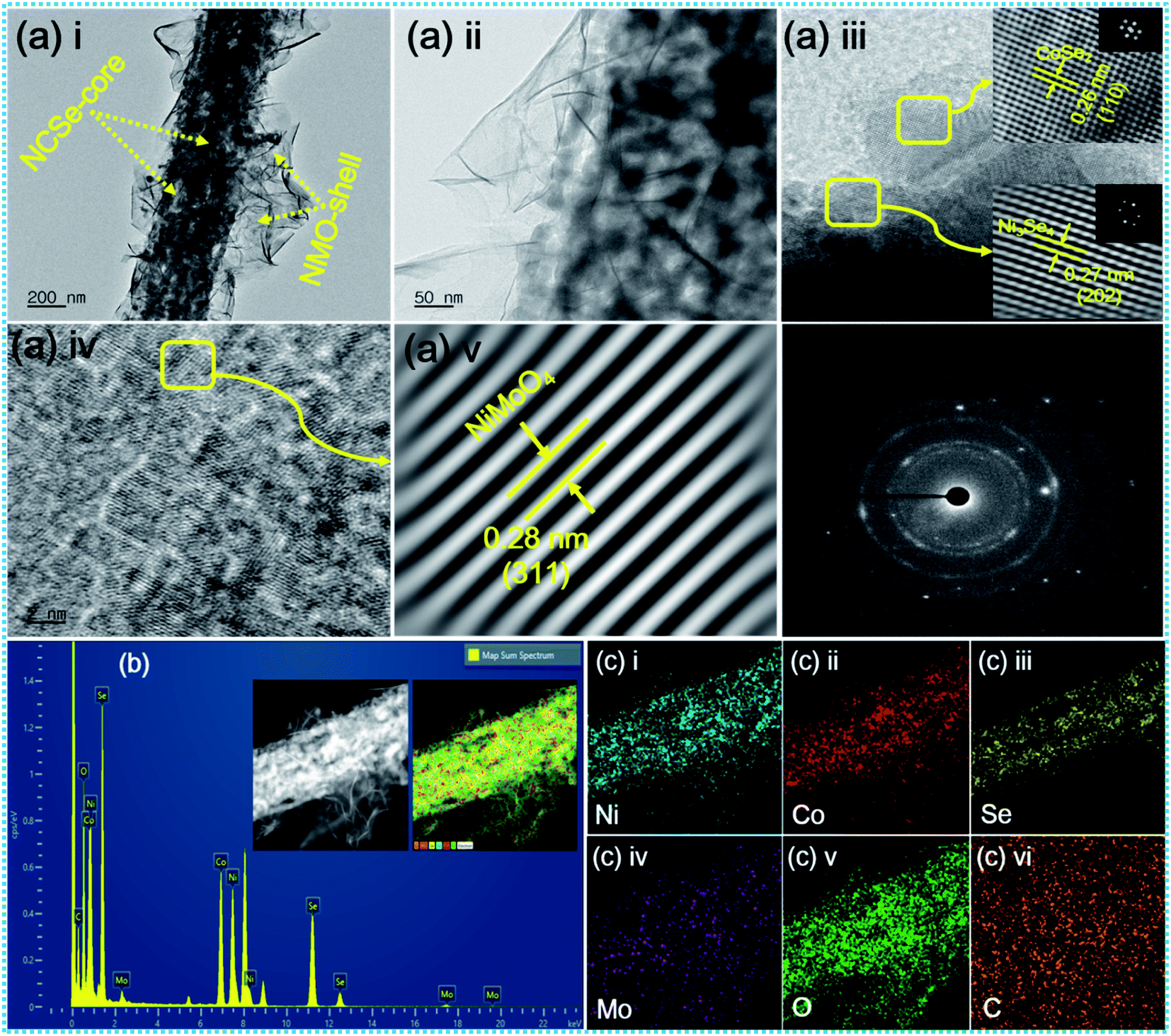
The inherent structural features of as-prepared NCSe@NMO@rGO–NF were explored by transmission electron microscopy (TEM) analysis. As can be recognized from Fig. 3a(i), NCSe nanorods can be seen to be cojoined with NMO nanosheets. Moreover, the high-magnification image in Fig. 3a(ii) shows the good interconnection of hollow NCSe nanorods with NMO nanosheets, corroborating the formation of a hierarchical NCSe@NMO@rGO–NF core–shell structure. The HRTEM images for NCSe@NMO@rGO–NF are shown in Fig. 3a(iii–v). As depicted in Fig. 3a(iii), the HRTEM images of the NCSe nanorods and the corresponding inverse fast Fourier transform (IFFT) pattern (inset) show an interplanar spacing of 0.27 nm, which can be ascribed to the crystal plane of (202) for Ni3Se4, whilst the lattice fringe spacing of 0.26 nm affirms the presence of a (110) crystal plane for CoSe2.46,47 Similarly, the HRTEM image (Fig. 3a(iv)) and IFFT pattern (Fig. 3a(v)) show an interplanar spacing of 0.28 nm, which can be attributed to the (311) crystal planes of the NMO nanosheets in the sample.48 The selected area electron diffraction (SAED) pattern shows diffraction rings coupled with bright spots, manifesting the polycrystalline nature of the as-fabricated sample (Fig. 3a(vi)). Energy-dispersive spectroscopy (EDS) analysis was assigned to testify the elements presented in the NCSe@NMO@rGO–NF sample. As depicted in Fig. 3b, the EDS spectrum of the NCSe@NMO@rGO–NF core–shell structure divulges the peaks assigned to Ni, Co, Se, Mo, O and C, corroborating the perfect engineering of Ni–Co–Se as a core and NMO as a shell in the single framework (the inset depicts the area taken for EDS analysis). Elemental distribution in NCSe@NMO@rGO–NF core–shell structure was scrutinized from the elemental mapping displayed in Fig. 3c(i–vi). The uniform distribution of the various elements, including Ni, Co, Se, Mo, O and C, in the sample endorsed the successful formation of the NCSe@NMO@rGO–NF core–shell structure.
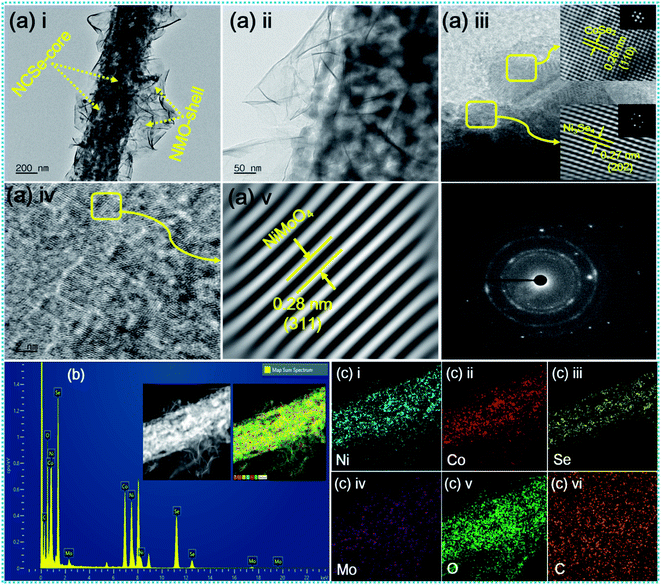 |
| | Fig. 3 (a) TEM and HR-TEM images of NCSe@NMO@rGO–NF (i–v), SAED pattern (vi), (b) EDS spectrum of NCSe@NMO@rGO–NF (inset shows the area selected for EDS elemental mapping), and (c) EDS elemental mapping images of Ni, Co, Se, Mo, O and C (i–vi). | |
X-ray diffraction (XRD) patterns were recorded to examine the crystalline phase of the as-prepared samples (Fig. 4a). The XRD pattern shows three prominent peaks at diffraction angles (2θ) of 43.4°, 43.4°, and 74.3°, which can be attributed to the characteristic peaks of the NF substrate. While, several well-defined peaks can be observed at 23.5°, 29.1°, 30.7°, 37.8°, 47.5°, 53.4° and 63.2° assigned to orthorhombic CoSe2 with crystal planes of (110), (011), (101), (111), (200), (211), (031), and (122) (JCPDS #53-0449), respectively.46 The diffraction peaks located at 17.1°, 33.6°, 45.3°, 50.1°, and 53.8° correspond to the (002), (202), (204), (020), and (002) crystal planes of monoclinic Ni3Se4 (JCPDS Card #13-0300), respectively.49 Furthermore, the characteristic peaks at diffraction angles (2θ) of 14.2°, 28.9°, 32.8°, 43.8°, 47.8°, 53.4°, 58.9°, 62.1° can be well assigned to the (110), (220), (022), (330), (−204), (510), (024), (−622) crystal planes of monoclinic NiMoO4 (JCPDS #86-0361), respectively.50 These results confirm the successful fabrication of a hierarchical NCSe@NMO@rGO–NF core–shell structure with optimal phase purity. The disappearance of rGO peaks occurs due to the minimum deposition of the rGO layer on the Ni foam compared to the deposition of the bulk sample (NCSe@NMO). Fig. 4b–h show the X-ray photoelectron spectroscopy (XPS) data of the NCSe@NMO@rGO–NF core–shell structure, recorded to investigate the surface elements and oxidation state of NCSe@NMO@rGO–NF. The full survey XPS spectra of the NCSe@NMO@rGO–NF core–shell structure displayed in Fig. 4b show signals for Ni, Co, Mo, Se, O and C, manifesting the presence of various elements without any redundant impurities, which are in good accordance with the EDS analysis. The valence states of the various elements were analyzed by recording high-resolution (HR) Ni 2p, Co 2p, Mo 3d, Se 3d, O 1s, and C 1s spectra. The HR Ni 2p spectrum shows the two primary peaks positioned at 855.3 eV (Ni 2p3/2) and 879.2 eV (Ni 2p1/2), respectively, which are further deconvoluted into four peaks (Fig. 4c). The satellite (“Sate.”) peaks of Ni 2p3/2 and Ni 2p1/2 appear at 854.4 and 872.4 eV, respectively. The presence of all of these peaks are intense enough to proclaim the presence of Ni2+ as well Ni3+ oxidation states in the NCSe@NMO@rGO–NF core–shell structure.51Fig. 4d shows the deconvolution of the Co 2p spectrum into two primary spin–orbit doublets of Co 2p3/2 (780.1 eV) and Co 2p1/2 (795.2 eV), which are further deconvoluted into four peaks. The other peaks at binding energies of 786.2 and 803.7 eV were assigned to the satellite peaks of Co 2p3/2 and Co 2p1/2, respectively. The scrutiny of all these peaks was sufficient to ascertain the presence of the Co2+ and Co3+ oxidation states.32 The HR spectrum of Mo 3d is displayed in Fig. 4e. The two deconvoluted peaks at binding energies of 232.1 and 235.5 eV with an energy gap of 3.4 eV were ascribed to the Mo 3d5/2 and Mo 3d3/2 energy levels, attributed to the Mo6+ valence state.52 Furthermore, as displayed in Fig. 4f, the HR spectrum of Se 3d is further deconvoluted into two peaks. The peaks positioned at 55.4 and 59.2 eV can be attributed to Se 3d5/2 and Se 3d3/2 respectively, which are recognized as typical peaks of a metal-selenium linkage.49 The HR spectrum of O 1s (Fig. 4g) displays two main peaks positioned at 530.3 and 531.3 eV, suggesting the presence of metal oxygen (M–O–M) bonding onto the NiMoO4 shell material.53,54 The core-level spectrum of C 1s (Fig. 4h) shows the prominent single peak accompanied by other secondary peaks positioned at binding energy values of 284.6, 285.7 and 287.8 eV, representing various functional groups and suggesting the successful deposition of rGO onto the Ni foam substrate.43,55 The specific surface area coupled with pore size distribution (PSD) of the as-prepared NCSe@NMO@rGO–NF core–shell structure were analyzed by N2 adsorption and desorption experiments using Brunauer–Emmett–Teller (BET) analysis, as shown in Fig. 4i. The isotherm of the NCSe@NMO@rGO–NF nanostructure shows a typical type IV curve with notable hysteresis loops, suggesting the presence of an abundance of mesopores on the surface of the core–shell structure.43 The PSD curve (inset of Fig. 4i) further demonstrates the presence of ample mesopores with a pore size of 2–30 nm, which are highly beneficial for the diffusion of electrolyte ions during the redox reaction. The BET surface area of the as-prepared NCSe@NMO@rGO–NF core–shell structure was measured as 114 m2 g−1. The high specific surface area of the NCSe@NMO@rGO–NF nanostructure shows copious electrochemical active sites that promote ion-electron transportation, ensuring the superb electrochemical performance of the material. Also, the pore volume of the NCSe@NMO@rGO–NF core–shell structure was evaluated using the Barrett–Joyner–Halenda method to be 0.42 cm3 g−1. The presence of such large pore volume provides a high specific surface area for the construction of a hierarchical NCSe@NMO@rGO–NF core–shell structure. The mesoporous features of the NCSe@NMO@rGO–NF core–shell with a high specific surface area and pore volume mean that the material exhibits superb electrochemical features, making it a strong candidate as an electrode material for use in SC applications.
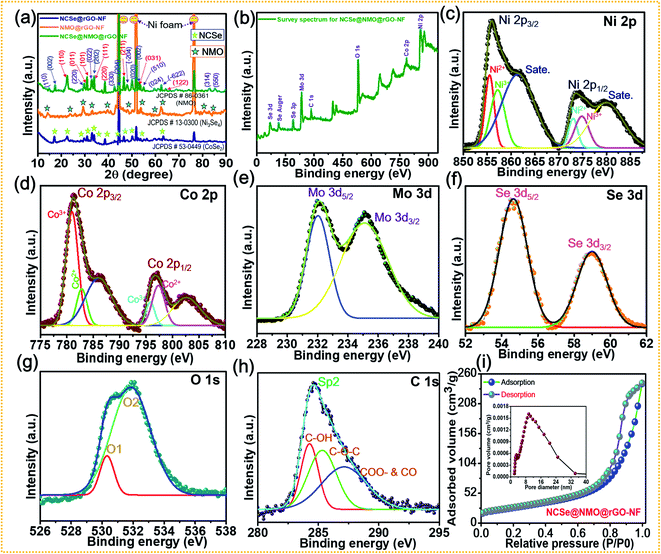 |
| | Fig. 4 (a) XRD patterns of NCSe@rGO–NF, NMO@rGO–NF, and NCSe@NMO@rGO–NF. (b) XPS survey spectra of NCSe@NMO@rGO–NF. Core-level spectra of various elements: (c) Ni 2p, (d) Co 2p, (e) Mo 3d, (f) Se 3d, (g) O 1s, (h) C 1s in NCSe@NMO@rGO–NF. (i) BET analysis showing a typical N2 adsorption–desorption isotherm and pore size distribution curve (inset) of the NCSe@NMO@rGO–NF core–shell structure. | |
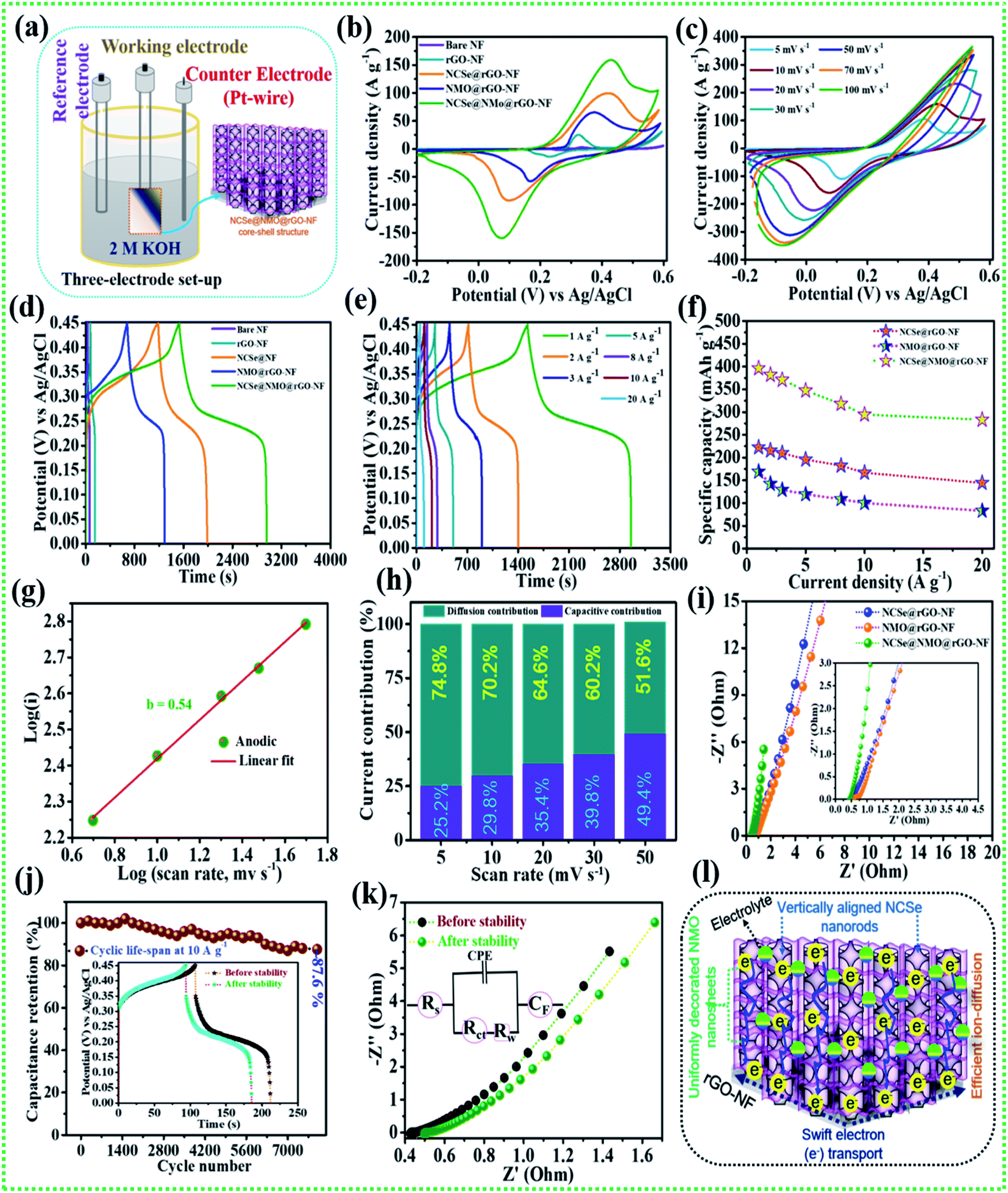
The comparative electrochemical properties of as-synthesized electrode materials, including bare NF, rGO–NF, Ni–Co–Se@rGO–NF, NMO@rGO–NF, and NCSe@NMO@rGO–NF, were evaluated by discerning the cyclic voltammetry (CV), galvanostatic charge discharge (GCD) and electrochemical impedance spectroscopy (EIS) data in a conventional three-electrode set-up. As depicted in Fig. 5a, the three-electrode set-up consists of Ag/AgCl, the as-prepared electrode and platinum wire as reference, working and counter electrodes, respectively. CV profiles of the bare NF, rGO–NF and as-prepared NCSe@rGO–NF, NMO@rGO–NF, NCSe@NMO@rGO–NF electrodes were recorded at a constant rate of 10 mV s−1 by sweeping the potential range of −0.2 to 0.6 V. The CV profiles of bare NF and rGO–NF clearly demonstrated their roles in enhancing the electrochemical performance of the as-fabricated hierarchical core–shell structure. The deposition of rGO enhances the conductivity of the 3D NF, which was further endorsed from the Nyquist plot of bare NF and the rGO–NF (Fig. S3†). Importantly, all of the CV curves display a pair of well-defined redox peaks in the forward and reverse sweeps, conveying strong evidence of the battery-type features of the as-prepared materials. Instead, a distinct difference in the CV area and intensity of the redox peaks in the CV profiles of various electrodes (NCSe@rGO–NF, NMO@rGO–NF, and NCSe@NMO@rGO–NF) can be apprehended from Fig. 5b. In particular, the NCSe@NMO@rGO–NF electrode shows dominant redox peaks and an optimal current response compared to NCSe@rGO–NF and NMO@rGO–NF. This might be attributed to the presence of more electroactive sites and charge storage capacity in the hybrid core–shell structures of these materials, tendered by the synergistic effect of the core (NCSe@rGO–NF) and shell (NMO@rGO–NF) materials in a single framework. Fig. 5c depicts the CV curves of the NCSe@NMO@rGO–NF electrode recorded at different scan rates (5 to 100 mV s−1). As predicted, a pair of noticeable redox peaks in the forward and reverse sweeps were observed, suggesting that the charge storage performance in the electrode is primarily governed by a battery-type mechanism. Additionally, on increasing the scan rate, the anodic and cathodic peaks linearly increase in the forward and reverse directions without apparent distortion, revealing the polarization effect, good reversibility and enhanced rate performance of the NCSe@NMO@rGO–NF electrode. The battery-type mechanism tendered by the faradaic reactions in the hybrid core–shell electrode can be expressed as follows.56,57
| | | (Ni, Co)Se2 + 2OH− ↔ NiSeOH + CoSeOH + 2e− | (5) |
| | | NiSeOH + 2OH− ↔ NiSeO + H2O + e− | (6) |
| | | CoSeOH + 2OH− ↔ CoSeO + H2O + e− | (7) |
| | | NiMoO4 + OH− ↔ NiMoO4OH + e− | (8) |
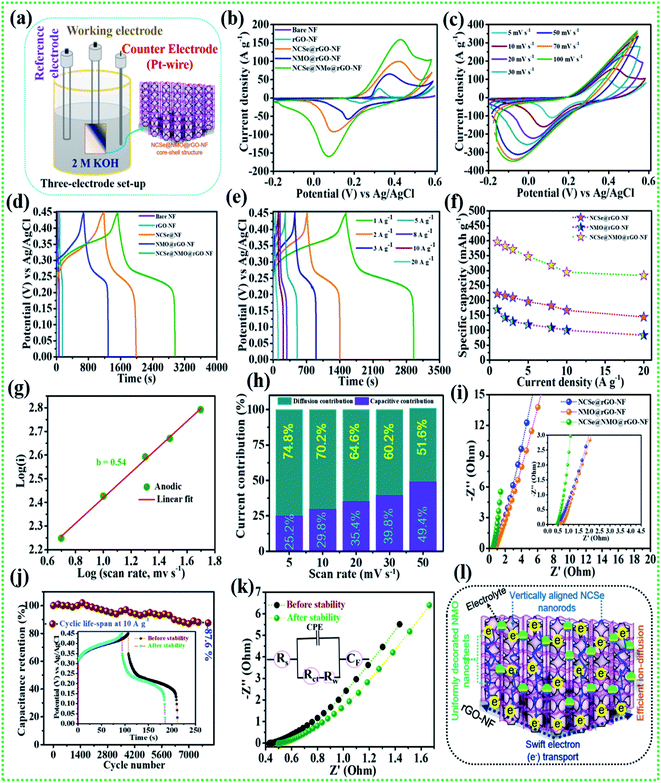 |
| | Fig. 5 (a) Schematic representation of the three-electrode system, (b) CV performance of various electrodes at a scan rate of 10 mV s−1, (c) CV performance of the NCSe@NMO@rGO–NF at different scan rates, (d) GCD performance of various electrodes at a current density of 1 A g−1, (e) GCD performance of NCSe@NMO@rGO–NF at different current densities, (f) rate capability curves, (g) relationship between log(i) vs. log(v) of the NCSe@NMO@rGO–NF electrode, (h) diffusion and capacitive current contributions of the NCSe@NMO@rGO–NF electrode, (i) EIS curves of the as-fabricated various electrodes, (j) cycling performance of the NCSe@NMO@rGO–NF electrode at a current density of 1 A g−1, (k) EIS in the form of a Nyquist plot of the NCSe@NMO@rGO–NF electrode before and after cycling, and (l) schematic representation of the electrochemical mechanism of the NCSe@NMO@rGO–NF electrode. | |
Similarly, the comparative CV curves of NCSe@rGO–NF and NMO@rGO–NF are displayed in Fig. S4a and S5a,† respectively, divulging the pseudocapacitive features of the electrode materials. The comparative GCD measurement of various electrodes at a current density of 1 A g−1 are presented in Fig. 5c. Analogous with the CV results, the discharge time of the rGO–NF is longer than that of the bare NF, thanks to the conductivity of the chemically reduced rGO sheets. As depicted in Fig. 5b, the NCSe@NMO@rGO–NF electrode exhibits the longest discharge time compared to NCSe@rGO–NF, and NMO@rGO–NF, suggesting that the former electrode accumulates plentiful charge and delivers optimal capacity. This might be attributed to the participation of the copious electroactive sites in reversible faradaic reactions on the surface of the hybrid core–shell electrode with electrolyte ions. Based on eqn (1), the maximum specific capacity of the NCSe@NMO@rGO–NF electrode was measured as 396.1 mA h g−1 at a current density of 1 A g−1, which is much higher than those of NCSe@rGO–NF (222.5 mA h g−1) and NMO@rGO–NF (169.1 mA h g−1) at the identical current density. Fig. 5d shows the GCD plot of the NCSe@NMO@rGO–NF electrode at various current densities of 1 to 20 A g−1. On enhancing the current density, the discharge time is shortened without much alteration in the shape of the GCD curve, reflected in the admirable rate performance and great reversibility of the hybrid core–shell electrode. The GCD profiles of individual NCSe@rGO–NF, and NMO@rGO–NF materials are displayed in Fig. S4b and S5b,† respectively. The observed specific capacity values of the NCSe@NMO@rGO–NF electrode are 396.1, 381.6, 370.8, 347.2, 317.7, 294.4, and 283.3 mA h g−1, corresponding to current densities of 1, 2, 3, 5, 8, 10, and 20 A g−1, respectively. Similarly, the specific capacities of NCSe@rGO–NF, and NMO@rGO–NF were calculated and are shown in Fig. 5e. The NCSe@NMO@rGO–NF hybrid core–shell electrode retained 71.5% of its specific capacity, even at a high current density of 20 A g−1, corroborating its excellent rate performance compared to the NCSe@rGO–NF (64.8%) and NMO@rGO–NF (49.2%) electrodes. The charge storage phenomenon of the NCSe@NMO@rGO–NF electrode was scrutinized using the power law:58
where
i represents the peak current (A),
v signifies the scan rate (mV s
−1), and
a and
b are adaptable parameters. The charge storage mechanism of the electrode can be evaluated by estimating the
b value. Values of
b close to 0.5 and 1 signify battery-type and capacitive mechanisms, respectively. The value of
b can be obtained by plotting the log(peak current)
versus the log(scan rate). The value of
b for the anodic graph was evaluated to be 0.54, showing the battery-type behavior of the NCSe@NMO@rGO–NF electrode (
Fig. 5g). Furthermore, the power law and its modified versions were employed to reveal the exact battery-type and capacitive type contributions:
43By plotting
i/
v1/2versus v1/2, the deductible parameters,
k1 and
k2, can be obtained from the slope of the linearly fitted line and
Y-axis intercept, respectively. The obtained
k1 and
k2 values can be calculated using
eqn (5) or
(6) to evaluate the diffusive (
k2v) and capacitive (
k1v) contributions, with the results shown in
Fig. 5h. As can be inspected from the results, the diffusion-controlled current contribution is dominant at a lower scan rate with 74.8% contribution at 5 mV s
−1, indicating the deep and spontaneous ion-penetration on the surface of the electrode. Instead, the capacitive contribution is superior at a high scan rate, with 51.6% contribution at 50 mV s
−1. Thus, it can be emphasized that the capacitive contribution increases with an enhancement in the scan rate, promoting the sectional interaction of electrolytic ions towards the surface of the electrode. In order to gain further insights, including electro-kinetics and electrochemical conductivity, EIS analysis was carried out for the as-prepared various electrodes by employing a perturbed potential of 5 mV within a frequency range of 0.01–100 kHz.
Fig. 5i (inset shows magnified view) shows the Nyquist plots of the NCSe@rGO–NF, NMO@rGO–NF, and NCSe@NMO@rGO–NF electrodes. The Nyquist plot shows two primary regions, including a linear portion in the low-frequency region and semicircular portion in the high-frequency region.
59,60 The point of intersection in the high-frequency region of the Nyquist plot signifies the solution resistance (
Rs), which corresponds to the contact resistance of the electrode interfaces and intrinsic resistance of the electrolyte. Similarly, the semicircle in the high-frequency region represents the charge transfer resistance (
Rct), which is associated with electronic resistance induced
via the electrode/electrolyte interface. The
Rs andRct values of the NCSe@NMO@rGO–NF electrode were evaluated to be 0.39 and 0.07 Ω, which are much lower than those of the NCSe@rGO–NF (
Rs = 0.47 Ω,
Rct = 0.12 Ω) and NMO@rGO–NF (
Rs = 0.54 Ω,
Rct = 0.22 Ω) electrodes, promoting rapid electron transfer between the electrode and electrolyte materials as a result of the better electrochemical activity of the hybrid core–shell electrode. Also, the vertical line of the NCSe@NMO@rGO–NF electrode appears to be steeper than those of the NCSe@rGO–NF and NMO@rGO–NF electrodes, suggesting ultra-fast electron transportation and feasible ion penetration on the surface of the electrode.
Cycling performance is one of the imperative parameters to evaluate the feasibility of the fabricated electrode materials to determine their practical implementation. Accordingly, cycling tests were carried out on the NCSe@NMO@rGO–NF electrode at a fixed current density of 10 A g−1. As displayed in Fig. 5j (inset depicts the GCD curve before and after the cycling tests), the NCSe@NMO@rGO–NF electrode shows 87.6% capacity retention after 8000 consecutive charge–discharge cycles. Fig. 5k demonstrates the EIS results before and after the cycling tests in the form of a Nyquist plot (the inset depicts the equivalent circuit diagram derived from EIS analysis). As depicted in the figure, there were no obvious changes in the Rs and Rct values after the cycling tests, which could be attributed to the fabrication of binder-free electrode materials that eradicate “dead mass”, leading to ameliorated electronic conductivity and swift charge transportation. Furthermore, the morphology of the NCSe@NMO@rGO–NF electrode after successive 8000 GCD cycles is displayed in Fig. S6.† As shown in the figure, there are no obvious morphological changes and the NMO nanosheets are still firmly attached to the NCSe nanorods, manifesting in the robust mechanical strength and remarkable structural stability of the electrode. This result suggests that the NMO nanosheets act as a protective shell layer on the surface of the porous Ni–Co–Se nanorods to achieve a hierarchical hybrid NCSe@NMO@rGO–NF core–shell nanoarchitecture. Fig. 5l shows a schematic diagram of the hierarchical NCSe@NMO@rGO–NF hybrid core–shell electrode with a lucid demonstration of its morphological features. Importantly, the integration of the 1D nanorods with the 2D nanosheets with enhanced porous features in a single framework provides ample reaction sites and accessible ion-electron transport roadways, resulting in amplified electrochemical kinetics and superlative capacitive performance. The 1D Ni–Co–Se nanorods with enhanced porosity serve as the swift channel for the transport of electrons, thereby improving the specific capacity of the electrode materials. The well-garnished shell material (NiMoO4 nanosheets) with open spaces enables facile penetration and easy diffusion of electrolyte ions into the surface of the electrode, which help to amplify the overall specific capacity and reinforce the structural durability during the long-term charge–discharge cycling of the electrode. Additionally, the coexistence of NCSe and NMO with multiple electronic states furnishes synergistic redox reactions for superlative electrochemical properties. Furthermore, the 3D rGO–NF eradicates the use of an organic binder, which curtails dead mass, providing minimal interfacial resistance. Meanwhile, the embellishing of rGO is beneficial to ameliorating the electronic conductivity and structural stability of the hybrid core–shell structure. Endowed with all of the above features, the NCSe@NMO@rGO–NF electrode exhibits excellent capacity performance, which is superior to those of previously reported metal selenide-based active materials, as illustrated in Table 1.
Table 1 Comparison of the specific capacity performance of the NCSe@NMO@rGO–NF electrode with those of various previously reported core–shell electrodes
| Active electrode materials |
Current collector |
Electrolyte |
Testing condition |
Mass loading (mg cm−2) |
Specific capacity (mA h g−1) |
Ref. |
| (Ni,Co)Se2/Ni–Co-LDH core–shell |
Carbon cloth |
3 M KOH |
2 A g−1 |
— |
∼170 |
61
|
| Hierarchical NiCu(OH)2@Ni–Cu–Se nanoparticles |
Ni foam |
1 M KOH |
1 A g−1 |
∼8.4 |
158.95 |
62
|
| ZnO/C@(Ni–Co)Se2 core–shell |
Carbon cloth |
6 M KOH |
1 A g−1 |
∼3 to 3.2 |
164.18 |
63
|
| CuCo2S4@Co(OH)2 core–shell nanoarrays |
Ni foam |
6 M KOH |
1 A g−1 |
∼2 |
258 |
64
|
| Ni–Mn–Co–S@Co(OH)2 core–shell nanoarrays |
Ni foam |
6 M KOH |
2 A g−1 |
∼2.8 |
284.85 |
65
|
| FeCo2S4@Ni(OH)2 core–shell |
Carbon paper |
6 M KOH |
1 A g−1 |
∼1.2 |
239.72 |
66
|
| Mo–NiS2@Ni–Co-LDH nanosheet to nanosheet arrays |
Nickel foam |
6 M KOH |
1 A g−1 |
2.35 |
325.6 |
67
|
| CoFeP@nickel–manganese sulfide core–shell nanoarrays |
Carbon cloth |
2 M KOH |
1 A g−1 |
3.3 |
260.7 |
68
|
| 3D NS@NCCH tree-like core–shell |
Carbon cloth |
1 M KOH |
4 mA cm−2 |
— |
228 |
69
|
|
Hierarchical NCSe@NMO@rGO–NF core–shell electrode
|
Ni foam
|
2 M KOH
|
1 A g
−1
|
2.3
|
396.1
|
This work
|
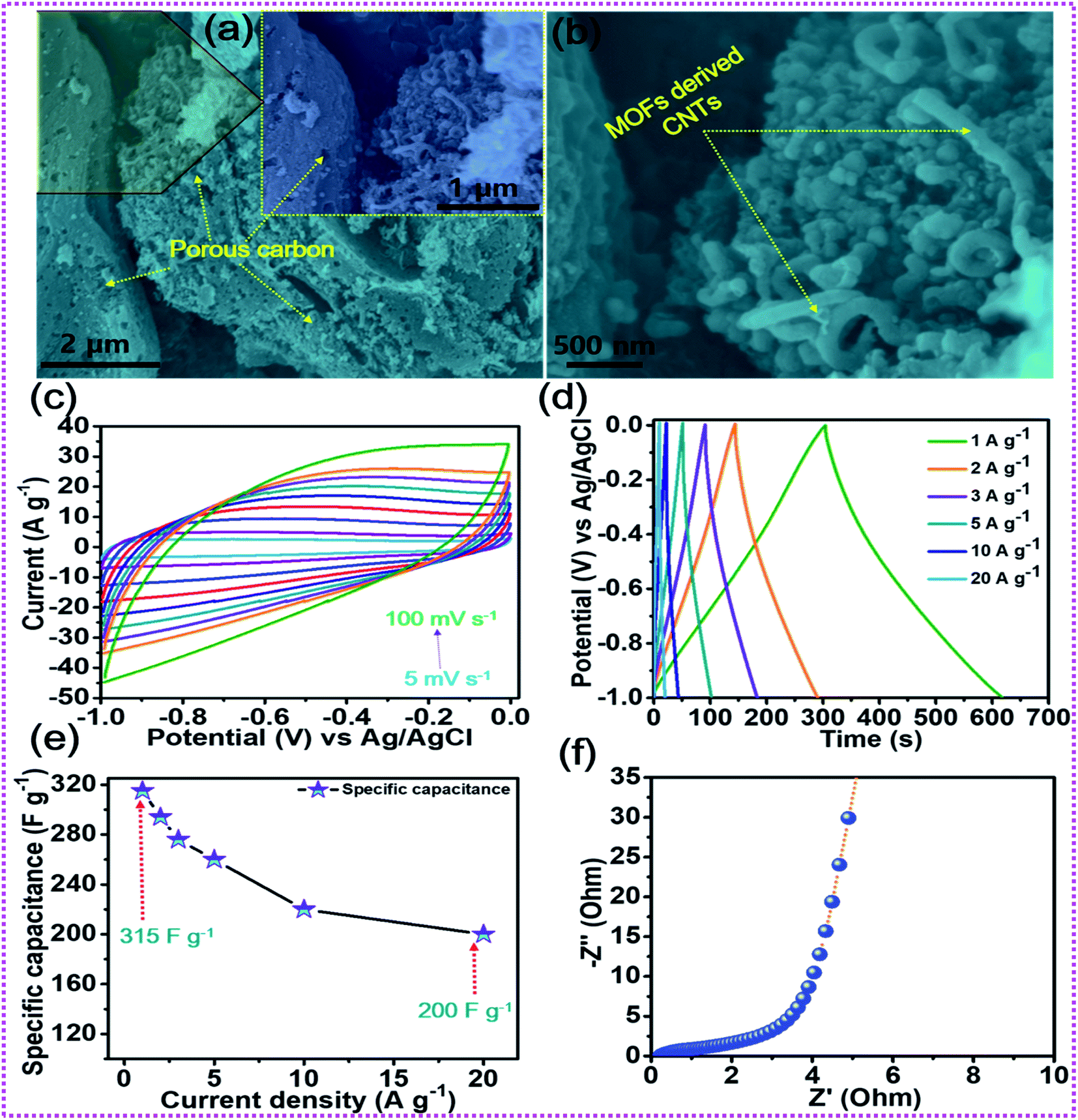
After the elaboration of the positive electrode, the in-depth electrochemical characterization of the oxalic acid and MOF-derived porous carbon decorated with carbon nanotubes (OA-MOF-PC-CNTs) was performed and employed as a negative electrode. The surface morphology of the OA-MOF-PC-CNTs (Fig. 6a and b) exhibits enhanced porous features with good decoration of conductive CNTs. Such a porous nanoarchitecture with ample hierarchical pores, high specific surface area (SSA) and conductivity provides a facile pathway for the spontaneous diffusion and transport of electrolytic ions throughout the electrode, thereby providing a consecutive conductive network for rapid expeditious electron transfer.70,71 As a result of these advantages, the OA-MOF-PC-CNT electrode delivers superlative electrochemical performance in terms of high specific capacitance as well as exceptional rate capability, which makes it an auspicious candidate as a negative electrode for the assembly of a high-performance HSC. Fig. 6c depicts the CV analysis of the OA-MOF-PC-CNTs under various scan rates in the range of 5–100 mV s−1, which clearly demonstrates the electric double layer (EDLC) behavior, as evident from the parallelogram appearance of the CV curves. Even when the scan rate was enhanced by 20 times, there were no changes in the nature of the CV curves, suggesting the great reversibility and excellent rate performance of the electrode. The GCD tests of the OA-MOF-PC-CNT electrode at various current densities in the range of 1–20 A g−1 are displayed in Fig. 6d. The symmetric and linear shape of the GCD curves, even at a high current density, provides supplemental evidence for the high rate performance and great reversibility of the as-fabricated electrode. The specific capacitance (CSP) of the OA-MOF-PC-CNTs electrode was calculated using eqn (2) and the values are displayed in Fig. 6e. The maximum CSP value of the OA-MOF-PC-CNTs electrode of 315 F g−1 was achieved at a current density of 1 A g−1, together with a capacitance retention of 63.49% of its initial capacitance, even when the current density was increased by 20 fold. The EIS testing of the OA-MOF-PC-CNT electrode in the form of Nyquist plot is presented in Fig. 6f. The internal resistance (Rs) and charge transfer resistance (Rct) values of the OA-MOF-PC-CNT electrode were evaluated to be 0.18 and 2.73 Ω, respectively. These results certainly prove that the as-fabricated OA-MOF-PC-CNT electrode is a great contender as a negative electrode for the assembly of a HSC device.
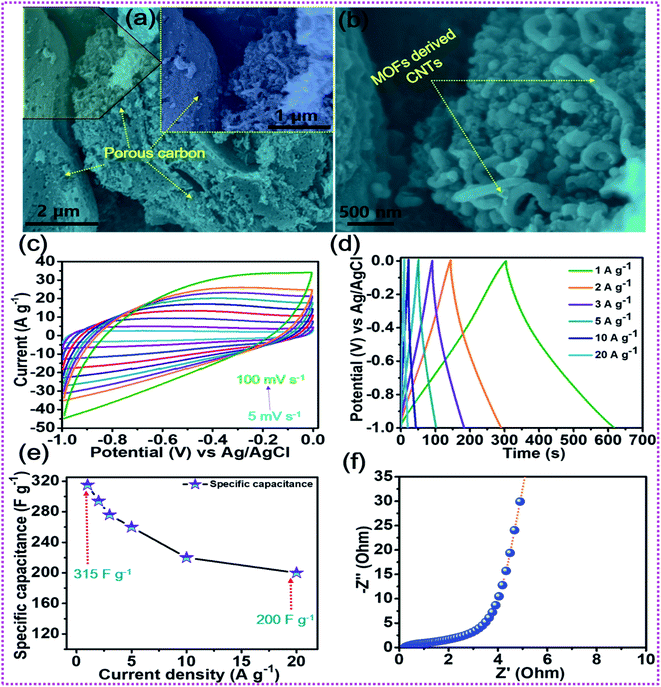 |
| | Fig. 6 (a, b) FESEM images of the OA-MOF-PC-CNTs, (c) CV tests, (d) GCD tests, (e) rate capability curves, and (f) EIS tests of the OA-MOF-PC-CNTs. | |
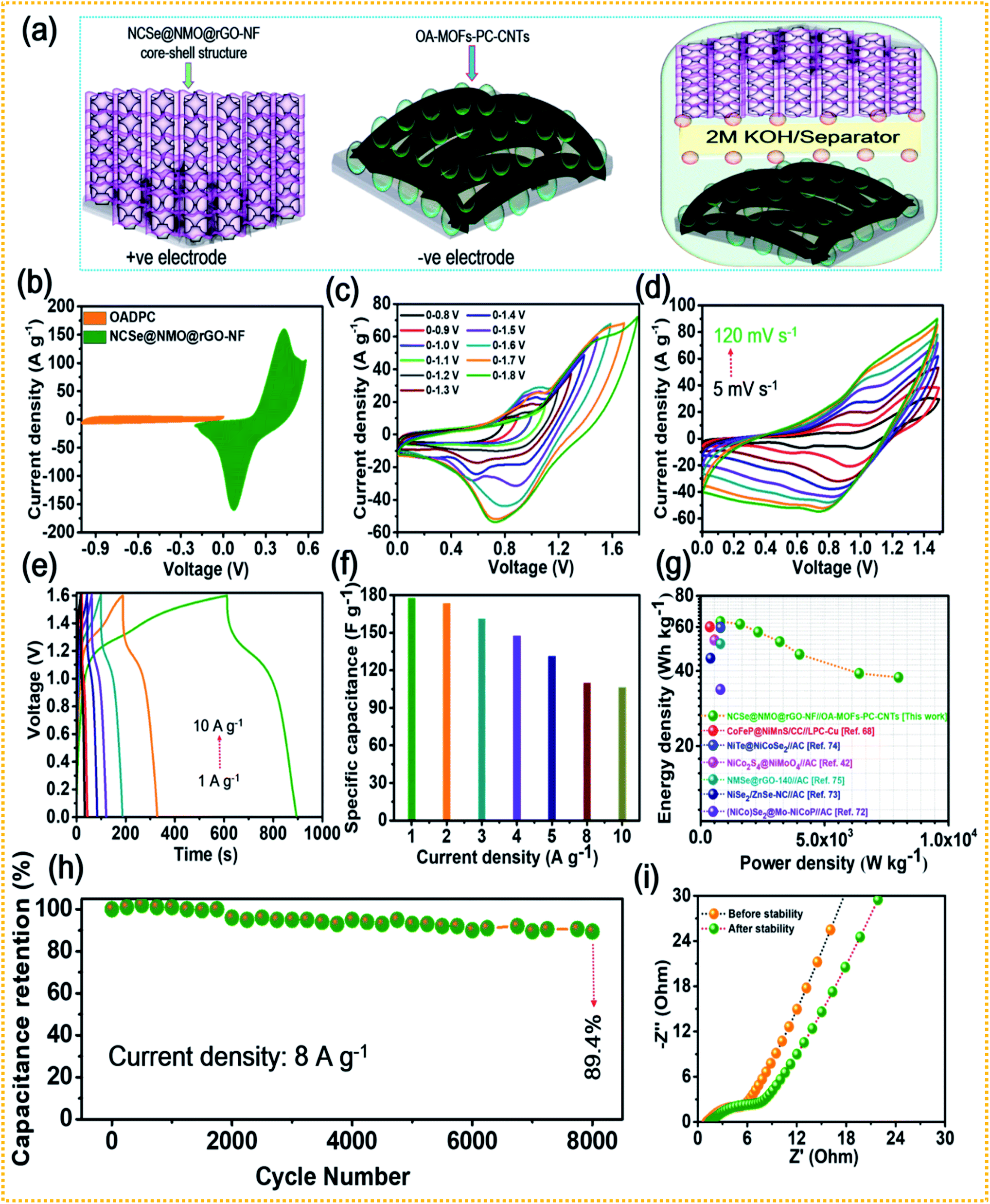
The combination of battery-type and EDLC-type electrodes to prepare sandwich HSCs is considered to be an effective approach to widen the voltage range of the device, thereby enhancing its energy density without compromising its power density and cycling life span. Thus, a HSC device was assembled using battery-type NCSe@NMO@rGO–NF (positive electrode) and EDLC type OA-MOF-PC-CNTs@NF (negative electrode) materials, with cellulose membrane (CHMLAB GROPU, grade F1001, medium size, weight: 85 g m−2, thickness of 180 μm, retention range of 10–13 μm, ASH content of <0.06%) employed as a separator and 2 M KOH used as the electrolyte. The two as-prepared electrodes and separator were soaked in electrolyte before the construction of the HSC device. A schematic presentation of the HSC device is displayed in Fig. 7a. The corresponding mass balancing of the active material plays a vital role in enhancing the performance of the HSC device. Hence, the mass ratio of the positive and negative electrodes was optimized using the following equation:2
| | | m+/m− = C−SP × ΔV−/Q+ | (12) |
where
m+ and
Q+ represent the mass and specific capacity of the positive electrode, and Δ
V−,
m− and
C−SP signify the working voltage, mass and specific capacitance of the negative electrode, respectively. After corresponding mass balancing, the approximated mass loading ratio of the positive to negative electrode was evaluated to be 0.79. Meanwhile, a total mass of the electrode of 5.2 mg was achieved during the construction of the HSC device. The CV curves of the positive (NCSe@NMO@rGO–NF) and negative (OA-MOF-PC-CNTs) electrodes at a scan rate of 10 mV s
−1 are displayed in
Fig. 7b. To validate the working voltage of the HSC device, CV curves recorded at various potentials at a constant scan rate (10 mV s
−1) are displayed in
Fig. 7c. When the potential was increased from 0 to 0.8 V and 0 to 1.6 V, there was no appearance of any remarkable hydrogen (H
2)/oxygen (O
2) evolution reaction, manifesting the better electrochemical operation stability and optimal voltage window of the HSC device when set to 1.6 V. Instead, beyond the voltage limit of 1.6 V, there is sudden rise in current suggesting the occurrence of the O
2 evolution reaction. Owing to these results, the suitable potential window for assembled HSC device was selected to be 1.6 V, and the electrochemical characterization was performed accordingly.
Fig. 7d shows the CV curves of the HSC device recorded at various scan rates of 5 to 100 mV s
−1. It can be validated from the figure that the HSC device exhibits a faradaic nature, even at a high scan rate, without any distortion in the nature of the CV curve, suggesting the good electrochemical reversibility and excellent rate performance of the HSC device.
Fig. 7e presents the GCD profiles of the HSC device recorded at various current densities at a fixed voltage of 1.6 V. As displayed in the figure, even at a higher current density, the HSC device displays a non-linear GCD curve, exhibiting its reversible charge-storage properties. The specific capacitance (
CSP) values of the HSC device at different current densities were calculated using
eqn (2) and the results are shown in
Fig. 7f. The obtained
CSP values for the HSC device are 177.5, 173.5, 161.2, 147.5, 131.2, 110, and 106.2 F g
−1 at applied current densities of 1, 2, 3, 4, 5, 8, and 10 A g
−1, respectively. The energy and power densities of the HSC device were calculated using
eqn (3) and
(4), respectively.
Fig. 7g displays the relationship between the energy and power densities in the form of a Ragone plot. The HSC device exhibited an energy density of 63.1 W h kg
−1 at a power density of 799.8 W kg
−1. Furthermore, the HSC device delivered a good energy density of 37.7 W h kg
−1, even at a high current density of 10 A g
−1, with an optimal power density of 7983.5 W kg
−1. The energy and power densities of the proposed HSC device were comparable and even higher than the values of previously reported transition metal chalcogenide-based supercapacitors.
42,68,72–75 As a pivotal parameter in measuring its practical applicability, long-term cycling tests were performed on the NCSe@NMO@rGO–NF//OA_MOFs-PC-CNTs HSC device.
Fig. 7h shows the cycling tests of the HSC device at a current density of 8 A g
−1 for up to 8000 consecutive GCD cycles. The HSC device exhibited good stability, with 89.4% capacitance retention after 8000 GCD cycles. EIS tests were further performed for the HSC device and the resultant Nyquist plot is shown in
Fig. 7i. Before the cycling stability tests, the
Rs and
Rct values of 1.1 and 3.6 Ω were measured from the EIS plots of the device, respectively. However, after the cycling performance, there were slight changes in the
Rs and
Rct values, suggesting the good electrochemical conductivity of the HSC device, even after 8000 consecutive GCD cycles.
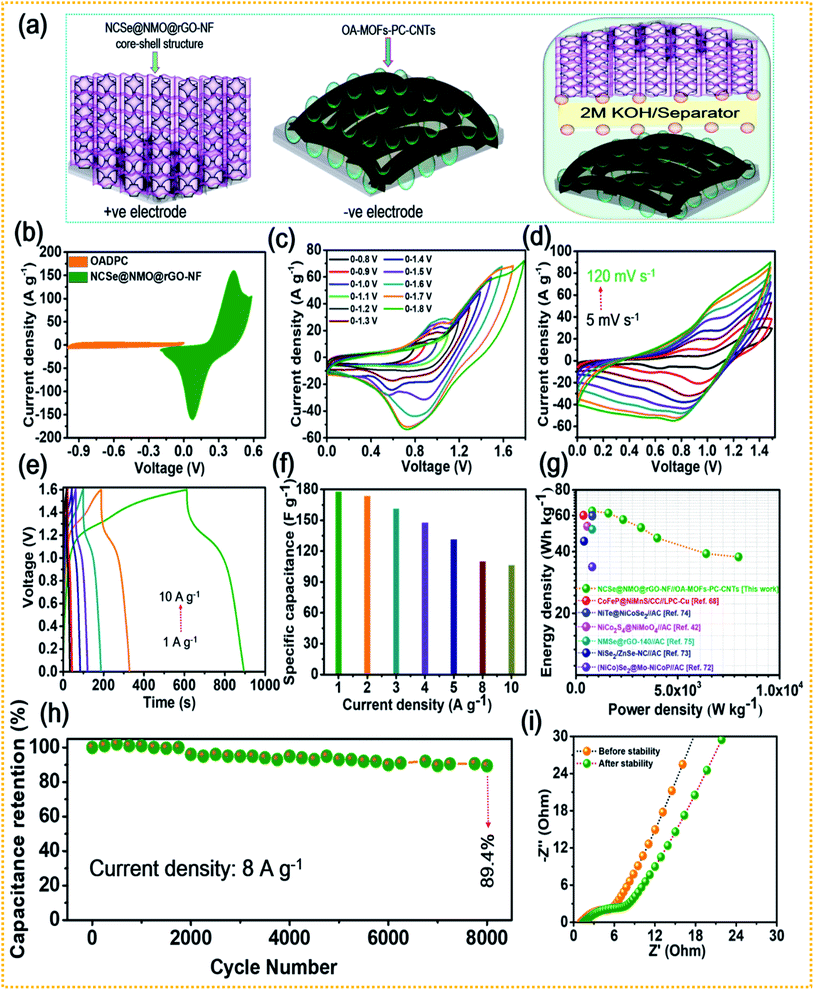 |
| | Fig. 7 (a) Schematic representation of the HSC device, (b) CV curves of the NCSe@NMO@rGO–NF and OA-MOF-PC-CNTs electrodes, (c) CV curves of the HSC device at different potentials, (d) CV curves recorded at different scan rates, (e) GCD curves recorded at various current densities, (f) rate capability data of the HSC device, (g) Ragone plot showing the energy and power densities of the HSC device, (h) cycling stability of the HSC, and (i) EIS curves of the HSC device tested before and after the cycling stability tests. | |
4. Conclusions
In summary, a hierarchical hybrid core–shell structure NCSe@NMO@rGO–NF was fabricated via an oxalic acid templated selenization process, followed by hydrothermal technique to limit “dead mass”, which was subsequently employed as a battery-type cathode material in a HSC. The absolute phase and optimal purity of NCSe@NMO@rGO–NF were analyzed via structural and compositional scrutiny. The successful growth of vertically aligned NCSe nanorods on rGO–NF and NMO decoration to achieve a NCSe@NMO@rGO–NF core–shell structure were clearly demonstrated from surface morphological analysis. Additionally, the uniform distribution of the NMO nanosheets over the porous surface of NCSe nanorods to achieve a hierarchical structure was also realized. The in-depth electrochemical investigations of the prepared electrode revealed that the NCSe@NMO@rGO–NF electrode exhibits a high specific capacity of 396.1 mA h g−1 at a current density of 1 A g−1. An identical electrode shows superb stability, with 87.6% capacity retention after 8000 consecutive GCD cycles. The HSC device was sandwiched with battery-type NCSe@NMO@rGO–NF (positive electrode) and EDLC type OA-MOF-PC-CNTs@NF (negative electrode) materials in 2 M KOH electrolyte. The as-assembled HSC device delivers a high energy density of 63.1 W h kg−1 and a high power density of 7983.5 W kg−1 at current densities of 1 and 10 A g−1, respectively. Moreover, 89.4% capacity of the HSC device was retained after 8000 GCD cycles. As a result of this study, it is anticipated that the nano-hybridization of a redox-enriched metal chalcogenide-metal oxide based nanoarchitecture directly on a carbonaceous decorated current collector using a cost-effective protocol could amplify the energy storage performance of electrochemical systems.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This research was supported by the Traditional Culture Convergence Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT and Future Planning (2018 M3C1B5052283). This work was also supported by the National Research Foundation of Korea (NRF) grant funded by the Korean Government (MSIT) (No. NRF-2019R1A2C1004467).
References
- G. Nagaraju, S. C. Sekhar, B. Ramulu and J. S. Yu, Energy Storage Materials, 2021, 35, 750–760 CrossRef.
- G. Nagaraju, S. C. Sekhar and J. S. Yu, Adv. Energy Mater., 2018, 8, 1702201 CrossRef.
- Z. Hao, X. He, H. Li, D. Trefilov, Y. Song, Y. Li, X. Fu, Y. Cui, S. Tang, H. Ge and Y. Chen, ACS Nano, 2020, 14, 12719–12731 CrossRef CAS PubMed.
- S. K. Hussain, G. Nagaraju, S. Chandra Sekhar and J. S. Yu, Energy Storage Materials, 2020, 27, 405–417 CrossRef.
- L. Wan, Y. Wang, Y. Zhang, C. Du, J. Chen, M. Xie, Z. Tian and W. Zhang, J. Power Sources, 2021, 506, 230096 CrossRef CAS.
- S. C. Sekhar, B. Ramulu, S. J. Arbaz, S. K. Hussain and J. S. Yu, Small Methods, 2021, 5, 2100335 CrossRef CAS PubMed.
- L. Naderi and S. Shahrokhian, Chem. Eng. J., 2020, 392, 124880 CrossRef CAS.
- L. Chen, Q. Zhang, H. Xu, X. Hou, L. Xuan, Y. Jiang and Y. Yuan, J. Mater. Chem. A, 2015, 3, 1847–1852 RSC.
- B. Huang, W. Wang, T. Pu, J. Li, C. Zhao, L. Xie and L. Chen, Chem. Eng. J., 2019, 375, 121969 CrossRef CAS.
- Y. Jiang, L. Chen, H. Zhang, Q. Zhang, W. Chen, J. Zhu and D. Song, Chem. Eng. J., 2016, 292, 1–12 CrossRef CAS.
- S. Liang, H. Wang, Y. Li, H. Qin, Z. Luo and L. Chen, Appl. Surf. Sci., 2021, 567, 150809 CrossRef CAS.
- J. Zhu, D. Song, T. Pu, J. Li, B. Huang, W. Wang, C. Zhao, L. Xie and L. Chen, Chem. Eng. J., 2018, 336, 679–689 CrossRef CAS.
- J. Y. Hwang, M. Li, M. F. El-Kady and R. B. Kaner, Adv. Funct. Mater., 2017, 27, 1605745 CrossRef.
- C. Miao, C. Zhou, H.-E. Wang, K. Zhu, K. Ye, Q. Wang, J. Yan, D. Cao, N. Li and G. Wang, J. Power Sources, 2021, 490, 229532 CrossRef CAS.
- H. Tian, K. Zhu, W. Li, L. Wang, Z. Yu, Y. Lai and Y. He, Electrochim. Acta, 2021, 377, 138087 CrossRef CAS.
- Y. Zeng, J. Liao, B. Wei, Z. Huang, W. Zhu, J. Zheng, H. Liang, Y. Zhang and Z. Wang, Chem. Eng. J., 2021, 409, 128297 CrossRef CAS.
- J. Ren, M. Shen, Z. Li, C. Yang, Y. Liang, H.-E. Wang, J. Li, N. Li and D. Qian, J. Power Sources, 2021, 501, 230003 CrossRef CAS.
- Y. Ren, T. Zhu, Y. Liu, Q. Liu and Q. Yan, Small, 2021, 17, 2008047 CrossRef CAS PubMed.
- J. Zhao, G. Wang, K. Cheng, K. Ye, K. Zhu, J. Yan, D. Cao and H.-E. Wang, J. Power Sources, 2020, 451, 227737 CrossRef CAS.
- T. Zhu, Z. He, Y. Ren, W. Zeng, J. Mao and L. Zhu, Sol. RRL, 2021, 5, 2100021 CrossRef CAS.
- T. Zhu, Y. Xiao, Y. Ren, W. Zeng, A. Pan, Y. Zheng and Q. Liu, ACS Appl. Energy Mater., 2021, 4, 2976–2982 CrossRef CAS.
- H. Liu, X. Liu, S. Wang, H.-K. Liu and L. Li, Energy Storage Materials, 2020, 28, 122–145 CrossRef.
- H. Hosseini and M. Roushani, Chem. Eng. J., 2020, 402, 126174 CrossRef CAS.
- A. Sobhani and M. Salavati-Niasari, Adv. Colloid Interface Sci., 2021, 287, 102321 CrossRef CAS PubMed.
- Z. Tian, Z. Zhao, X. Wang, Y. Chen, D. Li, Y. Linghu, Y. Wang and C. Wang, Nanoscale, 2021, 13, 6489–6498 RSC.
- L. Zhang, H. Dong, H. Wei, E. H. Ang, J. Yang, X. Miao, H. Geng and X. Zuo, J. Power Sources, 2021, 506, 230216 CrossRef CAS.
- Y. Wang, W. Zhang, X. Guo, K. Jin, Z. Chen, Y. Liu, L. Yin, L. Li, K. Yin, L. Sun and Y. Zhao, ACS Appl. Mater. Interfaces, 2019, 11, 7946–7953 CrossRef CAS PubMed.
- K. Xiao, L. Zhou, M. Shao and M. Wei, J. Mater. Chem. A, 2018, 6, 7585–7591 RSC.
- M. Sakthivel, R. Sukanya, S.-M. Chen, K. Pandi and K.-C. Ho, Renewable Energy, 2019, 138, 139–151 CrossRef CAS.
- T. T. Nguyen, J. Balamurugan, V. Aravindan, N. H. Kim and J. H. Lee, Chem. Mater., 2019, 31, 4490–4504 CrossRef CAS.
- X. Shi, H. Wang, S. Ji, V. Linkov, F. Liu and R. Wang, Chem. Eng. J., 2019, 364, 320–327 CrossRef CAS.
- X. Yun, T. Lu, R. Zhou, Z. Lu, J. Li and Y. Zhu, Chem. Eng. J., 2021, 426, 131328 CrossRef CAS.
- B. Zhao, L. Zhang, Q. Zhang, D. Chen, Y. Cheng, X. Deng, Y. Chen, R. Murphy, X. Xiong, B. Song, C.-P. Wong, M.-S. Wang and M. Liu, Adv. Energy Mater., 2018, 8, 1702247 CrossRef.
- Q. Zong, H. Yang, Q. Wang, Q. Zhang, Y. Zhu, H. Wang and Q. Shen, Chem. Eng. J., 2019, 361, 1–11 CrossRef CAS.
- B. Pant, G. P. Ojha, J. Acharya and M. Park, Int. J. Hydrogen Energy, 2021, 46, 2774–2782 CrossRef CAS.
- T. Xing, Y. Ouyang, Y. Chen, L. Zheng, H. Shu, C. Wu, B. Chang and X. Wang, Electrochim. Acta, 2020, 351, 136447 CrossRef CAS.
- Y. Feng, L. Liu, J. Liang, W. Yao, B. Tian, C. Jiang and W. Wu, J. Power Sources, 2019, 433, 126676 CrossRef CAS.
- S. Peng, L. Li, H. B. Wu, S. Madhavi and X. W. Lou, Adv. Energy Mater., 2015, 5, 1401172 CrossRef.
- C. Chen, S. Wang, X. Luo, W. Gao, G. Huang, Y. Zeng and Z. Zhu, J. Power Sources, 2019, 409, 112–122 CrossRef.
- J. Hong, Y.-W. Lee, D. Ahn, S. Pak, J. Lee, A. R. Jang, S. Lee, B. Hou, Y. Cho, S. M. Morris, H. S. Shin, S. Cha, J. I. Sohn and J. M. Kim, Nano Energy, 2017, 39, 337–345 CrossRef CAS.
- P. Bandyopadhyay, G. Saeed, N. H. Kim and J. H. Lee, Chem. Eng. J., 2020, 384, 123357 CrossRef CAS.
- S. Chen, Z. Zhang, W. Zeng, J. Chen and L. Deng, ChemElectroChem, 2019, 6, 590–597 CrossRef CAS.
- J. Acharya, B. Pant, G. P. Ojha, H.-S. Kong and M. Park, J. Colloid Interface Sci., 2021, 602, 573–589 CrossRef CAS PubMed.
- C. Xu, X. Shi, A. Ji, L. Shi, C. Zhou and Y. Cui, PLoS One, 2015, 10, e0144842 CrossRef PubMed.
- K. K. H. De Silva, H. H. Huang, R. K. Joshi and M. Yoshimura, Carbon, 2017, 119, 190–199 CrossRef CAS.
- X. Chen, J. Ding, Y. Li, Y. Wu, G. Zhuang, C. Zhang, Z. Zhang, C. Zhu and P. Yang, RSC Adv., 2018, 8, 26047–26055 RSC.
- C. Wang, Z. Song, H. Wan, X. Chen, Q. Tan, Y. Gan, P. Liang, J. Zhang, H. Wang, Y. Wang, X. Peng, P. A. van Aken and H. Wang, Chem. Eng. J., 2020, 400, 125955 CrossRef CAS.
- Y. Liu, Z. Ma, N. Xin, Y. Ying and W. Shi, J. Colloid Interface Sci., 2021, 601, 793–802 CrossRef CAS PubMed.
- J. Ding, H. Xu and X. Chen, RSC Adv., 2018, 8, 28710–28715 RSC.
- C. Chen, D. Yan, X. Luo, W. Gao, G. Huang, Z. Han, Y. Zeng and Z. Zhu, ACS Appl. Mater. Interfaces, 2018, 10, 4662–4671 CrossRef CAS PubMed.
- X. Xiong, G. Waller, D. Ding, D. Chen, B. Rainwater, B. Zhao, Z. Wang and M. Liu, Nano Energy, 2015, 16, 71–80 CrossRef CAS.
- H. Zhang, C. Lu, H. Hou, Y. Ma and S. Yuan, Chem. Eng. J., 2019, 370, 400–408 CrossRef CAS.
- J. Acharya, B. G. S. Raj, T. H. Ko, M.-S. Khil, H.-Y. Kim and B.-S. Kim, Int. J. Hydrogen Energy, 2020, 45, 3073–3085 CrossRef CAS.
- G. Prasad Ojha, A. Muthurasu, A. Prasad Tiwari, B. Pant, K. Chhetri, T. Mukhiya, B. Dahal, M. Lee, M. Park and H.-Y. Kim, Chem. Eng. J., 2020, 399, 125532 CrossRef CAS.
- L. G. Beka, X. Bu, X. Li, X. Wang, C. Han and W. Liu, RSC Adv., 2019, 9, 36123–36135 RSC.
- H. Zhao, Z. Zhang, C. Zhou and H. Zhang, Appl. Surf. Sci., 2021, 541, 148458 CrossRef CAS.
- Q. Zong, Y. Zhu, Q. Wang, H. Yang, Q. Zhang, J. Zhan and W. Du, Chem. Eng. J., 2020, 392, 123664 CrossRef CAS.
- S. C. Sekhar, G. Nagaraju, B. Ramulu, S. J. Arbaz, D. Narsimulu, S. K. Hussain and J. S. Yu, Nano Energy, 2020, 76, 105016 CrossRef CAS.
- D. Kong, Y. Wang, S. Huang, J. Hu, Y. V. Lim, B. Liu, S. Fan, Y. Shi and H. Y. Yang, Energy Storage Materials, 2019, 23, 653–663 CrossRef.
- G. P. Ojha, B. Pant, S.-J. Park, M. Park and H.-Y. Kim, J. Colloid Interface Sci., 2017, 494, 338–344 CrossRef CAS PubMed.
- X. Li, H. Wu, C. Guan, A. M. Elshahawy, Y. Dong, S. J. Pennycook and J. Wang, Small, 2019, 15, 1803895 Search PubMed.
- V. T. Chebrolu, B. Balakrishnan, V. Raman, I. Cho, J.-S. Bak, K. Prabakar and H.-J. Kim, Appl. Surf. Sci., 2020, 506, 145015 CrossRef CAS.
- W. Liu, F. Zhu, B. Ge, L. Sun, Y. Liu and W. Shi, Chem. Eng. J., 2022, 427, 130788 CrossRef CAS.
- X. Sun, X. Sun, S. Zhou, G. Luo, R. Liu, S. Li, A. Li and X. Zhu, ChemistrySelect, 2019, 4, 7751–7758 CrossRef CAS.
- R. Arian, A. M. Zardkhoshoui and S. S. Hosseiny Davarani, ChemElectroChem, 2020, 7, 2816–2825 CrossRef CAS.
- S. Zhou, Y. Liu, M. Yan, L. Sun, B. Luo, Q. Yang and W. Shi, Electrochim. Acta, 2020, 349, 136337 CrossRef CAS.
- M. Shi, M. Zhao, L. Jiao, Z. Su, M. Li and X. Song, J. Power Sources, 2021, 509, 230333 CrossRef CAS.
- Y. Wang, Y. Zhang, C. Du, J. Chen, Z. Tian, M. Xie and L. Wan, Dalton Trans., 2021, 50, 17181–17193 RSC.
- H. Gu, Y. Zeng, S. Wan, S. Zhang, Q. Zhong and Y. Bu, J. Mater. Chem. A, 2021, 9, 16099–16107 RSC.
- M. S. Javed, N. Shaheen, S. Hussain, J. Li, S. S. A. Shah, Y. Abbas, M. A. Ahmad, R. Raza and W. Mai, J. Mater. Chem. A, 2019, 7, 946–957 RSC.
- W. Ma, M. Li, X. Zhou, J. Li, Y. Dong and M. Zhu, ACS Appl. Mater. Interfaces, 2019, 11, 9283–9290 CrossRef CAS PubMed.
- W. Du, Q. Zong, J. Zhan, H. Yang and Q. Zhang, ACS Appl. Energy Mater., 2021, 4, 6667–6677 CrossRef CAS.
- R. Xu, K. Chi, J. Wu, L. Wang, J. Lin and S. Wang, Electrochem. Sci. Adv., 2021, e2100047 Search PubMed.
- B. Ye, S. Xiao, X. Cao, J. Chen, A. Zhou, Q. Zhao, W. Huang and J. Wang, J. Power Sources, 2021, 506, 230056 CrossRef CAS.
- G. Zhang, H. Xuan, J. Yang, R. Wang, Z. Xie, X. Liang, P. Han and Y. Wu, J. Power Sources, 2021, 506, 230255 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ta11063b |
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 ab and
Mira
Park
ab and
Mira
Park