
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHighly efficient on-DNA amide couplings promoted by micelle forming surfactants for the synthesis of DNA encoded libraries†
James H.
Hunter
a,
Matthew J.
Anderson
a,
Isaline F. S. F.
Castan
a,
Jessica S.
Graham
a,
Catherine L. A.
Salvini
a,
Harriet A.
Stanway-Gordon
a,
James J.
Crawford
 b,
Andrew
Madin
c,
Garry
Pairaudeau
d and
Michael J.
Waring
*a
b,
Andrew
Madin
c,
Garry
Pairaudeau
d and
Michael J.
Waring
*a
aCancer Research UK Newcastle Drug Discovery Unit, Chemistry, School of Natural and Environmental Sciences, Newcastle University, Bedson Building, Newcastle upon Tyne, NE1 7RU, UK. E-mail: mike.waring@ncl.ac.uk
bDepartment of Discovery Chemistry, Genentech Inc., 1 DNA Way, South San Francisco, CA 94080, USA
cHit Discovery, Discovery Sciences, R&D, AstraZeneca, Cambridge, CB4 0WG, UK
dExscientia, Schrödinger Building, Oxford Science Park, Oxford, OX4 4GE, UK
First published on 22nd June 2021
Abstract
DNA encoded libraries (DELs) represent powerful new technology for finding small molecule ligands for proteins and are increasingly being applied to hit finding in medicinal chemistry. Crucial to the synthesis of high quality DELs is the identification of chemical reactions for their assembly that proceed with very high conversion across a range of different substrates, under conditions compatible with DNA-tagged substrates. Many current chemistries used in DEL synthesis do not meet this requirement, resulting in libraries of low fidelity. Amide couplings are the most commonly used reaction in synthesis of screening libraries and also in DELs. The ability to carry out highly efficient, widely applicable amide couplings in DEL synthesis would therefore be highly desirable. We report a method for amide coupling using micelle forming surfactants, promoted by a modified linker, that is broadly applicable across a wide range of substrates. Most significantly, this works exceptionally well for coupling of DNA-conjugated carboxylic acids (N-to-C) with amines in solution, a procedure that is currently very inefficient. The optimisation of separate procedures for coupling of DNA-conjugated acids and amines by reagent screening and statistically driven optimisation is described. The generality of the method is illustrated by the application to a wide range of examples with unprecedented levels of conversion. The utility of the (N-to-C) coupling of DNA-conjugated acids in DEL synthesis is illustrated by the three cycle synthesis of a fully DNA-encoded compound by two cycles of coupling of an aminoester, with intermediate ester hydrolysis, followed by capping with an amine. This methodology will be of great utility in the synthesis of high fidelity DELs.
Introduction
DNA-encoded libraries (DELs) are an emerging technology for the discovery of chemical start points for drug discovery and for probe molecules in chemical biology.1–3 Typically, screening libraries are constructed from a DNA-conjugated monomer, often termed the “headpiece”, which is elaborated using multiple rounds of split-pool parallel synthesis. Each step is coupled with the appending of a DNA-codon, unique to each building block, such that every final compound in the library is conjugated to a unique DNA sequence, which serves as a barcode for the chemical structure.4 By carrying out multiple cycles of synthesis, with hundreds of parallel steps in each cycle, libraries of millions to billions of compounds can be prepared, something that is beyond the capacity of traditional chemical synthesis. These libraries can be screened against a protein target by affinity selection followed by PCR amplification and DNA sequencing to identify binders, which are then synthesised “off-DNA".5–10A significant limitation of the approach currently is the range and efficiency of the chemical reactions that can be carried out on DNA-tagged molecules, i.e. reaction media (traditionally aqueous) must be compatible with DNA and must not employ reagents that react with DNA.11 As a result, acids, strong bases, oxidants and reactive alkylating/acylating agents are typically excluded. Most reported methods for on-DNA synthesis, whilst having been proven to be effective, typically only proceed with moderate efficiency across a range of substrates. In most cases, the reaction efficiencies of individual transformations, although referred to as yields, actually correspond to percentage conversion from starting material to product, neglecting the common formation of side products. Even then, the level of conversion is typically moderate. Hence, there is a significant interest in new technological approaches for DEL synthesis.12–14
We have recently reported a procedure for the on-DNA Suzuki–Miyaura coupling, which is promoted by the vitamin E derived micelle-forming surfactant TPGS-750-M.15 The reaction proceeds with unprecedented levels of conversion (>98% on average across a range of substrates) with minimal side reactions. We hypothesised that the micellar surfactant would promote the reaction by solubilising the organic reactants and potentially protect the DNA by localising the reactants to the organic portion of the molecule. With the successful application of the micellar surfactants to Suzuki couplings, we were interested in exploring the application of micellar promoted on-DNA chemistry to other reaction types.
Amide coupling is the most commonly employed reaction in the synthesis of drug-like screening libraries16,17 due to the number and diversity of building blocks that are readily available, coupled with the desirable properties imparted by amide functionality in the products.
Amide bond formations have been reported under micellar conditions off-DNA.18 Amide couplings on-DNA have been reported but, at least in solution phase synthesis, exhibit the moderate conversions that are typical of standard DEL chemistry. In perhaps the most extensive study reported, it was shown that in the coupling of a primary amine-DNA conjugate with 543 carboxylic acids, a relatively small proportion met the moderate success criterion of >75% conversion (44% for DMTMM and 78% for EDC/HOAt).19 Moreover, the coupling is almost always reported with the amine component attached to the DNA (representing synthesis in the “C-to-N” direction for peptide couplings) with the acid monomers employed in large excess.10,20–24 Reports of couplings of DNA-conjugated carboxylates to amines building blocks (N-to-C) are severely limited25 and, in our hands, we have found extension to drug-like amine building blocks to be very challenging. A recent study showed that for a range of amines, on-DNA N-to-C amide couplings only gave >70% conversion in 30% of cases.26 Accordingly, we selected amide couplings between DNA-tagged acids and amine monomers as a highly desirable but suitably challenging application of micelle-promoted DEL synthesis.
Results and discussion
Initial investigations
Our initial investigations focused on carboxylic acid bearing headpiece 1 (Fig. 1), which was synthesised by coupling of bis-acetic acid substituted PEG-4 with a 5′-amino-substituted oligonucleotide. Coupling of 1 with four representative amines (glycine ethyl ester, aniline, 2-aminothiazole and 2-aminoimidazole) using HATU and DIPEA in 2% aqueous TPGS-750-M, resulted in no conversion to product for any of the amines.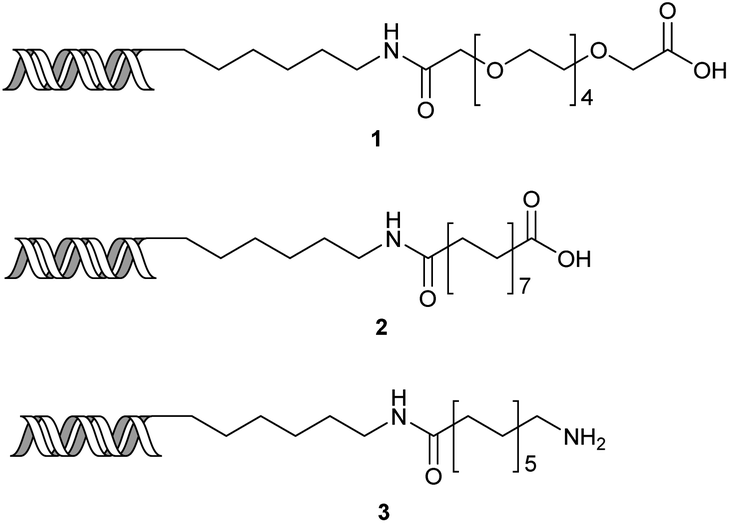 | ||
| Fig. 1 Structures of headpieces: carboxy-PEG4-hexylamido-DNA 1, carboxy-C14-hexylamidoDNA 2 and amino-C11-hexylamidoDNA 3. | ||
We considered the possibility that the PEG linker of 1 disfavoured association with the surfactant due to repulsion between the polar oxygen atoms of the linker and either the apolar head groups of the surfactant or its PEG side chain; and that a more lipophilic linker may promote reaction within the micelles. Accordingly, we synthesised the more hydrophobic hexadecanoic acid linked derivative 2 and investigated its coupling to glycine ethyl ester, aniline and 2-aminothiazole (Table 1). Coupling of glycine ethyl ester, aniline and 2-aminothiazole with 2 using HATU27 showed a clear improvement in reactivity, with formation of the desired amides in 57%, 15% and 45% respectively. In contrast, use of EDC/HOAt28 or DMT-MM,29 which are preferred reagents for amide couplings on-DNA,4,19,30 resulted in no conversion. With HATU, conversion could be improved by heating at 40 °C for glycine ethyl ester and 2-aminothiazole (90% conversion in both cases) but led to a slight reduction for aniline. At this stage, we also investigated the coupling of the less nucleophilic 2-aminoimidazole, which also resulted in 10% conversion.
|
|
||||||
|---|---|---|---|---|---|---|
| Coupling agent | Base | Temp/°C | % Product | |||
| a | b | c | d | |||
| HATU | DIPEA | 20 | 57 | 15 | 45 | — |
| EDC/HOAt | DIPEA | 20 | 0 | 0 | 0 | — |
| DMT-MM | DIPEA | 20 | 0 | 0 | 0 | — |
| HATU | DIPEA | 40 | 90 | 10 | 90 | 10 |
| HATU | Lutidine | 40 | 97 | 92 | 56a | 42b |
These results were encouraging, showing for the first time that a DNA-conjugated acid could be successfully coupled to amines under micellar conditions. For the procedure to be useful for the synthesis of drug-like DELs, it would ideally need give high conversion across a range of substrates, including electron deficient anilines and heteroarylamines.31 Hence, we investigated further optimisation of the reaction conditions. Screening of 12 different bases (Table S1†) with glycine ethyl ester and 2-aminoimidazole revealed that 2,6-lutidine was most effective, leading to 90% and 42% product respectively. Use of 2,6-lutidine also led to efficient coupling of aniline (92%) but was less effective for 2-aminothiazole under these conditions (Table 1).
The conditions were further optimised using factorial experimental design,15 exploring the effect of temperature (40–60 °C), surfactant (2–5%) and base concentration (0.5–1.5 M) for amines glycine ethyl ester and 2-aminoimidazole (Fig. 2). This revealed maxima within the specified ranges for temperature and surfactant strength but a suggestion that the reaction conversion continued to increase at base concentrations above 1.5 M. The design was augmented to extend the range of base concentration up to 2.5 M, resulting in a maximum within the experimental range. The experiments revealed interesting 2-dimensional relationships. For example, high conversion was favoured at low temperature with low base concentration or at high temperature and high base concentration (Fig. 2c). Significant reduction in reaction conversion could be observed within the explored parameters, in extreme cases resulting in no conversion at all (low base, low surfactant strength, high temperature), highlighting the value of using factorial design to simultaneously optimise multiple parameters for these types of reactions. The optimal conditions for glycine a were 45 °C, 3.5% TPGS and 1.5 M base, whereas 2-aminoimidazole d favoured higher temperature and base concentration with maxima at ∼55 °C and 2.0 M.
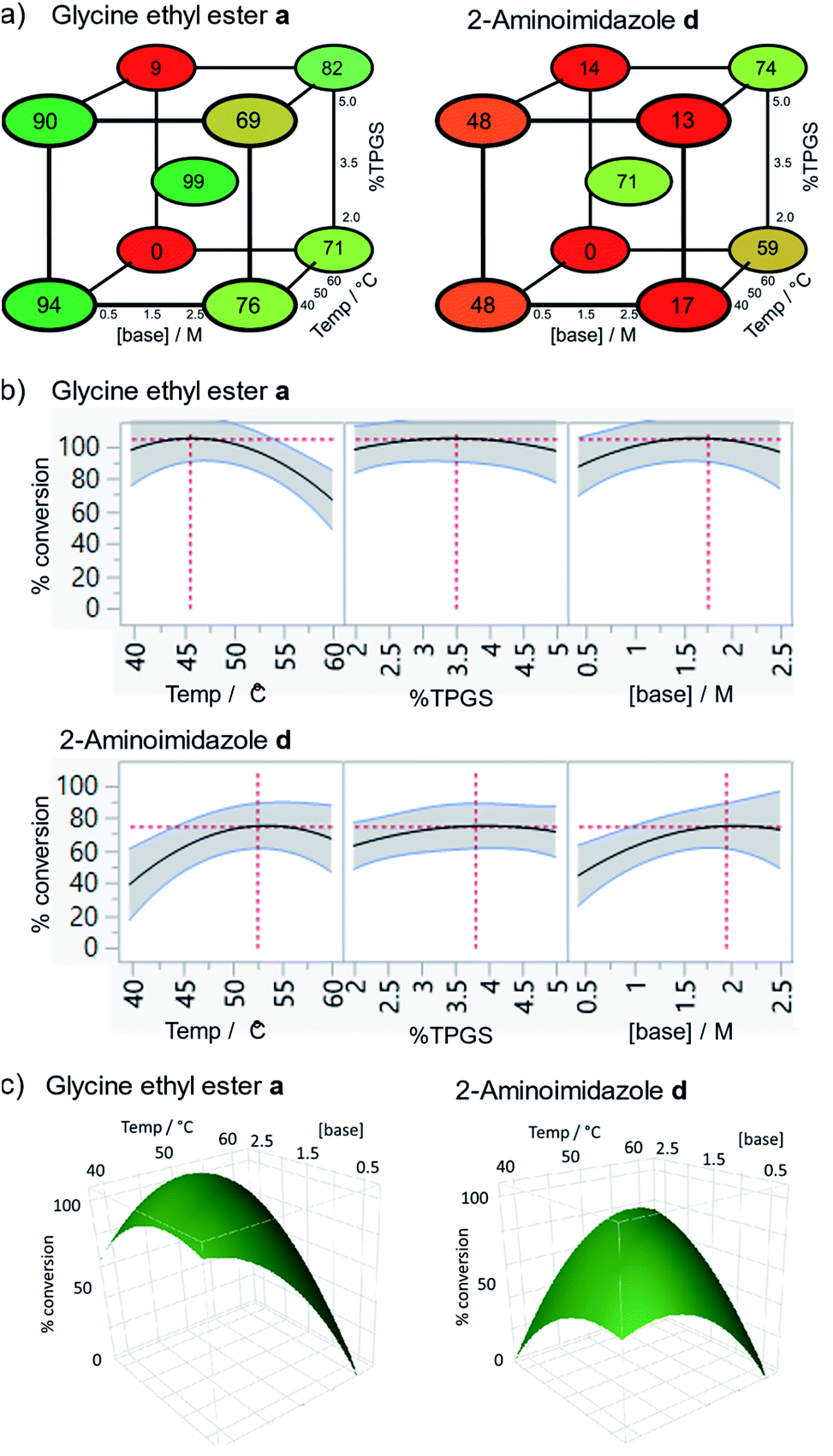 | ||
| Fig. 2 Optimisation of temperature, surfactant strength (% TPGS) and base concentration by factorial experimental design, conditions: 2 (10 nmol), 2,6-lutidine, HATU (0.5 M), 30 μl total volume, 16 h; (a) cube plots showing modelled conversions; (b) % conversion responses for each parameter; (c) response surfaces showing the 2-dimensional relationship between conversion and temperature/base concentration. For full results see Table S2.† | ||
To achieve the optimal conversion across a wider range of amines, conditions intermediate between the optima for the two amines (45 °C, 3.5% TPGS, 2 M base) were selected. The coupling conditions were tested across a range of amines of differing reactivity (Table S3†). The coupling of ester derivatives of canonical amino acids (glycine, alanine, D-phenylalanine, leucine and tyrosine) and cyclic amines (azetidine, morpholine and 4-piperidine acetic acid methyl ester) all coupled with >95% conversion. Tyrosine (entry 5) also coupled with 96% conversion without additional protection of the phenol. The conditions worked less well for benzylamines, anilines and heterocyclic amines (conversions ranging from 14 to 93%) with appreciable starting acid remaining and formation of the dimethylamide as a side product, which we postulate originates from the use of excess HATU.
Coupling of DNA conjugated amine to acids
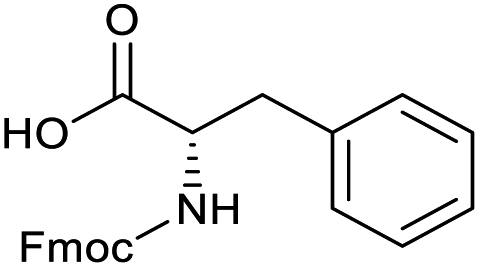
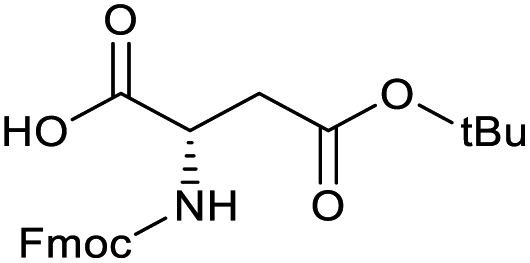
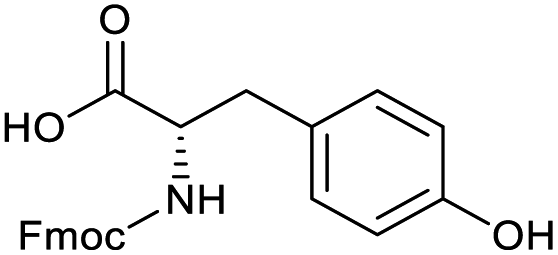
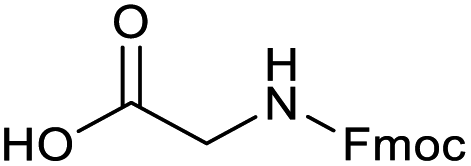
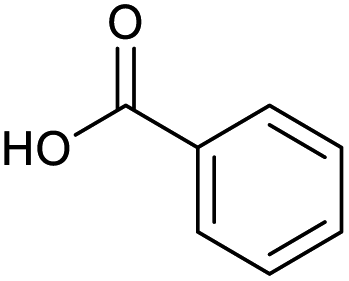
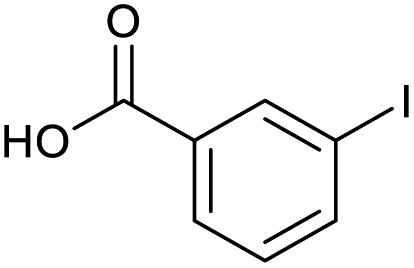
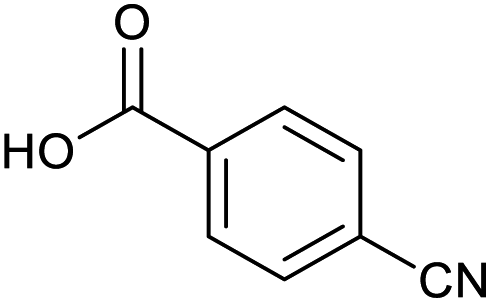
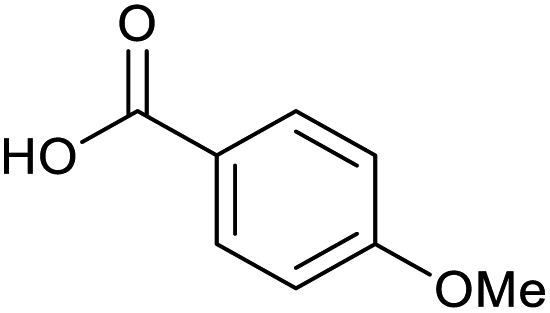
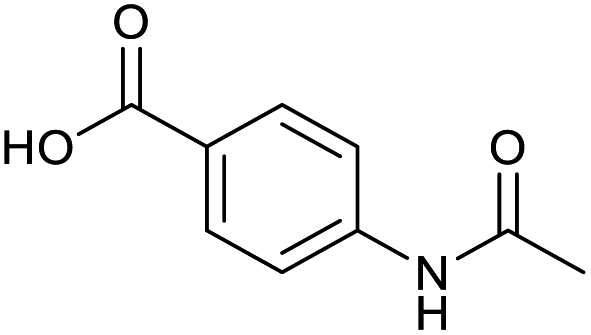
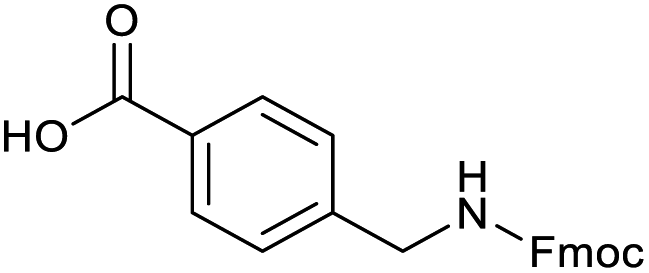
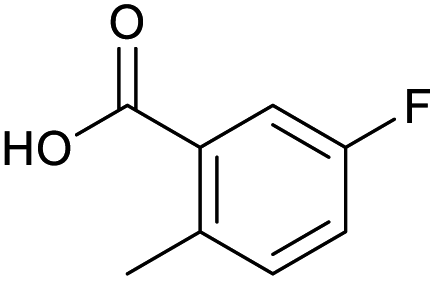
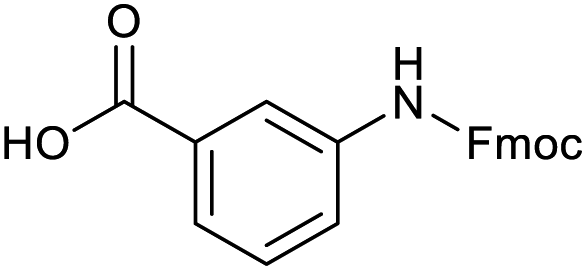
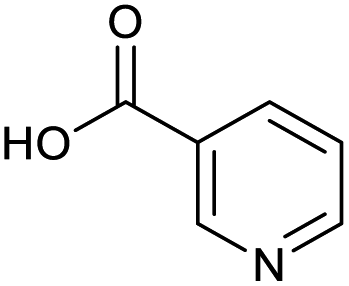
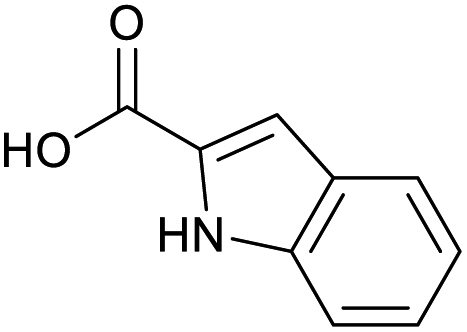
Applying the same conditions to the coupling reaction with the amine component on DNA (C-to-N) using the substrate amino-C14-hexylamidoDNA 3 proved far more general, leading to high conversion and product purity (>90% in all cases) (Table 2). Fmoc protected amino acids alanine, phenylalanine, t-butyl aspartate, tyrosine and sarcosine (entries 1–5) all coupled with 93–100% conversion. Benzoic acid and substituted derivatives 3-iodo-, 4-cyano, 4-methoxy, 4-acetamido-, 4-Fmoc-aminomethyl and 2-methyl-5-fluoro- (entries 6–12) all coupled with 100% conversion. 3-Fmoc-aminobenzoic acid coupled with 94% conversion (entry 13). Heterocyclic acids nicotinic acid and indole-2-carboxylic acid gave 100% conversion (entries 14 and 15).| Acids | % Conversion | % Product | |
|---|---|---|---|
| 1 |
|
100 | 100 |
| 2 |
|
100 | 98 |
| 3 |
|
97 | 94 |
| 4 |
|
93 | 93 |
| 5 |
|
100 | 100 |
| 6 |
|
100 | 98 |
| 7 |
|
100 | 97 |
| 8 |
|
100 | 98 |
| 9 |
|
100 | 97 |
| 10 |
|
100 | 98 |
| 11 |
|
100 | 89 |
| 12 |
|
100 | 100 |
| 13 |
|
94 | 94 |
| 14 |
|
100 | 95 |
| 15 |
|
100 | 100 |
The micelle promoted amide coupling performs exceptionally well for a wide range of relevant substrates and is insensitive to steric and electronic effects. This level of efficiency is at least as good as those reported previously as optimised conditions.19
Carrying out the same reactions on carboxy-PEG4-hexylamido-DNA performed less well. Fmoc protected alanine and phenyl alanine coupled with 86% and 67% conversion respectively, compared to 100% with the C11 linker 3 (Table S4†). Reaction with benzoic acid formed 14% of a side product (2% with 3), but 3-iodobenzoic acid and nicotinic acid still coupled well.
The examples studies here, which give >90% conversion in all cases, provide evidence that this method is advantageous over previous methods (cf. the published results that give >75% conversion in 78% of cases19). The comparison with the results obtained with the PEG-linked substrate demonstrate the enhanced level of reaction efficiency that is imparted by the use of the more hydrophobic linker.
Coupling of DNA conjugated acid to amines
Whilst these results suggested a highly efficient protocol for acylation of DNA-tagged amines, the outcome was considered inadequate for DNA-tagged carboxylates (N-to-C). Further coupling agents were screened in the coupling of 2 with 2-aminoimidazole and 2-aminopyridine, which had performed relatively poorly under the conditions tested previously (Table S5†).32 COMU promoted coupling, selected due to its effectiveness of off-DNA micellar amide coupling,18 failed to give the desired product; resulting only in unreacted 2 and formation of the morpholine amide. BOP and PYBOP also failed to give desired product with BOP forming appreciable amounts of the dimethylamide of 2. EDC coupling failed to give any product but more lipophilic carbodimides DCC33 and DIC,34 in the presence of HOAt did show product formation (DCC 23%, DIC 64% and DCC 57%, DIC 74% for 2-aminoimidazole and 2-aminopyridine respectively). This increase in reactivity was hypothesised to be due to increased partitioning of the less hydrophilic coupling agents and/or resulting active esters into the hydrophobic environment of the micelle. In the case of DIC, the level of conversion was slightly better than that observed for the optimised HATU conditions and so this coupling agent was selected for further optimisation.The DIC/HOAt mediated reaction was investigated using a further factorial experimental design, assessing the effect of temperature (40–60 °C), surfactant (2–5%) and base concentration (0.5–1.5 M) (Fig. 3). To determine the best conditions across the full substrate scope, these conditions were explored with both glycine ethyl ester and 2-aminoimidazole as used previously. The most significant influences on reaction conversion were observed for temperature, choice of the amine and a second order effect between temperature and base concentration (p values 0.00057, 0.00096, 0.0039 respectively). Parabolic effects on reaction conversion were seen for both temperature (peaks occurring at 45 and 52 °C respectively) and surfactant concentration (peaks at 4.5%). The effect of base concentration increased linearly across the design space. The sensitivity of the reaction to changes in parameters is striking, with some of the combination of conditions (high temperature and low base concentration) showing very little conversion for either amine, demonstrating the clear advantage of factorial experimental design to optimise these types of reactions. Because the temperature optima were different for the two amines, it was not possible to select a value for temperature that would be expected to be universal across the substrate scope. It was reasoned that in most cases, full conversion for the more reactive amines would be most desirable, meaning that a temperature of 45 °C along with 4.5% TPGS-750-M and 1.5 M lutidine represented the most universal conditions to employ in DEL synthesis and these were used to assess the substrate scope (see below).
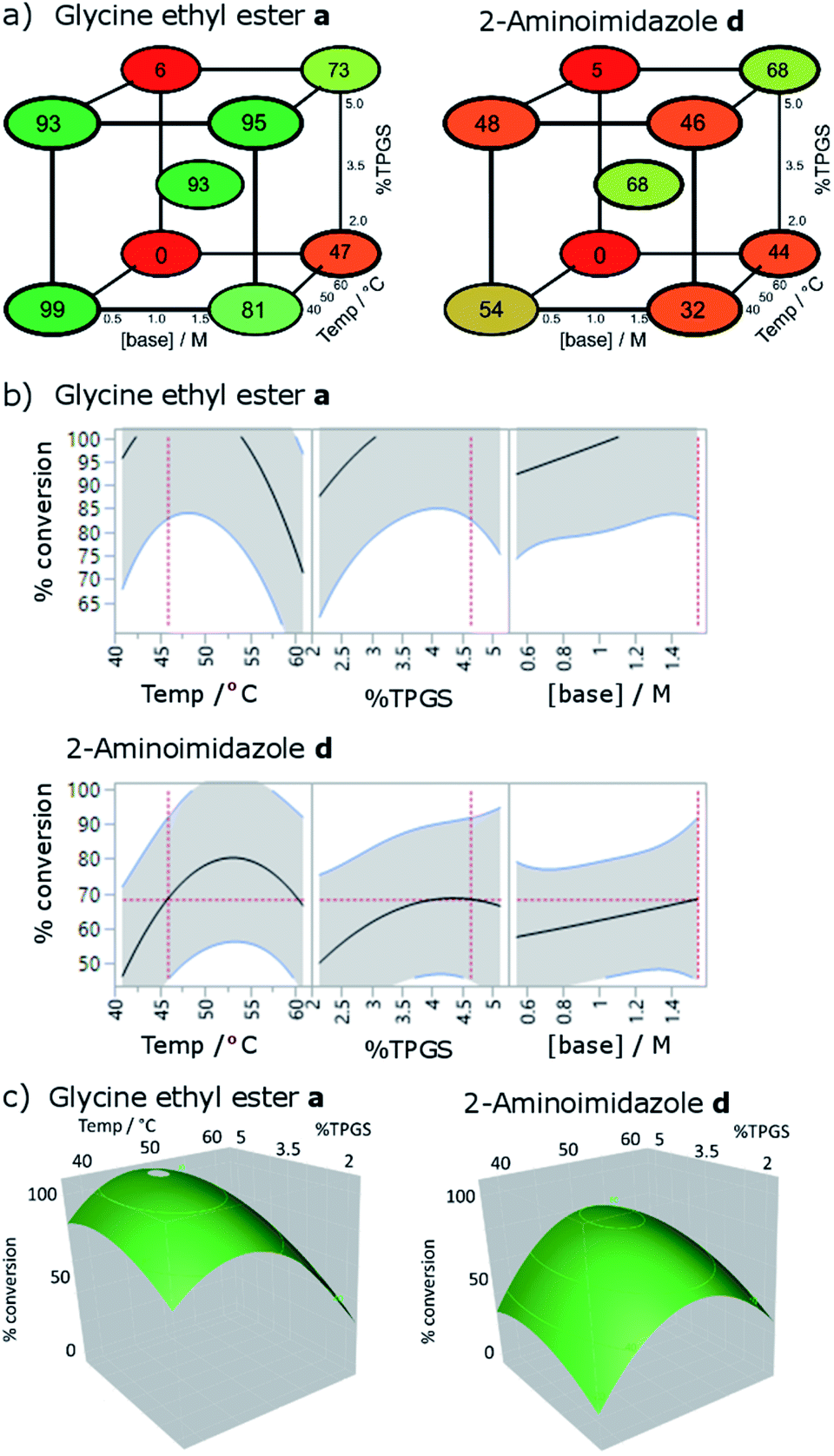 | ||
| Fig. 3 Optimisation of temperature, surfactant strength (% TPGS) and base concentration by factorial experimental design, conditions: 2 (10 nmol), amine (0.5 M), 2,6-lutidine, DIC (0.5 M), 30 μl total volume, 3 h; (a) cube plots showing modelled conversions; (b) % conversion responses for each parameter; (c) response surfaces showing the 2-dimensional effect of temperature and surfactant concentration on conversion. Data shown are fitted using a least squares model (r2 = 0.95, RMSE = 12). For full results see Table S6.† | ||
In all cases for which full conversion was not achieved, the only other DNA-containing species observed were the starting material and a side product of 13 Da higher mass than the starting acid, consistent with either the methyl amide or the acylnitroso derivatives of 2. No modification of the DNA strands was detected. Complete conversion to the same species was achieved by subjecting 2 to the reaction conditions in the absence of amine (Fig. S1†), suggesting that it arises from the coupling agent rather than the amine. We are not aware of previous reports of this side product in DIC mediated couplings. It is likely that it results from employing the reagents in large excess in aqueous solution during DEL synthesis either from decomposition or trace impurities (although further purification of the DIC did not alter the outcome).
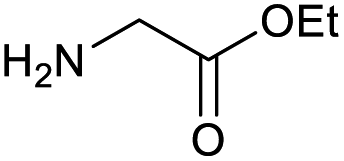
The optimised conditions performed exceptionally well across a wide range of amine substrates (Table 3). Amino acid esters glycine, alanine, D-phenyl alanine, valine, tyrosine, and asparagine coupled with 100% efficiency (entries 1–6). Primary and secondary aliphatic amines benzylamine, 4-chlorobenzylamine, cyclohexylmethylamine, cyclopropyl methylamine, azetidine, 1-methyl-3-aminoazetidine, piperidine, 4-dimethylaminopiperidine, N-cyclopropylamidopiperazine and aminoacetonitrile (entries 7–16) also showed 100% product. Aniline and p-fluoroaniline gave 100% product (entries 17–18); o- and p-anisidine and 3-chloroaniline gave 100% conversion, with the methylamide observed as the only other species (entries 19–21). Electron poor 5- and 6-membered heteroarylamines 2-aminoimidazole, 1-methyl-3-aminopyrazole and 2- and 3-aminopyridine performed similarly well (entries 22–25). Heteroarylmethylamines (2- ,3- and 4-pyridyl, pyrazinyl and pyrimidin-2-yl, entries 26–30) also gave >90% conversion and product. More complex heteroarylamines 6-aminoindazole, 2-methoxy-5-aminopyridine, methyl-5-aminonicotinate and methyl-6-aminopyridazine-3-carboxylate proceeded with 100% conversion (entries 31–34). The by-product described above was the only other detectable species. The most electron poor amines tended to be those that showed a larger amount of the by-product, presumably because the desired coupling is slow with these less reactive substrates.
| Amine | % Conversion | % Product | Amine | % Conversion | % Product | ||
|---|---|---|---|---|---|---|---|
| 1 |
|
100 | 100 | 18 |
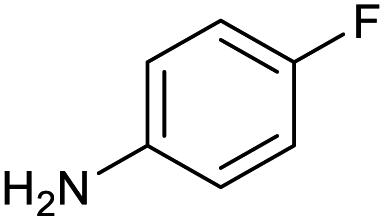
|
100 | 100 |
| 2 |
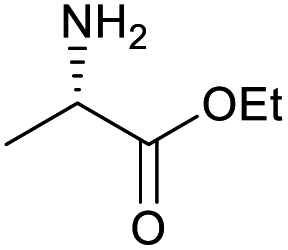
|
100 | 100 | 19 |
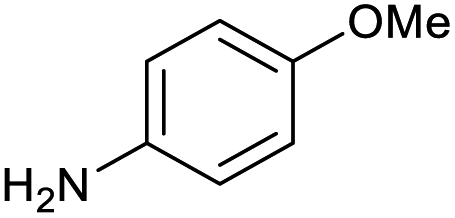
|
100 | 95 |
| 3 |
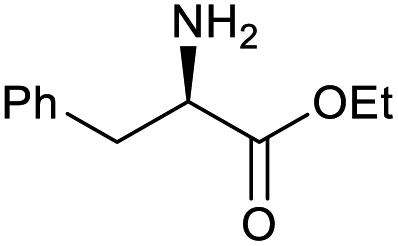
|
100 | 100 | 20 |
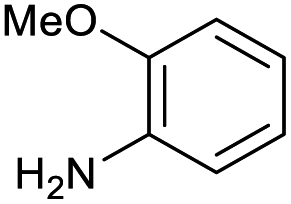
|
100 | 86 |
| 4 |
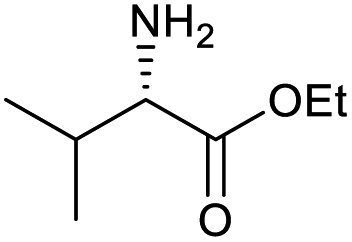
|
100 | 96 | 21 |
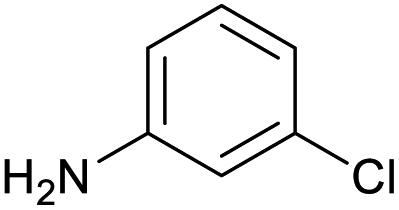
|
100 | 85 |
| 5 |
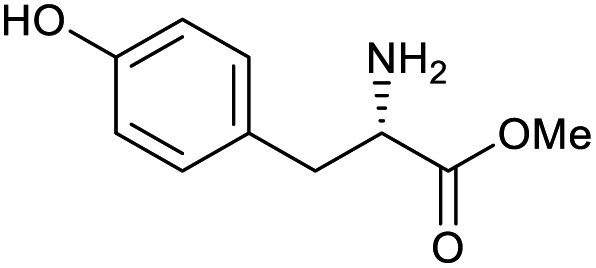
|
100 | 100 | 22 |
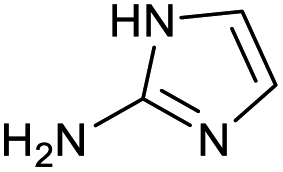
|
100 | 58 |
| 6 |
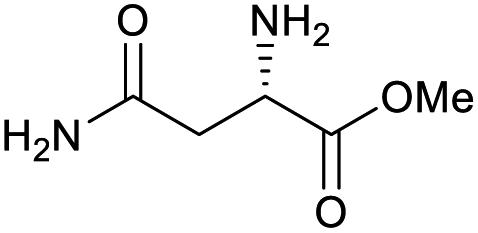
|
100 | 98 | 23 |
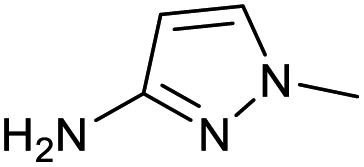
|
98 | 84 |
| 7 |
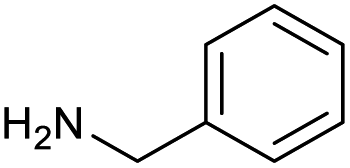
|
100 | 100 | 24 |
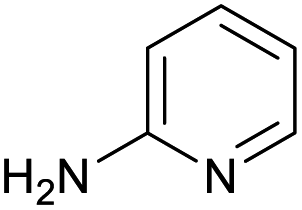
|
91 | 86 |
| 8 |
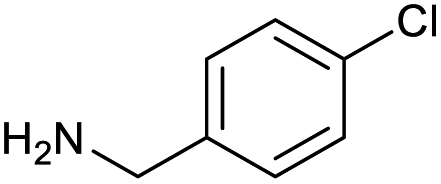
|
100 | 100 | 25 |
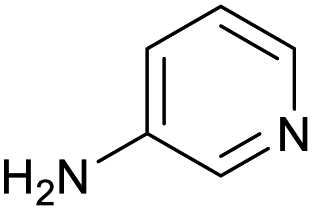
|
98 | 94 |
| 9 |
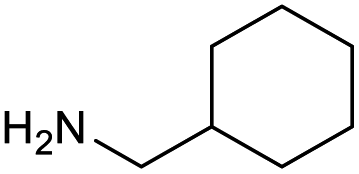
|
100 | 100 | 26 |
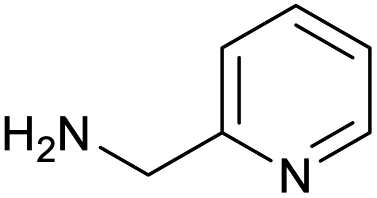
|
93 | 93 |
| 10 |
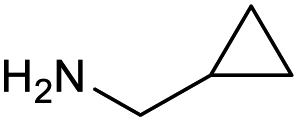
|
100 | 100 | 27 |
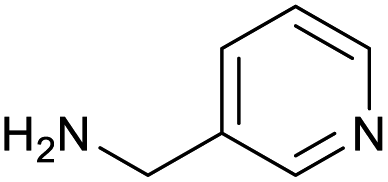
|
98 | 98 |
| 11 |
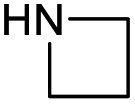
|
100 | 100 | 28 |
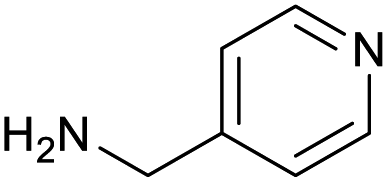
|
98 | 98 |
| 12 |
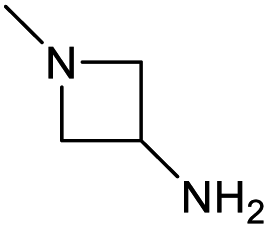
|
100 | 100 | 29 |
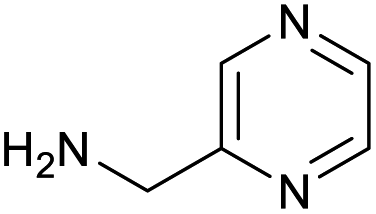
|
94 | 94 |
| 13 |
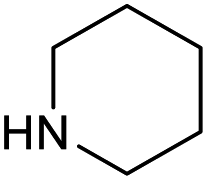
|
100 | 100 | 30 |
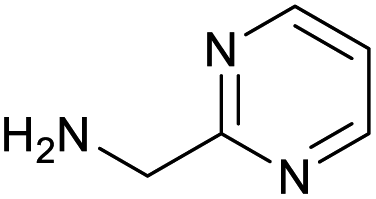
|
98 | 97 |
| 14 |
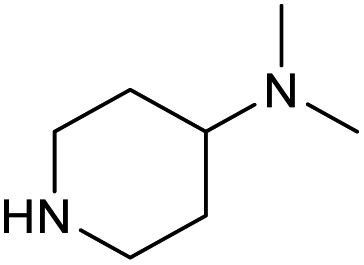
|
100 | 100 | 31 |
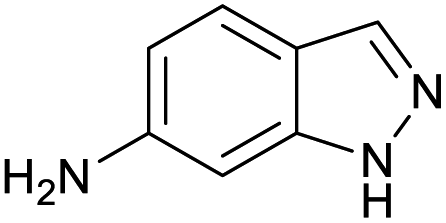
|
100 | 87 |
| 15 |
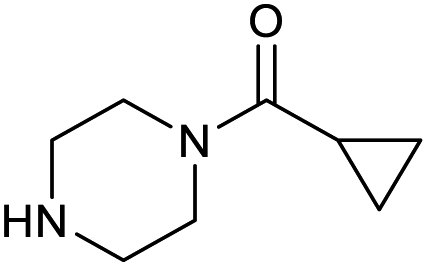
|
100 | 100 | 32 |
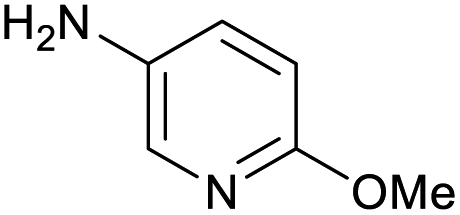
|
100 | 95 |
| 16 |
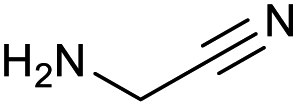
|
100 | 100 | 33 |
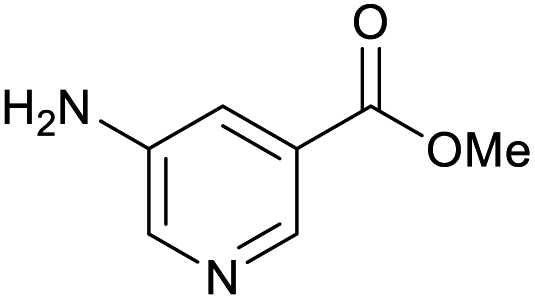
|
100 | 35 |
| 17 |

|
100 | 100 | 34 |
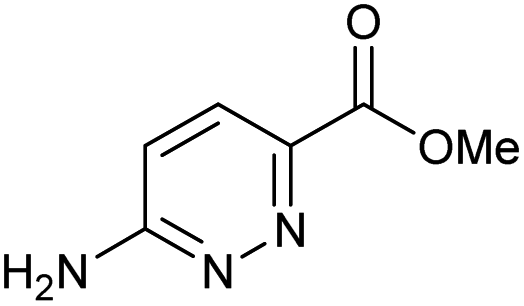
|
100 | 58 |
Overall, this represents an exceptional level of conversion across a wide range of substrates of spanning the full range of reactivity and polarity desired for library synthesis. This method would be expected to be broadly applicable to high-fidelity DEL synthesis. A number of these results indicate that the method is superior to previously published methods, for example, 3-pyridylmethylamine, which gave 98% product (Table 3, entry 27), was shown to fail completely using optimised literature conditions.26 The observation of the methylamide as the only by-product is also advantageous in DEL synthesis as it results in an in situ capping, thus preventing reaction of the carboxylate in subsequent steps. In the event that higher conversions for electron deficient heteroarylamines alone were required, the couplings could be carried out at higher temperature (52 °C), as suggested by the FED.
Carrying out an analogous reaction in the absence of TPGS performed less well, coupling of benzylamine resulted in only 46% and glycine ethyl ester gave 45% conversion with the formation of a significant amount of side products (Fig. S2†). Both substrates gave clean conversion to 100% product in the presence of surfactant. These results prove that the surfactant is essential for high conversions and suggest that the reaction takes place within the micelles.
Application of the optimised DIC coupling conditions to the C-to-N amide coupling were inferior to the optimised HATU method (data not shown). This is consistent with our rationale: the formation of a more hydrophobic active ester is most relevant for carboxy-linked DNA substrates, in which it is advantageous to protect the active ester (limiting reagent) from the water. It is presumably less important for amino-linked DNA in which the active ester is in large excess and a degree of hydrolysis can be tolerated.
Association of DNA conjugates with micelles
Transmission electron microscopy images of the reaction solution were collected. Formation of micellar structures was apparent from a sample obtained from 2.5% TPGS-750-M, which showed the appearance of nanoparticles of ca. 50 nm diameter consistent with those reported for TPGS-750-M micelles (Fig. 4a).35 Images of DNA conjugate 2 in pure water showed species of sizes consistent with those of the DNA conjugate, which were white in appearance (Fig. 4b). Samples of the mixture of 3 with 3.5% TPGS-750-M showed the formation of larger conglomerates of approximately 200 nm in size and white in appearance (Fig. 4c). These images provide evidence that DNA conjugate 3 interacts with the micelle forming surfactant and suggest that the micelles alter their size and shape as a result. Taken together with the inferior reaction outcomes in the absence of surfactant and those obtained with PEG-linked 1, we believe this provides evidence that the high levels of conversion obtained in these procedures are attributable to the association of hydrophobic linked species with the surfactant. Similar phenomena have been described previously, for instance the association of metal nanoparticles with TPGS-750-M micelles, referred to as the nano to nano effect.36,37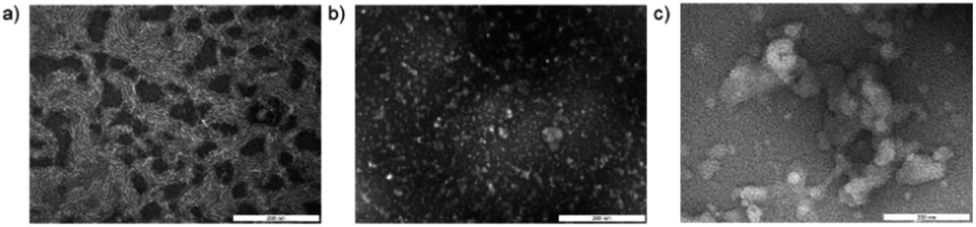 | ||
| Fig. 4 Characterisation of reaction media by negatively stained transmission electron microscopy. (a) 2.5% aqueous TPGS-750-M; (b) 0.1 mM amino-C11-hexylamidoDNA 3 in water; (c) 0.1 mM amino-C11-hexylamidoDNA 3 in 3.5% aqueous TPGS-750-M. | ||
Synthesis of a lead-like compound by three sequential couplings
The N-to-C amide coupling is of great potential in the synthesis of lead-like and peptidic libraries by sequential amide couplings. Carrying out the synthesis in this direction potentially allows the use of simple, more readily available aminoester building blocks, obviating the need to N-protecting groups, such as Fmoc, which are necessary for C-to-N coupling. To illustrate this concept, a representative compound was prepared through sequential coupling of glycine, threonine and 3-bromopropargylamine (Scheme 1).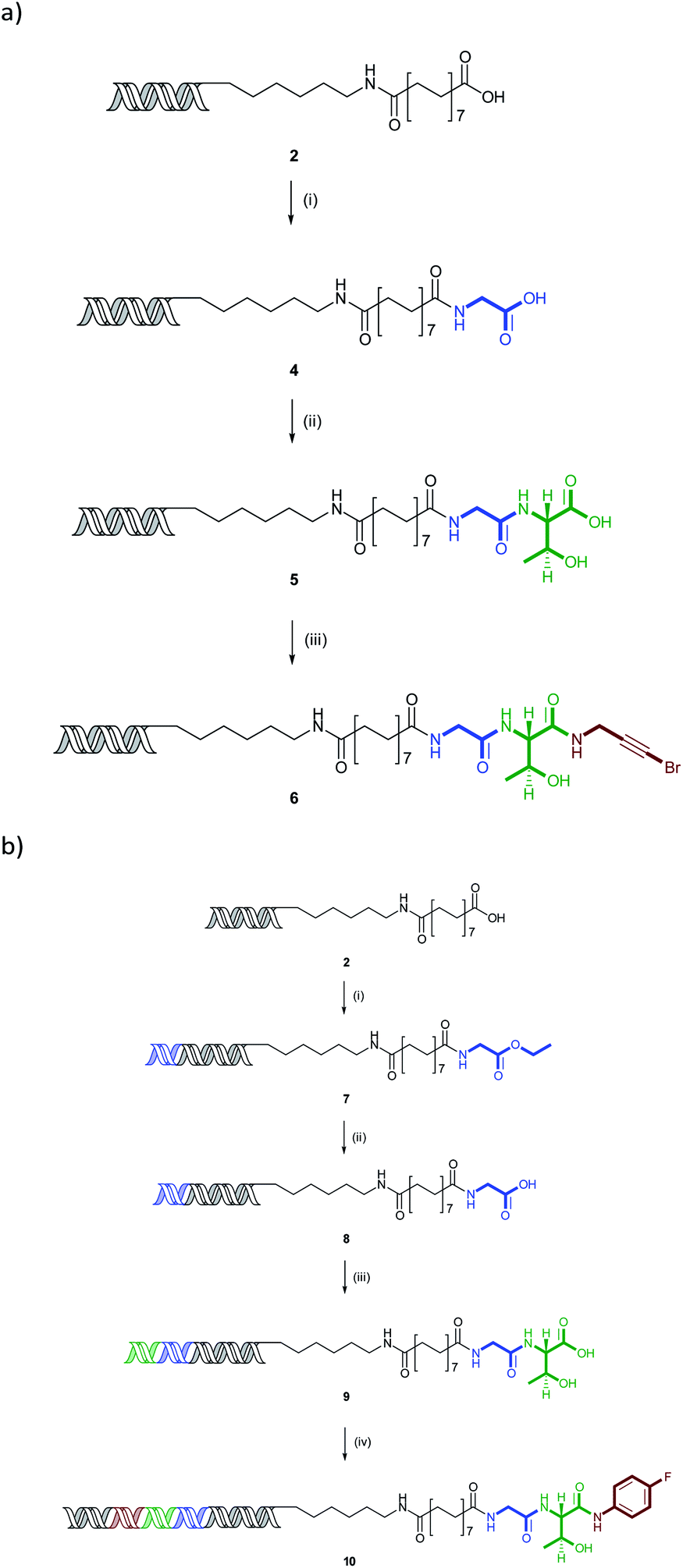 | ||
| Scheme 1 (a) Synthesis of representative encoded compound using 3 cycles of sequential amide couplings. Conditions: (i) glycine ethyl ester (0.5 M), 2,6-lutidine (1.5 M), DIC (0.5 M), HOAt (0.5 M), 4.5% TPGS, (30 μl), 45 °C, 3 h, then 0.25 M LiOH (0.25 M), 1 h, 63% overall yield; (ii) threonine methyl ester (0.5 M), 2,6-lutidine (1.5 M), DIC (0.5 M), HOAt (0.5 M), 4.5% TPGS, (30 μl), 45 °C, 3 h, then LiOH (0.25 M), 1 h, 27% overall yield; (iii) 3-bromopropargylamine (0.5 M), 2,6-lutidine (1.5 M), DIC (0.5 M), HOAt (0.5 M), 4.5% TPGS, (30 μl), 45 °C, 3 h, 75%. (b) Synthesis of representative encoded compound using 3 cycles of sequential amide couplings. Conditions: (i) ligation (primer and BB1 codon), then glycine ethyl ester (0.5 M), 2,6-lutidine (1.5 M), DIC (0.5 M), HOAt (0.5 M), 4.5% TPGS, (30 μl), 45 °C, 3 h, 94% yield for 2 steps; (ii) 0.25 M LiOH (0.25 M), 1 h, 100% yield; (iii) ligation (BB2 codon), then threonine methyl ester (0.5 M), 2,6-lutidine (1.5 M), DIC (0.5 M), HOAt (0.5 M), 4.5% TPGS, (30 μl), 45 °C, 3 h, then LiOH (0.25 M), 1 h, 51% yield for 3 steps; (iv) ligation (BB3 codon and closing primer sequence), then 4-fluoroaniline (0.5 M), 2,6-lutidine (1.5 M), DIC (0.5 M), HOAt (0.5 M), 4.5% TPGS, (30 μl), 45 °C, 3 h, 63% yield for 2 steps. Yields determined by Nanodrop™ spectrophotometry. | ||
Coupling of glycine ethyl ester followed by lithium hydroxide mediated ester hydrolysis gave the intermediate glycine conjugate 4 in 63% yield for the two steps. This was coupled with threonine methyl ester and saponified to give 5 (27%) and final amide coupling with 3-bromopropargyl amine gave the final product 6 in 75% yield (13% overall). In all cases, the couplings proceeded with complete conversion with the desired products being the only species detectable by QTOF mass spectrometry. The low yield of the threonine coupling is most likely due to losses in purification of the more polar intermediate 5, rather than the reaction itself.
Three-cycle synthesis of a fully encoded compound
To demonstrate the applicability of the reaction to library synthesis, a fully-DNA encoded compound was prepared in a manner analogous to that which would be used in a library synthesis. One obvious application of the amide coupling in library synthesis is the sequential coupling of amino acid derivatives akin to peptide synthesis. An illustrative 3-cycle synthesis involving the sequential coupling and ester hydrolysis steps as used for 6 with glycine ethyl ester, threonine methyl ester and 4-fluoroaniline was carried out (Scheme 1). In this case, at each step, a DNA sequence consisting of an 8 base-pair building block codon and 4 base-pair overhang was ligated and the overall sequence closed by the addition of a PCR primer recognition sequence to allow amplification.Initial DNA ligation and coupling of glycine ethyl ester to 2 using optimised conditions resulted in amide 7 in 94% yield (Scheme 1). A single band on gel electrophoresis indicating DNA ligation had occurred efficiently (see ESI Experimental section†). Ester hydrolysis with lithium hydroxide revealed acid 8 in quantitative yield. Repeating this with a second ligation followed by coupling of threonine methyl ester and subsequent hydrolysis gave bisamide 9 in 51% overall yield and as a major band on gel electrophoresis. Final ligation of the third codon and closing primer sequence, followed by coupling with 4-fluoroaniline resulted in the final triamide 10 in 63% yield. Hence, the three-cycle coded substrate was prepared in 30% overall yield.
Gel electrophoresis indicated a major band consistent with the intended coding sequence of 97 base pair in length. PCR amplification (40 cycles) of a sample of the resulting product with NGS elongation primers resulted in a major band of the expected 148 base pair length, showing that the DNA amplifies efficiently after the synthesis sequence. Finally, next generation sequencing of the amplified sample confirmed the integrity of the coding DNA sequence (>80% of 77![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 reads corresponded exactly to the expected sequence).
000 reads corresponded exactly to the expected sequence).
Experimental
Factorial experimental designs and analyses were carried out using SAS JMP v.14, SAS Institute Inc.Coupling of acids to amine headpiece 3
An aliquot of acid solution (60 μl, 0.25 M in NMP) was added to a 50 μl glass insert for a Para-dox™ 96-well micro photoredox/optimisation plate. The NMP was then removed at 55 °C in a Genevac for 60 min. To this solution was added 74 the amine headpiece 3 (30 μl, 5 nmol in 3.5% TPGS-750-M in water), 2,6-lutidine (6.92 μl, 0.06 mmol) and HATU (5.7 mg, 0.015 mmol). The vials were vortexed for 30 seconds each to enhance mixing. The samples were then heated in a Para-dox™ 96-well micro photoredox/optimisation plate at 45 °C overnight. Mass spectrometry was used to analyse reactions. Samples prepared by adding reaction mixture (1 μl) to water (20 μl) and filtered through a hydrophilic PTFE filter. To purify each sample, they were diluted with water (50 μl), DCM (2 × 100 μl) was added to each and the vial vortexed. If an emulsion remained, the sample was centrifuged to aid separation. The organics were removed, and aqueous washed with ethyl acetate (2 × 100 μl). Aqueous sodium chloride (8 μl, 4 M) and ethanol (264 μl) were added and the mixture incubated at −78 °C for 1 hour. The mixture was then centrifuged and the ethanol layer removed. The pellet of DNA was then dissolved in water to give a 1 mM solution.Coupling of amines to acid headpiece 2
An aliquot of amine solution (60 μl, 0.25 M in NMP) and HOAT (20 μl, 10 mg per 100 μl in NMP) were added to a 50 μl glass insert for a Para-dox™ 96-well micro photoredox/optimisation plate. The NMP was then removed at 55 °C in a Genevac for 60 min. To this solution was added the acid headpiece 2 (30 μl, 5 nmol in 4.5% TPGS-750-M in water), 2,6-lutidine (5.2 μl, 0.045 mmol) and DIC (2.2 μl, 0.015 mmol). The vials were vortexed for 30 seconds each to enhance mixing. The samples were then heated in a Para-dox™ 96-well micro photoredox/optimisation plate at 45 °C for 5 hours. Mass spectrometry was used to analyse reactions. Samples prepared by adding reaction mixture (1 μl) to water (20 μl) and filtered through a hydrophilic PTFE filter. To purify each sample, they were diluted with water (50 μl), DCM (2 × 100 μl) was added to each and the vial vortexed. If an emulsion remained, the sample was centrifuged to aid separation. The organics were removed, and aqueous washed with ethyl acetate (2 × 100 μl). Aqueous sodium chloride (8 μl, 4 M) and ethanol (264 μl) were added and the mixture incubated at −78 °C for 1 hour. The mixture was then centrifuged and the ethanol layer removed. The pellet of DNA was then dissolved in water to give a 1 mM solution.Conclusions
The methods developed here provide a highly efficient and generally applicable method for on-DNA amide coupling. This will be of great utility in the preparation of high fidelity DELs, especially those based on peptides and drug-like small molecules. The benefit of the application of micellar technology to DELs is demonstrated clearly by this work. The enhancement in reaction conversion and product purity using micelle forming surfactants in DEL synthesis will be of significant benefit to the field. The combination of more hydrophobic linkers with micellar conditions demonstrates an additional improvement in reaction efficiency and provides evidence that reactions can be improved by increasing the affinity of DNA-linked substrates for hydrophobic micelles.The ability to carry out efficient amide couplings, the most commonly used reaction in DEL synthesis and in medicinal chemistry more generally, will lead to a large number of higher quality DELs with wide substrate scope. The development of an efficient method for N-to-C on-DNA coupling allows the synthesis of peptide-type libraries from simple amino esters, obviating the need for Fmoc-protected amino acids that are required for C-to-N synthesis, thus greatly the accessible scope of this type of library.
This work provides further evidence for the benefits and wider applicability of micellar conditions in DEL synthesis more generally and suggests it could be of further utility for other chemistries.
Data availability
The datasets supporting this article have been uploaded as part of the supplementary material.Author contributions
JHH conceived of the project, designed and carried out the reaction screening, optimisation, substrate exemplification reactions and carried out the encoded compound synthesis, MJA provided additional examples, ISFSC generated the microscopy images, JSG prepared starting materials, CLAS carried out comparator reactions, HAS-G prepared starting materials, JJC supervised CLAS, AM and GP supervised JHH, MJW conceived of the project, supervised JHH, MJA, IFSFC, JSG, CLAS, HAS-G and wrote the manuscript. All authors proof-read the final manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Louis P. Valente for advice on factorial experimental design, Hilal Sarac and Tom McAllister for help with PCR and sequencing and Tracey Davey for carrying out the transmission electron microscopy. We thank AstraZeneca (studentship awards to JHH and IFSFC), EPSRC MoSMed CDT (EP/S022791/1, studentship awards to MJA and JSG), Genentech (studentship award to CLAS), EPSRC DTP (EP/R51309X/1, studentship to HAS-G), Cancer Research UK (C2115/A21421, Newcastle Drug Discovery Unit Programme Grant) and BBSRC (BB/R013942/1, transmission electron microscopy) for funding.References
- S. Brenner and R. A. Lerner, Proc. Natl. Acad. Sci., 1992, 89, 5381–5383 CrossRef CAS PubMed
.
- R. A. Goodnow, C. E. Dumelin and A. D. Keefe, Nat. Rev. Drug Discovery, 2016, 16, 131–147 CrossRef PubMed
-
D. R. Witty and B. Cox, Progress in Medicinal Chemistry, Elsevier Science, 2020 Search PubMed
- M. A. Clark, R. A. Acharya, C. C. Arico-Muendel, S. L. Belyanskaya, D. R. Benjamin, N. R. Carlson, P. A. Centrella, C. H. Chiu, S. P. Creaser, J. W. Cuozzo, C. P. Davie, Y. Ding, G. J. Franklin, K. D. Franzen, M. L. Gefter, S. P. Hale, N. J. V. Hansen, D. I. Israel, J. Jiang, M. J. Kavarana, M. S. Kelley, C. S. Kollmann, F. Li, K. Lind, S. Mataruse, P. F. Medeiros, J. A. Messer, P. Myers, H. O'Keefe, M. C. Oliff, C. E. Rise, A. L. Satz, S. R. Skinner, J. L. Svendsen, L. Tang, K. van Vloten, R. W. Wagner, G. Yao, B. Zhao and B. A. Morgan, Nat. Chem. Biol., 2009, 5, 647–654 CrossRef CAS PubMed
- H. H. Soutter, P. Centrella, M. A. Clark, J. W. Cuozzo, C. E. Dumelin, M.-A. Guie, S. Habeshian, A. D. Keefe, K. M. Kennedy, E. A. Sigel, D. M. Troast, Y. Zhang, A. D. Ferguson, G. Davies, E. R. Stead, J. Breed, P. Madhavapeddi and J. A. Read, Proc. Natl. Acad. Sci., 2016, 113, E7880–E7889 CrossRef CAS PubMed
- H. Salamon, M. Klika Škopić, K. Jung, O. Bugain and A. Brunschweiger, ACS Chem. Biol., 2016, 11, 296–307 CrossRef CAS PubMed
- R. M. Franzini, T. Ekblad, N. Zhong, M. Wichert, W. Decurtins, A. Nauer, M. Zimmermann, F. Samain, J. Scheuermann, P. J. Brown, J. Hall, S. Gräslund, H. Schüler and D. Neri, Angew. Chem., Int. Ed., 2015, 54, 3927–3931 CrossRef CAS PubMed
- S. L. Belyanskaya, Y. Ding, J. F. Callahan, A. L. Lazaar and D. I. Israel, ChemBioChem, 2017, 18, 837–842 CrossRef CAS PubMed
- Y. Ding, J. Chai, P. A. Centrella, C. Gondo, J. L. DeLorey and M. A. Clark, ACS Comb. Sci., 2018, 20, 251–255 CrossRef CAS PubMed
- J. M. Rectenwald, S. K. R. Guduru, Z. Dang, L. B. Collins, Y.-E. Liao, J. L. Norris-Drouin, S. H. Cholensky, K. W. Kaufmann, S. M. Hammond, D. B. Kireev, S. V. Frye and K. H. Pearce, Molecules, 2020, 25, 979 CrossRef CAS
- M. L. Malone and B. M. Paegel, ACS Comb. Sci., 2016, 18, 182–187 CrossRef CAS PubMed
- M. K. Škopić, H. Salamon, O. Bugain, K. Jung, A. Gohla, L. J. Doetsch, D. dos Santos, A. Bhat, B. Wagner and A. Brunschweiger, Chem. Sci., 2017, 8, 3356–3361 RSC
- M. K. Škopić, K. Götte, C. Gramse, M. Dieter, S. Pospich, S. Raunser, R. Weberskirch and A. Brunschweiger, J. Am. Chem. Soc., 2019, 141, 10546–10555 CrossRef PubMed
- D. T. Flood, S. Asai, X. Zhang, J. Wang, L. Yoon, Z. C. Adams, B. C. Dillingham, B. B. Sanchez, J. C. Vantourout, M. E. Flanagan, D. W. Piotrowski, P. Richardson, S. A. Green, R. A. Shenvi, J. S. Chen, P. S. Baran and P. E. Dawson, J. Am. Chem. Soc., 2019, 141, 9998–10006 CrossRef CAS PubMed
- J. H. Hunter, L. Prendergast, L. F. Valente, A. Madin, G. Pairaudeau and M. J. Waring, Bioconjugate Chem., 2020, 31, 149–155 CrossRef CAS PubMed
- S. D. Roughley and A. M. Jordan, J. Med. Chem., 2011, 54, 3451–3479 CrossRef CAS PubMed
- D. G. Brown and J. Boström, J. Med. Chem., 2016, 59, 4443–4458 CrossRef CAS PubMed
- C. M. Gabriel, M. Keener, F. Gallou and B. H. Lipshutz, Org. Lett., 2015, 17, 3968–3971 CrossRef CAS
- Y. Li, E. Gabriele, F. Samain, N. Favalli, F. Sladojevich, J. Scheuermann and D. Neri, ACS Comb. Sci., 2016, 18, 438–443 CrossRef CAS
- P.-P. Kung, P. Bingham, B. J. Burke, Q. Chen, X. Cheng, Y.-L. Deng, D. Dou, J. Feng, G. M. Gallego, M. R. Gehring, S. K. Grant, S. Greasley, A. R. Harris, K. A. Maegley, J. Meier, X. Meng, J. L. Montano, B. A. Morgan, B. S. Naughton, P. B. Palde, T. A. Paul, P. Richardson, S. Sakata, A. Shaginian, W. K. Sonnenburg, C. Subramanyam, S. Timofeevski, J. Wan, W. Yan and A. E. Stewart, ACS Med. Chem. Lett., 2020, 11, 1175–1184 CrossRef CAS PubMed
- L. H. Yuen, S. Dana, Y. Liu, S. I. Bloom, A.-G. Thorsell, D. Neri, A. J. Donato, D. Kireev, H. Schüler and R. M. Franzini, J. Am. Chem. Soc., 2019, 141, 5169–5181 CrossRef CAS PubMed
- M. K. Škopić, O. Bugain, K. Jung, S. Onstein, S. Brandherm, T. Kalliokoski and A. Brunschweiger, RSC Med. Chem., 2016, 7, 1957–1965 RSC
- S. Ahn, A. W. Kahsai, B. Pani, Q.-T. Wang, S. Zhao, A. L. Wall, R. T. Strachan, D. P. Staus, L. M. Wingler, L. D. Sun, J. Sinnaeve, M. Choi, T. Cho, T. T. Xu, G. M. Hansen, M. B. Burnett, J. E. Lamerdin, D. L. Bassoni, B. J. Gavino, G. Husemoen, E. K. Olsen, T. Franch, S. Costanzi, X. Chen and R. J. Lefkowitz, Proc. Natl. Acad. Sci., 2017, 114, 1708–1713 CrossRef CAS PubMed
- H. H. Soutter, P. Centrella, M. A. Clark, J. W. Cuozzo, C. E. Dumelin, M.-A. Guie, S. Habeshian, A. D. Keefe, K. M. Kennedy, E. A. Sigel, D. M. Troast, Y. Zhang, A. D. Ferguson, G. Davies, E. R. Stead, J. Breed, P. Madhavapeddi and J. A. Read, Proc. Natl. Acad. Sci., 2016, 113, E7880–E7889 CrossRef CAS PubMed
- Z. Wu, T. L. Graybill, X. Zeng, M. Platchek, J. Zhang, V. Q. Bodmer, D. D. Wisnoski, J. Deng, F. T. Coppo, G. Yao, A. Tamburino, G. Scavello, G. J. Franklin, S. Mataruse, K. L. Bedard, Y. Ding, J. Chai, J. Summerfield, P. A. Centrella, J. A. Messer, A. J. Pope and D. I. Israel, ACS Comb. Sci., 2015, 17, 722–731 CrossRef CAS PubMed
- N. Favalli, G. Bassi, D. Bianchi, J. Scheuermann and D. Neri, Bioorg. Med. Chem., 2021, 116206 CrossRef CAS PubMed
- L. A. Carpino, J. Am. Chem. Soc., 1993, 115, 4397–4398 CrossRef CAS
- J. C. Sheehan and J. J. Hlavka, J. Org. Chem., 1956, 21, 439–441 CrossRef CAS
- M. Kunishima, C. Kawachi, F. Iwasaki, K. Terao and S. Tani, Tetrahedron Lett., 1999, 40, 5327–5330 CrossRef CAS
- X. Wang, J. Liu, Z. Yan, X. Liu, S. Liu, Y. Suo, W. Lu, J. Yue, K. Chen, H. Jiang, Y. Zhao, M. Zheng, D. Dai and X. Lu, Chem. Sci., 2021, 12, 2841–2847 RSC
- A. Nadin, C. Hattotuwagama and I. Churcher, Angew. Chem., Int. Ed., 2012, 51, 1114–1122 CrossRef CAS
- F. Albericio and A. El-Faham, Org. Process Res. Dev., 2018, 22, 760–772 CrossRef CAS
- Y. S. Klausner and M. Bodansky, Synthesis, 1972, 1972, 453–463 CrossRef
- A. Verma, Synlett, 2012, 23, 1099–1100 CrossRef
- B. H. Lipshutz, S. Ghorai, A. R. Abela, R. Moser, T. Nishikata, C. Duplais, A. Krasovskiy, R. D. Gaston and R. C. Gadwood, J. Org. Chem., 2011, 76, 4379–4391 CrossRef CAS PubMed
- B. H. Lipshutz, Johnson Matthey Technol. Rev., 2017, 61, 196–202 CrossRef CAS
- S. Handa, Y. Wang, F. Gallou and B. H. Lipshutz, Science, 2015, 349, 1087–1091 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc03007h |
| This journal is © The Royal Society of Chemistry 2021 |