
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMultipronged diagnostic and therapeutic strategies for Alzheimer's disease
Madhu
Ramesh
 and
Thimmaiah
Govindaraju
*
and
Thimmaiah
Govindaraju
*
Bioorganic Chemistry Laboratory, New Chemistry Unit, Jawaharlal Nehru Centre for Advanced Scientific Research, Jakkur P.O., Bengaluru, Karnataka 560064, India. E-mail: tgraju@jncasr.ac.in
First published on 14th October 2022
Abstract
Alzheimer's disease (AD) is a progressive neurodegenerative disorder and a major contributor to dementia cases worldwide. AD is clinically characterized by learning, memory, and cognitive deficits. The accumulation of extracellular amyloid β (Aβ) plaques and neurofibrillary tangles (NFTs) of tau are the pathological hallmarks of AD and are explored as targets for clinical diagnosis and therapy. AD pathology is poorly understood and there are no fully approved diagnosis and treatments. Notwithstanding the gap, decades of research in understanding disease mechanisms have revealed the multifactorial nature of AD. As a result, multipronged and holistic approaches are pertinent to targeting multiple biomarkers and targets for developing effective diagnosis and therapeutics. In this perspective, recent developments in Aβ and tau targeted diagnostic and therapeutic tools are discussed. Novel indirect, combination, and circulating biomarkers as potential diagnostic targets are highlighted. We underline the importance of multiplexing and multimodal detection of multiple biomarkers to generate biomarker fingerprints as a reliable diagnostic strategy. The classical therapeutics targeting Aβ and tau aggregation pathways are described with bottlenecks in the strategy. Drug discovery efforts targeting multifaceted toxicity involving protein aggregation, metal toxicity, oxidative stress, mitochondrial damage, and neuroinflammation are highlighted. Recent efforts focused on multipronged strategies to rationally design multifunctional modulators targeting multiple pathological factors are presented as future drug development strategies to discover potential therapeutics for AD.
 Madhu Ramesh | Madhu Ramesh received his BVSc & AH in 2015 from Veterinary College, Bidar, Karnataka and MVSc in Biochemistry from Indian Veterinary Research Institute (IVRI), Bareilly, Uttar Pradesh, India. He joined JNCASR in 2017 for the PhD programme under the supervision of Prof. T Govindaraju. His research interests include understanding the pathophysiology and developing diagnostic and multifunctional therapeutic agents for Alzheimer's disease. |
 Thimmaiah Govindaraju | Thimmaiah Govindaraju is a Professor at the Bioorganic Chemistry Laboratory, New Chemistry Unit, JNCASR, Bengaluru, India. He received his MSc (2000) from Bangalore University and PhD in Chemistry (2006) from the National Chemical Laboratory and University of Pune, India. He carried out postdoctoral research at the University of Wisconsin-Madison, USA (2005–2006) and the Max Planck Institute of Molecular Physiology, Dortmund, Germany (2006–2008) as an Alexander von Humboldt postdoctoral fellow. His research interests are at the interface of chemistry, biology and biomaterials science, including Alzheimer's disease, peptide chemistry, molecular probes, theranostics, molecular architectonics, nanoarchitectonics, and silk and cyclic dipeptide derived biomimetics. |
1. Introduction
Dementia is a major cause of death globally and 70–80% of all cases are linked to AD.1 There are more than 55 million people suffering from dementia worldwide, which are expected to grow to 139 million by 2050.2 Over the decades, the number of deaths by leading diseases show a decreasing trend owing to the availability of reliable diagnostic and therapeutic interventions, while the deaths from AD increased by more than 145%.1,2 Clinically AD patients show learning and memory impairment, language problems, and cognitive deficits leading to fatality within 5 to 12 years of disease diagnosis based on behavioural and cognitive symptoms.3 Pathologically, AD is characterized by the extracellular Aβ senile plaques and NFTs of hyperphosphorylated tau protein, associated neurodegeneration, and brain atrophy.4 AD etiopathology has been described by the cholinergic hypothesis, amyloid hypothesis, and tau hypothesis over the last three decades. Recent discoveries have uncovered the complex pathobiology and showed the multifactorial nature of AD (Fig. 1).4–9 The accumulating evidence demonstrates the role of metal ion dyshomeostasis, reactive oxygen species (ROS), oxidative stress, mitochondrial damage, and neuroinflammation in the pathology of AD.10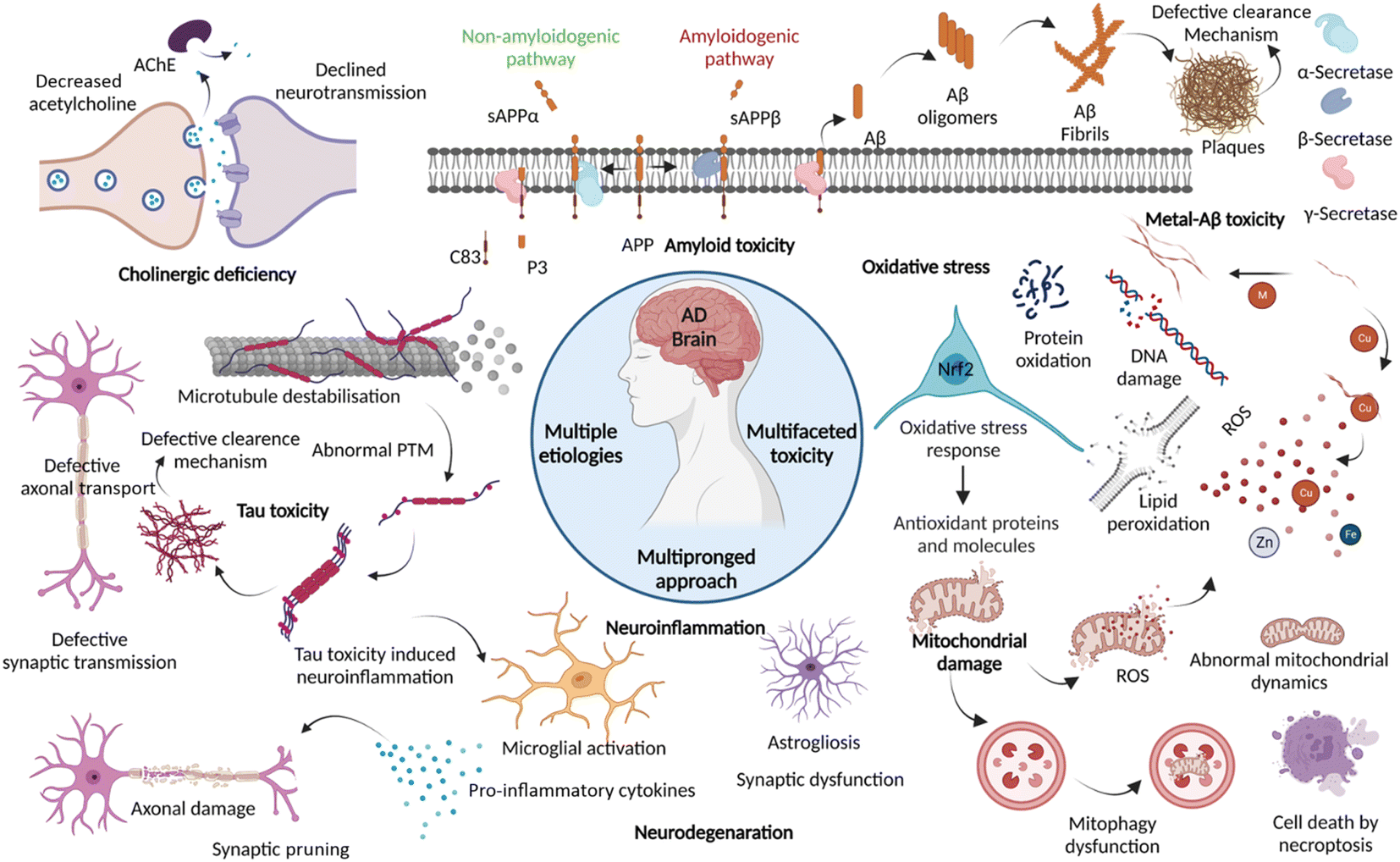 | ||
| Fig. 1 Schematic representation of multiple aetiologies and multifaceted toxicity of AD, which emphasize the need to adopt multipronged approaches for the management of diagnosis and treatment (created with http://BioRender.com). | ||
Aβ and tau aggregation species are the hallmarks of AD and targeted for diagnosis over the last two decades using different chemical tools with positron emission tomography (PET), magnetic resonance imaging (MRI), and fluorescence imaging techniques. The advancements in PET probes and the technique allow the clinical detection of Aβ and tau biomarkers.11 Aβ and tau PET imaging suffer from a few limitations like the requirement of clinical experts, cost, sophisticated instrumentation, radiation hazard, differential diagnosis, and failure to provide definite disease diagnosis at the early stage. Recently, the National Institute on Aging and Alzheimer's Association (NIA-AA) has set a framework, wherein the use of definitive core biomarkers like Aβ (A), tau (T), and neurodegeneration (N) is advocated for AD diagnosis.12 The proposed biomarker list is left open-ended to allow the addition of newly validated biomarkers. The multiple pathological pathways of AD emphasise the potential of considering novel and multiple biomarkers associated with disease pathology for early and accurate diagnosis. The disease-associated markers circulating in the fluids like cerebrospinal fluid (CSF), blood, saliva, and urine hold potential for disease diagnosis. Thus, circulating biomarkers have been explored in recent years owing to numerous advantages over imaging techniques.13 Recently, many multicentric clinical studies have identified promising circulating biomarkers in CSF and blood for their utility in routine screening in large clinical cases. We proposed multiplexed detection of multiple biomarkers using multimodal imaging and detection techniques to generate a signature fingerprint of biomarkers. The signature fingerprint aids early diagnosis and categorises different clinical stages with high accuracy for personalised medication and effective therapeutic intervention.14
AD drug developments have been revolving around cholinergic and amyloid hypothesis over the last three decades.15 Currently, drugs available to treat AD provide only symptomatic relief and do not directly target the underlying disease mechanisms. The therapeutic targeting of Aβ met with failures due to intervention at advanced stages and the multifactorial nature of AD. Aducanumab, a monoclonal antibody (mAb), has been conditionally approved for therapeutic targeting of Aβ in AD patients with mixed output.16 Similarly, targeting tau aggregation, modifications and clearance have drawn the attention of therapeutics development. Many of the tau-targeted therapeutic candidates are in clinical trials and their success is yet to be revealed.8 Recent discoveries on AD etiopathology uncovered many tangible drug targets, which are anticipated to drive drug development in a faster mode. The lesson learned from the failures of Aβ and tau-targeted drug developments are a guide to developing multifunctional molecules to tackle multiple AD pathologies.4,8,17–19 We propose rational development of multifunctional modulators targeting multiple disease mechanisms as a future therapeutics strategy to tackle multifactorial AD. In this perspective, we present multipronged diagnostic and therapeutic approaches, multiplexed and multimodal diagnosis, and rational design of multifunctional modulators of AD with a future outlook.
2. Diagnostic strategies
2.1 Clinical diagnosis of AD
Over the past two decades, there have been notable developments in identifying and validating reliable diagnostic methods and biomarkers for AD. The international working group (IWG) established a clinical-biological definition of AD, wherein the clinical phenotypes of cognitive impairment and biological biomarkers detected through in vivo imaging augment the diagnosis of AD. The clinical assessment of progressive decline in memory, impaired episodic memory, and cognitive changes along with support from biomarker positivity (ATN) has been employed for AD diagnosis.3 The clinical symptoms are assessed using memory and cognitive tests like Montreal Cognitive Assessment (MoCA), Mini-Mental State Exam (MMSE) and Mini Cog by clinical experts. Various chemical probes and techniques like near infrared-fluorescence (NIRF), PET, and MR imaging are being utilised for ATN biomarker imaging (Tables 1 and 2).| Sl. no. | Diagnostic markers and techniques | Chemical probe/techniques | Characteristic features | Model tested | Ref. | ||||
|---|---|---|---|---|---|---|---|---|---|
| a Tg M – transgenic mouse model. b Antibody like selectivity and sensitivity to Aβ plaques with potential for differential diagnosis of AD from other tauopathies and neurodegenerative diseases as in the case of mixed dementia. | |||||||||
| 1 | Aβ targeted diagnostics | ||||||||
| NIRF imaging | λ ex | λ em | ϕ | Fold | K d | ||||
| Oligomers | BD-Oligo | 530 | — | — | 6 | 0.48 μM | Tg M | 20 | |
| F-SLOH | — | 650 | — | — | 1.13 μM | Tg M | 21 | ||
| PTO-29 | 570 | 680 | 60 | 0.25 μM | Tg M | 22 | |||
| Soluble aggregates/protofibrils | DCM-AN | 500 | 661 | 0.015 | — | 0.85 μM | Tg M | 26 | |
| CRANAD102 | 580 | 700 | 0.018 | 68 | 7.5 nM | Tg M | 28 | ||
| Fibrils | TC | 537 | 638 | 0.4 | 30 | 58.4 nM | In vitro | 23 | |
| CQb | 516 | 664 | 0.36 | 100 | 84 nM | Human | 24 | ||
| PHC4 | — | 741 | 0.78 | 62.2 | 14.1 nM | Tg M/human | 27 | ||
| QM-FN-SO3 | 500 | 720 | — | — | 170 nM | Tg M | 30 | ||
| ADlumin1 | — | 540 | — | 100 | 2.1 μM | Tg M | 31 | ||
| 18 | — | 618 | 0.31 | 223 | 43.1 nM | Tg M | 32 | ||
| Different alloforms | QAD1 | Binds to monomers, oligomers, and fibrils | Tg M | 25 | |||||
| Combinatorial molecular sensor | Differentiate different alloforms like monomer, oligomer, LMW, and HMW aggregates | In vitro | 29 | ||||||
| PET imaging | |||||||||
| Fibrils | 11C-PiB | ThT based PET probe, approved for clinical use and a gold standard test | Human | 33 | |||||
| 18F-flutemetamol | Fluorine-labelled analogue of PiB, approved for clinical use | Human | 34 | ||||||
| 18F-florbetaben | Stilbene–polyethylene glycol conjugate, selective labelling, approved for clinical use | Human | 35 | ||||||
| 18F-florbetapir | Highly selective, low background, good pharmacokinetics and correlates with post-mortem Aβ deposition | Human | 36 | ||||||
| 18F-FIBT | Imidabenzothiazole-based, high contrast and better than the reported probes | Tg M | 37 | ||||||
| 64Cu-HYR17 | Benzothiazole-based, Cu chelating probe with a better half-life | Tg M | 38 | ||||||
| MRI | |||||||||
| Fibrils | USPIO-Aβ1–42 | Aβ conjugated to iron oxide NPs, micron-scale resolution, and map Aβ deposition | Tg M | 39 | |||||
| HMON-abAβ40 | Aβ antibody conjugated with manganese NPs, image and monitor Aβ load | Tg M | 40 | ||||||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
|||||||||
| 2 | Tau targeted diagnostics | ||||||||
| NIRF imaging | λ ex | λ em | ϕ | Fold | K d | ||||
| Fibrils | PBB3 | 405 | 520 | — | — | 2.55 nM | Tg M/human | 42 | |
| 2e | 550 | 660 | — | 310 | 0.77 μM | Tg M | 43 | ||
| BD-tau | 525 | 590 | 0.3 | 3.2 | 0.89 μM | Tg M | 44 | ||
| Tau 1 | 633 | 742 | 0.92b | 6.4 | 2.77 μM | Tg M | 45 | ||
| Tau 2 | 607 | 723 | 0.84b | 9.3 | 6.18 μM | Tg M | |||
| 2c | 502 | 632 | 0.82 | 44 | 6.06 nM | Tg M | 46 | ||
| Q-tau 4 | 424 | 630 | 0.01 | 3.82 | 16.6 nM | Human | 47 | ||
| 18 | — | 711 | 0.019 | 50.2 | 33.2 nM | Tg M | 32 | ||
| Soluble aggregates | pTP-TFE | 450 | 520 | 0.27 | — | 66 nM | Tg M | 48 | |
| PET imaging | |||||||||
| Fibrils | 18F-THK5117 | Arylquinoline based probe, good pharmacokinetics | Human | 49 | |||||
| 18F-flortaucipir (Tauvid) | Indole based probe, selective labelling and approved for clinical use | Human | 50 | ||||||
| 18F-JNJ64349311 | Performs better than 18F-flortaucipir | Tg M | 51 | ||||||
| 18F-GTP1 | Highly selective with no MAO binding | Human | 52 | ||||||
| 18F-PI-2620 | High contrast, signal to noise ratio, accurately distinguish AD from healthy | Human | 53 | ||||||
| MRI | |||||||||
| Tau tangles and tau positive cells | Shiga-X35 | 18F-MRI agent targeting tau tangles | Tg M | 54 | |||||
| Tau-X | Aptamer based nanoformulation targeting hyperphosphorylated tau positive neurons | Tg M | 56 | ||||||
|
|||||||||
| 3 | Neurodegeneration | ||||||||
| PET imaging | 18FDG–PET | Imaging metabolic activity of the brain | Human | 59 | |||||
| MRI | sMRI | Assessment of brain atrophy | Human | 61 | |||||
| fMRI | Mapping functional connectivity of the brain | Human | 64 | ||||||
|
|||||||||
| 4 | Indirect biomarker targeted diagnostics | ||||||||
| TSPO PET imaging | 11C-PK11195 | First in vivo clinical TSPO PET imaging | Human | 67 | |||||
| 11C-PBR28 | Correlates with Aβ and tau accumulation | Human | 68 | ||||||
| SV2A PET imaging | 11C-UCB-J | First in vivo PET probe with biocompatibility | Human | 69 | |||||
| 18F-UCB-H | Correlates with cognitive decline and Aβ load | Human | 70 | ||||||
| 18F-SynVesT-1 | Better pharmacokinetics and binding | Human | 71 | ||||||
| 18F-SynVesT-2 | High brain uptake, fast kinetics, and better binding | Human | 72 | ||||||
| HOCl fluorescence imaging | CM2 | Fluorescent probe detects selectively HOCl, demonstrates elevated levels and proximal localisation of HOCl with Aβ in the AD brain | Tg M | 73 | |||||
| Metal ion MRI | Intrinsic Fe | Elevated Fe levels in the AD brain serve as a MRI contrast agent and its quantification | Human | 76 | |||||
| Sl. no. | Biomarker class | Target biomarkers | Remarks | Specificity (%) | Sensitivity (%) | Sample size | Ref. |
|---|---|---|---|---|---|---|---|
| a p-tau: phosphorylated tau and t-tau: total tau. | |||||||
| 1 | CSF and blood | ||||||
| Aβ | Aβ42/40 | Decreased significantly, correlates with Aβ positivity | 100 | 84 | 45 | 91 | |
| Composite model | Model from Aβ42, 40 and ratio diagnose disease with 90% accuracy | 81 | 96.7 | N =121 and N = 252 | 92 | ||
| Tau | t-tau and p-tau | Increased | 80 | 80 | 97 | 94 | |
| p-tau-181 | Increased, discriminate MCI and AD. Correlates with tau PET positivity, atrophy, and CSF biomarkers | 87 | 92 | N1 = 182 | 95 | ||
| N2 = 344 | |||||||
| p-tau-217 | Increased 6-fold in AD correlates with Aβ and tau positivity, early diagnosis of AD | 91 | 91 | 194 + 32 | 96 | ||
| p-tau-231 | Increased in the early stage, outperforms p-tau-181, correlates with Aβ and tau deposition | — | — | 38 + 313 | 99 | ||
| Protein biomarkers | Neurogranin | Increased in CSF and correlates with CSF biomarkers, atrophy and brain Aβ load | 60 | 79 | 302 | 100 | |
| Synaptotagmin | Increased, discriminate MCI and AD | — | — | 39 + 78 | 102 | ||
| sTREM | Increased, correlates with CSF Aβ and tau biomarkers, atrophy and brain Aβ load | — | — | 155 + 93 | 103 | ||
| YKL-40 | Increased, correlates with Aβ deposition and memory deficits | 85 | 85 | 318 | 104 | ||
| Nf-L | Increased, correlates with CSF t-tau and p-tau, cortical atrophy, Aβ deposition and brain metabolism. Discriminate MCI and AD cases | CSF-187 | 106 | ||||
| Ser-405 | |||||||
| N = 196 | |||||||
| RNA biomarkers | BACE1 lcnRNA miRNA panels | Increased in plasma | 61.3 | 87.5 | 134 | 109 | |
| Exosome 7 miRNA panel diagnoses with 89% accuracy (miR-185-5p, miR-342-3p, miR-141-3p, miR342-5p, miR-23b-3p, miR-338-3p and miR-3613-3p) | — | — | 70 | 110 | |||
| Plasma 6 miRNA panel (miR-185-5p, miR-342-3p, miR-141-3p, miR342-5p, miR-23b-3p, miR-338-3p and miR-3613-3p) | 78 | 75 | 50 | 111 | |||
| CSF 6–7 miRNA panel, correlates with CSF biomarkers and the cognitive score | — | — | 118 | 112 | |||
| Protein and metabolite panel | Multiple protein panel in blood | 18 biomolecule alteration was identified (2 cohorts) | 93 | 85 | N1 = 961 | 113 | |
| 85 | 80 | N2 = 170 | |||||
| 3 protein panel | Plasma Aβ42/40, pTau-217 and Nf-L diagnose AD and correlates with cognitive changes | — | — | 435 | 114 | ||
| 4 protein panel | Blood exosomal GAP43, neurogranin, SNAP25 and synaptotagmin-1 are early diagnosis biomarkers | — | — | 320 | 116 | ||
| Spingomyelin metabolites | 26 metabolites, diagnose AD with an accuracy of 83.33% | 80 | 86.67 | N1 = 44 | 117 | ||
| N2 = 767 | |||||||
| N3 = 207 | |||||||
|
|||||||
| 2 | Saliva | ||||||
| Aβ | Aβ42 | Increased and specific to AD over PD | — | — | 22 | 118 | |
| Tau | p-tau/t-tau | Increased | — | — | 59 | 119 | |
| p-tau-396/t-tau | Increased, no correlation with other biomarkers | 50 | 73 | 148 | 120 | ||
| Lf | Full length Lf | Decreased, outperforms CSF biomarkers. Other studies contradict and changes are inconsistent | 100 | 100 | N1 = 274 | 121 | |
| 98.6 | 100 | N2 = 127 | |||||
|
|||||||
| 3 | Urine | ||||||
| Protein | SPP1, GSN and IGFBP7 | Increased, MS techniques were used to screen and confirmed by ELISA | — | — | 40 | 125 | |
|
|||||||
| 4 | Tear | ||||||
| Protein | Lipocalin-1, dermcidin, lysozyme-C and lacritin | Proteins are significantly altered in AD over healthy individuals | 77 | 81 | 23 | 124 | |
| Significant increase in the tear total protein and flow rate | |||||||
2.2 Aβ (A) targeted diagnosis
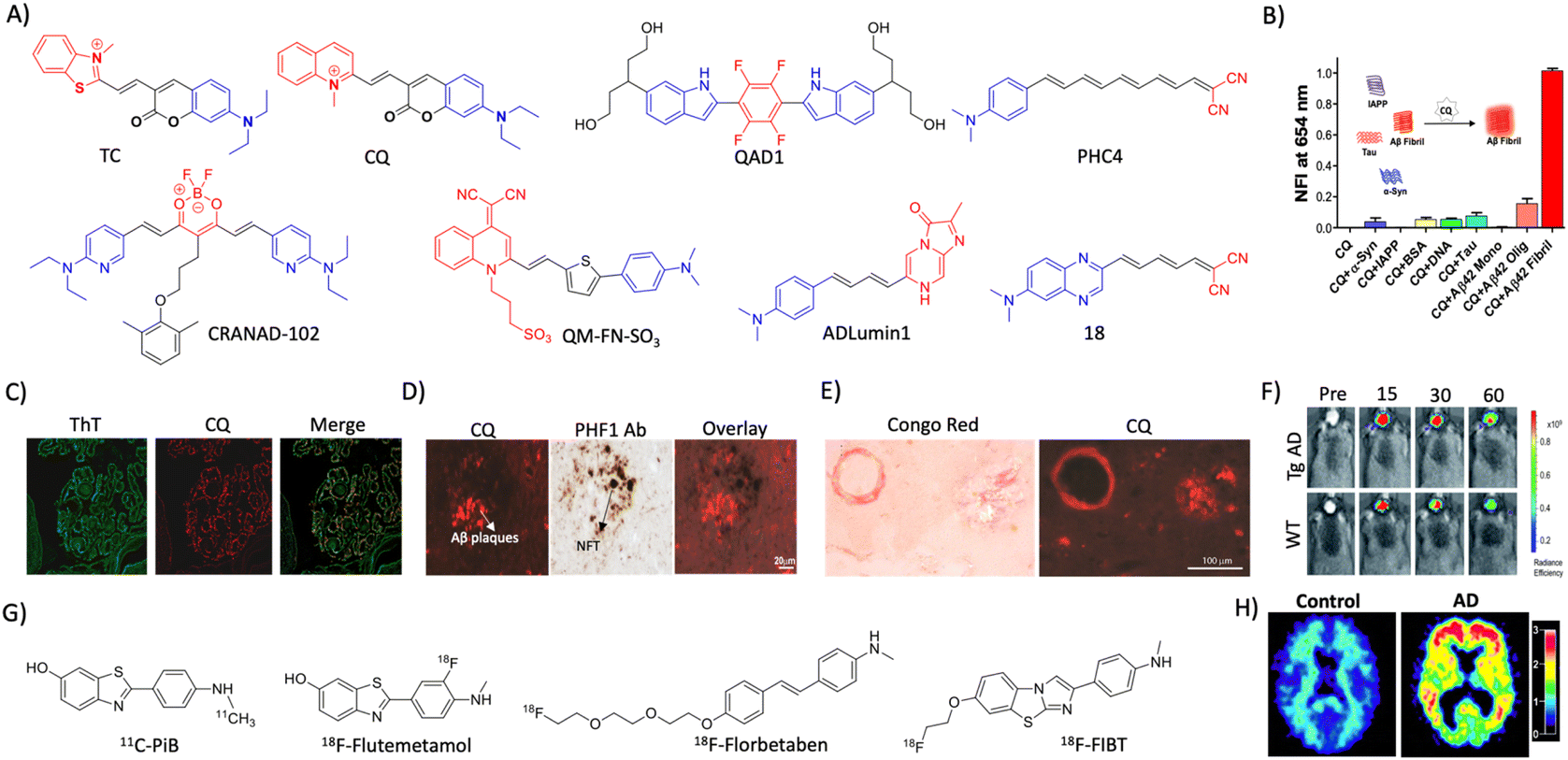 | ||
| Fig. 2 Molecular probe targeting Aβ. (A) NIR fluorescent probes targeting Aβ fibrils (blue and red represent donor and acceptor moieties, respectively). (B) Selective fluorescence response of the CQ probe to Aβ aggregates in comparison with BSA and other amyloid protein aggregates. (C) Fluorescence staining of human brain tissue with CQ and ThT shows selective Aβ aggregate staining by CQ. (D) Immunofluorescence staining of human AD brain tissue shows the selective binding of CQ with Aβ aggregates over tau aggregates as reflected by poor colocalisation with PHF1 tau antibody staining. (E) CQ stain congophilic angiopathy similar to Congo red staining. (B)– (E) Reproduced from ref. 24 with permission from Elsevier, copyright 2017. (F) NIRF imaging of amyloid aggregates in the Tg AD mice model using CRANAD102 at different time interval (min) distinguish AD from WT. Reproduced from ref. 28 with permission from the Royal Society of Chemistry, copyright 2017. (G) Aβ fibril targeting PET probes. (H) PiB PET images of human healthy and AD brains. Reproduced from ref. 33 with permission from the American Neurological Association, copyright 2003. | ||
A photoinduced electron transfer (PIET) quenched NIRF probe QAD1 was designed with Bodipy as the fluorophore and tetrahydroxyquinoxaline as the quenching moiety.25 The probe exhibits turn-on fluorescence upon interaction with different Aβ alloforms, which was used to detect and monitor Aβ load in a 6-month-old APP/PSEN1 Tg mouse model. Recently, DCM-AN was designed by combining the dicyanomethylene (DCM) skeleton with an Aβ targeting aminonaphthalene (AN) moiety, which showed selectivity towards protofibrils.26 Molecular dynamics (MD) simulation of the probe with trimer (oligomers), dodecamer (protofibrils) and fibrillar structures revealed strong binding towards the dodecamer, which suggests selectivity towards protofibrils. Upon binding to protofibrils, the rotation of both ethylene and piperidine groups of the probe is restrained, which results in fluorescence enhancement. The probe detects protofibrils in the ex vivo brain sections at different stages in the Tg AD mouse model. The donor–acceptor (D–A) architecture was expanded by playing around with the π-bridge, donor aromatic moieties, and dicyano moiety as the acceptor.27 Among them, PHC4 exhibits favourable properties with in vivo NIRF imaging to distinguish between WT and Tg AD mice. Ran et al. tuned the steriohindrance of curcumin at the phenoxy alkyl chain to make the probes selective to soluble Aβ aggregates.28 Among them, CRANAD102 exhibited selectivity (68-fold over insoluble aggregates) to soluble aggregates with a strong binding affinity (Kd = 7.5 nM). The probe was successfully utilised for in vivo NIRF imaging of the soluble aggregates in the early stage (4 months) of the Tg AD mouse model, which allowed monitoring of the changes over 4 to 12 months (Fig. 2F).
Margulies et al. have reported a combinatorial sensor for the detection of various Aβ alloforms.29 The sensor was constructed by conjugating three fluorescent probes ThT, sulforhodamine B, and sulfo Cy5 onto a proline scaffold with a KLVFF moiety. The sensor was employed to detect different alloforms viz., monomers, low molecular weight (LMW) and higher molecular weight (HMW) oligomers, and aggregates, based on differential fluorescence response due to varied intramolecular FRET among fluorophores. Recently, an aggregation-induced emission (AIE) based probe QM-FN-SO3 was developed by connecting DCM-N with dimethylaminobenzene through a π-conjugated thiophene bridge and introduced a sulphonate moiety as a substitution that keeps the probe in the off state.30 The thiophene bridge with π-conjugation retains the lipophilicity to enhance the BBB permeability and NIR emission of the probe. QM-FN-SO3 aggregates on the hydrophobic surface of Aβ aggregates and exhibits a turn-on fluorescence response. The AIE probe was BBB permeable and mapped Aβ aggregates with high fidelity in the Tg AD mouse model. A novel turn-on chemiluminescent probe ADlumin1 was designed to target Aβ aggregates.31 The discrimination of AD from WT was improved by dually amplifying the signal via chemiluminescence resonance energy transfer (DUS-CRET) between CRANAD-3 and ADlumin1 in a 5-month-old 5xFAD mouse model. The simultaneous detection of both Aβ and tau aggregates would enhance the accuracy of AD diagnosis. D–A based probe 18 with environmental sensitivity can differentially emit fluorescence in the presence of brain-derived Aβ and tau aggregates.32 Probe 18 successfully discriminates Aβ and tau aggregates in Tg mouse models and measure the load in vivo through NIRF imaging.
2.3 Tau (T) targeted diagnosis
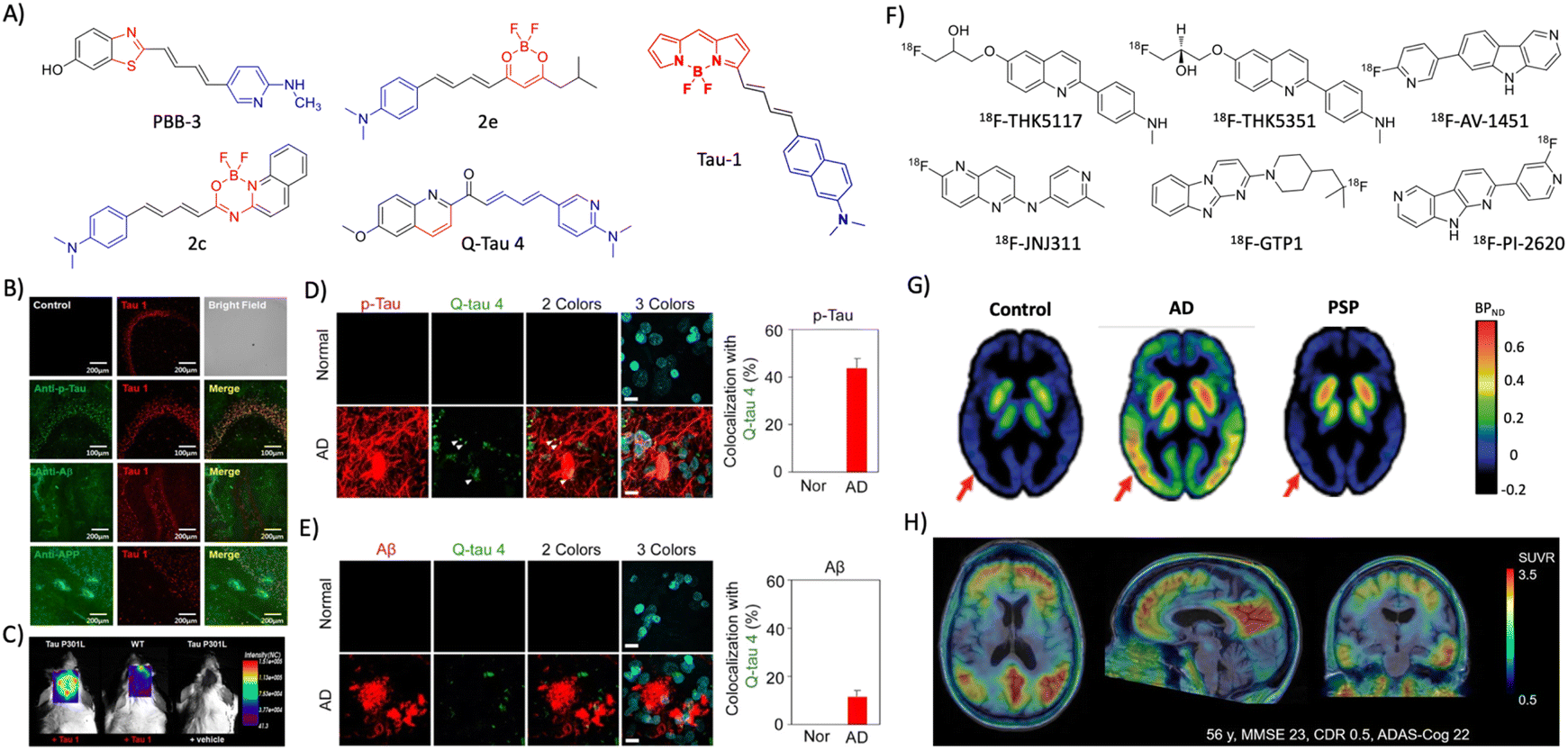 | ||
| Fig. 3 Molecular probes targeting tau. (A) NIR fluorescent probes targeting tau fibrils (blue and red represent donor and acceptor moieties, respectively). (B) Immunofluorescence imaging in a 3xTg mice brain with antibodies targeting tau aggregates, Aβ, and APP protein shows the selective tau labelling by tau 1 (scale bar 200 μm). (C) NIRF imaging of tau aggregates by tau 1 in a Tg mouse after 30 min of injection. (B) and (C) Reproduced from ref. 45 with permission from the American Chemical Society, copyright 2017. (D) Ex vivo immunofluorescence imaging of normal heathy and AD human brain tissues with Q-tau 4 and its colocalisation with the p-tau antibody (scale bar 20 μm). (E) Ex vivo fluorescence imaging of normal healthy and AD human brain tissues with Q-tau 4 and Aβ antibodies. Quantification shows the poor colocalisation indicating the selective tau labelling by the probe (scale bar 20 μm). (D) and (E) Reproduced from ref. 47 with permission from the American Chemical Society, copyright 2021. (F) Tau targeting PET probes. (G) PET images of control, AD, and PSP human subjects obtained by using the 18F-AV-1451 probe. Reproduced from ref. 50 with permission from Oxford University Press, copyright 2017. (H) PET images of an AD human brain acquired with 18F-PI-2620 exhibit a high contrast and signal-to-noise ratio. Reproduced from ref. 53 with permission from SNMMI, copyright 2020. | ||
The availability of MRI probes targeting tau pathology are limited, and recently few probes are developed and evaluated in Tg mouse models. A novel buta-1,3-diene derived 19F-MRI probe Shiga-X35 was developed targeting tau tangles for MR imaging.54Ex vivo immunofluorescence and in vivo19F-MR imaging in a rTg4510 mouse model revealed that Shiga-X35 colocalises with tau tangles and accumulates in the forebrain of the Tg AD mouse. The in vivo19F-MRI signal from the AD mouse brain was significantly higher which distinguishes AD from the WT mouse. The accumulation of hyperphosphorylated tau inside the neuronal cells is an early event of tau pathology in AD that possibly changes the cell surface markers. In this context, an aptamer-based nanoparticle MRI contrast agent tau-X targeting the neurons with hyperphosphorylated tau was developed for evaluating tau pathology by MRI.55 An aptamer that selectively binds to the neuronal cells with intracellular hyperphosphorylated tau was evolved by systematic evolution of ligands by the exponential enrichment (SELEX) method. A lipid nanoformulation tau-X was prepared using the aptamer and Gd-DOTA contrast agent. The developed tau-X MRI agent was evaluated in a 2 month old P301S Tg mouse model and age matched control, which showed a higher MRI signal in the AD mouse that developed tau accumulation compared to WT. Recently, improved aptamers were evolved and prepared tau-X nanoformulation, and evaluated for their MRI in a Tg mouse model.56 The results showed that tau-X based MRI successfully detects tau pathology in an early stage (2 months) in the Tg mouse model and may have implications for early detection of tau pathology in clinical cases.
2.4 Neurodegeneration (N)
The correlation of neurodegeneration with cognitive decline, Aβ, and tau made it one of the reliable biomarkers in the ATN framework. The assessment of brain metabolism and atrophy is performed by PET and MR imaging. In a multicentric study, fluorodeoxyglucose–PET (18FDG–PET) was employed to measure brain metabolism as a biomarker in AD and other dementia cases.57 The standardised signal pattern for each disease was developed and the PET scans diagnosed AD with 95% accuracy and differentiate from mild cognitive impairment (MCI) and other dementia cases viz., dementia with Lewy bodies (DLB), frontotemporal dementia (FTD) and normal individuals. Meta-analysis has suggested that FDG–PET can diagnose AD with 91% sensitivity and 78% specificity.58 Recently, in a large clinical study FDG–PET imaging was employed on amyloid and tau positive (A+T+) patients to evaluate the ability of FDG–PET for AD diagnosis.59 Study revealed that FDG–PET (F+) has differentiated AD from dementia and suggests F+ as an independent biomarker for AD. FDG–PET was considered for differential diagnosis of AD from other forms of dementia and the output is inconsistent and needs to be revisited before recommending it for AD diagnosis.60 There is a need for developing robust artificial intelligence (AI) and machine learning approaches for defining the patterns of brain hypometabolism for differential diagnosis of dementia.Structural MR imaging assesses brain tissue damage and atrophy evident in AD. Brain regiospecific volume measurements by MRI scans revealed volume reduction in various regions of the AD brain compared to healthy controls and the changes track the progression of AD.61 The brain volumetric measurements carried out using MRI of MCI in a longitudinal study demonstrate that MCI cases converted to AD had lower volumes compared to controls.62 The analysis of atrophy in the brains of DLB, AD, and controls by MRI demonstrates the bilateral damage of cornu ammonis and subiculum in AD, whereas it was intact in DBL.63 The study showed the ability of structural MRI for differential diagnosis and its potential application in other neurodegenerative disorders. Loss in neuronal function and degeneration was expected to affect the functional connectivity within the AD brain. The characterisation of brain network alterations by functional MRI in MCI and AD with disease progression in the longitudinal study suggests a gradual decline in the functional networks of AD compared to healthy controls.64 Studies have found that the connectivity alterations are specific to the disease and need to be characterized for accurate discrimination. A recent study by resting-state functional magnetic resonance imaging (rs-fMRI) revealed a decline in the connectivity between the posterior cingulate cortex to the whole brain for AD subjects.65 The functional connectivity changes need detailed characterization in the AD brain to use as a diagnostic marker.
2.5 Indirect biomarkers
Complex etiopathologies associated with AD were unrevealed in the last decade and many pathological events occur in preclinical stages. Oxidative stress, neuroinflammation, and synaptic damage are among the major early events and there are many associated biomolecules and biomolecular events that indirectly influence AD pathology, which are considered potential indirect biomarkers for AD diagnosis. These indirect biomarkers are promising, and their identification and validation may find a place in the NIA-AA framework along with ATN as additional biomarkers for early and accurate diagnosis.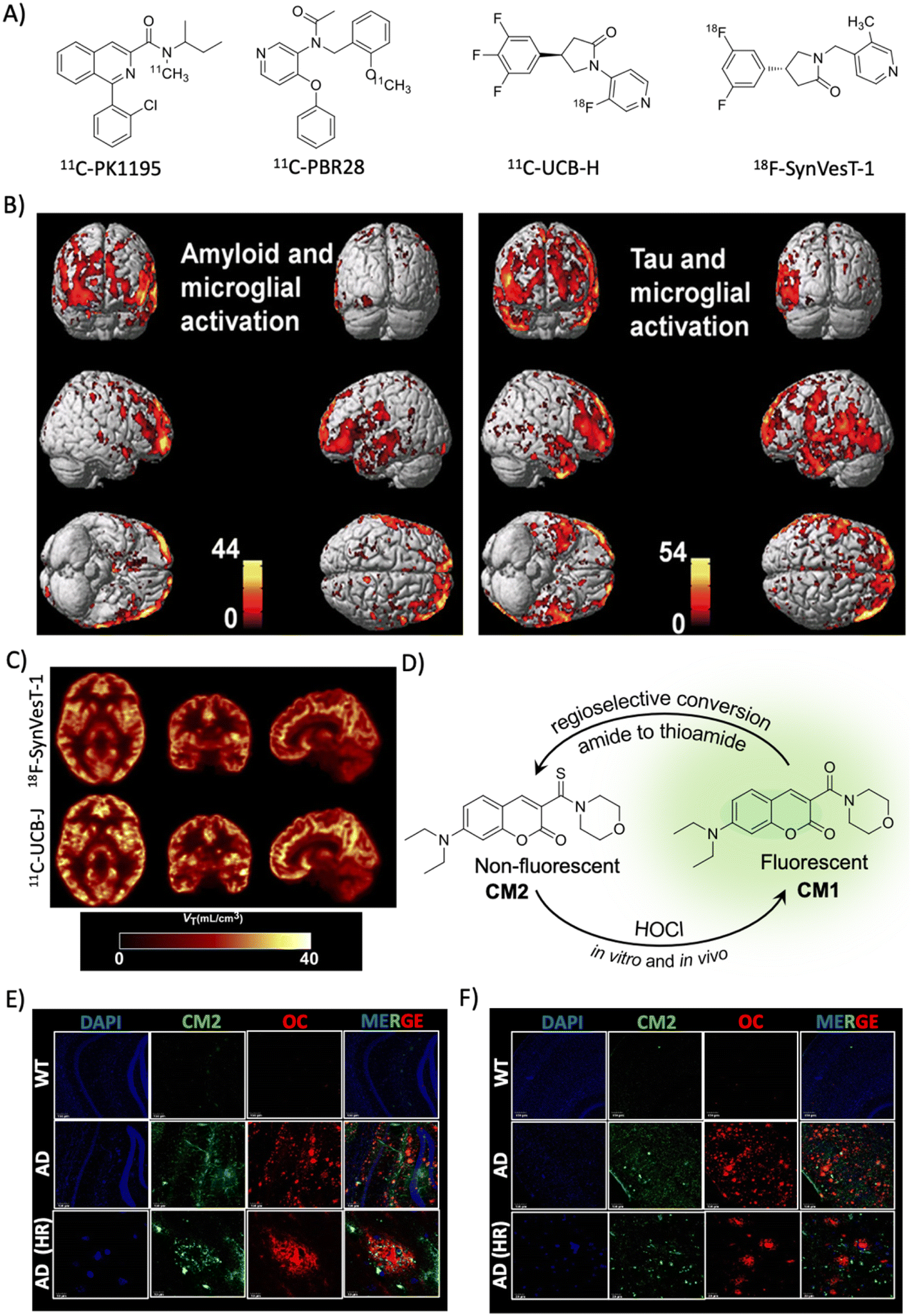 | ||
| Fig. 4 Targeting indirect biomarkers of potential diagnostic importance. (A) PET probes targeting TSPO and SV2A. (B) PET imaging of microglial activation in the AD human brain using the PBR28 probe. The accumulation of the probe in the brain correlates with Aβ and tau deposition as revealed by PET imaging. Reproduced from ref. 68 with permission from Oxford University Press, copyright 2018. (C) PET imaging of synaptic density in the AD brain targeting SV2A protein using 18F-SynVesT-1 and 11C-UCB-J. Reproduced from ref. 71 with permission from SNMMI, copyright 2020. (D) Regioselective conversion of a non-fluorescent CM2 probe to fluorescent CM1 in the presence of HOCl under in vitro and in vivo conditions. Elevated levels of HOCl proximally localised with Aβ aggregates (OC) detected by the CM2 probe in the cortex (E) and hippocampus (F) of the APP/PSEN1 Tg AD mice brain (scale bar 20 μm). Reproduced from ref. 73 with permission from the American Chemical Society, copyright 2019. | ||
2.6 Circulating biomarkers
Imaging the pathological lesions in the brain encounters limitations of exposure to radiation, complicated procedures, instrumentations, and the need for clinical experts. Disease associated alterations in the brain are expected to be reflected in the circulating fluids like CSF and blood. These circulating biomarkers have the advantage of being minimally invasive or non-invasive, simple, and cost-effective. In recent years, circulating biomarkers have been explored and potential biomarkers are identified for early and accurate diagnosis of AD (Fig. 5 and Table 2).89,90 Many biomarkers associated with AD pathology are altered in CSF and blood in the early stage, and correlate with ATN and cognitive decline. Due to non-invasiveness saliva and urine are also explored and their potential is yet to be proven.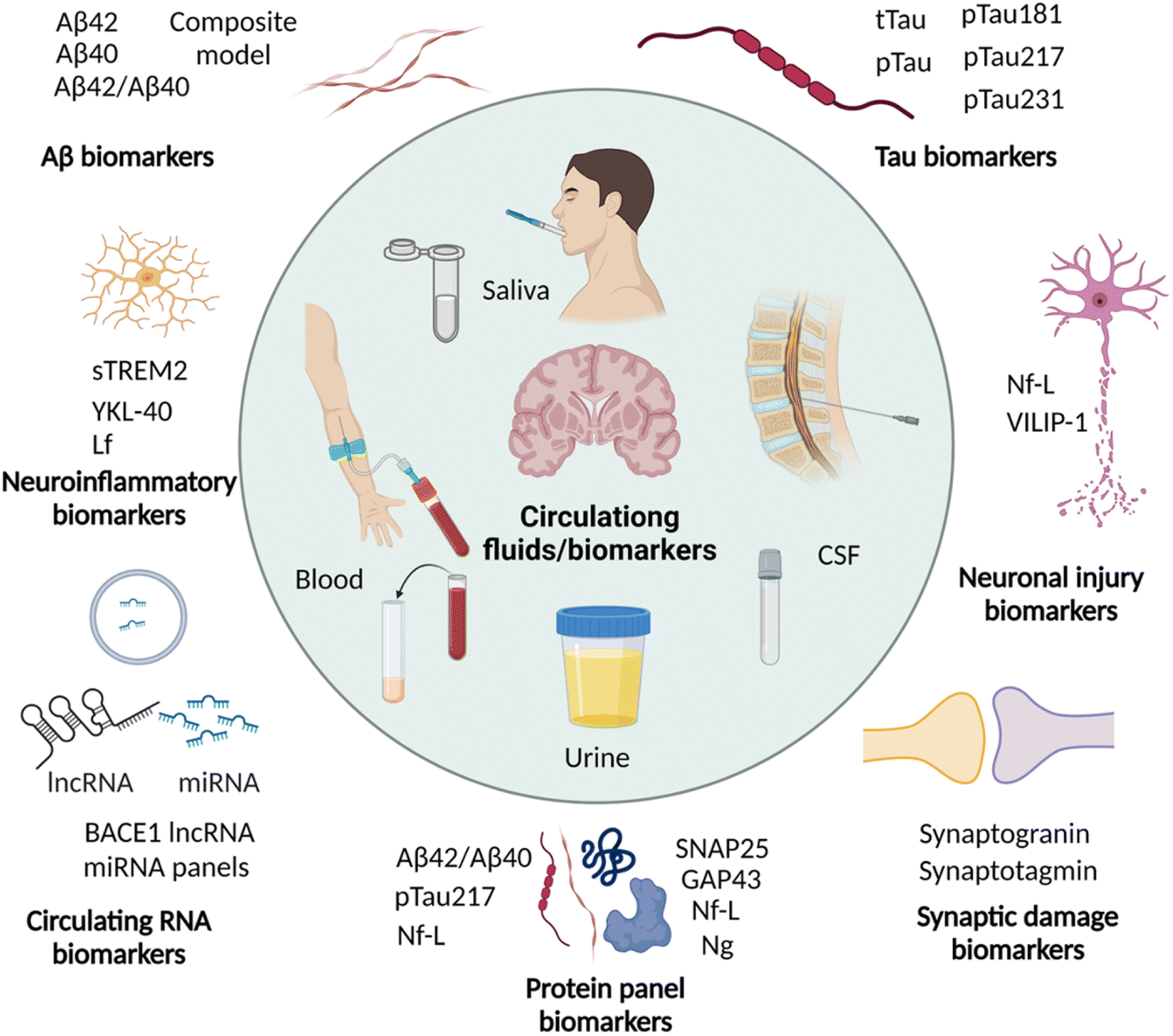 | ||
| Fig. 5 Circulating biomarkers in CSF, blood, saliva, and urine for the diagnosis of AD (created with http://BioRender.com). | ||
The pathological phosphorylation of tau (p-tau) is evident in AD and p-tau variants are anticipated to serve as biomarkers for diagnosis. A recent study has shown significantly elevated levels of CSF tau, CSF pTau, and plasma tau in AD compared to healthy controls.94 A large cohort study of p-tau-181 in CSF and plasma demonstrated high levels in preclinical dementia cases, which further increased in MCI and AD.95 The plasma level of p-tau-181 was correlated with CSF levels and tau PET positivity, and accurately differentiate dementia cases of non-AD subjects, MCI, and AD. A recent cohort study to analyse p-tau isoforms in CSF by the sensitive MS technique demonstrated that the CSF p-tau-217 isoform outperforms p-tau-181 for the diagnosis of PET confirmed AD cases.96 It was supported by another cohort and longitudinal study and demonstrated that p-tau-217 levels strongly correlate with tau and Aβ PET results and accurately diagnose AD and distinguish it from other dementia. The analysis of plasma p-tau levels in the cohorts of Aβ and tau PET confirmed preclinical and prodromal AD cases, and there was an association of plasma p-tau-217 with CSF p-tau-217 levels and tau PET positivity.97 Plasma and CSF p-tau-217 have increased significantly in the early stage of disease, wherein tau and Aβ deposition was not significant. These observations suggest p-tau-217 as one of the potential biomarkers for early diagnosis. In a retrospective study, plasma p-tau-181 and p-tau-217 showed excellent diagnostic performance and distinguish AD from other disease conditions.98 Recently p-tau-231 has emerged as a potential blood biomarker for early and accurate diagnosis of AD.99 The increased p-tau-231 distinguishes MCI and AD from healthy individuals outperforming p-tau-181 and correlates with Aβ and tau deposition. These circulating p-tau isoforms hold high diagnostic potential that needs to be established by multicentric longitudinal and cross-sectional clinical studies with large cohorts.
2.7 Multiplexing and multimodal diagnosis – a future approach for AD diagnosis
Chemical tools for NIRF, PET, and MR imaging targeting different core and indirect biomarkers have been established for the diagnosis of AD. Each of these tools and techniques holds merits and limitations. Developing chemical probes for multimodal imaging overcomes the limitations and enhances the sensitivity, specificity, and resolution of imaging that aids the accurate diagnosis of AD. Simultaneous detection of multiple diagnostic biomarkers by multiplexing is another approach to improve the accuracy and reliability of diagnosis. Multiplexed detection of Aβ, tau, and neurodegeneration by multimodal imaging using different probes targeting multiple biomarkers or with a single probe targeting different biomarkers is anticipated to gain utmost importance soon (Fig. 6).14,126 Multiplexed detection of multiple circulating biomarkers using multiple assays or integrated microarrays can be considered as the future direction for research on AD diagnostics. Protein, RNA, and metabolite biomarkers in circulating fluids can be detected by multiplexing with different modes of detection like MS, ELISA, microarrays, and paper-based sensors. In general, the detection of multiple biomarkers with multiplexed and multimodal approaches to develop the signature fingerprint is the future of AD diagnostics (Fig. 6).14,126 Such characteristic fingerprints hold potential for early diagnosis and accurate categorisation of clinical stages for better management and personalised detection and medication for AD patients.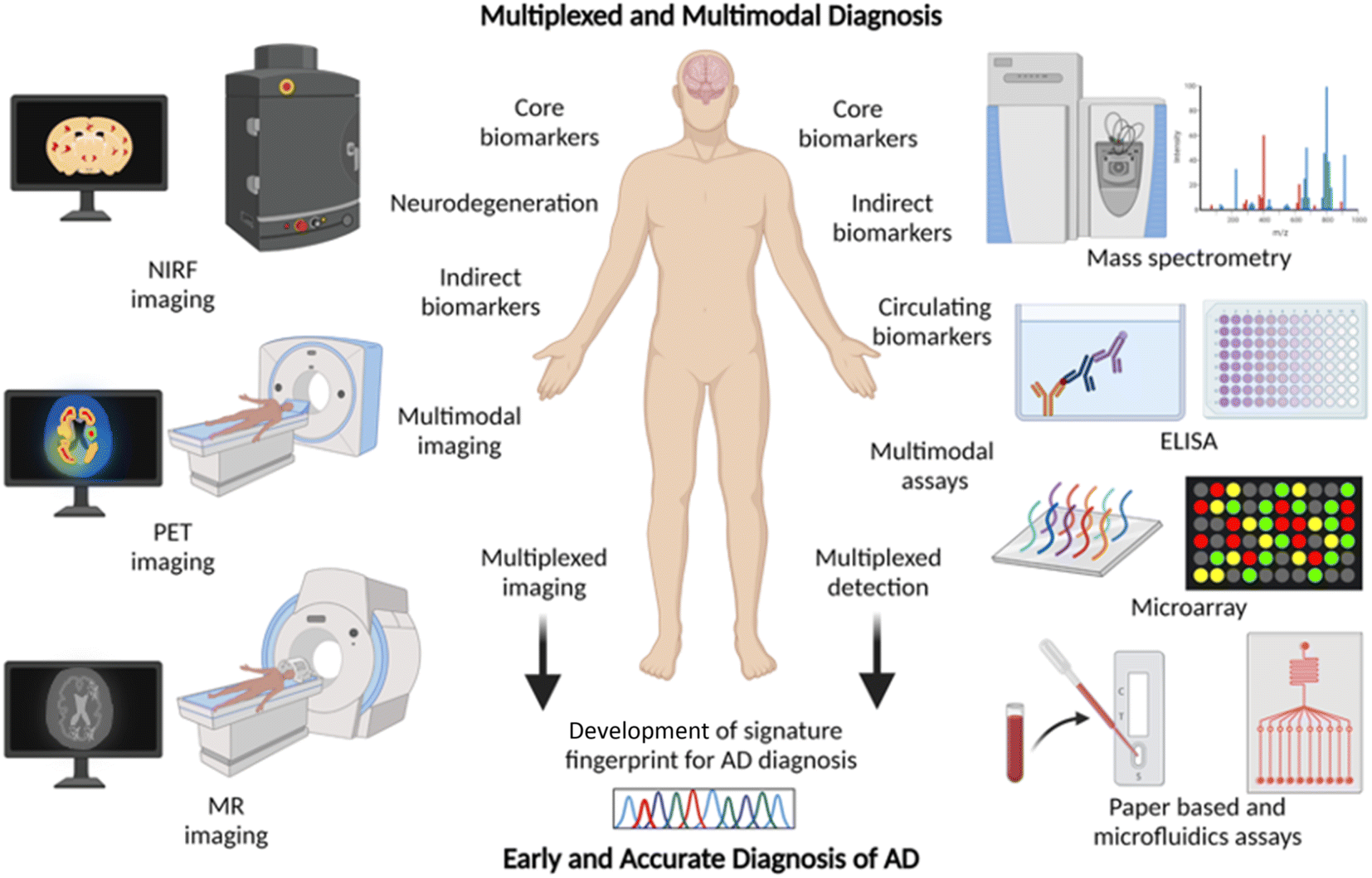 | ||
| Fig. 6 Multiplexing and multimodal approach for early and accurate diagnosis of AD (created with http://BioRender.com). | ||
3. Therapeutic strategies
3.1 AChE and N-methyl-D-aspartate (NMDA) receptor targeted therapies
AD drug discovery initially focused on the amyloid pathway and cholinergic deficiency as therapeutic targets. The efforts were made to target acetylcholine esterase (AChE) to improve the acetylcholine (ACh) levels and restore neuronal function. NMDA receptor signalling is involved in brain activity and abnormal brain activity is evident in AD. NMDA receptor antagonists were developed to overcome the abnormal brain activity that restores learning and memory functions. AChE acts in the synapse to cleave ACh into acetate and choline, and inhibition of the enzyme was expected to increase ACh levels in synapse and improve memory deficits (Fig. 7A). In this direction many AChE inhibitors have been developed and assessed in preclinical and clinical trials for therapeutic effects.127 Among the approved candidates for AD treatment mostly they are AChE inhibitors like tacrine, donepezil, rivastigmine, and galantamine.128 Another approach of counteracting the cholinergic deficiency is to treat with cholinergic precursors like choline or choline alphoscerate that stimulate the cholinergic system. The association between the cholinergic precursors and AChE inhibitors results in effective combination treatment. The treatment of rats with rivastigmine in association with choline or choline alphoscerate increased ACh levels, inhibited AChE and restored cholinergic transmission.129 In a double blinded clinical trial, donepezil and choline alphoscerate treated AD patients with ischemia showed improvements in behavioural and cognitive functions compared to control and donepezil treated patients.130 In a recent clinical study, AD patients treated with donepezil and choline alphoscerate showed improvement of depression symptoms.131 Cholinergic stimulation with donepezil along with a cholinergic precursor choline alphoscerate was effective in mild to moderate AD cases. Many NMDA receptor antagonists were developed and among them memantine was beneficial and approved for clinical use. They all are proven to improve the clinical conditions of AD but fail to address the root cause and cure the disease. They were tried in combination with many other drugs and analogues are in assessment for therapeutic benefits.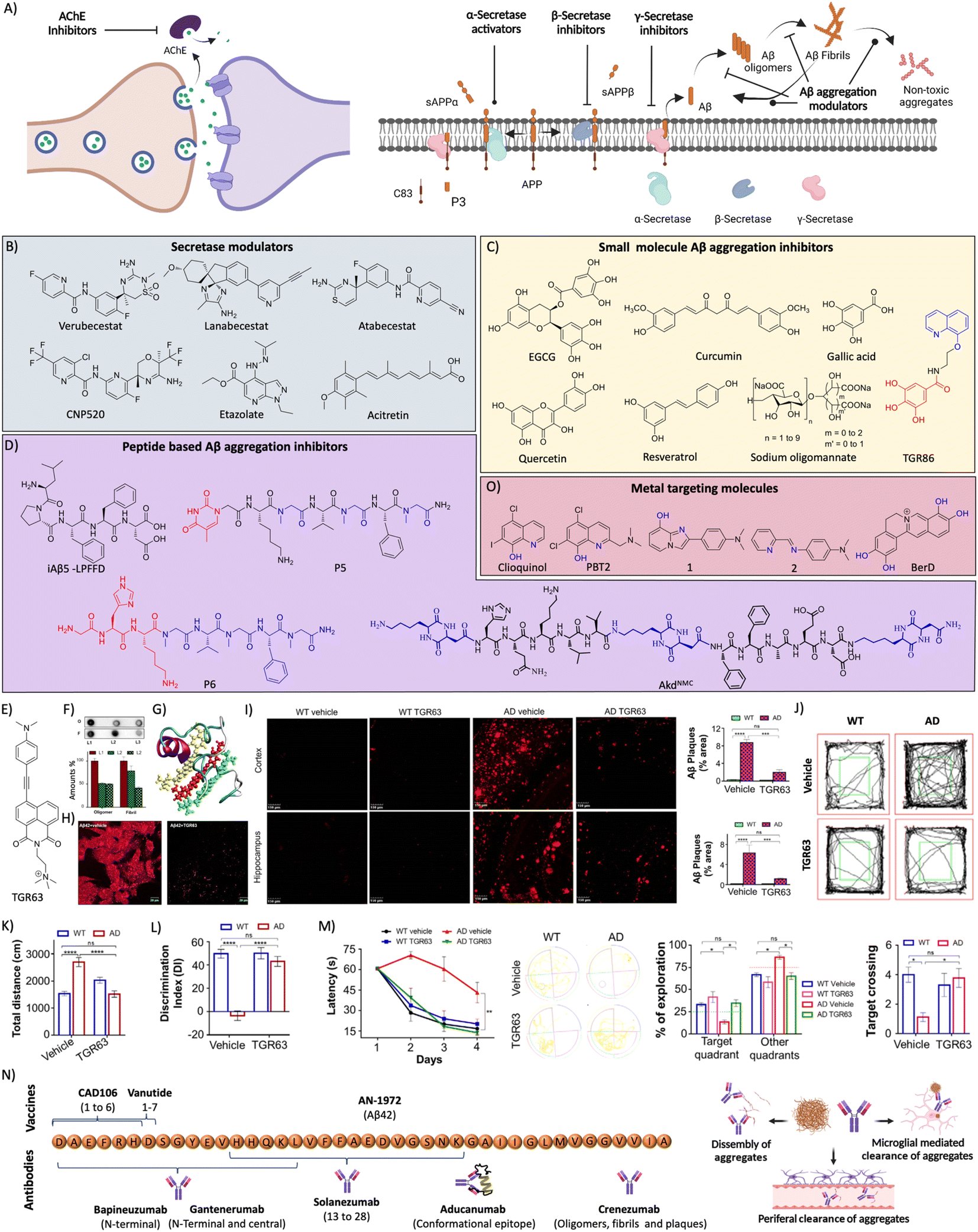 | ||
| Fig. 7 (A) Therapeutic approaches to target cholinergic and amyloid pathways. Therapeutics targeting (B) secretases, and (C) small molecules for Aβ aggregation (blue: quinoline moiety from clioquinol and red: phenolic moiety from EGCG in TGR86). (D) Peptides and peptidomimetics for Aβ aggregation (blue: sarcosine moiety and red: thymine in P5; blue: hybrid peptoid and red: GHK tripeptide in P6; blue: cyclic dipeptide kd in AkdNMC). (E) Chemical structure of TGR63. (F) Dot blot assay of Aβ oligomer and fibril inhibition by TGR63 and its quantification in the absence (L1) and presence of TGR63 at two different molar ratios 1:1 (L2) and 1:5 (L3). (G) Binding of TGR63 with Aβ42 by MD simulation. (H) Reduction of the membrane toxicity of Aβ fibrils by TGR63 revealed by immunofluorescence with fibril antibodies. (I) Immunofluorescence images show the amelioration of amyloid load in the APP/PSEN1 Tg mice model with the treatment of TGR63 and its quantification. (J) Tracing of control and TGR63 treated mice in an open field test and (K) quantification of the total distance travelled by subjects. (L) Novel object recognition by control and treated animals as a measure of the discrimination index (DI). (M) TGR63 treatment rescue learning and memory deficits as revealed by improvement in the latency period, exploration and target crossing from the MWM test. (N) Schematic representation of fragments for Aβ vaccines, the target site of mAb and mechanism of action of mAb in Aβ reduction. (O) Therapeutics targeting metal ion toxicity (blue: metal binding moiety). (F)–(M) Reproduced from ref. 150 with permission from Wiley-VCH, copyright 2021. | ||
3.2 Aβ targeted therapies
Aβ peptide is generated from amyloid precursor protein (APP) by enzymatic cleavage of β- and γ-secretase through the amyloidogenic pathway. The action of α-secretase and γ-secretase follows the non-amyloidogenic pathway that generates physiologically non-toxic peptides. Amyloidogenic processing results in the generation of Aβ peptides that misfold and aggregate to form toxic oligomers, protofibrils, and fibrils. Amyloid-targeted drugs were developed to modulate secretase enzymes and Aβ aggregation (Fig. 7A).Recently, we have designed a set of small molecules based on a naphthalene monoimide (NMI) core functionalised with N,N,N-trimethylethylenediamine as the imide substituent and electron rich N,N-dimethylamine, ethynylbenzene, and 4-ethynyl-N,N-dimethylaniline moieties to fine-tune the hydrophobicity and target Aβ.150In vitro studies demonstrated that the lead compound TGR63 has the potential to modulate Aβ aggregation and dissolve preformed aggregates (Fig. 7E and F). Further in silico studies to understand the mode of interaction showed TGR63 binding to surface and core binding sites mostly driven by electrostatic and van der Waals interactions. TGR63 also binds to the cryptic sites with a reduction of total hydrogen bonding and salt bridge interactions. Aβ42 fibrils consist of 81 intermolecular hydrogen bonds and 48 salt bridges that reduced to 75 and 41, respectively, in the presence of TGR63. There are two modes of binding with fibrils, core binding and surface binding. The ligand and Aβ42 fibril interaction is largely driven by electrostatic and van der Waals interactions with the latter being superior due to fact that electrostatic interactions are largely suppressed by polar solvation free energies. The interaction of TGR63 and Aβ42 monomer is mediated by three low energy binding modes (Fig. 7G). In the presence of TGR63, the α-helix content of Aβ42 effectively was reduced resulting in the formation of nontoxic globular structures. Further nuclear magnetic resonance (NMR) studies revealed the interaction of NMI and aniline aromatic protons with Aβ and the strong interaction of ethylene protons. Cellular studies showed that TGR63 rescues cells from Aβ toxicity and ameliorates membrane toxicity (Fig. 7H). Further in vivo studies showed that TGR63 is non-toxic (LD50 of 157.9 mg Kg−1 BW), stable in serum (24 h), and crosses BBB. TGR63 has effectively reduced Aβ load in the AD phenotypic mice brain (APP/PSEN1 Tg mouse model) as revealed by immunofluorescence data (Fig. 7I). TGR63 rescues AD phenotypic mice from learning, memory, and cognitive deficits as revealed by different behavioural tests (Fig. 7J–M). The memory processing and explorative behavioural rescue was demonstrated by the novel object identification (NOI) test. The learning and memory improvement in TGR63 treated AD mice was observed as evident from decreased latency time and increased exploration in the target platform quadrant in the Morris water maze (MWM) test (Fig. 7M). These in vivo results confirmed the significant improvement in cognitive and memory deficits by TGR63 that underscore its clinical implications and is currently under consideration for further clinical studies.
Bapineuzumab was the first mAb developed against the N-terminal of Aβ42 that selectively binds to oligomers and fibrils. A phase 3 clinical trial showed poor therapeutic effects indicating the limited success of the antibody and there is a need to look for better immunotherapeutics.154 Solanezumab was developed targeting the Aβ13-28 segment that was safe and efficient in clearing Aβ from the brain in preclinical studies. Clinical studies revealed that antibody treatment is safe and shows dose-dependent reduction of Aβ load but failed to rescue from memory deficits.155 Gantenerumab is another mAb developed targeting the N-terminal and central region of Aβ. The clinical trials gave mixed output with therapeutic benefits and safety concerns.156 Clinical studies with a larger sample size are underway. Crenezumab is an immunoglobulin G (IgG) mAb that binds to oligomers, fibrils, and plaques to inhibit the Aβ aggregation and dissemble fibrils. The clinical studies revealed that the mAb is safe at a lower dose and induces microhemorrhages at higher doses.157 Aducanumab is a potent mAb that targets conformational epitope of Aβ and binds to fibrillar aggregates, which has received conditional FDA approval for AD treatment. The approval was controversial with mixed opinions attributed to its moderate clinical benefits and side effects. Clinical studies demonstrated a beneficial effect with an improvement of cognitive score and change in biomarkers.158 The therapeutic efficacy and safety profile need to be established by advanced clinical trials.
3.3 Metal targeted therapeutics
Metal homeostasis is disturbed in the AD brain with increased metal levels (Cu, Zn, Fe and Al) that strongly bind to Aβ peptide and result in elevated toxicity. These metal ions are known to enhance the aggregation and formation of toxic species. The Aβ-metal ion complex results in oxidative stress and membrane damage, and among them Aβ-copper complexes exacerbate the production of ROS and induce biomolecular damage. The molecules possessing strong metal chelation properties and Aβ interacting ability were explored as AD therapeutics (Fig. 7O).159 Initially, clioquinol (Clq) was utilised that chelates copper and zinc and exhibit in vitro and in vivo beneficial effects. Further clinical studies revealed toxic effects and no improvement in the disease condition that results in the failure of the drug.160 A Clq derivative, PBT2, was developed as a metal-protein attenuator drug candidate for AD. PBT2 inhibits Aβ-metal interaction and reduces Aβ deposition and improves the cognitive performance in a Tg AD mice model.161 The clinical trial results were ambiguous with safety issues and insignificant therapeutic benefits reported. EGCG is a polyphenolic compound with Aβ aggregation inhibition, antioxidant and anti-inflammatory properties. The EGCG was also found to interact with metal-Aβ species and form unstructured aggregates and reduce toxicity.162 DP-109 is a lipophilic metal chelator that strongly binds to metal ions and reduces the Aβ burden in the Tg mouse model.163 The treatment with DP-109 has reduced insoluble Aβ aggregates and supports that the Aβ aggregation is driven by metal ions. Lim and co-workers have developed small molecules based on Clq and stilbene that target metals and Aβ, respectively.164 The molecules were designed by introducing nitrogen and/or oxygen donor atoms into Aβ interacting molecules to generate effective bifunctional molecules. Molecules 1 and 2 were potent copper chelators, inhibit Aβ aggregation and ameliorate Cu-Aβ mediated neuronal toxicity. A series of compounds were designed based on a selegiline core to target MAO and metal chelation.165 Among them, compound 8a displays good MAO inhibition, antioxidant activity, and chelation of biometals (Cu, Zn and Fe). Peptidomimetic P6 developed with GHK tripeptide has displayed good Cu chelation and Aβ aggregation inhibition.144 P6 sequesters Cu from Aβ peptide and maintains it in a redox dormant state to prevent ROS generation providing an antioxidant effect and protecting biomolecules from oxidative damage. A novel concept of the multipronged drug design strategy was introduced, wherein structural and functional components of known or failed drugs and natural products were integrated to develop hybrid multifunctional modulators (HMMs) to tackle the multifaceted toxicity of AD. TGR86 integrated with structural and functional components of Clq (modulate metal and Aβ toxicity) and EGCG (antioxidant and Aβ modulation) was found to reduce Cu dependent Aβ aggregation and ROS generation by preventing the redox cycle.140 The molecule successfully inhibits biomolecule and mitochondrial damage and effectively modulates multifaceted Aβ toxicity. We have modified berberine, an isoquinoline natural product, to multifunctional Ber-D, which binds to Cu and forms a ternary complex to prevent ROS generation and exerts antioxidant properties (Fig. 7O).166 Ber-D inhibits amyloid toxicity and rescues neuronal cells from apoptotic cell death.3.4 Tau targeted therapies
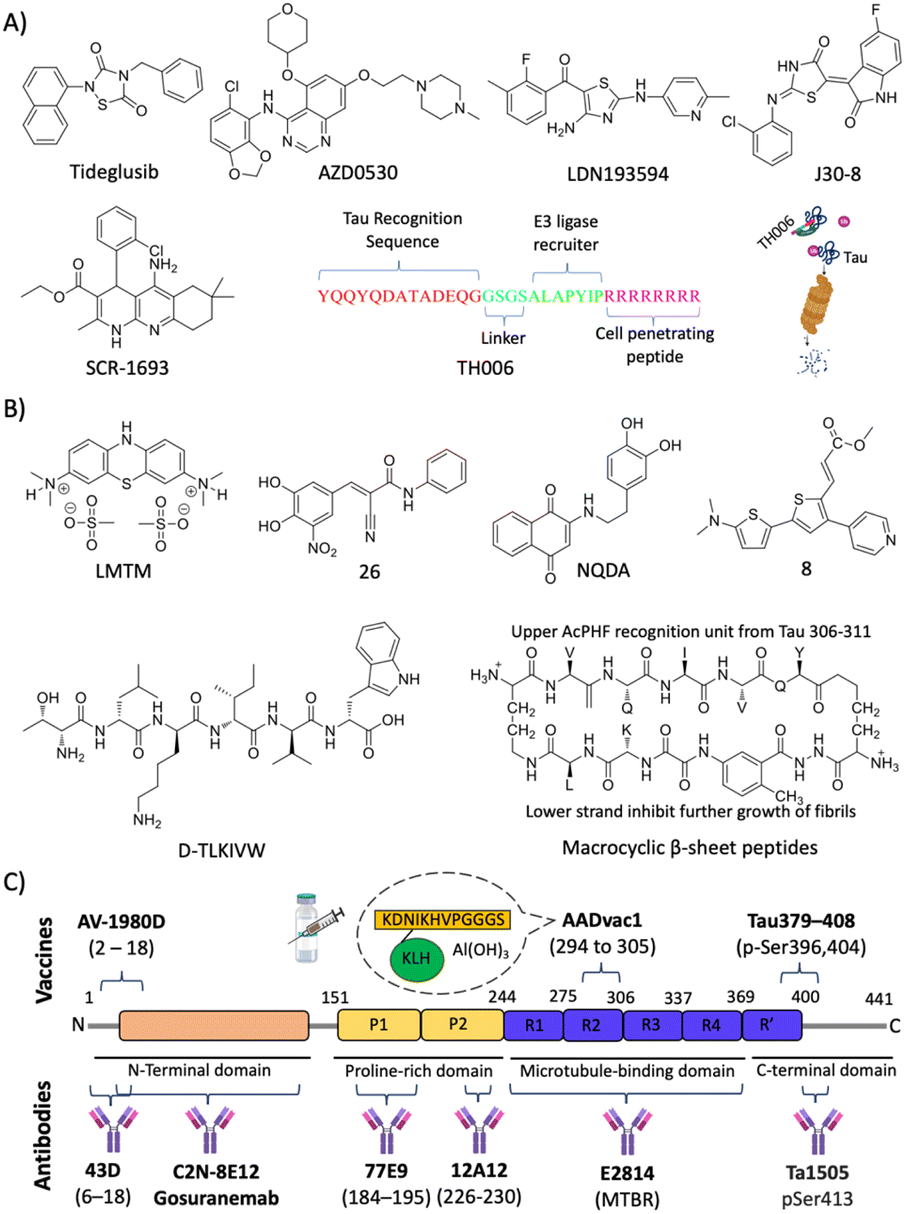 | ||
| Fig. 8 Tau targeted therapies for AD. (A) Small molecules and peptides targeting post translation modifications. (B) Small molecules and peptide-based molecules for tau aggregation inhibition. (C) Tau targeted active and passive immunotherapeutics. | ||
Eisenberg and co-workers have designed structure-based peptides of unnatural amino acids and identified an all D-peptide (D-TLKIVW) inhibitor of tau aggregation (Fig. 8B).181 The peptide targets the steric zipper motif of tau and successfully inhibits seeded and non-seeded tau aggregation. Macrocyclic β-sheet peptides were designed with an upper strand of a pentapeptide with two delta linkers of the ornithine moiety and a lower strand composed of 2 residues and a β sheet peptidomimetic Hao template (Fig. 8B).182 The designed peptidomimetics adopt β-sheet conformation and effectively inhibit the aggregation of PHF motif peptides. Structure-based short peptides from the VQIINK aggregation driver were designed and these peptides effectively inhibit the aggregation of full-length tau protein and the seeding effect of exogenous tau fibrils.183 We have reported peptoids derived from the KLVVF motif of Aβ and their effect on tau aggregation along with controls LPFFD and KLVFF.184 Hybrid peptoids P4 and P5 retained the tau in its random coil state as revealed by CD spectroscopy, inhibiting tau aggregation and rescuing neruro2a cells from tau toxicity.
A humanised anti-tau antibody C2N-8E12 (ABBV-8E12) was developed, which reduces tau burden and rescues cognitive impairments.189 Phase 1 clinical investigation revealed the safety, good pharmacokinetics, and brain uptake with poor immunogenicity. BIIB092 (Gosuranemab, BMS-986168) is a mAb selectively targeting the N-terminal fragment of tau.190 Preclinical studies are promising, and the clinical safety studies showed tolerability up to 2100 mg dose. The treatment of a Tg mouse model with mAb Ta1505 developed against pSer413 has reduced tau burden and improved synaptic density and cognitive deficits.191 Preclinical evaluation of two mAbs 43D (against tau 6–18) and 77 × 109 (against tau 184–195) revealed a reduction of tau burden and rescued from cognitive deficits.192 Six doses of 43D mAb effectively reduced total tau, hyperphosphorylated tau and rescued the 3xTg mouse model from spatial and short-term memory. Interestingly, a reduction in Aβ peptides and aggregates in the hippocampus was observed, which iterates the therapeutic potential of 43D mAb. Recently, 12A12 mAb that selectively binds to a pathologically relevant neurotoxic NH226-230 fragment has been explored for its beneficial effect in two different Tg mouse models.193 The treatment has neutralised the target tau, reduced tau and Aβ burden, and improved learning and memory deficits. The antibody reduced gliosis and rescued dendritic spine connectivity and hippocampal long-term potentiation (LTP). A high affinity mAb E2814 that binds to the microtubule binding region (MTBR) was developed (Kd = 88 pM).194 The results are promising in the Tg mouse model with mAb reducing the seeding, transmission, and tau burden. Many passive immunisations using novel mAb are in clinical trials and show promising results in early clinical studies. The success of mAb therapy in phase 3/4 clinical trials is yet to be disclosed.
3.5 Targeting ROS and oxidative stress
Elevated levels of ROS and oxidative stress in the AD brain compelled researchers to consider antioxidant molecules as therapeutic candidates to combat the disease pathology (Fig. 9A). There is an inter-relation between Aβ-metal ions in the production of ROS and oxidative stress.4,8,159,195 Aβ peptide has metal binding sites and the redox metal ions bound to the peptide generate excess ROS. Copper is a strong redox metal ion that in complexation with Aβ results in excess ROS generation causing membrane (lipid), DNA, and protein damage. These events elevate the oxidative stress of neuronal cells which leads to neurodegeneration. Vitamin E and selenium are natural bioavailable antioxidants and show beneficial effects in in vitro and in vivo studies. Dietary supplementation individually and in combination to AD patients has minimal beneficial effects and the overall results are ambiguous.196 Glutathione (GSH) is a natural tripeptide (γ-L-glutamyl-L-cysteinyl-glycine) produced in the body that has antioxidant activity. GSH acts as a cytoprotective molecule by effectively scavenging ROS, thereby reducing biomolecular damage and oxidative stress. The brain GSH level is reduced in the case of AD causing pronounced oxidative stress, and hence therapeutics elevating GSH levels is a rational approach to treat AD. N-Acetyl-L-cysteine (NAC) treatment is known to elevate the levels of GSH and imparts an antioxidant effect. The treatment of aging rat models with NAC increases the antioxidant enzymes and molecules.197 Recently, a study uncovered that patients with flavanol intake as part of their diet had a reduced risk of AD.198 These results suggest the possible therapeutic potential of flavonols and further need to explore the effect of dietary flavanols for AD treatment. Resveratrol is a natural antioxidant utilised as an AD therapeutic with beneficial effects.199 The beneficial effect of resveratrol was shown in many in vitro and preclinical study and clinical trial results revealed mixed output. Fisetin was assessed for its antioxidant and anti-inflammatory effect in an aging rat model.200 The alleviation of the oxidative stress and neuroprotective effect of the compound underscore fisetin as a potential anti-AD drug candidate. Nuclear factor-erythroid factor 2-related factor 2 (Nrf2) is a transcription factor that is activated to induce the expression of many antioxidant genes to counteract oxidative damage. Sulforaphane was reported to upregulate Nrf2 expression by decreasing methylation in the promoter region and rescue neuronal cells (Fig. 9A).201 The compound reduced Aβ levels and inflammatory mediators and increased cellular antioxidants in a cell model. Caffeic acid phenethyl ester (CAPE) is a potent antioxidant and anti-inflammatory agent assessed for its activity against Aβ oligomers and induced toxicity in the Tg mouse model.202 Treatment induces the expression of Nrf2 and counteracts oxidative stress, apoptosis, neuroinflammation, and rescue memory and cognitive deficits. We have developed GHK-based small molecules that target Aβ and metals, and reduce ROS to rescue from oxidative stress as evident from the emulation of Nrf2 action.203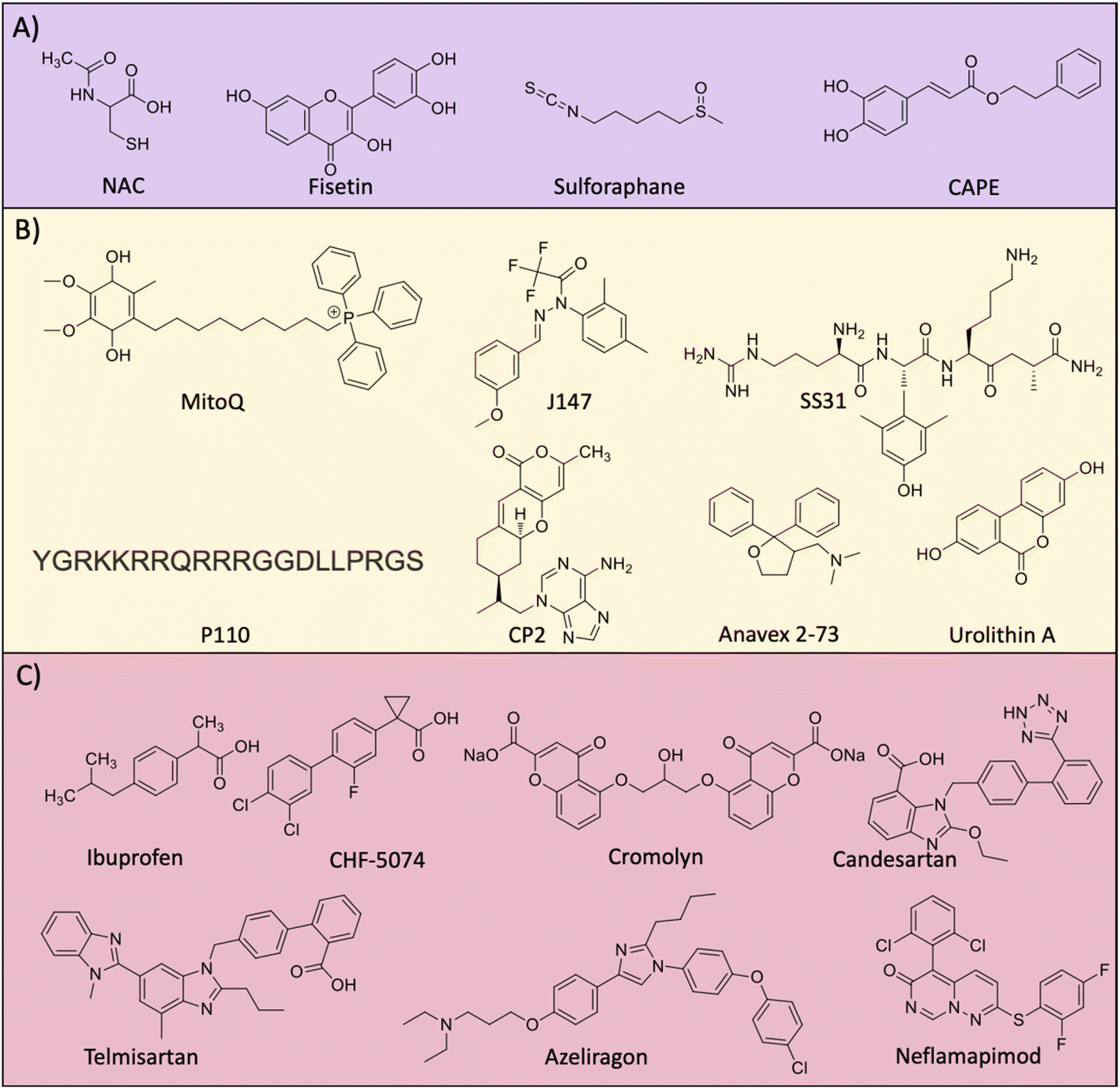 | ||
| Fig. 9 Molecules targeting (A) oxidative stress, (B) mitochondrial damage and (C) neuroinflammation. | ||
3.6 Mitochondrial dysfunction
The accumulation of Aβ and tau aggregates, ROS and oxidative stress results in mitochondrial damage and many therapeutics have been developed targeting mitochondria (Fig. 9B). MitoQ was developed by integrating a mitochondria targeting moiety (PPh3) with a hydroquinone moiety as a potent antioxidant to reduce the mitochondrial damage.204 The treatment with MitoQ in a Tg AD mouse model for a longer period was safe, reduced Aβ load, oxidative stress, and neuroinflammation and alleviated cognitive decline. The design strategy of integrating a mitochondria targeting moiety with diagnostic and therapeutic molecules has potential for AD diagnostics and therapeutics.204,205 J147 is a curcumin analogue targeting mitochondria to improve memory and cognitive deficits in a Tg AD mouse model.206 Mechanistic study reveals that J147 targets ATP synthase in mitochondria and increases the intracellular calcium that leads to the activation of the AMPK/mTOR pathway for improvement in longevity. In AD, multiple pathological factors trigger mitochondrial damage and result in the alteration of mitochondrial dynamics with decreased fusion, increased fission and biogenesis.207 The mitochondrial fission is mediated by Drp1 and Fis1 proteins that interact to fragment the damaged mitochondria. The beneficial effects of tetrapeptide SS31 were assessed in a Tg AD mouse model and it was found that it crosses BBB, inhibits mitochondrial fission, reduces soluble Aβ and enhances synaptic function.208 Resveratrol was shown to act on mitochondrial fission and biogenesis to maintain mitochondrial homeostasis.209 The compound also positively influences energy metabolism with glucose utilisation and ATP production. The rationally designed peptide-based inhibitor of mitochondrial fission P110 inhibits Drp1/Fis1 interaction.210 P110 treatment in Tg AD effectively reduced mitochondrial fission and Aβ accumulation, and rescued from energy imbalance and oxidative stress that underscores the possible clinical implications. Targeting complex 1 of mitochondria to suppress its activity was achieved by small molecule CP2.211 It was evident that treatment with CP2 reduced Aβ and tau burden in the Tg mouse model. CP2 also reduces the activity of GSK3β and restores axonal transport with multiple beneficial effects. The sigma 1 receptor (S1R) is expressed in the endoplasmic reticulum and its function is altered in AD.212 It is a ligand operated receptor that mediates protein homeostasis, synaptic plasticity, and neuroprotection. Anavex 2–73 is a small molecule agonist for S1R that is known to induce the misfolding protein rescue response.213 The compound was safe and improve cognition in a dose dependent manner in mild to moderate AD cases. The abnormal mitophagy is evident in the hippocampus of the human AD brain and induction of mitophagy by urolithin A reduces Aβ and tau burden and rescues memory and cognitive deficits in the Tg mouse model.214 The work demonstrated the role of mitophagy and mitophagy inducers as potential therapeutic candidates for AD. TGR86 that reduced ROS production and Aβ aggregation effectively prevents Aβ induced mitochondrial damage.140 BerD exhibits mitochondrial protection as shown by the rescue of cells from Aβ induced mitochondrial membrane potential (MMP) disruption and Cyt c mediated apoptotic cell death.1663.7 Neuroinflammation
Neuroinflammation is a pathological event that occurs in the early stage of AD. Therapeutic developments targeting neuroinflammation using novel drugs and repurposing of available anti-inflammatory drugs have been explored (Fig. 9C).215 Non-steroid anti-inflammatory drugs (NSAIDs) like ibuprofen, tarenflurbil, and CHF5074 were assessed for their anti-neuroinflammatory properties and encouraging results in vitro and in vivo preclinical studies were found.216 Despite having good safety profiles, most of them failed to improve cognition deficits in clinical studies. Combinational therapy of ibuprofen and cromolyn (ALZT-OP1) is under a phase 3 clinical trial (NCT02547818) and results are yet to be disclosed. Sargramostim is a synthetic granulocyte-macrophage colony-stimulating factor that stimulates the innate immune system to increase activated microglia and reduction in Aβ load, and increased synaptic area and cognitive performance. Clinically, the drug was safe and improve cognitive performance with a better MMSE score.217 Angiotensin1 receptor (AT1R) activation expressed on microglial cells and astrocytes induces neuroinflammation. Candesartan, a potent AT1R blocker exhibits anti-neuroinflammatory effects in the Tg AD mouse model.218 The compound shifted microglial activation towards the neuroprotective phenotype, alleviates lipopolysaccharide (LPS) treated neuroinflammation and significantly reduces the Aβ burden in a Tg AD mouse model. Telmisartan is another AT1R blocker developed as intranasal administration for the amelioration of neuroinflammation.219 Treatment with telmisartan reduces Aβ burden, microglial activation, and neuronal loss and improves the spatial memory of a 5xFAD Tg mouse leading to evaluation in clinical trials. GC021109 is a novel compound reported to target directly microglial cells on the purinergic P2Y6 receptor. The treatment modulates microglial cells to clear Aβ aggregates and reduced the release of proinflammatory mediators and a clinical study to assess the safety and efficacy was undertaken and the results are yet to be disclosed (NCT02386306).220 Receptors for advanced glycation end products (RAGE) are expressed on glial cells and RAGE activation was shown to play a role in AD pathology.221,222 Azeliragon is an orally active small molecule developed as a RAGE inhibitor, which showed to reduce Aβ burden, neuroinflammatory mediators and cognitive performance.223 The drug was effective in mild AD cases and is currently under a phase 3 clinical trial. Tumor necrosis factor alpha (TNFα) is one of the major mediators of neuroinflammation and downstream of the TNFα signalling pathway, p38a regulates the pathway. Neflamapimod is a small molecule inhibitor of p38a that improves synaptic dysfunction and reverses memory deficits. Clinical evaluation in mild to moderate AD cases showed that the drug is safe and well tolerated, but there was no improvement in episodic memory.224 Significant alteration was observed for CSF t-tau, p-tau and neurogranin, which indicates that the higher dose and longer duration of treatment possibly benefit the AD patients. We recently reported a multifunctional small molecule M3 that effectively reduced microglial activation and neuroinflammation.225 M3 effectively inhibits NF-κβ mediated neuroinflammation and reduces TNFα levels in Aβ activated microglial cells.3.8 Multifunctional modulators – future of AD drug discovery
Drugs developed against different individual targets encountered failure and some are in clinical trials. Aβ targeted drug discovery over the last two decades encountered limited success and search for novel tangible targets is necessary. In addition to Aβ, tau is considered a potential target and many therapeutic candidates are developed. Among them, immunotherapeutics are on the road to success in early clinical studies. Many other targets that were considered in the last decade are discussed vide supra. The multifactorial nature of AD and failures in drugs targeting a single pathological target emphasise the need for multifunctional therapeutics to tackle multiple disease causative factors. Recent understandings of various aetiological factors associated with AD helps to design chemical tools to target multiple disease pathologies.226 Natural products have been tuned to develop multifunctional molecules to target metal dependent and independent Aβ toxicity.227 Molecular architecture has been fine tuned to generate Aβ, metal and ROS targeting small molecules.228 Few combination therapies and dual targeting molecules were developed with better therapeutic benefits. We propose multifunctional small molecules that target multiple disease targets as potential future drug candidates for AD (Fig. 10).4,8,9,17 These molecules are anticipated to synergistically alleviate complex AD pathology and future therapeutics for AD.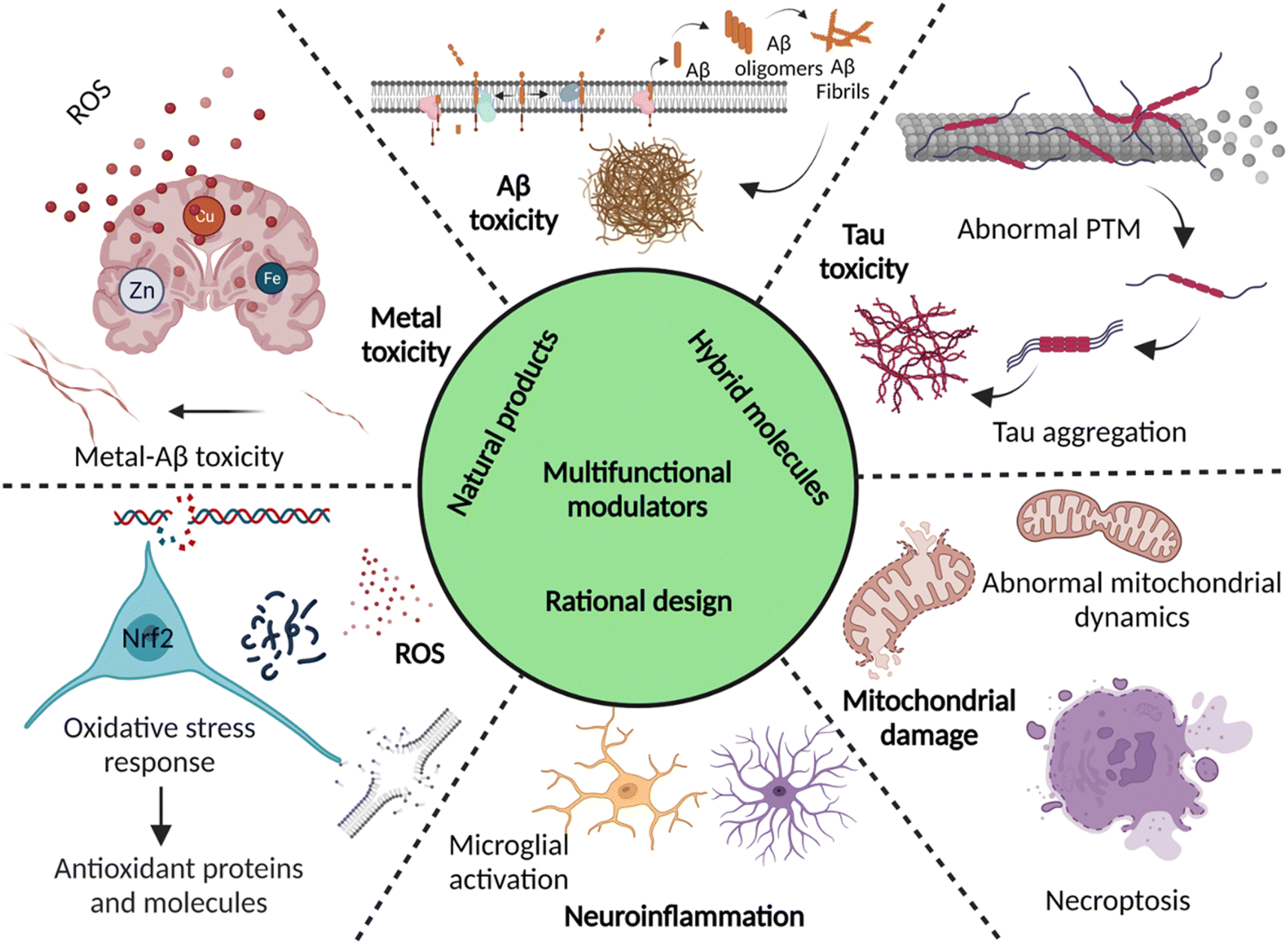 | ||
| Fig. 10 Multifunctional modulators targeting multiple disease pathological pathways of AD (created with http://BioRender.com). | ||
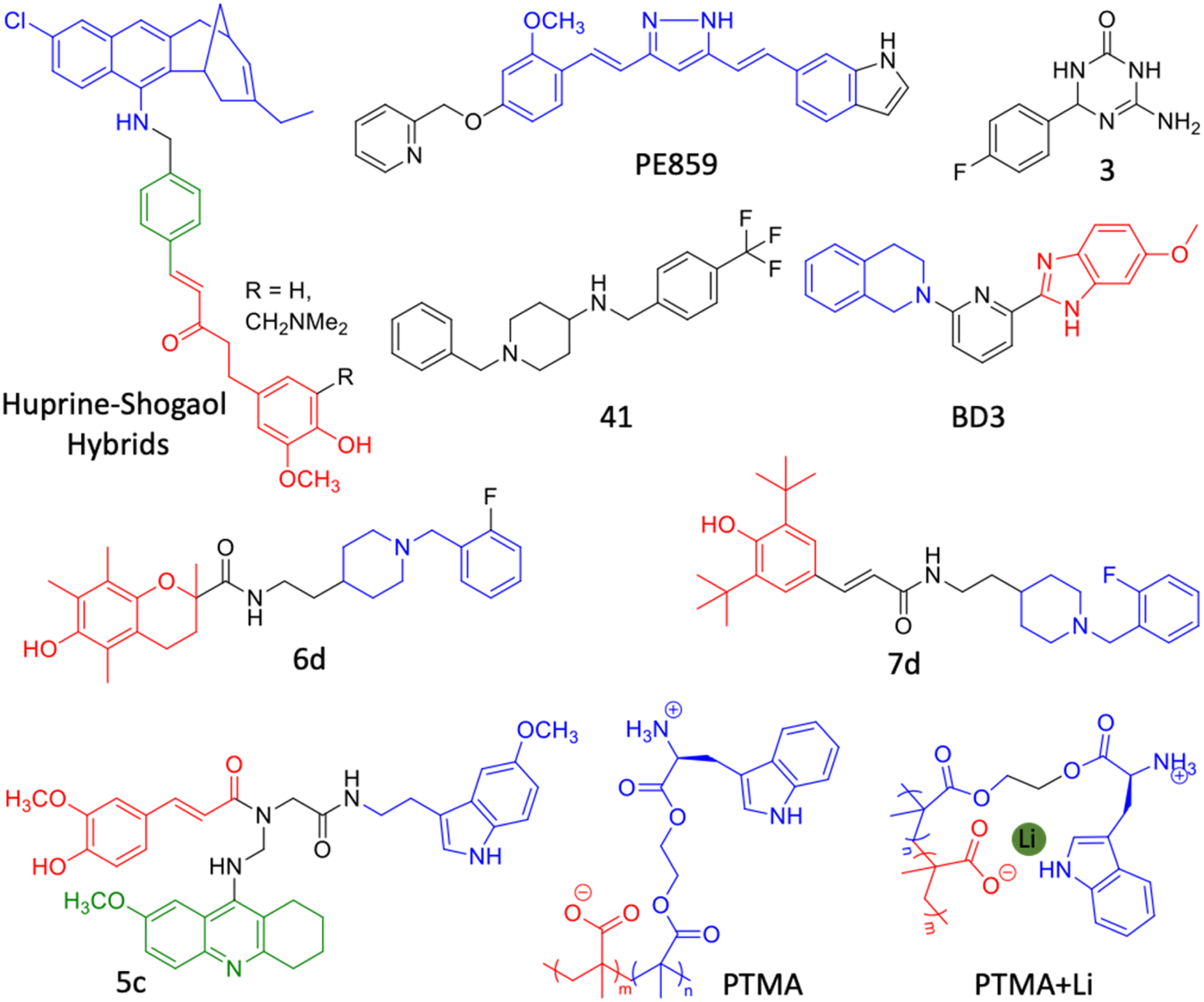 | ||
| Fig. 11 Hybrid multifunctional modulators targeting multiple therapeutic targets of AD (blue: huprine moiety, red: shogaol moiety and green: conjugated benzene ring for Aβ and tau aggregation inhibition in the huprine–shogaol hybrid; blue: curcumin moiety in PE859; blue: tetrahydroisoquinoline moiety and red: benzimidazole moiety in BD3; blue: donepezil moiety and red: trolox moiety in 6d; blue: donepezil moiety and red: butylated hydroxytoluene moiety in 7d; blue: melatonin moiety, red: feruloyl moiety and green: tacrine moiety in 5c; blue: tryptophan based polymer and red: polymethyl acrylate in PTMA). | ||
The development of dual inhibitor targeting of BACE1 and GSK3β responsible for Aβ and tau accumulation is a tangible approach. In this direction, triazinone derivative 3 is reported as an effective dual enzymes inhibitor (Fig. 11).237 Compound 3 showed a neuroprotective effect in cellular models and crossed BBB in the mouse. Hybrid conjugate 6 of tacrine and valmerin linked through a triazole linker inhibits AChE and GSK3β activity with nanomolar IC50, is nontoxic to neuronal cells and crosses BBB (Fig. 11).238 We have designed a diblock copolymer-based polyampholyte (PTMA) with tryptophan to impart biocompatibility, lithium (Li) encapsulation and intrinsic fluorescence to monitor the binding and Li release (Fig. 11).239 The designed PTMA effectively inhibits Aβ aggregation and dissolves the preformed fibrillar aggregates. PTMA is biocompatible with minimal toxicity up to 100 μM and rescues neuronal cells from Aβ toxicity. PTMA effectively encapsulates Li, delivers to cells and releases at acidic pH (endosome) in a stimuli responsive and controlled manner, which can be monitored by intrinsic fluorescence off-on modulation. Li therapy is considered viable to treat AD through possible inhibition of GSK3β by electrostatic and cation–π interactions. The Aβ aggregation modulation and effective Li delivery of PTMA underscore its potential for combinational therapy for AD and other neurological disorders.
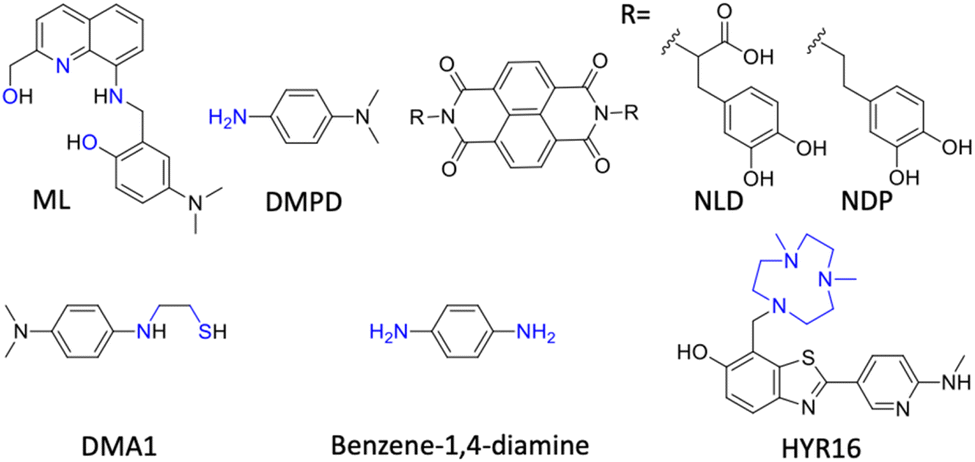 | ||
| Fig. 12 Multifunctional modulators (MFMs) targeting multiple pathological facets of AD (blue: metal chelating moiety in structures). | ||
Han et al. fine-tuned an N,N-dimethylaniline (DMA) moiety to develop bidentate ligands of different oxidation potential, with antioxidant and metal chelation properties.243 DMA1 with the lowest oxidation potential (Epa = 0.22 V) displays noticeable modulation of metal dependent and independent Aβ aggregation with radical scavenging activity (Fig. 12). DMA2 with moderate oxidation potential (Epa = 0.54/0.80 V) exhibits significant inhibition of Cu(II)-Aβ aggregation, whereas DMA3 with higher oxidation potential (Epa = 0.75 V) shows a poor modulatory effect. They further explored the phenylene moiety to design simple molecules with a minimalistic approach based on redox principles to target multiple disease targets.244 Benzene-1,4-diamine exhibits reactivity with free radicals and Aβ in free as well as metal bound states to retard aggregation (Fig. 12). Mechanistic studies revealed that the redox properties of the molecule favour the chemical modification of Aβ peptide by the formation of a chemical adduct that inhibits Aβ aggregation. In vivo treatment with compound results in reduced Aβ load and rescues from memory and cognitive deficits in a Tg AD mouse model. Mirica and the group developed MFMs with therapeutic benefits and imaging abilities (Fig. 12).38 MFMs were designed by combining benzothiazole, an Aβ binding moiety, with a strong Cu chelating 1,4-dimethyl-1,4,7-triazacyclononane (tacn) group. Among the MFMs, HYR-16 showed the prevention of Cu-mediated toxic oligomer formation, metal chelation and ROS scavenging. Ber-D with polyphenolic groups imparts better Cu chelation and antioxidant properties and reduces toxicity by reducing the interaction of Aβ aggregation species with a mitochondrial membrane.166 Ber-D effectively reduces metal independent and dependent Aβ aggregation. MD studies revealed the formation of a BerD-Aβ-Cu tetrahedral cooperative complex that ameliorates Cu-Aβ toxicity. The interaction analysis of Aβ42 monomer and Ber-D revealed that the Aβ42:Ber-D complex is stabilised by hydrogen bonding with His6 and His14 residues and two hydrogen bonds with Asp7 residue. In vitro studies showed antioxidant activity, reduced ROS and oxidative stress, and inhibited DNA and protein damage. Ber-D rescues PC12 neuronal cells from Aβ toxicity and apoptotic cell death. This natural product-derived molecular platform can be explored to develop MFMs for multifaceted AD pathology.
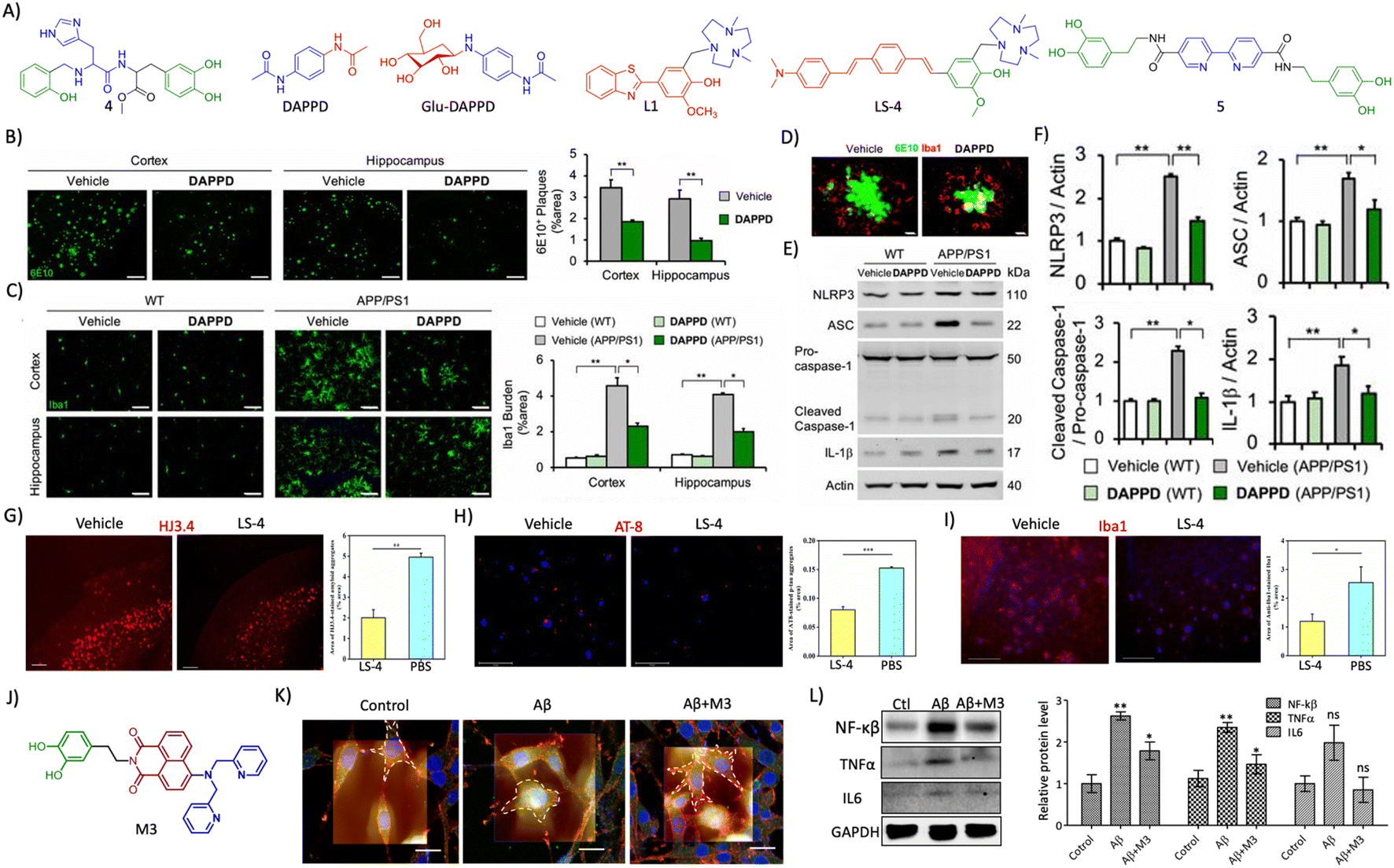 | ||
| Fig. 13 (A) Rationally designed multifunctional modulators targeting amyloid associated toxicity and neuroinflammation (blue: metal targeting moiety and green: L-dopa for antioxidant and anti-inflammatory activity in 4; blue: phenylacetamide moiety for neuroprotection and red: acetamide moiety for lipophilicity and stability in DAPPD; blue: DAPPD moiety and red: glucose moiety for BBB crossing in Glu-DAPPD; blue: triazacyclononane metal chelating moiety and red: 2-phenylbenzothiazole based Aβ targeting moiety in L1; blue: triazacyclononane metal chelating moiety, red: distyrylbenzene Aβ targeting moiety and green: vanillin based antioxidant moiety in LS-4; blue: 2,2′-bipyridine moiety for metal chelation and green: dopamine for antioxidant activity in 5). (B) Reduction of amyloid plaques (6E10 staining, scale bar 200 μm) and (C) microglial activation (Iba1, scale bar 50 μm) by DAPPD treatment in WT and APP/PSEN1 Tg mice. (D) Colocalisation of Aβ plaques with microglial cells in DAPPD treated cells (scale bar 10 μm). (E) Western blot analysis of NLRP3 and associated neuroinflammatory proteins and (F) its quantification in DAPPD treated WT and APP/PSEN1 mice brain samples. (B)– (F) Reproduced from ref. 245 with permission from PNAS, copyright 2019. (G) Immunofluorescence images of brain sections of 5xFAD mice treated with LS-4 stained for Aβ and quantification shows the reduction in Aβ load (scale bar 500 μm). (H) Reduction of tau aggregates by treatment with LS-4 as shown by AT8 staining and quantification (scale bar 125 μm). (I) LS-4 treatment ameliorates microglial activation as revealed by Iba1 immunofluorescence and its quantification (scale bar 125 μm). (G)–(I) Reproduced from ref. 248 with permission from the American Chemical Society, copyright 2021. (J) Structure of multifunctional modulator M3 (blue: DPA moiety for metal chelation, red: NMI for Aβ targeting and green: dopamine for antioxidant and anti-inflammatory activity). (K) Bio-AFM characterization of microglial activation and its reduction by M3 (scale bar 20 μm). (L) Western blot analysis of NF-κβ, TNFα and IL6 and its quantification in M3 treatment for its anti-neuroinflammatory effect. (K) and (L) Reproduced from ref. 225 with permission from the American Chemical Society, copyright 2022. | ||
4. Conclusion and future prospects
High prevalence of the disease and the absence of reliable diagnostics and therapeutics reiterates the need for adopting holistic approaches towards identifying better disease management strategies. The clinically approved PET and MRI imaging of core biomarkers (ATN) for AD diagnosis suffer from reliability, high cost, exceptionally sophisticated and expensive instrumentation, need for clinical expertise, radiation exposure, and low resolution. Notably, there are no approved early and differential diagnostic methods for the detection of AD in the case of mixed dementia. Over the last decade, many NIRF probes have been developed with high selectivity, sensitivity and accurate detection of different biomarkers with the potential for definitive and differential diagnosis of AD from other neurodegenerative disorders. Advancement in NIR imaging technology is not yet available for clinical use. Chemical tools for NIRF are available and the NIRF-based imaging techniques are anticipated to revolutionise AD diagnosis in the near future. The accumulation of different alloforms of Aβ and tau is associated with early and progressive stages of disease, and there is a need for developing chemical probes targeting these different alloforms. Recent and future advancements in NMR and cryo-EM to elucidate high resolution 3D structures of distinct pathologically relevant alloforms of Aβ and tau can foster the development of selective and sensitive probes. Although FDG–PET and MRI have been explored to assess neurodegeneration, they lack characteristic patterns and accuracy of diagnosis. A functional MRI technique augmented with better contrast agents is required to achieve required sensitivity. The development of an atlas encomapsing the structural and functional changes occur in the AD brain at different stages of disease aid the accuracy of early diagnosis and prognosis. In addition to core biomarkers (ATN), indirect biomarkers are emerging as potential targets for early and accurate diagnosis of AD. For instance, neuroinflammation and synaptic damage are evident in advance to clinical symptoms. Although there are potential biomarkers associated with AD pathologies identified, there is ample scope to find new and potentially relevant biomarkers. There are limited chemical probes to target indirect biomarkers and there is a need to develop selective and sensitive probes that can be used in combination with core biomarkers through multiplexing and multimodal imaging and detection for accurate diagnosis. Circulating biomarkers are simple and cost-effective targets for minimally or non-invasive AD diagnosis. Recently, many blood-based biomarkers are identified and validated with high diagnostic value in clinical cases. Among them, Aβ and tau biomarkers are tested in large clinical samples, which correlate with cognitive decline. These biomarkers in blood were analysed using standard ELISA and MS-based techniques. Future research must be aimed at developing simple, sensitive, and reliable chemical probes for the rapid detection of biomarkers in circulating fluids. The panel of circulating biomarkers (protein and RNA) enhances the sensitivity and specificity, through multiplexing and multimodal detection techniques. MS is a powerful technique that needs to be exploited to assess the protein and metabolite biomarker panel for accurate and early diagnosis. The multimodal imaging and multiplexed detection of various reliable biomarkers generating a signature fingerprint provide information on disease onset and progression which can be used for prognosis, early diagnosis, clinical staging and management of personalised medication (Fig. 6). AI and machine learning are highly valuable tools to develop molecular probes and signature fingerprints through integrated efforts by scientific, clinical and technology research communities.Over three decades of therapeutics targeting Aβ yielded just one conditionally approved drug that alters the disease pathology. In recent times, tau has been explored as a potential target with mixed outputs. Many immunotherapeutics targeting both Aβ and tau are successful in preclinical and initial clinical trials. Aβ antibody therapies are facing failures due to poor cognitive improvements, while the fate of tau targeted immunotherapeutics is yet to be fully assessed in large clinical trials. There is ample scope to develop antibody-drug conjugates (ADC) to tackle complex AD pathologies. The active drug aimed at one of the disease mechanisms or targets can be conjugated with mAbs targeting another target to synergistically ameliorate complex disease pathologies. Understanding of complex pathology of AD pushed the drug research from classical Aβ and tau targeting towards other tangible targets by adopting inclusive and holistic approaches. Among them, metal dyshomeostasis oxidative stress, mitochondrial damage and neuroinflammation need special consideration. Neuroinflammation plays a major role in AD and the therapeutics developed to curb neuroinflammation are promising. The treatment of complex multifactorial AD is possible through multipronged drug development strategies that synergistically address multiple targets. In recent years, many groups are actively developing hybrid and multifunctional molecules (HMMs and MFMs) that target two or more disease aetiologies and the results are promising. Utmost care must be taken to the design of HMMs and MFMs that synergistically target multiple disease targets to manage and cure AD. These rational strategies explore small molecules, natural products and their derivatives, and hybrid conjugates to discover multifunctional drug candidates through multiplexed high throughput screening platforms to find the magic bullets for AD (Fig. 10). Advanced computational approaches including AI and machine learning are anticipated to play key roles in the design, screening, and validation of novel drug candidates. The drugs developed in vitro are further assessed in Tg mouse models. The development of viable 3D cell and Tg mouse models inclusive of different AD pathological aspects is necessary to test the true efficacy of HMM and MFM therapeutic candidates.250 Over the years, several studies have showed the role of gender, ethnic and racial differences in AD pathology. AD drug discovery must take these factors into account in preclinical and clinical trials for better and personalised medication.251 Overall, multipronged strategies targeting multiple biomarkers and targets with synergistic action are indispensable in the development of early diagnostics and potent therapeutics to tackle multifactorial AD, which in turn ease the burden on global public health and economy.
Author contributions
TG conceived the topic and outline for the perspective. MR prepared the draft under the guidance of TG. TG and MR finalised the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank JNCASR, CEFPRA (IFCPAR/CEFIPRA-62T10-3), the Department of Science and Technology (DST), New Delhi, India, core grant (CRG/2020/004594), and Science and Engineering Research Board (SERB), New Delhi, India, and MR thanks UGC for a student fellowship and lab members for their inputs.References
- Alzheimer's disease: recent findings in pathophysiology, diagnostic and therapeutic modalities, ed. T. Govindaraju, Royal Society Chemistry, 2021 Search PubMed.
- Alzheimers Association, Alzheimer's Dementia, 2021, 17, 327–406 CrossRef PubMed.
- B. Dubois, H. H. Feldman, C. Jacova and S. T. DeKosky, et al. , Lancet Neurol., 2007, 6, 734–746 CrossRef PubMed.
- K. Rajasekhar, M. Chakrabarti and T. Govindaraju, Chem. Commun., 2015, 51, 13434–13450 RSC.
- S. Samanta, M. Ramesh and T. Govindaraju, Alzheimer's is a multifactorial disease in Alzheimer's disease: recent findings in pathophysiology, diagnostic and therapeutic modalities, Royal Society of Chemistry, 2022, pp. 1–34 Search PubMed.
- P. H. Axelsen, H. Komatsu and I. V. Murray, Physiology, 2011, 26, 54–69 CrossRef CAS PubMed.
- Y. Huang and L. Mucke, Cell, 2012, 148, 1204–1222 CrossRef CAS PubMed.
- K. Rajasekhar and T. Govindaraju, RSC Adv., 2018, 8, 23780–23804 RSC.
- M. Ramesh, P. Gopinath and T. Govindaraju, ChemBioChem, 2020, 21, 1052–1079 CrossRef CAS PubMed.
- J. M. Long and D. M. Holtzman, Cell, 2019, 179, 312–339 CrossRef CAS PubMed.
- M. A. DeTure and D. W. Dickson, Mol. Neurodegener., 2019, 14, 32 CrossRef PubMed.
- C. R. Jack Jr, D. A. Bennett, K. Blennow and M. C. Carrillo, et al. , Alzheimer's Dementia, 2018, 14, 535–562 CrossRef PubMed.
- T. Lashley, J. M. Schott, P. Weston, C. E. Murray, H. Wellington, A. Keshavan, S. C. Foti, M. Foiani, J. Toombs, J. D. Rohrer, A. Heslegrave and H. Zetterberg, Dis. Models Mech., 2018, 5, 11 Search PubMed.
- H. Arora, M. Ramesh, K. Rajasekhar and T. Govindaraju, Bull. Chem. Soc. Jpn., 2020, 93, 507–546 CrossRef CAS.
- P. C. Ke, R. Zhou, L. C. Serpell, R. Riek, T. P. J. Knowles, H. A. Lashuel, E. Gazit, I. W. Hamley, T. P. Davis, M. Fändrich, D. E. Otzen, M. R. Chapman, C. M. Dobson, D. S. Eisenberg and R. Mezzenga, Chem. Soc. Rev., 2020, 49, 5473–5509 RSC.
- S. Woloshin and A. S. Kesselheim, JAMA Intern. Med., 2022, 182, 892 CrossRef PubMed.
- T. Mondal, S. Samanta, A. Kumar and T. Govindaraju, Multifunctional inhibitors of multifaceted Aβ toxicity of Alzheimer's disease in Alzheimer's disease: recent findings in pathophysiology, diagnostic and therapeutic modalities, Royal Society of Chemistry, 2022, pp. 455–486 Search PubMed.
- M. G. Savelieff, G. Nam, J. Kang, H. J. Lee, M. Lee and M. H. Lim, Chem. Rev., 2019, 119, 1221–1322 CrossRef CAS PubMed.
- D. Padhi and T. Govindaraju, J. Med. Chem., 2022, 65, 7088–7105 CrossRef CAS PubMed.
- C. L. Teoh, D. Su, S. Sahu, S.-W. Yun, E. Drummond, F. Prelli, S. Lim, S. Cho, S. Ham, T. Wisniewski and Y.-T. Chang, J. Am. Chem. Soc., 2015, 137, 13503–13509 CrossRef CAS PubMed.
- Y. Li, D. Xu, A. Sun, S.-L. Ho, C.-Y. Poon, H.-N. Chan, O. T. W. Ng, K. K. L. Yung, H. Yan, H.-W. Li and M. S. Wong, Chem. Sci., 2017, 8, 8279–8284 RSC.
- J. Yang, F. Zeng, X. Li, C. Ran, Y. Xu and Y. Li, Chem. Commun., 2020, 56, 583–586 RSC.
- K. Rajasekhar, N. Narayanaswamy, N. A. Murugan, G. Kuang, H. Ågren and T. Govindaraju, Sci. Rep., 2016, 6, 23668 CrossRef CAS PubMed.
- K. Rajasekhar, N. Narayanaswamy, N. A. Murugan, K. Viccaro, H.-G. Lee, K. Shah and T. Govindaraju, Biosens. Bioelectron., 2017, 98, 54–61 CrossRef CAS PubMed.
- W. Ren, J. Zhang, C. Peng, H. Xiang, J. Chen, C. Peng, W. Zhu, R. Huang, H. Zhang and Y. Hu, Bioconjugate Chem., 2018, 29, 3459–3466 CrossRef CAS PubMed.
- G. Lv, A. Sun, M. Wang, P. Wei, R. Li and T. Yi, Chem. Commun., 2020, 56, 1625–1628 RSC.
- K. Zhou, H. Bai, L. Feng, J. Dai and M. Cui, Anal. Chem., 2017, 89, 9432–9437 CrossRef CAS PubMed.
- Y. Li, J. Yang, H. Liu, J. Yang, L. Du, H. Feng, Y. Tian, J. Cao and C. Ran, Chem. Sci., 2017, 8, 7710–7717 RSC.
- J. Hatai, L. Motiei and D. Margulies, J. Am. Chem. Soc., 2017, 139, 2136–2139 CrossRef CAS PubMed.
- W. Fu, C. Yan, Z. Guo, J. Zhang, H. Zhang, H. Tian and W.-H. Zhu, J. Am. Chem. Soc., 2019, 141, 3171–3177 CrossRef CAS PubMed.
- J. Yang, W. Yin, R. Van, K. Yin, P. Wang, C. Zheng, B. Zhu, K. Ran, C. Zhang, M. Kumar, Y. Shao and C. Ran, Nat. Commun., 2020, 11, 4052 CrossRef CAS PubMed.
- K. Zhou, C. Yuan, B. Dai, K. Wang, Y. Chen, D. Ma, J. Dai, Y. Liang, H. Tan and M. Cui, J. Med. Chem., 2019, 62, 6694–6704 CrossRef CAS PubMed.
- W. E. Klunk, H. Engler, A. Nordberg and Y. Wang, et al. , Ann. Neurol., 2004, 55, 306–319 CrossRef CAS PubMed.
- C. Curtis, J. E. Gamez, U. Singh and C. H. Sadowsky, et al. , JAMA Neurology, 2015, 72, 287–294 CrossRef PubMed.
- O. Sabri, M. N. Sabbagh, J. Seibyl and H. Barthel, et al. , Alzheimer's Dementia, 2015, 11, 964–974 CrossRef PubMed.
- C. M. Clark, M. J. Pontecorvo, T. G. Beach and B. J. Bedell, et al. , Lancet Neurol., 2012, 11, 669–678 CrossRef CAS PubMed.
- T. Grimmer, K. Shi, J. Diehl-Schmid, B. Natale, A. Drzezga, S. Förster, H. Förstl, M. Schwaiger, I. Yakushev, H.-J. Wester, A. Kurz and B. H. Yousefi, Mol. Psychiatry, 2020, 25, 2608–2619 CrossRef CAS PubMed.
- Y. Huang, H.-J. Cho, N. Bandara, L. Sun, D. Tran, B. E. Rogers and L. M. Mirica, Chem. Sci., 2020, 11, 7789–7799 RSC.
- J. Yang, Y. Zaim Wadghiri, D. Minh Hoang, W. Tsui, Y. Sun, E. Chung, Y. Li, A. Wang, M. de Leon and T. Wisniewski, NeuroImage, 2011, 55, 1600–1609 CrossRef PubMed.
- J.-H. Kim, T. L. Ha, G. H. Im, J. Yang, S. W. Seo, J. J. Chung, S. Y. Chae, I. S. Lee and J. H. Lee, NeuroReport, 2014, 25 Search PubMed.
- A. Petiet, M. Santin, A. Bertrand, C. J. Wiggins, F. Petit, D. Houitte, P. Hantraye, J. Benavides, T. Debeir, T. Rooney and M. Dhenain, Neurobiol. Aging, 2012, 33, 1533–1544 CrossRef CAS PubMed.
- M. Maruyama, H. Shimada, T. Suhara and H. Shinotoh, et al. , Neuron, 2013, 79, 1094–1108 CrossRef CAS PubMed.
- K.-s. Park, M. K. Kim, Y. Seo, T. Ha, K. Yoo, S. J. Hyeon, Y. J. Hwang, J. Lee, H. Ryu, H. Choo and Y. Chong, ACS Chem. Neurosci., 2017, 8, 2124–2131 CrossRef CAS PubMed.
- S. Lim, M. M. Haque, D. Su, D. Kim, J.-S. Lee, Y.-T. Chang and Y. K. Kim, Chem. Commun., 2017, 53, 1607–1610 RSC.
- P. Verwilst, H.-R. Kim, J. Seo, N.-W. Sohn, S.-Y. Cha, Y. Kim, S. Maeng, J.-W. Shin, J. H. Kwak, C. Kang and J. S. Kim, J. Am. Chem. Soc., 2017, 139, 13393–13403 CrossRef CAS PubMed.
- Y. Chen, C. Yuan, T. Xie, Y. Li, B. Dai, K. Zhou, Y. Liang, J. Dai, H. Tan and M. Cui, Chem. Commun., 2020, 56, 7269–7272 RSC.
- A. A. Elbatrawy, S. J. Hyeon, N. Yue, E. E. A. Osman, S. H. Choi, S. Lim, Y. K. Kim, H. Ryu, M. Cui and G. Nam, ACS Sens., 2021, 6, 2281–2289 CrossRef CAS PubMed.
- Y. Zhao, O. Tietz, W.-L. Kuan, A. K. Haji-Dheere, S. Thompson, B. Vallin, E. Ronchi, G. Tóth, D. Klenerman and F. I. Aigbirhio, Chem. Sci., 2020, 11, 4773–4778 RSC.
- T. J. Betthauser, P. J. Lao, D. Murali, T. E. Barnhart, S. Furumoto, N. Okamura, C. K. Stone, S. C. Johnson and B. T. Christian, J. Nucl. Med., 2017, 58, 996–1002 CrossRef CAS PubMed.
- L. Passamonti, P. Vázquez Rodríguez, Y. T. Hong and K. S. J. Allinson, et al. , Brain, 2017, 140, 781–791 Search PubMed.
- L. Declercq, F. Rombouts, M. Koole, K. Fierens, J. Mariën, X. Langlois, J. I. Andrés, M. Schmidt, G. Macdonald, D. Moechars, W. Vanduffel, T. Tousseyn, R. Vandenberghe, K. Van Laere, A. Verbruggen and G. Bormans, J. Nucl. Med., 2017, 58, 975–981 CrossRef CAS PubMed.
- S. Sanabria Bohórquez, J. Marik, A. Ogasawara and J. N. Tinianow, et al. , Eur. J. Nucl. Med. Mol. Imaging, 2019, 46, 2077–2089 CrossRef PubMed.
- A. Mueller, S. Bullich, O. Barret and J. Madonia, et al. , J. Nucl. Med., 2020, 61, 911–919 CrossRef CAS PubMed.
- D. Yanagisawa, N. F. Ibrahim, H. Taguchi, S. Morikawa, T. Kato, K. Hirao, N. Shirai, T. Sogabe and I. Tooyama, J. Neurosci. Res., 2018, 96, 841–851 CrossRef CAS PubMed.
- A. Badachhape, P. A. Parekh, Q. Mu, R. Bhavane, M. Srivastava, I. Stupin, P. Bhandari, L. Devkota, E. Tanifum, K. Ghaghada and A. Annapragada, Alzheimer's Dementia, 2020, 16, e041080 Search PubMed.
- P. Parekh, Q. Mu, A. Badachhape, R. Bhavane, M. Srivastava, L. Devkota, X. Sun, P. Bhandari, J. L. Eriksen, E. Tanifum, K. Ghaghada and A. Annapragada, Theranostics, 2022, 12, 5504–5521 CrossRef CAS PubMed.
- L. Mosconi, W. H. Tsui, K. Herholz and A. Pupi, et al. , J. Nucl. Med., 2008, 49, 390–398 CrossRef PubMed.
- L. M. Bloudek, D. E. Spackman, M. Blankenburg and S. D. Sullivan, J. Alzheimer's Dis., 2011, 26, 627–645 CAS.
- Y.-N. Ou, W. Xu, J.-Q. Li, Y. Guo, M. Cui, K.-L. Chen, Y.-Y. Huang, Q. Dong, L. Tan and J.-T. Yu, Alzheimer's Res. Ther., 2019, 11, 57 CrossRef CAS PubMed.
- P. J. Nestor, D. Altomare, C. Festari and A. Drzezga, et al. , Eur. J. Nucl. Med. Mol. Imaging, 2018, 45, 1509–1525 CrossRef PubMed.
- I. Kilimann, M. Grothe, H. Heinsen and E. J. L. Alho, et al. , J. Alzheimer's Dis., 2014, 40, 687–700 Search PubMed.
- M. Nesteruk, T. Nesteruk, M. Styczyńska, A. Barczak, M. Mandecka, J. Walecki and M. Barcikowska-Kotowicz, Neurol. Neurochir. Pol., 2015, 49, 349–353 CrossRef PubMed.
- S. Delli Pizzi, R. Franciotti, G. Bubbico, A. Thomas, M. Onofrj and L. Bonanni, Neurobiol. Aging, 2016, 40, 103–109 CrossRef PubMed.
- J. S. Damoiseaux, K. E. Prater, B. L. Miller and M. D. Greicius, Neurobiol. Aging, 2012, 33, 828.e819–828.e830 CrossRef PubMed.
- E. Yu, Z. Liao, D. Mao, Q. Zhang, G. Ji, Y. Li and Z. Ding, Curr. Alzheimer Res., 2017, 14, 628–635 CrossRef CAS PubMed.
- R. Zhou, B. Ji, Y. Kong, L. Qin, W. Ren, Y. Guan and R. Ni, Front. Immunol., 2021, 12, 739130 CrossRef CAS PubMed.
- P. Parbo, R. Ismail, K. V. Hansen and A. Amidi, et al. , Brain, 2017, 140, 2002–2011 CrossRef PubMed.
- M. Dani, M. Wood, R. Mizoguchi, Z. Fan, Z. Walker, R. Morgan, R. Hinz, M. Biju, T. Kuruvilla, D. J. Brooks and P. Edison, Brain, 2018, 141, 2740–2754 Search PubMed.
- S. J. Finnema, N. B. Nabulsi, T. Eid, K. Detyniecki, S.-f. Lin, M.-K. Chen, R. Dhaher, D. Matuskey, E. Baum, D. Holden, D. D. Spencer, J. Mercier, J. Hannestad, Y. Huang and R. E. Carson, Sci. Transl. Med., 2016, 8, 348ra396 Search PubMed.
- C. Bastin, M. A. Bahri, F. Meyer, M. Manard, E. Delhaye, A. Plenevaux, G. Becker, A. Seret, C. Mella, F. Giacomelli, C. Degueldre, E. Balteau, A. Luxen and E. Salmon, Eur. J. Nucl. Med. Mol. Imaging, 2020, 47, 390–402 CrossRef CAS PubMed.
- M. Naganawa, S. Li, N. Nabulsi, S. Henry, M.-Q. Zheng, R. Pracitto, Z. Cai, H. Gao, M. Kapinos, D. Labaree, D. Matuskey, Y. Huang and R. E. Carson, J. Nucl. Med., 2021, 62, 561–567 CrossRef CAS PubMed.
- Z. Cai, L. Drake, M. Naganawa and S. Najafzadeh, et al. , J. Nucl. Med., 2020, 61, 462 CrossRef PubMed.
- S. Samanta and T. Govindaraju, ACS Chem. Neurosci., 2019, 10, 4847–4853 CrossRef CAS PubMed.
- Y. Liu, M. Nguyen, A. Robert and B. Meunier, Acc. Chem. Res., 2019, 52, 2026–2035 CrossRef CAS PubMed.
- S. A. Virk and G. D. Eslick, J. Alzheimer's Dis., 2015, 47, 629–638 CAS.
- A. Damulina, L. Pirpamer, M. Soellradl, M. Sackl, C. Tinauer, E. Hofer, C. Enzinger, B. Gesierich, M. Duering, S. Ropele, R. Schmidt and C. Langkammer, Radiology, 2020, 296, 619–626 CrossRef PubMed.
- J. B. Torres, E. M. Andreozzi, J. T. Dunn, M. Siddique, I. Szanda, D. R. Howlett, K. Sunassee and P. J. Blower, J. Nucl. Med., 2016, 57, 109–114 CrossRef CAS PubMed.
- D. Maity, A. K. Manna, D. Karthigeyan, T. K. Kundu, S. K. Pati and T. Govindaraju, Chem.–Eur. J., 2011, 17, 11152–11161 CrossRef CAS PubMed.
- D. Maity, D. Karthigeyan, T. K. Kundu and T. Govindaraju, Sens. Actuators, B, 2013, 176, 831–837 CrossRef CAS.
- L. Wang, Y.-L. Yin, X.-Z. Liu, P. Shen, Y.-G. Zheng, X.-R. Lan, C.-B. Lu and J.-Z. Wang, Transl. Neurodegener., 2020, 9, 10 CrossRef PubMed.
- D. Maity and T. Govindaraju, Chem. Commun., 2012, 48, 1039–1041 RSC.
- N. Narayanaswamy and T. Govindaraju, Sens. Actuators, B, 2012, 161, 304–310 CrossRef CAS.
- D. Maity and T. Govindaraju, Eur. J. Inorg. Chem., 2011, 2011, 5479–5485 CrossRef CAS.
- D. Maity, V. Kumar and T. Govindaraju, Org. Lett., 2012, 14, 6008–6011 CrossRef CAS PubMed.
- D. Maity and T. Govindaraju, Inorg. Chem., 2010, 49, 7229–7231 CrossRef CAS PubMed.
- J. M. Jung, J. H. Kang, J. Han, H. Lee, M. H. Lim, K. T. Kim and C. Kim, Sens. Actuators, B, 2018, 267, 58–69 CrossRef CAS.
- J. B. Chae, D. Yun, S. Kim, H. Lee, M. Kim, M. H. Lim, K. T. Kim and C. Kim, Spectrochim. Acta, Part A, 2019, 219, 74–82 CrossRef CAS PubMed.
- M. Yang, S. C. Lee, M. Kim, M. H. Lim and C. Kim, Spectrochim. Acta, Part A, 2021, 245, 118899 CrossRef CAS PubMed.
- T. O. Klyucherev, P. Olszewski, A. A. Shalimova, V. N. Chubarev, V. V. Tarasov, M. M. Attwood, S. Syvänen and H. B. Schiöth, Transl. Neurodegener., 2022, 11, 25 CrossRef CAS PubMed.
- S. Nagaraj, M. Ramesh and T. Govindaraju, Circulating biomarkers for the diagnosis of Alzheimer's disease in Alzheimer's disease: recent findings in pathophysiology, diagnostic and therapeutic modalities, Royal Society of Chemistry, 2022, pp. 415–441 Search PubMed.
- K.-Y. Tzen, S.-Y. Yang, T.-F. Chen, T.-W. Cheng, H.-E. Horng, H.-P. Wen, Y.-Y. Huang, C.-Y. Shiue and M.-J. Chiu, ACS Chem. Neurosci., 2014, 5, 830–836 CrossRef CAS PubMed.
- A. Nakamura, N. Kaneko, V. L. Villemagne and T. Kato, et al. , Nature, 2018, 554, 249–254 CrossRef CAS PubMed.
- V. Pérez-Grijalba, J. Arbizu, J. Romero and E. Prieto, et al. , Alzheimer's Res. Ther., 2019, 11, 96 CrossRef PubMed.
- S. Fossati, J. Ramos Cejudo, L. Debure, E. Pirraglia, J. Y. Sone, Y. Li, J. Chen, T. Butler, H. Zetterberg, K. Blennow and M. J. de Leon, Alzheimer's Dementia, 2019, 11, 483–492 CrossRef PubMed.
- S. Janelidze, N. Mattsson, S. Palmqvist and R. Smith, et al. , Nat. Med., 2020, 26, 379–386 CrossRef CAS PubMed.
- N. R. Barthélemy, R. J. Bateman, C. Hirtz, P. Marin, F. Becher, C. Sato, A. Gabelle and S. Lehmann, Alzheimer's Res. Ther., 2020, 12, 26 CrossRef PubMed.
- S. Janelidze, D. Berron, R. Smith, O. Strandberg, N. K. Proctor, J. L. Dage, E. Stomrud, S. Palmqvist, N. Mattsson-Carlgren and O. Hansson, JAMA Neurology, 2021, 78, 149–156 CrossRef PubMed.
- E. H. Thijssen, R. La Joie, A. Strom and C. Fonseca, et al. , Lancet Neurol., 2021, 20, 739–752 CrossRef CAS PubMed.
- N. J. Ashton, T. A. Pascoal, T. K. Karikari and A. L. Benedet, et al. , Acta Neuropathol., 2021, 141, 709–724 CrossRef CAS PubMed.
- R. Tarawneh, G. D'Angelo, D. Crimmins, E. Herries, T. Griest, A. M. Fagan, G. J. Zipfel, J. H. Ladenson, J. C. Morris and D. M. Holtzman, JAMA Neurology, 2016, 73, 561–571 CrossRef PubMed.
- E. A. J. Willemse, A. Sieben, C. Somers, Y. Vermeiren, N. De Roeck, M. Timmers, C. Van Broeckhoven, B. De Vil, P. Cras, P. P. De Deyn, J.-J. Martin, C. E. Teunissen, S. Engelborghs and M. Bjerke, Neurobiol. Aging, 2021, 108, 99–109 CrossRef CAS PubMed.
- A. Öhrfelt, A. Brinkmalm, J. Dumurgier, G. Brinkmalm, O. Hansson, H. Zetterberg, E. Bouaziz-Amar, J. Hugon, C. Paquet and K. Blennow, Alzheimer's Res. Ther., 2016, 8, 41 CrossRef PubMed.
- E. Morenas-Rodríguez, Y. Li, B. Nuscher and N. Franzmeier, et al. , Lancet Neurol., 2022, 21, 329–341 CrossRef.
- A. Vergallo, S. Lista, P. Lemercier and P. A. Chiesa, et al. , Neurobiol. Aging, 2020, 96, 22–32 CrossRef CAS PubMed.
- K. Wilczyńska, M. Maciejczyk, A. Zalewska and N. Waszkiewicz, Front. Psychiatry, 2021, 12, 725511 CrossRef PubMed.
- O. Preische, S. A. Schultz, A. Apel and J. Kuhle, et al. , Nat. Med., 2019, 25, 277–283 CrossRef CAS PubMed.
- N. J. Ashton, S. Janelidze, A. Al Khleifat and A. Leuzy, et al. , Nat. Commun., 2021, 12, 3400 CrossRef CAS PubMed.
- M. Babić Leko, F. Borovečki, N. Dejanović, P. R. Hof and G. Šimić, J. Alzheimer's Dis., 2016, 50, 765–778 Search PubMed.
- L. Feng, Y.-T. Liao, J.-C. He, C.-L. Xie, S.-Y. Chen, H.-H. Fan, Z.-P. Su and Z. Wang, BMC Neurol., 2018, 18, 4 CrossRef PubMed.
- G. Lugli, A. M. Cohen, D. A. Bennett, R. C. Shah, C. J. Fields, A. G. Hernandez and N. R. Smalheiser, PLoS One, 2015, 10, e0139233 CrossRef PubMed.
- S. Nagaraj, K. Laskowska-Kaszub, K. J. Dębski, J. Wojsiat, M. Dąbrowski, T. Gabryelewicz, J. Kuźnicki and U. Wojda, Oncotarget, 2017, 8, 16122–16143 CrossRef PubMed.
- J. T. Wiedrick, J. I. Phillips, T. A. Lusardi, T. J. McFarland, B. Lind, U. S. Sandau, C. A. Harrington, J. A. Lapidus, D. R. Galasko, J. F. Quinn and J. A. Saugstad, J. Alzheimer's Dis., 2019, 67, 875–891 CAS.
- J. D. Doecke, S. M. Laws, N. G. Faux and W. Wilson, et al. , Arch. Neurol., 2012, 69, 1318–1325 CrossRef PubMed.
- N. C. Cullen, A. Leuzy, S. Janelidze, S. Palmqvist, A. L. Svenningsson, E. Stomrud, J. L. Dage, N. Mattsson-Carlgren and O. Hansson, Nat. Commun., 2021, 12, 3555 CrossRef CAS PubMed.
- M. Milà-Alomà, A. Brinkmalm, N. J. Ashton and H. Kvartsberg, et al. , Neurology, 2021, 97, e2065–e2078 CrossRef PubMed.
- L. Jia, M. Zhu, C. Kong, Y. Pang, H. Zhang, Q. Qiu, C. Wei, Y. Tang, Q. Wang, Y. Li, T. Li, F. Li, Q. Wang, Y. Li, Y. Wei and J. Jia, Alzheimer's Dementia, 2021, 17, 49–60 CrossRef CAS PubMed.
- V. R. Varma, A. M. Oommen, S. Varma and R. Casanova, et al. , PLoS Med., 2018, 15, e1002482 CrossRef PubMed.
- M. N. Sabbagh, J. Shi, M. Lee, L. Arnold, Y. Al-Hasan, J. Heim and P. McGeer, BMC Neurol., 2018, 18, 155 CrossRef PubMed.
- M. Shi, Y.-T. Sui, E. R. Peskind, G. Li, H. Hwang, I. Devic, C. Ginghina, J. S. Edgar, C. Pan, D. R. Goodlett, A. R. Furay, L. F. Gonzalez-Cuyar and J. Zhang, J. Alzheimer's Dis., 2011, 27, 299–305 CAS.
- H. Pekeles, H. Y. Qureshi, H. K. Paudel, H. M. Schipper, M. Gornistky and H. Chertkow, Alzheimer's Dementia, 2018, 10, 53–60 CrossRef PubMed.
- E. Carro, F. Bartolomé, F. Bermejo-Pareja, A. Villarejo-Galende, J. A. Molina, P. Ortiz, M. Calero, A. Rabano, J. L. Cantero and G. Orive, Alzheimer's Dementia, 2017, 8, 131–138 CrossRef PubMed.
- M. Ramesh and T. Govindaraju, Lactoferrin: a potential theranostic candidate for Alzheimer's disease in Alzheimer's disease: recent findings in pathophysiology, diagnostic and therapeutic modalities, Royal Society of Chemistry, 2022, pp. 442–454 Search PubMed.
- H. S. Gleerup, C. S. Jensen, P. Høgh, S. G. Hasselbalch and A. H. Simonsen, EBioMedicine, 2021, 67, 103361 CrossRef CAS PubMed.
- G. Kalló, M. Emri, Z. Varga, B. Ujhelyi, J. Tőzsér, A. Csutak and É. Csősz, PLoS One, 2016, 11, e0158000 CrossRef PubMed.
- F. Yao, X. Hong, S. Li, Y. Zhang, Q. Zhao, W. Du, Y. Wang and J. Ni, J. Alzheimer's Dis., 2018, 65, 421–431 CAS.
- M. Ramesh, S. Samanta and T. Govindaraju, Molecular probes for the diagnosis of Alzheimer's disease with implications for multiplexed and multimodal strategies in Alzheimer's disease: recent findings in pathophysiology, diagnostic and therapeutic modalities, Royal Society of Chemistry, 2022, pp. 377–414 Search PubMed.
- J. S. Birks, Cochrane Database of Systematic Reviews, 2006, 1, CD005593 Search PubMed.
- G. Marucci, M. Buccioni, D. D. Ben, C. Lambertucci, R. Volpini and F. Amenta, Neuropharmacology, 2021, 190, 108352 CrossRef CAS PubMed.
- F. Amenta, S. K. Tayebati, D. Vitali and M. A. Di Tullio, Mech. Ageing Dev., 2006, 127, 173–179 CrossRef CAS PubMed.
- F. Amenta, A. Carotenuto, A. M. Fasanaro, R. Rea and E. Traini, J. Alzheimer's Dis., 2014, 42, S281–S288 Search PubMed.
- A. Carotenuto, A. M. Fasanaro, V. Manzo, F. Amenta and E. Traini, Journal of Alzheimer's Disease Reports, 2022, 6, 235–243 Search PubMed.
- E. McDade, I. Voytyuk, P. Aisen, R. J. Bateman, M. C. Carrillo, B. De Strooper, C. Haass, E. M. Reiman, R. Sperling, P. N. Tariot, R. Yan, C. L. Masters, R. Vassar and S. F. Lichtenthaler, Nat. Rev. Neurol., 2021, 17, 703–714 CrossRef PubMed.
- U. Neumann, M. Ufer, L. H. Jacobson and M.-L. Rouzade-Dominguez, et al. , EMBO Mol. Med., 2018, 10, e9316 Search PubMed.
- J. E. Luo and Y.-M. Li, Cell Biosci., 2022, 12, 2 CrossRef CAS PubMed.
- B. Vellas, O. Sol, P. J. Snyder, P. J. Ousset, R. Haddad, M. Maurin, J. C. Lemarie, L. Desire and M. P. Pando, Curr. Alzheimer Res., 2011, 8, 203–212 CrossRef CAS PubMed.
- K. Endres, F. Fahrenholz, J. Lotz, C. Hiemke, S. Teipel, K. Lieb, O. Tüscher and A. Fellgiebel, Neurology, 2014, 83, 1930–1935 CrossRef CAS PubMed.
- Study of APH-1105 in Patients with Mild to Moderate Alzheimer's Disease, ClinicalTrials.gov identifier: NCT03806478, Updated July 27, 2021, https://clinicaltrials.gov/ct2/show/NCT03806478.
- Y. Y. Syed, Drugs, 2020, 80, 441–444 CrossRef PubMed.
- S. Samanta, M. Ramesh, A. Kumar and T. Govindaraju, The role of gut microbiome in Alzheimer's disease and therapeutic strategies in Alzheimer's disease: recent findings in pathophysiology, diagnostic and therapeutic modalities, Royal Society of Chemistry, 2022, pp. 354–376 Search PubMed.
- K. Rajasekhar, K. Mehta and T. Govindaraju, ACS Chem. Neurosci., 2018, 9, 1432–1440 CrossRef CAS PubMed.
- C. Soto, E. M. Sigurdsson, L. Morelli, R. Asok Kumar, E. M. Castaño and B. Frangione, Nat. Med., 1998, 4, 822–826 CrossRef CAS PubMed.
- L. O. Tjernberg, J. Näslund, F. Lindqvist, J. Johansson, A. R. Karlström, J. Thyberg, L. Terenius and C. Nordstedt, J. Biol. Chem., 1996, 271, 8545–8548 CrossRef CAS PubMed.
- K. Rajasekhar, S. N. Suresh, R. Manjithaya and T. Govindaraju, Sci. Rep., 2015, 5, 8139 CrossRef CAS PubMed.
- K. Rajasekhar, C. Madhu and T. Govindaraju, ACS Chem. Neurosci., 2016, 7, 1300–1310 CrossRef CAS PubMed.
- S. Pellegrino, N. Tonali, E. Erba, J. Kaffy, M. Taverna, A. Contini, M. Taylor, D. Allsop, M. L. Gelmi and S. Ongeri, Chem. Sci., 2017, 8, 1295–1302 RSC.
- S. L. Griner, P. Seidler, J. Bowler, K. A. Murray, T. P. Yang, S. Sahay, M. R. Sawaya, D. Cascio, J. A. Rodriguez, S. Philipp, J. Sosna, C. G. Glabe, T. Gonen and D. S. Eisenberg, eLife, 2019, 8, e46924 CrossRef PubMed.
- M. Konar, D. Ghosh, S. Samanta and T. Govindaraju, RSC Chem. Biol., 2022, 3, 220–226 RSC.
- C. Madhu, C. Voshavar, K. Rajasekhar and T. Govindaraju, Org. Biomol. Chem., 2017, 15, 3170–3174 RSC.
- C. Balachandra, D. Padhi and T. Govindaraju, ChemMedChem, 2021, 16, 2558–2587 CrossRef CAS PubMed.
- S. Samanta, K. Rajasekhar, M. Ramesh, N. A. Murugan, S. Alam, D. Shah, J. P. Clement and T. Govindaraju, Adv. Ther., 2021, 4, 2000225 CrossRef CAS.
- C. Holmes, D. Boche, D. Wilkinson, G. Yadegarfar, V. Hopkins, A. Bayer, R. W. Jones, R. Bullock, S. Love, J. W. Neal, E. Zotova and J. A. R. Nicoll, Lancet, 2008, 372, 216–223 CrossRef CAS.
- F. Pasquier, C. Sadowsky, A. Holstein, G. L. P. Leterme, Y. Peng, N. Jackson, N. C. Fox, N. Ketter, E. Liu, J. M. Ryan and ACC-001 (QS-21) Study Team, J. Alzheimer's Dis., 2016, 51, 1131–1143 CAS.
- M. R. Farlow, N. Andreasen, M.-E. Riviere, I. Vostiar, A. Vitaliti, J. Sovago, A. Caputo, B. Winblad and A. Graf, Alzheimer's Res. Ther., 2015, 7, 23 CrossRef PubMed.
- S. Salloway, R. Sperling, N. C. Fox, K. Blennow, W. Klunk, M. Raskind, M. Sabbagh, L. S. Honig, A. P. Porsteinsson, S. Ferris, M. Reichert, N. Ketter, B. Nejadnik, V. Guenzler, M. Miloslavsky, D. Wang, Y. Lu, J. Lull, I. C. Tudor, E. Liu, M. Grundman, E. Yuen, R. Black and H. R. Brashear, N. Engl. J. Med., 2014, 370, 322–333 CrossRef CAS PubMed.
- L. S. Honig, B. Vellas, M. Woodward and M. Boada, et al. , N. Engl. J. Med., 2018, 378, 321–330 CrossRef CAS PubMed.
- S. Ostrowitzki, R. A. Lasser, E. Dorflinger and P. Scheltens, et al. , Alzheimer's Res. Ther., 2017, 9, 95 CrossRef PubMed.
- H. Guthrie, L. S. Honig, H. Lin, K. M. Sink, K. Blondeau, A. Quartino, M. Dolton, M. Carrasco-Triguero, Q. Lian, T. Bittner, D. Clayton, J. Smith and S. Ostrowitzki, J. Alzheimer's Dis., 2020, 76, 967–979 CAS.
- S. Budd Haeberlein, J. O'Gorman, P. Chiao, T. Bussière, P. von Rosenstiel, Y. Tian, Y. Zhu, C. von Hehn, S. Gheuens, L. Skordos, T. Chen and A. Sandrock, Journal of Prevention of Alzheimer's Disease, 2017, 4, 255–263 CAS.
- C. Hureau, Role of metal ions in Alzheimer's disease: mechanistic aspects contributing to neurotoxicity in Alzheimer's disease: recent findings in pathophysiology, diagnostic and therapeutic modalities, Royal Society of Chemistry, 2022, pp. 170–192 Search PubMed.
- Y.-H. Zhang, J. Raymick, S. Sarkar, D. K. Lahiri, B. Ray, D. Holtzman, M. Dumas and L. C. Schmued, Curr. Alzheimer Res., 2013, 10, 494–506 CrossRef CAS PubMed.
- A. I. Bush and R. E. Tanzi, Neurotherapeutics, 2008, 5, 421–432 CrossRef CAS PubMed.
- S.-J. Hyung, A. S. DeToma, J. R. Brender, S. Lee, S. Vivekanandan, A. Kochi, J.-S. Choi, A. Ramamoorthy, B. T. Ruotolo and M. H. Lim, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 3743–3748 CrossRef CAS PubMed.
- J.-Y. Lee, J. E. Friedman, I. Angel, A. Kozak and J.-Y. Koh, Neurobiol. Aging, 2004, 25, 1315–1321 CrossRef CAS PubMed.
- S. S. Hindo, A. M. Mancino, J. J. Braymer, Y. Liu, S. Vivekanandan, A. Ramamoorthy and M. H. Lim, J. Am. Chem. Soc., 2009, 131, 16663–16665 CrossRef CAS PubMed.
- S. Xie, J. Chen, X. Li, T. Su, Y. Wang, Z. Wang, L. Huang and X. Li, Bioorg. Med. Chem., 2015, 23, 3722–3729 CrossRef CAS PubMed.
- K. Rajasekhar, S. Samanta, V. Bagoband, N. A. Murugan and T. Govindaraju, iScience, 2020, 23, 101005 CrossRef CAS PubMed.
- D. Padhi, M. Ramesh and T. Govindaraju, Post-translational modifications and Alzheimer's disease in Alzheimer's disease: recent findings in pathophysiology, diagnostic and therapeutic modalities, Royal Society of Chemistry, 2022, pp. 255–286 Search PubMed.
- S. Lovestone, M. Boada, B. Dubois, M. Hüll, J. O. Rinne, H.-J. Huppertz, M. Calero, M. V. Andrés, B. Gómez-Carrillo, T. León, T. del Ser and ARGO investigators, J. Alzheimer's Dis., 2015, 45, 75–88 CAS.
- C. H. van Dyck, H. B. Nygaard, K. Chen and M. C. Donohue, et al. , JAMA Neurology, 2019, 76, 1219–1229 CrossRef PubMed.
- X. Zhang, I. Hernandez, D. Rei, W. Mair, J. K. Laha, M. E. Cornwell, G. D. Cuny, L.-H. Tsai, J. A. J. Steen and K. S. Kosik, J. Biol. Chem., 2013, 288, 22042–22056 CrossRef CAS PubMed.
- X. Dou, H. Huang, Y. Li, L. Jiang, Y. Wang, H. Jin, N. Jiao, L. Zhang, L. Zhang and Z. Liu, J. Med. Chem., 2019, 62, 6645–6664 CrossRef CAS PubMed.
- X.-L. Wang, Y. Xiong, Y. Yang, Q.-z. Tuo, X.-c. Wang, R. Chen, Q. Tian, Z.-p. Zhang, X. Yan, Z.-y. Yang, J.-Z. Wang and R. Liu, Eur. J. Pharmacol., 2015, 754, 134–139 CrossRef CAS PubMed.
- T.-T. Chu, N. Gao, Q.-Q. Li, P.-G. Chen, X.-F. Yang, Y.-X. Chen, Y.-F. Zhao and Y.-M. Li, Cell Chem. Biol., 2016, 23, 453–461 CrossRef CAS PubMed.
- B. Zhang, J. Carroll, J. Q. Trojanowski, Y. Yao, M. Iba, J. S. Potuzak, A.-M. L. Hogan, S. X. Xie, C. Ballatore, A. B. Smith, V. M.-Y. Lee and K. R. Brunden, J. Neurosci., 2012, 32, 3601–3611 CrossRef CAS PubMed.
- R. M. Tsai, Z. Miller, M. Koestler and J. C. Rojas, et al. , JAMA Neurology, 2020, 77, 215–224 CrossRef PubMed.
- P. Gopinath, M. Ramesh and T. Govindaraju, Tau-targeting therapeutic strategies for Alzheimer's disease in Alzheimer's disease: recent findings in pathophysiology, diagnostic and therapeutic modalities, Royal Society of Chemistry, 2022, pp. 487–514 Search PubMed.
- G. K. Wilcock, S. Gauthier, G. B. Frisoni and J. Jia, et al. , J. Alzheimer's Dis., 2018, 61, 435–457 CAS.
- T. Silva, T. Mohamed, A. Shakeri, P. P. N. Rao, P. Soares da Silva, F. Remião and F. Borges, Eur. J. Med. Chem., 2019, 167, 146–152 CrossRef CAS PubMed.
- A. Paul, G. K. Viswanathan, A. Huber, E. Arad, H. Engel, R. Jelinek, E. Gazit and D. Segal, FEBS J., 2021, 288, 4267–4290 CrossRef CAS PubMed.
- M. Ramesh, A. Acharya, N. A. Murugan, H. Ila and T. Govindaraju, ChemBioChem, 2021, 22, 3348–3357 CrossRef CAS PubMed.
- S. A. Sievers, J. Karanicolas, H. W. Chang, A. Zhao, L. Jiang, O. Zirafi, J. T. Stevens, J. Münch, D. Baker and D. Eisenberg, Nature, 2011, 475, 96–100 CrossRef CAS PubMed.
- J. Zheng, C. Liu, M. R. Sawaya, B. Vadla, S. Khan, R. J. Woods, D. Eisenberg, W. J. Goux and J. S. Nowick, J. Am. Chem. Soc., 2011, 133, 3144–3157 CrossRef CAS PubMed.
- P. M. Seidler, D. R. Boyer, J. A. Rodriguez, M. R. Sawaya, D. Cascio, K. Murray, T. Gonen and D. S. Eisenberg, Nat. Chem., 2018, 10, 170–176 CrossRef CAS PubMed.
- N. V. Gorantla, L. P. Sunny, K. Rajasekhar, P. G. Nagaraju, P. P. Cg, T. Govindaraju and S. Chinnathambi, ACS Omega, 2021, 6, 11131–11138 CrossRef CAS PubMed.
- E. Kontsekova, N. Zilka, B. Kovacech, P. Novak and M. Novak, Alzheimer's Res. Ther., 2014, 6, 44 CrossRef PubMed.
- P. Novak, B. Kovacech, S. Katina and R. Schmidt, et al. , Nature Aging, 2021, 1, 521–534 CrossRef.
- H. Rajamohamedsait, S. Rasool, W. Rajamohamedsait, Y. Lin and E. M. Sigurdsson, Sci. Rep., 2017, 7, 17034 CrossRef PubMed.
- H. Davtyan, W. W. Chen, K. Zagorski, J. Davis, I. Petrushina, K. Kazarian, D. H. Cribbs, M. G. Agadjanyan, M. Blurton-Jones and A. Ghochikyan, Vaccine, 2017, 35, 2015–2024 CrossRef CAS PubMed.
- T. West, Y. Hu, P. B. Verghese, R. J. Bateman, J. B. Braunstein, I. Fogelman, K. Budur, H. Florian, N. Mendonca and D. M. Holtzman, Journal of Prevention of Alzheimer's Disease, 2017, 4, 236–241 CAS.
- A. L. Boxer, I. Qureshi, M. Ahlijanian and M. Grundman, et al. , Lancet Neurol., 2019, 18, 549–558 CrossRef CAS PubMed.
- T. Umeda, H. Eguchi, Y. Kunori, Y. Matsumoto, T. Taniguchi, H. Mori and T. Tomiyama, Ann. Clin. Transl. Neurol., 2015, 2, 241–255 CrossRef CAS PubMed.
- C.-l. Dai, Y. C. Tung, F. Liu, C.-X. Gong and K. Iqbal, Alzheimer's Res. Ther., 2017, 9, 1 CrossRef PubMed.
- V. Corsetti, A. Borreca, V. Latina and G. Giacovazzo, et al. , Brain Commun., 2020, 2, fcaa039 CrossRef PubMed.
- M. Roberts, I. Sevastou, Y. Imaizumi and K. Mistry, et al. , Acta Neuropathol. Commun., 2020, 8, 13 CrossRef CAS PubMed.
- A. S. Pithadia and M. H. Lim, Curr. Opin. Chem. Biol., 2012, 16, 67–73 CrossRef CAS PubMed.
- R. J. Kryscio, E. L. Abner, A. Caban-Holt, M. Lovell, P. Goodman, A. K. Darke, M. Yee, J. Crowley and F. A. Schmitt, JAMA Neurology, 2017, 74, 567–573 CrossRef PubMed.
- G. Garg, S. Singh, A. K. Singh and S. I. Rizvi, Can. J. Physiol. Pharmacol., 2018, 96, 1189–1196 CrossRef CAS PubMed.
- T. M. Holland, P. Agarwal, Y. Wang, S. E. Leurgans, D. A. Bennett, S. L. Booth and M. C. Morris, Neurology, 2020, 94, e1749–e1756 CrossRef CAS PubMed.
- C. Sawda, C. Moussa and R. S. Turner, Ann. N. Y. Acad. Sci., 2017, 1403, 142–149 CrossRef CAS PubMed.
- S. Singh, A. K. Singh, G. Garg and S. I. Rizvi, Life Sci., 2018, 193, 171–179 CrossRef CAS PubMed.
- F. Zhao, J. Zhang and N. Chang, Eur. J. Pharmacol., 2018, 824, 1–10 CrossRef CAS PubMed.
- F. Morroni, G. Sita, A. Graziosi, E. Turrini, C. Fimognari, A. Tarozzi and P. Hrelia, Aging Dis., 2018, 9, 605–622 CrossRef PubMed.
- S. Samanta, K. Rajasekhar, V. Babagond and T. Govindaraju, ACS Chem. Neurosci., 2019, 10, 3611–3621 CrossRef CAS PubMed.
- M. J. McManus, M. P. Murphy and J. L. Franklin, J. Neurosci., 2011, 31, 15703–15715 CrossRef CAS PubMed.
- M. Ramesh, K. Rajasekhar, K. Gupta, V. Babagond, D. K. Saini and T. Govindaraju, Org. Biomol. Chem., 2021, 19, 801–808 RSC.
- J. Goldberg, A. Currais, M. Prior, W. Fischer, C. Chiruta, E. Ratliff, D. Daugherty, R. Dargusch, K. Finley, P. B. Esparza-Moltó, J. M. Cuezva, P. Maher, M. Petrascheck and D. Schubert, Aging Cell, 2018, 17, e12715 CrossRef PubMed.
- X. Zhu, G. Perry, M. A. Smith and X. Wang, J. Alzheimer's Dis., 2013, 33, S253–S262 Search PubMed.
- P. H. Reddy, M. Manczak and R. Kandimalla, Hum. Mol. Genet., 2017, 26, 1483–1496 CrossRef CAS PubMed.
- T. Ahmed, S. Javed, S. Javed, A. Tariq, D. Šamec, S. Tejada, S. F. Nabavi, N. Braidy and S. M. Nabavi, Mol. Neurobiol., 2017, 54, 2622–2635 CrossRef CAS PubMed.
- A. U. Joshi, N. L. Saw, M. Shamloo and D. Mochly-Rosen, Oncotarget, 2018, 9, 6128–6143 CrossRef PubMed.
- L. Zhang, S. Zhang, I. Maezawa, S. Trushin, P. Minhas, M. Pinto, L. W. Jin, K. Prasain, T. D. Nguyen, Y. Yamazaki, T. Kanekiyo, G. Bu, B. Gateno, K. O. Chang, K. A. Nath, E. Nemutlu, P. Dzeja, Y. P. Pang, D. H. Hua and E. Trushina, EBioMedicine, 2015, 2, 294–305 CrossRef PubMed.
- D. A. Ryskamp, S. Korban, V. Zhemkov, N. Kraskovskaya and I. Bezprozvanny, Front. Neurosci., 2019, 13, 862 CrossRef PubMed.
- S. Macfarlane, M. Cecchi, D. Moore, P. Maruff, T. Zografidis and C. Missling, Alzheimer's Dementia, 2016, 12, P1174 Search PubMed.
- E. F. Fang, Y. Hou, K. Palikaras and B. A. Adriaanse, et al. , Nat. Neurosci., 2019, 22, 401–412 CrossRef CAS PubMed.
- D. V. Hansen, B. K. Cullimore, J. C. Johanson and W. Cheng, Microglial blockade of the amyloid cascade: a new therapeutic frontier in Alzheimer's disease: recent findings in pathophysiology, diagnostic and therapeutic modalities, Royal Society of Chemistry, 2022, pp. 193–254 Search PubMed.
- J. Wang, L. Tan, H. F. Wang, C. C. Tan, X. F. Meng, C. Wang, S. W. Tang and J. T. Yu, J. Alzheimer's Dis., 2015, 44, 385–396 CAS.
- H. Potter, J. H. Woodcock, T. D. Boyd and C. M. Coughlan, et al. , Alzheimer's Dementia, 2021, 7, e12158 Search PubMed.
- N. Torika, K. Asraf, R. N. Apte and S. Fleisher-Berkovich, CNS Neurosci. Ther., 2018, 24, 231–242 CrossRef CAS PubMed.
- N. Torika, K. Asraf, H. Cohen and S. Fleisher-Berkovich, Brain, Behav., Immun., 2017, 64, 80–90 CrossRef CAS PubMed.
- Study Evaluating Safety, Tolerability, and PK of Multiple Ascending Doses of GC021109 in Subjects with Mild to Moderate Alzheimer's Disease, ClinicalTrials.gov identifier: NCT02386306, Updated February 3, 2016, https://clinicaltrials.gov/ct2/show/NCT02386306.
- Z. Cai, N. Liu, C. Wang, B. Qin, Y. Zhou, M. Xiao, L. Chang, L. J. Yan and B. Zhao, Cell. Mol. Neurobiol., 2016, 36, 483–495 CrossRef CAS PubMed.
- A. Kumar, L. P. Datta, S. Samanta, H. Arora and T. Govindaraju, Isr. J. Chem., 2021, 61, 222–230 CrossRef CAS.
- A. H. Burstein, M. Sabbagh, R. Andrews, C. Valcarce, I. Dunn and L. Altstiel, Journal of Prevention of Alzheimer's Disease, 2018, 5, 149–154 CAS.
- N. D. Prins, J. E. Harrison, H.-M. Chu and K. Blackburn, et al. , Alzheimer's Res. Ther., 2021, 13, 106 CrossRef CAS PubMed.
- M. Ramesh, C. Balachandra, P. Andhare and T. Govindaraju, ACS Chem. Neurosci., 2022, 13, 2209–2221 CrossRef CAS PubMed.
- J. Han, Z. Du and M. H. Lim, Acc. Chem. Res., 2021, 54, 3930–3940 CrossRef CAS PubMed.
- S. Park, Y. Yi and M. H. Lim, Bull. Korean Chem. Soc., 2021, 42, 17–24 CrossRef CAS.
- M. Kim and M. H. Lim, Bull. Korean Chem. Soc., 2021, 42, 1272–1280 CrossRef CAS.
- M. Okuda, Y. Fujita, I. Hijikuro, M. Wada, T. Uemura, Y. Kobayashi, T. Waku, N. Tanaka, T. Nishimoto, Y. Izumi, T. Kume, A. Akaike, T. Takahashi and H. Sugimoto, J. Alzheimer's Dis., 2017, 59, 313–328 CAS.
- P. Lv, C.-L. Xia, N. Wang, Z.-Q. Liu, Z.-S. Huang and S.-L. Huang, Bioorg. Med. Chem., 2018, 26, 4693–4705 CrossRef CAS PubMed.
- F. J. Pérez-Areales, O. Di Pietro, A. Espargaró and A. Vallverdú-Queralt, et al. , Bioorg. Med. Chem., 2014, 22, 5298–5307 CrossRef PubMed.
- P. Sharma, A. Tripathi, P. N. Tripathi, S. K. Prajapati, A. Seth, M. K. Tripathi, P. Srivastava, V. Tiwari, S. Krishnamurthy and S. K. Shrivastava, Eur. J. Med. Chem., 2019, 167, 510–524 CrossRef CAS PubMed.
- Y. Fang, H. Zhou, Q. Gu and J. Xu, Eur. J. Med. Chem., 2019, 167, 133–145 CrossRef CAS PubMed.
- P. Cai, S.-Q. Fang, X.-L. Yang, J.-J. Wu, Q.-H. Liu, H. Hong, X.-B. Wang and L.-Y. Kong, ACS Chem. Neurosci., 2017, 8, 2496–2511 CrossRef CAS PubMed.
- P. Cai, S. Q. Fang, H. L. Yang, X. L. Yang, Q. H. Liu, L. Y. Kong and X. B. Wang, Eur. J. Med. Chem., 2018, 157, 161–176 CrossRef CAS PubMed.
- M. Benchekroun, A. Romero, J. Egea, R. León, P. Michalska, I. Buendía, M. L. Jimeno, D. Jun, J. Janockova, V. Sepsova, O. Soukup, O. M. Bautista-Aguilera, B. Refouvelet, O. Ouari, J. Marco-Contelles and L. Ismaili, J. Med. Chem., 2016, 59, 9967–9973 CrossRef CAS PubMed.
- F. Prati, A. De Simone, P. Bisignano and A. Armirotti, et al. , Angew. Chem., Int. Ed., 2015, 54, 1578–1582 CrossRef CAS PubMed.
- K. Oukoloff, N. Coquelle, M. Bartolini and M. Naldi, et al. , Eur. J. Med. Chem., 2019, 168, 58–77 CrossRef CAS PubMed.
- L. P. Datta, S. Samanta and T. Govindaraju, ACS Chem. Neurosci., 2020, 11, 2812–2826 CrossRef CAS PubMed.
- S. Lee, X. Zheng, J. Krishnamoorthy, M. G. Savelieff, H. M. Park, J. R. Brender, J. H. Kim, J. S. Derrick, A. Kochi, H. J. Lee, C. Kim, A. Ramamoorthy, M. T. Bowers and M. H. Lim, J. Am. Chem. Soc., 2014, 136, 299–310 CrossRef CAS PubMed.
- J. S. Derrick, R. A. Kerr, Y. Nam, S. B. Oh, H. J. Lee, K. G. Earnest, N. Suh, K. L. Peck, M. Ozbil, K. J. Korshavn, A. Ramamoorthy, R. Prabhakar, E. J. Merino, J. Shearer, J.-Y. Lee, B. T. Ruotolo and M. H. Lim, J. Am. Chem. Soc., 2015, 137, 14785–14797 CrossRef CAS PubMed.
- M. Ramesh, P. Makam, C. Voshavar, H. Khare, K. Rajasekhar, S. Ramakumar and T. Govindaraju, Org. Biomol. Chem., 2018, 16, 7682–7692 RSC.
- J. Han, H. J. Lee, K. Y. Kim, S. J. C. Lee, J.-M. Suh, J. Cho, J. Chae and M. H. Lim, ACS Chem. Neurosci., 2018, 9, 800–808 CrossRef CAS PubMed.
- M. Kim, J. Kang, M. Lee, J. Han, G. Nam, E. Tak, M. S. Kim, H. J. Lee, E. Nam, J. Park, S. J. Oh, J.-Y. Lee, J.-Y. Lee, M.-H. Baik and M. H. Lim, J. Am. Chem. Soc., 2020, 142, 8183–8193 CrossRef CAS PubMed.
- M. H. Park, M. Lee, G. Nam, M. Kim, J. Kang, B. J. Choi, M. S. Jeong, K. H. Park, W. H. Han, E. Tak, M. S. Kim, J. Lee, Y. Lin, Y.-H. Lee, I.-S. Song, M.-K. Choi, J.-Y. Lee, H. K. Jin, J.-s. Bae and M. H. Lim, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 23426–23436 CrossRef CAS PubMed.
- M. Kim, M. H. Park, G. Nam, M. Lee, J. Kang, I.-S. Song, M.-K. Choi, H. K. Jin, J.-s. Bae and M. H. Lim, Mol. Pharm., 2021, 18, 101–112 CrossRef CAS PubMed.
- H.-J. Cho, A. K. Sharma, Y. Zhang, M. L. Gross and L. M. Mirica, ACS Chem. Neurosci., 2020, 11, 1471–1481 CrossRef CAS PubMed.
- L. Sun, H.-J. Cho, S. Sen, A. S. Arango, T. T. Huynh, Y. Huang, N. Bandara, B. E. Rogers, E. Tajkhorshid and L. M. Mirica, J. Am. Chem. Soc., 2021, 143, 10462–10476 CrossRef CAS PubMed.
- D. Padhi, C. Balachandra, M. Ramesh and T. Govindaraju, Chem. Commun., 2022, 58, 6288–6291 RSC.
- A. Mishra, V. Tiwari, S. Singh and S. Shukla, Experimental models to study Alzheimer's disease in Alzheimer's disease: recent findings in pathophysiology, diagnostic and therapeutic modalities, Royal Society of Chemistry, 2022, pp. 591–607 Search PubMed.
- P. Rekulapally, L. Garimella, S. Krishnan, N. Ashwin Kumar and S. N. Suresh, Ethnic and racial differences in the pathophysiology of Alzheimer's disease in Alzheimer's disease: recent findings in pathophysiology, diagnostic and therapeutic modalities, Royal Society of Chemistry, 2022, pp. 608–659 Search PubMed.
| This journal is © The Royal Society of Chemistry 2022 |