
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Addressing big data challenges in mass spectrometry-based metabolomics
Jian
Guo
,
Huaxu
Yu
,
Shipei
Xing
and
Tao
Huan
 *
*
Department of Chemistry, University of British Columbia, 2036 Main Mall, Vancouver, BC Canada, V6T 1Z1, Canada. E-mail: thuan@chem.ubc.ca
First published on 16th August 2022
Abstract
Advancements in computer science and software engineering have greatly facilitated mass spectrometry (MS)-based untargeted metabolomics. Nowadays, gigabytes of metabolomics data are routinely generated from MS platforms, containing condensed structural and quantitative information from thousands of metabolites. Manual data processing is almost impossible due to the large data size. Therefore, in the “omics” era, we are faced with new challenges, the big data challenges of how to accurately and efficiently process the raw data, extract the biological information, and visualize the results from the gigantic amount of collected data. Although important, proposing solutions to address these big data challenges requires broad interdisciplinary knowledge, which can be challenging for many metabolomics practitioners. Our laboratory in the Department of Chemistry at the University of British Columbia is committed to combining analytical chemistry, computer science, and statistics to develop bioinformatics tools that address these big data challenges. In this Feature Article, we elaborate on the major big data challenges in metabolomics, including data acquisition, feature extraction, quantitative measurements, statistical analysis, and metabolite annotation. We also introduce our recently developed bioinformatics solutions for these challenges. Notably, all of the bioinformatics tools and source codes are freely available on GitHub (https://www.github.com/HuanLab), along with revised and regularly updated content.
 Jian Guo | Jian Guo obtained his BSc in materials engineering from Zhejiang University in 2012 and MSc in chemical engineering from the University of Alberta in 2014. He then worked in the oil and gas industry for four years. Currently, he is a PhD candidate in the Department of Chemistry at the University of British Columbia, supervised by Prof. Tao Huan. His research focuses on the development of data acquisition modes and data processing programs for untargeted metabolomics and their applications in systems biology. He has 16 publications with more than 300 citations and an h-index of 8. |
 Huaxu Yu | Huaxu Yu received his BSc in Chemistry at Zhejiang University in 2018. With a passion for mass spectrometry and metabolomics, he joined Prof. Tao Huan's group in 2019 to pursue a PhD in analytical chemistry. His research focuses on quantitative metabolomics. By developing novel bioanalytical workflows, bioinformatics software, and statistical methods, he seeks to facilitate fundamental and applied aspects of mammalian, plant, and microbial research. He is the author of 14 publications and the developer of two R packages. |
 Shipei Xing | Shipei Xing obtained his BSc in Chemistry at Zhejiang University (Chu Kochen Honors College) in 2018. Currently, he is a 4th-year PhD candidate in Dr Tao Huan's group at the University of British Columbia. His research interests focus on developing novel bioinformatic solutions addressing annotation, classification, and purification of tandem mass spectra in untargeted metabolomics. |
 Tao Huan | Dr Tao Huan is an Assistant Professor in the Department of Chemistry at the University of British Columbia (Vancouver, Canada). Research in the Huan lab is focused on the development and application of mass spectrometry-based metabolomics. As of August 2022, he has 66 peer-reviewed publications in high-impact journals, including Nature Methods, Nature Protocols, and Analytical Chemistry, garnering more than 2700 citations and an h-index of 24. Dr Huan is also an affiliated faculty member of the UBC Social Exposome Cluster, the Graduate Program in Bioinformatics, the Genome Science and Technology program, the Djavad Mowafaghian Centre for Brain Health, and the Cluster for Microplastics, Health and the Environment. |
Introduction
Small molecule metabolites play critical roles in numerous cellular activities and provide both direct and indirect readouts of various phenotypes.1–3 The study of the entire collection of metabolites in a given biological system is termed metabolomics. Over the past few decades, metabolomics has been developed as a powerful and indispensable biotechnology in the postgenomic era of biology. In particular, metabolomics has been in demand across many research disciplines to gain a global view of metabolic changes for biomarker discovery and mechanistic understandings.4–7 Given the wide chemical coverage, metabolomics has also been demonstrated as an important tool in exposome research to study the concurrent exposure to a wide variety of xenobiotics and understand their combined toxic effects in health and disease.8–10 Among the various analytical instruments used to perform untargeted metabolomics, mass spectrometry (MS) is the most prominent choice. In particular, liquid chromatography (LC) coupled to high-resolution MS systems can routinely detect and quantify thousands of metabolic features from as little as 10 mg of tissue, 50 μL of urine, and half a million cells.7,11,12 The high sensitivity and throughput of MS also generate a large amount of data. For instance, a typical LC-MS-based metabolomics study can generate over 10 GB of data in a 30-sample analysis. The gigantic amount of metabolomic information cannot be manually processed and interpreted as we usually do in traditional analytical chemistry. Furthermore, the large amount of data makes it challenging to perform metabolomics data acquisition, feature extraction, quantification, statistical analysis, metabolite annotation, data sharing, meta-analysis, and others. Fig. 1 summarizes the common big data challenges in mass spectrometry-based untargeted metabolomics. Addressing these big data challenges is vital to improving metabolomics data quality, obtaining confident biological insights, and minimizing biased biological claims. This Feature Article focuses on four major aspects of big data challenges in LC-MS-based metabolomics, including (1) data acquisition, (2) feature extraction, (3) quantitative and statistical analysis, and (4) metabolite annotation. It also reviews our recent developments and publications addressing these big data challenges from the past two years (2020–2022) (Table 1).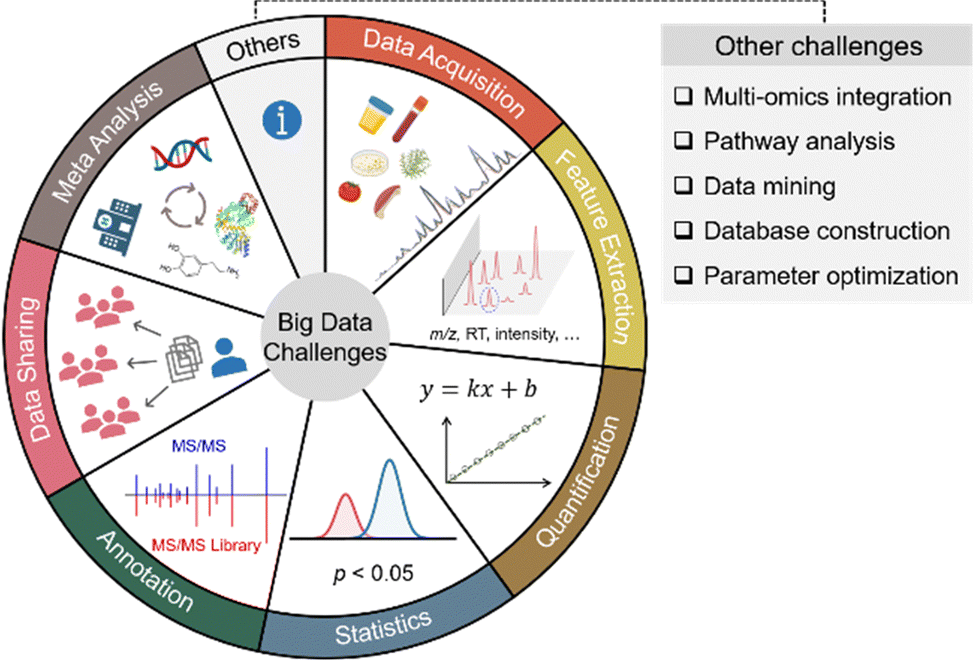 | ||
| Fig. 1 Overview of the big data challenges in mass spectrometry-based untargeted metabolomics. | ||
| Category | Software name | Purpose |
|---|---|---|
| Data acquisition | DaDIA13 | Combining DDA and DIA modes for metabolomics data acquisition |
| Feature extraction | Paramounter14 | Directly measuring the optimal feature extraction parameters |
| Integrated Feature Extraction,15 JPA16 | Extracting metabolic features of both high and low confidence | |
| EVA17 | Evaluating feature fidelity using chromatographic peak shapes | |
| ISFrag18 | De novo annotation of false positive metabolic features generated from in-source fragmentation | |
| Quantitative comparison and statistical analysis | MRC19 | Correcting fold change compression and inflation in MS-based metabolomics |
| MAFFIN20 | Post-acquisition sample normalization | |
| PHPA_precision21 | Correcting computational variation caused by peak height or peak area-based quantification | |
| PowerU22 | Improving the statistical power of MS-based metabolomics | |
| ABC Transformation23 | Improving data normality with feature-specific data transformation | |
| Metabolite annotation | HNL, CSS, and McSearch24 | Concept, algorithm, and web platform to perform spectral similarity analysis and molecular networking |
| MS2Purifier25 | Recognizing and removing contamination fragment ions in experimental MS/MS spectra | |
| SteroidXtract26 | Extracting steroid-like metabolic features based on their unique MS/MS patterns |
Data acquisition
There are three common strategies to collect metabolomics data from MS: full-scan, data-dependent acquisition (DDA), and data-independent acquisition (DIA).27–29 Full-scan mode acquires MS spectra composed of mass-to-charge ratios (m/z) and signal intensities of ions. Since the entire acquisition time is assigned to obtaining MS1 data, full-scan mode provides the detailed chromatographic peak shape and is suitable for quantification. However, full-scan cannot generate tandem MS (MS/MS) data, thus making confident metabolite annotation impossible. In this regard, DDA and DIA modes were developed to obtain the MS/MS spectra after each MS1 spectrum. Particularly, DDA and DIA modes differ in ion isolation windows, which select ions for fragmentation. DDA opens narrow m/z windows (usually 1–5 Da) to acquire the MS/MS spectra for the most intense metabolic ions (i.e., precursor ions), producing a pure MS/MS spectrum for each selected precursor ion, but often lacks the MS/MS acquisition of low-abundant metabolic features. However, MS/MS spectra collection takes time away from MS1 data collection and reduces the number of MS1 data points for constructing chromatographic peaks. Moreover, in the analysis of complex biological samples, the MS/MS acquisition speed is insufficient for comprehensive MS/MS coverage of all detected metabolic features. Many strategies have been developed to improve the efficiency of MS/MS collection, such as iterative MS/MS, gas-phase fractionation, and data-set-dependent acquisition, among others.30–33 In comparison to DDA, DIA opens wide m/z windows (usually >100 Da) to obtain the fragments of multiple precursor ions, covering all metabolic ions, but requires sophisticated spectrum deconvolution tools to assign fragments to their parent ions. Although the pros and cons of each acquisition mode are intuitively known in metabolomics, there are few systematic comparisons to guide metabolomics practitioners to determine which mode fits a given study the best.34,35To address the knowledge gap, we systematically compared the three abovementioned data acquisition modes, focusing on their performance in metabolomics profiling36 and ability to identify statistically significant features.37 In the comparison of metabolomics profiling, we assessed the number of features, MS/MS spectra coverage and quality, quantitative precision, and data processing convenience (Fig. 2). Our results show that the most metabolic features are extracted from full-scan data, which is 53.7% and 64.8% more than DIA and DDA, respectively. In terms of MS/MS spectra, DDA generates higher quality MS/MS spectra that match MS/MS libraries better, whereas DIA has higher MS/MS spectral coverage. Regarding the quantitative precision, no significant difference was observed among these three acquisition modes. In the comparison of significant features discovered across these acquisition modes, we concluded that the consistently discovered ones are mostly true positive features (i.e., real metabolic features).37 They have a strong correlation in abundance among all three modes and present similar statistical performance. On the other side, many uniquely discovered significant features are false positive features from background noise and system contamination.37 Although DDA slightly underperforms full-scan and DIA in significant metabolic feature discovery, it is the most convenient method to obtain high-quality MS/MS spectra for metabolite annotation.
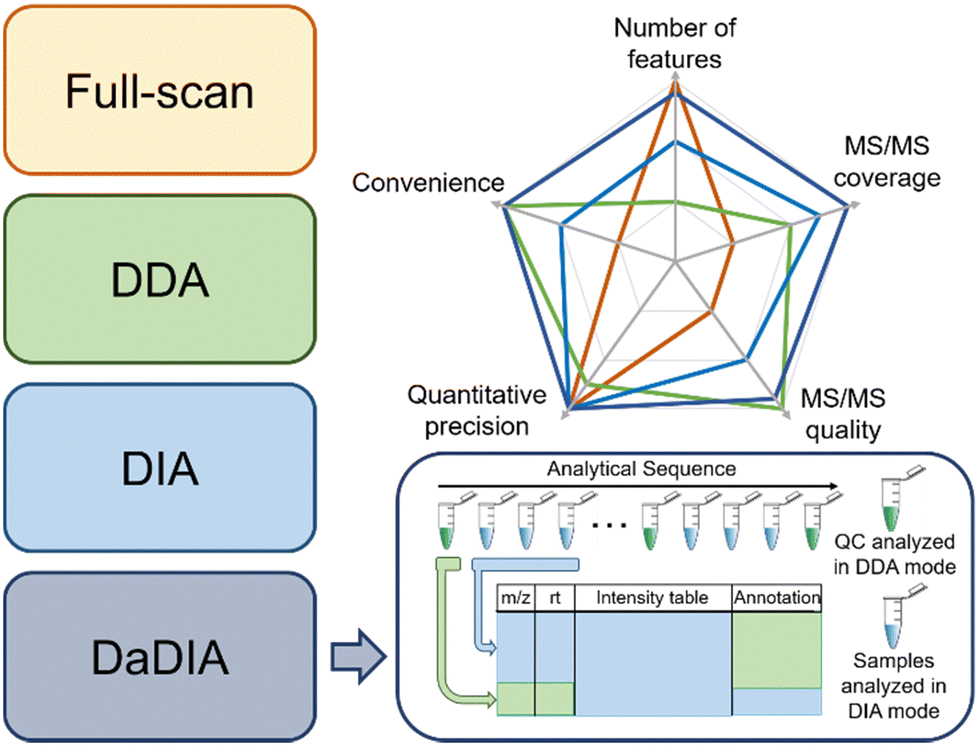 | ||
| Fig. 2 Summary of the commonly used metabolomics data acquisition modes, including full-scan, DDA, DIA, and our recently developed DaDIA workflow. The comparison of their performance is presented in the top-right corner. The schematic workflow of DaDIA is presented in the bottom-right corner. | ||
Following the comparison of these data acquisition modes, we believe that a better data acquisition strategy that integrates the advantages of the existing methods is essential for advancing LC-MS-based metabolomics. We thus developed data-dependent assisted data-independent acquisition (DaDIA).13 The DaDIA workflow performs DIA analyses of biological samples and DDA analyses of the pooled quality control (QC) samples analysed at regular intervals between biological samples throughout the analytical sequence (Fig. 2). The DIA analyses provide high coverage of metabolic features and MS/MS spectra, and the DDA analyses generate high-quality MS/MS spectra to improve the overall confidence of metabolite annotation. We further developed an R package, DaDIA.R, to automate the data processing and metabolite annotation of DaDIA data. Since DaDIA takes full advantage of DDA and DIA, it achieves a much higher coverage of metabolic features with better spectral quality. DaDIA was applied to a study comparing the metabolic alteration in the plasma of leukemia patients before and after receiving chemotherapy. Our results demonstrated that the DaDIA workflow can efficiently detect and annotate approximately four times more significantly altered metabolites than the conventional DDA workflow.
Feature extraction
Extracting metabolic features from raw LC-MS data is a long-standing challenge in untargeted metabolomics. The key is to accurately recognize the chromatographic peaks of real metabolic features and also efficiently clean up the false positive metabolic features coming from the background noise and artificial contaminants.38 Our lab developed a suite of bioinformatics tools to make this process convenient and intuitive (Fig. 3). Over the past few decades, various metabolic feature extraction algorithms, including centWave,39 GridMass,40 and others,41,42 have been developed to automatically recognize metabolic features in raw LC-MS data. These algorithms have also been implemented in commonly used open-source data processing software, such as XCMS, MS-DIAL, MZmine 2, OpenMS, El-MAVEN, and others.43–48 Although the feature extraction process is automated, properly setting over a dozen different feature extraction parameters is difficult. Conventionally, the strategy of design of experiments (DOE) has been implemented to optimize feature extraction parameters. However, DOE-based optimization requires the testing of many parameter combinations, which is time-consuming and ineffective.49–54 After reviewing the well-established metabolomics data processing software, we concluded that four universal chromatographic peak attributes are critical to feature extraction: mass tolerance, peak height, peak width, and instrumental shift. By measuring these peak attributes directly from the raw LC-MS data, it is possible to attain optimal peak picking parameters defined as universal parameters. This is facilitated by the development of the novel concepts of rank-based intensity sorting, zone of interest, and many others. These concepts were then implemented into Paramounter, an R program that automatically and accurately extracts the distributions of these universal parameters from the raw LC-MS-based metabolomics data before feature extraction.14 Our results showed that Paramounter-based direct measurement of feature extraction parameters performs better than conventional DOE-based approaches. It is also more efficient and convenient to use. The proposed universal parameters and the development of Paramounter address a critical need in metabolomics data processing. It is important to note that this work can potentially extend to optimizing parameters for gas chromatography-mass spectrometry (GC-MS)-based metabolomics data.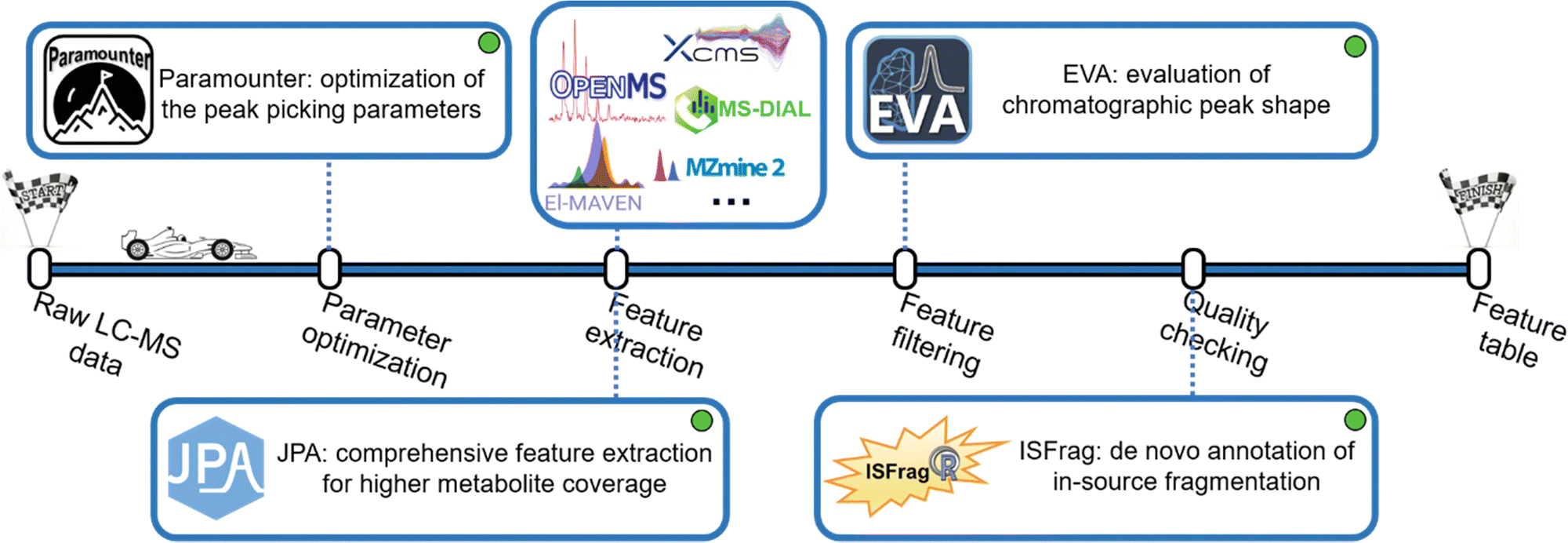 | ||
| Fig. 3 The pipeline of metabolic feature extraction. A suite of bioinformatics tools has been developed in our lab to address the challenges of feature extraction, including optimizing feature extraction parameters, extracting low-quality metabolic features, evaluating feature quality, and removing in-source fragment features. | ||
Another notable challenge in feature extraction is that conventional peak picking algorithms are unable to completely extract features with low abundance or poor chromatographic peak shapes. In particular, many real metabolic features have valid MS/MS spectra but cannot be extracted by conventional peak picking algorithms. In this regard, we designed a peak picking algorithm that can directly extract metabolic features based on their available MS/MS spectra in the raw DDA-based LC-MS data. We also combined this MS/MS spectra-based peak picking algorithm with conventional peak shape-based peak picking to build an integrated workflow for a more comprehensive extraction of metabolic features.15 The proposed integrated feature extraction algorithm extracted 25% more metabolic features from a human urine sample than the conventional centWave-based feature extraction algorithm with the same parameter settings. Furthermore, we created a targeted feature extraction algorithm for use with a targeted list of metabolites with known m/z and retention time. Combining the peak shape-, MS/MS spectra- and targeted list-based peak picking strategies, we constructed JPA (short for joint metabolic feature extraction and automated metabolite annotation), an R package that not only extracts the metabolic features using the integrated strategy but also performs the automated metabolite annotation. When the three algorithms were applied together on a mixture of 134 endogenous metabolite standards, JPA demonstrated superior feature detection sensitivity by reaching a limit of detection (LOD) thousands of times lower than the conventional centWave peak picking algorithm. Moreover, JPA also surpassed the conventional centWave algorithm by detecting 2.3-fold more exposure chemicals from a standard mixture containing 505 drugs and pesticides.16
On the other hand, enhancing feature extraction sensitivity usually comes with an increase in false positive features. False positive metabolic features can reduce the confidence of downstream statistical and biological interpretations.38 A common practice to find false positive features is to manually check the peak shapes of the extracted ion chromatograms (EICs) of metabolic features. Typically, real metabolites are more likely to have Gaussian-shaped chromatographic peaks. Manual checking of EICs is very effective in filtering out false positive noise peaks, as an experienced analytical chemist can easily differentiate between true and false positive features by simply looking at their peak shapes. However, metabolomics data contains thousands of metabolic features, and manual inspection of their EICs is extremely labour-intensive and time-consuming. Previous work developed a strategy to send the data to a smartphone application so that users can manually check the peak shapes, but the time spent on manual checking was not clearly reduced.55 To replace the tedious process with a labour-free task, we developed an artificial intelligence-based program using a convolutional neural network (CNN) model, well-known for its efficient performance in image classification.56 CNN is a type of deep learning algorithm, along with artificial neural networks (ANN), recurrent neural networks (RNN), and many others.57 Compared to traditional machine learning (e.g., Support Vector Machine and Random Forest), the biggest advantage of these deep learning algorithms is that they can learn high-level features from data without manual feature extraction and are very efficient with large-scale data sets.58 Notably, the metabolomics community has recognized the potential of deep learning in metabolomics.58,59 Previous research efforts have incorporated deep learning in metabolic feature extraction, metabolite annotation, and the predictions of retention time and collision cross section.60–69 Regarding classifying good and bad chromatographic peak shapes, a previous work applied CNN to the classification of GC-MS chromatographic peaks.70 In another study, CNN was used to determine the chromatographic peak shapes in LC-MS-based metabolomics data.71 However, that workflow involves multiple R and Python scripts that users have to run individually. Due to their small training data size, users would also need to retrain the model for different LC-MS conditions, such as different spectra acquisition rates.
In our work, we aimed to develop a robust and easy-to-use CNN model for chromatographic peak recognition. To minimize data overfitting and ensure the robustness of the model, we trained our CNN model with over 25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 manually inspected plots of true and false chromatographic peaks generated from 22 different LC-MS-based metabolomics studies. Furthermore, we created a Windows application named EVA (short for evaluation of chromatographic peak shapes) for the convenience of metabolomics researchers with limited programming experience.17 Evaluated using metabolomics data from different MS instruments and acquisition rates, EVA was proved to achieve over 90% classification accuracy when referenced against manual checking results. Notably, another work was published later following a similar CNN strategy. Future work is needed to make performance comparisons.72
000 manually inspected plots of true and false chromatographic peaks generated from 22 different LC-MS-based metabolomics studies. Furthermore, we created a Windows application named EVA (short for evaluation of chromatographic peak shapes) for the convenience of metabolomics researchers with limited programming experience.17 Evaluated using metabolomics data from different MS instruments and acquisition rates, EVA was proved to achieve over 90% classification accuracy when referenced against manual checking results. Notably, another work was published later following a similar CNN strategy. Future work is needed to make performance comparisons.72
Removing false positive features with poor peak shapes is not the last stop, as metabolic features with good EIC peak shapes still might not be real metabolites. Another common type of false positive feature originates from in-source fragmentation (ISF). In LC-MS analysis, ions generated during electrospray ionization (ESI) are always accompanied by ion fragmentation, which leads to ISF ions. ISF is a naturally occurring and inevitable phenomenon that is independent of ionization voltage.73 Annotating ISF features as real metabolites by mistake is detrimental to the downstream biological interpretation. Previous works have developed strategies to recognize ISF via manual efforts, stable isotopes, or reference standards.74–78 However, many metabolic features have diverse ISF patterns and might not have a standard MS/MS spectrum available for such manual checking. To provide an automated workflow for de novo recognition of ISF features, we developed the MS/MS library-free R package ISFrag to seek ISF features based on three patterns: (1) ISF ions coelute with their precursor ion, (2) the m/z of ISF ions appear in the MS/MS spectrum of their precursor ion, and (3) ISF ions and their precursor ion are similar in fragmentation patterns and thus have highly correlated MS/MS spectra.18 Notably, ISFrag can be used on LC-MS data generated from full-scan, DIA, and DDA modes as long as at least one DDA analysis is performed to provide the MS/MS spectra required by ISFrag. Our results show that ISFrag achieves 100% accuracy in recognizing the ISF features that fit all three abovementioned patterns from the data of a standard mixture containing 125 endogenous metabolites. ISFrag allowed us to successfully recognize falsely annotated metabolites in a human urine dataset, determining them to be in-source fragments.
Quantitative measurement and statistical analysis
Besides the number of detected metabolic features, quantitative accuracy and precision are additional key drivers for delivering successful metabolomics analyses. Our lab has developed a set of bioinformatics tools to improve quantitative accuracy, precision, and statistical performance (Fig. 4). In untargeted metabolomics, the absolute quantification of every detected metabolic feature is not possible. Instead, the relative MS signal ratio, or signal fold change, between biological sample groups (e.g., normal vs. diseased) is widely used for quantitative comparison. In this case, quantitative accuracy in untargeted metabolomics is more about whether the concentration ratio can be accurately determined. Our recent works discovered that the MS signal intensity ratio can have clear quantitative biases.19,79 Particularly for the ESI-MS analytical platform, the measured MS signal intensity ratio can be significantly lower or higher than the concentration ratio, which are termed fold change compression and inflation, respectively (Fig. 4a). Mechanistically, fold change compression and inflation are caused by the non-negligible intercept values of the linear calibration curves. Our urine metabolomics study showed that 72% of metabolic features have compressed fold changes and 16% of features have inflated fold changes. Surprisingly, only 12% of features possess unbiased MS signal intensity ratios with 10% relative error or lower. Even worse, these biased ratios exist in the linear range of ESI responses and cannot be corrected even after careful injection volume optimization. In this respect, we developed the metabolic ratio correction (MRC) workflow, an integrated analytical workflow with automated data processing tools to correct the biased MS signal intensity ratios. In addition to the routine analytical sequence, MRC workflow analyses serial diluted QC samples to construct calibration curves for each metabolic feature (Fig. 4a). The measured MS signal intensities in biological samples are then converted to QC loading amounts for downstream quantitative comparison and statistical analysis.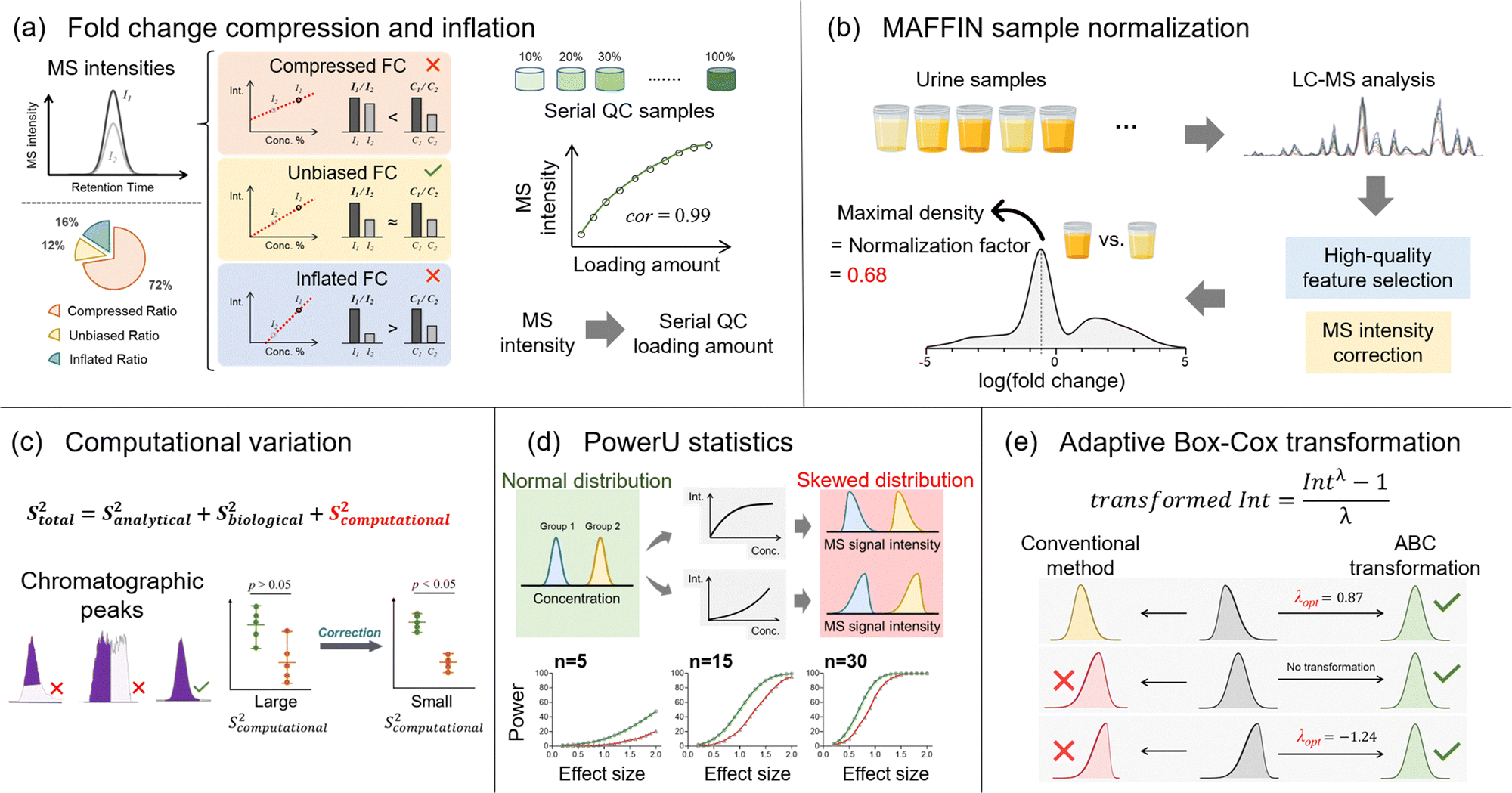 | ||
| Fig. 4 Summary of bioinformatics tools and developments that address the big data challenges of quantitative comparison and statistical analysis. (a) The underrated fold change compression and inflation in the linear ESI ranges; (b) the development of the MAFFIN sample normalization workflow to achieve post-acquisition normalization; (c) the proposal of computational variation on top of conventional analytical and biological variations; (d) the assessment of the diminished statistical power caused by nonlinear ESI responses; (e) adaptive Box-Cox transformation enables the conversion of non-normal metabolic data distributions into normal distributions for statistical analysis. | ||
The fair comparison of samples with equal amounts or concentrations is also critical to quantitative accuracy, which is achieved by sample normalization. Sample normalization is especially important for biological samples with significant biological dilution effects, such as urine, saliva, and feces.80–83 In general, sample normalization can be applied either before or after data acquisition. Pre-acquisition sample normalization measures a certain quantity that reflects the total sample amount or metabolite concentration. The samples are then reconstituted to appropriate final volumes based on the measured quantities to make the total concentration consistent between the samples. For example, creatinine level generally reflects the urine concentration and is commonly used for normalizing urine samples.80,84,85 However, many biological sample types lack reliable quantities that represent the total sample amount for normalization.84
Post-acquisition sample normalization is an alternate strategy that is data-driven and does not require reliable quantities for different sample types. Certain assumptions are made about the metabolomics data structure, and then normalization factors are calculated for adjusting the measured signal intensities. In essence, the accuracy of the assumption determines the post-acquisition sample normalization performance. For instance, the mass spectrum total useful signal (MSTUS) algorithm assumes equal total signal intensities among samples. However, the MS signal intensities of different metabolic features can vary by several magnitudes owing to different concentrations and ionization efficiencies. Therefore, the MSTUS algorithm can be dominated by high-intensity metabolic features and fail to reflect the change in the total metabolome. Probabilistic quotient normalization (PQN) addresses the issue of drastically different signal intensities and thus can be more useful. In the PQN-based workflow, quotients of all metabolic features are calculated against the reference sample, and the median of the calculated quotients is used as a normalization factor.86 After quotient calculation, all metabolic features are equally considered for normalization regardless of their original MS intensities. However, the median quotient only correctly represents the normalization factor when the numbers of up- and down-regulated metabolic features are equal.20 However, it is quite common to see different numbers of up- and down-regulated metabolic features in metabolomics. In addition, given the above-mentioned issue of signal ratio bias, calculating quotients directly using MS signals might not represent metabolic concentration changes accurately. Even so, the detected metabolic features are not pre-processed before normalization in conventional post-acquisition normalization algorithms, which causes significant bias.
In this regard, we developed MAFFIN (short for maximal density fold change normalization with high-quality metabolic features and corrected signal intensities), an accurate and robust post-acquisition sample normalization workflow for MS-generated metabolomics data that is independent of sample type (Fig. 4b).20 MAFFIN first selects high-quality metabolic features by evaluating multiple orthogonal quantification criteria and then corrects their MS signal intensities for normalization. Then, we created an efficient method to calculate normalization factors, which is based on the maximal density fold change (MDFC) computed by a kernel density approach.87 Unlike the PQN algorithm, which relies on balanced up- and down-regulated metabolic features, MDFC normalization assumes that the unchanged metabolic features dominate the fold change frequency. Hence, it is not influenced by the balance of up- and down-regulated metabolic features. Using simulated data, we show that as long as the percentage of unchanged metabolic features is larger than 25%, MDFC is a good representation of the true normalization factor.20 Using twenty publicly available and two in-house metabolomics data sets, we confirmed that MAFFIN outperforms four commonly used post-acquisition normalization methods, including total intensity, median intensity, PQN, and quantile normalizations, in terms of reducing intragroup variations. The biological application of MAFFIN on a human saliva metabolomics study reduces the unwanted variation introduced by the biological dilution effect, leading to better data separation in principal component analysis (PCA) and more significantly altered metabolic features.
Quantitative precision is another key factor for a successful metabolomics study. Our recent work recognizes that besides the well-recognized analytical and biological variations, untargeted metabolomics encounters additional quantitative variation, termed computational variation.21 The computational variation is caused by automated computational data processing steps, where the software cannot accurately determine chromatographic peak heights/areas for metabolic features with poor chromatographic peak shapes (Fig. 4c).16 Using various biological sample types, we systematically investigated how sample concentration, LC separation conditions, and data processing software contribute to computational variation. Our results suggest that the computational variation is largely determined by the data processing software. In addition, the magnitude of the computational variation is consistent across different samples when their metabolic concentrations are similar. We further developed PHPA_precision, a tool to minimize the computational variation in metabolomics studies by properly selecting between peak height or area for the peak intensity calculation method. This bioinformatics solution helped reduce the computational variation of 71% (652/915) of metabolic features, and over 31% (206/652) of the corrected features showed distinctly changed statistical significance.
Following the quantitative comparison, our lab also attempted to understand metabolomics data distributions in order to improve the performance of statistical analyses. Currently, parametric statistical models, such as Student's t-test, are widely used to extract the significantly changed metabolites. However, the requirements on data normality for these statistical analyses are often violated due to the nonlinear ESI responses in MS-based metabolomics. As a result, the statistical power can be reduced and some significantly changed metabolites are thus missed. Although nonlinear ESI response has been well-known for decades, its impact on data distribution and statistical analysis has not been systematically studied. To address this knowledge gap, we used both Monte Carlo simulations and real metabolomics data sets to quantitatively assess the diminished statistical power caused by nonlinear ESI responses (Fig. 4d). Our urine metabolomics data demonstrated that over 80% of metabolic features present nonlinear ESI response patterns, causing either left-skewed or right-skewed MS signal distributions.22 In addition, clear relationships between the degree of reduced statistical power and sample size/effect size were observed. To address this issue, we developed PowerU, a data processing tool to minimize the non-normality induced by nonlinear ESI response.22 Applying PowerU to a metabolomics study of mouse gut microbiome led to 105 extra metabolic features being discovered as significant, which largely reduces the chance of missing important biomarkers.
Besides nonlinear signal response, many other factors contribute to the overall non-normal metabolomics data distribution, including intrinsically non-normally distributed concentration data, sample collection, and sample preparation. As a result, the metabolomics data distributions are often diverse and complicated. However, despite the thousands of metabolomics publications every year, the study of metabolomics data distribution is limited. Additionally, in routine metabolomics practice, data transformation is commonly used to shape the various non-normal data distributions for statistical analysis. However, the most popular transformation approaches, log and square root transformations,88 do not consider the data structure and treat all the metabolic features equally. Therefore, there is no guarantee that the data normality can be improved after applying those transformations. Recently, our work explored and modeled the metabolic feature intensity distributions using three large and publicly available data sets, which confirmed that the non-normal distribution is common and varied in untargeted metabolomics research. The metabolomics data were modeled into nine types of beta distributions, among which two low-normality types are particularly common. Given the diverse data distributions, we proposed adaptive Box-Cox (ABC) transformation, a feature-specific data transformation approach for improving data normality (Fig. 4e).23 A power parameter, lambda, is tuned based on the data structure of each metabolic feature to ensure improved data normality after transformation. Tested on a series of Monte Carlo simulations, ABC transformation outperforms the two abovementioned conventional data transformation methods for both positively and negatively skewed data distributions. However, it is important to recognize that any nonlinear data transformation will change feature-to-feature relationships. For the correlation analysis of a metabolic feature pair, it is recommended to use the original quantitative data rather than the transformed data. Additionally, data transformation methods can alter the overall data distribution pattern. Especially in our feature-specific data transformation workflow, different features can be subjected to different transformation functions. Consequently, the visualization of the overall metabolic changes (e.g., principal component analysis) might be distorted.23
Metabolite annotation
Metabolite annotation is the last step before sending biological researchers a list of significantly changed metabolites for biological interpretation. According to the metabolite annotation confidence levels proposed by the Metabolomics Standards Initiative (MSI), level 1 identifications refer to metabolite structures confirmed by chemical standards with MS1, MS/MS, and retention time matched.89 However, due to the limited chemical standards available, the majority of metabolic features remain unannotated in MS-based metabolomics studies, forming the “dark matter” in untargeted metabolomics.90,91 To properly annotate unrecognized metabolites, there are generally three complementary strategies, including (1) known-to-unknown-based propagated annotation,92 (2) de novo annotation,93 and (3) in silico fragmentation-based annotation.94 Our lab recognizes the limitations of each strategy and develops bioinformatics solutions to address them (Fig. 5).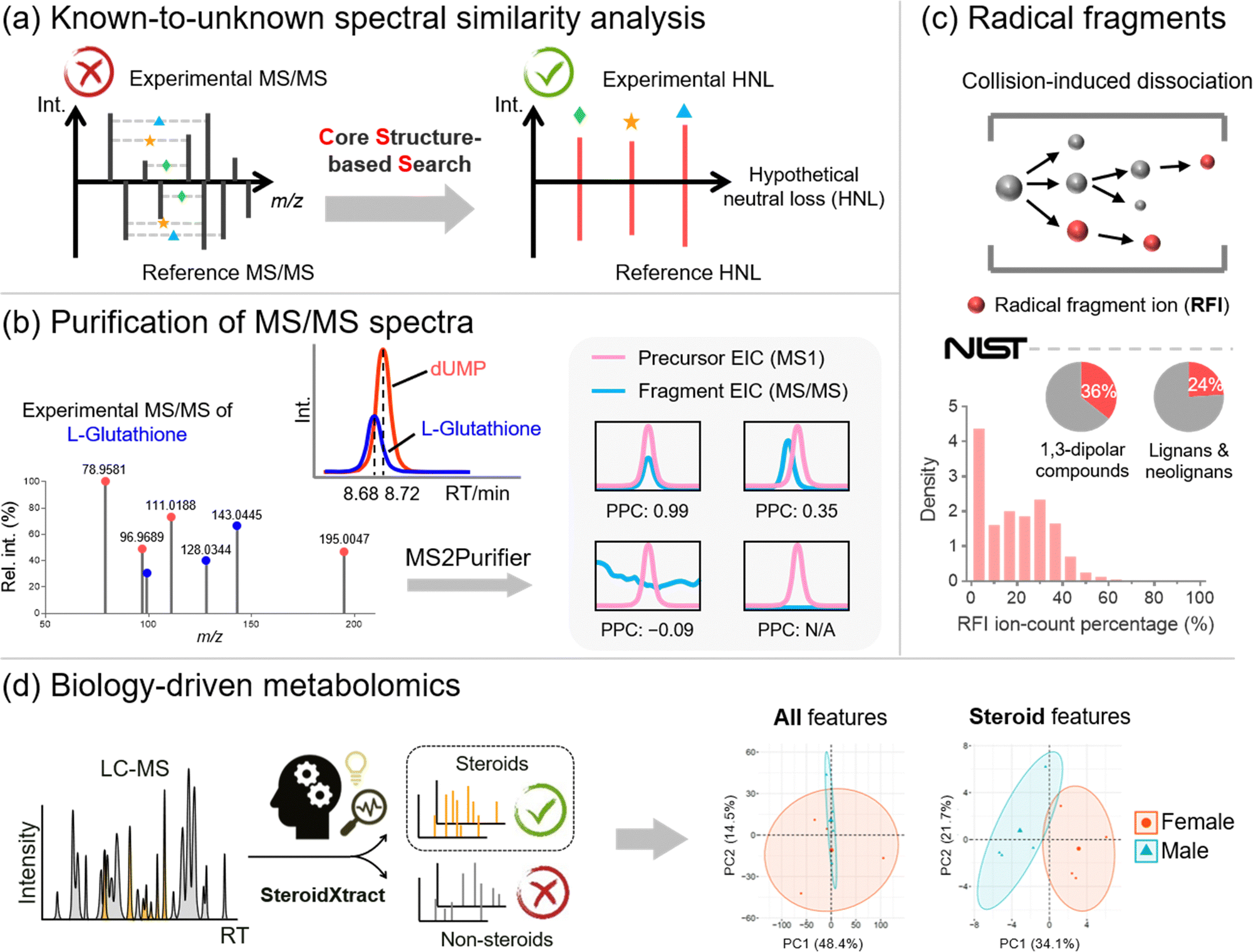 | ||
| Fig. 5 Summary of metabolite annotation developments. (a) The implementation of core structure-based search to improve performance of known-to-unknown spectral similarity analysis; (b) the purification of experimental MS/MS spectra by using MS2Purifier to remove contamination fragment ions; (c) the presence of radical fragment ions in collision-induced dissociation-based MS/MS spectra; (d) biology-driven metabolomics enables the targeted study of both known and unknown steroid features using a high-coverage untargeted metabolomics approach. | ||
First of all, annotation of unrecognized metabolites often relies on searching for known metabolites with similar chemical structures. The structural similarity can be reflected by MS/MS spectral similarity, which is the key in known-to-unknown based metabolite annotation. Therefore, the development of a proper algorithm to compute spectral similarity is of great importance. Previous developments of Global Natural Products Social Molecular Networking (GNPS) and NIST Hybrid Similarity Search (HSS), among others, have been proposed.95–97 These algorithms consider the matching of both the m/z of fragment ions and the m/z differences between fragment ions and their precursors (i.e., neutral losses). They can reflect a certain degree of spectral similarity between metabolites and their one-step reaction biotransformed derivatives. However, these conventional algorithms show limited capability in capturing the common core structural component embedded in the metabolites, as the core structural information cannot be captured using fragment ions or neutral losses.
To create a spectral similarity algorithm that considers the core structural information, we proposed the concept of hypothetical neutral loss (HNL), which is defined as the mass difference between a pair of fragment ions in an MS/MS spectrum (Fig. 5a).24 These mass differences are hypothetical as (1) some HNL values of an experimental spectrum may not represent real metabolite substructures but are merely arbitrary values; and (2) some HNLs are not even generated during the fragmentation process. We demonstrated that HNL values contain core structural information that can improve access to shared structural units between two MS/MS spectra. We thus developed the Core Structure-based Search (CSS) algorithm, which considers conventional fragment ions, neutral losses, and more importantly, HNL values. Compared to existing spectral comparison algorithms, CSS shows a significantly improved correlation between spectral and structural similarities, paving the way for more accurate and informative molecular networking analysis. Furthermore, by combining the CSS algorithm, an HNL library, and a biotransformation database, we developed Metabolite core structure-based Search (McSearch), a web-based platform to facilitate the annotation of unknown metabolites by referencing the MS/MS spectra of their structural analogs.
During spectral similarity analysis, as well as de novo spectra interpretation, the spectral quality of experimental MS/MS matters. However, MS/MS data collected from LC-MS analyses are often contaminated because the selection of precursor ions is based on a low-resolution quadrupole mass filter. A consequence of the wide m/z isolation window is that precursor ions of other chemicals with similar m/z values can also get through the mass filter into the collision cell for fragmentation. The fragmentation of unwanted precursor ions generates contamination fragmentation ions (CFIs), which show up with true fragmentation ions (TFIs) from the targeted precursor ions, leading to “chimeric” MS/MS spectra. This issue has been recognized in metabolomics with the development of RAMSY.98 To recognize and remove CFIs in experimental MS/MS spectra, we proposed a peak correlation-based approach (Fig. 5b).25 The primary premise is that TFIs should coelute with their parent ions with highly correlated LC chromatographic patterns, but CFIs do not necessarily follow the patterns. On top of that, we developed MS2Purifier, a machine learning-assisted solution that removes CFIs from experimental MS/MS spectra and improves MS/MS spectral quality for more confident metabolite identification and MS/MS interpretation. Our work was published at a similar time as another library-based MS/MS cleaning platform, DecoID.99 These two approaches use complementary algorithms to remove contamination fragment ions, and the combined usage may lead to better spectra purification.
On the other hand, in silico fragmentation is a powerful solution that generates predicted MS/MS spectra for a broad range of chemicals without reference standards.94,100–106 Particularly, combining in silico structural databases with machine learning approaches further enhances the confidence of unknown identification.93,107,108 To achieve in silico MS/MS prediction, fragmentation rules are usually implemented, of which an important one is the even-electron rule. It states that even-electron precursor ions should follow heterolytic cleavages and predominately generate even-electron fragment ions with very few radical fragment ions (RFIs).109 However, our study of over one million low-energy collision-induced dissociation (CID) MS/MS spectra for 27,613 unique chemical compounds in the NIST20 MS/MS spectral library shows that over 60% of MS/MS spectra of even-electron precursors contain at least 10% RFIs by ion-count (total number of ions) in positive and negative ESI modes (Fig. 5c).110 This work indicates that the even-electron rule is widely disobeyed, and strictly following the even-electron rule may lead to the non-comprehensive prediction of MS/MS spectra.
Last but not least, in many metabolomics studies, biological researchers are interested in not the entire metabolome but specific classes of chemicals that are essential to the biological process. For instance, steroids are a class of molecules that play a critical role in many physiological systems and diseases, yet many steroids are unrecognized and unreported in the literature. The ability to unbiasedly and accurately detect and quantify both known and unknown steroids is of great significance. However, the recognition of unknown steroids is a big challenge. To address this question, our lab proposed a biology-driven solution.26 In that work, we developed a CNN-based bioinformatics tool, SteroidXtract, to recognize steroid molecules in MS-based untargeted metabolomics using their unique MS/MS spectral patterns (Fig. 5d). Our results demonstrate that SteroidXtract can confidently identify a broad range of both known and unknown steroids in biological samples, greatly accelerating a variety of steroid-focused life science research. Compared to conventional statistics-driven untargeted metabolomics data interpretation, our work offers a novel automated biology-driven approach that prioritizes biologically significant molecules with high throughput and sensitivity.
In general, the prediction of chemical classes directly from MS/MS data alone does not work well for all chemical classes. This is mainly due to the limited reference spectra available, which leads to the problem of compound class imbalance. Our SteroidXtract work addressed this issue by data augmentation, the creation of artificial training data from the existing steroid MS/MS spectra.26 However, achieving a system-level chemical classification using data augmentation has not been tested. Moreover, many chemical classes do not have clear or specific MS/MS spectral patterns, lowering the prediction sensitivity and specificity. As such, achieving generic chemical class prediction requires other structural and spectral information. The recent publication of CANOPUS (class assignment and ontology prediction using mass spectrometry) makes it possible to perform system-level compound class predictions directly from molecular fingerprints.111 CANOPUS was trained using support vector machine and deep learning algorithms to build the connections between fragmentation patterns, molecular fingerprints, and chemical classes. A key advantage of this design is that it separates the prediction of fingerprints using MS/MS spectra and the prediction of chemical classes using fingerprints. Therefore, these two models can be trained using separate datasets. This allows the prediction of chemical class using fingerprints, not limited to the data that have available reference MS/MS spectra, and it can utilize the entire chemical database for training. For the application of CANOPUS, an inputted MS/MS spectrum is processed to generate a fragmentation tree and predicted molecular fingerprints that are then used to predict the hierarchical compound class of the represented metabolite. There are also other structural classification approaches that rely on MS/MS clustering or chemical database searching.112,113 Future research may go towards in-depth global metabolite annotation and structural analog discovery with the aid of compound class-enhanced molecular networking. Additionally, comparative metabolomics on the compound class level may also provide a more comprehensive and intuitive mechanistic insight behind biological questions.111,114
Conclusion and future perspectives
In this Feature Article, we elaborated on our bioinformatics solutions that address the major big data challenges regarding data acquisition, feature extraction, quantitative and statistical analysis, and metabolite annotation. A successful metabolomics study depends on the careful consideration of all these challenges given a data acquisition platform. Therefore, it is important to have a metabolomics data processing workflow that reasonably combines the newly developed computational tools. In addition, more advanced data acquisition techniques come with new data challenges. For instance, ion mobility-mass spectrometry (IM-MS) adds collision cross section (CCS) as another dimension of data complexity, imaging MS generates metabolomics data with spatial distribution in tissue samples (i.e., spatial omics), and so on. Hence, new bioinformatics tools are needed to reveal the meaningful biological information hidden in those high-dimensional data. Finally, tools for multi-omics data integration and visualization are still greatly needed. We hope that this paper can help researchers become more aware of big data challenges in MS-based metabolomics and encourage the metabolomics community to further develop bioinformatics solutions to address them.Author contributions
T. Huan, J. Guo, H. Yu, and S. Xing wrote the manuscript jointly.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was funded by the University of British Columbia Start-up Grant (F18-03001), Canada Foundation for Innovation (CFI 38159), the UBC Support for Teams to Advance Interdisciplinary Research Award (F19-05720), the New Frontiers in Research Fund/Exploration (NFRFE-2019-00789), the Natural Sciences and Engineering Research Council of Canada (NSERC) Discovery Grant (RGPIN-2020-04895). We also thank Alisa Hui for proofreading this manuscript.Notes and references
- J. H. Low, P. Li, E. G. Y. Chew, B. Zhou, K. Suzuki, T. Zhang, M. M. Lian, M. Liu, E. Aizawa and C. R. Esteban, Cell Stem Cell, 2019, 25, 373–387.e379 CrossRef CAS PubMed.
- M. M. Rinschen, J. Ivanisevic, M. Giera and G. Siuzdak, Nat. Rev. Mol. Cell Biol., 2019, 20, 353–367 CrossRef CAS PubMed.
- M. Yang, T. Soga and P. J. Pollard, J. Clin. Invest., 2013, 123, 3652–3658 CrossRef CAS PubMed.
- G. J. Patti, O. Yanes and G. Siuzdak, Nat. Rev. Mol. Cell Biol., 2012, 13, 263–269 CrossRef CAS PubMed.
- C. H. Johnson, J. Ivanisevic and G. Siuzdak, Nat. Rev. Mol. Cell Biol., 2016, 17, 451–459 CrossRef CAS PubMed.
- L. Li, X. Zheng, Q. Zhou, N. Villanueva, W. Nian, X. Liu and T. Huan, Sci. Rep., 2020, 10, 1–12 CrossRef CAS PubMed.
- C. H. Johnson, C. M. Dejea, D. Edler, L. T. Hoang, A. F. Santidrian, B. H. Felding, J. Ivanisevic, K. Cho, E. C. Wick and E. M. Hechenbleikner, Cell Metab., 2015, 21, 891–897 CrossRef CAS PubMed.
- B. Warth, S. Spangler, M. Fang, C. H. Johnson, E. M. Forsberg, A. Granados, R. L. Martin, X. Domingo-Almenara, T. Huan and D. Rinehart, Anal. Chem., 2017, 89, 11505–11513 CrossRef CAS PubMed.
- Y.-M. Go, D. I. Walker, Y. Liang, K. Uppal, Q. A. Soltow, V. Tran, F. Strobel, A. A. Quyyumi, T. R. Ziegler and K. D. Pennell, Toxicol. Sci, 2015, 148, 531–543 CrossRef CAS PubMed.
- D. I. Walker, D. Valvi, N. Rothman, Q. Lan, G. W. Miller and D. P. Jones, Curr. Epidemiol. Rep., 2019, 6, 93–103 CrossRef PubMed.
- H. Yu, N. Villanueva, T. Bittar, E. Arsenault, B. Labonté and T. Huan, Anal. Chim. Acta, 2020, 1136, 168–177 CrossRef CAS PubMed.
- B. Warth, P. Raffeiner, A. Granados, T. Huan, M. Fang, E. M. Forsberg, H. P. Benton, L. Goetz, C. H. Johnson and G. Siuzdak, Cell Chem. Biol., 2018, 25, 291–300.e293 CrossRef CAS PubMed.
- J. Guo, S. Shen, S. Xing and T. Huan, Anal. Chem., 2021, 93, 2669–2677 CrossRef CAS PubMed.
- J. Guo, S. Shen and T. Huan, Anal. Chem., 2022, 94, 4260–4268 CrossRef CAS PubMed.
- Y. Hu, B. Cai and T. Huan, Anal. Chem., 2019, 91, 14433–14441 CrossRef CAS PubMed.
- J. Guo, S. Shen, M. Liu, C. Wang, B. Low, Y. Chen, Y. Hu, S. Xing, H. Yu and Y. Gao, Metabolites, 2022, 12, 212 CrossRef CAS PubMed.
- J. Guo, S. Shen, S. Xing, Y. Chen, F. Chen, E. M. Porter, H. Yu and T. Huan, Anal. Chem., 2021, 93, 12181–12186 CrossRef CAS PubMed.
- J. Guo, S. Shen, S. Xing, H. Yu and T. Huan, Anal. Chem., 2021, 93, 10243–10250 CrossRef CAS PubMed.
- H. Yu and T. Huan, Anal. Chem., 2021, 93, 2254–2262 CrossRef CAS PubMed.
- H. Yu and T. Huan, Bioinformatics, 2022, 38, 3429–3437 CrossRef PubMed.
- H. Yu, Y. Chen and T. Huan, Anal. Chem., 2021, 93, 8719–8728 CrossRef CAS PubMed.
- H. Yu and T. Huan, Anal. Chim. Acta, 2022, 1200, 339614 CrossRef CAS PubMed.
- H. Yu, P. Sang and T. Huan, Anal. Chem., 2022, 94, 8267–8276 CrossRef CAS PubMed.
- S. Xing, Y. Hu, Z. Yin, M. Liu, X. Tang, M. Fang and T. Huan, Anal. Chem., 2020, 92, 14476–14483 CrossRef CAS PubMed.
- S. Xing, H. Yu, M. Liu, Q. Jia, Z. Sun, M. Fang and T. Huan, J. Am. Soc. Mass Spectrom., 2021, 32, 2296–2305 CrossRef CAS PubMed.
- S. Xing, Y. Jiao, M. Salehzadeh, K. K. Soma and T. Huan, Anal. Chem., 2021, 93, 5735–5743 CrossRef CAS PubMed.
- H. P. Benton, J. Ivanisevic, N. G. Mahieu, M. E. Kurczy, C. H. Johnson, L. Franco, D. Rinehart, E. Valentine, H. Gowda and B. K. Ubhi, Anal. Chem., 2015, 87, 884–891 CrossRef CAS PubMed.
- A. T. Roemmelt, A. E. Steuer, M. Poetzsch and T. Kraemer, Anal. Chem., 2014, 86, 11742–11749 CrossRef CAS PubMed.
- H. L. Röst, G. Rosenberger, P. Navarro, L. Gillet, S. M. Miladinović, O. T. Schubert, W. Wolski, B. C. Collins, J. Malmström and L. Malmström, Nat. Biotechnol., 2014, 32, 219–223 CrossRef PubMed.
- C. D. Broeckling, E. Hoyes, K. Richardson, J. M. Brown and J. E. Prenni, Anal. Chem., 2018, 90, 8020–8027 CrossRef CAS PubMed.
- M. R. Hoopmann, G. E. Merrihew, P. D. von Haller and M. J. MacCoss, J. Proteome Res., 2009, 8, 1870–1875 CrossRef CAS PubMed.
- J. P. Koelmel, N. M. Kroeger, E. L. Gill, C. Z. Ulmer, J. A. Bowden, R. E. Patterson, R. A. Yost and T. J. Garrett, J. Am. Soc. Mass Spectrom., 2017, 28, 908–917 CrossRef CAS PubMed.
- M. N. Calderón-Santiago, F. Priego-Capote and M. D. Luque de Castro, Anal. Chem., 2014, 86, 7558–7565 CrossRef PubMed.
- P. Barbier Saint Hilaire, K. Rousseau, A. Seyer, S. Dechaumet, A. Damont, C. Junot and F. Fenaille, Metabolites, 2020, 10, 158 CrossRef PubMed.
- X. Zhu, Y. Chen and R. Subramanian, Anal. Chem., 2014, 86, 1202–1209 CrossRef CAS PubMed.
- J. Guo and T. Huan, Anal. Chem., 2020, 92, 8072–8080 CrossRef CAS PubMed.
- J. Guo and T. Huan, Anal. Chim. Acta, 2020, 1137, 37–46 CrossRef CAS PubMed.
- M. Sindelar and G. J. Patti, J. Am. Chem. Soc., 2020, 142, 9097–9105 CrossRef CAS PubMed.
- R. Tautenhahn, C. Böttcher and S. Neumann, BMC Bioinf., 2008, 9, 1–16 CrossRef PubMed.
- V. Treviño, I. L. Yañez-Garza, C. E. Rodriguez-López, R. Urrea-López, M. L. Garza-Rodriguez, H. A. Barrera-Saldaña, J. G. Tamez-Peña, R. Winkler and R. I. Díaz de-la-Garza, J. Mass Spectrom., 2015, 50, 165–174 CrossRef PubMed.
- H. Tsugawa, M. Kanazawa, A. Ogiwara and M. Arita, Bioinformatics, 2014, 30, 2379–2380 CrossRef CAS PubMed.
- V. P. Andreev, T. Rejtar, H.-S. Chen, E. V. Moskovets, A. R. Ivanov and B. L. Karger, Anal. Chem., 2003, 75, 6314–6326 CrossRef CAS PubMed.
- H. Tsugawa, T. Cajka, T. Kind, Y. Ma, B. Higgins, K. Ikeda, M. Kanazawa, J. VanderGheynst, O. Fiehn and M. Arita, Nat. Methods, 2015, 12, 523–526 CrossRef CAS PubMed.
- C. A. Smith, E. J. Want, G. O'Maille, R. Abagyan and G. Siuzdak, Anal. Chem., 2006, 78, 779–787 CrossRef CAS PubMed.
- R. Tautenhahn, G. J. Patti, D. Rinehart and G. Siuzdak, Anal. Chem., 2012, 84, 5035–5039 CrossRef CAS PubMed.
- T. Pluskal, S. Castillo, A. Villar-Briones and M. Orešič, BMC Bioinf., 2010, 11, 1–11 CrossRef PubMed.
- M. F. Clasquin, E. Melamud and J. D. Rabinowitz, Curr. Protoc. Bioinf., 2012, 37, 14.11.11–14.11.23 Search PubMed.
- E. Melamud, L. Vastag and J. D. Rabinowitz, Anal. Chem., 2010, 82, 9818–9826 CrossRef CAS PubMed.
- L. Eriksson, E. Johansson, N. Kettaneh-Wold, C. Wikström and S. Wold, Principles and Applications, Learn ways AB, Stockholm, 2000 Search PubMed.
- H. Zheng, M. R. Clausen, T. K. Dalsgaard, G. Mortensen and H. C. Bertram, Anal. Chem., 2013, 85, 7109–7116 CrossRef CAS PubMed.
- M. Eliasson, S. Rännar, R. Madsen, M. A. Donten, E. Marsden-Edwards, T. Moritz, J. P. Shockcor, E. Johansson and J. Trygg, Anal. Chem., 2012, 84, 6869–6876 CrossRef CAS PubMed.
- G. Libiseller, M. Dvorzak, U. Kleb, E. Gander, T. Eisenberg, F. Madeo, S. Neumann, G. Trausinger, F. Sinner and T. Pieber, BMC Bioinf., 2015, 16, 1–10 CrossRef CAS PubMed.
- O. E. Albóniga, O. González, R. M. Alonso, Y. Xu and R. Goodacre, Metabolomics, 2020, 16, 1–12 CrossRef PubMed.
- C. McLean and E. B. Kujawinski, Anal. Chem., 2020, 92, 5724–5732 CrossRef CAS PubMed.
- J. R. Montenegro-Burke, T. Phommavongsay, A. E. Aisporna, T. Huan, D. Rinehart, E. Forsberg, F. L. Poole, M. P. Thorgersen, M. W. Adams and G. Krantz, Anal. Chem., 2016, 88, 9753–9758 CrossRef CAS PubMed.
- K. M. Mendez, D. I. Broadhurst and S. N. Reinke, Metabolomics, 2019, 15, 1–14 CrossRef PubMed.
- I. Goodfellow, Y. Bengio and A. Courville, Deep learning, MIT Press, 2016 Search PubMed.
- P. Sen, S. Lamichhane, V. B. Mathema, A. McGlinchey, A. M. Dickens, S. Khoomrung and M. Orešič, Briefings Bioinf., 2021, 22, 1531–1542 CrossRef PubMed.
- Y. Pomyen, K. Wanichthanarak, P. Poungsombat, J. Fahrmann, D. Grapov and S. Khoomrung, Comput. Struct. Biotechnol. J., 2020, 18, 2818–2825 CrossRef CAS PubMed.
- A. D. Melnikov, Y. P. Tsentalovich and V. V. Yanshole, Anal. Chem., 2019, 92, 588–592 CrossRef PubMed.
- M. Li and X. R. Wang, J. Chromatogr. A, 2019, 1604, 460476 CrossRef CAS PubMed.
- H. Ji, Y. Xu, H. Lu and Z. Zhang, Anal. Chem., 2019, 91, 5629–5637 CrossRef CAS PubMed.
- F. Allen, A. Pon, R. Greiner and D. Wishart, Anal. Chem., 2016, 88, 7689–7697 CrossRef CAS PubMed.
- P. Bonini, T. Kind, H. Tsugawa, D. K. Barupal and O. Fiehn, Anal. Chem., 2020, 92, 7515–7522 CrossRef CAS PubMed.
- Z. Zhou, X. Shen, J. Tu and Z.-J. Zhu, Anal. Chem., 2016, 88, 11084–11091 CrossRef CAS PubMed.
- P.-L. Plante, É. Francovic-Fontaine, J. C. May, J. A. McLean, E. S. Baker, F. Laviolette, M. Marchand and J. Corbeil, Anal. Chem., 2019, 91, 5191–5199 CrossRef CAS PubMed.
- Z. Zhou, J. Tu, X. Xiong, X. Shen and Z.-J. Zhu, Anal. Chem., 2017, 89, 9559–9566 CrossRef CAS PubMed.
- S. M. Colby, J. R. Nuñez, N. O. Hodas, C. D. Corley and R. R. Renslow, Anal. Chem., 2019, 92, 1720–1729 CrossRef PubMed.
- X. Domingo-Almenara, C. Guijas, E. Billings, J. R. Montenegro-Burke, W. Uritboonthai, A. E. Aisporna, E. Chen, H. P. Benton and G. Siuzdak, Nat. Commun., 2019, 10, 1–9 CrossRef PubMed.
- A. B. Risum and R. Bro, Talanta, 2019, 204, 255–260 CrossRef CAS PubMed.
- E. D. Kantz, S. Tiwari, J. D. Watrous, S. Cheng and M. Jain, Anal. Chem., 2019, 91, 12407–12413 CrossRef CAS PubMed.
- Y. Gloaguen, J. A. Kirwan and D. Beule, Anal. Chem., 2022, 94(12), 4930–4937 CrossRef CAS PubMed.
- V. Gabelica and E. D. Pauw, Mass Spectrom. Rev., 2005, 24, 566–587 CrossRef CAS PubMed.
- C. Hu, W. Luo, J. Xu and X. Han, Mass Spectrom. Rev., 2022, 41, 15–31 CrossRef CAS PubMed.
- Z. He, L. Zhao, X. Liu and Y. Xu, J. Chromatogr. A, 2020, 1633, 461637 CrossRef CAS PubMed.
- A. Criscuolo, M. Zeller and M. Fedorova, J. Am. Soc. Mass Spectrom., 2019, 31, 463–466 CrossRef PubMed.
- X. Domingo-Almenara, J. R. Montenegro-Burke, C. Guijas, E. L.-W. Majumder, H. P. Benton and G. Siuzdak, Anal. Chem., 2019, 91, 3246–3253 CrossRef CAS PubMed.
- N. G. Mahieu, J. L. Spalding, S. J. Gelman and G. J. Patti, Anal. Chem., 2016, 88, 9037–9046 CrossRef CAS PubMed.
- H. Yu, S. Xing, L. Nierves, P. F. Lange and T. Huan, Anal. Chem., 2020, 92, 7011–7019 CrossRef CAS PubMed.
- B. M. Warrack, S. Hnatyshyn, K.-H. Ott, M. D. Reily, M. Sanders, H. Zhang and D. M. Drexler, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2009, 877, 547–552 CrossRef CAS PubMed.
- D. Z. Bliss, K. Savik, H. Jung, L. Jensen, M. LeMoine and A. Lowry, J. Wound Ostomy Continence Nurs., 1999, 26, 137–141 CAS.
- N. Karu, L. Deng, M. Slae, A. C. Guo, T. Sajed, H. Huynh, E. Wine and D. S. Wishart, Anal. Chim. Acta, 2018, 1030, 1–24 CrossRef CAS PubMed.
- L. P. Silva, P. L. Lorenzi, P. Purwaha, V. Yong, D. H. Hawke and J. N. Weinstein, Anal. Chem., 2013, 85, 9536–9542 CrossRef CAS PubMed.
- S. S. Waikar, V. S. Sabbisetti and J. V. Bonventre, Kidney Int., 2010, 78, 486–494 CrossRef CAS PubMed.
- P. Jatlow, S. McKee and S. S. O’Malley, Clin. Chem., 2003, 49, 1932–1934 CrossRef CAS PubMed.
- F. Dieterle, A. Ross, G. Schlotterbeck and H. Senn, Anal. Chem., 2006, 78, 4281–4290 CrossRef CAS PubMed.
- S. J. Sheather and M. C. Jones, J. R. Stat. Soc. Series B Stat. Methodol., 1991, 53, 683–690 Search PubMed.
- R. A. van den Berg, H. C. Hoefsloot, J. A. Westerhuis, A. K. Smilde and M. J. van der Werf, BMC Genomics, 2006, 7, 1–15 CrossRef PubMed.
- E. L. Schymanski, J. Jeon, R. Gulde, K. Fenner, M. Ruff, H. P. Singer and J. Hollender, Environ. Sci. Technol., 2014, 48, 2097–2098 CrossRef CAS PubMed.
- R. R. da Silva, P. C. Dorrestein and R. A. Quinn, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 12549–12550 CrossRef CAS PubMed.
- R. A. Quinn, A. V. Melnik, A. Vrbanac, T. Fu, K. A. Patras, M. P. Christy, Z. Bodai, P. Belda-Ferre, A. Tripathi and L. K. Chung, Nature, 2020, 579, 123–129 CrossRef CAS PubMed.
- M. Wang, J. J. Carver, V. V. Phelan, L. M. Sanchez, N. Garg, Y. Peng, D. D. Nguyen, J. Watrous, C. A. Kapono and T. Luzzatto-Knaan, Nat. Biotechnol., 2016, 34, 828–837 CrossRef CAS PubMed.
- K. Dührkop, M. Fleischauer, M. Ludwig, A. A. Aksenov, A. V. Melnik, M. Meusel, P. C. Dorrestein, J. Rousu and S. Böcker, Nat. Methods, 2019, 16, 299–302 CrossRef PubMed.
- T. Huan, C. Tang, R. Li, Y. Shi, G. Lin and L. Li, Anal. Chem., 2015, 87, 10619–10626 CrossRef CAS PubMed.
- A. S. Moorthy, W. E. Wallace, A. J. Kearsley, D. V. Tchekhovskoi and S. E. Stein, Anal. Chem., 2017, 89, 13261–13268 CrossRef CAS PubMed.
- J. A. Falkner, J. W. Falkner, A. K. Yocum and P. C. Andrews, J. Proteome Res., 2008, 7, 4614–4622 CrossRef CAS PubMed.
- J. Watrous, P. Roach, T. Alexandrov, B. S. Heath, J. Y. Yang, R. D. Kersten, M. van der Voort, K. Pogliano, H. Gross and J. M. Raaijmakers, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, E1743–E1752 CrossRef CAS PubMed.
- H. Gu, G. N. Gowda, F. C. Neto, M. R. Opp and D. Raftery, Anal. Chem., 2013, 85, 10771–10779 CrossRef CAS PubMed.
- E. Stancliffe, M. Schwaiger-Haber, M. Sindelar and G. J. Patti, Nat. Methods, 2021, 18, 779–787 CrossRef CAS PubMed.
- C. Ruttkies, E. L. Schymanski, S. Wolf, J. Hollender and S. Neumann, J. Cheminf., 2016, 8, 1–16 Search PubMed.
- Y. Djoumbou-Feunang, A. Pon, N. Karu, J. Zheng, C. Li, D. Arndt, M. Gautam, F. Allen and D. S. Wishart, Metabolites, 2019, 9, 72 CrossRef CAS PubMed.
- K. Dührkop, H. Shen, M. Meusel, J. Rousu and S. Böcker, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 12580–12585 CrossRef PubMed.
- L. Ridder, J. J. van der Hooft, S. Verhoeven, R. C. de Vos, R. J. Bino and J. Vervoort, Anal. Chem., 2013, 85, 6033–6040 CrossRef CAS PubMed.
- L. Ridder, J. J. van der Hooft, S. Verhoeven, R. C. de Vos, R. van Schaik and J. Vervoort, Rapid Commun. Mass Spectrom., 2012, 26, 2461–2471 CrossRef CAS PubMed.
- S. Wolf, S. Schmidt, M. Müller-Hannemann and S. Neumann, BMC Bioinf., 2010, 11, 1–12 CrossRef PubMed.
- Y. Wang, G. Kora, B. P. Bowen and C. Pan, Anal. Chem., 2014, 86, 9496–9503 CrossRef CAS PubMed.
- J. J. J. van Der Hooft, J. Wandy, M. P. Barrett, K. E. Burgess and S. Rogers, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 13738–13743 CrossRef PubMed.
- F. Huber, L. Ridder, S. Verhoeven, J. H. Spaaks, F. Diblen, S. Rogers and J. J. Van Der Hooft, PLoS Comput. Biol., 2021, 17, e1008724 CrossRef CAS PubMed.
- M. Karni and A. Mandelbaum, Org. Mass Spectrom, 1980, 15, 53–64 CrossRef CAS.
- S. Xing and T. Huan, Anal. Chim. Acta, 2022, 339613 CrossRef CAS PubMed.
- K. Dührkop, L.-F. Nothias, M. Fleischauer, R. Reher, M. Ludwig, M. A. Hoffmann, D. Petras, W. H. Gerwick, J. Rousu and P. C. Dorrestein, Nat. Biotechnol., 2021, 39, 462–471 CrossRef PubMed.
- H. Tsugawa, R. Nakabayashi, T. Mori, Y. Yamada, M. Takahashi, A. Rai, R. Sugiyama, H. Yamamoto, T. Nakaya and M. Yamazaki, Nat. Methods, 2019, 16, 295–298 CrossRef CAS PubMed.
- M. Ernst, K. B. Kang, A. M. Caraballo-Rodríguez, L.-F. Nothias, J. Wandy, C. Chen, M. Wang, S. Rogers, M. H. Medema and P. C. Dorrestein, Metabolites, 2019, 9, 144 CrossRef CAS PubMed.
- K. Peters, H. Treutler, S. Döll, A. S. Kindt, T. Hankemeier and S. Neumann, Metabolites, 2019, 9, 222 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2022 |