
Chromium complexes supported by NNO-tridentate ligands: an unprecedented activity with the requirement of a small amount of MAO†
Jiliang
Tian
a,
Xingwang
Zhang
a,
Shaofeng
Liu
 *a and
Zhibo
Li
ab
*a and
Zhibo
Li
ab
aKey Laboratory of Biobased Polymer Materials, College of Polymer Science and Engineering, Qingdao University of Science and Technology, Qingdao, 266042, China. E-mail: shaofengliu@qust.edu.cn
bCollege of Chemical Engineering, Qingdao University of Science and Technology, Qingdao 266042, China
First published on 4th March 2022
Abstract
The development of metal catalysts with high activity and thermal stability but with the requirement of a small amount of MAO as a cocatalyst is highly desired for polyolefin industrial application. In this contribution, a series of phenoxy-imine-amine compounds were prepared and used as NNO-tridentate ligands (LH1–LH6: LH = 2,6-(R1)2–C6H3–NH–C6H4–N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C-3,5–(R2)2–C6H2–OH; LH1: R1 = iPr, R2 = tBu; LH2: R1 = Me, R2 = tBu; LH3: R1 = H, R2 = tBu; LH4: R1 = F, R2 = tBu; LH5: R1 = Me, R2 = H; LH6: R1 = Me, R2 = Cl) to support chromium complexes CrCl2L(THF) (Cr1(THF)–Cr6(THF)). Upon activation with only 200 equivalents of Al(MAO), these Cr complexes exhibited extremely high activity toward ethylene polymerization and an unprecedented activity as high as 1.18 × 108 g(PE) mol−1(Cr) h−1 was obtained using Cr2(THF)/MAO. Moreover, these Cr catalysts exhibited high thermal stability, with respect to both the activity and the molecular weight. At 100 °C, Cr2(THF)/MAO showed a remarkable activity of 1.01 × 108 g(PE) mol−1(Cr) h−1 and produced polyethylene with a high molecular weight of 24.6 × 104 g mol−1. Therefore, the newly developed NNO-tridentate Cr complexes exhibiting unprecedented activity and excellent thermal stability but only requiring a small amount of MAO (as low as Al/Cr = 100) as a cocatalyst are very promising in the polyolefin industry.
C-3,5–(R2)2–C6H2–OH; LH1: R1 = iPr, R2 = tBu; LH2: R1 = Me, R2 = tBu; LH3: R1 = H, R2 = tBu; LH4: R1 = F, R2 = tBu; LH5: R1 = Me, R2 = H; LH6: R1 = Me, R2 = Cl) to support chromium complexes CrCl2L(THF) (Cr1(THF)–Cr6(THF)). Upon activation with only 200 equivalents of Al(MAO), these Cr complexes exhibited extremely high activity toward ethylene polymerization and an unprecedented activity as high as 1.18 × 108 g(PE) mol−1(Cr) h−1 was obtained using Cr2(THF)/MAO. Moreover, these Cr catalysts exhibited high thermal stability, with respect to both the activity and the molecular weight. At 100 °C, Cr2(THF)/MAO showed a remarkable activity of 1.01 × 108 g(PE) mol−1(Cr) h−1 and produced polyethylene with a high molecular weight of 24.6 × 104 g mol−1. Therefore, the newly developed NNO-tridentate Cr complexes exhibiting unprecedented activity and excellent thermal stability but only requiring a small amount of MAO (as low as Al/Cr = 100) as a cocatalyst are very promising in the polyolefin industry.
1. Introduction
Polyolefins have become the largest synthetic polymer materials that are still most commercially produced using the heterogeneous Ziegler–Natta catalyst1 and Phillips catalyst.2,3 However, the mounting demand for high-value-added polyolefins in recent years has spurred extensive interest in both industry and academia to develop homogeneous metal catalysts for olefin polymerization,4–10 which are also beneficial for better understanding the polymerization mechanism and, in turn, are greatly helpful in developing new-generation catalysts with high-performances.11–13 In the past few decades, a series of homogeneous metal catalysts, including metallocene14 and non-metallocene catalysts,15–21 have been developed and applied to synthesize a variety of high-value-added polyolefins, such as polar functionalized polyethylene,22–25 ultrahigh molecular weight polyethylene (UHMWPE),26,27 syndiotactic polypropylene (sPP),28 syndiotactic polystyrene (sPS),29 polyolefin elastomers (POE),30 and cyclic olefin copolymers (COC).31,32Although the great successes as mentioned above have been achieved, homogeneous metal catalysts still have many unexpected defects. For example, it is well known that a large amount of MAO is generally required as a cocatalyst for homogeneous metallocene or non-metallocene catalysts to achieve high polymerization activity33 which is highly expensive and leads to the unexpected ash-content in the production.34 Thus, it is an urgent desire to develop new metal catalysts that can offer high catalytic activity but need less expensive MAO as a cocatalyst. On the other hand, the operative temperature range for most industrial polyolefin production is 70–110 °C because of the highly exothermic nature of the polymerization process.35 However, many homogeneous metal catalysts suffer from poor thermal stability and undergo fast decomposition with increasing reaction temperature, which greatly limited their potential applications in industry. Moreover, the molecular weights of resultant polymers are significantly reduced at high temperature, because of increased chain transfer and β-H elimination reactions.36,37 Therefore, the development of new metal catalysts having high activity and thermal stability but requiring a small amount of MAO as a cocatalyst is highly desired but also challenging.
Phenoxy-imine compounds are simple, readily accessible and amenably modifiable, and thus have been widely employed as ligands to support both early- and late-transition metal complexes for olefin polymerization.7,9 For example, the early-transition metal titanium and zirconium phenoxyiminato catalysts reported by Fujita38 and Coates39 show high activity, living polymerization behavior, and stereoregularity control to synthesize novel polyolefins,9 while the late-transition metal nickel phenoxyiminato catalysts developed by Grubbs40 exhibit great powers of endurance toward polar monomers to prepare functional polyolefins.7 Chromium-based metal complexes, both heterogeneous and homogeneous, are among the most important polyolefin catalysts and have received increasing attention because of their wide use in both ethylene polymerization and oligomerization.41–52 However, Cr complexes bearing phenoxy-imine ligands are rare and underdeveloped. Previously, Gibson explored this ligand family for Cr-based polymerization catalysts, but only low to modest activity (usually less than 105 g(PE) mol−1(Cr) h−1) was obtained.53,54 Later, the same group extended this NO-bidentate ligand to the NNO-tridentate system and discovered an exceptionally active NNO-Cr catalyst by the High Throughput Screening (HTS) method.55 The NNO-Cr complex with the bulky triptycenyl substituent and an additional pyridyl-N donor exhibited an activity of 6.97 × 106 g(PE) mol−1(Cr) h−1 bar−1, which was comparable to that of the most active homogeneous chromium olefin polymerization catalysts.56 Nevertheless, a large amount of MAO (2200 equivalents of Al(MAO) relative to Cr) was needed to maintain the high activity and only low molecular weight polyethylenes (Mw ≈ 1200 g mol−1) were produced at 50 °C. In this study, a series of phenoxy-imine-amine compounds were designed, synthesized, and used as a new type of NNO-tridentate ligand. The corresponding chromium complexes were also reported and used as catalysts toward ethylene polymerization. The influence of the ligand and various reaction conditions, such as the nature and amount of the cocatalyst and the polymerization temperature and pressure, was systematically investigated. It is remarkable that these Cr complexes were extremely active with only a very small amount of MAO, and an unprecedented activity over 108 g(PE) mol−1(Cr) h−1 can be obtained using 100 equivalents of Al(MAO) as a cocatalyst.
2. Results and discussion
2.1. Synthesis and characterization of ligands and chromium complexes
The NNO-tridentate ligands LH1–LH6 were prepared by a successive three-step method including cross-coupling, reduction and condensation reactions as shown in Scheme 1. In toluene, palladium-catalyzed coupling of 1-bromo-2-nitrobenzene with 1 equivalent of 2,6-disubstituted aniline using CsCO3 as a base afforded intermediates N-(2-nitrophenyl)-2,6-(R1)2-aniline.57 The nitro group could be efficiently reduced to an amino group using SnCl2 in the presence of an acid. The condensation reaction was catalyzed by p-TsOH in methanol at room temperature to give the desired NNO-tridentate ligands LH1–LH6 in high yields (77–95%).58,59 All the organic ligands were characterized by 1H and 13C NMR (Fig. S1–S12†), and elemental analysis. Treatment of CrCl3(THF)3 with one equivalent of the sodium salt of ligands LH1–LH6 in THF at room temperature afforded the brown chromium dichloride complexes Cr1(THF)–Cr6(THF) in high yields (88–95%; Scheme 1). These complexes are air-sensitive solids and the mass spectra of complexes showed the main fragments of [M − 2Cl − THF + OH + Na]+ ions (Fig. S13–S18†). | ||
| Scheme 1 Synthesis of ligands and chromium complexes. | ||
In order to firmly establish the coordination mode of the NNO-tridentate ligands, single crystals of chromium complexes Cr2(THF) and Cr4(THF) were grown through slow diffusion of n-hexane into their CH2Cl2 solutions at room temperature. The molecular structures of Cr2(THF) and Cr4(THF) by X-ray diffractions are given in Fig. 1 and 2 with the selected bond lengths and angles in the captions. As shown in Fig. 1, Cr2(THF) has a distorted octahedral coordination geometry around the Cr center. The coordination sphere is comprised of one NNO-tridentate ligand, two chlorides and one THF molecule. The tridentate ligand is situated around the Cr center in a meridional manner (N^N^O). The two chelating rings (Cr1–N2–C5–C4–N1, Cr1–N1–C3–C2–C1–O1) are almost coplanar and the corresponding dihedral angle defined is 9.81°. The Cr atom and the mutually trans-disposed chlorides are almost in one line [Cl1–Cr1–Cl2 = 177.50(5)°] with O1, N1, N2 and O2 atoms locating in the equatorial plane. The sum of the bond angles (N1–Cr1–N2 80.67(13)°, O1–Cr1–N1 91.83(13)°, O1–Cr1–O2 92.67(12)°, and O2–Cr1–N2 94.57(13)°) around the Cr1 center in this equatorial plane is 359.74°, indicating that atoms O1, N1, N2, O2 and Cr1 are essentially coplanar. Complex Cr4(THF) (Fig. 2) shows similar coordination features to Cr2(THF), suggesting that all Cr complexes should have similar coordination geometries.
 | ||
| Fig. 1 ORTEP of the molecular structure of Cr2(THF). Ellipsoids at the 50% probability level. Hydrogen atoms except for H2 are omitted for clarity. Selected distances (Å) and angles (deg): Cr1–Cl1 2.3756(13), Cr1–Cl2 2.2912(13), Cr1–O1 1.894(3), Cr1–O2 2.071(3), Cr1–N1 1.999(3), Cr1–N2 2.170(3), N1–C3 1.297(5), N1–C4 1.422(5); Cl2–Cr1–Cl1 177.50(5), O1–Cr1–Cl1 91.99(10), O1–Cr1–Cl2 90.35(10), O1–Cr1–O2 92.67(12), O1–Cr1–N1 91.83(13), O1–Cr1–N2 170.99(13), O2–Cr1–Cl1 88.29(9), O2–Cr1–Cl2 90.72(9), O2–Cr1–N2 94.57(13), N1–Cr1–Cl1 88.57(10), N1–Cr1–Cl2 92.23(11), N1–Cr1–O2 174.60(12), N1–Cr1–N2 80.67(13), N2–Cr1–Cl1 82.89(11), N2–Cr1–Cl2 94.90(11). | ||
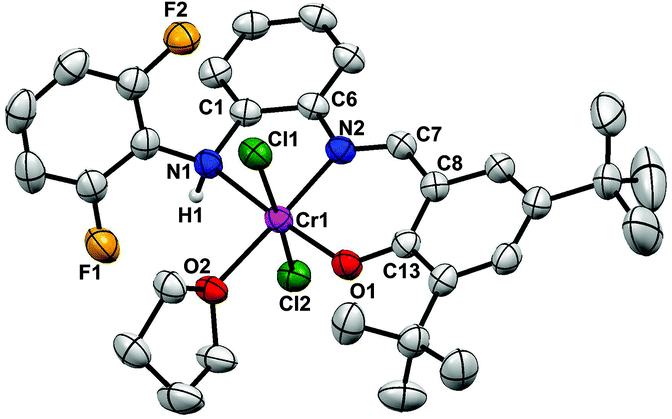 | ||
| Fig. 2 ORTEP of the molecular structure of Cr4(THF). Ellipsoids at the 50% probability level. Hydrogen atoms except for H1 are omitted for clarity. Selected distances (Å) and angles (deg): Cr1–Cl1 2.3068(8), Cr1–Cl2 2.3389(8), Cr1–O1 1.879(2), Cr1–O2 2.056(2), Cr1–N1 2.182(2), Cr1–N2 1.994(2), N2–C6 1.422(3), N2–C7 1.298(4); Cl1–Cr1–Cl2 172.48(3), O1–Cr1–Cl1 93.30(7), O1–Cr1–Cl2 94.04(7), O1–Cr1–O2 90.68(9), O1–Cr1–N1 173.30(9), O1–Cr1–N2 92.63(9), O2–Cr1–Cl1 91.98(6), O2–Cr1–Cl2 89.55(6), O2–Cr1–N1 94.52(9), N1–Cr1–Cl1 90.74(7), N1–Cr1–Cl2 81.79(7), N2–Cr1–Cl1 90.16(7), N2–Cr1–Cl2 87.90(7), N2–Cr1–O2 175.96(9), N2–Cr1–N1 82.01(9). | ||
2.2. Ethylene polymerization
| Run | Cocatalyst | Al/Cr | PE (mg) | Activityb |
|---|---|---|---|---|
| a Polymerization conditions: 2 μmol Cr2(THF), 2 min, 80 °C, 200 mL toluene, 5 atm of ethylene. b Activity in units of 106 g(PE) mol−1(Cr) h−1. c 1.2 equivalents of [Ph3C][B(C6F5)4] were added. | ||||
| 1 | MAO | 200 | 470 | 7.05 |
| 2 | AlMe3 | 200 | 8 | 0.12 |
| 3 | AlEtCl2 | 200 | 4 | 0.06 |
| 4 | AlEt2Cl | 200 | 5 | 0.07 |
| 5c | [Ph3C][B(C6F5)4]/AliBu3 | 50 | 90 | 1.35 |
| Run | P (atm) | T (°C) | Al/Cr | t (min) | PE (mg) | Act.b |
M
w![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) c (104 g mol−1) c (104 g mol−1) |
Đ |
T
md (°C) |
|---|---|---|---|---|---|---|---|---|---|
| a Polymerization conditions: 2 μmol Cr2(THF), 200 mL toluene. b Activity in units of 106 g(PE) mol−1(Cr) h−1. c Determined by GPC. d Melting temperature by DSC. | |||||||||
| 1 | 5 | 80 | 200 | 2 | 470 | 7.05 | 14.9 | 1.8 | 134 |
| 2 | 20 | 80 | 200 | 2 | 3300 | 49.5 | 20.0 | 1.8 | 134 |
| 3 | 40 | 80 | 200 | 2 | 7870 | 118 | 38.9 | 1.6 | 135 |
| 4 | 40 | 20 | 200 | 2 | 2700 | 40.5 | 1.50 | 3.1 | 135 |
| 5 | 40 | 60 | 200 | 2 | 4400 | 66.0 | 12.0 | 2.6 | 135 |
| 6 | 40 | 100 | 200 | 2 | 6700 | 101 | 24.6 | 1.8 | 135 |
| 7 | 40 | 120 | 200 | 2 | 4500 | 67.5 | 8.90 | 3.2 | 134 |
| 8 | 40 | 80 | 100 | 2 | 6850 | 103 | 38.3 | 1.9 | 134 |
| 9 | 40 | 80 | 300 | 2 | 7000 | 105 | 13.9 | 2.2 | 135 |
| 10 | 40 | 80 | 600 | 2 | 4850 | 72.8 | 4.40 | 2.9 | 135 |
| 11 | 40 | 80 | 200 | 5 | 14600 |
87.6 | 36.5 | 1.7 | 135 |
| 12 | 40 | 80 | 200 | 10 | 22000 |
66.0 | 37.1 | 1.6 | 135 |
As expected, the catalytic activity increased dramatically at high ethylene pressure (Table 2 runs 1–3). Remarkably, an extremely high activity of 1.18 × 108 g(PE) mol−1(Cr) h−1 was obtained with an Al(MAO)/Cr ratio of 200 at 80 °C and 40 atm of ethylene, and it is among the highest for Cr-mediated ethylene polymerization although performed under different conditions.41,42,60,61 The GPC analyses of the polymers obtained with Cr2(THF)/MAO at different pressures show unimodal and narrow distributions (Fig. 3A, Đ < 2.0), indicating the typical single site catalysis. The molecular weights of the resultant polyethylenes increase with the increase of polymerization pressure, suggesting fast chain propagation relative to chain termination/transfer reactions.
 | ||
| Fig. 3 GPC curves of polyethylenes prepared from Cr2(THF)/MAO under different (A) pressures (Table 2, runs 1–3); (B) temperatures (Table 2, runs 3–7); (C) Al(MAO)/Cr ratio (Table 2, runs 3 and 8–10). | ||
As shown in Table 2 runs 3–6, the reaction temperature (from 20 to 100 °C) significantly affected the activity and properties of products, in regard to the molecular weights and their distributions. The polymerization temperature was raised from 20 to 100 °C leading to a maximum peak in activity of 1.18 × 108 g(PE) mol−1(Cr) h−1 at 80 °C. Further increasing the temperature to 100 °C only resulted in a little degradation in activity (1.01 × 108 g(PE) mol−1(Cr) h−1 at 100 °C; Table 2 run 6). Because of the extremely high activity, the temperature at the end of polymerization in Table 2 run 6 could reach up to 110 °C, although the heat was continuously removed from the reactor through an internal cooling coil. For most industrial polyolefin production, the operative temperature range is 70–110 °C because of the highly exothermic nature of the polymerization process.35 Therefore, Cr2(THF)/MAO had an excellent thermal stability and showed great potential for practical application in the polyolefin industry. It is interesting that the molecular weights of the resultant polyethylenes increased with the increasing polymerization temperature from 20 to 80 °C (Fig. 3B, black, red and blue curves), suggesting the relatively slow chain termination/transfer reactions and further confirming the high thermal stability of the Cr2(THF)/MAO system. We also carried out polymerization at an even higher start temperature (T = 120 °C, Table 2 run 7) with Cr2(THF)/MAO, and a temperature over 130 °C was observed at the end of polymerization. The activity was 6.75 × 107 g(PE) mol−1(Cr) h−1, which is lower than those at 80 or 100 °C (Table 2 runs 3 and 6). On the other hand, the Mw of the resultant polymer decreased and the distribution became relatively broad (Mw = 8.90 × 104 g mol−1, Đ = 3.2; Fig. 3B, purple curve), indicating the partial decomposition of the catalyst over 120 °C.
Besides, the catalytic performances were also significantly dependent upon the amount of MAO used (Table 2 runs 3 and 8–10). On elevating the molar ratio of Al(MAO)/Cr from 100 to 600, the best activity of 1.18 × 108 g(PE) mol−1(Cr) h−1 was achieved with Al(MAO)/Cr = 200 at 80 °C (Table 2 run 3). However, it is surprising that only 100 equivalents of Al(MAO) relative to the Cr catalyst also resulted in an activity as high as 1.03 × 108 g(PE) mol−1(Cr) h−1 (Table 2 run 8). It is well known that a large amount of MAO is generally required as a cocatalyst for homogeneous metallocene or non-metallocene catalysts in order to achieve a high polymerization activity,33 which is highly expensive and leads to the unexpected ash content in the products.34 Therefore, the Cr2(THF)/MAO system can achieve an activity of 1.03 × 108 g(PE) mol−1(Cr) h−1 with such a small amount of MAO (Al/Cr = 100). Further addition of MAO (Al/Cr = 600 in Table 2 run 10) led to a decrease in activity, probably because some impurities (such as AlMe3) present in the commercial MAO solution can react with Cr active species and result in deactivation, which was reported previously.60,62 With the increase of the Al(MAO)/Cr ratio, the molecular weights of the resultant products decreased significantly (Fig. 3C), e.g. polyethylene with a Mw of 4.40 × 104 g mol−1 was obtained with Al(MAO)/Cr = 600 (Table 2 run 10), indicative of predominant chain transfer to Al reactions. The chain transfer reaction to Al was further confirmed by high temperature 1H and 13C NMR spectroscopy (120 °C in 1,1,2,2-tetrachloroethane-d2, C2D2Cl4) of the polyethylene sample obtained with Al(MAO)/Cr = 600 (Table 2 run 10). There is no resonance between 5.0–6.0 ppm in the 1H NMR spectrum (Fig. S20†) and between 110–140 ppm in the 13C NMR spectrum (Fig. S21†), which are attributed to unsaturated end groups. Therefore, the β-H elimination reaction as the termination and chain transfer reaction to the monomer can be ruled out.
The polymerizations at 5 min and 10 min were also carried out and the results are shown in Table 2 runs 11 and 12. On increasing the polymerization time, the calculated activity decreased, probably because the large amount of the resultant polymers could retard the mass transfer of the monomer. However, much more polymers were obtained at a longer reaction time (7.87 g, 14.6 g, and 22.0 g for 2 min, 5 min, and 10 min, respectively) and similar Mws and distributions of the resultant polymers obtained at different times (Table 2, runs 3, 11 and 12), suggesting the high thermal stability and long lifetime of the catalytic system.
| Run | Complex | PE (mg) | Act.b |
M
wc (104 g mol−1) |
Đ |
T
md (°C) |
|---|---|---|---|---|---|---|
| a Polymerization conditions: 2 μmol Cr complexes, 40 atm of ethylene, 80 °C, 2 min, Al/Cr = 200, 200 mL toluene. b In units of 106 g(PE) mol−1(Cr) h−1. c Determined by GPC. d Determined by DSC. | ||||||
| 1 | Cr1(THF) | 1150 | 17.3 | 14.4 | 2.3 | 135 |
| 2 | Cr2(THF) | 7870 | 118 | 38.9 | 1.6 | 135 |
| 3 | Cr3(THF) | 1600 | 24.0 | 22.3 | 4.2 | 133 |
| 4 | Cr4(THF) | 1750 | 26.3 | 18.0 | 10 | 135 |
| 5 | Cr5(THF) | 190 | 2.85 | 45.6 | 19 | 133 |
| 6 | Cr6(THF) | 180 | 2.70 | 53.4 | 19 | 133 |
Substituents on both aniline (R1) and salicylaldehyde (R2) could remarkably affect the catalytic performances of the Cr complexes. Comparing the activity of Cr1(THF)–Cr4(THF) with the same R2-substituents on salicylaldehyde but different R1-substituents on aniline (Table 3 runs 1–4), Cr2(THF) exhibited the highest activity of 1.18 × 108 g(PE) mol−1(Cr) h−1, which was 6.8, 4.9 and 4.5 times higher than those of Cr1(THF), Cr3(THF) and Cr4(THF) under otherwise identical conditions. On the other hand, the R2-substituents on salicylaldehyde showed a greater influence on the catalytic activity than R1-substituents on aniline. Thus, Cr2(THF) also showed much higher activity than Cr5(THF) and Cr6(THF) that had the same R1-substituents on aniline but different R2-substituents on salicylaldehyde (Table 3 runs 2, 5 and 6). These results demonstrate that the delicate choice of substituents on the ligand that determine the coordination environments could largely affect the catalytic performances of the Cr complexes. Besides, the properties of the resultant polymers, regarding the molecular weights and distributions, also depended upon the coordination environment. Generally, the sterically bulkier substituents on the ligand could prevent the chain transfer reaction and β-H elimination reaction, and thus lead to the formation of polyethylene with a high molecular weight. In contrast, these bulky groups can also inhibit monomer-coordination and thus result in a low rate of chain propagation. The molecular weight values typically scale as the net rate of chain propagation divided by the net rate of all competing chain transfer/termination processes. As shown in Table 3 runs 1 and 2, the chain propagation rate (activity) by Cr1(THF) is significantly lower than that by Cr2(THF). Thus, it is not surprising that Cr2(THF) produced polyethylene with a higher molecular weight than that by Cr1(THF) under otherwise identical conditions. Regarding the molecular weight distributions, Cr1(THF) and Cr2(THF) produced polymers with unimodal and narrow distributions (Fig. S22 and S23†), indicating typical single site catalysis. In contrast, polyethylenes obtained by other Cr complexes all had multimodal distributions (Fig. S24–S27†).
To investigate the different catalytic properties of Cr complexes, Cr2(THF) and Cr6(THF), in the presence or absence of MAO, were characterized by the UV-vis-NIR spectroscopy technique, which have been widely used to assess the pre-catalyst (neutral metal complex) and cationic active species for polyolefin Cr catalysts.3,47Fig. 4A shows the UV-vis-NIR spectra of Cr2(THF) (black) and the Cr2(THF)/MAO system (red), while Fig. 4B shows the UV-vis-NIR spectra of Cr6(THF) (black) and the Cr6(THF)/MAO system (red). The spectrum of Cr2(THF) is characterized by well-defined bands at 29800 and 20100 cm−1. After contact with 10 equivalents of Al(MAO) at room temperature, the absorptions of two bands are significantly degraded and a dramatic bathochromic shift is observed for the later band (from 20100 cm−1 to 17700 cm−1; Fig. 4A). There is no additional band obtained by comparing the spectra of Cr2(THF) and the Cr2(THF)/MAO system. This result is consistent with the single-site catalysis by the Cr2(THF)/MAO system that produced polyethylene with unimodal and narrow distribution (Fig. S23†). In contrast, two weak bands at 23900 and 13600 cm−1 are observed in the spectrum of the Cr6(THF)/MAO system (Fig. 4B, red) in comparison to the spectrum of Cr6(THF) (Fig. 4B, black), suggesting that multi-site active species are present. Thus, the Cr6(THF)/MAO system produced polymers with multimodal distribution as shown in Table 3 run 6 and Fig. S27.†
 | ||
| Fig. 4 UV-vis-NIR spectra in toluene (Cr = 10−3 M in toluene, Al/Cr = 10), (A) Cr2(THF) (black) and Cr2(THF)/MAO (red); (B) Cr6(THF) (black) and Cr6(THF)/MAO (red). | ||
3. Conclusions
The chromium complexes CrCl2L(THF) (Cr1(THF)–Cr6(THF)) bearing phenoxy-imine-amine NNO-tridentate ligands (LH1–LH6; 2,6-(R1)2–C6H3–NH–C6H4–NC-3,5–(R2)2–C6H2–OH) have been successfully synthesized, characterized, and used for ethylene polymerization. The influence of the ligands and various reaction conditions, including the nature and amount of the cocatalyst and the polymerization temperature and pressure, was systematically investigated. These Cr complexes were superior toward ethylene polymerization: (1) these Cr complexes were extremely active and, particularly, Cr2(THF) with R1 = Me and R2 = tBu exhibited the highest activity of 1.18 × 108 g(PE) mol−1(Cr) h−1, and produced high molecular weight polyethylene with unimodal and narrow distribution (38.9 × 104 g mol−1, Đ = 1.6). (2) It is remarkable that an activity of 1.03 × 108 g(PE) mol−1(Cr) h−1 was obtained using Cr2(THF) even with a very small amount of MAO (Al/Cr = 100). (3) These Cr complexes exhibited high thermal stability, with respect to both the activity and the molecular weight. For example, Cr2(THF)/MAO showed an activity of 1.01 × 108 g(PE) mol−1(Cr) h−1 and produced polyethylene with a high molecular weight of 24.6 × 104 g mol−1 at 100 °C. In summary, the newly developed NNO-tridentate Cr complexes having unprecedented activity and excellent thermal stability but requiring only a very small amount of MAO as the cocatalyst are very promising for olefin polymerization.
4. Experimental section
4.1. General considerations
All oxygen/moisture sensitive compounds/reactions were handled and/or performed in a glovebox or using standard Schlenk techniques under nitrogen. Toluene, THF, n-hexane and CH2Cl2 were purified by first purging with dry nitrogen, followed by passing through columns of activated alumina. Ethylene was purified by passage through a supported MnO oxygen-removal column and an activated Davison 4A molecular sieve column. A 1.46 M Al(MAO) solution in toluene was purchased from Akzo Nobel Corp. The borate [Ph3C][B(C6F5)4] was purchased from Sigma Aldrich. AlEt2Cl (1.7 M in hexanes), AliBu3 (1.0 M in toluene), and AlMe3 (2.0 M in toluene) were purchased from Acros Chemicals. All other chemicals were purchased from commercial suppliers and used without further purification unless otherwise noted. Reaction temperatures were controlled using an IKA temperature modulator. NMR spectra were recorded on a Bruker AVANCE NEO 400 MHz NMR spectrometer (400 MHz for 1H NMR and 100 MHz for 13C NMR). Elemental analyses were performed on a Flash EA 1112 microanalyzer. Gel permeation chromatography (GPC) was carried out in 1,2,4-trichlorobenzene (stabilized with 125 ppm BHT) at 150 °C on a Agilent 1260 infinity II HT GPC instrument equipped with a set of three Agilent PL gel 10 μm mixed-BLS columns with differential refractive index, viscosity, and light scattering (15 and 90°) detectors. Data reported were determined via triple detection. Molecular weights were determined through universal calibration relative to polystyrene standards. Differential scanning calorimetry (DSC) was performed using a TA differential scanning calorimeter DSC 25 that was calibrated using high purity indium at a heating rate of 10 °C min−1. Melting points were determined from the second scan at a heating rate of 10 °C min−1 followed by a slow cooling rate of 10 °C min−1 to remove the influence of thermal history. N-(2,6-Diisopropylphenyl)benzene-1,2-diamine, N-(2,6-dimethylphenyl)benzene-1,2-diamine, N-phenyl-benzene-1,2-diamine, N-(2,6-difluorophenyl)benzene-1,2-diamine were prepared according to literature procedures.574.2. Synthesis of ligands LH1–LH6
CH), 7.49 (d, J = 2.3 Hz, 1 H, Ar–H), 7.35–7.29 (m, 2 H, Ar–H), 7.27 (d, J = 7.8 Hz, 2 H, Ar–H), 7.12 (dd, J = 7.8, 1.1 Hz, 1 H, Ar–H), 7.02 (t, J = 7.7 Hz, 1 H, Ar–H), 6.75 (t, J = 7.7 Hz, 1 H, Ar–H), 6.26 (d, J = 8.1 Hz, 1 H, Ar–H), 5.87 (s, 1 H, N–H), 3.28–3.14 (m, 2 H, CHMe2), 1.48 (s, 9 H, CMe3), 1.38 (s, 9 H, CMe3), 1.19 (dd, J = 11.9, 6.7 Hz, 12 H, CHMe2). 13C NMR (100 MHz, CDCl3): δ 163.36 (NCH), 158.14, 147.92, 142.21, 141.00, 137.20, 135.44, 134.59, 128.19, 127.95, 127.56, 126.92, 124.00, 118.89, 118.09, 117.66, 112.16, 35.29, 34.39, 31.65, 29.55, 28.51, 24.91, 23.12. Anal. Calcd for C33H44N2O: C, 81.77; H, 9.15; N, 5.78. Found: C, 81.56; H, 9.02; N, 5.56.
CH), 7.48 (d, J = 1.5 Hz, 1 H, Ar–H), 7.30 (d, J = 2.2 Hz, 1 H, Ar–H), 7.18–7.10 (m, 4 H, Ar–H), 7.03 (t, J = 7.7 Hz, 1 H, Ar–H), 6.77 (t, J = 7.5 Hz, 1 H, Ar–H), 6.25 (d, J = 8.1 Hz, 1 H, Ar–H), 5.84 (s, 1 H, N–H), 2.25 (s, 6 H, Ar–Me), 1.47 (s, 9 H, CMe3), 1.36 (s, 9 H, CMe3). 13C NMR (100 MHz, CDCl3): δ 163.58 (NCH), 158.10, 141.02, 140.48, 138.29, 137.22, 136.78, 135.29, 128.68, 128.21, 127.97, 127.00, 126.34, 118.90, 118.28, 117.99, 112.06, 35.29, 34.38, 31.64, 29.60, 18.51. Anal. Calcd for C29H36N2O: C, 81.27; H, 8.47; N, 6.54. Found: C, 81.35; H, 8.27; N, 6.43.
CH), 7.48 (d, J = 2.2 Hz, 1 H, Ar–H), 7.36–7.30 (m, 3 H, Ar–H), 7.27 (d, J = 7.7 Hz, 1 H, Ar–H), 7.22–7.11 (m, 4 H, Ar–H), 7.01 (t, J = 7.7 Hz, 1 H, Ar–H), 6.92 (t, J = 7.5 Hz, 1 H, Ar–H), 6.23 (s, 1 H, N–H), 1.47 (s, 9 H, CMe3), 1.35 (s, 9 H, CMe3). 13C NMR (100 MHz, CDCl3): δ 164.63 (NCH), 158.07, 142.27, 141.07, 137.72, 137.65, 137.21, 129.52, 128.42, 127.56, 127.08, 122.17, 120.33, 119.97, 119.09, 118.78, 115.05, 35.27, 34.36, 31.62, 29.56. Anal. Calcd for C27H32N2O: C, 80.96; H, 8.05; N, 6.99. Found: C, 80.79; H, 8.01; N, 6.74.
CH), 7.49 (d, J = 2.4 Hz, 1 H, Ar–H), 7.29 (d, J = 2.4 Hz, 1 H, Ar–H), 7.18–7.05 (m, 3 H, Ar–H), 6.99 (t, J = 7.9 Hz, 2 H, Ar–H), 6.93 (td, J = 7.7, 1.2 Hz, 1 H, Ar–H), 6.70 (dd, J = 8.1, 0.9 Hz, 1 H, Ar–H), 5.91 (s, 1 H, N–H), 1.49 (s, 9 H, CMe3), 1.36 (s, 9 H, CMe3). 13C NMR (100 MHz, CDCl3): δ 164.75 (NCH), 158.81 (d, J = 5.5 Hz), 158.13, 156.35 (d, J = 5.3 Hz), 141.05, 137.78, 137.19 (d, J = 8.0 Hz), 128.48, 127.55, 127.17, 124.37 (t, J = 9.6 Hz), 120.48, 119.00 (d, J = 15.5 Hz), 118.71 (d, J = 15.9 Hz), 113.93, 112.16 (d, J = 5.8 Hz), 111.99 (d, J = 5.8 Hz), 35.28, 34.37, 31.62, 29.58. Anal. Calcd for C27H30F2N2O: C, 74.29; H, 6.93; N, 6.42. Found: C, 74.35; H, 6.79; N, 6.33.
CH), 7.47 (dd, J = 7.7, 1.6 Hz, 1 H, Ar–H), 7.44–7.36 (m, 1 H, Ar–H), 7.18–7.08 (m, 4 H, Ar–H), 7.08–6.93 (m, 3 H, Ar–H), 6.78 (td, J = 7.6, 1.2 Hz, 1 H, Ar–H), 6.25 (dd, J = 8.1, 1.1 Hz, 1 H, Ar–H), 5.79 (s, 1H, N–H), 2.23 (s, 6 H, Ar–Me). 13C NMR (100 MHz, CDCl3): δ 162.21 (NCH), 160.93, 140.62, 138.04, 136.70, 134.92, 133.30, 132.41, 128.66, 128.38, 126.40, 119.77, 119.49, 118.22, 118.02, 117.29, 112.26, 18.42. Anal. Calcd for C21H20N2O: C, 79.72; H, 6.37; N, 8.85. Found: C, 79.55; H, 6.28; N, 8.71.
CH), 7.48 (s, 1 H, Ar–H), 7.37 (s, 1 H, Ar–H), 7.14 (s, 5 H, Ar–H), 6.80 (s, 1 H, Ar–H), 6.28 (s, 1 H, Ar–H), 5.73 (s, 1 H, N–H), 2.23 (s, 6 H, Me). 13C NMR (100 MHz, CDCl3): δ 159.46 (NCH), 155.47, 140.98, 137.55, 136.66, 133.38, 132.63, 129.80, 129.47, 128.71, 126.61, 123.92, 122.80, 120.96, 118.17, 112.84, 18.39. Anal. Calcd for C21H18Cl2N2O: C, 65.47; H, 4.71; N, 7.27. Found: C, 65.41; H, 4.53; N, 7.05.
4.3. Synthesis of chromium complexes Cr1(THF)–Cr6(THF)
4.4. X-ray crystallographic studies
Single crystals of Cr2(THF) and Cr4(THF) were obtained by slow diffusion of n-hexane into their CH2Cl2 solutions at room temperature. A single crystal X-ray diffraction study for Cr2(THF) and Cr4(THF) was carried out on a Bruker D8 Venture diffractometer using graphite-monochromated Ga-Kα radiation (λ = 1.34139 Å). Cell parameters were obtained by global refinement of the positions of all collected reflections. Intensities were corrected for Lorentz and polarization effects and empirical absorption. Using Olex2, the structures were solved with XS and refined with ShelXL (Sheldrick, 2015).63 Crystal data and processing parameters for Cr2(THF) and Cr4(THF) are summarized in Table S1.† CCDC reference numbers 2133043–2133044 are for complexes Cr2(THF) and Cr4(THF), respectively.†4.5. General procedures for polymerization in an autoclave reactor
A 1-liter steel reactor with a heating jacket and an internal cooling coil was used for the polymerizations. The stirred 1-liter reactor was charged with about 200 mL of toluene solvent and the desired amount of MAO. The reactor content was heated to the desired polymerization temperature. Once that temperature was reached, the reactor was filled with ethylene at 5 atm or 20 atm or 40 atm. Cr complexes in 5 mL of chlorobenzene were transferred directly to a catalyst addition tank through stainless steel tubing and injected into the reactor with a nitrogen overpressure. The polymerization conditions were maintained with ethylene added on demand. Although heat was continuously removed from the reaction vessel through an internal cooling coil, the temperature increased by 5 to 10 °C (dependent upon the activity) at the end of polymerization in comparison to the original temperature. The resulting solution was removed from the reactor and quenched with acidified methanol. The polymer was filtered off, washed with methanol, and then dried under vacuum overnight.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support by NSFC (No. 52073152, 21871157), the 111 Project (No. D17004), the Shandong Province Natural Science Foundation (No. 2021CXGC010902), the Shandong Provincial Education Department (No. 2019KJA001), and the Taishan Scholar Constructive Engineering Foundation (No. tsqn20161031) is gratefully acknowledged.Notes and references
- G. Wilke, Angew. Chem., Int. Ed., 2003, 42, 5000–5008 CrossRef CAS PubMed.
- K. C. Jayaratne, T. H. Cymbaluk and M. D. Jensen, ACS Catal., 2018, 8, 602–614 CrossRef CAS.
- E. Groppo, C. Lamberti, S. Bordiga, G. Spoto and A. Zecchina, Chem. Rev., 2005, 105, 115–183 CrossRef CAS PubMed.
- S.-F. Yuan, Y. Yan, G. A. Solan, Y. Ma and W.-H. Sun, Coord. Chem. Rev., 2020, 411, 213254 CrossRef CAS.
- Z. Wang, Q. Liu, G. A. Solan and W.-H. Sun, Coord. Chem. Rev., 2017, 350, 68–83 CrossRef CAS.
- D. Peng, X. Yan, C. Yu, S. Zhang and X. Li, Polym. Chem., 2016, 7, 2601–2634 RSC.
- H. Mu, L. Pan, D. Song and Y. Li, Chem. Rev., 2015, 115, 12091–12137 CrossRef CAS PubMed.
- M. C. Baier, M. A. Zuideveld and S. Mecking, Angew. Chem., Int. Ed., 2014, 53, 9722–9744 CrossRef CAS PubMed.
- H. Makio, H. Terao, A. Iwashita and T. Fujita, Chem. Rev., 2011, 111, 2363–2449 CrossRef CAS PubMed.
- M. Delferro and T. J. Marks, Chem. Rev., 2011, 111, 2450–2485 CrossRef CAS PubMed.
- C. Chen, Nat. Rev. Chem., 2018, 2, 6–14 CrossRef CAS.
- J. M. Eagan, J. Xu, R. Di Girolamo, C. M. Thurber, C. W. Macosko, A. M. La Pointe, F. S. Bates and G. W. Coates, Science, 2017, 355, 814–816 CrossRef CAS PubMed.
- D. J. Arriola, E. M. Carnahan, P. D. Hustad, R. L. Kuhlman and T. T. Wenzel, Science, 2006, 312, 714–719 CrossRef CAS PubMed.
- W. Kaminsky, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 3911–3921 CrossRef CAS.
- C. Bariashir, C. Huang, G. A. Solan and W.-H. Sun, Coord. Chem. Rev., 2019, 385, 208–229 CrossRef CAS.
- V. C. Gibson, C. Redshaw and G. A. Solan, Chem. Rev., 2007, 107, 1745–1776 CrossRef CAS PubMed.
- J. Chen, Y. Gao and T. J. Marks, Angew. Chem., Int. Ed., 2020, 59, 14726–14735 CrossRef CAS PubMed.
- C. Tan, C. Zou and C. Chen, J. Am. Chem. Soc., 2022, 144, 2245–2254 CrossRef CAS PubMed.
- M. Xu and C. Chen, Sci. Bull., 2021, 66, 1429–1436 CrossRef CAS.
- Q. Muhammad, C. Tan and C. Chen, Sci. Bull., 2020, 65, 300–307 CrossRef CAS.
- G. Wang, D. Peng, Y. Sun and C. Chen, CCS Chem., 2020, 2, 2025–2034 Search PubMed.
- T. Wang, C. Wu, X. Ji and D. Cui, Angew. Chem., Int. Ed., 2021, 60, 25735–25740 CrossRef CAS PubMed.
- M. Chen and C. Chen, Angew. Chem., Int. Ed., 2020, 59, 1206–1210 CrossRef CAS PubMed.
- K. Li, J. Ye, Z. Wang, H. Mu and Z. Jian, Polym. Chem., 2020, 11, 2740–2748 RSC.
- C. Du, L. Zhong, J. Gao, S. Zhong, H. Liao, H. Gao and Q. Wu, Polym. Chem., 2019, 10, 2029–2038 RSC.
- P. Kenyon, M. Wörner and S. Mecking, J. Am. Chem. Soc., 2018, 140, 6685–6689 CrossRef CAS PubMed.
- C. Feng, Q. Gou, S. Liu, R. Gao and Z. Li, Catalysts, 2021, 11, 276 CrossRef CAS.
- L. Resconi, L. Cavallo, A. Fait and F. Piemontesi, Chem. Rev., 2000, 100, 1253–1345 CrossRef CAS PubMed.
- Y. Zhong, Y. Wu and D. Cui, Macromolecules, 2021, 54, 1754–1759 CrossRef CAS.
- B. Han, Y. Liu, C. Feng, S. Liu and Z. Li, Organometallics, 2021, 40, 242–252 CrossRef CAS.
- H. Wang, H. Cheng, R. Tanaka, T. Shiono and Z. Cai, Polym. Chem., 2018, 9, 4492–4497 RSC.
- X. Li and Z. Hou, Coord. Chem. Rev., 2008, 252, 1842–1869 CrossRef CAS.
- E. Y. X. Chen and T. J. Marks, Chem. Rev., 2000, 100, 1391–1434 CrossRef CAS PubMed.
- T. Xu, Y. Mu, W. Gao, J. Ni, L. Ye and Y. Tao, J. Am. Chem. Soc., 2007, 129, 2236–2237 CrossRef CAS PubMed.
- L. Guo, W. Zhang, F. Cao, Y. Jiang, R. Zhang, Y. Ma, G. A. Solan, Y. Sun and W.-H. Sun, Polym. Chem., 2021, 12, 4214–4225 RSC.
- Q. Zhang, N. Wu, J. Xiang, G. A. Solan, H. Suo, Y. Ma, T. Liang and W.-H. Sun, Dalton Trans., 2020, 49, 9425–9437 RSC.
- J. C. Jenkins and M. Brookhart, J. Am. Chem. Soc., 2004, 126, 5827–5842 CrossRef CAS PubMed.
- S. Matsui, M. Mitani, J. Saito, Y. Tohi, H. Makio, N. Matsukawa, Y. Takagi, K. Tsuru, M. Nitabaru, T. Nakano, H. Tanaka, N. Kashiwa and T. Fujita, J. Am. Chem. Soc., 2001, 123, 6847–6856 CrossRef CAS.
- J. Tian, P. D. Hustad and G. W. Coates, J. Am. Chem. Soc., 2001, 123, 5134–5135 CrossRef CAS PubMed.
- T. R. Younkin, E. F. Connor, J. I. Henderson, S. K. Friedrich, R. H. Grubbs and D. A. Bansleben, Science, 2000, 287, 460–462 CrossRef CAS PubMed.
- G. Zanchin, A. Piovano, A. Amodio, F. De Stefano, R. Di Girolamo, E. Groppo and G. Leone, Macromolecules, 2021, 54, 1243–1253 CrossRef CAS.
- H.-B. Hansen, H. Wadepohl and M. Enders, Chem. – Eur. J., 2021, 27, 11084–11093 CrossRef CAS PubMed.
- S. Deng, Z. Liu, B. Liu and Y. Jin, ChemCatChem, 2021, 13, 2278–2292 CrossRef CAS.
- K. Wang, Y. Jin, B. Liu, R. Zhang, H. Ren, N. Zhao and X. He, Macromol. Chem. Phys., 2020, 221, 1900503 CrossRef CAS.
- O. L. Sydora, Organometallics, 2019, 38, 997–1010 CrossRef CAS.
- C. A. Cruz, M. M. Monwar, J. Barr and M. P. McDaniel, Macromolecules, 2019, 52, 5750–5760 CrossRef CAS.
- E. Groppo, G. A. Martino, A. Piovano and C. Barzan, ACS Catal., 2018, 8, 10846–10863 CrossRef.
- W. Luo, A. Li, S. Liu, H. Ye and Z. Li, Organometallics, 2016, 35, 3045–3050 CrossRef CAS.
- A. Fong, Y. Yuan, S. L. Ivry, S. L. Scott and B. Peters, ACS Catal., 2015, 5, 3360–3374 CrossRef CAS.
- D. S. McGuinness, Chem. Rev., 2011, 111, 2321–2341 CrossRef CAS PubMed.
- S. Liu, R. Pattacini and P. Braunstein, Organometallics, 2011, 30, 3549–3558 CrossRef CAS.
- S. Liu, R. Peloso, R. Pattacini and P. Braunstein, Dalton Trans., 2010, 39, 7881–7883 RSC.
- V. C. Gibson, S. Mastroianni, C. Newton, C. Redshaw, G. A. Solan, A. J. P. White and D. J. Williams, J. Chem. Soc., Dalton Trans., 2000, 1969–1971 RSC.
- V. C. Gibson, C. Newton and C. Redshaw, J. Chem. Soc., Dalton Trans., 1999, 827–830 RSC.
- D. J. Jones, V. C. Gibson, S. M. Green and P. J. Maddox, Chem. Commun., 2002, 1038–1039 RSC.
- M. Enders, P. Fernandez, G. Ludwig and H. Pritzkow, Organometallics, 2001, 20, 5005–5007 CrossRef CAS.
- J. Li, F. Liang, Y. Zhao, X.-Y. Liu, J. Fan and L.-S. Liao, J. Mater. Chem. C, 2017, 5, 6202–6209 RSC.
- S. Liu, Y. Xing, Q. Zheng, Y. Jia and Z. Li, Organometallics, 2020, 39, 3268–3274 CrossRef CAS.
- C. Feng, S. Zhou, D. Wang, Y. Zhao, S. Liu, Z. Li and P. Braunstein, Organometallics, 2021, 40, 184–193 CrossRef CAS.
- Q. Zheng, D. Zheng, B. Han, S. Liu and Z. Li, Dalton Trans., 2018, 47, 13459–13465 RSC.
- C. Huang, Y. Huang, Y. Ma, G. A. Solan, Y. Sun, X. Hu and W.-H. Sun, Dalton Trans., 2018, 47, 13487–13497 RSC.
- J. Zhang, A. F. Li and T. S. A. Hor, Organometallics, 2009, 28, 2935–2937 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra of organic compounds, the ESI spectra of Cr complexes, the characterization of polymer materials, crystal data and processing parameters. CCDC 2133043 and 2133044. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d2py00125j |
| This journal is © The Royal Society of Chemistry 2022 |