
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIndirect reduction of CO2 and recycling of polymers by manganese-catalyzed transfer hydrogenation of amides, carbamates, urea derivatives, and polyurethanes†
Xin
Liu
a and
Thomas
Werner
 *ab
*ab
aLeibniz-Institute for Catalysis, Albert-Einstein-Str. 29a, 18059 Rostock, Germany. E-mail: thomas.werner@catalysis.de
bDepartment of Chemistry, Paderborn University, Warburger Str. 100, 33098 Paderborn, Germany
First published on 29th June 2021
Abstract
The reduction of polar bonds, in particular carbonyl groups, is of fundamental importance in organic chemistry and biology. Herein, we report a manganese pincer complex as a versatile catalyst for the transfer hydrogenation of amides, carbamates, urea derivatives, and even polyurethanes leading to the corresponding alcohols, amines, and methanol as products. Since these compound classes can be prepared using CO2 as a C1 building block the reported reaction represents an approach to the indirect reduction of CO2. Notably, these are the first examples on the reduction of carbamates and urea derivatives as well as on the C–N bond cleavage in amides by transfer hydrogenation. The general applicability of this methodology is highlighted by the successful reduction of 12 urea derivatives, 26 carbamates and 11 amides. The corresponding amines, alcohols and methanol were obtained in good to excellent yields up to 97%. Furthermore, polyurethanes were successfully converted which represents a viable strategy towards a circular economy. Based on control experiments and the observed intermediates a feasible mechanism is proposed.
Introduction
The hydrogenation and transfer hydrogenation of polar bonds, in particular carbonyl groups, has attracted great attention during the past four decades.1–7 These reactions are of synthetic significance as they represent an environmentally benign approach to fundamental synthetic building blocks such as alcohols and amines.8–10 Significant progress has been made in the (transfer) hydrogenation of ketones11–14 and aldehydes15–18 as well as of more challenging substrates such as esters19–21 and amides.22–24 However, the (transfer) hydrogenation of organic urea derivatives and carbamates as well as the C–N bond cleavage in amides remain a major challenge. Indeed, to the best of our knowledge, catalytic transfer hydrogenations of these important compound classes have not been reported so far. Notably, amides,25–27 carbamates,28–30 urea derivatives31–33 as well as polyurethanes34,35 can be produced from CO2, and their hydrogenation offers a facile approach to the indirect reduction of CO2 to methanol.36 However, compared to other carbonyl derivatives these compounds are less reactive towards hydrogenation and nucleophilic attack to the carbonyl group (Fig. 1a).2 This can be ascribed to resonance stabilization (Fig. 1b). Amide resonance is leading to the delocalization of the nitrogen electronic lone pair which lowers the reactivity of the carbonyl group (Fig. 1b, I).37 Also, intermolecular hydrogen bonding between amide groups can increase their stability additionally (Fig. 1b, II).38 In comparison with amides, carbamates and urea derivatives are even less reactive, mainly due to the additional resonance stabilization by the second oxygen or nitrogen atom, respectively (Fig. 1b, III and IV).39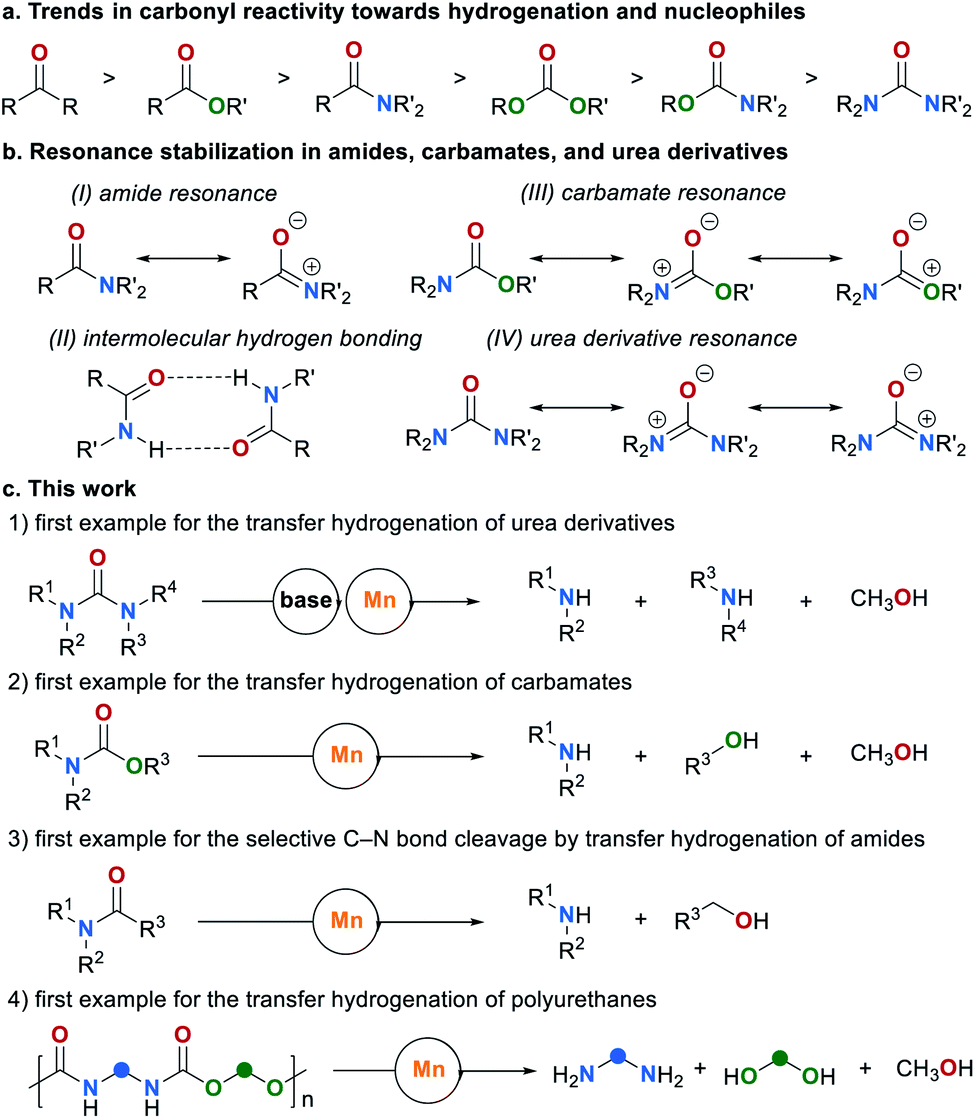 | ||
| Fig. 1 (a) Trends in carbonyl reactivity. (b) Resonance stabilization. (c) Manganese-catalyzed transfer hydrogenation of amides, carbamates, urea derivatives, and polyurethanes reported in this work. | ||
Numerous procedures have been developed for the reduction of amide-related substrates, e.g. hydrogenation,40 hydrosilylation41–43 and hydroborylation.44,45 Among them, hydrogenation methods stand out as a green approach. During the past decade, hydrogenation of amides through either C–N or C–O bond cleavage were widely studied. Surprisingly, only a few catalytic systems based on ruthenium have been reported for the hydrogenation for carbamates,36,46,47 and urea derivatives,48–51 respectively. Most recently, the group of Milstein reported a catalytic system based on earth abundant manganese for the hydrogenation for these compound classes.52 Direct hydrogenation and transfer hydrogenation are two parallel strategies for the reduction of carbonyl compounds.53,54 Complementary to the direct hydrogenation, transfer hydrogenation allows reductions to proceed without the need of pressurized H2 and special experimental settings. However, this strategy is mainly limited to the reduction of ketones55–57 and esters.21,58–60 Most recently, significant contributions were made in the transfer hydrogenation of amides.61–63 Klankermayer and co-workers reported an efficient Ru-catalyzed transfer hydrogenation of cyclic amides to cyclic tertiary amines,61 while the groups of Xu, Fan and Xiao reported the Ru- and B(C6F5)3- catalyzed reduction of aliphatic amides to amines.62,63 Notably, in all cases a selective C–O cleavage was observed over the possible C–N bond cleavage. Despite these advancements, methodologies to reduce carbamates and urea derivatives by transfer hydrogenation have not been reported. Based on our interest in the transfer hydrogenation64,65 and borrowing hydrogen reactions,66 we envisioned to overcome this limitation by using earth-abundant metal catalysts. Herein, we report the first examples of the transfer hydrogenation of carbamates, urea derivatives and amides under C–N bonds cleavage (Fig. 1c). Even polyurethanes can be reduced to the corresponding diols, amines and methanol, realizing a transfer hydrogenative degradation of commercial polyurethane materials into valuable products.
Results and discussion
Investigation of reaction conditions
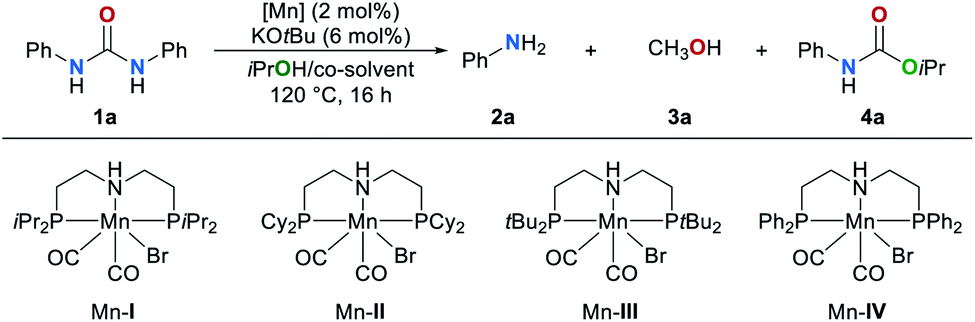
We began our studies by investigating the catalytic activities of a series of well-known manganese complexes67–73 in the transfer hydrogenation of 1,3-diphenylurea (1a) (Table 1). These reactions were performed in the presence of 2 mol% manganese complexes Mn–I–IV and 6 mol% of KOtBu (entries 1–4). We chose isopropanol (iPrOH) which is often used in transfer hydrogenation reactions as solvent and hydrogen donor. The manganese complex Mn–IV was found to be the most efficient catalyst (entry 4). In this case the amine 2a was obtained in 71% yield along with carbamate 4a in 54% and methanol in 45% yield, respectively. The formation of methanol clearly indicates that a transfer hydrogenation takes place. In the absence of catalyst Mn–IV the formation of methanol was not observed and the products 2a and 4a were obtained in 46% and 90% yield, respectively (entry 5). Other catalysts based on earth-abundant Co and Fe proved to be less efficient (for details, see ESI, Table S1†). Our group recently reported that cosolvents can increase the efficiency and selectivity of a hydrogen transfer reaction.64 Therefore, different solvents were evaluated as cosolvent for the reduction of 1a in the presence of catalyst Mn–IV (entries 6–9). An excellent yield of 2a (91%) and methanol 3a (89%) were obtained and no side product 4a was detected when toluene was used. Lower yields of both products were detected when the reaction temperature was reduced to 100 °C (entry 10). Other hydrogen donors, different bases, variation of the catalyst amount and reaction temperatures were also studied but did not lead to an improvement (for details, see ESI, Tables S2–S5†).|
|
|||||
|---|---|---|---|---|---|
| Entry | [Mn] | Co-solvent | 2a/% | 3a/% | 4a/% |
| a Reaction conditions: 1a (0.5 mmol), catalyst [Mn] (2 mol%), KOtBu (6 mol%), iPrOH (2 mL), 120 °C, 16 h. Yields were determined by GC using mesitylene as the internal standard. b iPrOH (1 mL) and cosolvent (1 mL). c 100 °C. | |||||
| 1 | Mn–I | — | 54 | 19 | 63 |
| 2 | Mn–II | — | 47 | 7 | 77 |
| 3 | Mn–III | — | 52 | — | 85 |
| 4 | Mn–IV | — | 71 | 45 | 54 |
| 5 | — | — | 46 | — | 90 |
| 6 | Mn–IV | CH2Cl2b | 39 | 5 | 35 |
| 7 | Mn–IV | THFb | 78 | 64 | 19 |
| 8 | Mn–IV | Tolueneb | 91 | 89 | — |
| 9 | Mn–IV | 1,4-Dioxaneb | 73 | 60 | 14 |
| 10c | Mn–IV | Tolueneb | 46 | 9 | 73 |
Substrate scope of urea derivatives
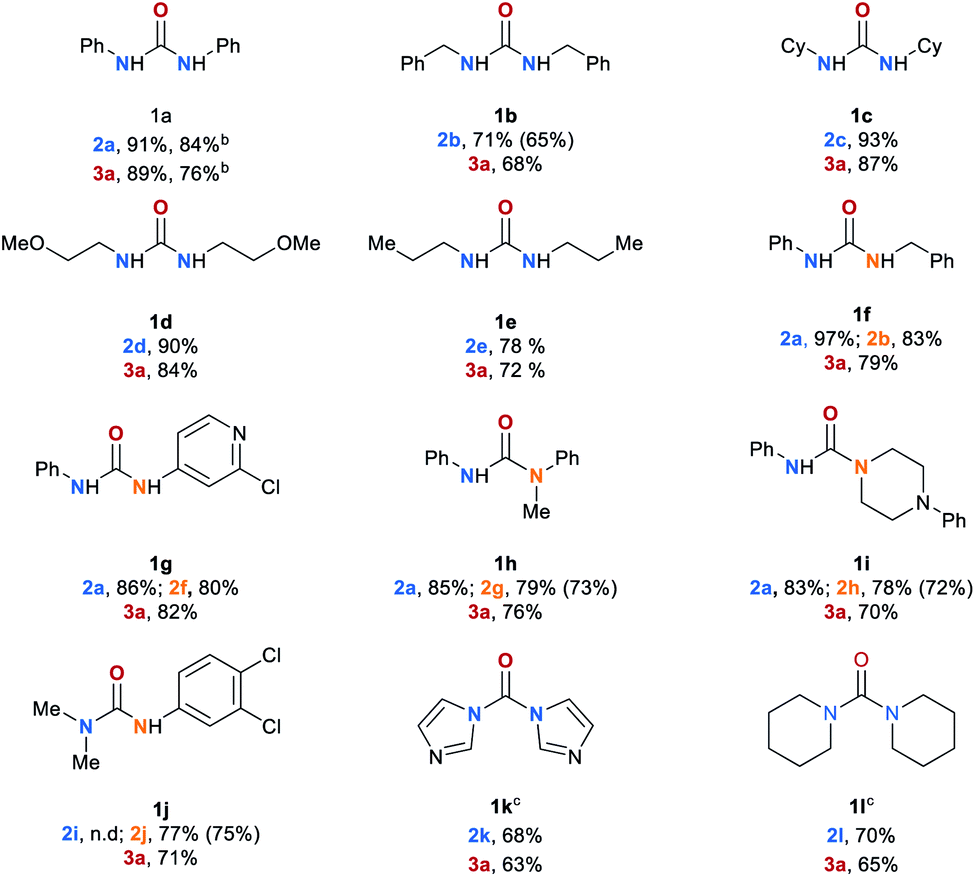
Having the optimized reaction conditions in hand, we explored the scope of the manganese-catalyzed transfer hydrogenation of different urea derivatives (Table 2). Under standard reaction conditions symmetrically substituted urea derivatives 1a–1e were converted to desired amines 2a–2e in up to 91% and methanol in up to 89% yield. The conversion of 1a was also performed on a 50 mmol scale leading to desired products, aniline (2a) and methanol (3a) in 84% and 76%, respectively. Unsymmetric urea derivatives 1f and 1g were converted to the respective amines 2a, 2b and 2f, in up to 97% yield. Notably, also trisubstituted urea derivatives 1h–1j as well as sterically hindered tetrasubstituted substrates 1k and 1l were selectively converted to methanol and the corresponding amines 2a, 2g–2j in good yields. Eventhough for the full conversion of 1k and 1l the reaction time had to be extended to 36 h.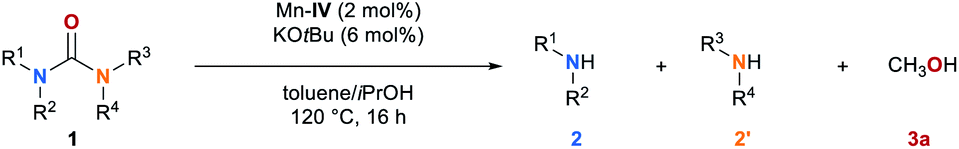
Mechanism study
Several control experiments were performed to shed light on the mechanism. Firstly, we monitored the conversion of the model substrate 1a under the standard reaction conditions (Fig. 2).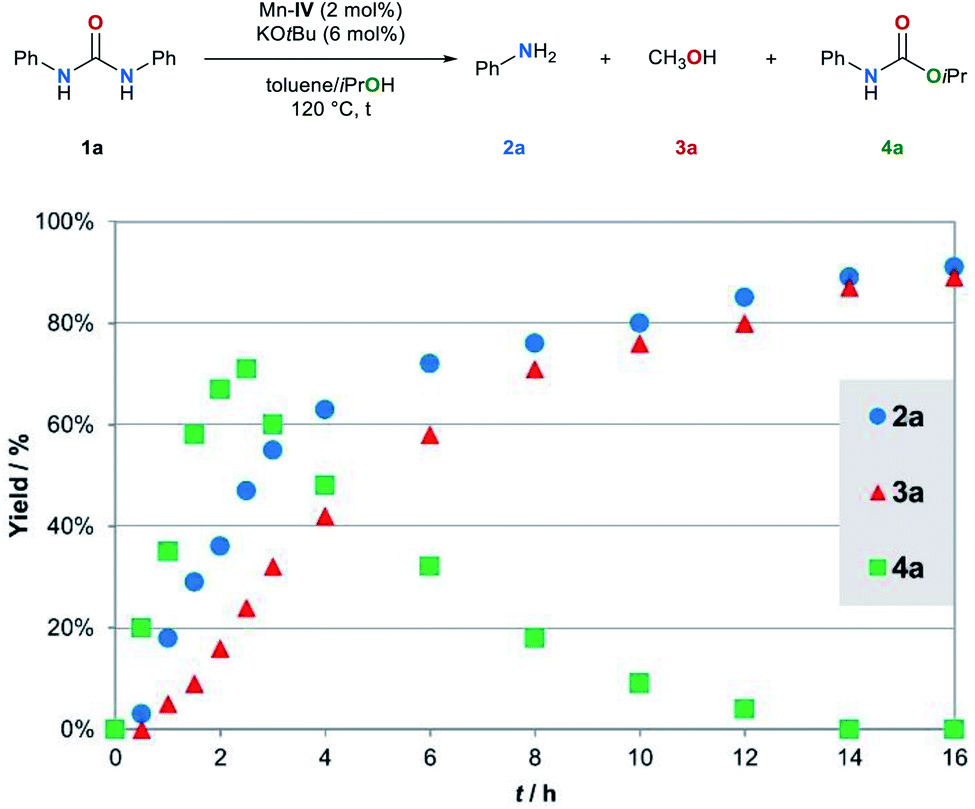 | ||
| Fig. 2 Kinetic profile for the transfer hydrogenation of 1a. Reaction conditions: 1a (0.5 mmol), Mn–IV (2 mol%), KOtBu (6 mol%), toluene (1 mL), iPrOH (1 mL), 120 °C, 16 h. The yield was determined by GC using mesitylene as the internal standard. | ||
The kinetic profile of the Mn–IV catalyzed transfer hydrogenation of 1a indicates that 1a is converted to 4a. Simultaneously, the formation of the amine 2a and methanol (3a) is observed. The yield of carbamate 4a reaches a maximum of 71% after 2.5 h. Subsequently, 4a is reduced to the amine 2a and methanol (3a). These results indicate that carbamate 4a is most likely a reaction intermediate in the transfer hydrogenation of urea derivatives. If carbamate 4a is converted under the standard reaction conditions full conversion is achieved after 12 h (for details, see ESI, Fig. S2†). The kinetic profile of this reaction also shows the simultaneous formation of amine 2a and methanol (3a).
Subsequently, a series of control experiments were performed (Fig. 3). In the absence of catalysts Mn–IV substrate 1a was fully converted in the presence of catalytic amounts of KOtBu (6 mol%) yielding the carbamate 4a in 90% along with the amine 2a in an expectable yield of 46% (Fig. 3a). Notably, the synthesis of carbamates from urea derivatives and alcohols typically requires high reaction temperatures and metal catalysts.74–76 To the best of our knowledge, a base-promoted conversion of urea derivatives to carbamates has not been reported so far. Other bases gave only poor or no conversion (for details, see ESI, Scheme S1†). In the absence of KOtBu no conversion of 1a was observed which indicates again that carbamate 4a is a reaction intermediate. If hydrogen (5 bar) is used instead of the hydrogen donor isopropanol no conversion was achieved which supports that indeed a transfer hydrogenation takes place under the standard conditions. In addition, the reaction of the proposed intermediate 4a under the standard reaction conditions, yields the corresponding amine 2a and methanol (3a) in excellent yields of 92% and 84%, respectively (Fig. 3b). formanilide (5a) was proposed by Milstein as an intermediate in the hydrogenation of carbamates and urea derivatives.52 The Formamide 5a was not observed under the standard conditions, but it was detected at lower base loading (4 mol% KOtBu), temperature (110 °C) and short reaction time (3 h) by GC and GCMS (for details, see ESI, Fig. S6†). Intermediate 5a was also successfully converted to 2a and methanol (3a) under our standard conditions (Fig. 3b and ESI, Fig. S3†).
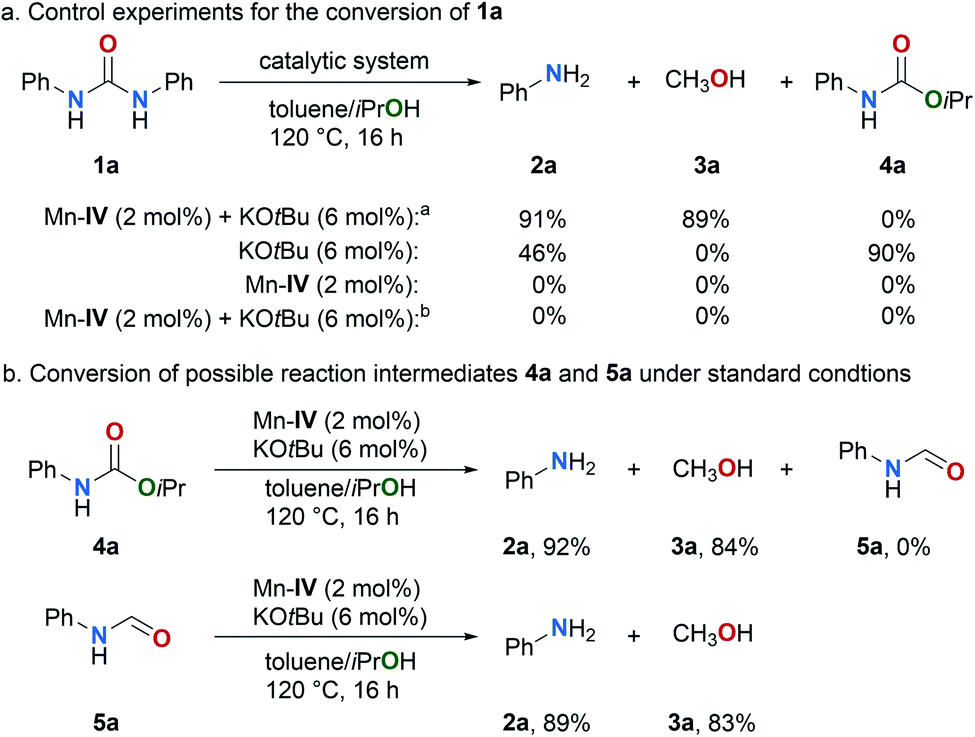 | ||
| Fig. 3 Control experiments for the conversion of 1a and possible intermediates 4a and 5a. a Result from Table 1, entry 8. b H2 (5 bar) was used instead of iPrOH. | ||
Considering these results and previous reports,36,48,52 we propose the catalytic cycle shown in Fig. 4. Initially, the pre-catalyst Mn–IV is deprotonated by KOtBu to afford the complex Mn–IV-1. Subsequently, dehydrogenation of iPrOH by Mn–IV-1 leads to complex Mn–IV-2. Hydrogen transfer from complex Mn–IV-2 to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O group of the carbamate intermediate 4a furnishes Mn–IV-3. The formation of 4a from 1a and iPrOH was demonstrated to be a catalytic process, and KOtBu has been proven as an efficient catalyst for this reaction (see above). Elimination of iPrOH, from complex Mn–IV-3 leads to Mn–IV-4 by metal–ligand cooperation. The release of formamide 5a regenerates catalyst Mn–IV-1. The reaction of 5a with Mn–IV-2 gives complex Mn–IV-5, which eliminates the final product, amine 2 and Mn–IV-6. Finally, formaldehyde (6) can be reduced to methanol (3a).
O group of the carbamate intermediate 4a furnishes Mn–IV-3. The formation of 4a from 1a and iPrOH was demonstrated to be a catalytic process, and KOtBu has been proven as an efficient catalyst for this reaction (see above). Elimination of iPrOH, from complex Mn–IV-3 leads to Mn–IV-4 by metal–ligand cooperation. The release of formamide 5a regenerates catalyst Mn–IV-1. The reaction of 5a with Mn–IV-2 gives complex Mn–IV-5, which eliminates the final product, amine 2 and Mn–IV-6. Finally, formaldehyde (6) can be reduced to methanol (3a).
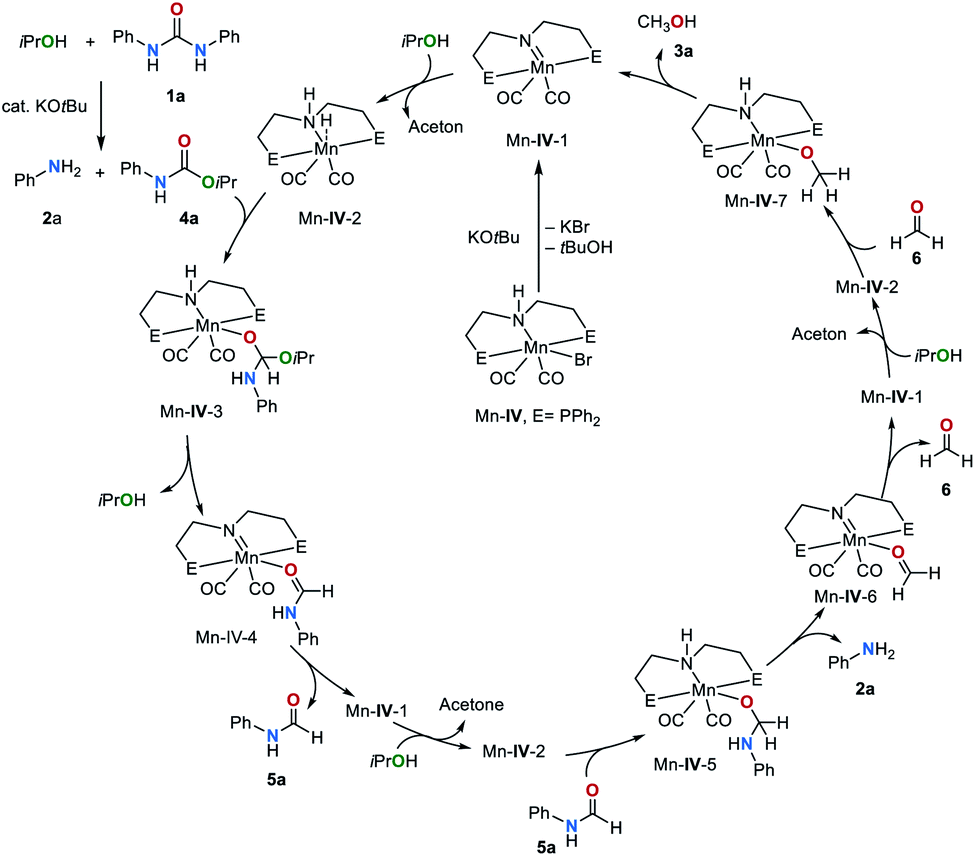 | ||
| Fig. 4 Proposed mechanism for the transfer hydrogenation of urea derivatives by catalyst Mn–IV. | ||
Substrate scope of carbamates
Encouraged by these results, we sought to extend the substrate scope to carbamates 4 (Table 3). Thus, carbamates bearing heteroaromatic substituents 4b–4d were successfully reduced to amines and methanol in excellent yields of up to 94% and 88% respectively. In an upscaling experiment 50 mmol of 4b were converted to 2a and methanol in 84% and 76%, respectively. Additionally, carbamates with different functional groups have been tested under standard conditions (for details, see ESI, Table S12†). Carbamates bearing aliphatic and benzylic substituents 4e–4h were also converted to the desired products in yields up to 80%. The conversion of sterically more demanding disubstituted carbamates 4i–4n also proceeded smoothly leading to the corresponding amines and methanol in yields up to 83% and 76%, respectively. Finally, we studied the reaction of carbamates with different alkoxy groups (4o–4q). The respective amines, alcohols and methanol were obtained in good yield even though in some cases the reaction temperature had to be increased to 140 °C.
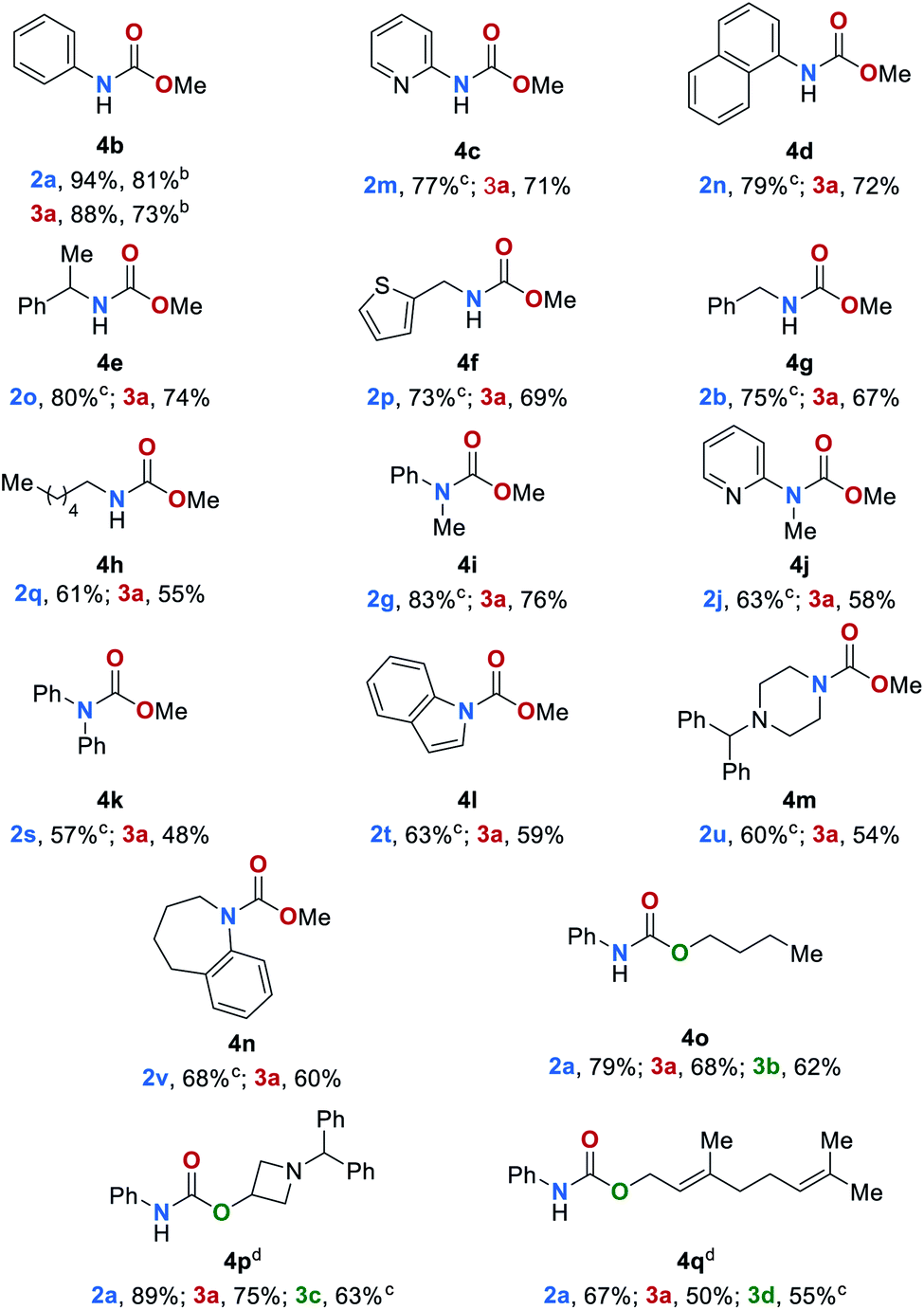
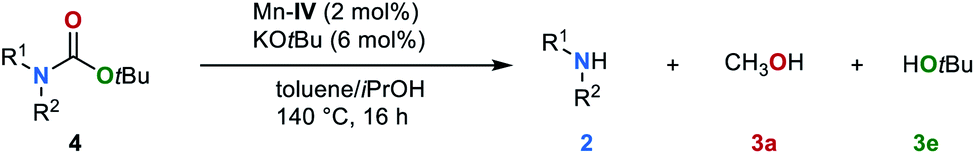
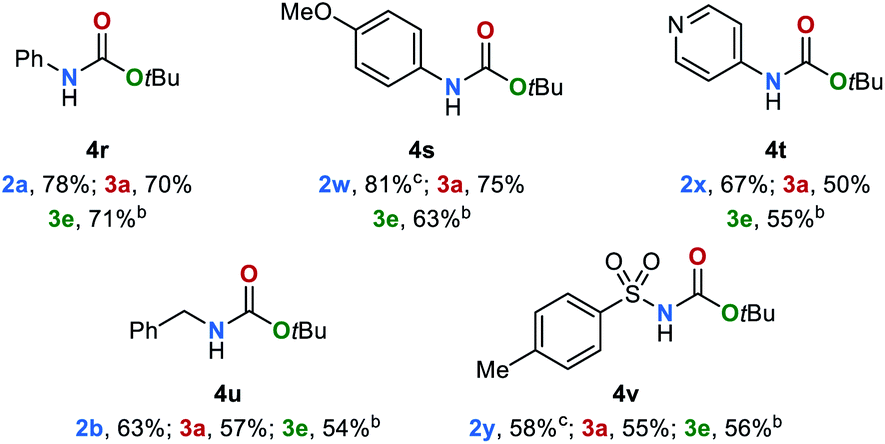
The tert-butyloxycarbonyl protecting group (Boc) is frequently used in organic synthesis to protect amines as their corresponding carbamates. The Boc group is stable to nucleophilic reagents, hydrogenolysis and base hydrolysis. Traditional approaches for N-Boc deprotection are based on Brønsted acids.77 We envisioned that our method might be suitable for the deprotection of N-Boc protected amines under transfer hydrogenation conditions. However, the reduction of tert-butyl phenylcarbamate (4r) gives low yields of the corresponding products under our standard conditions (for details, see ESI, Table S7†). To promote the reaction, we then evaluated the effect of temperature. By increasing the temperature to 140 °C good yields of aniline (2a), methanol (3a) and tert-butyl alcohol (3e) were observed (78%, 70% and 71%, respectively). Next, the scope for the conversion of N-Boc protected amines 4s–4v was explored (Table 4). The desired amines were obtained in yields up to 81% indicating the feasibility of this alternative deprotection approach.
Substrate scope of amides
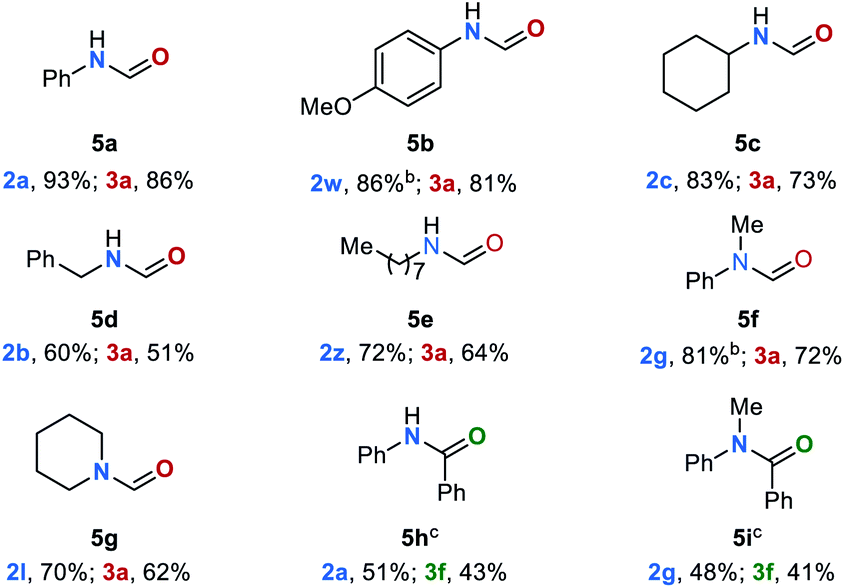
To the best of our knowledge, so far there is no precedence in the literature on C–N bond cleavage in amides via transfer hydrogenation. Encouraged by the above-mentioned observation of formamide 5a as an intermediate of the reduction of urea derivative 1a as well as the possibility of efficiently reducing 5a to the corresponding amine 2a and methanol under our standard conditions (Fig. 3b), the general potential of this reaction was evaluated. Various amides 5 in the presence of catalyst Mn–IV (2 mol%) and KOtBu (6 mol%) were converted (Table 5). Notably, in these cases a reaction temperature of 100 °C was sufficient (for details, see ESI, Table S6†). Aromatic formamides 5a and 5b gave excellent yields of 91% and 86%, respectively on the corresponding amine. The (cyclo-)aliphatic formamide derivatives 5c–5e were successfully converted to the respective amines yielding the desired products in up to 73%, while the disubstituted formamides 5f and 5g gave the corresponding products in 81% and 72% yield. To further extend the substrate scope, we evaluated the reaction of benzanilide (5h) and N-methylphenyl benzamide (5i). Under our standard reaction conditions C–N bond cleavage also occurred, leading to the corresponding amines and benzyl alcohol (3f) in moderate yields up to 51%.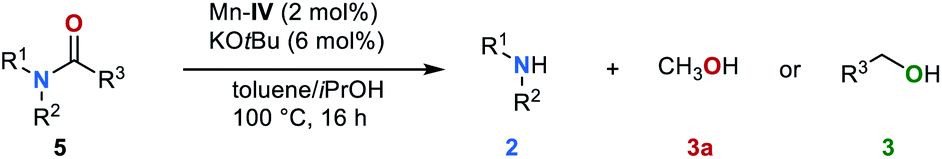
Recycling of polyurethanes to valuable products
Polyurethanes are the 6th most used class of polymers worldwide with an annual production of 18 million tons per year.78 The application of polyurethanes ranges from foams, varnishes and adhesives to insulation materials. Notably, polyurethanes can also be prepared from CO2.34In order to establish a circular economy, the development of new recycling methods is of particular interest. Most recently, Milstein et al. and Schaub et al. demonstrated that the hydrogenation of polyurethane is possible,79,80 and valuable chemicals such as diols, diamines and methanol can be obtained. Thus, this new strategy was extended to the transfer hydrogenation of polyurethanes (Fig. 5). Under the optimized reaction conditions (for details, see ESI, Table S8†), commercially available polyurethane 7a was successfully reduced to the corresponding amine 8a, diol 9a and methanol in moderate yields of 65%, 52% and 47%, respectively. Furthermore, polyurethanes 7b and 7c were also depolymerized smoothly. In the case of the depolymerisation of 7b the diamine 8b and diol 9b were obtained in isolated yields of 41 and 34%, respectively.
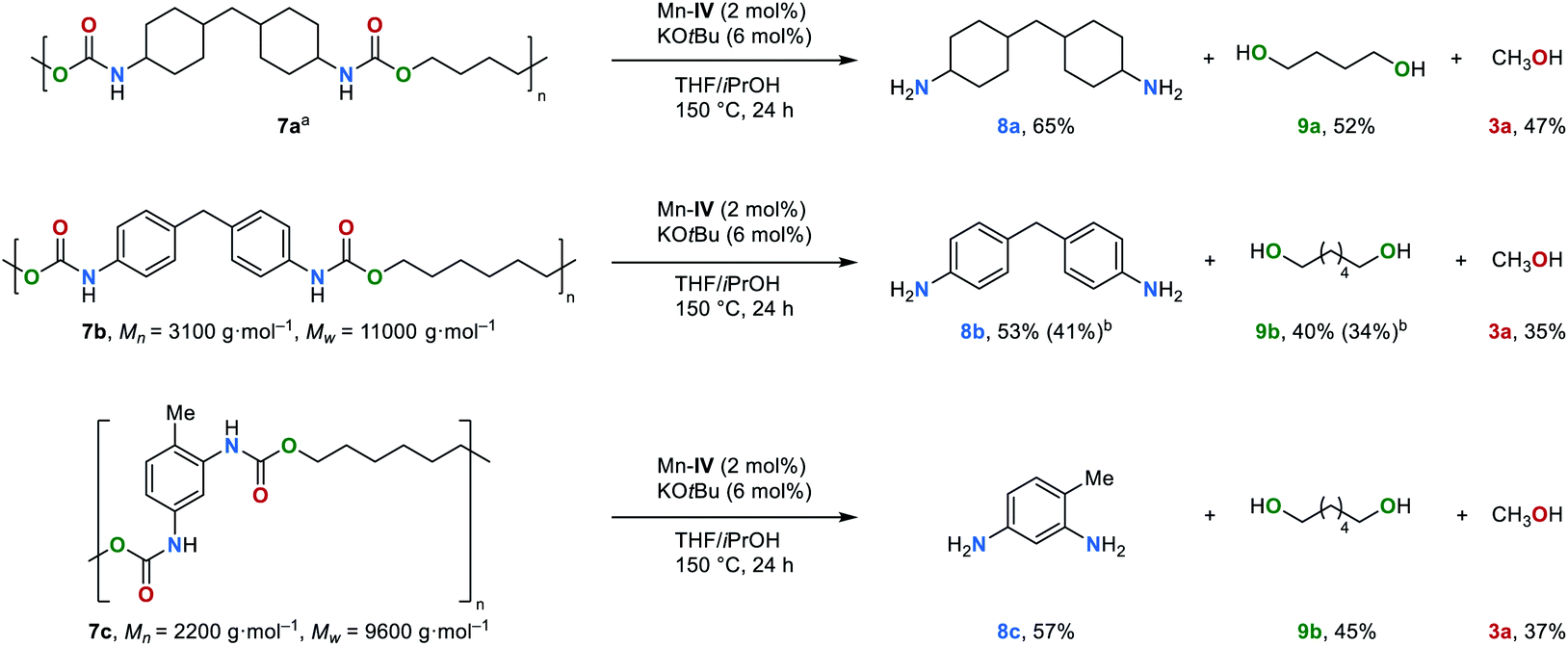 | ||
| Fig. 5 First example on the transfer hydrogenation of polyurethanes 7 (0.5 mmol of substrate was used according to the repeating unit of polyurethanes). aMolecular weight could not be determined.79bIsolated yield. | ||
Conclusions
The reported method is a highly efficient transfer hydrogenation protocol for the reduction of amides, carbamates, urea derivatives and even polyurethane using a pincer catalyst based on earth-abundant manganese and catalytic amounts of KOtBu. This catalytic system allowed for the transfer hydrogenation of a variety of simple as well as highly substituted carbamate and urea derivatives in good to excellent yields. Thus, this method represents a facile possibility for the indirect reduction of CO2. The conversion of carbamates was extended to N-Boc protected amines which highlights the potential use of transfer hydrogenation as an alternative method for the deprotection of N-Boc protected amines. Moreover, a polyurethane was successfully converted into valuable diols, amines and methanol. This represents a viable strategy for a circular plastic economy for polyurethane.Data availability
The ESI include experimental detail, NMR data and HRMS data.Author contributions
X. L. conceptualized and performed the investigations, analysed and validated the obtained data. He also wrote the first draft of the manuscript. T. W. supervised and conceptualized the project. He reviewed and edited the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Xin Liu is grateful for the financial support of the Chinese Scholarship Council (CSC, 201708530236). This work was conducted in the scope of the Leibniz Science Campus Phosphorus Research. We also thank Libo Yao, Dr Lars Longwitz, Dr Wu Li and Dr Duo Wei as well as Jan Tönjes for helpful discussions.Notes and references
- P. Mäki-Arvela, J. Hájek, T. Salmi and D. Y. Murzin, Appl. Catal., A, 2005, 292, 1–49 CrossRef.
- P. A. Dub and T. Ikariya, ACS Catal., 2012, 2, 1718–1741 CrossRef CAS.
- A. M. Smith and R. Whyman, Chem. Rev., 2014, 114, 5477–5510 CrossRef CAS PubMed.
- S. Werkmeister, K. Junge and M. Beller, Org. Process Res. Dev., 2014, 18, 289–302 CrossRef CAS.
- D. Wang and D. Astruc, Chem. Rev., 2015, 115, 6621–6686 CrossRef CAS PubMed.
- A. Volkov, F. Tinnis, T. Slagbrand, P. Trillo and H. Adolfsson, Chem. Soc. Rev., 2016, 45, 6685–6697 RSC.
- J. R. Cabrero-Antonino, R. Adam, V. Papa and M. Beller, Nat. Commun., 2020, 11, 1–18 Search PubMed.
- S. Das, B. Join, K. Junge and M. Beller, Chem. Commun., 2012, 48, 2683–2685 RSC.
- S. Das, Y. Li, L. Q. Lu, K. Junge and M. Beller, Chem.–Eur. J., 2016, 22, 7050–7053 CrossRef CAS PubMed.
- S. Das, Y. Li, C. Bornschein, S. Pisiewicz, K. Kiersch, D. Michalik, F. Gallou, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2015, 127, 12566–12570 CrossRef.
- R. H. Morris, Chem. Soc. Rev., 2009, 38, 2282–2291 RSC.
- R. Malacea, R. Poli and E. Manoury, Coord. Chem. Rev., 2010, 254, 729–752 CrossRef CAS.
- R. Langer, G. Leitus, Y. Ben-David and D. Milstein, Angew. Chem., Int. Ed., 2011, 50, 2120–2124 CrossRef CAS PubMed.
- Y.-Y. Li, S.-L. Yu, W.-Y. Shen and J.-X. Gao, Acc. Chem. Res., 2015, 48, 2587–2598 CrossRef CAS.
- J. W. Yang, M. T. Hechavarria Fonseca, N. Vignola and B. List, Angew. Chem., Int. Ed., 2005, 44, 108–110 CrossRef CAS PubMed.
- X. Wu, J. Liu, X. Li, A. Zanotti-Gerosa, F. Hancock, D. Vinci, J. Ruan and J. Xiao, Angew. Chem., Int. Ed., 2006, 45, 6718–6722 CrossRef CAS PubMed.
- S. Mazza, R. Scopelliti and X. Hu, Organometallics, 2015, 34, 1538–1545 CrossRef CAS.
- X. Lan and T. Wang, ACS Catal., 2020, 10, 2764–2790 CrossRef CAS.
- J. Zhang, G. Leitus, Y. Ben-David and D. Milstein, Angew. Chem., Int. Ed., 2006, 118, 1131–1133 CrossRef.
- L. A. Saudan, C. M. Saudan, C. Debieux and P. Wyss, Angew. Chem., Int. Ed., 2007, 119, 7617–7620 CrossRef.
- R. A. Farrar-Tobar, B. Wozniak, A. Savini, S. Hinze, S. Tin and J. G. de Vries, Angew. Chem., Int. Ed., 2019, 58, 1129–1133 CrossRef CAS PubMed.
- S. E. Clapham, A. Hadzovic and R. H. Morris, Coord. Chem. Rev., 2004, 248, 2201–2237 CrossRef CAS.
- A. A. N. Magro, G. R. Eastham and D. J. Cole-Hamilton, Chem. Commun., 2007, 3154–3156 RSC.
- E. Balaraman, B. Gnanaprakasam, L. J. Shimon and D. Milstein, J. Am. Chem. Soc., 2010, 132, 16756–16758 CrossRef CAS PubMed.
- X. F. Liu, X. Y. Li, C. Qiao, H. C. Fu and L. N. He, Angew. Chem., Int. Ed., 2017, 56, 7425–7429 CrossRef CAS PubMed.
- X. Y. Mak, R. P. Ciccolini, J. M. Robinson, J. W. Tester and R. L. Danheiser, J. Org. Chem., 2009, 74, 9381–9387 CrossRef CAS PubMed.
- S. Bongarzone, A. Runser, C. Taddei, A. H. Dheere and A. Gee, Chem. Commun., 2017, 53, 5334–5337 RSC.
- W. Schilling and S. Das, ChemSusChem, 2020, 13, 6246–6258 CAS.
- S. L. Peterson, S. M. Stucka and C. J. Dinsmore, Org. Lett., 2010, 12, 1340–1343 CrossRef CAS PubMed.
- R. Yousefi, T. J. Struble, J. L. Payne, M. Vishe, N. D. Schley and J. N. Johnston, J. Am. Chem. Soc., 2018, 141, 618–625 CrossRef PubMed.
- M. Xu, A. R. Jupp, M. S. Ong, K. I. Burton, S. S. Chitnis and D. W. Stephan, Angew. Chem., Int. Ed., 2019, 58, 5707–5711 CrossRef CAS PubMed.
- F. Shi, Y. Deng, T. SiMa, J. Peng, Y. Gu and B. Qiao, Angew. Chem., Int. Ed., 2003, 115, 3379–3382 CrossRef.
- A. Ion, V. Parvulescu, P. Jacobs and D. De Vos, Green Chem., 2007, 9, 158–161 RSC.
- J. Langanke, A. Wolf, J. Hofmann, K. Böhm, M. Subhani, T. Müller, W. Leitner and C. Gürtler, Green Chem., 2014, 16, 1865–1870 RSC.
- O. Ihata, Y. Kayaki and T. Ikariya, Angew. Chem., Int. Ed., 2004, 116, 735–737 CrossRef.
- E. Balaraman, C. Gunanathan, J. Zhang, L. J. Shimon and D. Milstein, Nat. Chem., 2011, 3, 609–614 CrossRef CAS PubMed.
- L. Pauling, J. Am. Chem. Soc., 1931, 53, 1367–1400 CrossRef CAS.
- E. S. Eberhardt and R. T. Raines, J. Am. Chem. Soc., 1994, 116, 2149–2150 CrossRef CAS PubMed.
- D. Kaur, P. Sharma and P. V. Bharatam, J. Mol. Struct., 2005, 757, 149–153 CrossRef CAS.
- G. J. Kubas, Chem. Rev., 2007, 107, 4152–4205 CrossRef CAS PubMed.
- S. Das, S. Zhou, D. Addis, S. Enthaler, K. Junge and M. Beller, Top. Catal., 2010, 53, 979–984 CrossRef CAS.
- D. Addis, S. Das, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2011, 50, 6004–6011 CrossRef CAS PubMed.
- A. Chardon, E. Morisset, J. Rouden and J. Blanchet, Synthesis, 2018, 50, 984–997 CrossRef CAS.
- N. L. Lampland, M. Hovey, D. Mukherjee and A. D. Sadow, ACS Catal., 2015, 5, 4219–4226 CrossRef CAS.
- M. Magre, M. Szewczyk and M. Rueping, Org. Lett., 2020, 22, 3209–3214 CrossRef CAS PubMed.
- M. Ito, L. W. Koo, A. Himizu, C. Kobayashi, A. Sakaguchi and T. Ikariya, Angew. Chem., Int. Ed., 2009, 48, 1324–1327 CrossRef CAS PubMed.
- J. M. John, R. Loorthuraja, E. Antoniuk and S. H. Bergens, Catal. Sci. Technol., 2015, 5, 1181–1186 RSC.
- E. Balaraman, Y. Ben-David and D. Milstein, Angew. Chem., Int. Ed., 2011, 123, 11906–11909 CrossRef.
- T. Miura, I. E. Held, S. Oishi, M. Naruto and S. Saito, Tetrahedron Lett., 2013, 54, 2674–2678 CrossRef CAS.
- T. vom Stein, M. Meuresch, D. Limper, M. Schmitz, M. Hölscher, J. Coetzee, D. J. Cole-Hamilton, J. Klankermayer and W. Leitner, J. Am. Chem. Soc., 2014, 136, 13217–13225 CrossRef CAS PubMed.
- Y. Xie, P. Hu, Y. Ben-David and D. Milstein, Angew. Chem., Int. Ed., 2019, 58, 5105–5109 CrossRef CAS PubMed.
- U. K. Das, A. Kumar, Y. Ben-David, M. A. Iron and D. Milstein, J. Am. Chem. Soc., 2019, 141, 12962–12966 CrossRef CAS PubMed.
- G. Brieger and T. J. Nestrick, Chem. Rev., 1974, 74, 567–580 CrossRef CAS.
- C. Zheng and S.-L. You, Chem. Soc. Rev., 2012, 41, 2498–2518 RSC.
- S. Hashiguchi, A. Fujii, J. Takehara, T. Ikariya and R. Noyori, J. Am. Chem. Soc., 1995, 117, 7562–7563 CrossRef CAS.
- G. Zhang and S. K. Hanson, Chem. Commun., 2013, 49, 10151–10153 RSC.
- M. Perez, S. Elangovan, A. Spannenberg, K. Junge and M. Beller, ChemSusChem, 2017, 10, 83–86 CrossRef CAS PubMed.
- S. H. Lee and G. I. Nikonov, ChemCatChem, 2015, 7, 107–113 CrossRef CAS.
- A. Dubey and E. Khaskin, ACS Catal., 2016, 6, 3998–4002 CrossRef CAS.
- C. L. Oates, M. B. Widegren and M. L. Clarke, Chem. Commun., 2020, 56, 8635–8638 RSC.
- S. Westhues, M. Meuresch and J. Klankermayer, Angew. Chem., Int. Ed., 2016, 55, 12841–12844 CrossRef CAS PubMed.
- Y. Pan, Z. Luo, X. Xu, H. Zhao, J. Han, L. Xu, Q. Fan and J. Xiao, Adv. Synth. Catal., 2019, 361, 3800–3806 CrossRef CAS.
- Y. Pan, Z. Luo, J. Han, X. Xu, C. Chen, H. Zhao, L. Xu, Q. Fan and J. Xiao, Adv. Synth. Catal., 2019, 361, 2301–2308 CrossRef CAS.
- X. Liu, J. G. de Vries and T. Werner, Green Chem., 2019, 21, 5248–5255 RSC.
- X. Liu, L. Longwitz, B. Spiegelberg, J. Tönjes, T. Beweries and T. Werner, ACS Catal., 2020, 10, 13659–13667 CrossRef CAS.
- X. Liu and T. Werner, Adv. Synth. Catal., 2021, 363, 1096–1104 CrossRef CAS.
- S. Elangovan, C. Topf, S. Fischer, H. Jiao, A. Spannenberg, W. Baumann, R. Ludwig, K. Junge and M. Beller, J. Am. Chem. Soc., 2016, 138, 8809–8814 CrossRef CAS PubMed.
- S. Elangovan, J. Neumann, J.-B. Sortais, K. Junge, C. Darcel and M. Beller, Nat. Commun., 2016, 7, 1–8 Search PubMed.
- S. Fu, Z. Shao, Y. Wang and Q. Liu, J. Am. Chem. Soc., 2017, 139, 11941–11948 CrossRef CAS PubMed.
- A. Kaithal, L.-L. Gracia, C. m. Camp, E. A. Quadrelli and W. Leitner, J. Am. Chem. Soc., 2019, 141, 17487–17492 CrossRef CAS PubMed.
- Y. Wang, L. Zhu, Z. Shao, G. Li, Y. Lan and Q. Liu, J. Am. Chem. Soc., 2019, 141, 17337–17349 CrossRef CAS PubMed.
- J. C. Borghs, Y. Lebedev, M. Rueping and O. El-Sepelgy, Org. Lett., 2018, 21, 70–74 CrossRef PubMed.
- A. Kaithal, M. Hölscher and W. Leitner, Angew. Chem., Int. Ed., 2018, 57, 13449–13453 CrossRef CAS PubMed.
- P. Wang, Y. Ma, S. Liu, F. Zhou, B. Yang and Y. Deng, Green Chem., 2015, 17, 3964–3971 RSC.
- M. Peña-López, H. Neumann and M. Beller, ChemSusChem, 2016, 9, 2233–2238 CrossRef PubMed.
- Q. Li, P. Wang, S. Liu, Y. Fei and Y. Deng, Green Chem., 2016, 18, 6091–6098 RSC.
- T. W. Greene and P. G. Wuts, Protective groups in organic synthesis, Wiley, 1999 Search PubMed.
- D. Simón, A. Borreguero, A. De Lucas and J. Rodríguez, Waste Manage., 2018, 76, 147–171 CrossRef PubMed.
- A. Kumar, N. von Wolff, M. Rauch, Y.-Q. Zou, G. Shmul, Y. Ben-David, G. Leitus, L. Avram and D. Milstein, J. Am. Chem. Soc., 2020, 142, 14267–14275 CrossRef CAS PubMed.
- W. Zhou, P. Neumann, M. Al Batal, F. Rominger, A. S. K. Hashmi and T. Schaub, ChemSusChem, 2020, 13, 1–6 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc02663a |
| This journal is © The Royal Society of Chemistry 2021 |