
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLeveraging an enzyme/artificial substrate system to enhance cellular persulfides and mitigate neuroinflammation†
Prerona
Bora‡
 a,
Suman
Manna‡
a,
Mrutyunjay A.
Nair‡§
a,
Rupali R. M.
Sathe
b,
Shubham
Singh
b,
Venkata Sai
Sreyas Adury
a,
Kavya
Gupta
c,
Arnab
Mukherjee
a,
Deepak K.
Saini
c,
Siddhesh S.
Kamat
b,
Amrita B.
Hazra
*ab and
Harinath
Chakrapani
*a
a,
Suman
Manna‡
a,
Mrutyunjay A.
Nair‡§
a,
Rupali R. M.
Sathe
b,
Shubham
Singh
b,
Venkata Sai
Sreyas Adury
a,
Kavya
Gupta
c,
Arnab
Mukherjee
a,
Deepak K.
Saini
c,
Siddhesh S.
Kamat
b,
Amrita B.
Hazra
*ab and
Harinath
Chakrapani
*a
aDepartment of Chemistry, Indian Institute of Science Education and Research Pune, Dr. Homi Bhabha Road, Pashan, Pune 411 008, Maharashtra, India. E-mail: amrita@iiserpune.ac.in; harinath@iiserpune.ac.in
bDepartment of Biology, Indian Institute of Science Education and Research Pune, Dr. Homi Bhabha Road, Pashan, Pune 411 008, Maharashtra, India
cDepartment of Molecular Reproduction, Development and Genetics, Indian Institute of Science, Bangalore 560012, Karnataka, India
First published on 24th August 2021
Abstract
Persulfides and polysulfides, collectively known as the sulfane sulfur pool along with hydrogen sulfide (H2S), play a central role in cellular physiology and disease. Exogenously enhancing these species in cells is an emerging therapeutic paradigm for mitigating oxidative stress and inflammation that are associated with several diseases. In this study, we present a unique approach of using the cell's own enzyme machinery coupled with an array of artificial substrates to enhance the cellular sulfane sulfur pool. We report the synthesis and validation of artificial/unnatural substrates specific for 3-mercaptopyruvate sulfurtransferase (3-MST), an important enzyme that contributes to sulfur trafficking in cells. We demonstrate that these artificial substrates generate persulfides in vitro as well as mediate sulfur transfer to low molecular weight thiols and to cysteine-containing proteins. A nearly 100-fold difference in the rates of H2S production for the various substrates is observed supporting the tunability of persulfide generation by the 3-MST enzyme/artificial substrate system. Next, we show that the substrate 1a permeates cells and is selectively turned over by 3-MST to generate 3-MST-persulfide, which protects against reactive oxygen species-induced lethality. Lastly, in a mouse model, 1a is found to significantly mitigate neuroinflammation in the brain tissue. Together, the approach that we have developed allows for the on-demand generation of persulfides in vitro and in vivo using a range of shelf-stable, artificial substrates of 3-MST, while opening up possibilities of harnessing these molecules for therapeutic applications.
Introduction
Hydrogen sulfide (H2S) and its redox congeners, persulfides (HSSH) and polysulfides (H2Sn) act as mediators of several intracellular signaling processes.1,2 Persulfides and polysulfides, collectively known as the sulfane sulfur pool, along with H2S are summoned in response to oxidative stress in cells, and dysfunctional sulfur metabolism is implicated in neurodegeneration,3,4 cardiovascular disease5,6 and antibiotic resistance.7,8 To regulate the sulfane sulfur pool, one of the strategies used by cells is to employ protein persulfidation, a post-translational modification where the thiol group of a reactive cysteine (Cys-SH) is modified to a persulfide (Cys-SSH).9 The persulfidated protein mediates S-transfer to appropriate biomolecular targets, and not only provides protection against irreversible oxidation of cysteine residues but also regulates cellular signaling and sulfur homeostasis.10,11 Persulfidation has been linked to modulation of the catalytic activity of key proteins such as parkin and GSK-3β that are involved in neurodegenerative diseases, underscoring the therapeutic relevance of exogenously controlling this post-translational modification by administering persulfide/H2S.4,9 Here, we report a novel approach of developing artificial/unnatural substrates for persulfide-generating enzymes in vivo as a tool to study and tune sulfur metabolism in cells with applications in mitigating neuroinflammation.In this study, we leverage natural biochemical mechanisms for generating persulfides by the development of a new class of substrates for 3-mercaptopyruvate sulfurtransferase (3-MST), a central persulfide/polysulfide generating enzyme that has been implicated in the mitigation of oxidative stress.12–14 3-MST operates by generating its own persulfide by the activation of a hyper-reactive active site cysteine with the sulfur atom of 3-mercaptopyruvate (3-MP) and produces pyruvate as the byproduct (Fig. 1A).15 The persulfidated 3-MST (3-MST-SS−) can transfer the sulfur to low molecular weight thiols such as glutathione (GSH) to produce a persulfide (GSSH), which in turn persulfidates proteins (Fig. 1B).16 Alternately, 3-MST-SS− can produce a polysulfide species (3-MST-SnS−) which subsequently persulfidates low molecular weight thiols and other proteins in the cell.2 3-MST-SS− has been found to be involved in mitochondrial respiration and fatty acid metabolism,17,18 synthesis of thiouridine in tRNA,19 iron–sulfur cluster formation,20 and in cyanide detoxification.21 Also, 3-MST is among the major H2S-generating enzymes in the brain and is thus implicated in regulation of the sulfane sulfur pool and sulfur trafficking in neuronal cells.14 Therefore, a methodology that generates 3-MST-SS− in a specific, controlled and catalytic manner would be advantageous for understanding the mechanisms of sulfur metabolism, and its role in signaling and disease.
 | ||
| Fig. 1 (A) Catalytic cycle of 3-MST: the sulfur of 3-MP is transferred to 3-MST to produce 3-MST-SS− and pyruvate. (B) 3-MST-SS− reacts with reducing agents containing two cysteine residues such as thioredoxin (Trx) to generate H2S. 3-MST-SS− can also generate a protein persulfide through protein–protein interaction or transfer sulfur to low molecular weight thiols such as GSH to produce glutathione persulfide (GSSH). (C) Thioacetate 1 is expected to be cleaved by esterase (ES) to produce the designed 3-MST substrate 2. This thiol is positioned to undergo a sulfur transfer reaction to produce 3-MST-SS− and a ketone 3 as the byproduct. | ||
The natural substrate 3-MP can, in principle, be used for this purpose. However, previous reports suggest that 3-MP produces H2S even in the absence of 3-MST in the cell, thus, using it may have limitations.22 Hence, our strategy of developing a suite of artificial substrates specific for 3-MST as a methodology for the controlled generation of its persulfide is unique with diverse implications. Since cellular signaling is highly dependent on the concentration and rate of generation of the signaling species, it is important that the new methodology developed contains in-built characteristics for tuning these parameters. We expect that the systematic functionalization of the artificial substrates will alter their binding to the enzyme, as well as the rate of persulfide generation (Fig. 1C). Together, these factors will consequently allow for tuning the sulfur transfer reaction. Finally, since thiols are prone to oxidation, it was envisaged that the unnatural substrate could be generated in situ from the corresponding thioesters through ester hydrolysis that is catalyzed by a widely prevalent esterase enzyme (Fig. 1C).
Results and discussions
Docking studies, synthesis, enzymology, detection of persulfide and H2S
Molecular docking studies of the crystal structure of the human homolog of 3-MST (h3-MST, PDB ID: 4JGT) with the natural substrate 3-MP were conducted (Fig. 2A). The most energetically favorable conformation (−4.2 kcal mol−1) revealed interactions with arginine residues R188 and R197 that are consistent with previous accounts of these residues aiding in anchoring the substrate carboxylate and carbonyl groups.23,24 The cysteine C248 residue was proximal to the reactive sulfhydryl group of 3-MP at a distance of 4.4 Å.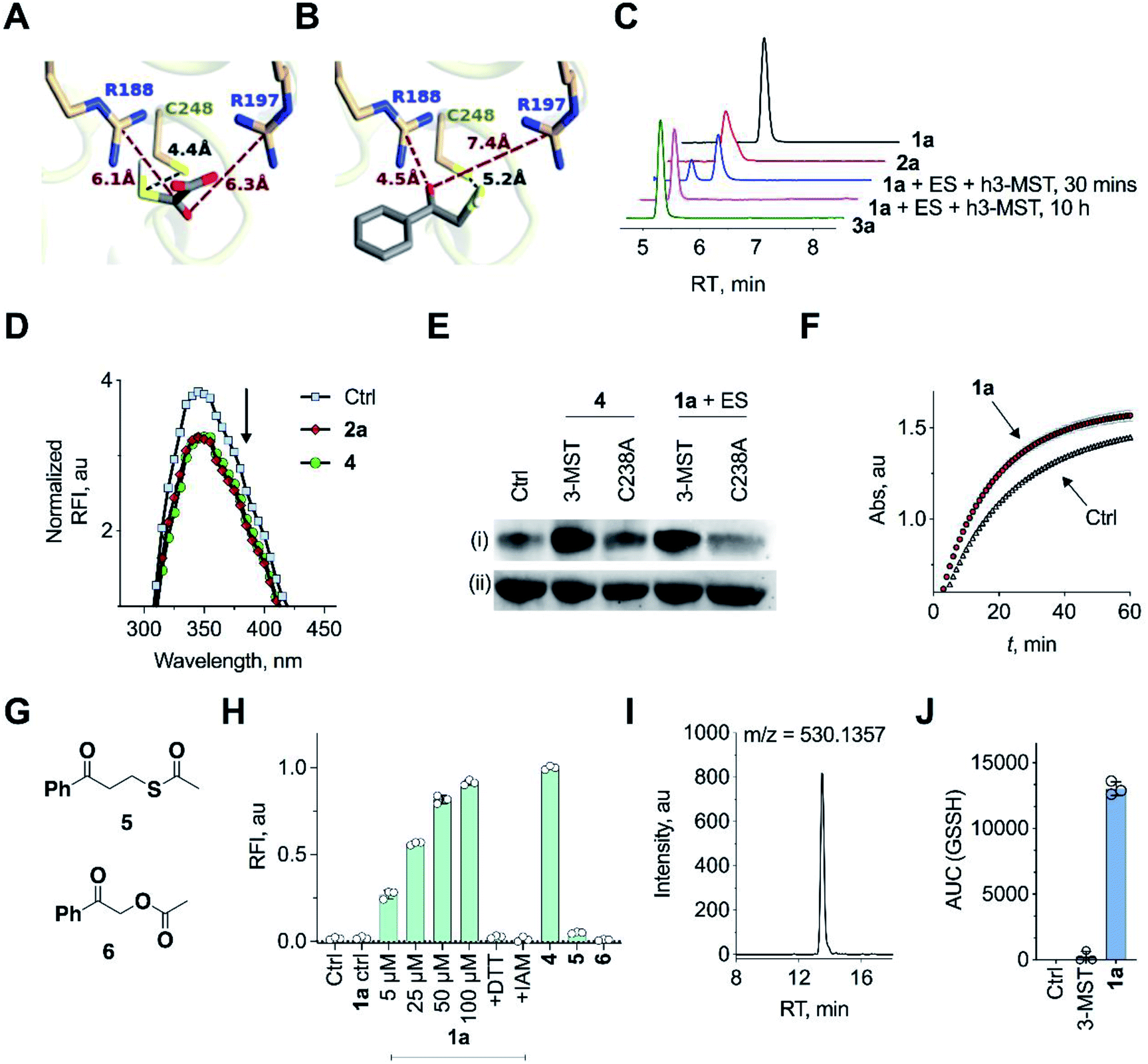 | ||
| Fig. 2 (A) Docking analysis of the active site of h3-MST with 3-MP shows a favorable conformation with the S–S bond distance as 4.4 Å. The R188 residue is 6.1 Å from the carboxyl group whereas the R197 residue is at a distance of 6.3 Å from the carbonyl group. (B) Docking analysis of the designed substrate 2a (R1 = Ph, R2 = H in Fig. 1C) reveals a similar anchoring of the substrate by the two arginine residues and the S–S bond distance was found as 5.2 Å. (C) HPLC analysis of 1a + ES in the presence of h3-MST shows the formation of the thiol 2a which is then subsequently converted to acetophenone 3a during 10 h. (D) Intrinsic fluorescence assay on 3-MST with dimer of ethyl 3-mercaptopyruvate (E3-MP) 4 shows a decrease in fluorescence intensity which is consistent with the generation of 3-MST-SS−. A similar result was observed with the unnatural substrate 2a. Ctrl refers to 3-MST alone. (E) Detection of 3-MST-SS− using the modified tag-switch technique (Fig. S7a†), conducted with 4 and 1a + ES; The C238A 3-MST mutant treated under similar conditions showed a diminished band corresponding to the formation of 3-MST-SS−: (i) detection of 3-MST persulfide (ii) loading control. (F) Effect of persulfidation on the activity of GAPDH: GAPDH upon treatment with 1a + ES + 3-MST enhances its activity compared to GAPDH alone presumably due to the formation of the persulfide of GAPDH. Ctrl refers GAPDH + ES + 3-MST; 1a refers to co-incubation of 1a + ES + 3-MST followed by addition of GAPDH (absorbance 340 nm). (G) Structures of compounds 5 and 6. (H) Persulfide/polysulfide detection using SSP-2: Ctrl refers to 3-MST alone and 1a ctrl refers to 1a + ES only (100 μM); 1a refers to co-incubation of varying concentrations of 1a, ES and 3-MST; +DTT: addition of DTT; +IAM: addition of iodoacetamide, an electrophile that reacts with thiols; 4 refers to incubation of the compound with 3-MST; 5 and 6 refers to the incubation of the compounds with ES followed by treatment with 3-MST. (I) Extracted ion chromatogram from a mass spectrometry-based analysis of reaction products formed upon incubation of 1a and 3-MST in the presence of ES followed by addition of GSH as the thiol acceptor. Reaction of the reactive sulfur species (GSSH) with an electrophile monobromobimane (mBBr) was employed. LC/MS analysis revealed the formation of the GSS-bimane adduct (expected m/z = 530.1379; observed m/z = 530.1357) when 1a was incubated with ES and 3-MST. (J) Area under the curve (AUC) for the peak corresponding to GSS-bimane (Fig. 2I); Ctrl refers to 1a alone while 3-MST refers to 3-MST alone and 1a refers to 1a + ES + 3-MST. | ||
When a similar study was conducted with the proposed aryl substrate, this compound 2a (R1 = Ph, R2 = H in Fig. 1C) was well accommodated in the active site and key interactions of 2a with R188 and R197 were preserved (Fig. 2B). The distance between the C248 and the sulfur of 2a was 5.2 Å, which was comparable with the lowest energy conformation of 3-MP in the active site of 3-MST. The binding energy of 2a was nearly identical with 3-MP, −4.2 kcal mol−1.
We cloned and purified h3-MST and a bacterial 3-MST (b3-MST) from Escherichia coli (Table S3, Fig. S1†) for subsequent in vitro assays.23,25 Compound 1a (Fig. 1C) was synthesized by the treatment of 2-bromoacetophenone with potassium thioacetate using a reported protocol.26 This compound should produce the thiol 2a upon reaction with an esterase enzyme (ES). Compound 1a was incubated in the presence of ES and h3-MST enzymes. Gradual disappearance of 1a and concomitant formation of the thiol 2a was observed (Fig. 2C and S2†). After 10 hours, complete disappearance of 2a along with the formation of acetophenone 3a was recorded. These observations suggest that after 1a produces 2a upon ester hydrolysis, it is utilized by 3-MST to produce 3a. Under the same conditions, b3-MST also shows complete conversion to 3a in 2 h (Fig. S3†).
The next series of experiments were designed to probe the intermediates during this transformation. The first intermediate that is expected to be formed in this reaction is 3-MST-SS−. The formation of 3-MST-SS− was monitored by measurement of the intrinsic fluorescence of this enzyme using a reported protocol.27 Upon incubation of 3-MST with 2a, a significant quenching of intrinsic fluorescence of the enzyme as compared to the apo-protein was observed suggesting the formation of 3-MST-SS− (Fig. 2D and S4†).
Compound 4, which exists as a dimer and dissociates in buffer to produce the ethyl 3-mercaptopyruvate, E3-MP, was synthesized and used as a substrate for 3-MST (Scheme 1).21 Molecular docking analysis gave a binding energy for this ester as −4.2 kcal mol−1 (Table S4†), identical to that of 3-MP. When 3-MST was incubated with 4, quenching of fluorescence was observed as before supporting the generation of the protein persulfide (Fig. 2D and S4†). A similar set of results was also recorded for b3-MST (Fig. S5†). The compounds themselves did not show significant fluorescence under these conditions (Fig. S6†).
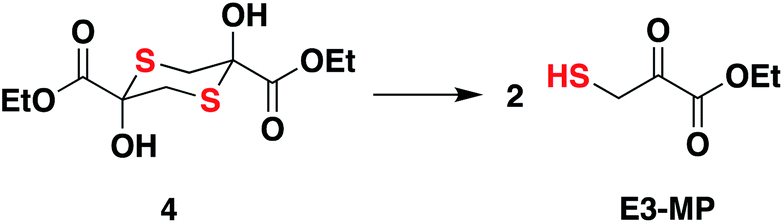 | ||
| Scheme 1 Compound 4 is the dimer of E3-MP and in pH 7.4 buffer, dissociates to produce the E3-MP. | ||
A reported tag-switch assay (Fig. S7†) was used for detecting persulfidated proteins (Fig. 2E).28 When 3-MST was reacted with 4 or with 1a and ES, as expected, this assay confirmed the formation of 3-MST-SS− (Fig. 2E). In contrast, under similar conditions, the mutant of 3-MST where the catalytic cysteine was replaced with alanine (3-MST C238A) showed no significant increase in the signal corresponding to the 3-MST-SS− (Fig. 2E and S7†). This result is consistent with a previous report of a similar lack of activity of the h3-MST cysteine mutant.29
Next, we assayed the ability of the 3-MST-SS− to transfer the sulfide to another protein. GAPDH is a redox-sensitive protein involved in the glycolytic cycle whose active site cysteine residue is susceptible to redox fluctuations and its function is influenced by this modification.30 Accordingly, 3-MST-SS− was prepared by treating 3-MST with 1a in presence of ES, following which GAPDH was added and incubated for 30 min. The activity of the enzyme was then estimated using a previously reported protocol.9 Under these conditions, the GAPDH activity was found to be significantly enhanced as compared to a GAPDH only control (Fig. 2F, S8a). Treatment with dithiothreitol (DTT), which is expected to cleave the persulfide and produce the native enzyme, resulted in an activity profile that was comparable with the control GAPDH (Fig. S8b†). Thus, 3-MST-SS−, through sulfur trafficking, regulates the activity of key enzymes such as GAPDH.
The formation of persulfide/polysulfide was then assessed using SSP-2, a fluorescence turn-on probe that is sensitive to these species.31 The probe was validated using GSnSH (prepared using a reported protocol) (Fig. S9†).10 When exposed to 1a + ES, a distinct increase in fluorescence was recorded (Fig. 2H and S10a†). When the standard reaction mixture was treated with the reducing agent DTT, a decrease in signal, likely due to the cleavage of persulfide by DTT, was observed (Fig. 2H and S10b†). Pre-treatment with iodoacetamide (IAM), a thiol alkylating agent, is expected to inactivate the enzyme. When IAM-pre-treated 3-MST was reacted with 1a + ES, nearly complete abrogation of signal attributable to persulfide/polysulfide generation was observed (Fig. 2H). This result is consistent with the previous observation in the tag-switch persulfide assay where we found no evidence for persulfide generation in the catalytically dead 3-MST C238A mutant. Notably, compound 4 generated persulfide at levels comparable with 1a (Fig. S11†). Next, two control compounds – 5, the thioacetate with an additional methylene group and 6, the analogous ester where sulfur was replaced with oxygen – were prepared (Fig. 2G). Both these analogues should not produce persulfide under the standard assay conditions. Generation of persulfide was assessed by the tag-switch assay (Fig. S12†) as well as by the fluorescence turn-on assay and no evidence for generation of persulfide from 5 or 6 was found (Fig. 2H).
The formation of persulfide/polysulfide was next studied by leveraging the ability of 3-MST-SS− to transfer its sulfane sulfur to an acceptor thiol. GSH, a thiol found abundantly in biological systems, was reacted with the 3-MST-SS− formed from the reaction of 1a + ES with 3-MST. The formation of various reactive sulfur species was analysed using LC/MS, where monobromobimane was used as an alkylating agent (Scheme S4†).10 LC/MS analysis showed the formation of the GSS-bimane adduct only in the presence of 1a + ES and 3-MST (Fig. 2I, J and S13†). In addition, we found evidence for the formation of GSSSG and H2S as its bis-S-bimane adduct (Fig. S14 and S15†). Together, these data support the ability of our unnatural substrate–enzyme system to transfer S to low molecular weight thiols.
A standard methylene blue colorimetric assay for the detection of H2S was conducted in the presence of DTT (Fig. 1B).23,32 This assay revealed the formation of H2S during incubation of 1a with 3-MST in the presence of DTT (Fig. 3A). H2S release from compound 4 when treated with 3-MST was also observed (Fig. S16†). Based on our observation that the thioacetate 1a could be cleaved by DTT even in the absence of the esterase to produce 2a, the remaining experiments were conducted with 1a + 3-MST + DTT (Fig. S17†). The rate constant kh for H2S generation from 1a when treated with h3-MST in the presence of DTT was 1.48 h−1 (Table 1). A similar rate constant kb was observed with b3-MST (Table 1).
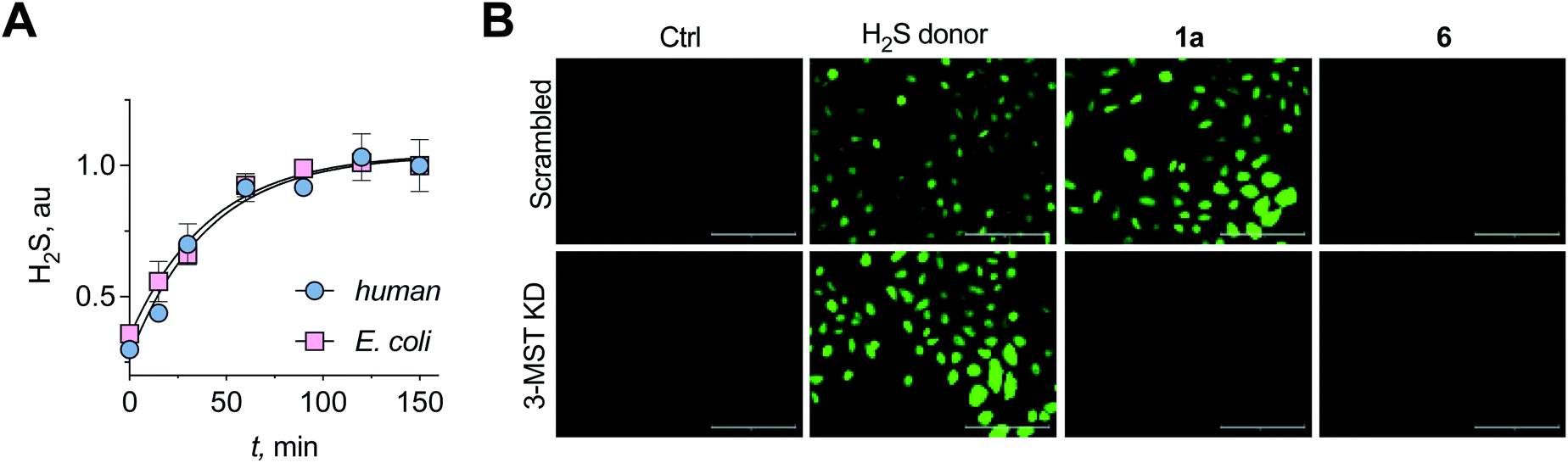 | ||
| Fig. 3 (A) A methylene blue assay was used to measure the rate of H2S generation with 1a in the presence of h3-MST or b3-MST and DTT as the reducing agent. Nearly identical rates of H2S generation were observed. (B) 3-MST KD refers to knock-down of expression of 3-MST in A549 cells while scrambled refers to A549 cells containing non-targeting scrambled shRNA. The H2S donor used is an esterase-sensitive COS/H2S donor that has been previously characterized. H2S levels were assessed using a previously reported dye NBD-fluorescein (see ESI, Fig. S20†). Ctrl refers to untreated cells. Scale bar represents 200 μm. | ||
|
|
||||||
|---|---|---|---|---|---|---|
| Compd | X | Y | h3-MST | b3-MST | k b/kh | |
| k h (h−1) | Rel. rateb | k b (h−1) | ||||
| a Reaction conditions: compound + 3-MST + DTT. H2S was monitored using a methylene blue assay. b Calculated based on normalizing the rate constant with respect to 1k. | ||||||
| 1a | H | H | 1.48 | 30 | 1.45 | 1.0 |
| 1b | NO2 | H | 11.13 | 223 | 9.27 | 0.8 |
| 1c | CN | H | 2.5 | 50 | 4.9 | 2.0 |
| 1d | CF3 | H | 7.51 | 150 | 5.83 | 0.8 |
| 1e | OCF3 | H | 4.48 | 90 | 2.29 | 0.5 |
| 1f | F | H | 1.11 | 22 | 1.54 | 1.4 |
| 1g | Me | H | 1.02 | 20 | 0.97 | 1.0 |
| 1h | OMe | H | 0.39 | 8 | 0.37 | 0.9 |
| 1i | H | F | 1.68 | 34 | 3.85 | 2.5 |
| 1j | H | OMe | 0.08 | 2 | 0.99 | 12.5 |
| 1k | H | Me | 0.05 | 1 | 1.22 | 25.0 |
| 1l | R1 = Ph; R2 = Me | Slow | — | 0.19 | — | |
| 1m | R1 = 1-naphthyl; R2 = H | 0.23 | 5 | 2.87 | 12.5 | |
| 1n | R1 = 2-naphthyl; R2 = H | 0.50 | 10 | 1.98 | 4.0 | |

Having established that the substrates we designed could be used to generate persulfides as well as H2S in vitro, their ability to permeate cells to function as substrates for cellular 3-MST was studied. A human lung carcinoma A549 cell line with 3-MST knocked down (3-MST KD) was first generated (Fig. S18†). This cell line was used in conjunction with the corresponding scrambled cell line where 3-MST levels are not perturbed (Fig. S18†). When treated with an esterase-activated H2S donor32 (Fig. S19†), significant enhancement in H2S levels in both these cell lines was observed as measured by the H2S-sensitive dye NBD-fluorescein (Fig. 3B).33 However, in the presence of 1a, when the 3-MST KD cells were imaged, a significantly diminished signal for intracellular H2S levels was observed (Fig. 3B). In the scrambled cell line, however, where endogenous 3-MST levels are not perturbed, an enhanced signal corresponding to intracellular H2S was seen (Fig. 3B). Compound 6, which lacks a sulfur, failed to enhance H2S in both cell lines (Fig. 3B, S20 and S21†). Together, these data suggest that 1a is a cell-permeable persulfidating agent that is activated by 3-MST within cells to produce H2S, putatively via an intracellular persulfidation mechanism, as confirmed by a separate tag-switch fluorescence reporter technique34 (Fig. S22†).
Based on the above observations, we put forth the following mechanism for persulfide generation from this unnatural substrate – 3-MST system (Scheme 2). Cleavage of 1a by esterase produces the thiol 2a, which then reacts with 3-MST to produce the 3-MST-SS− and an enol(ate), which is expected to tautomerize to the ketone 3a. In the absence of a reducing agent, 3-MST-SS− appears to catalyze the turnover of 2a to produce 3a and produces polysulfide during this transformation (Schemes 2 and S5; Fig. S15†). Under reducing conditions (DTT), 3-MST-SS− is cleaved to produce H2S (Scheme 2, Fig. 3A). HPLC analysis of the reaction mixture containing 2a, 3-MST and DTT showed gradual disappearance of 2a and the rate constant k1 was found to be 0.99 h−1. The formation of 3a was observed and a rate constant k2 of 0.75 h−1 was obtained (Fig. 4A and S23†). The rate of disappearance of 2a and formation of 3a was nearly identical and both parameters are comparable with the rate of H2S generation under these conditions (Table 1, see h3-MST data). In the absence of 3-MST, compound 2a is prone to oxidation to its disulfide PhCOCH2S–SCH2COPh under ambient aerobic conditions (Fig. S24†). The conversion of 1a to 2a (and its disulfide) occurs in a nearly quantitative yield. Hence, our overall analysis demonstrates that compound 1a is an excellent substrate for 3-MST and produces a persulfide intermediate, which is cleaved under reducing conditions to produce H2S. In the absence of esterase, 1a is gradually consumed in the presence of 3-MST and the formation of 3a is observed after several hours (Fig. S25 and S26†). Also, persulfide/polysulfide were detected using the probe SSP-2 under similar conditions (Fig. S27†). We find no evidence for the formation of the thiol 2a. This may be explained by the formation of 3-MST thioacetate as a possible intermediate during the transformation of 1a to 3a (Scheme S5†).
 | ||
| Scheme 2 Proposed mechanism: thioacetate 1 is cleaved by esterase or DTT (HPLC in Fig. S23, ESI†) to produce the thiol 2, which is then turned over by 3-MST to produce 3-MST-SS− and an enol(ate). The enol(ate) in aqueous buffer is rapidly converted to the ketone 3. 3-MST-SS− in the presence of a reducing agent produces H2S. 3-MST-SS− can react with low molecular weight thiols such as GSH to produce GSSH. Under non-reducing conditions, 3-MST-SS− can further turn over 2, generating the ketone 3; the likely byproduct of this reaction is polysulfide. | ||
Substrate scope
In order to assess the unnatural substrate scope and the possibility of tuning sulfur transfer using this newly developed protocol, we synthesized a series of analogues. Arginine residues in the active site interact with the carbonyl group of 3-MP and are implicated in stabilizing the pyruvate enolate.23,24,27 In order to test if the stability of the incipient enolate played a role in the rates of H2S production, compounds 1b–1h with electron-withdrawing or electron-donating groups were synthesized. Molecular docking analysis with the corresponding thiols 2b–2h showed nearly identical conformations (Table S5†). The rate constants of H2S release ranged from 0.39 to 7.51 h−1 (Table 1, Fig. S28†). Hammett analysis of rate constants gave a positive ρ value of +1.11, that is consistent with a partial negative charge developing in the transition state in the rate limiting step of the reaction (Fig. 4B).Due to the proximity of a substituent at the ortho position of the aryl ring, enolate formation is likely to be affected by stereoelectronic effects. In order to investigate these effects, compounds 1i–1k were prepared and their H2S release was recorded (Table 1, Fig. S29†). While the rate of H2S release from 1i was comparable with 1a, compounds 1j and 1k were substantially slower in generating H2S when compared with 1a. Molecular docking studies with the 2-fluorophenyl derivative 2i showed a conformation and S–S distance comparable with that of 2a bound to the active site (Table S6†). However, the lowest energy conformation of the thiol 2k was significantly removed from the active site with the S–S bond distance of 10.7 Å (Fig. 4C). Unlike 2a, the interaction of 2k with the active site cysteine is restricted by steric clashes of A185 and R188 with its ortho-Me group, as illustrated in a higher energy conformation (Fig. 4D). A similar result was recorded for the 2-methoxy derivative 2j (Tables S6 and S7†). These studies provide a molecular basis for the diminished H2S release rates from these analogues.
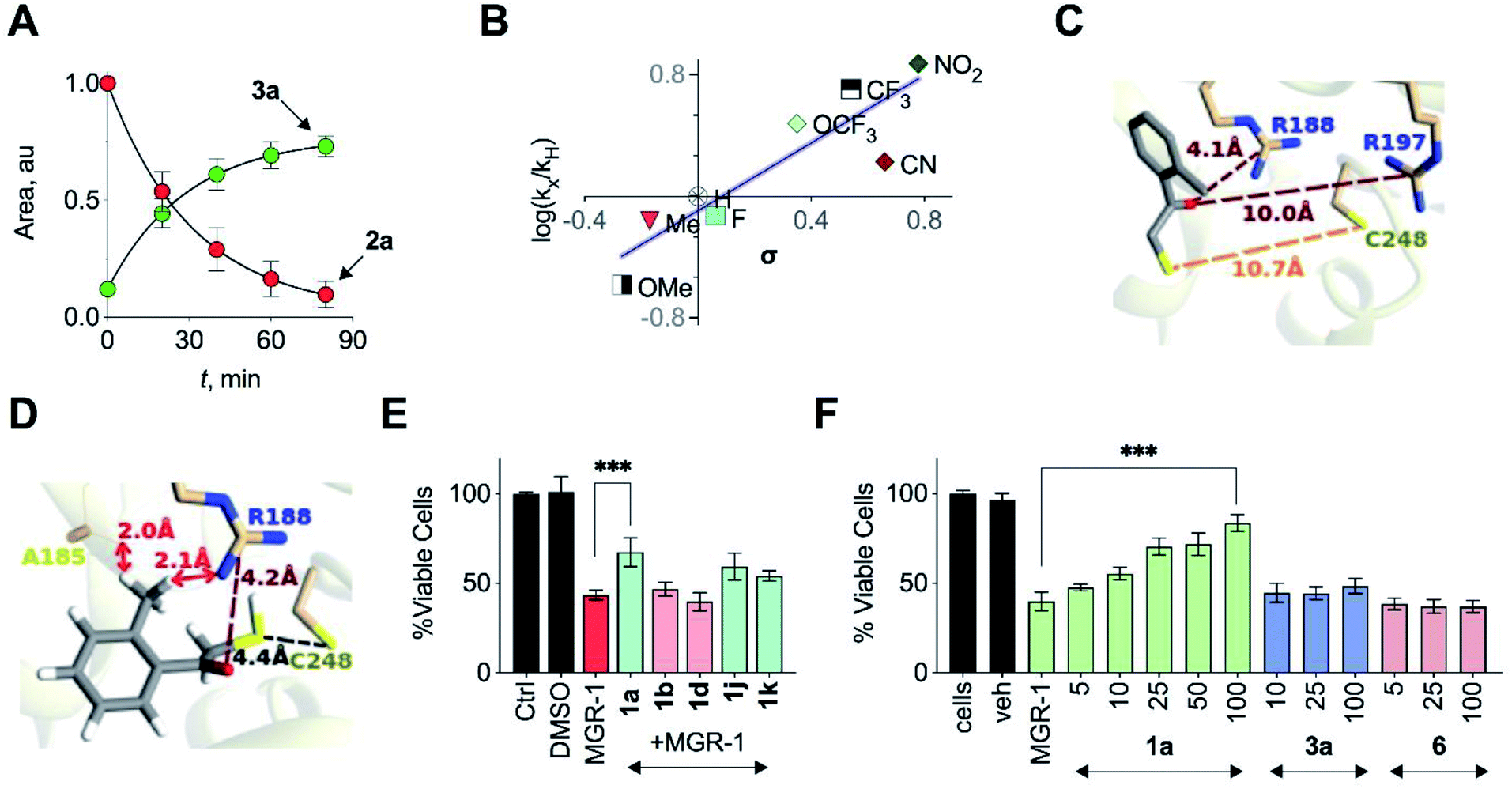 | ||
| Fig. 4 (A) HPLC analysis of a reaction mixture containing 2a and 3-MST in the presence of DTT showed the gradual disappearance of 2a and concomitant formation of 3a. Curve fitting to first order kinetics gave rate constants of 0.99 h−1 and 0.75 h−1 for the disappearance of 2a and the formation of 3a, respectively. (B) Hammett analysis of rate constants of H2S generation from unnatural substrates (see Table 1) with wt h3-MST. Linear regression analysis yielded a slope of +1.11 (R2 = 0.8). (C) Docking of 2k in the active site of h3-MST shows S–S bond distance of 10.7 Å which is substantially larger when compared with the low energy conformation of 2a (5.2 Å). (D) Docking of 2k in the active site of h3-MST shows steric clashes of ortho-Me group of 2k with A185 and R188 residues in a higher energy conformation. (E) Cell viability assay conducted on N2a cells: Cells were pre-treated with 25 μM of compounds 1a, 1b, 1d, 1j and 1k for 12 h and then exposed to MGR-1 (25 μM) for 4 h. Ctrl refers to untreated cells. Cell viability was determined using a standard MTT assay. Results are expressed as mean ± SD (n = 3). ***p < 0.001 vs. MGR-1. (F) Cell viability assay conducted on N2a cells: cells that were pre-treated with 1a, 3a or 6 were then treated with a cell permeable ROS generator MGR-1. A dose-dependent protection of cells from MGR-1 induced cell death by 1a was observed. The byproduct ketone 3a or the negative control 6 did not show any protection against the cytotoxic effects of MGR-1. All data are presented as mean ± SD (n = 3 per group). ***p < 0.001 vs. MGR-1. | ||
In addition to electronics, enolate stability and reactivity can also be affected by sterics. In order to study the effects of an added substituent, compound 1l which has an α-methyl substituent was synthesized. Docking analysis of 2l with 3-MST revealed a docking score as well as a conformation that was comparable with 2a (Table S6†). H2S release from 1l was, however, found to be extremely slow (Table 1, Fig. S30†). The formation of the enolate appears to be more sensitive to steric effects than electronic effects. Recently, inhibitors for 3-MST were developed and one of these inhibitors had a naphthyl ring.35 Taking this cue, compounds 1m and 1n were synthesized (Table S2†). Our assays confirmed that these compounds were substrates for 3-MST, and H2S generation rates were moderately higher when compared with 1a (Table 1, Fig. S30†) suggesting that a combination of binding and turnover is essential for tuning 3-MST-mediated sulfur transfer reactions.
Normalizing the rate constants with respect to 1k revealed more than a 100-fold difference in relative rates for H2S release (Table 1). This observation suggests that this series of substrates can be used to tune rates of persulfide/polysulfide generation. Next, bacterial 3-MST was used to study the rates of H2S generation from the analogues and first-order rate constants were calculated (Table 1). Linear free energy relationship study of H2S generation rates of 1a–1h with b3-MST gave a slope of +1.13 (Fig. S31†), comparable with the data from h3-MST. Control compounds 5 and 6 do not produce H2S under these conditions, as expected (Fig. S32†). Under the standard reaction conditions with 3-MST homologs, we found no major difference in rates of H2S release from 4 (Fig. S16†). While a majority of the substrates tested showed similar rates, compounds 1j, 1k and 1m whose kb/kh > 10, were notable exceptions. These results indicate that our approach differentiates between human and bacterial 3-MST homologs, and is capable of achieving selective H2S generation between species. To the best of our knowledge, this is the first example of tuning persulfide/polysulfide generation, with the added advantage of species selectivity.
Antioxidant activity
Persulfides are widely prevalent in cells as protein persulfides as well as other low molecular weight persulfide species. For example, glutathione persulfide/polysulfide has excellent in vitro ROS-scavenging activity when compared with glutathione or H2S alone.10 These species have been previously shown to have potent antioxidant activity likely through the Nrf2-KEAP1 pathway, imparting protection from ROS-induced injury.36,37 Selected analogues with varying relative rates of H2S generation (in the range of 1 to 223) were next tested to study their protective ability against oxidative stress-induced cell death. A mouse neuroblastoma cell line N2a was used in this experiment. MGR-1, a known cell-permeable ROS generator, was used to induce oxidative stress and cell viability was determined.38 N2a cells were first independently treated with fast H2S generators 1b and 1d (relative rate > 50), 1a (relative rate = 30) as well as slow generators 1j and 1k (relative rate < 2). At 25 μM, these compounds showed no significant effect on the growth of cells as determined by a cell viability assay (Fig. S33†). Cells that were independently pre-incubated with these compounds (25 μM) for 12 h were next exposed to MGR-1. Viable cells were determined using a standard cell viability assay. The results of this assay indicate that the fast generators did not significantly protect cells from oxidative stress-induced cell death while the other analogues 1a, 1j and 1k showed remarkable protective effects (Fig. 4E). From this initial screen, we identified compound 1a for further evaluation. The compound 1a itself was well tolerated by cells (Fig. S34†) and the byproduct of turnover of 1a is 3a, which is classified as Generally Recognized as Safe (GRAS) by the Food and Drug Administration.The lead compound 1a was next tested against N2a cells using MGR-1, and a dose-dependent protection from lethality was recorded (Fig. 4F). The control compound 6 and the acetophenone 3a tested under similar conditions failed to show any protective effects (Fig. 4F). To further corroborate our results, menadione, another known ROS generator was used to induce lethality. Again, a dose-dependent protection was recorded when cells were pre-treated with 1a (Fig. S35†). In a separate experiment with mouse embryonic fibroblasts (MEF), 1a was similarly found to have significant protective effects (Fig. S36†). Together, these data illustrate the cytoprotective effect of the cell-permeable persulfide generator 1a.
To validate the molecular basis of this result, the effect of 1a in reducing hydrogen peroxide (H2O2) levels in cells was studied. Again, MGR-1 was used to increase H2O2 as determined by the TCF-B fluorescent dye in A549 cells (Fig. 5A).39 When cells pre-treated with 1a were exposed to MGR-1, a significant decrease in H2O2 levels was observed (Fig. 5A). Under similar conditions, control compound 6 did not affect H2O2 levels in cells, suggesting that the ROS-quenching effects were directly mediated by persulfide/H2S generated by 1a (Fig. S37†).
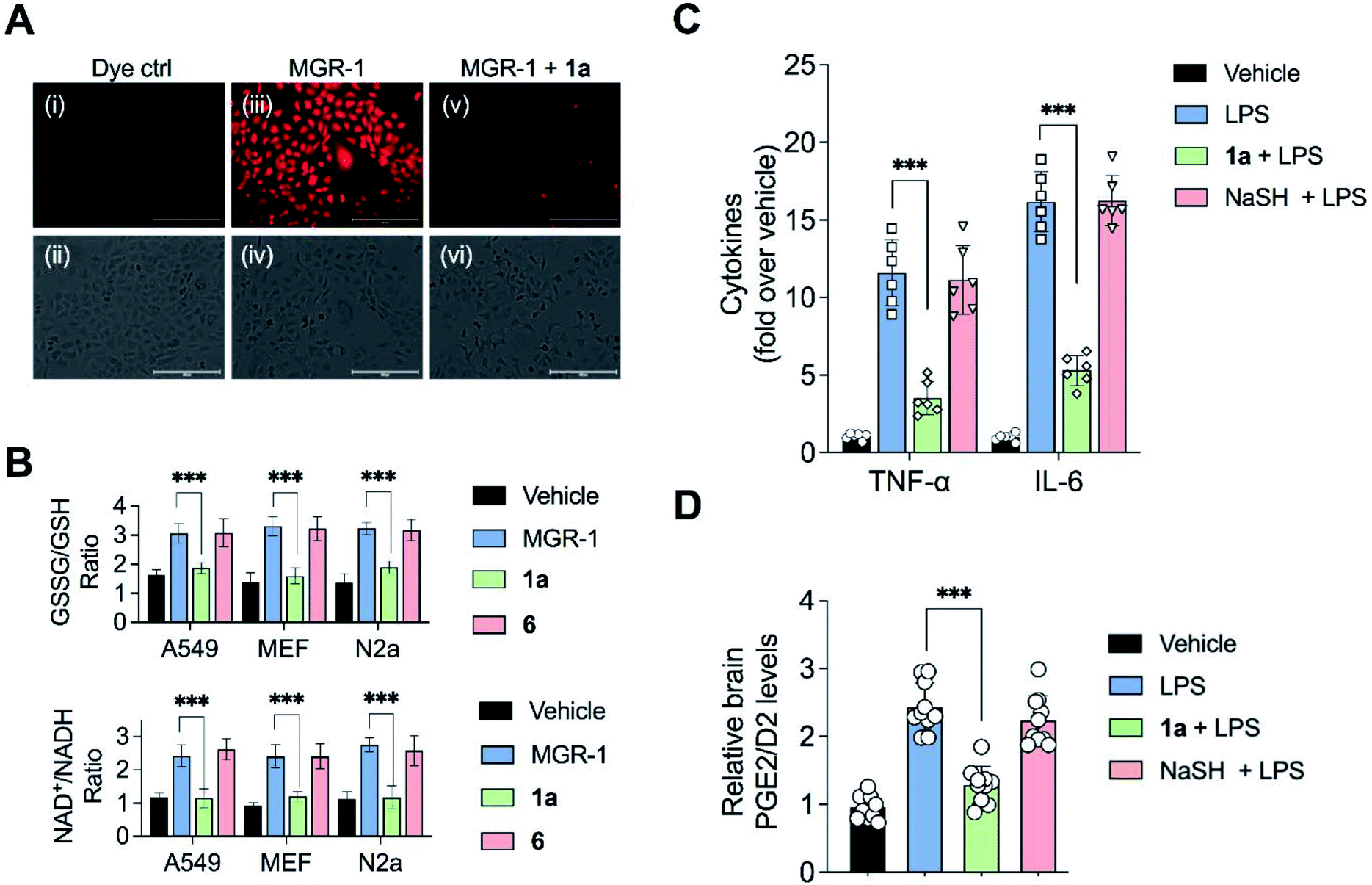 | ||
| Fig. 5 (A) Effects of 1a on quenching hydrogen peroxide (H2O2) generated by MGR1:A549 cells treated with: (i and ii) dye control; (iii and iv) 25 μM MGR-1; (v and vi) 25 μM of 1a for 12 h followed by addition of the 25 μM MGR-1 for 1 h. Intracellular H2O2 was detected using a reported H2O2-sensor TCF-B (25 μM). Scale bar represents 200 μm. (B) Biomarkers for oxidative stress: Three cell lines (A549, mouse embryonic fibroblasts (MEF) and N2a) were pre-treated with vehicle, 1a or 6 and exposed to MGR-1 following which NAD+/NADH ratio and GSSG/GSH ratio were determined using an ELISA assay. All data are presented as mean ± SD (n = 5 per group). ***p < 0.001 vs. MGR-1. (C and D) Mouse endotoxin shock model: animals were treated with 1a (20 mg kg−1) or NaSH (20 mg kg−1) 4 h prior to treatment with lipopolysaccharide (LPS, 5 mg kg−1). 30 min post-administration of LPS, another dose of 1a or NaSH was given. The brain tissue samples were harvested followed by measurement of: (C) Pro-inflammatory cytokines, TNF-α and IL-6 using a standard ELISA assay. All data are presented as mean ± SD (n = 6 per group). ***p < 0.001 vs. LPS; and (D) prostaglandins PGE2/D2 using LC/MS. All data are presented as mean ± SD (n = 10 per group). ***p < 0.001 vs. LPS. | ||
Elevation of ROS in cells leads to perturbation of redox homeostasis and alteration of levels of various components of cellular antioxidant response. NAD+/NADH and GSSG/GSH ratio serve as reliable markers of oxidative stress.40 Three cell lines (A549, MEF and N2a) were exposed to MGR-1 and enhanced NAD+/NADH as well as GSSG/GSH ratios were recorded (Fig. 5B). Pre-treatment of cells with 1a followed by incubation with MGR-1 showed restoration of nearly normal ratios, and the control compound 6 was found to be ineffective in doing so. Taken together, these studies conclusively establish the efficacy of the artificial substrate 1a that we designed to mitigate oxidative stress in both human as well as mouse-derived cell lines (Fig. 5B).35
Mitigation of neuroinflammation
Recently, a polysulfide donor was used to study the effect of enhanced endogenous polysulfide levels on innate immune response using a mouse endotoxin shock model.41 Lipopolysaccharides (LPS), a common constituent of bacterial cell walls, are recognized by toll-like receptor 4 (TLR4) and trigger the innate immune response leading to the activation of macrophages to generate pro-inflammatory cytokines that contribute to clearance of bacteria (Fig. S38†). The major finding of this study was that elevated polysulfides can negatively regulate TLR4-mediated pro-inflammatory signaling and the polysulfide donor protects against this endotoxin shock. Taking this cue, we evaluated the ability of 1a to mitigate neuroinflammation in a similar mouse model system. First, C57BL/6J mice were intraperitoneally injected once daily with compound 1a over a period of seven days, and it was found to be well tolerated. Next, mice were treated with LPS alone and increased levels of pro-inflammatory cytokines TNF-α and IL-6 in the brain were observed as compared with vehicle-treated mice (Fig. 5C).In a separate experiment, mice which were treated with LPS and 1a were found to have significantly lower levels of TNF-α and IL-6 in the brain, suggesting that 1a suppressed the inflammatory response triggered by LPS. No evidence for reduction of pro-inflammatory cytokines in mice treated with LPS and NaHS was observed, consistent with the previous report that NaHS had little or no effect on the reduction of cytokines.41 LPS is also known to induce the formation of pro-inflammatory prostaglandins in the brain. Prostaglandin E2 (PGE2) has a variety of functions in the nervous system. Interaction of PGE2 with prostaglandin EP3 receptors leads to an increase in body temperature and inflammation (Fig. S39†). The use of nonsteroidal anti-inflammatory drugs (NSAIDs) blocks the activity of cyclooxygenase-2 (COX-2) which decreases PGE2 production, resulting in remediation of fever and inflammation.42 Hence, measurement of PGE2/D2 levels is a reliable inflammation biomarker. LC/MS analysis of brain homogenates was carried out to measure PGE2/D2 levels. LPS-treated mice were found to have significantly higher levels of PGE2/D2 when compared with vehicle-treated mice. Animals which were exposed to both LPS and 1a were found to have significantly lower levels of PGE2/D2, reiterating the ability of 1a to reduce neuroinflammation (Fig. 5D). Again, NaSH-treated mice that were exposed to LPS did not show any effect on PGE2/D2 levels. Taken together, these assays conclusively demonstrate the ability of 1a to act as an antioxidant as well as an anti-inflammatory agent.
Our data shows that the unnatural substrate is able to transfer sulfur to low molecular weight thiols via 3-MST, and induce protein persulfidation in cells. Other strategies for generating persulfides in cells have relied on installing the persulfide/polysulfide functional group in the donor.5,6,41,43–53 These strategies work very well but have limitations in the ease of synthesis, poor shelf-life due to susceptibility towards decomposition, and heterogeneity of the persulfide source due to the ambiguity in the number of sulfurs in the donor. The protocol developed here involves the synthesis of a thioacetate, which is easy to prepare with high reproducibility and is stable for extended periods. For in vitro experiments to generate persulfide, the addition of enzymes that are readily available (esterase and 3-MST) is necessary while for cellular experiments to enhance persulfide, addition of the compound is sufficient. The substrates we have developed allow for systematic study of cellular sulfur transfer, and can be used to investigate the persulfide proteome which has been elusive till date.11
Conclusions
The study of reactive sulfur species is often complicated by poor detection techniques, artefacts and uncharacterized cellular interactions. Thus, several facets of sulfur metabolism remain to be studied in molecular-level detail. The use of selective tools developed herein allow for systematic study of redox biology through the lens of an important enzyme, 3-MST. Our data underscores the potential of this approach as a novel therapeutic paradigm in mitigating inflammation. The importance of persulfides in ameliorating symptoms associated with neurodegenerative disorders as well as favourably impacting behavior in animals are some of the possible applications of the strategy developed here.4 The tunability of rates of persulfide generation using the class of compounds that we have developed adds a new dimension to persulfide donors that has hitherto not been studied. Projecting forward, further structural optimization of the substrate would be necessary to fully exploit the translational potential of this approach. The deficits associated with diminished expression of 3-MST will likely need mapping out the 3-MST-SS− proteome and identification of proteins that depend on this enzyme for persulfidation. Given the selectivity of the substrate towards 3-MST in cells, the use of our compounds as probes for studying sulfur trafficking will provide insights into molecular mechanisms associated with the dysfunction or deficiency of 3-MST in neuronal diseases, such as intellectual disability and Down's syndrome.54,55Data availability
Experimental protocols, characterization and all data pertaining to the manuscript have been uploaded in the ESI.†Author contributions
ABH and HC wrote the manuscript with inputs from all authors. PB, SM, MN, RRMS, SS, VSSA and KG carried out the experiments and computation under the supervision of AM, DKS, SSK, ABH and HC.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the SERB, DST (CRG/2019/002900) and IISER Pune is acknowledged. This work was supported by the DBT-Ramalingaswami Re-entry Fellowship (grant number BT/RLF/Re-entry/12/2014) awarded to ABH. DBT/Wellcome Trust India Alliance Fellowship (grant number IA/I/15/2/502058) awarded to SSK. DST Fund for Improvement of S&T Infrastructure (SR/FST/LSII-043/2016) to the IISER Pune Biology Department for setting up the Biological Mass Spectrometry Facility. The National Facility for Gene Function in Health and Disease at IISER Pune is thanked for maintaining and providing mice for this study (DBT: BT/INF/22/SP17358/2016). Research fellowship for PB (DST-INSPIRE), SM (UGC) and RRMS (CSIR) are acknowledged. The authors thank Dr Rajesh Viswanathan, IISER Tirupati and Dr Sridhar Rajaram, JNCASR Bengaluru for their critical reading of the manuscript.Notes and references
- M. R. Filipovic, J. Zivanovic, B. Alvarez and R. Banerjee, Chem. Rev., 2018, 118, 1253–1337 CrossRef CAS PubMed.
- T. V. Mishanina, M. Libiad and R. Banerjee, Nat. Chem. Biol., 2015, 11, 457 CrossRef CAS PubMed.
- M. S. Vandiver, B. D. Paul, R. Xu, S. Karuppagounder, F. Rao, A. M. Snowman, H. Seok Ko, Y. Il Lee, V. L. Dawson, T. M. Dawson, N. Sen and S. H. Snyder, Nat. Commun., 2013, 4, 1626 CrossRef PubMed.
- D. Giovinazzo, B. Bursac, J. I. Sbodio, S. Nalluru, T. Vignane, A. M. Snowman, L. M. Albacarys, T. W. Sedlak, R. Torregrossa, M. Whiteman, M. R. Filipovic, S. H. Snyder and B. D. Paul, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2017225118 CrossRef CAS PubMed.
- Y. Zheng, B. Yu, Z. Li, Z. Yuan, C. L. Organ, R. K. Trivedi, S. Wang, D. J. Lefer and B. Wang, Angew. Chem., Int. Ed., 2017, 56, 11749–11753 CrossRef CAS PubMed.
- V. S. Khodade, B. M. Pharoah, N. Paolocci and J. P. Toscano, J. Am. Chem. Soc., 2020, 142, 4309–4316 CrossRef CAS PubMed.
- K. Shatalin, E. Shatalina, A. Mironov and E. Nudler, Science, 2011, 334, 986–990 CrossRef CAS PubMed.
- P. Shukla, V. S. Khodade, M. SharathChandra, P. Chauhan, S. Mishra, S. Siddaramappa, B. E. Pradeep, A. Singh and H. Chakrapani, Chem. Sci., 2017, 8, 4967–4972 RSC.
- A. K. Mustafa, M. M. Gadalla, N. Sen, S. Kim, W. Mu, S. K. Gazi, R. K. Barrow, G. Yang, R. Wang and S. H. Snyder, Sci. Signaling, 2009, 2, ra72 Search PubMed.
- T. Ida, T. Sawa, H. Ihara, Y. Tsuchiya, Y. Watanabe, Y. Kumagai, M. Suematsu, H. Motohashi, S. Fujii, T. Matsunaga, M. Yamamoto, K. Ono, N. O. Devarie-Baez, M. Xian, J. M. Fukuto and T. Akaike, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 7606–7611 CrossRef CAS PubMed.
- J. Zivanovic, E. Kouroussis, J. B. Kohl, B. Adhikari, B. Bursac, S. Schott-Roux, D. Petrovic, J. L. Miljkovic, D. Thomas-Lopez, Y. Jung, M. Miler, S. Mitchell, V. Milosevic, J. E. Gomes, M. Benhar, B. Gonzales-Zorn, I. Ivanovic-Burmazovic, R. Torregrossa, J. R. Mitchell, M. Whiteman, G. Schwarz, S. H. Snyder, B. D. Paul, K. S. Carroll and M. R. Filipovic, Cell Metab., 2019, 30, 1152–1170 CrossRef CAS PubMed.
- N. Nagahara and A. Katayama, J. Biol. Chem., 2005, 280, 34569–34576 CrossRef CAS PubMed.
- N. Nagahara, Br. J. Pharmacol., 2018, 175, 577–589 CrossRef CAS PubMed.
- Y. Kimura, S. Koike, N. Shibuya, D. Lefer, Y. Ogasawara and H. Kimura, Sci. Rep., 2017, 7, 10459 CrossRef PubMed.
- N. Nagahara and T. Nishino, J. Biol. Chem., 1996, 271, 27395–27401 CrossRef CAS PubMed.
- M. Libiad, P. K. Yadav, V. Vitvitsky, M. Martinov and R. Banerjee, J. Biol. Chem., 2014, 289, 30901–30910 CrossRef CAS PubMed.
- K. Módis, C. Coletta, K. Erdélyi, A. Papapetropoulos and C. Szabo, FASEB J., 2013, 27, 601–611 CrossRef PubMed.
- M. Li, C. Xu, J. Shi, J. Ding, X. Wan, D. Chen, J. Gao, C. Li, J. Zhang, Y. Lin, Z. Tu, X. Kong, Y. Li and C. Yu, Gut, 2018, 67, 2169–2180 CrossRef CAS PubMed.
- P. M. Palenchar, C. J. Buck, H. Cheng, T. J. Larson and E. G. Mueller, J. Biol. Chem., 2000, 275, 8283–8286 CrossRef CAS PubMed.
- E. G. Mueller, Nat. Chem. Biol., 2006, 2, 185 CrossRef CAS PubMed.
- H. T. Nagasawa, D. J. W. Goon, D. L. Crankshaw, R. Vince and S. E. Patterson, J. Med. Chem., 2007, 50, 6462–6464 CrossRef CAS PubMed.
- C. Coletta, K. Módis, B. Szczesny, A. Brunyánszki, G. Oláh, E. C. S. Rios, K. Yanagi, A. Ahmad, A. Papapetropoulos and C. Szabo, Mol. Med., 2015, 21, 1–14 CAS.
- P. K. Yadav, K. Yamada, T. Chiku, M. Koutmos and R. Banerjee, J. Biol. Chem., 2013, 288, 20002–20013 CrossRef CAS PubMed.
- G.-T. Huang and J.-S. K. Yu, J. Phys. Chem. B, 2016, 120, 4608–4615 CrossRef CAS PubMed.
- F. van den Ent and J. Löwe, J. Biochem. Biophys. Methods, 2006, 67, 67–74 CrossRef CAS PubMed.
- A. A. Heredia, S. M. Soria-Castro, L. M. Bouchet, G. Oksdath-Mansilla, C. A. Barrionuevo, D. A. Caminos, F. R. Bisogno, J. E. Argüello and A. B. Peñéñory, Org. Biomol. Chem., 2014, 12, 6516–6526 RSC.
- J.-C. Lec, S. Boutserin, H. Mazon, G. Mulliert, S. Boschi-Muller and F. Talfournier, ACS Catal., 2018, 8, 2049–2059 CrossRef CAS.
- D. Zhang, I. Macinkovic, N. O. Devarie-Baez, J. Pan, C.-M. Park, K. S. Carroll, M. R. Filipovic and M. Xian, Angew. Chem., Int. Ed., 2014, 53, 575–581 CrossRef CAS PubMed.
- N. Shibuya, M. Tanaka, M. Yoshida, Y. Ogasawara, T. Togawa, K. Ishii and H. Kimura, Antioxid. Redox Signaling, 2009, 11, 703–714 CrossRef CAS PubMed.
- C. Tristan, N. Shahani, T. W. Sedlak and A. Sawa, Cell. Signalling, 2011, 23, 317–323 CrossRef CAS PubMed.
- W. Chen, C. Liu, B. Peng, Y. Zhao, A. Pacheco and M. Xian, Chem. Sci., 2013, 4, 2892 RSC.
- P. Chauhan, P. Bora, G. Ravikumar, S. Jos and H. Chakrapani, Org. Lett., 2017, 19, 62–65 CrossRef CAS PubMed.
- C. Wei, Q. Zhu, W. Liu, W. Chen, Z. Xi and L. Yi, Org. Biomol. Chem., 2014, 12, 479–485 RSC.
- R. Wedmann, C. Onderka, S. Wei, I. A. Szijártó, J. L. Miljkovic, A. Mitrovic, M. Lange, S. Savitsky, P. K. Yadav, R. Torregrossa, E. G. Harrer, T. Harrer, I. Ishii, M. Gollasch, M. E. Wood, E. Galardon, M. Xian, M. Whiteman, R. Banerjee and M. R. Filipovic, Chem. Sci., 2016, 7, 3414–3426 RSC.
- K. Hanaoka, K. Sasakura, Y. Suwanai, S. Toma-Fukai, K. Shimamoto, Y. Takano, N. Shibuya, T. Terai, T. Komatsu, T. Ueno, Y. Ogasawara, Y. Tsuchiya, Y. Watanabe, H. Kimura, C. Wang, M. Uchiyama, H. Kojima, T. Okabe, Y. Urano, T. Shimizu and T. Nagano, Sci. Rep., 2017, 7, 40227 CrossRef CAS PubMed.
- G. Yang, K. Zhao, Y. Ju, S. Mani, Q. Cao, S. Puukila, N. Khaper, L. Wu and R. Wang, Antioxid. Redox Signaling, 2012, 18, 1906–1919 CrossRef PubMed.
- S. Koike, Y. Ogasawara, N. Shibuya, H. Kimura and K. Ishii, FEBS Lett., 2013, 587, 3548–3555 CrossRef CAS PubMed.
- D. S. Kelkar, G. Ravikumar, N. Mehendale, S. Singh, A. Joshi, A. K. Sharma, A. Mhetre, A. Rajendran, H. Chakrapani and S. S. Kamat, Nat. Chem. Biol., 2019, 15, 169–178 CrossRef CAS PubMed.
- A. C. Sedgwick, H.-H. Han, J. E. Gardiner, S. D. Bull, X.-P. He and T. D. James, Chem. Commun., 2017, 53, 12822–12825 RSC.
- F. Q. Schafer and G. R. Buettner, Free Radical Biol. Med., 2001, 30, 1191–1212 CrossRef CAS PubMed.
- T. Zhang, K. Ono, H. Tsutsuki, H. Ihara, W. Islam, T. Akaike and T. Sawa, Cell Chem. Biol., 2019, 26, 686–698 CrossRef CAS PubMed.
- J. R. Vane, Nat. New Biol., 1971, 231, 232–235 CrossRef CAS PubMed.
- P. Bora, P. Chauhan, S. Manna and H. Chakrapani, Org. Lett., 2018, 20, 7916–7920 CrossRef CAS PubMed.
- A. Chaudhuri, Y. Venkatesh, J. Das, M. Gangopadhyay, T. K. Maiti and N. D. P. Singh, J. Org. Chem., 2019, 84, 11441–11449 CrossRef CAS PubMed.
- R. A. Hankins, S. I. Suarez, M. A. Kalk, N. M. Green, M. N. Harty and J. C. Lukesh, Angew. Chem., 2020, 132, 22422–22429 CrossRef.
- V. S. Khodade, S. C. Aggarwal, B. M. Pharoah, N. Paolocci and J. P. Toscano, Chem. Sci., 2021, 12, 8252–8259 RSC.
- B. Yu, Y. Zheng, Z. Yuan, S. Li, H. Zhu, L. K. De La Cruz, J. Zhang, K. Ji, S. Wang and B. Wang, J. Am. Chem. Soc., 2018, 140, 30–33 CrossRef CAS PubMed.
- Z. Yuan, Y. Zheng, B. Yu, S. Wang, X. Yang and B. Wang, Org. Lett., 2018, 20, 6364–6367 CrossRef CAS PubMed.
- J. Kang, S. Xu, M. N. Radford, W. Zhang, S. S. Kelly, J. J. Day and M. Xian, Angew. Chem., Int. Ed., 2018, 57, 5893–5897 CrossRef CAS PubMed.
- C. R. Powell, M. K. Dillon, Y. Wang, R. J. Carrazzone and J. B. Matson, Angew. Chem., Int. Ed., 2018, 57, 6324–6328 CrossRef CAS PubMed.
- K. M. Dillon, R. J. Carrazzone, Y. Wang, C. R. Powell and J. B. Matson, ACS Macro Lett., 2020, 9, 606–612 CrossRef CAS PubMed.
- Y. Wang, K. M. Dillon, Z. Li, E. W. Winckler and J. B. Matson, Angew. Chem., Int. Ed., 2020, 59, 16698–16704 CrossRef CAS PubMed.
- V. S. Khodade and J. P. Toscano, J. Am. Chem. Soc., 2018, 140, 17333–17337 CrossRef CAS PubMed.
- Y. Suwanai, N. Nagahara, Z. Naito and H. Orimo, Adv. Tech. Biol. Med., 2016, 4, 167 Search PubMed.
- T. Panagaki, E. B. Randi and C. Szabo, Biomolecules, 2020, 10, 653 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc03828a |
| ‡ These authors contributed equally. |
| § Presently at Department of Chemistry, The Pennsylvania State University, USA. |
| This journal is © The Royal Society of Chemistry 2021 |