
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCarbonate formation lowers the electrocatalytic activity of perovskite oxides for water electrolysis†
Christoph
Baeumer
 *abcd,
Allen Yu-Lun
Liang
be,
Urška
Trstenjak‡
f,
Qiyang
Lu
abg,
Rainer
Waser
cf,
J. Tyler
Mefford
ab,
Felix
Gunkel
f,
Slavomír
Nemšák
*fg and
William C.
Chueh
ab
*abcd,
Allen Yu-Lun
Liang
be,
Urška
Trstenjak‡
f,
Qiyang
Lu
abg,
Rainer
Waser
cf,
J. Tyler
Mefford
ab,
Felix
Gunkel
f,
Slavomír
Nemšák
*fg and
William C.
Chueh
ab
aDepartment of Materials Science and Engineering, Stanford University, Stanford, CA 94305, USA. E-mail: c.baeumer@fz-juelich.de
bStanford Institute for Materials and Energy Science, SLAC National Accelerator Laboratory, Menlo Park, CA 94025, USA
cInstitute of Electronic Materials (IWE2) and JARA-FIT, RWTH Aachen University, 52074 Aachen, Germany
dMESA+ Institute for Nanotechnology, University of Twente, Faculty of Science and Technology, 7500 AE Enschede, Netherlands
eDepartment of Chemistry, Stanford University, Stanford, CA 94305, USA
fPeter Gruenberg Institute and JARA-FIT, Forschungszentrum Juelich GmbH, 52425 Juelich, Germany
gAdvanced Light Source, Lawrence Berkeley National Laboratory, Berkeley, California 94720, USA. E-mail: SNemsak@lbl.gov
First published on 30th June 2021
Abstract
The study of oxide electrocatalysts is often complicated by the formation of complex and unknown surface species as well as the interaction between the catalysts and common support materials. Because unknown surface species may result from air exposure, we developed a clean transfer system for the air-free electrochemical investigation of epitaxial thin films fabricated under typical surface science conditions. LaNiO3 electrocatalysts exposed to ambient air exhibit a lower activity towards the oxygen evolution reaction than samples probed without air exposure. We demonstrate that this decrease in activity is connected to an alteration of the chemical environment of the electrocatalytically active sites through carbonate formation on exposure to CO2. Our study therefore shows that (1) the effects of air exposure must be considered for transition metal oxide catalysts and (2) that for the perovskite oxide LaNiO3 the clean surface is more active than the air-exposed surface.
Introduction
The field of electrocatalysis has experienced a resurgence due to its potential for applications in climate-neutral energy storage and conversion and chemical feedstock production.1 Noble metal-free transition metal oxides, often in the form of perovskites (ABO3), are of special interest because of high activities in alkaline media. Catalysts from earth-abundant materials now outperform precious metal benchmarks for the oxygen evolution reaction (OER), a bottleneck for various electrochemical energy transformation technologies.2–7 Due to the complexity of the active surface under operation conditions, further optimization of catalyst activity and stability makes use of single crystalline catalyst surfaces to derive structure–function relationships between the reactivity and the atomic-level surface structure and composition.8–10Epitaxial thin films allow investigation of oxide catalysts fabricated with unit-cell precision.11 Recently, we used epitaxial LaNiO3 layers to demonstrate the important role of the surface terminating layer composition,12 an activity descriptor that is difficult to recognize in catalysts fabricated using traditional routes.13,14 Beyond the intrinsic oxide surface composition, surface contaminants may alter the catalyst surface composition and electronic properties, as has been demonstrated extensively at solid/gas interfaces, where trace amounts of CO, CO2, and H2S are “poisonous” for fuel cell operation.15–17 For the technologically important solid/liquid interface, typical investigation pathways always involve exposure to ambient air after synthesis and before testing the electrocatalytic performance. Surface contaminants induced by simple exposure to air are well recognized for Li-ion batteries, where the formation of lithium and nickel carbonates results in degradation, especially for Ni-rich cathode materials.18,19 For electrocatalysts, the possible formation of such contaminants has been recognized,20 but their effect on electrocatalytic performance has yet to be explored.
Here, we investigate the influence of air exposure and isolate the role of CO2 contamination on the surface chemistry of atomically-defined LaNiO3 catalyst layers for OER. Inspired by the recent report by Faisal et al.,21 we developed a clean transfer method that bridges surface science fabrication and analysis techniques with electrochemical characterization of the solid/liquid interface without exposure to ambient air. Making use of this clean transfer, we find that preventing contamination from ambient air can lead to improvements in OER overpotential of 90 mV at 1 mA cm−2 compared to electrodes exposed to air. Using near-ambient pressure XPS, we demonstrate that the local chemical environment of the active Ni sites changes on exposure to gaseous CO2, due to the formation of surface carbonate groups. These carbonate groups are thermodynamically stable,22 block the active sites, and ultimately prevent the formation of the active Ni oxyhydroxide surfaces.12 Understanding the deactivation of surfaces by air exposure has important implications for technological applications of transition metal oxide catalysts.
Results and discussion
UHV characterization of LaNiO3 thin films
We fabricated epitaxial 20 nm LaNiO3 thin films on (001)-oriented Nb:SrTiO3 substrates with predominant Ni-termination using pulsed laser deposition (PLD) guided by in situ reflection high-energy electron diffraction (RHEED) (see Methods and ref. 12 for details). The growth process proceeds in a two-dimensional manner, resulting in smooth, step-terraced surface morphologies with ∼200 pm RMS roughness (Fig. 1). X-ray diffraction of the (002)pc peak (using the pseudocubic description of the unit cell) reveals a single-phase perovskite layer with pronounced Kiessig oscillations, indicative of coherent thickness and smooth interfaces. XPS investigation after storage in UHV (ptot ≤ 2 × 10−9 mbar) for ∼40 h and UHV transfer to the analysis chamber confirms that the LaNiO3 thin films are Ni-terminated (see ref. 12) and free of adsorbed contaminants except for a thin layer of adventitious carbon, as expected for a UHV transfer system (Fig. 2).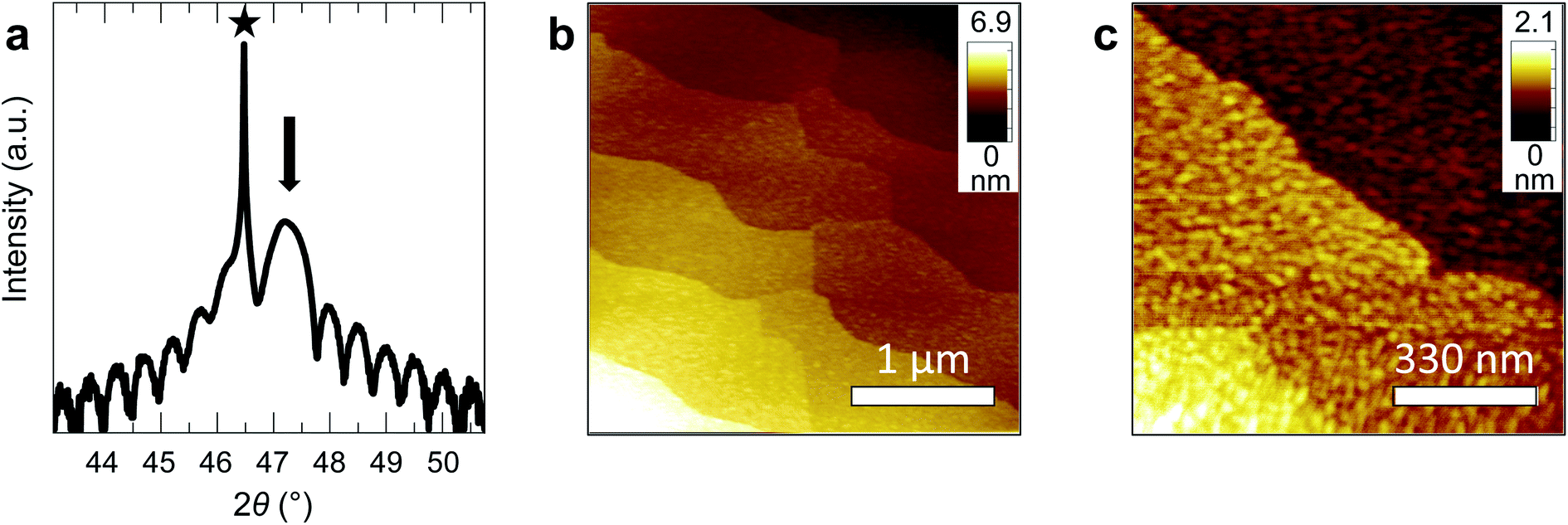 | ||
| Fig. 1 (a) X-ray diffractogram of a 20 nm LaNiO3 film on Nb:SrTiO3. The (002) substrate peak is marked with a star, the (002)pc film peak with an arrow. (b and c) Atomic force microscopy (AFM) of a 20 nm LaNiO3 film with different magnification. AFM was performed after UHV transfer from the deposition tool to the analysis chamber. | ||
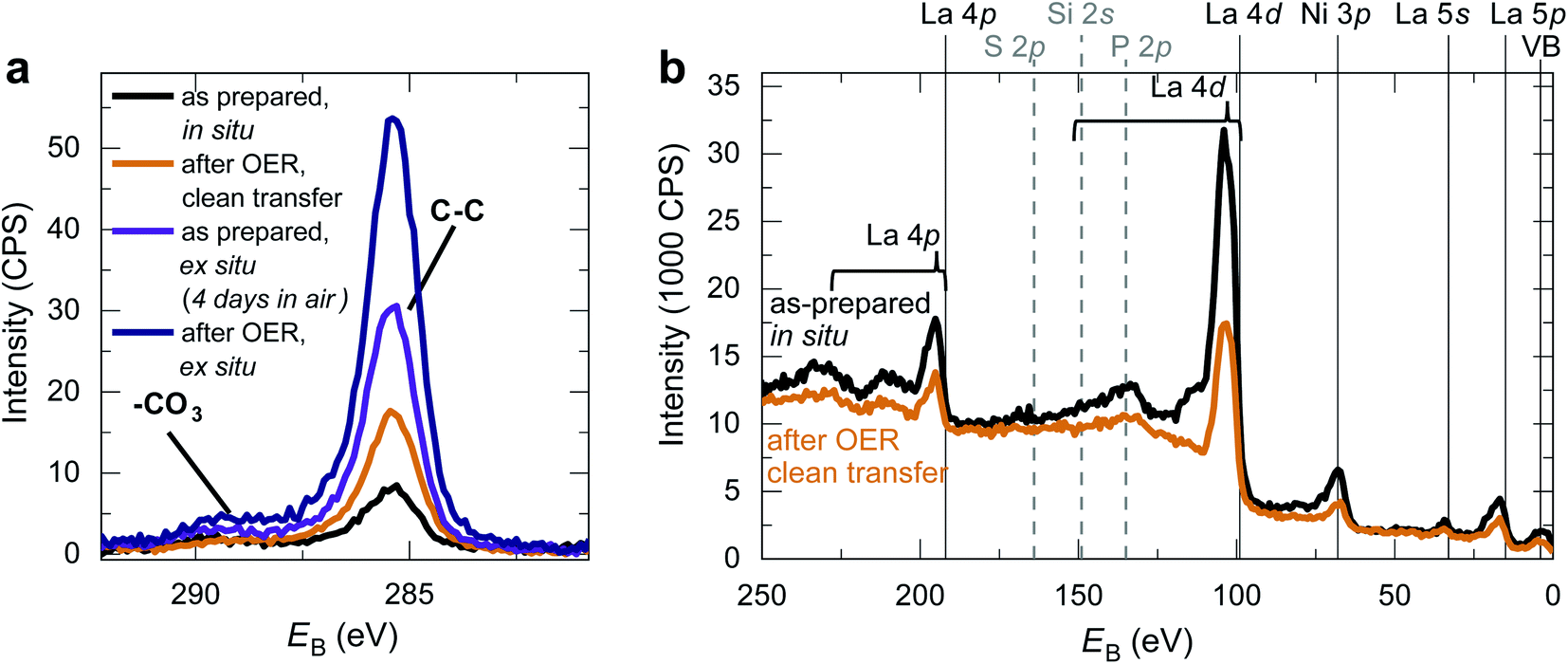 | ||
| Fig. 2 (a) C 1s XPS spectra for LaNiO3 surfaces in various conditions. The C–C peak indicates the presence of adventitious carbon and the –CO3 peak indicates the presence of surface carbonates. Both peaks exhibit increasing intensity for increased exposure to ambient air. For the clean transfer method, only the C–C peak but not the –CO3 peak increase compared to the as-prepared state. (b) XPS survey spectra in the as-prepared state after in situ transfer and after OER using the novel clean transfer method. Expected core level and possible contaminants are marked as solid lines at the literature main peak position. The La core levels have a complex shape and satellite structure spanning tens of eV as indicated by the brackets. In both cases, only the LaNiO3-related core levels are detectable. | ||
Clean transfer systems bridging surface science characterization and liquid electrochemistry
To avoid possible contaminants from air-exposure when examining the electrocatalytic properties, we developed a clean transfer system. A UHV compatible clean transfer vessel was attached to our UHV synthesis and characterization cluster setup. After pulsed laser deposition and surface characterization including AFM, XPS, and RHEED, the sample is brought into the clean transfer vessel, which can be detached and inserted into a dedicated glove box for electrochemical characterization. This glove box contained a N2 atmosphere (CO2 partial pressure ∼5 ppm) with a constant stream of pure N2 gas and hosted a rotating disc electrode setup specifically modified for the study of single crystalline samples.12,23 After electrochemical characterization, the sample was thoroughly rinsed with deionized water (Milli-Q, R > 18 MΩ cm), dried, remounted into the UHV sample holder and inserted into the clean transfer vessel. Importantly, the samples are mounted without the frequently applied silver paste and tested electrochemically without the need for epoxy coverage,24,25 further minimizing possible contamination sources. After attachment to UHV chamber, the transfer vessel was evacuated, and the samples were further characterized.To test our clean transfer between UHV characterization and electrochemical testing, we first considered the electrochemical oxidation of Pt as a benchmark based on the report by Faisal et al.21 Transferring a 50 nm Pt thin film on a Si/SiO2 substrate back and forth between the glove box-based electrochemical setup and an XPS tool, we found the expected reversible oxidation and reduction of the Pt layer as evidenced by cyclic voltammetry and XPS analysis (supplementary Fig. 2†). This test indicates that our tool yields the desired combination of surface-science characterization with electrochemical investigation.
Next, we characterized LaNiO3 epitaxial thin films as electrocatalysts using the clean transfer method. AFM after the electrochemical treatment revealed a smooth and step-terraced morphology even after tens of cycles during cyclic voltammetry, similar to the as-prepared state (supplementary Fig. 1†), confirming the suitability of our model-system approach.
The effect of ambient air for LaNiO3 electrocatalytic performance
We now consider the impact of air exposure on our LaNiO3 model electrocatalysts. For this purpose, we tested three nominally identical sets of three 20 nm LaNiO3 thin films with Ni termination. All films were deposited on the same day and under identical conditions. The first set of LaNiO3 thin films was transferred to the electrochemical setup immediately after growth without air exposure, with approximately 10 min exposure to the N2 atmosphere in the glove box after leaving UHV environment. The other two sets of LaNiO3 thin films were exposed to ambient air for 2 and 4 days, respectively, before undergoing identical electrochemical treatment. The first cyclic voltammetry sweeps showed that the samples transferred without air exposure (“quasi in situ”) showed no Ni oxidation and reduction peaks, indicative of a pristine LaNiO3 surface (Fig. 3a). Our prior work showed that the often-observed Ni oxidation and reduction peaks at 1.44 and 1.39 V vs. RHE indicate the formation of a (oxy-)hydroxide on the perovskite surface.12,26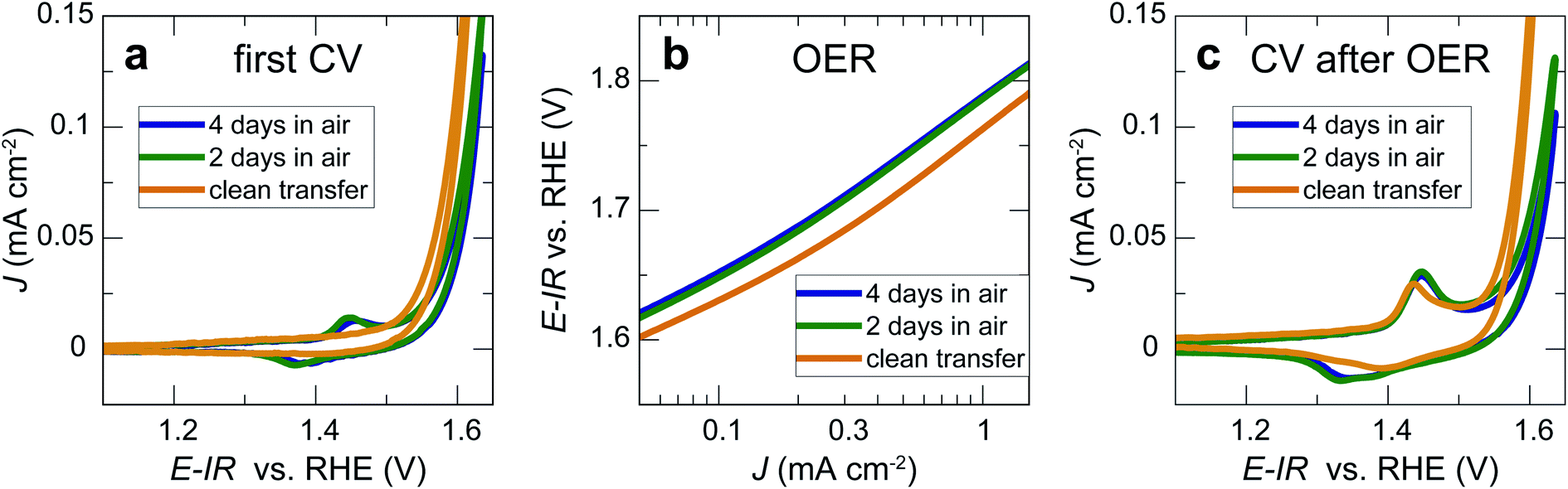 | ||
| Fig. 3 (a) First sweep during cyclic voltammetry in the Ni redox region with representative 20 nm LaNiO3 thin film electrocatalysts. (b) Cyclic voltammetry in the OER potential regime. The average between anodic and cathodic sweep of the second cycle is shown. (c) First sweep during cyclic voltammetry in the Ni redox region after two sweeps to OER potentials. Scan rate was 10 mV s−1 for all panels. | ||
In contrast, the samples exposed to air showed these peaks in the first cycle, presumably due to reaction with water vapor in ambient air. On investigation of the OER activity (Fig. 3b), we found that the activity of the films exposed to ambient air is generally lower than for samples transferred to the electrochemistry setup without air exposure, but the activity differences are relatively small for the initial cyclic voltammetry sweeps. After two sweeps to ∼1.9 V vs. RHE, the Ni oxidation and reduction peaks increased for all samples. For the samples after clean transfer, the typical oxidation and reduction peaks at 1.44 and 1.39 V vs. RHE appeared, indicating the formation of an OER-active oxyhydroxide-type surface layer.12 For the samples exposed to air, the anodic peak at 1.45 V vs. RHE increased. In the cathodic sweep, a second peak at 1.33 V vs. RHE appeared. These redox features indicate the formation of new surface phases formed during OER, a point that we will return to below.
For the same films, we also performed chronopotentiometry measurements for current densities in the range of 0.06 to 1 mA cm−2 (Fig. 4a). During chronopotentiometry, the behavior of the differently treated films is noticeably different. While samples directly transferred without air exposure show the highest activity, samples exposed to air for 2 and 4 days exhibit higher overpotentials for each current density. The overpotential is higher for films exposed to air for 4 days than for 2 days, with total overpotential differences of ∼90 mV for current densities of 1 mA cm−2, as can be seen in the Tafel plot in Fig. 4b. The Tafel slope at high current densities is similar (≥120 mV dec−1), but for low current densities, air-exposed samples exhibit an inactive Tafel slope of ≥120 mV dec−1 while samples without air exposure show a wider potential range with a more active Tafel slope of ≈80 mV dec−1. These trends were found throughout the set of three times three samples, verifying the robustness of our observations.
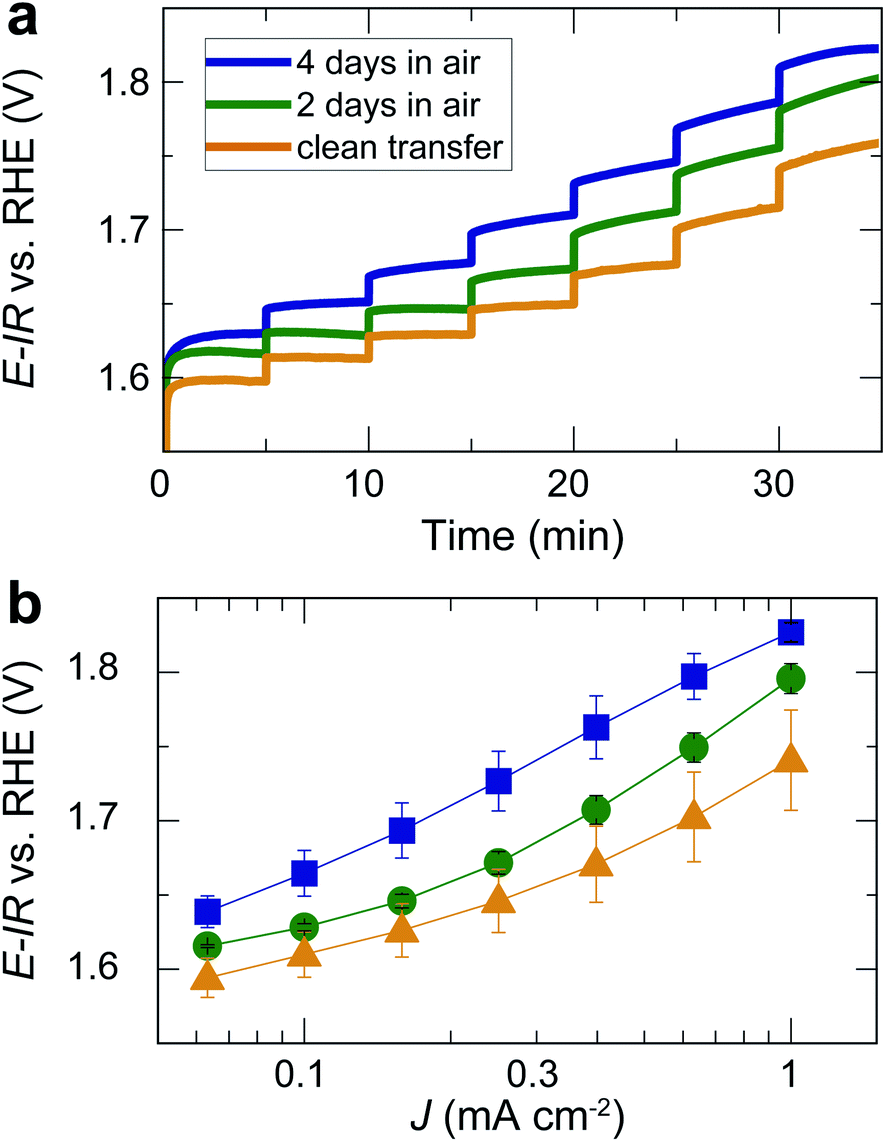 | ||
| Fig. 4 (a) Chronopotentiometry of the same films as in Fig. 3 for different current densities applied for 5 min each. Note that the current density did not reach a steady state for all potentials. The time for each step was kept short to avoid long-term degradation effects. (b) Tafel plot based on the measurement in (b) and additional, nominally identical samples. Error bars indicate the standard error for three nominally identical samples treated in the same way. | ||
Making use of our clean transfer approach, we performed an XPS investigation of the LaNiO3 surface after OER with and without air exposure. As Fig. 2 shows, the C 1s spectra after OER reveal an increase in adventitious carbon for all samples. For the samples without air exposure, this increase is markedly smaller than for samples exposed to air. An increase in adventitious carbon is expected even for a clean transfer due to the unavoidable storage times in the N2-glove box and non-UHV conditions during pump-down of the transfer vessel. Importantly, the C 1s spectra of samples transferred without air exposure do not show an increase in the coverage of carbonates at the catalyst surface before and after OER, and a survey spectrum confirms the absence of other typical contaminants. In contrast, samples exposed to air show a distinct shoulder at a binding energy of ∼289 eV, indicating the formation of carbonate groups on the surface. These groups form on the surface during air exposure and remain on the surface even after using LaNiO3 to catalyze the OER for the conditions investigated here. For the clean transfer samples, they do not occur before or after OER.
The effect of CO2 exposure traced in near ambient pressure XPS analysis
The formation of carbonates is likely an effect of exposure to CO2 in the ambient atmosphere, possibly in combination with other gases like water vapor. To clarify the effect of exposure to CO2 selectively, we employed near ambient pressure X-ray photoelectron spectroscopy (NAP-XPS)27 at beamline 9.3.2 at the LBNL Advanced Light Source (see Methods for details). After annealing in an atmosphere of 10 mTorr O2 at 300 °C, the surface is clean; no contamination peaks are detectable in an XPS survey scan within the detection limit.28 The Ni 3p and La 4d level obtained at 300 °C in O2 are typical for LaNiO3 (Fig. 5a–c). Ni 3p can be fitted with an asymmetric Doniach-Sunjic doublet, characteristic of the envelope for the complex multiplet splitting in a transition metal compound, which we described in more detail elsewhere.12 The La 4d level contains a doublet related to the bulk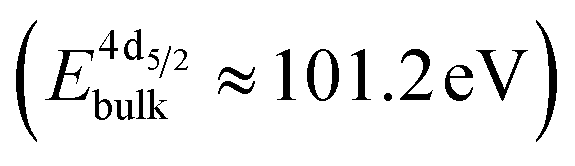 , a second doublet related to surface species
, a second doublet related to surface species 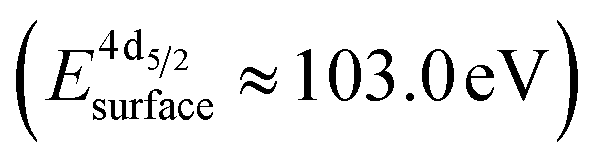 , and a satellite doublet at
, and a satellite doublet at 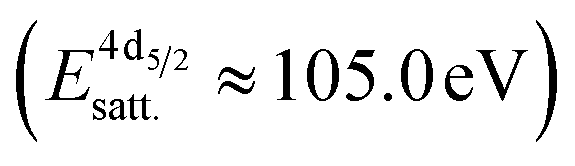 , similar to earlier reports.12,29
, similar to earlier reports.12,29
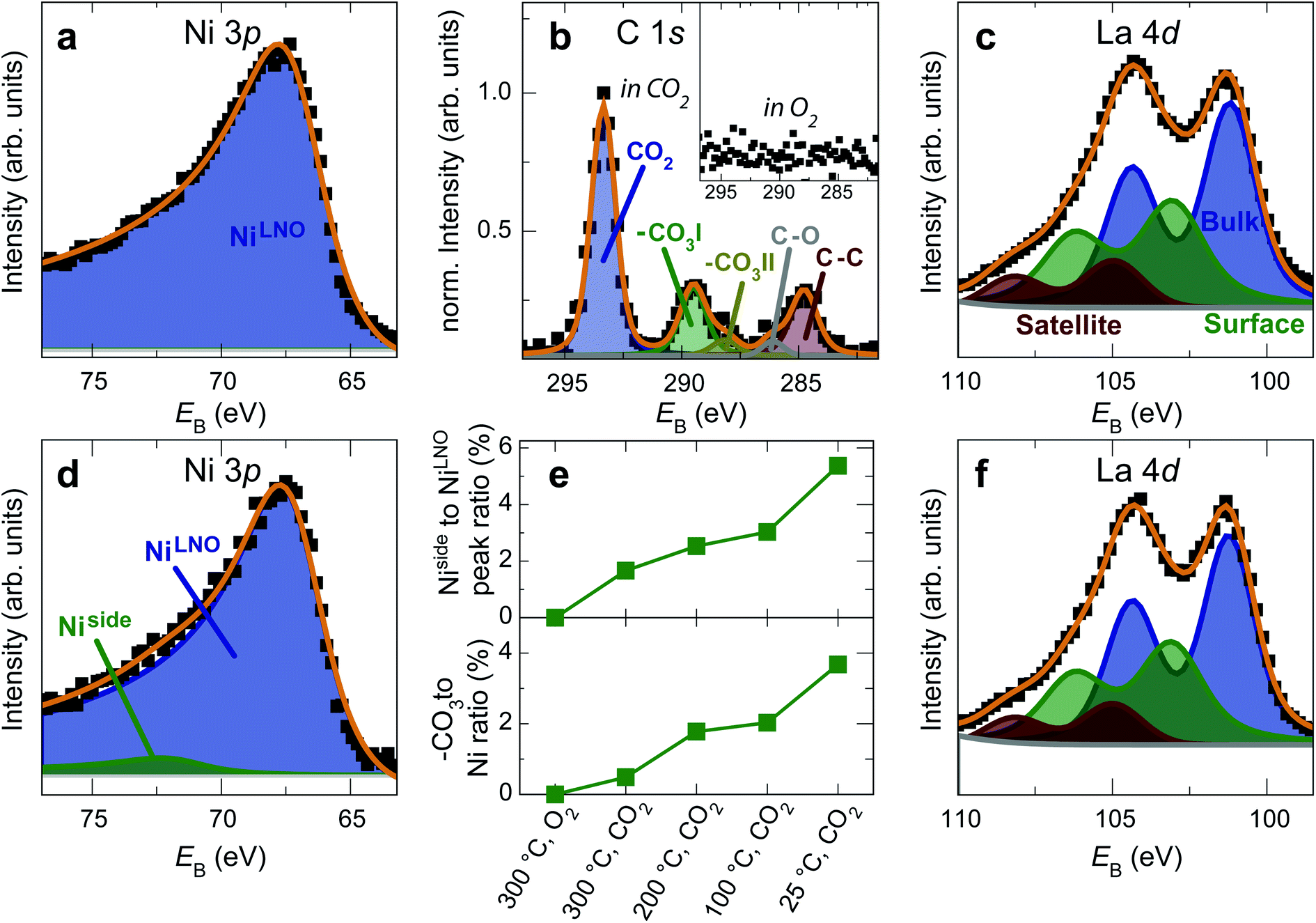 | ||
| Fig. 5 (a) Ni 3p, (b, inset) C 1s and (c) La 4d spectra of a LaNiO3 film in O2 at 300 °C. The surface is clean and the spectra are typical for as-prepared LaNiO3. (d) Ni 3p, (b) C 1s and (f) La 4d spectra of the same film after cooling down to room temperature in CO2. Fits in panel b were obtained according to ref. 30. (e) Top panel: Intensity ratio of the Ni side peak and LNO-bulk peak. Bottom panel: Intensity ratio of the C 1sCO3 peak and the total Ni 3p intensity. The energy was calibrated through a reference measurement on a Au foil. | ||
After exposure to 150 mtorr CO2 and cooling down to room temperature, the surface is covered by –CO3 carbonate species and adventitious carbon, as evidenced in the C 1s spectrum (Fig. 5b). The O 1s spectrum also indicates carbonate formation in the CO2 atmosphere (supplementary Fig. 4†). At the same time, a high-binding energy shoulder arises in the Ni 3p level with 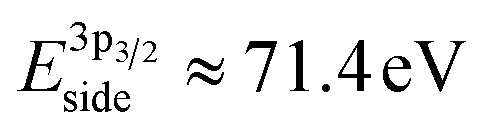 , while the La 4d level does not change appreciably (Fig. 5d and f). The Ni 3pside shoulder contributes ≈6% of the total Ni 3p intensity and comparison to experimental spectra obtained from several reference measurements including NiCO3·2Ni(OH)2·xH2O powder
, while the La 4d level does not change appreciably (Fig. 5d and f). The Ni 3pside shoulder contributes ≈6% of the total Ni 3p intensity and comparison to experimental spectra obtained from several reference measurements including NiCO3·2Ni(OH)2·xH2O powder 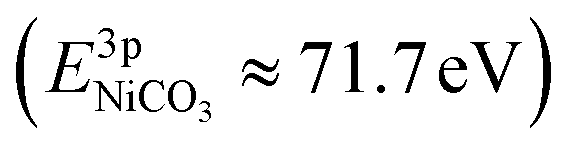 suggests that the side peak occurring after exposure to CO2 may be related to the formation of NiCO3 surface species. This assignment is substantiated as the Ni 3p side peak increases monotonically with the increasing C 1sCO3 and O 1sCO3 intensity during cooldown in CO2 (Fig. 5e and supplementary Fig. 4†). The formation of carbonate species on the surface is particularly favorable at room temperature, as would be expected from adsorption thermodynamics. Our NAP-XPS analysis therefore indicates that exposure to common contaminants like CO2 present in ambient air leads to a change in the surface chemistry of the electrocatalyst layer. In particular, the local binding environment of the Ni species – which we assume are the active sites for electrocatalysis – changes appreciably.
suggests that the side peak occurring after exposure to CO2 may be related to the formation of NiCO3 surface species. This assignment is substantiated as the Ni 3p side peak increases monotonically with the increasing C 1sCO3 and O 1sCO3 intensity during cooldown in CO2 (Fig. 5e and supplementary Fig. 4†). The formation of carbonate species on the surface is particularly favorable at room temperature, as would be expected from adsorption thermodynamics. Our NAP-XPS analysis therefore indicates that exposure to common contaminants like CO2 present in ambient air leads to a change in the surface chemistry of the electrocatalyst layer. In particular, the local binding environment of the Ni species – which we assume are the active sites for electrocatalysis – changes appreciably.
Discussion
Our results show that the OER overpotential during CV sweeps is ∼20 mV higher for LaNiO3 electrocatalysts exposed to air than for a clean LaNiO3 surface. This overpotential difference goes along with an increase in carbonate groups contaminating the perovskite surface, as revealed by NAP-XPS. Interestingly, during extended use for the chronopotentiometry (CP) measurement, the overpotential for the clean-transfer samples decreased, with an initial overpotential of 530 mV for 1 mA cm−2 in the second CV cycle (Fig. 3b) and 510 mV observed during CP (Fig. 4b). For samples exposed to air for 2 days, this activity enhancement is smaller, and for samples exposed to air for 4 days, the activity measured in CP is even lower than during the CV sweep (overpotentials of 560 mV for 1 mA cm−2 in the CV, and 600 mV in the CP). Therefore, the chronopotentiometry results (Fig. 4b) show larger activity differences than the cyclic voltammetry results (Fig. 3b). Note that the current density did not reach a steady state for all potentials. The time for each step was kept to five minutes to avoid long-term degradation effects.We discuss these observations in the context of our recent demonstration that Ni-oxide-terminated 20 nm LaNiO3 thin films undergo an electrochemically-driven surface phase change to a disordered Ni oxyhydroxide-type surface layer supported by the perovskite lattice, which in turn gives rise to the high OER activity compared to the La-oxide-termination.12 The decrease in OER activity with increasing exposure to ambient air indicates that the surface contaminants typically resulting from air exposure alter the electrochemical surface phase transformation of the catalyst surface. The contaminated surfaces also undergo a surface transformation, but the presence of carbonate species leads to a less active phase than observed for the air-free samples, where we suspect a Ni oxyhydroxide-type surface layer. It is known from the study of alkaline batteries that carbonate formation on Ni surfaces leads to an impediment of surface oxidation towards trivalent Ni species and the appearance of a shoulder in the Ni reduction peak.31 This corresponds well to our observation of a higher oxidation potential and the lower reduction potential (including the formation of a shoulder-peak) for the air exposed samples compared to the clean-transfer samples (Fig. 3c). In other words, the clean transfer samples exhibit a clean, perovskite-type surface before OER and an active oxyhydroxide-type surface after OER, while air-exposed samples show a hydroxylated- and carbonate-contaminated surface even before OER, and these species remain on the surface during OER, as confirmed in our laboratory XPS analysis. Due to the carbonate coverage, the oxidation and reduction of the surface hydroxyl groups is still possible but requires larger overpotentials and the entirety of the electrochemical data suggests that the presence of carbonate species results in an altered surface composition under reaction conditions, which results in an activity decrease. The NAP-XPS analysis suggests that carbonate formation occurs on the active sites (forming Ni carbonate species). The contaminants, accordingly, either block the active sites for the OER or lead to a less active surface phase due to different mechanisms than the samples transferred without air exposure. For the CV OER data, we tracked the ratio of the current densities for air-exposed and clean-transfer samples, revealing that air exposure leads to a decrease of the current density by ∼20–30% for the potential range of 1.55 to 1.85 V vs. RHE, which is relatively independent of the potential (supplementary Fig. 3†). It is therefore conceivable that indeed a blocking mechanism decreases the number of active sites, and the decrease is on the same order of magnitude as the number of surface Ni sites bonding to carbonate species on exposure to CO2, which can be estimated to ∼15%, using the results from Fig. 5 and the inelastic mean free path of ∼6 Å.
It is also conceivable that other typical contaminants like Si, S, and the extended exposure to water in the ambient atmosphere may play a role for this less active surface,27 but Si and S were not observable in XPS. In contrast, we found small concentrations of F and Ca on the surface for the samples exposed to ambient air, indicating that the surface contamination process during exposure to ambient air is complex and possibly involves multiple chemical species. Nonetheless, the examination of the surface transferred using our clean transfer vessel did not show any contaminations, suggesting an intrinsic surface phase transformation of our LaNiO3 thin film.
Conclusions
In this study, we developed a clean transfer system for the combination of surface-science-related and electrochemical investigation of epitaxial thin films. We found that exposure of electrocatalysts to ambient air results in a deterioration of the OER activity, resulting from common contaminants such as carbonate groups. These alter the chemical environment of the active sites, thus preventing the formation of the active surface phase. The OER-active surface phase forming on clean Ni-terminated LaNiO3 thin films shows higher activity compared to air-exposed samples, which remain covered with carbonate species. Our study therefore shows that effects of air exposure must be considered for applications of transition metal oxide electrocatalysts and we provide an experimental pathway for such analysis.Methods
Thin film preparation
Thin films were grown by pulsed-laser epitaxy using the PLD/MBE2300 setup (PVD Products, USA, at Stanford University) or the Laser MBE (Surface GmbH, Germany, at Forschungszentrum Jülich GmbH) using target-to-substrate distances of 83 mm and 48 mm, respectively. The growth was monitored in situ using a RHEED system (TorrRHEED™, Staib Instruments and kSA400, k-space Associates, Inc, respectively) operated at 35 kV (1.5 μA). The growth temperature was 550 °C. The laser fluence was 1.6 J cm−2. The oxygen pressure during deposition was 15 mtorr. After deposition, the samples were post-annealed at deposition temperature for 10 min in an atmosphere of 200 mtorr of oxygen and cooled down to temperatures below 300 °C within 5 minutes. All substrates were 10 × 10 mm2 (001) Nb-doped SrTiO3 (0.5 wt% doping) (Shinkosha Co., Japan). Prior to the growth, the substrates were first etched in buffered HF (pH = 4.5) and then annealed at 950 °C for 2 h. The commercial 99.95% elemental purity multiphase LaNiO3 target (La2NiO4 + NiO inclusions) was supplied by Toshima Manufacturing Co., Japan. Comparison to an in-lab sintered target fabricated with high purity (5 N) metal oxide precursors showed no differences in growth parameters or catalytic activity. In both experimental setups, the samples were mounted into sample carriers without silver paste.Clean transfer setups
Two separate setups were designed and tested, leading to equivalent results. For the setup at Stanford University, a custom-made N2-filled glove box manufactured by PVD Products was attached to the loadlock. The samples were inserted into air-tight containers and moved into a N2-filled glove box by MBraun for electrochemical characterization. For the setup at Forschungszentrum Jülich GmbH, a custom-made vacuum transfer vessel was attached to the synthesis and characterization UHV tool. The samples were transferred into this chamber using a wobble stick. The transfer vessel was detached from the UHV chamber and moved into a N2-filled custom-made glove box where the samples were retrieved and unmounted from the sample holder for electrochemical characterization. The N2 flow rate was ∼3 L min−1 and the CO2 partial pressure was measured using a pSense High Accuracy Portable CO2 Meter or K33 ELG CO2 + RH/T Data Logging Sensor (CO2METER.COM). For post-OER analysis, the samples were thoroughly cleaned in Milli-Q water, remounted into the sample carrier and inserted into the transfer vessel, which was now filled with N2. The transfer vessel was reattached to the UHV cluster and evacuated to p < 3 × 10−7 mbar, enabling sample transfer to the UHV analysis chambers. Available characterization tools include AFM/STM, XPS/UPS and PEEM instruments.Thin film characterization
X-ray diffractograms were collected using PANalytical X'Pert PRO diffractometer equipped with a double monochromator and operating in a parallel-beam geometry, using Cu Kα X-ray illumination. Atomic force microscopy was carried out without air exposure using an Omicron VT UHV SPM operated in contact mode with tips coated with boron-doped polycrystalline diamond. The cantilever tips had a nominal radius of less than 150 nm and a spring constant of 25 N m−1 (AppNano, DD-ACTA-10). The force setpoint was limited to 2 nN in order to minimize a possible effect of the contact mode measurements on the surface morphology. The samples were transferred from the analysis chamber or from the transfer vessel in ultra-high vacuum. XPS characterization with and without air exposure was performed with a PHI Versa Probe (Physical Electronics Inc., USA) with Al Kα X-ray illumination, a pass energy of 23.5 eV and at a photoemission angle of 45° without neutralization. The mean escape depth d = 1.6 nm is defined through the inelastic mean free path of photoelectrons λ = 2.2 nm (calculated via QUASES-IMFP-TPP2M)32 and the photoemission angle θ through d = λ × cos![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) θ.33 The energy scale of the instrument was calibrated on a Au foil.
θ.33 The energy scale of the instrument was calibrated on a Au foil.
AP XPS experiments
The measurements were carried out at the Ambient-Pressure Soft X-ray Photoelectron Spectroscopy beamline 9.3.2 at the LBNL Advanced Light Source.34 The photon energy was chosen to result in approximately 200 eV kinetic energy for each core level to ensure equal information depth throughout the measurements. The chamber was filled with 10 mtorr O2 and the sample was heated to 300 °C to remove any surface contaminations before exposing the sample to an atmosphere of 150 mtorr CO2. Spectral fitting was performed using KolXPD using the peak models described in the main text after subtraction of a Shirley background.Electrochemical characterization
To perform electrochemical experiments with epitaxial thin films on 10 × 10 × 0.5 mm3 single crystal substrates, we used a custom-made adapter to press the sample back side to the Pt plug of a rotating disk electrode (RDE, Pine Research). 50 nm Pt connections from the sample back side to the front side ensured ohmic contact to the LaNiO3 layer. On the front side, a film area of 0.75 mm diameter was exposed to the electrolyte and sealed using an O-ring (FFKM, Marco Rubber, USA). The RDE shaft was rotating at 1600 rpm. Electrochemical testing was performed using BioLogic VSP-300or VSP-150 potentiostats, in a 150 mL alkaline-resistant Teflon cell (Pine Research) with a Pt wire as a counter electrode. Electrochemical impedance spectroscopy (EIS) was conducted with the amplitude of 10 mV at open circuit potential and the correction for the cell resistance (IR correction, typically 50–60 Ω) was based on the high-frequency intercept of the real impedance. The electrolyte solution of 0.1 M KOH was prepared by dissolving KOH pellets (Sigma-Aldrich, 99.99%) in deionized water (Milli-Q, >18.2 M Ω cm). We degassed the electrolyte rigorously using N2 bubbling in an inert atmosphere before bringing it into the glove box. It was then O2-saturated prior to testing for at least 30 minutes and maintained under O2 atmosphere during testing. All electrochemical measurements were performed at room temperature. While higher electrolyte concentrations and temperatures can be used to test stability in application-near conditions,35 0.1 M KOH and room temperature represent the recommended best practices for comparison and benchmarking of model electrocatalyst systems.36 Potentials were referenced to a Hg/HgO reference electrode (CHI Instruments, USA), which was periodically calibrated to the reversible hydrogen electrode (HydroFlex, USA) in 0.1 M KOH with typical values of ∼880 mV. All OER testing was performed on a fresh electrode that had not undergone previous testing. Cyclic voltammetry was first performed in the pseudocapacitive redox phase change region (∼0.9 to 1.6 V vs. RHE) at scan rates between 10 and 500 mV s−1, followed by OER testing performed from 0.9 to 1.9 V vs. RHE at a scan rate of 10 mV s−1. The CV data was capacitance corrected through averaging the forward and backward scans. The second cycle is shown for each sample. Tafel plots were obtained from chronopotentiometry measurements by averaging the potential of the last 100 s of a 5 min hold at each current density step. After electrochemical characterization, the samples were checked by XPS to verify the absence of Fe-contamination in the electrode surface.Funding sources
This project has received funding from the European Union's Horizon 2020 research and innovation programme under the Marie Sklodowska-Curie grant agreement No 796142. C. B., A. Y. L. L., Q. L., J. T. M. and W. C. C. gratefully acknowledge financial support through the Department of Energy, Office of Basic Energy Sciences, Division of Materials Sciences and Engineering under contract no. DE-AC02-76SF00515. Part of this work was performed at the Stanford Nano Shared Facilities (SNSF)/Stanford Nanofabrication Facility (SNF), supported by the National Science Foundation under award ECCS-1542152. This research used beamline 9.3.2 of the Advanced Light Source, a U.S. DOE Office of Science User Facility under contract no. DE-AC02-05CH11231.Author contributions
C. B., S. N. and W. C. C. conceived and designed the experiments. C. B., A. Y. L. L., U. G. and Q. L. performed and analyzed the experiments. R. W., J. T. M., F. G., S. N. and W. C. C. supervised the research. C. B. wrote the manuscript with contributions from all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Stephan Masberg, Georg Pickartz and Jochen Friedrich for support in designing the clean transfer setup.References
- E. Fabbri, A. Habereder, K. Waltar, R. Kötz and T. J. Schmidt, Developments and Perspectives of Oxide-Based Catalysts for the Oxygen Evolution Reaction, Catal. Sci. Technol., 2014, 4(11), 3800–3821, 10.1039/c4cy00669k.
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Combining Theory and Experiment in Electrocatalysis: Insights into Materials Design, Science, 2017, 355(6321), eaad4998, DOI:10.1126/science.aad4998.
- J. Suntivich, K. J. May, H. A. Gasteiger, J. B. Goodenough and Y. Shao-Horn, A Perovskite Oxide Optimized for Oxygen Evolution Catalysis from Molecular Orbital Principles, Science, 2011, 334(6061), 1383–1385, DOI:10.1126/science.1212858.
- L. C. Seitz, C. F. Dickens, K. Nishio, Y. Hikita, J. Montoya, A. Doyle, C. Kirk, A. Vojvodic, H. Y. Hwang, J. K. Norskov and T. F. Jaramillo, A Highly Active and Stable IrOx/SrIrO3 Catalyst for the Oxygen Evolution Reaction, Science, 2016, 353(6303), 1011–1014, DOI:10.1126/science.aaf5050.
- A. Grimaud, O. Diaz-Morales, B. Han, W. T. Hong, Y.-L. Lee, L. Giordano, K. A. Stoerzinger, M. T. M. Koper and Y. Shao-Horn, Activating Lattice Oxygen Redox Reactions in Metal Oxides to Catalyse Oxygen Evolution, Nat. Chem., 2017, 9(5), 457–465, DOI:10.1038/nchem.2695.
- J. H. Montoya, L. C. Seitz, P. Chakthranont, A. Vojvodic, T. F. Jaramillo and J. K. Nørskov, Materials for Solar Fuels and Chemicals, Nat. Mater., 2017, 16(1), 70–81, DOI:10.1038/nmat4778.
- J. Suntivich, H. A. Gasteiger, N. Yabuuchi, H. Nakanishi, J. B. Goodenough and Y. Shao-Horn, Design Principles for Oxygen-Reduction Activity on Perovskite Oxide Catalysts for Fuel Cells and Metal–Air Batteries, Nat. Chem., 2011, 3(7), 546–550, DOI:10.1038/nchem.1069.
- V. R. Stamenkovic, D. Strmcnik, P. P. Lopes and N. M. Markovic, Energy and Fuels from Electrochemical Interfaces, Nat. Mater., 2017, 16(1), 57–69, DOI:10.1038/nmat4738.
- M. L. Weber and F. Gunkel, Epitaxial Catalysts for Oxygen Evolution Reaction: Model Systems and Beyond, JPhys Energy, 2019, 1(3), 031001, DOI:10.1088/2515-7655/ab1577.
- D. Antipin and M. Risch, Trends of Epitaxial Perovskite Oxide Films Catalyzing the Oxygen Evolution Reaction in Alkaline Media, JPhys Energy, 2020, 2(3), 032003, DOI:10.1088/2515-7655/ab812f.
- C. J. Eom, D.-Y. Y. Kuo, C. Adamo, E. J. Moon, S. J. May, E. J. Crumlin, D. G. Schlom and J. Suntivich, Tailoring Manganese Oxide with Atomic Precision to Increase Surface Site Availability for Oxygen Reduction Catalysis, Nat. Commun., 2018, 9(1), 4034, DOI:10.1038/s41467-018-06503-8.
- C. Baeumer, J. Li, Q. Lu, A. Y.-L. A. Y.-L. Liang, L. Jin, H. P. H. P. Martins, T. Duchoň, M. Glöβ, S. M. S. M. Gericke, M. A. M. A. Wohlgemuth, M. Giesen, E. E. E. E. Penn, R. Dittmann, F. Gunkel, R. Waser, M. Bajdich, S. Nemšák, J. T. T. Mefford and W. C. W. C. Chueh, Tuning Electrochemically Driven Surface Transformation in Atomically Flat LaNiO3 Thin Films for Enhanced Water Electrolysis, Nat. Mater., 2021, 20(5), 674–682, DOI:10.1038/s41563-020-00877-1.
- K. Chakrapani, G. Bendt, H. Hajiyani, I. Schwarzrock, T. Lunkenbein, S. Salamon, J. Landers, H. Wende, R. Schlögl, R. Pentcheva, M. Behrens and S. Schulz, Role of Composition and Size of Cobalt Ferrite Nanocrystals in the Oxygen Evolution Reaction, ChemCatChem, 2017, 9(15), 2988–2995, DOI:10.1002/cctc.201700376.
- H. Hajiyani and R. Pentcheva, Surface Termination and Composition Control of Activity of the CoxNi1−xFe2O4 (001) Surface for Water Oxidation: Insights from DFT+U Calculations, ACS Catal., 2018, 8(12), 11773–11782, DOI:10.1021/acscatal.8b00574.
- S. Sun and Z. Cheng, Effects of H2O and CO2 on Electrochemical Behaviors of BSCF Cathode for Proton Conducting IT-SOFC, J. Electrochem. Soc., 2017, 164(2), F81–F88, DOI:10.1149/2.0611702jes.
- M. Winter and R. J. Brodd, What Are Batteries, Fuel Cells, and Supercapacitors?, Chem. Rev., 2004, 104(10), 4245–4270, DOI:10.1021/cr020730k.
- F. A. de Bruijn, D. C. Papageorgopoulos, E. F. Sitters and G. J. M. Janssen, The Influence of Carbon Dioxide on PEM Fuel Cell Anodes, J. Power Sources, 2002, 110(1), 117–124, DOI:10.1016/s0378-7753(02)00227-6.
- J. Sicklinger, M. Metzger, H. Beyer, D. Pritzl and H. A. Gasteiger, Ambient Storage Derived Surface Contamination of NCM811 and NCM111: Performance Implications and Mitigation Strategies, J. Electrochem. Soc., 2019, 166(12), A2322–A2335, DOI:10.1149/2.0011912jes.
- R. Jung, R. Morasch, P. Karayaylali, K. Phillips, F. Maglia, C. Stinner, Y. Shao-Horn and H. A. Gasteiger, Effect of Ambient Storage on the Degradation of Ni-Rich Positive Electrode Materials (NMC811) for Li-Ion Batteries, J. Electrochem. Soc., 2018, 165(2), A132–A141, DOI:10.1149/2.0401802jes.
- T. Shen, L. Spillane, J. Vavra, T. H. M. Pham, J. Peng, Y. Shao-Horn and V. Tileli, Oxygen Evolution Reaction in Ba0.5Sr0.5Co0.8Fe0.2O3−δ Aided by Intrinsic Co/Fe Spinel-like Surface, J. Am. Chem. Soc., 2020, 142, 15876–15883, DOI:10.1021/jacs.0c06268.
- F. Faisal, C. Stumm, M. Bertram, F. Waidhas, Y. Lykhach, S. Cherevko, F. Xiang, M. Ammon, M. Vorokhta, B. Šmíd, T. Skála, N. Tsud, A. Neitzel, K. Beranová, K. C. Prince, S. Geiger, O. Kasian, T. Wähler, R. Schuster, M. A. Schneider, V. Matolín, K. J. J. Mayrhofer, O. Brummel and J. Libuda, Electrifying Model Catalysts for Understanding Electrocatalytic Reactions in Liquid Electrolytes, Nat. Mater., 2018, 17(7), 592–598, DOI:10.1038/s41563-018-0088-3.
- H. Gamsjäger, J. Bugajski, T. Gajda, R. J. Lemire and W. Preis, Chemical Thermodynamics, Volume 6, Chemical Thermodynamics of Nickel, North Holland Elsevier Science Publishers B. V., Amsterdam, The Netherlands, 2005, vol. 6 Search PubMed.
- A. R. Akbashev, L. Zhang, J. T. Mefford, J. Park, B. Butz, H. Luftman, W. C. Chueh and A. Vojvodic, Activation of Ultrathin SrTiO3 with Subsurface SrRuO3 for the Oxygen Evolution Reaction, Energy Environ. Sci., 2018, 136(5), 1012–1021, 10.1039/c8ee00210j.
- J. R. Petrie, H. Jeen, S. C. Barron, T. L. Meyer, H. N. Lee, V. R. Cooper, J. W. Freeland, T. L. Meyer, Z. Zhang, D. A. Lutterman and H. N. Lee, Enhanced Bifunctional Oxygen Catalysis in Strained LaNiO3 Perovskites, J. Am. Chem. Soc., 2016, 138(8), 2488–2491, DOI:10.1021/jacs.5b11713.
- L. Wang, K. A. Stoerzinger, L. Chang, J. Zhao, Y. Li, C. S. Tang, X. Yin, M. E. Bowden, Z. Yang, H. Guo, L. You, R. Guo, J. Wang, K. Ibrahim, J. Chen, A. Rusydi, J. Wang, S. A. Chambers and Y. Du, Tuning Bifunctional Oxygen Electrocatalysts by Changing the A-Site Rare-Earth Element in Perovskite Nickelates, Adv. Funct. Mater., 2018, 28(39), 1803712, DOI:10.1002/adfm.201803712.
- L. Trotochaud, J. K. Ranney, K. N. Williams and S. W. Boettcher, Solution-Cast Metal Oxide Thin Film Electrocatalysts for Oxygen Evolution, J. Am. Chem. Soc., 2012, 134(41), 17253–17261, DOI:10.1021/ja307507a.
- D. Zhang, M. L. Machala, D. Chen, Z. Guan, H. Li, S. Nemsak, E. J. Crumlin, H. Bluhm and W. C. Chueh, Hydroxylation and Cation Segregation in (La0.5Sr0.5)FeO3−δ Electrodes, Chem. Mater., 2020, 32(7), 2926–2934, DOI:10.1021/acs.chemmater.9b05151.
- M. Andrä, F. Dvořák, M. Vorokhta, S. Nemšák, V. Matolín, C. M. Schneider, R. Dittmann, F. Gunkel, D. N. Mueller and R. Waser, Oxygen Partial Pressure Dependence of Surface Space Charge Formation in Donor-Doped SrTiO3, APL Mater., 2017, 5(5), 056106, DOI:10.1063/1.4983618.
- K. A. Stoerzinger, W. T. Hong, E. J. Crumlin, H. Bluhm, M. D. Biegalski and Y. Shao-Horn, Water Reactivity on the LaCoO3 (001) Surface: An Ambient Pressure X-Ray Photoelectron Spectroscopy Study, J. Phys. Chem. C, 2014, 118(34), 19733–19741, DOI:10.1021/jp502970r.
- T. L. Barr and S. Seal, Nature of the Use of Adventitious Carbon as a Binding Energy Standard, J. Vac. Sci. Technol., A, 1995, 13(3), 1239–1246, DOI:10.1116/1.579868.
- V. S. Muralidharan, N. Jayalakshmi and P. Mageswari, Electrochemical Behaviour of Nickel and Nickel Oxyhydroxide in Alkaline Carbonate Solutions, Bull. Electrochem., 1991, 7(8), 355–361 CAS.
- S. Tanuma, C. J. Powell and D. R. Penn, Calculations of Electron Inelastic Mean Free Paths. V. Data for 14 Organic Compounds over the 50-2000 EV Range, Surf. Interface Anal., 1994, 21(3), 165–176, DOI:10.1002/sia.740210302.
- C. J. Powell, Practical Guide for Inelastic Mean Free Paths, Effective Attenuation Lengths, Mean Escape Depths, and Information Depths in x-Ray Photoelectron Spectroscopy, J. Vac. Sci. Technol., A, 2020, 38(2), 023209, DOI:10.1116/1.5141079.
- M. E. Grass, P. G. Karlsson, F. Aksoy, M. Lundqvist, B. Wannberg, B. S. Mun, Z. Hussain and Z. Liu, New Ambient Pressure Photoemission Endstation at Advanced Light Source Beamline 9.3.2, Rev. Sci. Instrum., 2010, 81(5), 053106, DOI:10.1063/1.3427218.
- D. S. Bick, A. Kindsmüller, G. Staikov, F. Gunkel, D. Müller, T. Schneller, R. Waser and I. Valov, Stability and Degradation of Perovskite Electrocatalysts for Oxygen Evolution Reaction, Electrochim. Acta, 2016, 218, 156–162, DOI:10.1016/j.electacta.2016.09.116.
- C. Wei, R. R. Rao, J. Peng, B. Huang, I. E. L. Stephens, M. Risch, Z. J. Xu and Y. Shao-Horn, Recommended Practices and Benchmark Activity for Hydrogen and Oxygen Electrocatalysis in Water Splitting and Fuel Cells, Adv. Mater., 2019, 31(31), 1806296, DOI:10.1002/adma.201806296.
Footnotes |
| † Electronic supplementary information (ESI) available: XPS, AFM and RHEED analysis of the samples: ex situ and in situ samples compared with clean-transfer samples. Testing the clean transfer system with Pt oxidation and reduction. Analysis of the activity enhancement. See DOI: 10.1039/d1ta03205d |
| ‡ Present address: Advanced Materials Department, Jožef Stefan Institute, SI-1000 Ljubljana, Slovenia. |
| This journal is © The Royal Society of Chemistry 2021 |