
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCopper-catalyzed asymmetric cyclization of alkenyl diynes: method development and new mechanistic insights†
Xin-Qi
Zhu‡
a,
Pan
Hong‡
a,
Yan-Xin
Zheng
a,
Ying-Ying
Zhen
a,
Feng-Lin
Hong
a,
Xin
Lu
 *a and
Long-Wu
Ye
*ab
*a and
Long-Wu
Ye
*ab
aState Key Laboratory of Physical Chemistry of Solid Surfaces, Key Laboratory of Chemical Biology of Fujian Province, College of Chemistry and Chemical Engineering, Xiamen University, Xiamen 361005, China. E-mail: longwuye@xmu.edu.cn; xinlu@xmu.edu.cn
bState Key Laboratory of Organometallic Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, Shanghai 200032, China
First published on 11th June 2021
Abstract
Metal carbenes have proven to be one of the most important and useful intermediates in organic synthesis, but catalytic asymmetric reactions involving metal carbenes are still scarce and remain a challenge. Particularly, the mechanistic pathway and chiral induction model in these asymmetric transformations are far from clear. Described herein is a copper-catalyzed asymmetric cyclization of alkenyl diynes involving a vinylic C(sp2)–H functionalization, which constitutes the first asymmetric vinylic C(sp2)–H functionalization through cyclopentannulation. Significantly, based on extensive mechanistic studies including control experiments and theoretical calculations, a revised mechanism involving a novel type of endocyclic copper carbene via remote-stereocontrol is proposed, thus providing new mechanistic insight into the copper-catalyzed asymmetric diyne cyclization and representing a new chiral control pattern in asymmetric catalysis based on remote-stereocontrol and vinyl cations. This method enables the practical and atom-economical construction of an array of valuable chiral polycyclic-pyrroles in high yields and enantioselectivities.
Introduction
In the past decades, metal carbenes have proven to be one of the most important and useful intermediates.1 However, catalytic asymmetric reactions involving metal carbene intermediates are relatively rare and remain a challenge.2 In particular, the mechanistic pathway and chiral induction model in these asymmetric systems are far from clear.3 Therefore, the exploration of novel enantioselective transformations based on metal carbenes, especially with high enantioselectivity and new chiral control pattern, is significantly important.Among the catalytic transformations of metal carbene species, transition-metal-catalyzed carbene insertion into C–H bonds has attracted much attention during the last decade.4 Compared to the well-established C(sp3)–H bond insertion and aromatic C(sp2)–H bond, carbene insertion into vinylic C(sp2)–H bonds remains much less developed, presumably due to the competing cyclopropanation of the alkene. Such a side reaction can be prohibited in intramolecular reactions. In this context, significant advances have been made in recent years on formal intramolecular carbene insertion into vinylic C(sp2)–H bonds via cyclopentannulation of metallahexatrienes, which consist of the aryl- or alkenyl-linked metal carbenes and alkenyl moieties, as elegantly exploited by the groups of Sarpong,5a R. Liu,5b Echavarren,5c Y. Liu,5d,e Davies,6a Xu,6b Wang6c and others.7 Here, the metal carbenes of metallahexatrienes could be trapped by the intramolecular alkenyl groups via a cyclopentannulation process but not the typical cyclopropanation, thus providing a novel way for rapid and efficient assembly of cyclopentadiene derivatives (Scheme 1a). However, these alkenyl carbene cyclizations have been mostly limited to noble metal catalysts, such as Pt, Au, Rh, Pd, Ru, etc. Furthermore, no direct catalytic asymmetric formal carbene insertion into vinylic C(sp2)–H bonds have been reported to the best of our knowledge.
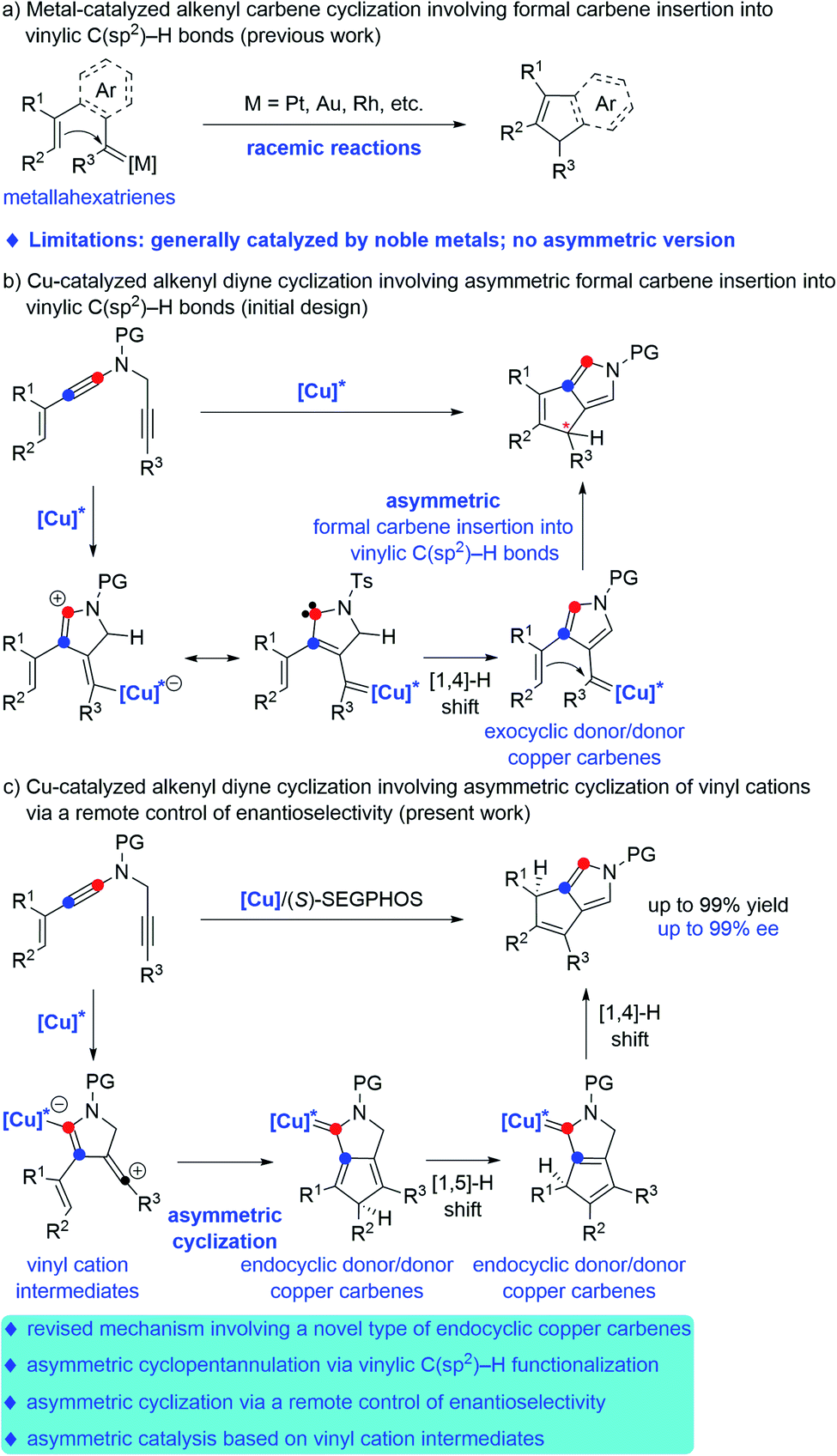 | ||
| Scheme 1 Transition-metal-catalyzed cyclopentannulation involving vinylic C(sp2)–H functionalization. | ||
In recent years, transition-metal catalyzed diyne cyclization has been of increasing importance for the rapid assembly of various synthetically useful cyclic molecules due to their high bond-forming efficiency and atom economy, but there remain significant challenges regarding the catalytic asymmetric diyne cyclization.8 In this respect, our group in 2019 reported an unprecedented copper-catalyzed diyne cyclization through presumable donor/donor carbenes,9 and importantly, the asymmetric version could be achieved by employing chiral copper catalysts, thus allowing the practical and divergent synthesis of various chiral polycyclic pyrroles.10a After that, our group very recently demonstrated a copper-catalyzed asymmetric reaction of alkenyl diynes with styrenes by formal [3 + 2] annulation, leading to a range of chiral pyrrole-fused bridged [2.2.1] skeletons.10b Encouraged by these results and our recent study on developing ynamide chemistry for heterocycle synthesis,11,12 we envisaged that the direct copper-catalyzed tandem cyclization of alkenyl N-propargyl ynamides might produce exocyclic donor/donor copper carbene intermediates, which would undergo further formal carbene insertion into vinylic C(sp2)–H bonds (Scheme 1b). On the basis of this assumption, we herein disclose a copper-catalyzed asymmetric cyclization of alkenyl N-propargyl ynamides involving a vinylic C(sp2)–H functionalization, which constitutes the first example of asymmetric vinylic C(sp2)–H functionalization through cyclopentannulation. Significantly, extensive mechanistic studies based on control experiments and density functional theory (DFT) calculations revealed that vinyl cations13 and endocyclic donor/donor copper carbenes are presumably involved as key intermediates, and remote-stereocontrol is established in this cyclopentannulation,14 thus providing new mechanistic insight into the copper-catalyzed asymmetric diyne cyclization (Scheme 1c). In particular, this type of endocyclic donor/donor copper carbenes can be regarded as Bertrand's cyclic (alkyl) (amino)carbene (CAAC),15 which now can be generated directly from alkynes. This method enables the practical and atom-economic synthesis of a diverse array of chiral dicyclic- and polycyclic-pyrroles in generally good to excellent yields (up to 99% yield) with high enantioselectivities (up to 99% ee). Notably, dicyclic- and polycyclic-pyrroles16 and their reductive derivatives, dicyclic- and polycyclic-pyrrolidines,17 are two kinds of important N-heterocycle skeletons found in a wide range of bioactive molecules and natural products.
Results and discussion
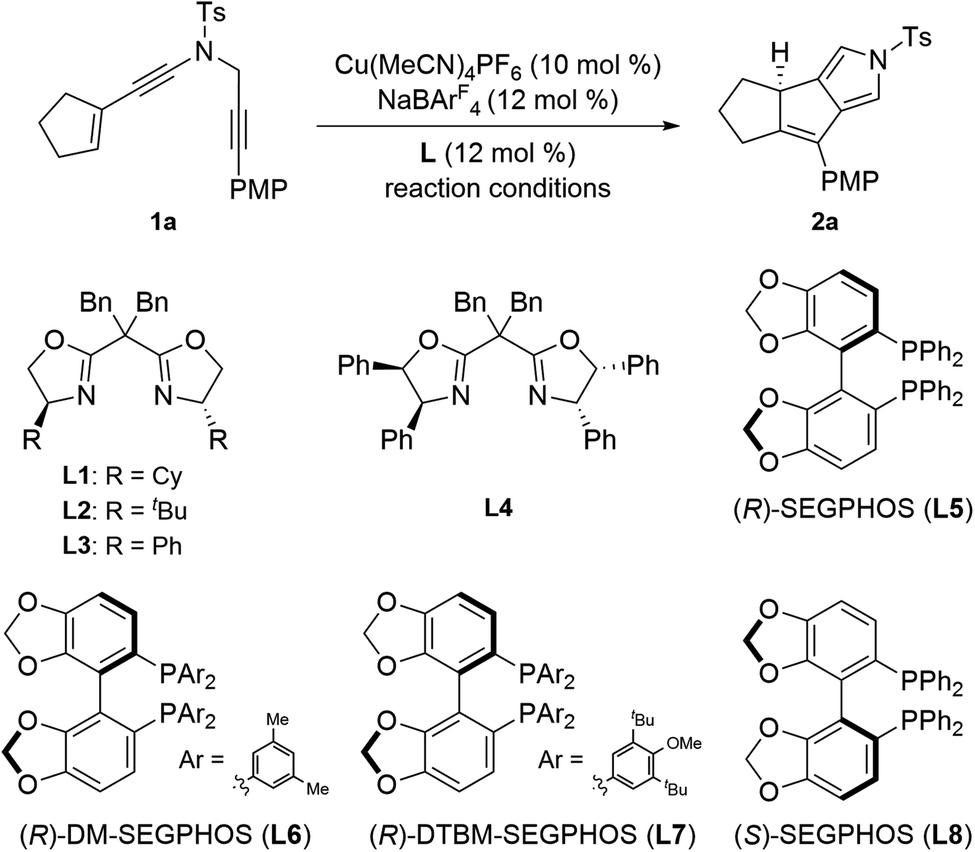
We commenced our investigation by using readily prepared cyclopentenyl N-propargyl ynamide181a as the model substrate under previous Cu-catalyzed reaction conditions. As shown in Table 1, at the outset, various substituted bisoxazoline (BOX) ligands L1–L4 (12 mol%) were employed as the chiral ligands in the presence of 10 mol% of Cu(CH3CN)4PF6 as the catalyst and 12 mol% of NaBArF4 as the additive, and the expected chiral tricyclic pyrrole 2a was furnished in excellent yields (>90%) but with low enantioselectivities (<40% ee, Table 1, entries 1–4). Inspired by these preliminary results, we next investigated different types of chiral biphosphine ligands. To our delight, the enantioselectivity was significantly improved by the use of (R)-SEGPHOS L5 as a ligand, and the corresponding chiral product 2a was formed in 92% yield with 85% ee (Table 1, entry 5). Further screening of SEGPHOS ligands L6 and L7 with large steric hindrance led to the decreased enantioselectivities (Table 1, entries 6 and 7). Interestingly, the enantioselectivity was slightly improved by employing opposite (S)-SEGPHOS L8 as a ligand, and the chiral 2a was formed in 91% yield with 87% ee (Table 1, entry 8). Afterward, solvent examination showed that other typical solvents such as DCE, THF and Et2O failed to further improve the enantioselectivity (Table 1, entries 9–11). Gratifyingly, an obvious temperature influence was observed (Table 1, entries 12 and 13), and decreasing the reaction temperature to 0 °C allowed for the formation of the desired 2a in 90% yield with 93% ee (Table 1, entry 13). Of note, the use of other copper catalysts such as CuOTf and CuI led to a comparable enantioselectivity but slightly decreased yields.|
|
||||
|---|---|---|---|---|
| Entry | L | Conditions | Yieldb (%) | Eec (%) |
| a Reaction conditions: 1a (0.05 mmol), Cu(CH3CN)4PF6 (0.005 mmol), L (0.006 mmol), NaBArF4 (0.006 mmol), solvent (1 mL), in Schlenk tubes. b Measured by 1H NMR using diethyl phthalate as the internal standard. c Determined by HPLC analysis. Ts = p-toluenesulfonyl, PMP = 4-methoxyphenyl, NaBArF4 = sodium tetrakis[3,5-bis(trifluoromethyl)phenyl]borate, DCE = 1,2-dichloroethane. | ||||
| 1 | L1 | Toluene, 35 °C, 2 h | 93 | 35 (+) |
| 2 | L2 | Toluene, 35 °C, 2 h | 90 | <5 (+) |
| 3 | L3 | Toluene, 35 °C, 2 h | 93 | <5 (+) |
| 4 | L4 | Toluene, 35 °C, 2 h | 92 | 21 (−) |
| 5 | L5 | Toluene, 35 °C, 2 h | 92 | 85 (+) |
| 6 | L6 | Toluene, 35 °C, 2 h | 86 | 81 (+) |
| 7 | L7 | Toluene, 35 °C, 5 h | 80 | 15 (+) |
| 8 | L8 | Toluene, 35 °C, 2 h | 91 | 87 (−) |
| 9 | L8 | DCE, 35 °C, 2 h | 82 | 83 (−) |
| 10 | L8 | THF, 35 °C, 12 h | 75 | 82 (−) |
| 11 | L8 | Et2O, 35 °C, 4 h | 72 | 82 (−) |
| 12 | L8 | Toluene, 20 °C, 12 h | 89 | 90 (−) |
| 13 | L8 | Toluene, 0 °C, 72 h | 90 | 93 (−) |
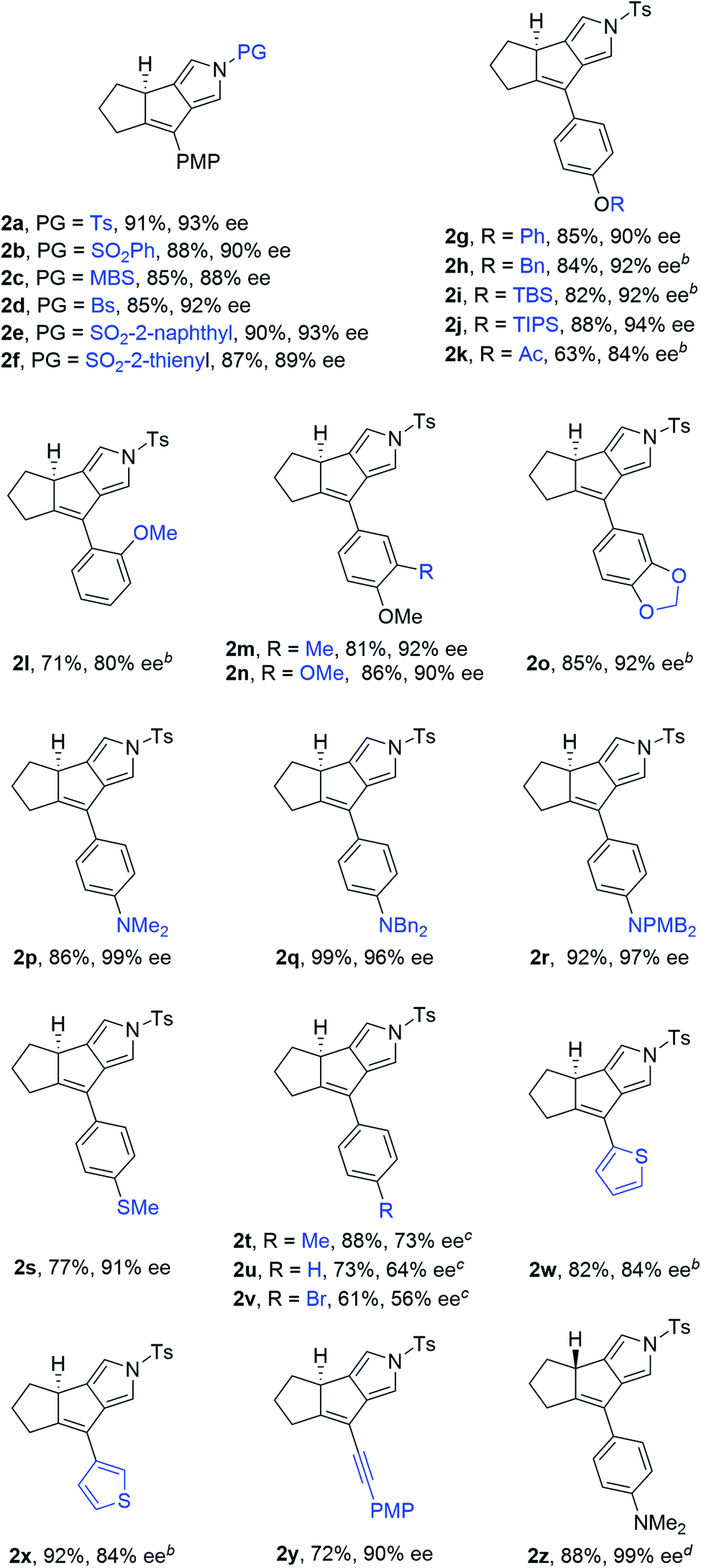
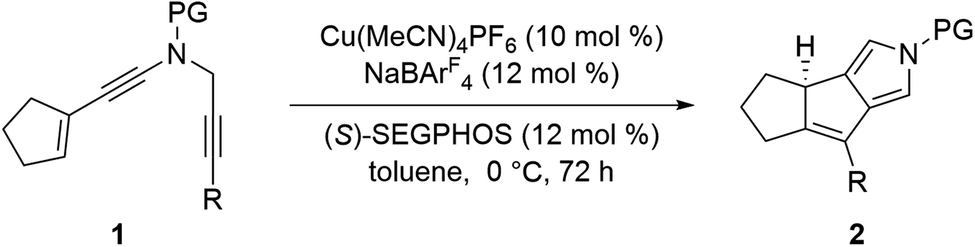
Having established the optimized reaction conditions (Table 1, entry 13), we next explored the substrate scope of this copper-catalyzed asymmetric annulation of alkenyl N-propargyl ynamides. As shown in Table 2, apart from the Ts-protected ynamide 1a, the reaction proceeded smoothly with various N-protected ynamides, including PhSO2-, MBS-, Bs-, 2-naphthyl-SO2- and 2-thienyl-SO2-protected alkenyl N-propargyl ynamides, providing the corresponding chiral tricyclic pyrroles 2b–2f in generally excellent yields and excellent enantioselectivities. Then, we changed PMP to other O-containing electron-donating groups (EDGs). For instance, different O-containing aryl-substituted alkenyl N-propargyl ynamides encompassing functional groups (e.g. phenyl, benzyl, silyl, Ac) were suitable substrates for this tandem annulation, affording the desired chiral pyrroles 2g–2k in 63–88% yields with 84–94% ees. The methoxyl group at the ortho position of the aryl ring led to slightly decreased yield and ee (71%, 80% ee). Besides mono-substituted aryl groups, di-substituted aromatic groups were also readily tolerated, leading to products 2m–2o in 81–86% yields with 90–92% ees. In addition, the reaction was also extended to heteroatom-containing electron-rich aryl groups, and we were pleased to find that NMe2-substituted ynamide 1p occurred efficiently under the standard conditions, delivering the expected tricyclic pyrrole 2p in 86% yield with 99% ee. Subsequently, other heteroatom-containing EDGs (e.g. NBn2, NPMB2, SMe) substituted diynes were also applicable substrates for this reaction to produce the desired pyrroles 2q–2s in excellent yields with 91–97% ees. We then attempted to decrease the electric density of the aryl groups, such as methyl- and bromo-substituted phenyl rings, and the apparent diminution occurred on both yields and enantioselectivities. In addition to the aryl-substituted diynes, heteroaryl-substituted alkenyl N-propargyl ynamides 1w and 1x were also tolerated, allowing the assembly of the desired products 2w (82%, 84% ee) and 2x (92%, 84% ee), respectively. Interestingly, the alkenyl substituted triyne substrate 1y was also compatible with this asymmetric annulation, and the corresponding chiral product 2y was formed in 72% yield and 90% ee, which has great potential in further derivatizations. Finally, the synthesis of tricyclic pyrrole 2z (88%, 99% ee) with the opposite enantioselectivity could also be achieved by employing the opposite (R)-SEGPHOS as a chiral ligand. Our attempts to extend the reaction to the alkenyl- and alkyl-substituted ynamides 1al–1am and terminal ynamide 1an have been unsuccessful as yet.19 The absolute configuration of product 2s was verified by X-ray crystallographic analysis (Fig. 1).20
|
|
|---|
| a Reaction conditions: 1 (0.2 mmol), Cu(MeCN)4PF6 (0.02 mmol), NaBArF4 (0.024 mmol), (S)-SEGPHOS (0.024 mmol), toluene (2 mL), 0 °C, 72 h, in Schlenk tubes; yields are those for the isolated products; ees are determined by HPLC analysis. b 10 °C, 72 h. c 20 °C, 5 d. d (R)-SEGPHOS as the ligand. MBS = 4-methoxybenzenesulfonyl, Bs = 4-bromobenzenesulfonyl, PMB = p-methoxybenzyl. |
|
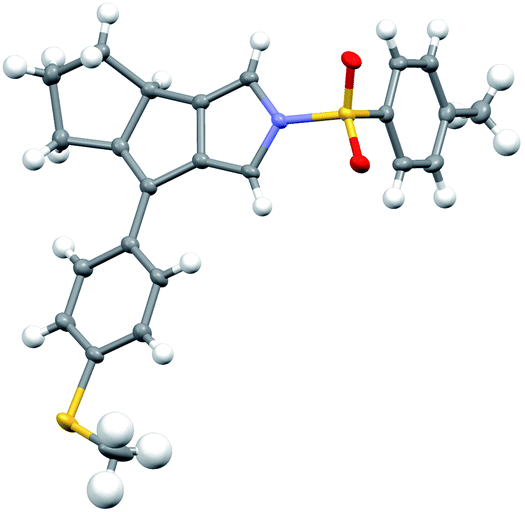 | ||
| Fig. 1 Structure of compound 2s in its crystal. | ||
Various alkenyl moieties of the diynes were then explored under the optimized conditions. As depicted in Table 3, we were delighted to observe that acyclic alkenyl N-propargyl ynamides also served as appropriate substrates to afford the corresponding enantioenriched dicyclic-pyrroles 2aa–2ac in good yields with the ees of 90–93%. Besides 5-membered alkenyl substitution, other ring systems on alkenyl N-propargyl ynamides (e.g. cyclohexenyl, cycloheptenyl, cyclooctenyl) were also suitable substrates for this annulation, furnishing the anticipated chiral tricyclic-pyrroles 2ad–2af in generally excellent yields and enantioselectivities. Of note, indenyl substituted diyne was able to undergo smooth asymmetric cyclization to provide the desired enantioenriched polycyclic-pyrrole 2ag efficiently (91%, 95% ee). Importantly, this asymmetric annulation could be extended to alkenyl diynes bearing the complex pharmaceutical molecule scaffolds. For example, the propofol and theobromine derived diynes were applicable substrates to generate the pharmaceutical derived pyrroles 2ah–2ai in moderate to good yields with acceptable enantioselectivities. In addition, more complex chiral polycyclic-pyrroles 2aj–2al could be obtained smoothly with excellent diastereoselectivities (d.r. > 20/1) from the corresponding complicated substrates bearing pharmaceutical moieties such as ethinylestradiol, ethisterone and diosgenin, and it is notable that low efficiency (<10% yield) was observed in these cases in the absence of chiral ligands.10b
|
|
|---|
| a Reaction conditions: 1 (0.2 mmol), Cu(MeCN)4PF6 (0.02 mmol), NaBArF4 (0.024 mmol), (S)-SEGPHOS (0.024 mmol), toluene (2 mL), 20 °C, 24 h, in Schlenk tubes; yields are those for the isolated products; ees are determined by HPLC analysis; d.r.s are determined by 1H NMR. b 2-Me-THF as the solvent. c DCE as the solvent, 0 °C. d DCE as the solvent, 40 °C. |

|
Further synthetic applications of the as-synthesized chiral tricyclic-pyrroles such as 2a and 2p were then investigated, as indicated in Scheme 2. First, preparative scales could be obtained for the synthesis of 2a (0.70 g, 86%, 92% ee) and 2p (0.65 g, 77%, 99% ee) respectively. Then, the treatment of 2a and 2p with NaBH3CN as the reductant led to tricyclic-dihydropyrroles 3aa and 3pa bearing three contiguous stereocenters in good yields with >20/1 d.r.s. (Scheme 2). Next, selective reduction of tricyclic-pyrroles 2a and 2p by H2 in the presence of Pd/C as the catalyst could allow the formation of the corresponding chiral pyrroles 3ab and 3pb containing three contiguous stereocenters, and tricyclic-pyrrolidines 3ac and 3pc encompassing five contiguous stereocenters in good yields with excellent diastereoselectivities, respectively. The absolute configuration of compound 3pc was confirmed by X-ray diffraction analysis (Fig. 2).20 The NMe2 group of pyrrole 2p could be easily transferred into the aryl group in 84% yield via a Pd-catalyzed cross coupling with the aryl Grignard reagent. Finally, products 3qa and 3qb with the free anilines could be readily obtained respectively from the NBn2-substituted pyrrole 2qvia a debenzylation process with Pd/C under a H2 atmosphere, which might undergo smooth further functionalization. Importantly, almost no erosion of the enantiopurity of the compounds was observed and excellent diastereoselectivities (d.r. > 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) were achieved in all these elaborations.
:1) were achieved in all these elaborations.
 | ||
| Scheme 2 Scale-up reaction and product elaboration. Reagents and conditions: (i) NaBH3CN (5 equiv.), TFA, rt, 1 h. (ii) Pd/C (10 mol%) H2 (1 atm), MeOH, rt, 2 h. (iii) Pd/C (10 mol%) H2 (4 MPa), MeOH/EA, 60 °C, 36 h. (iv) Pd/C (10 mol%) H2 (4 MPa), MeOH, 60 °C, 36 h. (v) PtO2 (10 mol%), H2 (4 MPa), AcOH, 80 °C, 72 h. (vi) MeOTf (1.2 equiv.), Et2O, 0 °C to rt, 2 h; Pd(PPh3)2Cl2 (5 mol%), PhMgBr (2 equiv.), THF, rt, 2 h. (vii) Pd/C (10 mol%) H2 (4 MPa), MeOH/EA, 60 °C, 72 h. (viii) Pd/C (20 mol%) H2 (5 MPa), EtOH/EA, 80 °C, 96 h. | ||
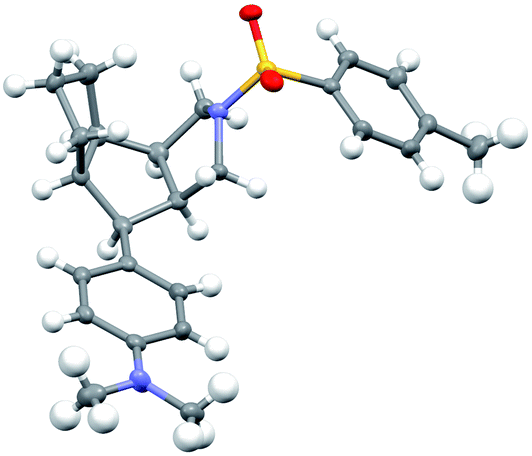 | ||
| Fig. 2 Structure of compound 3pc in its crystal. | ||
On the basis of our previous studies on copper-catalyzed diyne cyclization,10 a plausible mechanism for the synthesis of tricyclic pyrrole 2a is depicted in Scheme 3. The reaction commences with attacking of electron-rich ynamide moiety to the copper-activated C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C bond of N-propargyl moiety, providing the vinyl copper intermediate II or its resonance form III. Subsequent [1,4]-H shift generates donor/donor copper carbene IV. Then, this copper carbene may undergo two pathways. In path a, intramolecular nucleophilic attack of the electron-rich alkene moiety on the carbene carbon delivers the five-membered intermediate VI. This intermediate would go through a protodemetallation to afford the intermediate VII with the regeneration of the copper catalyst, followed by tautomerism to produce the final product 2a, which may be thermodynamically more stable than VII. Alternatively, in path b, direct intramolecular vinylic C(sp2)–H bond insertion into copper carbene may occur to deliver intermediate VIIviaTS-I,6c eventually leading to the desired 2a by a tautomerization process. However, the above proposed mechanism is difficult to explain the stereospecific [1,3]-H transfer in the tautomerism process, which is extremely rare and has to rely the use of extra base to promote the suprafacial [1,3]-hydrogen atom transfer.21 Furthermore, no formation of intermediate VII was observed by 1H NMR monitoring of this annulation under the standard conditions.
C bond of N-propargyl moiety, providing the vinyl copper intermediate II or its resonance form III. Subsequent [1,4]-H shift generates donor/donor copper carbene IV. Then, this copper carbene may undergo two pathways. In path a, intramolecular nucleophilic attack of the electron-rich alkene moiety on the carbene carbon delivers the five-membered intermediate VI. This intermediate would go through a protodemetallation to afford the intermediate VII with the regeneration of the copper catalyst, followed by tautomerism to produce the final product 2a, which may be thermodynamically more stable than VII. Alternatively, in path b, direct intramolecular vinylic C(sp2)–H bond insertion into copper carbene may occur to deliver intermediate VIIviaTS-I,6c eventually leading to the desired 2a by a tautomerization process. However, the above proposed mechanism is difficult to explain the stereospecific [1,3]-H transfer in the tautomerism process, which is extremely rare and has to rely the use of extra base to promote the suprafacial [1,3]-hydrogen atom transfer.21 Furthermore, no formation of intermediate VII was observed by 1H NMR monitoring of this annulation under the standard conditions.
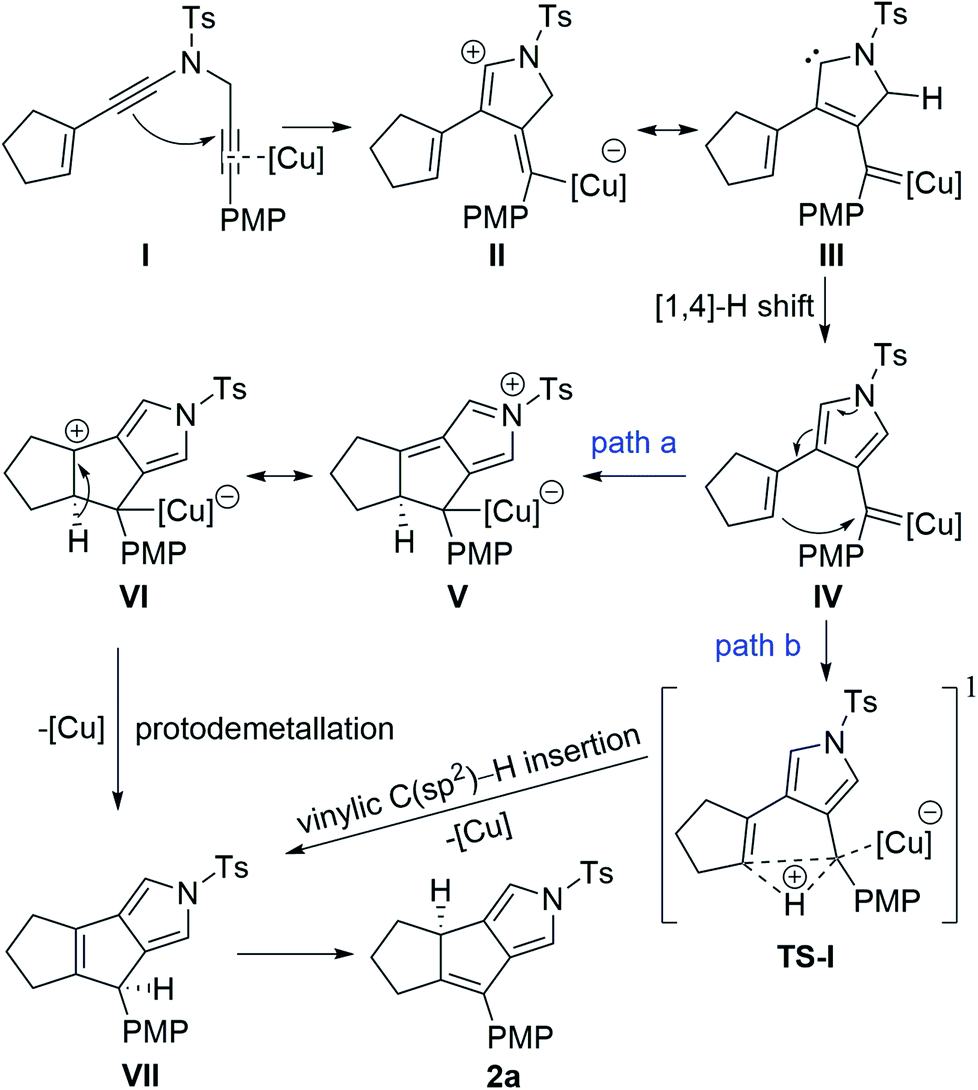 | ||
| Scheme 3 Original mechanism proposed for the formation of product 2a. | ||
To further probe the reaction mechanism, we carried out several control experiments. First, deuterium labeling experiment was performed with the substrate [D3]-1a. It was found that the deuterium atoms were thoroughly retained in [D3]-2a under the standard conditions (Scheme 4a). Then, we also subjected ynamide 1a with D2O or CD3OD (10 equiv.) under standard conditions, and found that <1% deuterium incorporation into the carbocycle partner of the desired product was observed (Scheme 4b). Thus, these results indicate that path a of Scheme 3 is unlikely, as H/D exchange should occur in the deprotonation/protonation sequence.6c Meanwhile, it is notable that significant deuterium incorporation into the pyrrole partner of 2a was detected. To confirm this, we also synthesized substrate [D2]-1a and subjected it under the standard conditions and in the presence of 10 equiv. of H2O, and found that only 30% deuterium and 6% deuterium were retained on one of the α-position pyrrole, respectively (Scheme 4c). Moreover, the use of 2,6-di-tert-butyl-4-methylpyridine as base with large steric hindrance substantially accelerated the reaction (rt: 1.5 h vs. 3 h).19 These results suggested that proton transfer but not the previously proposed hydride shift was presumably involved in the formation of pyrrole moiety, and the proton transfer could be assisted by proper organic base or water. In addition, it was found that the reaction of alkenyl diazo compound 1u′ under the current copper catalysis only led to the formation of the corresponding oxidized ketone 2u′ in 50% yield, and no desired cyclization product 2u was obtained (Scheme 4d). Finally, kinetic isotope effect (KIE) experiment was also conducted with the mixture of substrates 1a and [D3]-1a, and the KIE data (kH/kD = 1.5) suggest that the cleavage of vinylic C(sp2)–H bond is not the rate-determining step.19
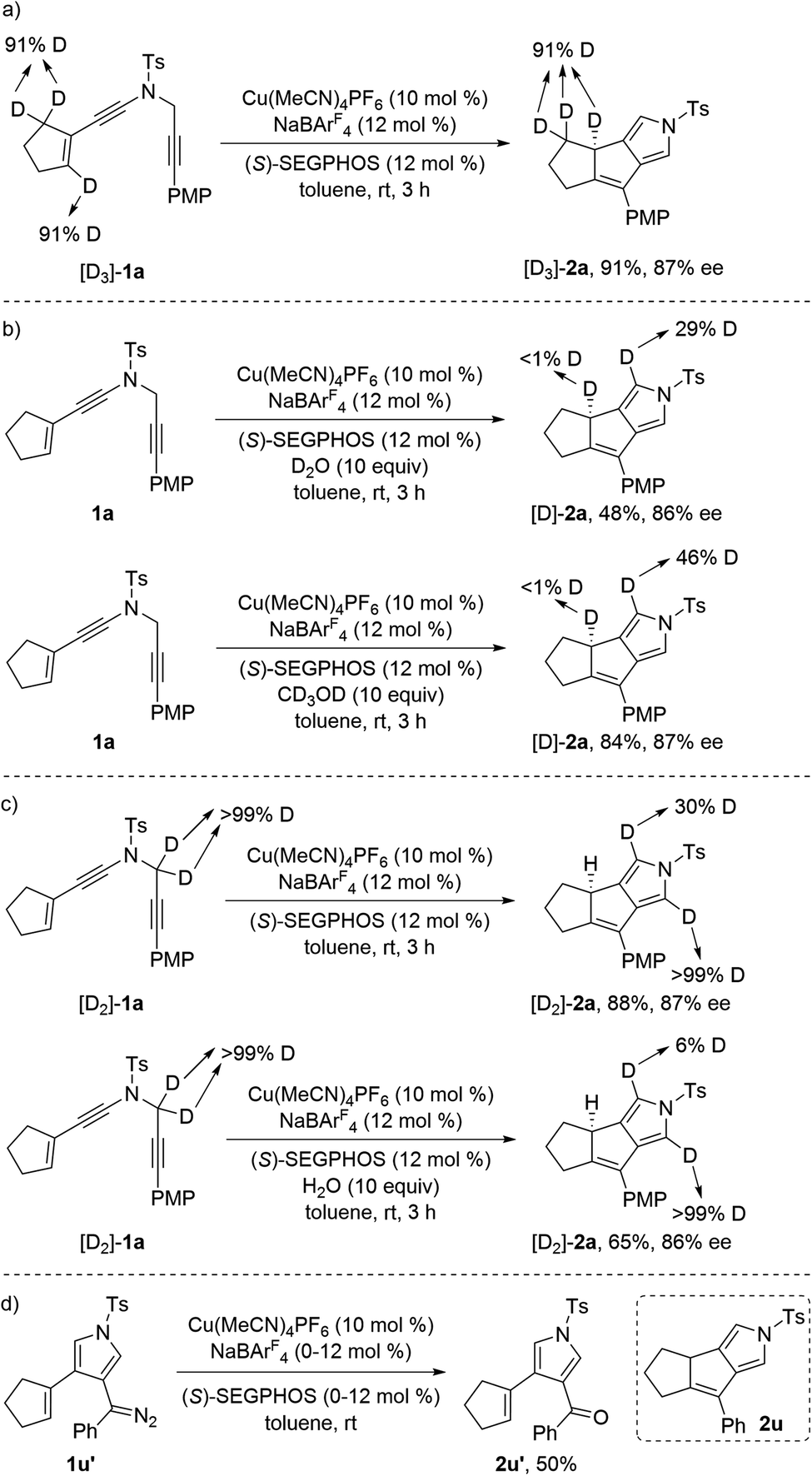 | ||
| Scheme 4 (a) The reaction of [D3]-1a under the standard conditions. (b) Cu-catalyzed cyclization of 1a in the presence of 10 equiv. of D2O or CD3OD. (c) The reaction of [D2]-1a under the standard conditions or in the presence of 10 equiv. of H2O. (d) Cu-catalyzed reaction of 1u′ under the standard conditions. | ||
Based on the above experimental observations, to clearly understand the mechanism for formation of the tricyclic pyrrole 2a from cyclopentenyl N-propargyl ynamide 1a, detailed DFT calculations were carried out with the achiral CuI catalyst and the plausible mechanism is shown in Scheme 5. Initially, the single catalytic CuI species is preferentially bound to the electron-richer amide-tethered CC bond of the substrate 1a, forming the precursor A and the possibility of dual-copper catalysis is completely excluded out.19 Subsequent electrophilic attack of the copper-activated CC bond to the N-propargyl moiety viaTSA with a free energy barrier of 9.4 kcal mol−1 gives the cyclized intermediate B. The vinyl-cation center of B is highly reactive and readily attacked by the electron-rich alkene group to form the cyclopentadiene intermediate C; this step is highly exergonic (ΔG = −41.0 kcal mol−1) with a free energy barrier of only 4.8 kcal mol−1. Then, a normal suprafacial [1,5]-H shift occurs within the cyclopentadiene ring with a free energy barrier of 16.8 kcal mol−1 to afford the second cyclopentadiene intermediate D. The remaining steps include [1,4]-H shift within the five-membered N-heterocycle of D and demetallation to afford the final product 2a. The [1,4]-H shift is found to be assisted by H2O via two consecutive steps of proton transfer (D → F → G) by overcoming an activation free energy of 28.0 kcal mol−1,19,22 or more readily assisted by such an organic base as 2,6-di-tert-butyl-4-methylpyridine with a lower activation free energy of 23.9 kcal mol−1. Such rate-limiting 1,4-proton transfer mechanism is in line with the aforementioned isotopic control experiments (Scheme 4) and the observation that the use of 2,6-di-tert-butyl-4-methylpyridine obviously accelerated the reaction.
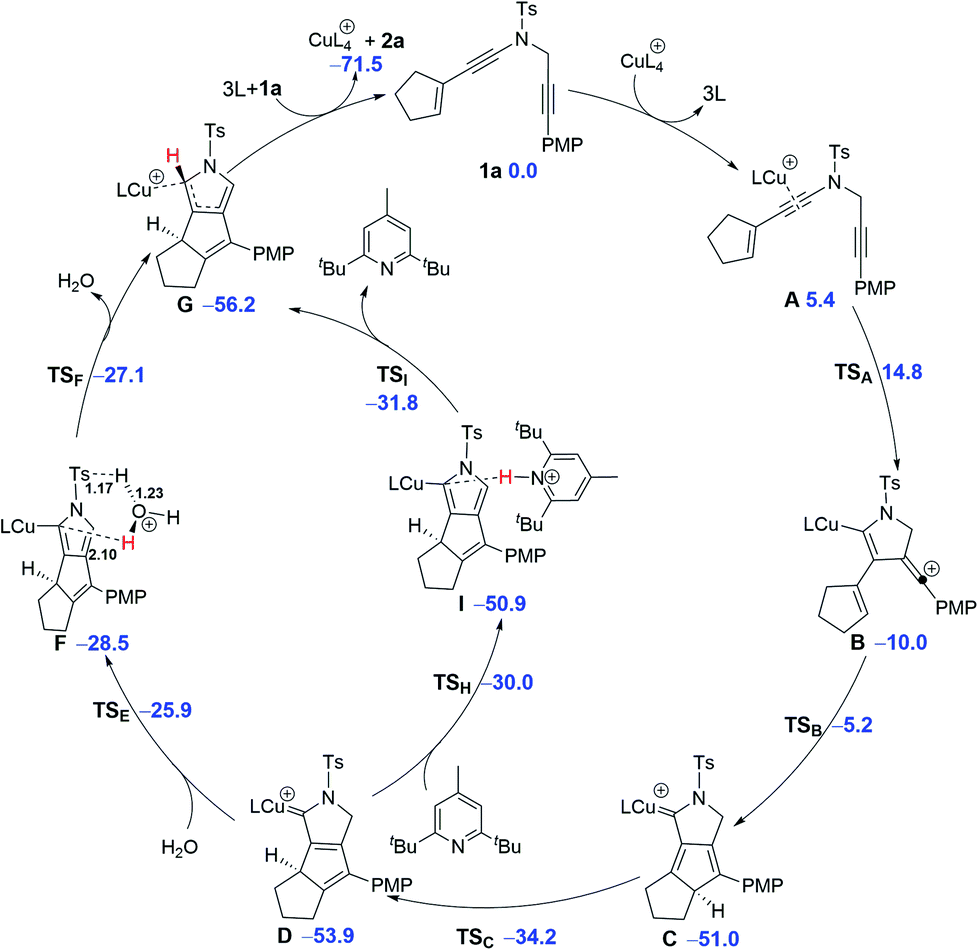 | ||
| Scheme 5 Revised mechanism for the formation of product 2a (CuL = CuMeCN). Relative free energies (ΔG, in kcal mol−1) of the key intermediates and transition states are calculated at the SMD(toluene)-M06/6-31G(d,p)/LANL2DZ level of theory at 298 K. Key bond lengths are shown in bold (unit: Å). | ||
To unravel the origin of enantioselectivity, the transition states of the second cyclization step, i.e., B → C, were explored with use of the chiral ligand L8 coordinated onto the Cu(I) center (Scheme 6), as the stereoselectivity of this step can be completely maintained in the subsequent stereospecific suprafacial [1,5]-H shift. The free energy of the transition state [CuL8]-R TSB is 2.0 kcal mol−1 lower than the [CuL8]-S TSB, agreeing well with the experimental ee value of 93%. Despite that the chiral ligand is far away from the highly active vinyl-cation center, the steric repulsion between the bulky chiral ligand L8 and the rigid cyclopentene ring is responsible for the stereoselectivity of such a bicyclization process (Scheme 6).
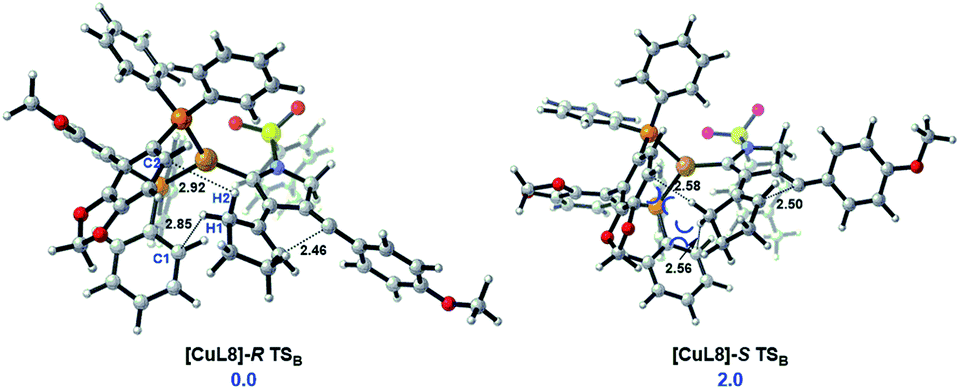 | ||
| Scheme 6 The geometries and relative free energies (ΔG, in kcal mol−1) of the transition state [CuL8]-R TSB and [CuL8]-S TSB with the chiral ligand L8 in the trans-formation of intermediate B to C with different orientations of hydrogen atom. Calculations at SMD(toluene)-M06-D3/Def2tzvp//M06/6-31G(d,p)/LANL2DZ level. Key bond lengths are shown in bold (unit: Å). | ||
Finally, we should mention that similar vinyl cation-involving mechanism supported by DFT computations19 also accounts for the copper-catalyzed diyne cyclizations reported previously.10
Conclusions
In summary, we have developed a novel copper-catalyzed asymmetric cyclization of alkenyl N-propargyl ynamides involving a vinylic C(sp2)–H functionalization, representing the first asymmetric such a vinylic C(sp2)–H functionalization through cyclopentannulation. This method enables the practical and atom-economical construction of a diverse array of valuable chiral dicyclic- and polycyclic-pyrroles in moderate to excellent yields with wide substrate compatibility and high enantioselectivities (up to 99% ee) under mild conditions. Significantly, based on detailed mechanistic studies including control experiments and DFT calculations, a revised mechanism involving a novel type of endocyclic copper carbenes, which constitutes the first generation of electron-rich heteroatom-substituted metal carbenes (CAAC type) from alkynes, and a remote control of enantioselectivity is proposed, Thus, this protocol provides new mechanistic insight into the copper-catalyzed asymmetric diyne cyclization and represents a new chiral control pattern in asymmetric catalysis based on remote-stereocontrol and vinyl cations. To our best knowledge, examples of the direct catalytic asymmetric reactions of vinyl cations have not been reported. We envision that the findings described in this paper will open up new horizons in the field of asymmetric diyne cyclizations, and catalytic asymmetric reactions involving metal carbenes, vinyl cations and remote-stereocontrol.Data availability
Data for the crystal structures reported in this paper have been deposited at the Cambridge Crystallographic Data Centre (CCDC) under the deposition numbers CCDC 2046032 (2s), 2046033 (3pc). All other data supporting the findings of this study, including experimental procedures and compound characterization, are available within the paper and its supplementary information files, or from the corresponding authors on request.Author contributions
X.-Q. Z., Y.-X. Z., Y.-Y. Z., F.-L. H. performed experiments. P. H., X. L. performed DFT calculations. L.-W. Y. conceived and directed the project and wrote the paper. All authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for financial support from the National Natural Science Foundation of China (92056104, 21772161 and 91545105), the China Postdoctoral Science Foundation (2020M680087), the Natural Science Foundation of Fujian Province of China (2019J02001), the President Research Funds from Xiamen University (20720180036), NFFTBS (J1310024), PCSIRT, and Science & Technology Cooperation Program of Xiamen (3502Z20183015). We thank Mr Zanbin Wei from Xiamen University for assistance with X-ray crystallographic analysis. Dedicated to the 100th anniversary of Xiamen University.Notes and references
- For recent selected reviews, see: (a) X. Tian, L. Song and A. S. K. Hashmi, Chem.–Eur. J., 2020, 26, 3197 CrossRef CAS; (b) Y. Xia, D. Qiu and J. Wang, Chem. Rev., 2017, 117, 13810 CrossRef CAS PubMed; (c) R. J. Harris and R. A. Widenhoefer, Chem. Soc. Rev., 2016, 45, 4533 RSC; (d) M. Jia and S. Ma, Angew. Chem., Int. Ed., 2016, 55, 9134 CrossRef CAS; (e) A. Padwa, Chem. Soc. Rev., 2009, 38, 3072 RSC; (f) H. M. L. Davies and J. R. Denton, Chem. Soc. Rev., 2009, 38, 3061 RSC.
- For recent selected reviews, see: (a) Q.-Q. Cheng, Y. Deng, M. Lankelma and M. P. Doyle, Chem. Soc. Rev., 2017, 46, 5425 RSC; (b) W. Zi and F. D. Toste, Chem. Soc. Rev., 2016, 45, 4567 RSC; (c) S.-F. Zhu and Q.-L. Zhou, Acc. Chem. Res., 2012, 45, 1365 CrossRef CAS PubMed; (d) H. M. L. Davies and J. S. Alford, Chem. Soc. Rev., 2014, 43, 5151 RSC.
- For recent selected examples, see: (a) X.-L. Xie, S.-F. Zhu, J.-X. Guo, Y. Cai and Q.-L. Zhou, Angew. Chem., Int. Ed., 2014, 53, 2978 CrossRef CAS PubMed; (b) B. Lu, Y. Li, Y. Wang, D. H. Aue, Y. Luo and L. Zhang, J. Am. Chem. Soc., 2013, 135, 8512 CrossRef CAS PubMed; (c) E. L. Noey, Y. Luo, L. Zhang and K. N. Houk, J. Am. Chem. Soc., 2012, 134, 1078 CrossRef CAS PubMed.
- For selected reviews, see: (a) X. Zhao, M. Rudolph and A. S. K. Hashmi, Chem. Commun., 2019, 55, 12127 RSC; (b) A. S. K. Hashmi, Acc. Chem. Res., 2014, 47, 864 CrossRef CAS PubMed; (c) H. M. Davies and L. D. Morton, Chem. Soc. Rev., 2011, 40, 1857 RSC; (d) M. P. Doyle, R. Duffy, M. Ratnikov and L. Zhou, Chem. Rev., 2010, 110, 704 CrossRef CAS PubMed; (e) H. M. L. Davies and J. R. Manning, Nature, 2008, 451, 417 CrossRef CAS PubMed.
- For the relevant Pt- or Au-catalyzed cyclopentannulation of metallahexatrienes, see: (a) B. A. B. Prasad, F. K. Yoshimoto and R. Sarpong, J. Am. Chem. Soc., 2005, 127, 12468 CrossRef CAS PubMed; (b) D. Vasu, H.-H. Hung, S. Bhunia, S. A. Gawade, A. Das and R.-S. Liu, Angew. Chem., Int. Ed., 2011, 50, 6911 CrossRef CAS PubMed; (c) Y. Wang, P. R. McGonigal, B. Herlé, M. Besora and A. M. Echavarren, J. Am. Chem. Soc., 2014, 136, 801 CrossRef CAS PubMed; (d) J. Liu, M. Chen, L. Zhang and Y. Liu, Chem.–Eur. J., 2015, 21, 1009 CrossRef CAS PubMed; (e) J. Zhao, S. Yang, X. Xie, X. Li and Y. Liu, J. Org. Chem., 2018, 83, 1287 CrossRef CAS PubMed.
- For the relevant Rh-catalyzed cyclopentannulation of metallahexatrienes, see: (a) J. F. Briones, J. Hansen, K. I. Hardcastle, J. Autschbach and H. M. L. Davies, J. Am. Chem. Soc., 2010, 132, 17211 CrossRef CAS PubMed; (b) Y. Zheng, J. Mao, Y. Weng, X. Zhang and X. Xu, Org. Lett., 2015, 17, 5638 CrossRef CAS PubMed; (c) Q. Zhou, S. Li, Y. Zhang and J. Wang, Angew. Chem., Int. Ed., 2017, 56, 16013 CrossRef CAS PubMed.
- For other relevant cyclopentannulations of metallahexatrienes: (a) H. Wang, S. Cai, W. Ai, X. Xu, B. Li and B. Wang, Org. Lett., 2020, 22, 7255 CrossRef CAS PubMed; (b) C. Pei, G.-W. Rong, Z.-X. Yu and X.-F. Xu, J. Org. Chem., 2018, 83, 13243 CrossRef CAS; (c) B. G. Das, A. Chirila, M. Tromp, J. N. H. Reek and B. de Bruin, J. Am. Chem. Soc., 2016, 138, 8968 CrossRef CAS PubMed; (d) X. Wang, Y. Zhou, L. Qiu, R. Yao, Y. Zheng, C. Zhang, X. Bao and X. Xu, Adv. Synth. Catal., 2016, 358, 1571 CrossRef CAS; (e) K. Chen, Z.-Z. Zhu, Y.-S. Zhang, X.-Y. Tang and M. Shi, Angew. Chem., Int. Ed., 2014, 53, 6645 CrossRef CAS PubMed; (f) J. Barluenga, A. Álvarez-Fernández, Á. L. Suárez-Sobrino and M. Tomás, Angew. Chem., Int. Ed., 2012, 51, 183 CrossRef CAS PubMed; (g) T. Bolaño, R. Castarlenas, M. A. Esteruelas and E. Oñate, J. Am. Chem. Soc., 2009, 131, 2064 CrossRef PubMed; (h) R. Sanz, D. Miguel and F. Rodríguez, Angew. Chem., Int. Ed., 2008, 47, 7354 CrossRef CAS PubMed; (i) S. Datta, A. Odedra and R.-S. Liu, J. Am. Chem. Soc., 2005, 127, 11606 CrossRef CAS PubMed; for early work on cyclopentannulation of metallahexatrienes, see: (j) I. Göttker-Schnetmann, R. Aumann and K. Bergander, Organometallics, 2001, 20, 3574 CrossRef; (k) V. Rautenstrauch, J. Org. Chem., 1984, 49, 950 CrossRef CAS.
- For recent reviews, see: (a) A. M. Asiri and A. S. K. Hashmi, Chem. Soc. Rev., 2016, 45, 4471 RSC; (b) D. P. Day and P. W. H. Chan, Adv. Synth. Catal., 2016, 358, 1368 CrossRef CAS; (c) A. S. K. Hashmi, Acc. Chem. Res., 2014, 47, 864 CrossRef CAS PubMed.
- For reviews, see: (a) B. D. Bergstrom, L. A. Nickerson, J. T. Shaw and L. W. Souza, Angew. Chem., Int. Ed., 2021, 60, 6864 CrossRef CAS PubMed; (b) D. Zhu, L. Chen, H. Fan, Q. Yao and S. Zhu, Chem. Soc. Rev., 2020, 49, 908 RSC; (c) L. Chen, K. Chen and S. Zhu, Chem, 2018, 4, 1208 CrossRef CAS.
- (a) F.-L. Hong, Z.-S. Wang, D.-D. Wei, T.-Y. Zhai, G.-C. Deng, X. Lu, R.-S. Liu and L.-W. Ye, J. Am. Chem. Soc., 2019, 141, 16961 CrossRef CAS PubMed; (b) F.-L. Hong, Y.-B. Chen, S.-H. Ye, G.-Y. Zhu, X.-Q. Zhu, X. Lu, R.-S. Liu and L.-W. Ye, J. Am. Chem. Soc., 2020, 142, 7618 CrossRef CAS PubMed.
- For recent reviews on ynamide reactivity, see: (a) Y.-C. Hu, Y. Zhao, B. Wan and Q.-A. Chen, Chem. Soc. Rev., 2021, 50, 2582 RSC; (b) Y.-B. Chen, P.-C. Qian and L.-W. Ye, Chem. Soc. Rev., 2020, 49, 8897 RSC; (c) C. C. Lynch, A. Sripada and C. Wolf, Chem. Soc. Rev., 2020, 49, 8543 RSC; (d) J. Luo, G.-S. Chen, S.-J. Chen, J.-S. Yu, Z.-D. Li and Y.-L. Liu, ACS Catal., 2020, 10, 13978 CrossRef CAS; (e) B. Zhou, T.-D. Tan, X.-Q. Zhu, M. Shang and L.-W. Ye, ACS Catal., 2019, 9, 6393 CrossRef CAS; (f) G. Evano, C. Theunissen and M. Lecomte, Aldrichimica Acta, 2015, 48, 59 CAS; (g) X.-N. Wang, H.-S. Yeom, L.-C. Fang, S. He, Z.-X. Ma, B. L. Kedrowski and R. P. Hsung, Acc. Chem. Res., 2014, 47, 560 CrossRef CAS PubMed; (h) K. A. DeKorver, H. Li, A. G. Lohse, R. Hayashi, Z. Lu, Y. Zhang and R. P. Hsung, Chem. Rev., 2010, 110, 5064 CrossRef CAS PubMed; (i) G. Evano, A. Coste and K. Jouvin, Angew. Chem., Int. Ed., 2010, 49, 2840 CrossRef CAS PubMed.
- For a review on the catalytic tandem reactions of ynamides by our group, see: (a) F.-L. Hong and L.-W. Ye, Acc. Chem. Res., 2020, 53, 2003 CrossRef CAS PubMed; for recent selected examples by our group, see: (b) Z.-S. Wang, Y.-B. Chen, H.-W. Zhang, Z. Sun, C. Zhu and L.-W. Ye, J. Am. Chem. Soc., 2020, 142, 3636 CrossRef CAS PubMed; (c) X. Liu, Z.-S. Wang, T.-Y. Zhai, C. Luo, Y.-P. Zhang, Y.-B. Chen, C. Deng, R.-S. Liu and L.-W. Ye, Angew. Chem., Int. Ed., 2020, 59, 17984 CrossRef CAS PubMed; (d) Y. Xu, Q. Sun, T.-D. Tan, M.-Y. Yang, P. Yuan, S.-Q. Wu, X. Lu, X. Hong and L.-W. Ye, Angew. Chem., Int. Ed., 2019, 58, 16252 CrossRef CAS PubMed; (e) B. Zhou, Y.-Q. Zhang, K. Zhang, M.-Y. Yang, Y.-B. Chen, Y. Li, Q. Peng, S.-F. Zhu, Q.-L. Zhou and L.-W. Ye, Nat. Commun., 2019, 10, 3234 CrossRef PubMed; (f) L. Li, X.-Q. Zhu, Y.-Q. Zhang, H.-Z. Bu, P. Yuan, J. Chen, J. Su, X. Deng and L.-W. Ye, Chem. Sci., 2019, 10, 3123 RSC.
- T. Wurm, J. Bucher, S. B. Duckworth, M. Rudolph, F. Rominger and A. S. K. Hashmi, Angew. Chem., Int. Ed., 2017, 56, 3364 CrossRef CAS PubMed.
- For recent selected examples on the remote-stereocontrol, see: (a) R. L. Reyes, M. Sato, T. Iwai, K. Suzuki, S. Maeda and M. Sawamura, Science, 2020, 369, 970 CrossRef CAS PubMed; (b) A. Seegerer, J. Hioe, M. M. Hammer, F. Morana, P. J. W. Fuchs and R. M. Gschwind, J. Am. Chem. Soc., 2016, 138, 9864 CrossRef CAS PubMed; (c) J. Mo, X. Chen and Y. R. Chi, J. Am. Chem. Soc., 2012, 134, 8810 CrossRef CAS PubMed.
- For recent selected examples, see: (a) R. Jazzar, M. Soleilhavoup and G. Bertrand, Chem. Rev., 2020, 120, 4141 CrossRef CAS PubMed; (b) M. Melaimi, R. Jazzar, M. Soleilhavoup and G. Bertrand, Angew. Chem., Int. Ed., 2017, 56, 10046 CrossRef CAS PubMed; (c) M. Soleilhavoup and G. Bertrand, Acc. Chem. Res., 2015, 48, 256 CrossRef CAS PubMed; (d) R. Hamze, J. L. Peltier, D. Sylvinson, M. Jung, J. Cardenas, R. Haiges, M. Soleilhavoup, R. Jazzar, P. I. Djurovich, G. Bertrand and M. E. Thompson, Science, 2019, 363, 601 CrossRef CAS PubMed.
- For selected examples, see: (a) S. Kultgen and A. G. Cole, PCT Int. Appl. WO2020023710A1, 2020; (b) B. O. Buckman, J. B. Nicholas, K. Emayan, S. D. Seiwert and S. Yuan, PCT Int. Appl. WO2014113485A1, 2014; (c) P. Silakari, S. D. Shrivastava, G. Silakari, D. V. Kohli, G. Rambabu, S. Srivastava, S. K. Shrivastava and O. Silakari, Eur. J. Med. Chem., 2008, 43, 1559 CrossRef CAS PubMed; (d) A. K. Chakraborti and R. Thilagavathi, Bioorg. Med. Chem., 2003, 11, 3989 CrossRef CAS PubMed; (e) B. Portevin, C. Tordjman, P. Pastoureau and J. Bonnet, J. Med. Chem., 2000, 43, 4582 CrossRef CAS PubMed.
- For recent selected examples, see: (a) C. L. Cioffi, B. Racz, A. Varadi, E. E. Freeman, M. P. Conlon, P. Chen, L. Zhu, D. B. Kitchen, K. D. Barnes, W. H. Martin, P. G. Pearson, G. Johnson, W. S. Blaner and K. Petrukhin, J. Med. Chem., 2019, 62, 5470 CrossRef CAS PubMed; (b) B. M. Williams and D. Trauner, Angew. Chem., Int. Ed., 2016, 55, 2191 CrossRef CAS PubMed; (c) A. A. Mazurov, D. C. Kombo, S. Akireddy, S. Murthy, T. A. Hauser, K. G. Jordan, G. J. Gatto and D. Yohannes, Bioorg. Med. Chem. Lett., 2013, 23, 3927 CrossRef CAS PubMed; (d) M. Rey-Carrizo, S. Gazzarrini, S. Llabrés, M. Frigolé-Vivas, J. Juárez-Jiménez, M. Font-Bardia, L. Naesens, A. Moroni, F. J. Luque and S. Vázquez, Eur. J. Med. Chem., 2015, 96, 318 CrossRef CAS PubMed; (e) J. M. LaLonde, M. A. Elban, J. R. Courter, A. Sugawara, T. Soeta, N. Madani, A. M. Princiotto, Y. D. Kwon, P. D. Kwong, A. Schoen, E. Freire, J. Sodroski and A. B. Smith, Bioorg. Med. Chem., 2011, 19, 91 CrossRef CAS PubMed; (f) L. Shao, M. C. Hewitt, S. C. Malcolm, F. Wang, J. Ma, U. C. Campbell, N. A. Spicer, S. R. Engel, L. W. Hardy, Z.-D. Jiang, R. Schreiber, K. L. Spear and M. A. Varney, J. Med. Chem., 2011, 54, 5283 CrossRef CAS PubMed.
- For recent selected examples on the yne-ynamide cyclization, see: (a) B. Prabagar, R. K. Mallick, R. Prasad, V. Gandon and A. K. Sahoo, Angew. Chem., Int. Ed., 2019, 58, 2365 CrossRef CAS PubMed; (b) S. Dutta, R. K. Mallick, R. Prasad, V. Gandon and A. K. Sahoo, Angew. Chem., Int. Ed., 2019, 58, 2289 CrossRef CAS PubMed; (c) Q. Zhao, D. F. L. Rayo, D. Campeau, M. Daenen and F. Gagosz, Angew. Chem., Int. Ed., 2018, 57, 13603 CrossRef CAS PubMed; (d) W. Xu, G. Wang, X. Xin and Y. Liu, Org. Lett., 2018, 20, 3273 CrossRef CAS PubMed; (e) W.-B. Shen, B. Zhou, Z.-X. Zhang, H. Yuan, W. Fang and L.-W. Ye, Org. Chem. Front., 2018, 5, 2468 RSC; (f) T. Wang and T. R. Hoye, J. Am. Chem. Soc., 2016, 138, 13870 CrossRef CAS PubMed; (g) T. Wang, D. Niu and T. R. Hoye, J. Am. Chem. Soc., 2016, 138, 7832 CrossRef CAS PubMed; (h) Y. Tokimizu, M. Wieteck, M. Rudolph, S. Oishi, N. Fujii, A. S. K. Hashmi and H. Ohno, Org. Lett., 2015, 17, 604 CrossRef CAS PubMed; for an asymmetric yne-ynamide cyclization, see: (i) J. Febvay, Y. Sanogo, P. Retailleau, M. P. Gogoi, A. K. Sahoo, A. Marinetti and A. Voituriez, Org. Lett., 2019, 21, 9281 CrossRef CAS PubMed.
- For details, see the ESI.†.
- CCDC 2046032 (2s) and 2046033 (3pc) contains the supplementary crystallographic data for this paper.†.
- For the relevant stereospecific 1,3-H transfer, see: (a) D. M. H. Ascough, F. Duarte and R. S. Paton, J. Am. Chem. Soc., 2018, 140, 16740 CrossRef CAS PubMed; (b) C. Hedberg and P. G. Andersson, Adv. Synth. Catal., 2005, 347, 662 CrossRef CAS; (c) W. M. Clark, A. M. Tickner-Eldridge, G. K. Huang, L. N. Pridgen, M. A. Olsen, R. J. Mills, I. Lantos and N. H. A. Baine, J. Am. Chem. Soc., 1998, 120, 4550 CrossRef CAS.
- C. M. Krauter, A. S. K. Hashmi and M. Pernpointner, ChemCatChem, 2010, 2, 1226 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2046032 and 2046033. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc02773e |
| ‡ X.-Q. Z. and P. H. contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |