
Planar defect-driven electrocatalysis of CO2-to-C2H4 conversion†
Zhengyuan
Li
a,
Yanbo
Fang
b,
Jianfang
Zhang
a,
Tianyu
Zhang
a,
Juan D.
Jimenez
 c,
Sanjaya D.
Senanayake
c,
Vesselin
Shanov
ab,
Shize
Yang
*d and
Jingjie
Wu
*a
c,
Sanjaya D.
Senanayake
c,
Vesselin
Shanov
ab,
Shize
Yang
*d and
Jingjie
Wu
*a
aDepartment of Chemical and Environmental Engineering, University of Cincinnati, Cincinnati, OH 45221, USA. E-mail: jingjie.wu@uc.edu
bDepartment of Mechanical and Materials Engineering, University of Cincinnati, Cincinnati, OH 45221, USA
cChemistry Division, Brookhaven National Laboratory, Upton, NY 11973, USA
dEyring Materials Center, Arizona State University, Tempe, AZ 85287, USA. E-mail: shize.yang@asu.edu
First published on 15th June 2021
Abstract
The selectivity towards a specific C2+ product, such as ethylene (C2H4), is sensitive to the surface structure of copper (Cu) catalysts in carbon dioxide (CO2) electro-reduction. The fundamental understanding of such sensitivity can guide the development of advanced electrocatalysts, although it remains challenging at the atomic level. Here we demonstrated that planar defects, such as stacking faults, could drive the electrocatalysis of CO2-to-C2H4 conversion with higher selectivity and productivity than Cu(100) facets in the intermediate potential region (−0.50 ∼ −0.65 V vs. RHE). The unique right bipyramidal Cu nanocrystals containing a combination of (100) facets and a set of parallel planar defects delivered 67% faradaic efficiency (FE) for C2H4 and a partial current density of 217 mA cm−2 at −0.63 V vs. RHE. In contrast, Cu nanocubes with exclusive (100) facets exhibited only 46% FE for C2H4 and a partial current density of 87 mA cm−2 at an identical potential. Both ex situ CO temperature-programmed desorption and in situ Raman spectroscopy analysis implied that the stronger *CO adsorption on planar defect sites facilitates CO generation kinetics, which contributes to a higher surface coverage of *CO and in turn an enhanced reaction rate of C–C coupling towards C2+ products, especially C2H4.
1. Introduction
The CO2 electro-reduction reaction (CO2RR) to value-added multi-carbon (C2+) products is a promising avenue for artificial carbon recycling using renewable energy sources.1,2 In particular, the conversion of CO2 to ethylene (C2H4), a major industrial feedstock with a large market size and relatively high market price,2 has received immense attention. Techno-economic analyses (TEAs) reveal that when (1) electricity costs fall below 4 cents kW−1 h−1, (2) C2H4 partial current density meets 450 mA cm−2, and (3) energy efficiency is at least 60%, the C2H4 generated from the CO2RR becomes competitive with current market prices for that derived from fossil fuel sources.2 So far, copper (Cu) and Cu-derived materials have been the most efficient electrocatalysts that can convert CO2 to C2+ products with appreciable reaction rates.3 However, the unsatisfactory selectivity of CO2 reduction towards a specific high-order product still impedes its large-scale implementation.3,4It has been demonstrated that the coupling of the key intermediate (e.g., surface adsorbed *CO) is a critical step in CO2-to-C2+ product conversion.5–8 Therefore, numerous strategies have been developed to maximize the utilization of *CO. Cascade catalysis involving tandem catalysts,9,10 tandem electrodes,11,12 and tandem reactors13 has been proposed to supply extra CO and thus to raise local CO concentration near the Cu surface for facilitating C–C coupling. Another widely investigated approach is to properly increase the *CO binding energy via tuning the surface structures of Cu, such as alloying,14 surface doping,15,16 crystal faceting,17,18 subsurface oxygen engineering,19 and surface reconstruction.20 It is worth noting that the CO generation rate can also be enhanced with the *CO binding energy since the activation energy for CO formation decreases according to the scaling relations and Brønsted–Evans–Polanyi (BEP) relations.21 Subsequently, the surface coverage of *CO on Cu can be simultaneously enhanced as *CO binding energy increases.5,22,23
It is well known that the Cu(100) facet with square atomic configurations favors the formation of C2H4, which benefits from the stronger *CO binding energy and subsequently lower *CO dimerization barrier compared to that on the Cu(111) facet.8 Meanwhile, low-coordinated defect sites, such as vacancies, steps, twin boundaries, and grain boundaries, arisen from in situ or ex situ growth on the metallic Cu surface were proposed to be responsible for the enhanced catalytic reactivity.23–26 Indeed, research on twin boundaries and grain boundaries has pointed out that the high activity and selectivity are correlated with the sites binding CO more strongly than low-index Cu facets, and the reactivity is linearly proportional to the density of such active sites.26 But most of these two-dimensional defects are associated with the Cu(111) facets or interfaces between (111) and (100) facets.23,27 A comprehensive comparison between (100) facets and planar defects, which is missing, could assist in designing electrocatalysts for further improvement of CO2-to-C2+ conversion.
Herein, we demonstrated that planar defects could drive the electrocatalysis of CO2-to-C2H4 conversion with higher selectivity and productivity than Cu low-index facets. To this end, we prepared a right bipyramidal Cu (Rbp-Cu) electrocatalyst with both exposed Cu(100) facets and a set of parallel planar defects (e.g., stacking faults). Rbp-Cu achieved a Faradaic efficiency (FE) of 67% and 89% for C2H4 and C2+ products, respectively, at a current density of over 325 mA cm−2 and a corresponding potential of −0.63 V vs. RHE. In contrast, Cu nanocubes (Cube-Cu) with exclusive Cu(100) facets exhibited only 46% FE for C2H4 and a partial current density of 87 mA cm−2 at an identical potential. The planar defect was discovered to show superior activity and selectivity towards C2H4 formation to Cu(100) by comparing the performance between Rbp-Cu and Cube-Cu. The reaction mechanism study using CO temperature-programmed desorption and in situ Raman spectroscopy implied that the enhancement of reactivity for CO2-to-C2H4 conversion originates from the stronger *CO adsorption energy on planar defects than Cu(100).
2. Experimental
2.1 Chemicals and materials
Copper(II) chloride dehydrate (CuCl2·2H2O, 99.0%), D-glucose (C6H12O6, 99.5%), hexadecylamine (HDA, 98%), sodium tetrachloropalladate(II) (Na2PdCl4, 98%), lead(II) perchlorate hydrate (Pb(ClO4)2·xH2O, 99.995%), perchloric acid (HClO4, 70%), α-alumina (α-Al2O3, 99.9%) and potassium hydroxide (KOH, 99.99%) were obtained from Sigma Aldrich and used as received.2.2 Synthesis of Cu nanocrystals
The synthetic methods are adapted from previous reports.28,29 In a standard synthesis of Rbp-Cu nanocrystals (NCs), 21 mg CuCl2·2H2O, 180 mg HDA, 50 mg glucose, 0.2 mg Na2PdCl4, and 10 ml water were mixed and magnetically stirred in a 20 ml vial at room temperature overnight. Then Ar was slowly bubbled through the solution for 15 min. After that, the vial was tightly capped and heated at 100 °C for 6 h under magnetic stirring. The as-prepared Rbp-Cu NCs were centrifuged and washed with hexane and ethanol several times. Cube-Cu NCs were prepared following the same procedure, except that the amounts of Na2PdCl4 and surfactant (HDA) were adjusted to 0 mg and 90 mg, respectively.2.3 Characterization
The morphology of the as-prepared Cu NCs was imaged by scanning electron microscopy (SEM, FEI Apreo LV-SEM) and transmission electron microscopy (TEM, FEI Titan ETEM). The crystalline structure was identified by X-ray diffraction (XRD, Rigaku D/MAX2500VL). The chemical compositions and surface valence states were determined by X-ray photoelectron spectroscopy (XPS, ESCALAB250Xi).CO temperature-programmed desorption (CO-TPD) was performed under a 10% CO/He atmosphere. The α-alumina support was applied to disperse 10 wt% Cu NCs by a simple impregnation method. 50 mg of Cu samples were first purged with 10% H2/He at 200 °C for 30 min to remove any impurity on the surface, and then adsorbed CO gas at 50 °C for 60 min. After purging again in pure He at 50 °C to remove physisorbed CO on the sample surface for 60 min, the temperature was raised to 500 °C at a ramp rate of 10 °C min−1. The gas flow rate was fixed at 50 sccm for all steps. The resulting species were traced by on-line mass spectrometry (SRS, RGA 100 Headgas Analyzer). The mass-to-charge (m/z) signal of 28 (CO) was monitored with a time interval of 2 s.
2.4 Electrochemical measurements
For each applied potential, an on-line gas chromatograph (GC, SRI Instruments MultipleGas#5) equipped with both a thermal conductivity detector and a flame ionization detector was used to monitor the gas products. To calibrate the outlet gas flow rate of CO2, Ar, as an internal standard, was fed at 10 sccm and mixed with the outlet gas stream from the flow cell before looping to the GC.11 The FEs for gas products were calculated using the following equation (eqn (1)):
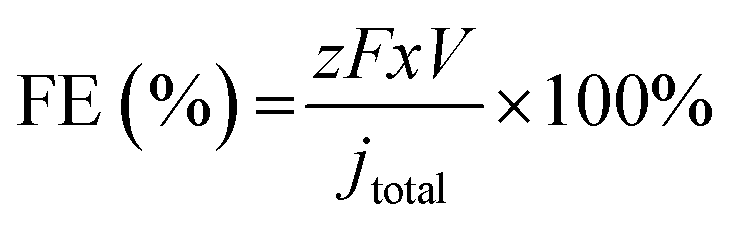 | (1) |
Meanwhile, the catholyte was collected after electrolysis followed by quantifying the liquid products via1H NMR (Bruker AV 400 MHz spectrometer). 500 μl of the catholyte was mixed with a 100 μl internal standard of 5 mM 3-(trimethylsilyl)propionic-2,2,3,3-d4 acid sodium salt in D2O. The standard deviations were calculated based on the measurements of three independent electrodes.
The total CO generation and dimerization rates were calculated according to the following two equations (eqn (2) and (3)):
 | (2) |
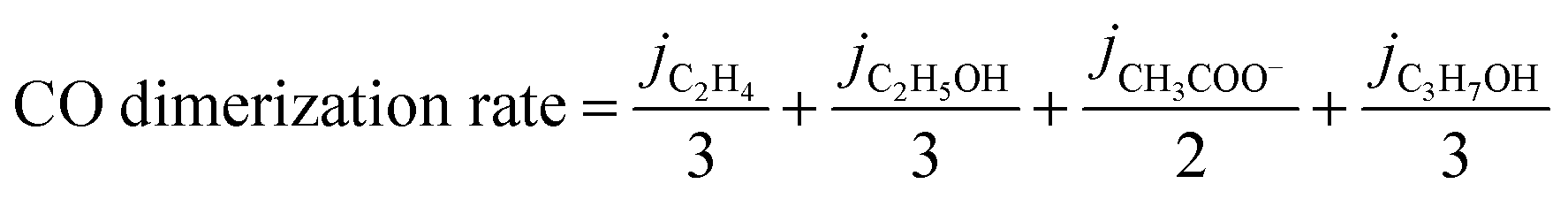 | (3) |
2.5 In situ electrochemical Raman spectroscopy measurements
In situ Raman spectroscopy measurements were carried out on a modified flow cell with a quartz window in front of the cathode GDE. Graphite and Ag/AgCl (3 M KCl) were used as counter and reference electrodes, respectively. The cathode and anode were separated by an anion exchange membrane (FAA-3-PK-75). Syringe pumps were used to pump 1 M KOH at 2 ml min−1 to both the cathode and the anode. CO2 gas was introduced via machined flow channels to the back of the GDE at a flow rate of 50 sccm. The Raman spectra were recorded by using a Renishaw inVia Raman microscope with a 785 nm laser. For each in situ Raman spectroscopy measurement, the acquisition time was 10 s, and the total accumulation of scans was 30. Cathode potentials were applied in the potentiostatic mode and converted to the RHE scale accordingly. The samples were initially activated under CO2RR conditions for 15 min before starting in situ Raman spectroscopy measurements.3. Results and discussion
The Rbp-Cu NCs, which contain Cu(100) facets and abundant planar defects exposed simultaneously, were synthesized by a one-step reduction process using HDA as the capping agent.28 In comparison, the Cube-Cu NCs with exclusive Cu(100) facet surfaces were prepared following the same procedure except for regulating the amount of reactants.29 As shown in SEM images (Fig. 1a), the Rbp-Cu NCs possess a particle size of 60–80 nm with several typical projected profiles, such as triangle, rhombus, and tetragon. Additional high-magnification SEM images and schematic models of individual Rbp-Cu in different orientations are shown in the ESI (Fig. S1†). TEM images unraveled the existence of parallel planar defects (e.g., stacking faults) on the Rbp-Cu surface (Fig. 1b, c and S2, ESI†). Alternating light/dark contrasts in the inset of Fig. 1b are typically a set of stacking faults.30 Analogous to the right bipyramid structures of Ag and Pd,31,32 each Rbp-Cu NC is bounded by six triangular (100) side facets (yellow in the Rbp model in the inset of Fig. 1a) and is mirror-symmetrically divided by a set of planar defects (green in the Rbp model in the inset of Fig. 1a). Benefiting from the regular morphologies of Rbp-Cu, the proportion of planar defects is calculated to be 20.1% based on the geometric dimensions for a typical Rbp-Cu NC (Fig. S2c†). As expected, the as-prepared Cube-Cu with a particle size of 80–120 nm, as a control sample, exhibits a regular cubic shape with (100) facets (Fig. S3†).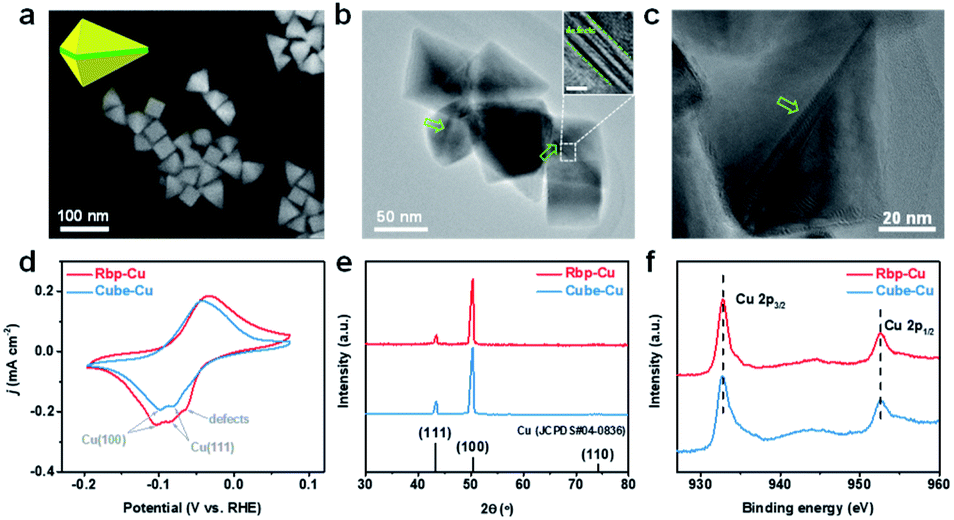 | ||
| Fig. 1 (a) SEM image of Rbp-Cu. Inset shows the schematic model of an individual Rbp-Cu. Yellow and green represent (100) facets and planar defects, respectively. (b) TEM image of Rbp-Cu. The arrows denote the planar defects. Inset shows a zoomed-in TEM image taken from the box region. Inset scale bar: 5 nm. (c) TEM image of a Rbp-Cu NC highlighting triangular (100) facets bisected by planar defects. (d) Pb-UPD profiles at a scan rate of 10 mV s−1, (e) XRD, and (f) Cu 2p XPS spectrum of Rbp-Cu and Cube-Cu. | ||
We used Pb underpotential deposition (UPD), a surface-sensitive electrochemical technique, to further probe the surface structures of Cu NCs. Different Cu facets exhibit distinguished Pb-UPD peaks in the CV curves.17,33 As displayed in Fig. 1d, the peaks located at ca. −0.10 V and −0.08 V can be assigned to Cu(100) and Cu(111), respectively.17 The peaks of Pb electrodeposition on Cu(100) are more prominent for both Rbp-Cu and Cube-Cu samples than those on Cu(111). An additional peak centered at a less cathodic potential (ca. −0.06 V) is only observed for Rbp-Cu, which is ascribed to the Pb-UPD on the low-coordinated sites from surface defects.33,34 Such a kind of phenomenon that the Pb over-layer on the face-centered cubic metal preferentially nucleates at the edges or steps and then expands to the terraces has been demonstrated by in situ scanning probe microscopy studies.35 OH− electro-adsorption was also performed to verify the predominant (100) facets for both Cu NCs (Fig. S4†). An additional shoulder peak at ca. 0.34 V was observed for Rbp-Cu, which is associated with surface defects.36 The percent of high-index surface defects on Rbp-Cu is estimated to be 17.3% according to the integral peak areas,37 which is close to the value obtained from TEM aforementioned. In agreement with TEM, Pb-UPD and OH− electro-adsorption results confirm the presence of defect sites on Rbp-Cu, although the exact facet index is hard to identify. XRD patterns show that both types of Cu NCs exhibit a much more pronounced (100) peak and a weaker (111) peak (Fig. 1e), which is consistent with electrochemical characterization results and confirms the uniform shape of the NCs. Moreover, other than Cu peaks, no other peaks associated with copper oxide or other impurities were observed. XPS analysis reveals that Rbp-Cu and Cube-Cu both present two main peaks at 932.8 eV and 952.6 eV in the Cu 2p spectrum (Fig. 1f), corresponding to Cu0 or Cu+ species.38 The Auger Cu LMM spectrum (Fig. S5†) shows that both Rbp-Cu and Cube-Cu mainly consist of Cu0 (918.6 eV) at the surface, with a minimal amount of Cu+ (916.6 eV). The formation of Cu+ is caused by the rapid oxidation of Cu0 in air and would be quickly in situ reduced to metallic Cu under CO2 electrolysis.22,39 Due to the introduction of a trace amount of Pd (<0.55 at%) as the seed during the synthesis of Rbp-Cu, Pd 3d XPS was carried out. There is no Pd element on the Rbp-Cu surface (Fig. S6†), suggesting the absence of Pd dopants or Pd–Cu bimetallic structures.
The CO2RR performance on Rbp-Cu and Cube-Cu based GDEs was measured in a customized flow cell using 1 M KOH as the catholyte (Fig. S7 and S8†). Rbp-Cu exhibited a higher selectivity towards C2H4 than Cube-Cu. The maximum FE of C2H4 for Rbp-Cu reached 67% at −0.63 V (vs. RHE, thereafter), at which the partial current density (jC2H4) reached 217 mA cm−2 (Fig. 2a and b). The overall performance of Rbp-Cu for CO2-to-C2H4 conversion is superior to that of most copper-based catalysts tested under identical conditions (Table S1†). Combined with C2+ liquid products analyzed by 1H NMR, including ethanol (C2H5OH), acetate (CH3COO−), and n-propone (C3H7OH), the FE and partial current density of C2+ products (jC2+) on Rbp-Cu reached 89% and 289 mA cm−2, respectively, at −0.63 V (Fig. 2c and d). In contrast, Cube-Cu showed only 46% FE for C2H4 and 68% FE for C2+ products at the same overpotential. Moreover, the FE ratio of C2H4 over C2H5OH was enhanced from 3.10 for Cube-Cu to 3.77 for Rbp-Cu at −0.63 V (Fig. S9†). Compared to Rbp-Cu, Cube-Cu manifested lower jC2H4 and jC2+ over the applied potential range (Fig. 2b and d). Interestingly, we found that Rbp-Cu showed a relatively much lower FE for CO than Cube-Cu (Fig. S10†), indicating the more efficient conversion of the *CO intermediate to hydrocarbons and oxygenates on Rbp-Cu.10 The improvement of activity and selectivity towards C2H4 and C2+ products suggests that the planar defect sites on Rbp-Cu may be responsible for the enhanced C–C coupling rate.
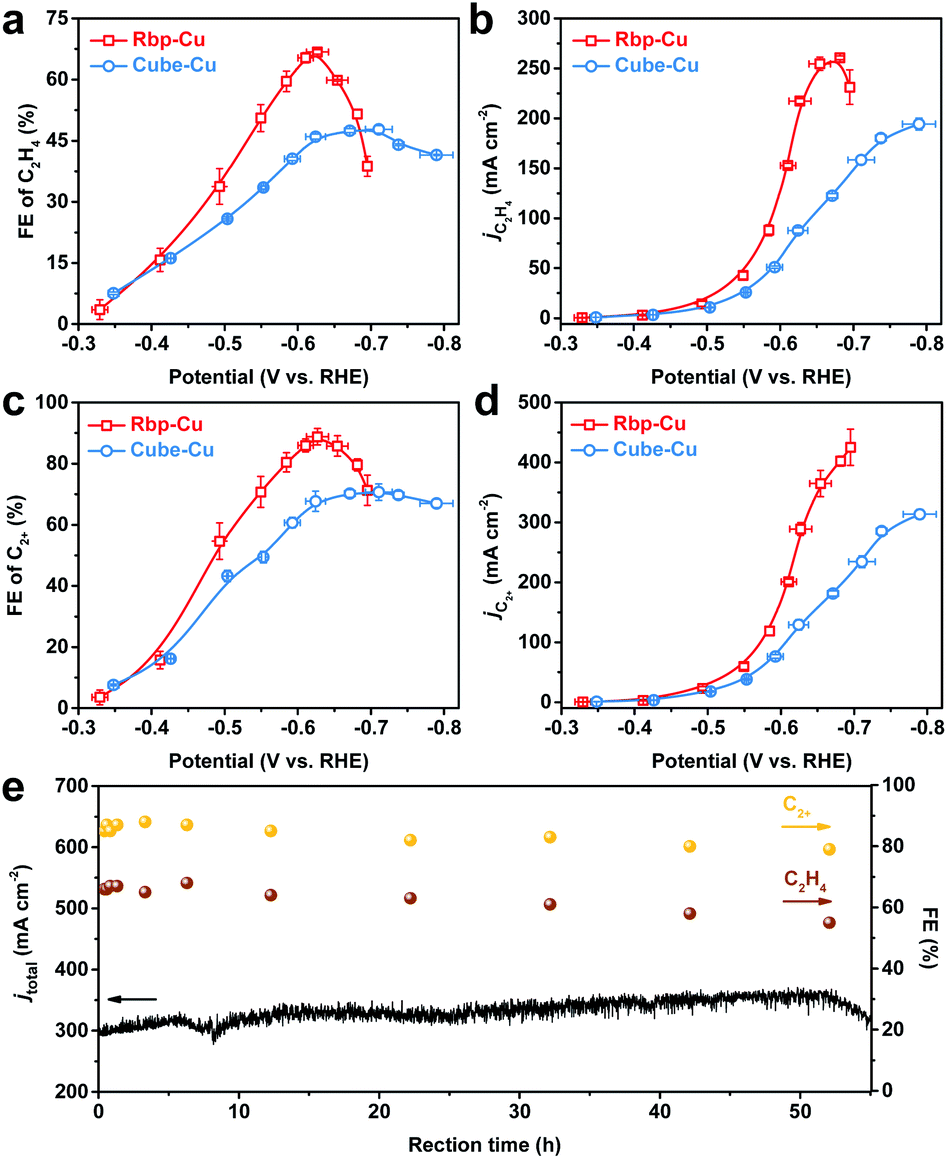 | ||
| Fig. 2 Comparison of the CO2RR performance of Rbp-Cu and Cube-Cu. (a–d) The potential-dependent (a) FE for C2H4, (b) jC2H4, (c) FE for C2+ products, and (d) jC2+. The error bars represent the standard deviation based on the measurements of three independent electrodes. (e) The stability of Rbp-Cu operated at −0.63 V vs. RHE. | ||
To exclude the possible effect of surface roughness, the electrochemical surface area (ECSA) was determined by using the double-layer capacitance (Cdl) method (Fig. S11†). The ECSA normalized current density of each product followed the same trend as the geometric current density for both Rbp-Cu and Cube-Cu (Fig. S12†). At an optimal potential of −0.63 V for C2H4 and C2+ product formation, the normalized jC2H4 and jC2+ for Rbp-Cu were 1.9-fold and 1.7-fold higher than those for Cube-Cu, respectively, indicating the excellent intrinsic activity of Rbp-Cu towards C2H4 and C2+ products. Considering a lower surface-to-volume ratio of Cube-Cu related to Rbp-Cu,28 the loading of Cube-Cu was increased from 0.30 to 0.42 mg cm−2, which increased the Cdl (proportional to the ECSA) to 0.57 mF cm−2 accordingly, similar to that of Rbp-Cu (0.56 mF cm−2) with a loading of 0.30 mg cm−2 (Fig. S13†). Although the current density increased with the catalyst loading, a similar product distribution was obtained for Cube-Cu with two different loadings. Especially, the FEs for C2H4 were almost identical in the investigated overpotential range (Fig. S13e†). Therefore, we infer that a higher ECSA is not the main factor for promoting selectivity towards C2H4.
Notably, Rbp-Cu exhibited activity towards C2H4 and C2+ products similar to Cube-Cu at potentials between −0.35 and −0.50 V (Fig. 2 and S12†), likely owing to the Cu(100) facet on both Cu NCs which facilitates the *COCO dimerization in such a low overpotential region.7,39 In the potential region of −0.50 ∼ −0.65 V, the planar defect sites on the Rbp-Cu surface play a more critical role in CO2-to-C2+ product conversion. The possible dimerization pathways in this overpotential range may involve coupling between *CO and reduced *CO species (i.e., *CHO and/or *COH),8,40 the reaction barrier of which could be decreased on low-coordinated defects.37,41,42 However, the selectivity towards C2H4 for Rbp-Cu decreased dramatically at higher cathodic potentials (−0.65 ∼ −0.70 V). Simultaneously, the activity and selectivity towards hydrogen (H2) and methane (CH4) rose accordingly (Fig. S10 and S12†). The upsurge of reactivity towards H2 arises from the stronger competitive hydrogen evolution reaction (HER) occurring on low-coordinated sites in the high cathodic potential region.30,43 To verify this, the HER was performed under an Ar atmosphere in a flow cell. Linear sweep voltammetry (LSV) showed higher electrocatalytic activity of Rbp-Cu for the HER than Cube-Cu as the potential was swept beyond −0.50 V (Fig. S14†), demonstrating that planar defects are more active for the HER as well as the CO2RR in the high cathodic potential region. At higher overpotentials, increased adsorption of hydrogen would suppress CO2-to-C2+ product conversion.44
To demonstrate a stable CO2-to-C2+ product conversion, a durability test for Rbp-Cu was conducted at −0.63 V, where optimal selectivity towards C2H4 and C2+ products was achieved. By using polytetrafluoroethylene-treated GDL to improve the hydrophobicity, a relatively stable FE of (61 ± 5)% for C2H4 was recorded while FE for C2+ products was maintained at (82 ± 4)% with a total current density of (330 ± 30) mA cm−2 during a 50 hour continuous test (Fig. 2e). As examined by TEM, the post-electrolysis Rbp-Cu retained the planar defects (Fig. S15†), although the corners or edges of the well-defined morphology were degraded to some extent.45,46 Surface adsorbed *CO, the key intermediate for CO2 reduction to hydrocarbons and oxygenates, may stabilize Cu defects in line with prior density functional theory (DFT) calculations.22
Given that the same Cu(100) facets were exposed for both Rbp-Cu and Cube-Cu samples, we envisioned that the superior CO2-to-C2H4 conversion is attributed to the introduction of the planar defects on the Rbp-Cu surface. These planar defects with a low coordination number enable stronger *CO adsorption that assists in building a sufficiently high surface coverage of *CO to direct the C–C coupling.6,23 To prove this hypothesis, an ex situ CO-TPD experiment and in situ electrochemical Raman spectroscopy measurements were conducted.
The monitored CO-TPD traces, which can give the amount and strength of *CO adsorption, are shown in Fig. 3a and S16.† For both types of Cu NCs, the onset CO desorption began at approximately 60 °C. The observation of a later CO desorption peak at 90 °C for Rbp-Cu indicates a stronger adsorption affinity for CO and CO-related intermediates, resulting from the presence of planar defects.36,47 Additionally, the larger CO desorption peak area for Rbp-Cu implies a larger surface coverage of *CO on Rbp-Cu than Cube-Cu.48 These CO-TPD results preliminarily support that both the adsorption energy and surface coverage of *CO are boosted on Rbp-Cu, benefiting from planar defect sites.
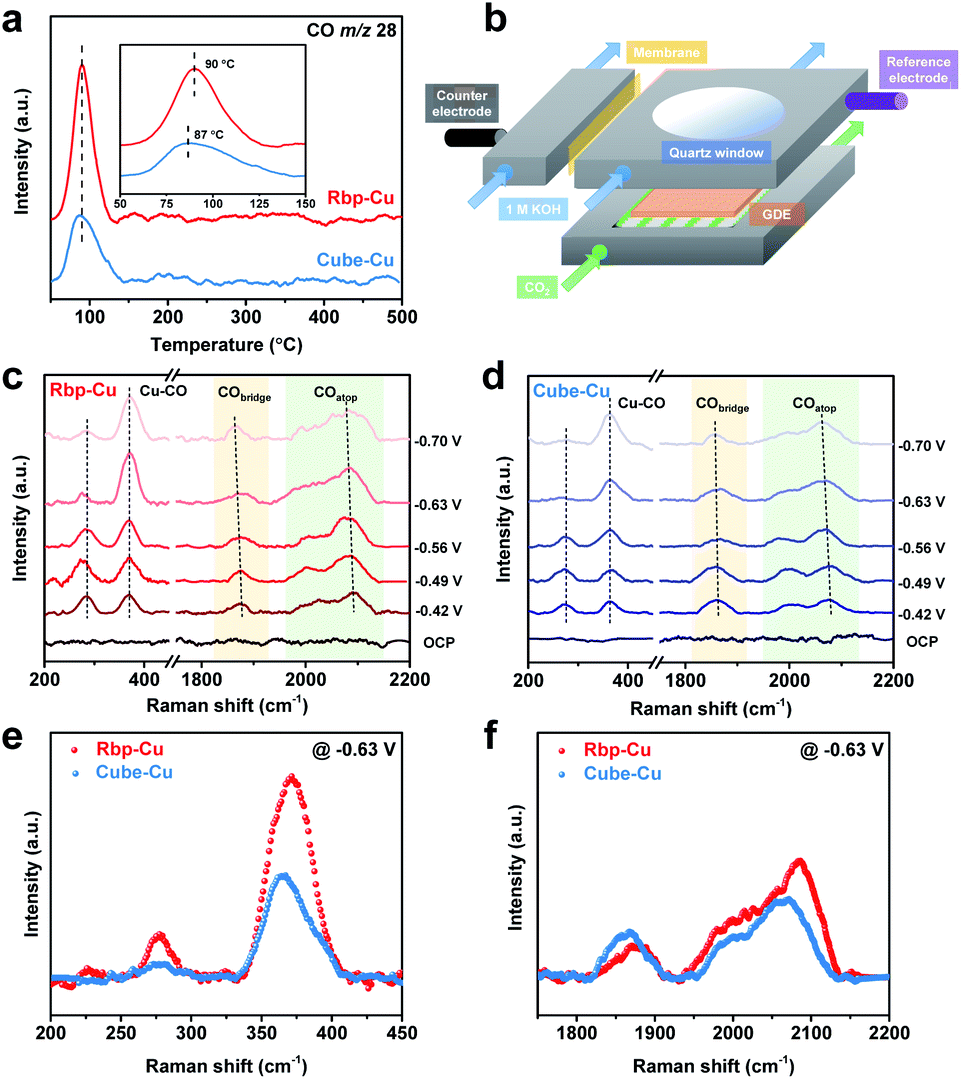 | ||
| Fig. 3 (a) CO-TPD profiles of Rbp-Cu and Cube-Cu. The desorption compound is monitored with an m/z ratio of 28 for CO. Inset shows the zoomed-in region from 50 to 150 °C. (b) Schematic of the in situ Raman spectroscopy flow cell. (c, d) In situ Raman spectroscopy of (c) Rbp-Cu and (d) Cube-Cu during the CO2RR at different applied potentials. All the given potentials are referred to the RHE. (e, f) Comparison of in situ Raman spectra for Rbp-Cu and Cube-Cu in the (e) low-frequency range and (f) high-frequency range at −0.63 V vs. RHE. | ||
To evaluate the *CO adsorption behaviors under CO2 electrocatalysis conditions, in situ Raman spectroscopy measurements were further carried out in a modified flow cell with a quartz window in front of the cathode GDE (Fig. 3b). The Raman spectra were recorded in a range of applied potentials from the open circuit potential (OCP) to −0.70 V for both types of Cu NCs (Fig. 3c and d). For Rbp-Cu, the low-frequency bands of *CO at ca. 282 and 371 cm−1 are characteristic of Cu–CO frustrated rotation and Cu–CO stretch, respectively (Fig. 3c).49 The high-frequency bands are associated with C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) O stretching vibrations with different adsorption configurations, including bridge-bound CO (CObridge, 1850–1880 cm−1) and atop-bound CO (COatop, 2000–2090 cm−1).49,50 Notably, the asymmetric broad band for COatop is likely due to the *CO adsorption on various Cu surface sites, which, in turn, may affect the activity and selectivity of the CO2RR.49,51 The difference in *CO adsorption behaviors between Rbp-Cu and Cube-Cu was assessed by comparing the in situ Raman spectra collected at the same applied potential of −0.63 V. First, a blue shift of the COatop sharp band suggests the stronger binding of *CO at low-coordinated defect sites for Rbp-Cu (2084 cm−1) relative to Cube-Cu (2067 cm−1) (Fig. 3f).49 Meanwhile, the band for the Cu–CO stretch also exhibited a blue shift on Rbp-Cu (371 cm−1) compared to that on Cube-Cu (365 cm−1) (Fig. 3e), confirming a stronger binding of *CO to the Rbp-Cu surface.52 Second, the integrated areas of bands (e.g., Cu–CO and COatop bands), which are proportional to the *CO surface coverage,49 increase for Rbp-Cu compared with Cube-Cu (Fig. 3e and f). Taken together, ex situ CO-TPD and the in situ Raman spectroscopy results unravel that Rbp-Cu promotes the adsorption and surface coverage of *CO, which could enhance the subsequent C–C coupling kinetics and thus the production of C2+ products.
O stretching vibrations with different adsorption configurations, including bridge-bound CO (CObridge, 1850–1880 cm−1) and atop-bound CO (COatop, 2000–2090 cm−1).49,50 Notably, the asymmetric broad band for COatop is likely due to the *CO adsorption on various Cu surface sites, which, in turn, may affect the activity and selectivity of the CO2RR.49,51 The difference in *CO adsorption behaviors between Rbp-Cu and Cube-Cu was assessed by comparing the in situ Raman spectra collected at the same applied potential of −0.63 V. First, a blue shift of the COatop sharp band suggests the stronger binding of *CO at low-coordinated defect sites for Rbp-Cu (2084 cm−1) relative to Cube-Cu (2067 cm−1) (Fig. 3f).49 Meanwhile, the band for the Cu–CO stretch also exhibited a blue shift on Rbp-Cu (371 cm−1) compared to that on Cube-Cu (365 cm−1) (Fig. 3e), confirming a stronger binding of *CO to the Rbp-Cu surface.52 Second, the integrated areas of bands (e.g., Cu–CO and COatop bands), which are proportional to the *CO surface coverage,49 increase for Rbp-Cu compared with Cube-Cu (Fig. 3e and f). Taken together, ex situ CO-TPD and the in situ Raman spectroscopy results unravel that Rbp-Cu promotes the adsorption and surface coverage of *CO, which could enhance the subsequent C–C coupling kinetics and thus the production of C2+ products.
To further explore the impact of the enhancement of *CO adsorption energy on the C–C coupling kinetics, we analyzed and compared the CO generation and dimerization rates (Fig. 4 and S17†).12 The CO generation rate was referred to as the summary of the normalized production rates of CO, CH4, and C2+ products (eqn (2) in the Experimental section). According to the scaling relations of adsorption energy between intermediates and BEP relations between the intermediate adsorption energy and activation barrier,21 the enhancement of *CO binding indicates stronger *COOH binding and a lower activation barrier in the elementary step of CO2-to-*COOH conversion, which leads to an accelerated CO generation rate. Fig. 4a shows that Rbp-Cu indeed provided more CO than Cube-Cu, manifesting that planar defects facilitate the CO generation rate compared to terraces such as (100) facets in our case. Meanwhile, the CO dimerization rate was derived based on the normalized production rates of C2+ products (eqn (3) in the Experimental section). Again, Rbp-Cu exhibited a much faster CO dimerization rate than Cube-Cu due partly to the increased surface coverage of *CO (Fig. 4b). For example, the CO dimerization rate of Rbp-Cu reached >2.2-times as high as that of Cube-Cu at around −0.65 V. It is also reasonable to propose that C–C coupling intermediates, such as *COCO, *COCHO, or *COCOH, can be efficiently stabilized on defect sites, which further facilitates the dimerization reaction.40,41 In short, compared to (100) facets, the stronger *CO adsorption on planar defect sites facilitates CO generation kinetics in the intermediate overpotential region, which contributes to a higher surface coverage of *CO and in turn an enhanced reaction rate of C–C coupling towards C2+ products, especially C2H4.
 | ||
| Fig. 4 Comparison of the (a) CO generation rate and (b) CO dimerization rate between Rbp-Cu and Cube-Cu at different applied potentials. | ||
4. Conclusions
In summary, compared to a regular Cube-Cu NC with exclusive (100) facets, Rbp-Cu, containing a combination of planar defects (e.g., stacking faults) and (100) facets, exhibits a higher selectivity to C2H4 (FE = 67%) and C2+ products (FE = 89%) with a corresponding jC2H4 of 217 mA cm−2 at −0.63 V. Using CO-TPD and in situ Raman spectroscopy, the introduction of planar defects on the Rbp-Cu surface is found to contribute to the increase of *CO adsorption energy and *CO surface coverage. Based on CO generation and dimerization rates analysis, a stronger *CO binding facilitates the *CO surface coverage and promotes the C–C coupling kinetics. Additionally, a durability test illustrates that defects are stabilized by *CO under CO2RR conditions. Further DFT investigations are expected to explore the relationship between specific active sites and favorable reaction pathways to either C2H4 or C2H5OH at the atomic level. Nevertheless, this work provides an in-depth insight into the reactivity comparison between planar defects and Cu(100) facets, which guides the design of advanced catalysts for efficient CO2-to-C2H4 conversion.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by NSF CBET-2033343. S. Y. acknowledges the use of facilities within the Eyring Materials Center at Arizona State University supported in part by NNCI-ECCS-1542160. J. J. and S. S. acknowledge that the research carried out at the Brookhaven National Laboratory (BNL) was supported by the Division of Chemical Science, Geoscience, and Bioscience, Office of Basic Energy Science of the US Department of Energy (DOE), under contract no. DE-SC0012704.Notes and references
- Y. Y. Birdja, E. Pérez-Gallent, M. C. Figueiredo, A. J. Göttle, F. Calle-Vallejo and M. T. M. Koper, Nat. Energy, 2019, 4, 732–745 CrossRef CAS.
- P. D. Luna, C. Hahn, D. Higgins, S. A. Jaffer, T. F. Jaramillo and E. H. Sargent, Science, 2019, 364, eaav3506 CrossRef PubMed.
- S. Nitopi, E. Bertheussen, S. B. Scott, X. Liu, A. K. Engstfeld, S. Horch, B. Seger, I. E. L. Stephens, K. Chan, C. Hahn, J. K. Nørskov, T. F. Jaramillo and I. Chorkendorff, Chem. Rev., 2019, 119, 7610–7672 CrossRef CAS PubMed.
- R. I. Masel, Z. Liu, H. Yang, J. J. Kaczur, D. Carrillo, S. Ren, D. Salvatore and C. P. Berlinguette, Nat. Nanotechnol., 2021, 16, 118–128 CrossRef CAS PubMed.
- Y. Huang, A. D. Handoko, P. Hirunsit and B. S. Yeo, ACS Catal., 2017, 7, 1749–1756 CrossRef CAS.
- R. B. Sandberg, J. H. Montoya, K. Chan and J. K. Nørskov, Surf. Sci., 2016, 654, 56–62 CrossRef CAS.
- J. H. Montoya, C. Shi, K. Chan and J. K. Nørskov, J. Phys. Chem. Lett., 2015, 6, 2032–2037 CrossRef CAS PubMed.
- F. Calle-Vallejo and M. T. M. Koper, Angew. Chem., Int. Ed., 2013, 52, 7282–7285 CrossRef CAS PubMed.
- D. Ren, B. S.-H. Ang and B. S. Yeo, ACS Catal., 2016, 6, 8239–8247 CrossRef CAS.
- Z. Li, R. M. Yadav, L. Sun, T. Zhang, J. Zhang, P. M. Ajayan and J. Wu, Appl. Catal., A, 2020, 606, 117829 CrossRef CAS.
- X. She, T. Zhang, Z. Li, H. Li, H. Xu and J. Wu, Cell Rep. Phys. Sci., 2020, 1, 100051 CrossRef.
- T. Zhang, Z. Li, J. Zhang and J. Wu, J. Catal., 2020, 387, 163–169 CrossRef CAS.
- A. Ozden, Y. Wang, F. Li, M. Luo, J. Sisler, A. Thevenon, A. Rosas-Hernández, T. Burdyny, Y. Lum, H. Yadegari, T. Agapie, J. C. Peters, E. H. Sargent and D. Sinton, Joule, 2021, 5, 706–719 CrossRef CAS.
- M. Zhong, K. Tran, Y. Min, C. Wang, Z. Wang, C.-T. Dinh, P. De Luna, Z. Yu, A. S. Rasouli, P. Brodersen, S. Sun, O. Voznyy, C.-S. Tan, M. Askerka, F. Che, M. Liu, A. Seifitokaldani, Y. Pang, S.-C. Lo, A. Ip, Z. Ulissi and E. H. Sargent, Nature, 2020, 581, 178–183 CrossRef CAS PubMed.
- Y. Zhou, F. Che, M. Liu, C. Zou, Z. Liang, P. De Luna, H. Yuan, J. Li, Z. Wang, H. Xie, H. Li, P. Chen, E. Bladt, R. Quintero-Bermudez, T.-K. Sham, S. Bals, J. Hofkens, D. Sinton, G. Chen and E. H. Sargent, Nat. Chem., 2018, 10, 974–980 CrossRef CAS PubMed.
- W. Ma, S. Xie, T. Liu, Q. Fan, J. Ye, F. Sun, Z. Jiang, Q. Zhang, J. Cheng and Y. Wang, Nat. Catal., 2020, 3, 478–487 CrossRef CAS.
- Y. Wang, Z. Wang, C.-T. Dinh, J. Li, A. Ozden, M. Golam Kibria, A. Seifitokaldani, C.-S. Tan, C. M. Gabardo, M. Luo, H. Zhou, F. Li, Y. Lum, C. McCallum, Y. Xu, M. Liu, A. Proppe, A. Johnston, P. Todorovic, T.-T. Zhuang, D. Sinton, S. O. Kelley and E. H. Sargent, Nat. Catal., 2020, 3, 98–106 CrossRef CAS.
- G. L. De Gregorio, T. Burdyny, A. Loiudice, P. Iyengar, W. A. Smith and R. Buonsanti, ACS Catal., 2020, 10, 4854–4862 CrossRef CAS PubMed.
- S.-C. Lin, C.-C. Chang, S.-Y. Chiu, H.-T. Pai, T.-Y. Liao, C.-S. Hsu, W.-H. Chiang, M.-K. Tsai and H. M. Chen, Nat. Commun., 2020, 11, 3525 CrossRef CAS PubMed.
- H. Jung, S. Y. Lee, C. W. Lee, M. K. Cho, D. H. Won, C. Kim, H.-S. Oh, B. K. Min and Y. J. Hwang, J. Am. Chem. Soc., 2019, 141, 4624–4633 CrossRef CAS PubMed.
- A. H. Motagamwala and J. A. Dumesic, Chem. Rev., 2021, 121, 1049–1076 CrossRef CAS PubMed.
- Z. Gu, H. Shen, Z. Chen, Y. Yang, C. Yang, Y. Ji, Y. Wang, C. Zhu, J. Liu, J. Li, T.-K. Sham, X. Xu and G. Zheng, Joule, 2021, 5, 429–440 CrossRef CAS.
- C. Choi, T. Cheng, M. F. Espinosa, H. Fei, X. Duan, W. A. Goddard and Y. Huang, Adv. Mater., 2019, 31, 1805405 CrossRef PubMed.
- B. Zhang, J. Zhang, M. Hua, Q. Wan, Z. Su, X. Tan, L. Liu, F. Zhang, G. Chen, D. Tan, X. Cheng, B. Han, L. Zheng and G. Mo, J. Am. Chem. Soc., 2020, 142, 13606–13613 CrossRef CAS PubMed.
- C. W. Li, J. Ciston and M. W. Kanan, Nature, 2014, 508, 504–507 CrossRef CAS PubMed.
- X. Feng, K. Jiang, S. Fan and M. W. Kanan, ACS Cent. Sci., 2016, 2, 169–174 CrossRef CAS PubMed.
- Y. Pang, J. Li, Z. Wang, C.-S. Tan, P.-L. Hsieh, T.-T. Zhuang, Z.-Q. Liang, C. Zou, X. Wang, P. De Luna, J. P. Edwards, Y. Xu, F. Li, C.-T. Dinh, M. Zhong, Y. Lou, D. Wu, L.-J. Chen, E. H. Sargent and D. Sinton, Nat. Catal., 2019, 2, 251–258 CrossRef CAS.
- Z. Lyu, M. Xie, K. D. Gilroy, Z. D. Hood, M. Zhao, S. Zhou, J. Liu and Y. Xia, Chem. Mater., 2018, 30, 6469–6477 CrossRef CAS.
- M. Jin, G. He, H. Zhang, J. Zeng, Z. Xie and Y. Xia, Angew. Chem., Int. Ed., 2011, 50, 10560–10564 CrossRef CAS PubMed.
- Z. Li, J.-Y. Fu, Y. Feng, C.-K. Dong, H. Liu and X.-W. Du, Nat. Catal., 2019, 2, 1107–1114 CrossRef CAS.
- J. Zhang, S. Li, J. Wu, G. C. Schatz and C. A. Mirkin, Angew. Chem., Int. Ed., 2009, 48, 7787–7791 CrossRef CAS PubMed.
- X. Xia, S.-I. Choi, J. A. Herron, N. Lu, J. Scaranto, H.-C. Peng, J. Wang, M. Mavrikakis, M. J. Kim and Y. Xia, J. Am. Chem. Soc., 2013, 135, 15706–15709 CrossRef CAS.
- C. Kim, T. Möller, J. Schmidt, A. Thomas and P. Strasser, ACS Catal., 2019, 9, 1482–1488 CrossRef CAS.
- S. Mezzavilla, S. Horch, I. E. L. Stephens, B. Seger and I. Chorkendorff, Angew. Chem., Int. Ed., 2019, 58, 3774–3778 CrossRef CAS PubMed.
- E. Herrero, L. J. Buller and H. D. Abruña, Chem. Rev., 2001, 101, 1897–1930 CrossRef CAS PubMed.
- D. Raciti, L. Cao, K. J. T. Livi, P. F. Rottmann, X. Tang, C. Li, Z. Hicks, K. H. Bowen, K. J. Hemker, T. Mueller and C. Wang, ACS Catal., 2017, 7, 4467–4472 CrossRef CAS.
- C. Choi, S. Kwon, T. Cheng, M. Xu, P. Tieu, C. Lee, J. Cai, H. M. Lee, X. Pan, X. Duan, W. A. Goddard and Y. Huang, Nat. Catal., 2020, 3, 804–812 CrossRef CAS.
- A. F. Carley, L. A. Dollard, P. R. Norman, C. Pottage and M. W. Roberts, J. Electron Spectrosc. Relat. Phenom., 1999, 98–99, 223–233 CrossRef CAS.
- T.-T. Zhuang, Y. Pang, Z.-Q. Liang, Z. Wang, Y. Li, C.-S. Tan, J. Li, C. T. Dinh, P. De Luna, P.-L. Hsieh, T. Burdyny, H.-H. Li, M. Liu, Y. Wang, F. Li, A. Proppe, A. Johnston, D.-H. Nam, Z.-Y. Wu, Y.-R. Zheng, A. H. Ip, H. Tan, L.-J. Chen, S.-H. Yu, S. O. Kelley, D. Sinton and E. H. Sargent, Nat. Catal., 2018, 1, 946–951 CrossRef CAS.
- J. D. Goodpaster, A. T. Bell and M. Head-Gordon, J. Phys. Chem. Lett., 2016, 7, 1471–1477 CrossRef CAS PubMed.
- Z. Chen, T. Wang, B. Liu, D. Cheng, C. Hu, G. Zhang, W. Zhu, H. Wang, Z.-J. Zhao and J. Gong, J. Am. Chem. Soc., 2020, 142, 6878–6883 CrossRef CAS.
- D. Cheng, Z.-J. Zhao, G. Zhang, P. Yang, L. Li, H. Gao, S. Liu, X. Chang, S. Chen, T. Wang, G. A. Ozin, Z. Liu and J. Gong, Nat. Commun., 2021, 12, 395 CrossRef CAS PubMed.
- R. Reske, H. Mistry, F. Behafarid, B. Roldan Cuenya and P. Strasser, J. Am. Chem. Soc., 2014, 136, 6978–6986 CrossRef CAS PubMed.
- X. Liu, P. Schlexer, J. Xiao, Y. Ji, L. Wang, R. B. Sandberg, M. Tang, K. S. Brown, H. Peng, S. Ringe, C. Hahn, T. F. Jaramillo, J. K. Nørskov and K. Chan, Nat. Commun., 2019, 10, 32 CrossRef CAS PubMed.
- J. Huang, N. Hörmann, E. Oveisi, A. Loiudice, G. L. De Gregorio, O. Andreussi, N. Marzari and R. Buonsanti, Nat. Commun., 2018, 9, 3117 CrossRef PubMed.
- M. de Jesus Gálvez-Vázquez, P. Moreno-García, H. Xu, Y. Hou, H. Hu, I. Z. Montiel, A. V. Rudnev, S. Alinejad, V. Grozovski, B. J. Wiley, M. Arenz and P. Broekmann, ACS Catal., 2020, 10, 13096–13108 CrossRef.
- S. Vollmer, G. Witte and C. Wöll, Catal. Lett., 2001, 77, 97–101 CrossRef CAS.
- J. Strunk, R. N. d‘Alnoncourt, M. Bergmann, S. Litvinov, X. Xia, O. Hinrichsen and M. Muhler, Phys. Chem. Chem. Phys., 2006, 8, 1556–1565 CAS.
- C. M. Gunathunge, X. Li, J. Li, R. P. Hicks, V. J. Ovalle and M. M. Waegele, J. Phys. Chem. C, 2017, 121, 12337–12344 CrossRef CAS.
- X. Chang, Y. Zhao and B. Xu, ACS Catal., 2020, 10, 13737–13747 CrossRef CAS.
- C. M. Gunathunge, V. J. Ovalle, Y. Li, M. J. Janik and M. M. Waegele, ACS Catal., 2018, 8, 7507–7516 CrossRef CAS.
- X. Wang, Z. Wang, F. P. García de Arquer, C.-T. Dinh, A. Ozden, Y. C. Li, D.-H. Nam, J. Li, Y.-S. Liu, J. Wicks, Z. Chen, M. Chi, B. Chen, Y. Wang, J. Tam, J. Y. Howe, A. Proppe, P. Todorović, F. Li, T.-T. Zhuang, C. M. Gabardo, A. R. Kirmani, C. McCallum, S.-F. Hung, Y. Lum, M. Luo, Y. Min, A. Xu, C. P. O'Brien, B. Stephen, B. Sun, A. H. Ip, L. J. Richter, S. O. Kelley, D. Sinton and E. H. Sargent, Nat. Energy, 2020, 5, 478–486 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ta02565a |
| This journal is © The Royal Society of Chemistry 2021 |