
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGeneral stereoretentive preparation of chiral secondary mixed alkylmagnesium reagents and their use for enantioselective electrophilic aminations†
Alexander
Kremsmair‡
,
Henrik R.
Wilke‡
,
Matthias M.
Simon
,
Quirin
Schmidt
,
Konstantin
Karaghiosoff
 and
Paul
Knochel
*
and
Paul
Knochel
*
Department of Chemistry, Ludwig-Maximilians-Universität München, Butenandtstraße 5-13, 81377 München, Germany. E-mail: paul.knochel@cup.uni-muenchen.de
First published on 19th October 2021
Abstract
A general preparation of enantiomerically and diastereomerically enriched secondary alkylmagnesium reagents was reported as well as their use for performing highly stereoselective transition-metal free electrophilic aminations leading to α-chiral amines in up to 97% ee. Thus, the reaction of t-BuLi (2.2 equiv.) with a mixture of chiral secondary alkyl iodides and the commercially available magnesium reagent Me3SiCH2MgCl in a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of pentane and diethyl ether at up to −50 °C provided optically enriched secondary mixed alkylmagnesium species of the type alkyl(Me)CHMgCH2SiMe3 with high retention of configuration (up to 99% ee). The resulting enantiomerically enriched dialkylmagnesium reagents were trapped with electrophiles such as non-enolizable ketones, aldehydes, acid chlorides, isocyanates, chlorophosphines and O-benzoyl hydroxylamines providing α-chiral tertiary alcohols, ketones, amides, phosphines and tertiary amines in up to 89% yield (over three reaction steps) and up to 99% ee.
:1 mixture of pentane and diethyl ether at up to −50 °C provided optically enriched secondary mixed alkylmagnesium species of the type alkyl(Me)CHMgCH2SiMe3 with high retention of configuration (up to 99% ee). The resulting enantiomerically enriched dialkylmagnesium reagents were trapped with electrophiles such as non-enolizable ketones, aldehydes, acid chlorides, isocyanates, chlorophosphines and O-benzoyl hydroxylamines providing α-chiral tertiary alcohols, ketones, amides, phosphines and tertiary amines in up to 89% yield (over three reaction steps) and up to 99% ee.
Introduction
Organomagnesium reagents are key intermediates in organic synthesis, which have found numerous applications.1 Nevertheless, a general and practical enantioselective preparation of secondary alkylmagnesium reagents is still pending. To date, only one example of a non-heteroatom-stabilized α-chiral Grignard reagent has been reported, which was prepared via a sulfoxide-magnesium exchange.2 Furthermore, deracemization of a mixture of endo- and exo- norbornylmagnesium bromide using benzophenone3 or exploiting the different reactivities of menthylmagnesium chloride epimers4 have been reported. Recently, we have shown that various enantiomerically enriched secondary alkyl iodides of type 1 underwent an I/Li-exchange5 at −100 °C in pentane:ether mixtures furnishing the corresponding chiral alkyllithiums of type 2 with retention of configuration (Scheme 1a).6 After transmetalations with appropriate ether soluble Cu and Zn salts at −100 °C, chiral organometallics of type 3 were obtained, which reacted with various electrophiles providing products of type 4 with high retention of configuration.7 Although chiral building blocks and natural products were available by these procedures,7d,g,h such reaction sequences required very low temperatures (−100 °C) due to the occurrence of configurationally labile alkyllithium reagents (2), which epimerized readily at temperatures above −100 °C (ref. 6b) and tolerated no sensitive functional groups. Thus, Barbier-conditions involving the generation of the organometallic species in the presence of an electrophile or transmetalation reagent might circumvent the configurational lability and high reactivity of these secondary alkyllithiums.8 Recently, we have shown that Barbier conditions were broadly applicable for the directed lithiation of (hetero)aromatics with TMPLi (TMP = 2,2,6,6,-tetramethylpiperidyl) in the presence of various salts.9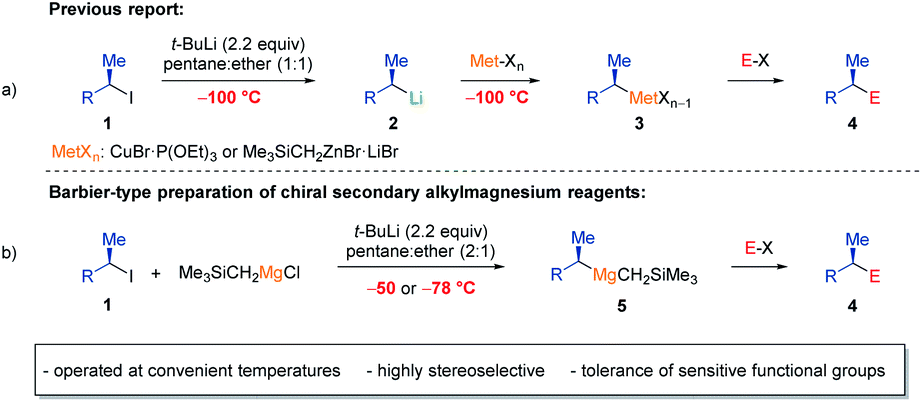 | ||
| Scheme 1 (a) Previous preparation of chiral secondary alkyl organometallics of type 3via I/Li-exchange and subsequent transmetalation at −100 °C; (b) Barbier-type preparation of chiral secondary alkylmagnesium reagents (5) via I/Li-exchange and in situ transmetalation with Me3SiCH2MgCl at −50 °C or −78 °C. | ||
With these previous reports in mind, we have envisioned Barbier conditions, that would overcome the use of unpractical very low temperatures for the preparation of chiral secondary alkylmagnesium reagents (5) by generating the chiral secondary alkyllithiums in the presence of an appropriate magnesium transmetalation reagent.
Herein, we report a general and practical preparation of non-stabilized enantiomerically enriched mixed alkylmagnesium reagents of type 5 from optically enriched secondary alkyl iodides in the presence of commercially available Me3SiCH2MgCl using t-BuLi at convenient temperatures of up to −50 °C (Scheme 1b). These enantiomerically enriched Grignard reagents (5) reacted with a range of eletrophiles including ketones, aldehydes, acid chlorides, isocyanates, S-methyl methanethiosulfonate, chlorophosphines and O-benzoyl hydroxylamines providing products of type 4 such as α-chiral tertiary alcohols, ketones, amides, thioethers, phosphines and tertiary amines in up to 89% yield (over 3 reaction steps) with high retention of configuration (up to 99% ee).
Results and discussion
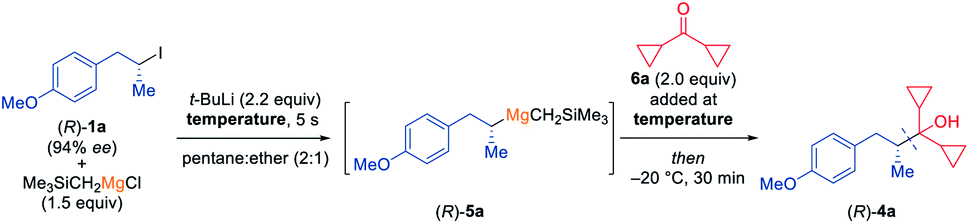
Thus, in preliminary experiments, we have mixed the chiral secondary alkyl iodide (R)-1a with Me3SiCH2MgCl and have subsequently added t-BuLi (2.2 equiv.) at various temperatures (see Table 1). The resulting secondary alkylmagnesium species (R)-5a was quenched with dicyclopropyl ketone (6a) and stirred at −20 °C for 30 min. The desired tertiary alcohol (R)-4a was obtained in 80% yield and 91% ee at a reaction temperature of −100 °C (entry 1). To our delight, the same enantioselectivity was achieved by performing the I/Li-exchange at −78 °C (entry 2). As expected, if the reaction was performed at −78 °C without Me3SiCH2MgCl, (R)-4a was obtained in comparable yield (78%) but only with 12% ee (entry 3). A slight erosion of enantioselectivity was observed if the reaction was carried out at −60 °C (73% yield; 90% ee, entry 4). However, higher temperatures led to a significant loss of enantiomeric purity as a reaction at −40 °C afforded (R)-4a in 71% yield and 82% ee (entry 5). Further raising the reaction temperature and performing the exchange at −20 °C gave (R)-4a in 59% yield with 52% ee (entry 6). Interestingly, using 2.0 equiv. of Me3SiCH2MgCl considerably reduced the racemization rate at −20 °C, since the tertiary alcohol (R)-4a was obtained in 78% ee and 52% yield (entry 7). Lowering the reaction temperature to −40 °C again and using 2.0 equiv. of Me3SiCH2MgCl did not give any improved result (67% yield, 84% ee, entry 8). Alternative transmetalating reagents may also be used for this reaction, however with less satisfactory results.10 Next, we have examined the configurational stability of such chiral secondary alkylmagnesium reagents of type 5. Therefore, we have generated the mixed dialkylmagnesium reagent (R)-5a at −78 °C and kept it at this temperature for 1 h before adding 6a, yielding the corresponding alcohol (R)-4a in 75% yield and 91% ee (entry 9). A detectable racemization was observed after stirring the reaction mixture of (R)-5a for 1 h (71% yield, 86% ee; entry 10) or 3 h (64% yield, 69% ee; entry 11) at −50 °C. Furthermore, a large loss of enantioselectivity was observed by keeping (R)-5a for 1 h at −20 °C (66% yield, 60% ee, entry 12). These results indicated, that mixed non-stabilized secondary alkylmagnesium reagents of type 5 were configurationally stable up to ca. −50 °C for ca. 1 h and in comparison with the corresponding alkyllithium reagents (configurationally stable only below −100 °C for some minutes)6 were better suited for synthetic applications.|
|
|||
|---|---|---|---|
| Entry | Temperature [°C] | Yield of (R)-4ab | ee of (R)-4ac |
| a Stereoselective preparation of (R)-5a at various reaction temperatures under Barbier conditions. b The yield was determined by GC-analysis of reaction aliquotes. c The enantiomeric excess (% ee) was determined by chiral HPLC-analysis. d Yield of analytically pure isolated product. e The reaction was performed without Me3SiCH2MgCl. f Me3SiCH2MgCl (2.0 equiv.) was used. g The chiral Grignard reagent (R)-5a was kept for 1 h at this temperature before quenching. h The chiral Grignard reagent (R)-5a was kept for 3 h at this temperature before quenching. | |||
| (a) Reaction condition optimization | |||
| 1 | −100 | 80% | 91% |
| 2 | −78 | 80% (75%) | 91% |
| 3 | −78 | 78%e | 12% |
| 4 | −60 | 73% | 90% |
| 5 | −40 | 71% | 82% |
| 6 | −20 | 59% | 52% |
| 7 | −20f | 52% | 78% |
| 8 | −40f | 67% | 84% |
| (b) Configurational stability evaluation of (R)- 5a | |||
| 9 | −78g | 75% | 91% |
| 10 | −50 | 71% | 86% |
| 11 | −50h | 64% | 69% |
| 12 | −20g | 66% | 60% |
With these optimized conditions in hand (Table 1, entry 2), we have prepared chiral Grignard reagents (5a–i) with high retention of configuration starting from the corresponding enantiomerically enriched secondary alkyl iodides (1a-I; Scheme 2a). Thus, using (S)-5a (ca. 92–94% ee (ref. 11) instead of (R)-5a described in Table 1) obtained from the alkyl iodide (S)-1a (95% ee), gave the alcohol (S)-4a after quenching with 6a in 81% yield and 90% ee. Related Grignard reagents such as (S)-5b (ca. 99% ee) or (S)-5c (ca. 98% ee) add similarly to 6a or 6b providing the alcohols (S)-4b and (S)-4c in 65–73% yield and 98–99% ee. Also, diastereomerically enriched Grignard reagents syn-5d (dr = 99:1) and anti-5d (dr = 1:99) provided after addition to 6a the alcohols syn- and anti-4d respectively in 74% yield and dr = 96:4 as well as 70% yield and dr = 5:95. Remarkably, functionalized alkylmagnesium species bearing a tert-butyl ester function such as (R)- and (S)-5e or an alkynyl group like (R)-5f reacted with excellent stereoretention with 6a giving the alcohols (R)-4e (65% yield, 93% ee) or (S)-4e (72% yield, 93% ee) and (R)-4f (73% yield, 93% ee). Although enolizable ketones did not give satisfactory results, addition of Grignard reagents (R)-5a, (R)- and (S)-5g and (R)-5h to aldehydes such as c-hexylcarboxaldehyde (6c), (S)-(−)-citronellal (6d) and (1R)-(−)-myrtenal (6e) afforded after Dess–Martin oxidation the corresponding ketones (R)-4g (68%, 89% ee), (2R,5S)-4h (74%, dr = 95:5), (2S,5S)-4h (75%, dr = 5:95) as well as (2R,1′R,5′S)-4i (46%, dr = 95:5) and (2S,1′R,5′S)-4i (49%, dr = 3:97). Performance of acylations did not require Weinreb amides as used for the acylation of chiral alkyllithiums,6c but commercially available acid chlorides were employed. Thus, (R)-5a reacted with (S)-tetrahydrofuran-2-carbonyl chloride (6f) affording (2S,2′R)-4j in 89% yield and dr = 11:89. Similarly, acylation of anti-5i with 3-fluorobenzoyl chloride (6g) led to anti-4k in 52% yield and dr = 5:95. Further functionalizations of these optically enriched Grignard reagents were realized by addition of (R)- and (S)-5g, (S)-5b and (S)-5e to cyclohexyl isocyanate (6h) and the commercial chiral (S)-(−)-α-methylbenzyl isocyanate (6i, 96% ee) or (R)-(−)-1-(1-naphthyl)ethyl isocyanate (6j, 95% ee) providing under our standard conditions, the chiral amides (R)-4l (80%, 94% ee) or (S)-4l (81%, 94% ee) as well as the diastereomerically enriched amides (2S,1′S)-4m (71%, dr = 96; 4) and (2R,1′R)-4n (53%, dr = 96:4). The enantioselective preparation of carbon-sulfur bonds was briefly examined since it proceeded also directly with chiral secondary alkyllithiums.6 We observed that the coupling of (R)- or (S)-5h and syn-5d with MeSO2SMe (6k) led to the thioethers (R)- or (S)-4o and syn-4p in up to 85% yield, but moderate stereoselectivity (up to 78% ee or dr = 93:7). However, phosphorus electrophiles such as Ph2PCl (6l) reacted with high stereoretention with chiral Grignard reagents. Thus, (R)- and (S)-5g or (R)-5e led, after protection with BH3·SMe2,12 to a practical synthesis of chiral phosphine BH3-complexes (R)- and (S)-4q as well as (R)-4r in up to 75% yield and up to 90% ee. These optically enriched phosphines could be of interest for asymmetric catalysis.
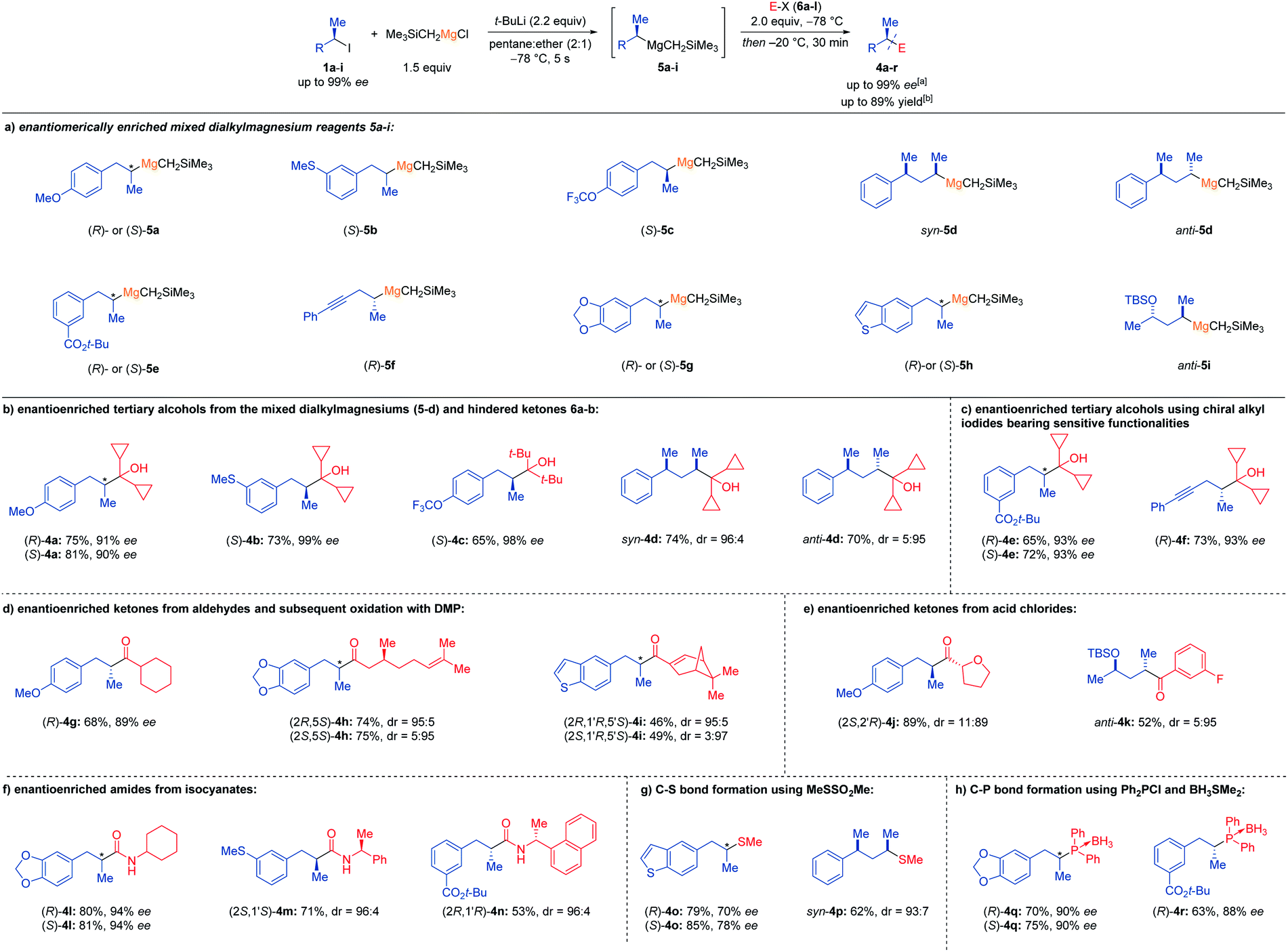 | ||
| Scheme 2 Scope of optically enriched secondary alkylmagnesium reagents 5a-i and subsequent reactions with electrophiles (6a–l). Enantiomerically and diastereomerically enriched secondary alkylmagnesium reagents 5a–i prepared by an I/Li-exchange in the presence of Me3SiCH2MgCl and their reactions with electrophiles (6a–l) leading to the corresponding optically and diastereomerically enriched products 4a–r. aThe enantiomeric excess (% ee) was determined by chiral HPLC-analysis. The diastereomeric ratio (dr; syn/anti ratio) was determined by 1H-NMR spectroscopy and GC-analysis. bYield refers to isolated analytically pure compounds. | ||
α-Chiral alkyl amines are of considerable interest due to their presence in natural products, pharmaceuticals and other biologically active molecules.13 Inspired by pioneering work of Johnson,14 we envisioned using O-benzoyl hydroxylamines15 as electrophilic amination reagents with chiral dialkyl Grignard reagents of type 5. After optimization, we have found that at a reaction temperature of −50 °C, the desired α-chiral amines 8a–j were obtained with high stereoretention (Scheme 3). Remarkably, all these reactions occurred chemoselectively without any transition metal additive and only minimal amounts of usual side-products (ketone)16 were observed. Thus, (R)- and (S)-5a reacted with 4-(pyrimidin-2-yl)piperazin-1-yl benzoate (7a) affording the corresponding α-chiral tertiary amines (R)- and (S)-8a in 73% yield and 88–91% ee. Furthermore, sertraline, a commercially available drug molecule,17 was successfully aminated via7b and (S)-5a using this procedure, giving (2′S,1S,4S)-8b in 52% yield and dr = 91:9. Also, the chiral Grignard reagents (R)- and (S)-5g were generated at −50 °C and trapped with 6,7-dihydrothieno[3,2-c]pyridin-5(4H)-yl benzoate (7c) and non-cyclic O-benzoyl-N,N-bis(2-methoxyethyl) hydroxylamine (7d) yielding (R)- and (S)-8c as well as (R)- and (S)-8d in 70–85% yield and 84–93% ee. The functionalized secondary alkylmagnesium reagent (R)-5e was also aminated with 7c providing (R)-8e in 68% yield and 83% ee. Additionally, the aminated benzothiophene derivatives (R)- and (S)-8f–g were prepared in up to 85% yield and up to 97% ee from optically enriched Grignard reagents (R)- and (S)-5h and the O-benzoyl hydroxylamines 7c and N-morpholino benzoate (7e).
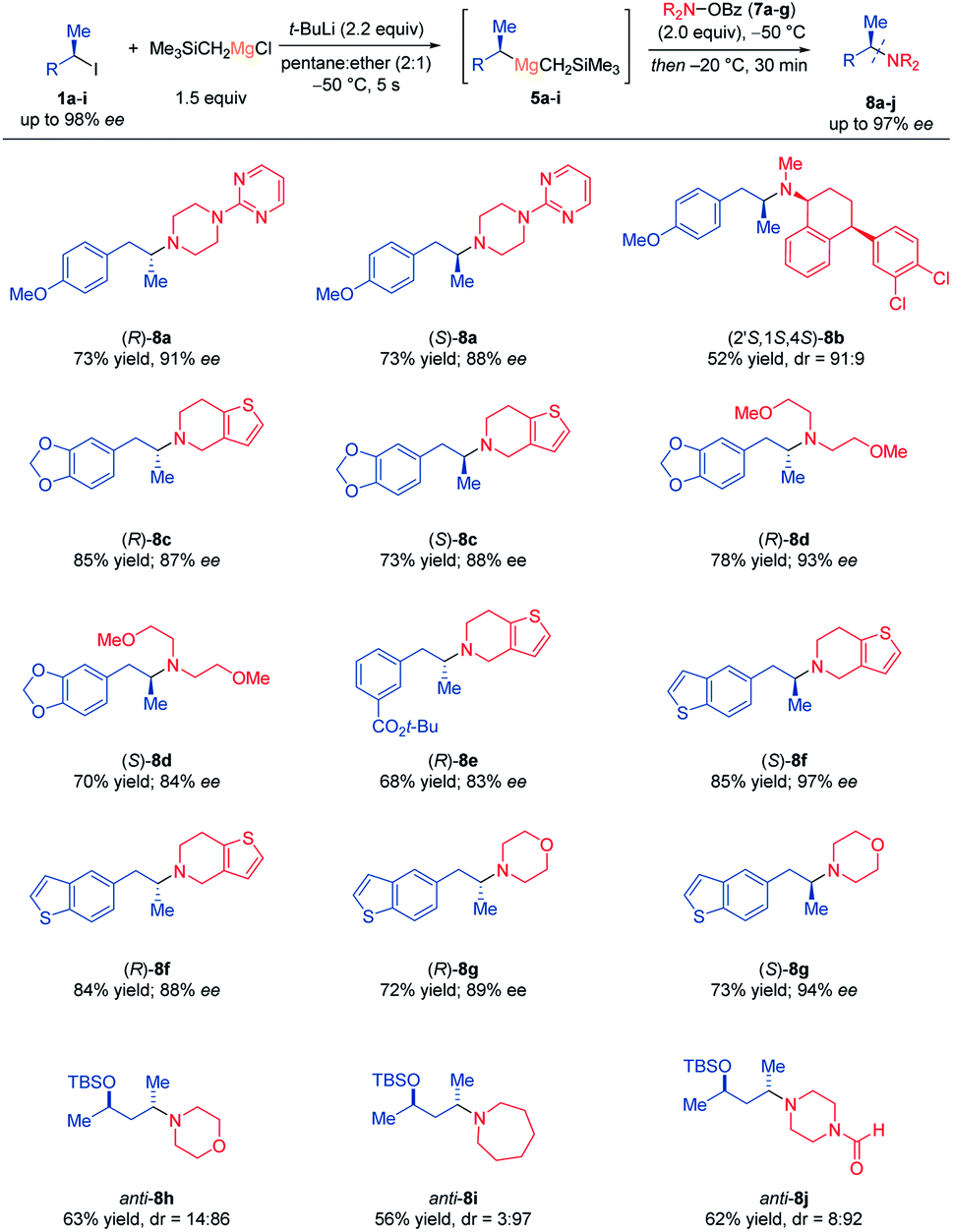 | ||
| Scheme 3 Scope of prepared enantiomerically and diastereomerically enriched α-chiral tertiary amines. Enantiomerically and diastereomerically enriched tertiary amines 8a–j obtained by electrophilic amination of secondary alkylmagnesium reagents 5a–i with O-benzoyl hydroxylamines (7a–g). The enantiomeric excess (% ee) was determined by chiral HPLC-analysis. The diastereomeric ratio (dr; syn/anti ratio) was determined by 1H-NMR spectroscopy and GC-analysis. | ||
Also, the diastereomerically enriched secondary alkylmagnesium reagent anti-5i was successfully quenched with 7e, azepan-1-yl benzoate (7f) and even the sensitive formamide 4-formylpiperazin-1-yl benzoate (7g) affording anti-8h–j in 56–63% yield and up to dr = 3:97. This preparation of tertiary amines was found to be superior to standard nucleophilic substitutions of chiral secondary alkyl iodides, phosphates and tosylates by metallic amides, which gave erratic results in our hands.18 The determination of the absolute configuration was made in the case of the amine hydrochloride derivative of (S)-8g using X-ray diffraction (Flack parameter method)19 confirming the (S)-configuration of 8g as well as the retention of configuration of this electrophilic amination (Fig. 1).
 | ||
| Fig. 1 X-ray structure of (S)-8g crystallized as the corresponding amine hydrochloride as a representative example of the overall stereoretention of the electrophilic amination. | ||
Conclusions
In summary, we have prepared a broad range of functionalized enantiomerically enriched non-stabilized secondary alkylmagnesium reagents from the corresponding alkyl iodides via I/Li-exchange and in situ transmetalation with Me3SiCH2MgCl at convenient temperatures. The resulting new chiral alkylmagnesium reagents of the type AlkCH(Me)MgCH2SiMe3 (5) were trapped with ketones, aldehydes, acid chlorides, isocyanates, S-methyl methanethiosulfonate and chlorophosphines providing the corresponding tertiary alcohols, ketones, amides, thioethers and phosphines with high retention of configuration (up to 99% ee). These mixed Grignard reagents were found to be configurationally stable up to −50 °C (for ca. 1 h) and allowed a convenient preparation of enantiomerically enriched α-chiral tertiary amines starting from O-benzoyl hydroxylamines in the absence of a transition metal catalyst, which demonstrated a valuable alternative to nucleophilic substitutions of secondary alkyl iodides, phosphates and tosylates with metallic amides. Further applications of these new chiral magnesium intermediates are currently underway.Data availability
All experimental and crystallographic data is available in the ESI.†Author contributions
A. K., H. R. W., M. M. S. and Q. S. performed and analyzed the experiments. K. K. measured and analyzed X-ray crystal structures. A. K., H. R. W. and P. K. designed the experiments. A. K., H. R. W. and P. K. prepared the manuscript with contributions of all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Deutsche Forschungsgemeinschaft (DFG) and the Ludwig-Maximilians-Universität München for financial support. We also thank Albemarle for the generous gift of chemicals. We thank Dr J. Skotnitzki for fruitful discussions and K. Kublik, Y. Gong and R. Traber for preliminary experiments.Notes and references
- (a) H. G. Richey, Grignard Reagents: New Developments, John Wiley & Sons, Ltd.New York, 1999 Search PubMed; (b) Z. Rappoport and I. Marek, PATAI's Chemistry of Functional Groups: The Chemistry of Organomagnesium Compounds, John Wiley & Sons, Ltd., New York, 2008 Search PubMed; (c) P. Knochel, W. Dohle, N. Gommermann, F. F. Kneisel, F. Kopp, T. Korn and I. Sapountzis, Angew. Chem., Int. Ed., 2003, 42, 4302–4320 CrossRef CAS PubMed; (d) B. Haag, M. Mosrin, H. Ila and V. Malakhov, Angew. Chem., Int. Ed., 2011, 50, 9794–9824 CrossRef CAS; (e) A. Kremsmair, J. H. Harenberg, K. Schwärzer, A. Hess and P. Knochel, Chem. Sci., 2021, 12, 6011–6019 RSC.
- (a) R. W. Hoffmann, B. Hölzer, O. Knopff and K. Harms, Angew. Chem., Int. Ed., 2000, 39, 3072–3074 CrossRef CAS; (b) R. W. Hoffmann and B. Hölzer, Chem. Commun., 2001, 491–492 RSC; (c) R. W. Hoffmann, B. Hölzer and O. Knopff, Org. Lett., 2001, 3, 1945–1948 CrossRef CAS; (d) R. W. Hoffmann and B. Hölzer, J. Am. Chem. Soc., 2002, 124, 4204–4205 CrossRef CAS; (e) B. Hölzer and R. W. Hoffmann, Chem. Commun., 2003, 732–733 RSC.
- (a) F. R. Jensen and K. L. Nakamaye, J. Am. Chem. Soc., 1966, 88, 3437–3438 CrossRef CAS; (b) J. San Filippo and J. W. Nicoletti, J. Org. Chem., 1977, 42, 1940–1944 CrossRef CAS.
- J. Beckmann, D. Dakternieks, M. Dräger and A. Duthie, Angew. Chem., Int. Ed., 2006, 45, 6509–6512 CrossRef CAS PubMed.
- We used 2.2 equiv. of t-BuLi for best results (formation of lithium reagent and formation of isobutylene and isobutene as side-products); see: M. Schlosser, Organometallics in Synthesis: Third Manual, John Wiley & Sons, Ltd., New York, 2013 Search PubMed.
- (a) S. Seel, G. Dagousset, T. Thaler, A. Frischmuth, K. Karaghiosoff, H. Zipse and P. Knochel, Chem.–Eur. J., 2013, 19, 4614–4622 CrossRef CAS PubMed; (b) G. Dagousset, K. Moriya, R. Mose, G. Berionni, K. Karaghiosoff, H. Zipse, H. Mayr and P. Knochel, Angew. Chem., Int. Ed., 2014, 53, 1425–1429 CrossRef CAS; (c) K. Moriya, D. Didier, M. Simon, J. M. Hamann, G. Berionni, K. Karaghiosoff, H. Zipse, H. Mayr and P. Knochel, Angew. Chem., Int. Ed., 2015, 54, 10963–10967 CrossRef CAS PubMed; (d) J. Skotnitzki, A. Kremsmair and P. Knochel, Synthesis, 2020, 52, 189–196 CrossRef CAS.
- For transmetalations to alkylcopper reagents: (a) K. Moriya, M. Simon, R. Mose, K. Karaghiosoff and P. Knochel, Angew. Chem., Int. Ed., 2015, 54, 10963–10967 CrossRef CAS PubMed; (b) V. Morozova, K. Moriya, P. Mayer and P. Knochel, Chem.–Eur. J., 2016, 22, 9962–9965 CrossRef CAS PubMed; (c) J. Skotnitzki, V. Morozova and P. Knochel, Org. Lett., 2018, 20, 2365–2368 CrossRef CAS PubMed; (d) V. Morozova, J. Skotnitzki, K. Moriya, K. Karaghiosoff and P. Knochel, Angew. Chem., Int. Ed., 2018, 57, 5515–5519 CrossRef; (e) J. Skotnitzki, A. Kremsmair, D. Keefer, F. Schüppel, B. Le Cacher de Bonneville, R. de Vivie-Riedle and P. Knochel, Chem. Sci., 2020, 11, 5328–5332 RSC; (f) A. Kremsmair, J. Skotnitzki and P. Knochel, Chem.–Eur. J., 2020, 26, 11971–11973 CrossRef CAS PubMedFor transmetalations to mixed alkylcopper-zinc reagents: (g) J. Skotnitzki, L. Spessert and P. Knochel, Angew. Chem., Int. Ed., 2019, 58, 1509–1514 CrossRef CAS; (h) J. Skotnitzki, A. Kremsmair, B. Kicin, R. Saeb, V. Ruf and P. Knochel, Synthesis, 2020, 52, 873–881 CrossRef CASFor transmetalations to alkylzinc reagents see: (i) J. Skotnitzki, A. Kremsmair, D. Keefer, Y. Gong, R. de Vivie-Riedle and P. Knochel, Angew. Chem., Int. Ed., 2020, 59, 320–324 CrossRef CAS PubMed.
- (a) C. Blomberg, The Barbier Reaction and Related One-Step Processes, Springer-Verlag, Berlin Heidelberg, 1993 CrossRef; (b) S. Goto, J. Velder, S. El Sheikh, Y. Sakamoto, M. Mitani, S. Elmas, A. Adler, A. Becker, J.-M. Neudörfl, J. Lex and H.-G. Schmalz, Synlett, 2008, 9, 1361–1365 Search PubMed.
- A. Frischmuth, M. Fernández, N. M. Barl, F. Achrainer, H. Zipse, G. Berionni, H. Mayr, K. Karaghiosoff and P. Knochel, Angew. Chem., Int. Ed., 2014, 53, 7928–7932 CrossRef CAS PubMed.
- For a detailed screening table, see ESI.†.
- The enantiopurity was estimated based on the enantiopurity of the obtained tertiary alcohols after reaction with the ketones 6a or 6b.
- F. Langer and P. Knochel, Tetrahedron Lett., 1995, 36, 4591–4594 CrossRef CAS.
- (a) N. E. Lee and S. L. Buchwald, J. Am. Chem. Soc., 1994, 116, 5985–5986 CrossRef CAS; (b) T. C. Nugent and M. El-Shazly, Adv. Synth. Catal., 2010, 352, 753–819 CrossRef CAS; (c) G.-H. Hou, J.-H. Xie, P.-C. Yan and Q.-L. Zhou, J. Am. Chem. Soc., 2009, 131, 1366–1371 CrossRef CAS PubMed; (d) D. Matheau-Raven, P. Gabriel, J. A. Leitch, Y. A. Almehmadi, K. Yamazaki and D. J. Dixon, ACS Catal., 2020, 10, 8880–8897 CrossRef CAS; (e) A. Trowbridge, S. M. Walton and M. J. Gaunt, Chem. Rev., 2020, 120, 2613–2692 CrossRef CAS PubMed.
- (a) A. M. Berman and J. S. Johnson, J. Am. Chem. Soc., 2004, 126, 5680–5681 CrossRef CAS PubMed; (b) A. M. Berman and J. S. Johnson, J. Org. Chem., 2005, 70, 364–366 CrossRef CAS; (c) A. M. Berman and J. S. Johnson, J. Org. Chem., 2006, 71, 219–224 CrossRef CAS.
- (a) S. L. McDonald, C. E. Hendrick and Q. Wang, Angew. Chem., Int. Ed., 2014, 53, 4667–4670 CrossRef CAS PubMed; (b) K. Shen and Q. Wang, Chem. Sci., 2015, 6, 4279–4283 RSC; (c) B. N. Hemric, K. Shen and Q. Wang, J. Am. Chem. Soc., 2016, 138, 5813–5816 CrossRef CAS; (d) C. E. Hendrick, K. J. Bitting, S. Cho and Q. Wang, J. Am. Chem. Soc., 2017, 139, 13110–13116 CrossRef; (e) Y.-H. Chen, S. Graßl and P. Knochel, Angew. Chem., Int. Ed., 2018, 57, 1108–1111 CrossRef CAS; (f) S. Graßl, Y.-H. Chen, C. Hamze, C. P. Tüllmann and P. Knochel, Org. Lett., 2019, 21, 494–497 CrossRef; (g) Z. Xiong, P. Cai, Y. Mei and J. Wang, RSC Adv., 2019, 9, 42072–42076 RSC; (h) V. A. Van der Puyl, J. Derosa and K. M. Engle, ACS Catal., 2019, 9, 224–229 CrossRef CAS; (i) S. Graßl and P. Knochel, Org. Lett., 2020, 22, 1947–1950 CrossRef PubMed; (j) J. He, Y. Xue, B. Han, C. Zhang, Y. Wang and S. Zhu, Angew. Chem., Int. Ed., 2020, 59, 2328–2332 CrossRef CAS PubMed; (k) Y. Kwon and Q. Wang, Org. Lett., 2020, 22, 4141–4145 CrossRef CAS PubMed; (l) B. N. Hemric, C. K. Ku and Q. Wang, Encyclopedia of Reagents for Organic Synthesis, Wiley VCH 2020, DOI:10.1002/047084289X.rn02290.
- M. J. Campbell and J. S. Johnson, Org. Lett., 2007, 9, 1521–1524 CrossRef CAS PubMed.
- M. W. Welch, A. R. Kraska, R. Sarges and B. K. Koe, J. Med. Chem., 1984, 27, 1508–1515 CrossRef.
- For a detailed comparison of electrophilic and nucleophilic aminations see ESI.†.
- (a) H. D. Flack, Acta Crystallogr., 1983, 39, 876–881 CrossRef; (b) E. C. Constable and C. E. Housecroft, Chemistry, 2020, 2, 759–776 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2110720. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc05315a |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2022 |