
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCatalytic enantioselective synthesis of benzocyclobutenols and cyclobutanols via a sequential reduction/C–H functionalization†
Jun
Chen
,
Zhan
Shi
,
Chunyu
Li‡
and
Ping
Lu
 *
*
Research Center for Molecular Recognition and Synthesis, Department of Chemistry, Fudan University, 220 Handan Lu, Shanghai 200433, P. R. China. E-mail: plu@fudan.edu.cn
First published on 5th July 2021
Abstract
We report here a sequential enantioselective reduction/C–H functionalization to install contiguous stereogenic carbon centers of benzocyclobutenols and cyclobutanols. This strategy features a practical enantioselective reduction of a ketone and a diastereospecific iridium-catalyzed C–H silylation. Further transformations have been explored, including controllable regioselective ring-opening reactions. In addition, this strategy has been utilized for the synthesis of three natural products, phyllostoxin (proposed structure), grandisol and fragranol.
Molecules with inherent ring strain have gained considerable interest in the synthetic community.1 Among them, four-membered ring molecules have been recognized as powerful building blocks in organic synthesis.2 Driven by ring strain releasing, the reactions of carbon–carbon bond cleavage have been extensively studied in recent years.3 Meanwhile, cyclobutane motifs represent important structural units in natural product and bioactive molecules as well (Scheme 1).4 Therefore, a general and robust method to constitute four-membered ring derivatives is of great value, especially in an enantiomerically pure form.5
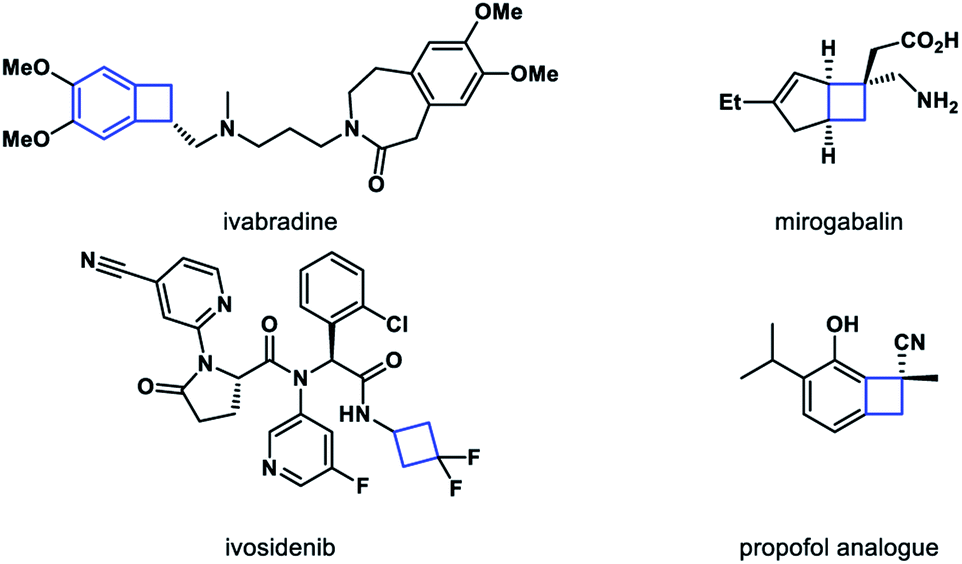 | ||
| Scheme 1 Representative cyclobutane-containing bioactive molecules. | ||
[2 + 2]-Cycloaddition6 and the skeleton rearrangement reaction7 are two primary methods to prepare chiral cyclobutane derivatives. Recently, the precision modification of four-membered ring skeletons to access enantioenriched cyclobutane derivatives has attracted emerging attention. Several strategies have been developed, including allylic alkylation,8 α-functionalization,9 conjugate addition10 and C–H functionalization11 of prochiral or racemic cyclobutane derivatives (Scheme 2a).12 However, the enantioselective synthesis of chiral benzocyclobutene derivatives is still underdeveloped.13 Although two efficient palladium-catalyzed C–H activation strategies have been developed by Baudoin14 and Martin15 groups via similar intermediate five-membered palladacycles, no enantioenriched benzocyclobutene derivative has been prepared by employing the above two methods. In 2017, Kawabata reported an elegant example of asymmetric intermolecular α-arylation of enantioenriched amino acid derivatives to afford benzocyclobutenones with tetrasubstituted carbon via memory of chirality (Scheme 2b).16 In 2018, Zhang reported an iridium-catalyzed asymmetric hydrogenation of α-alkylidene benzocyclobutenones in good enantioselectivities (3 examples, 83–88% ee).12c To the best of our knowledge, there is no report on enantioselective synthesis of benzocyclobutene derivatives with all-carbon quaternary centers.
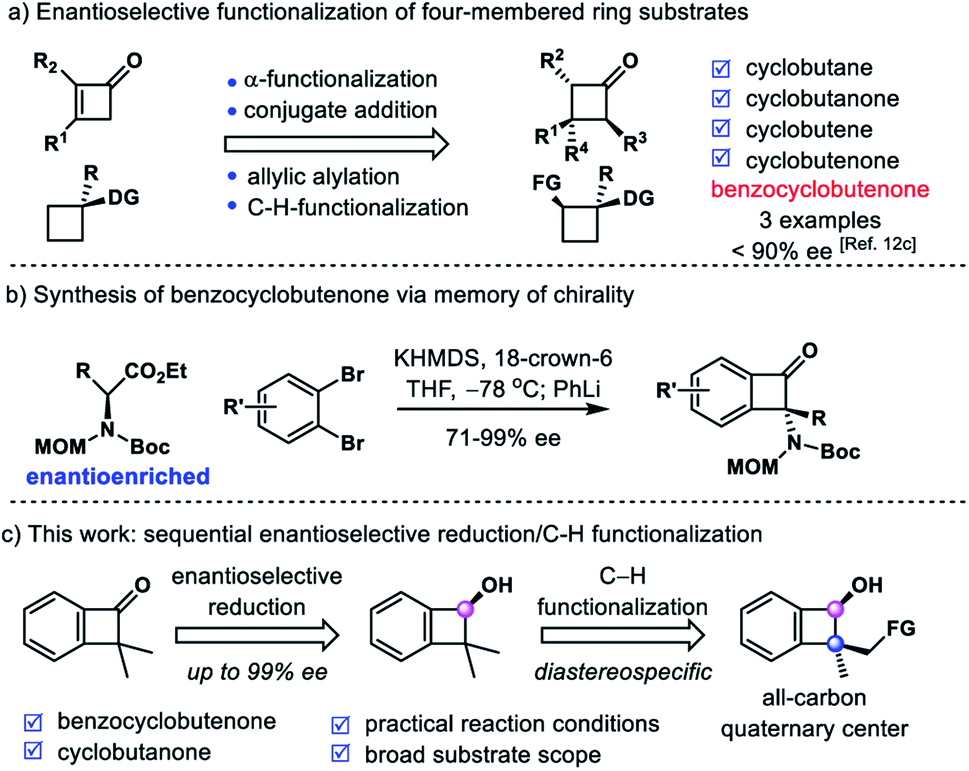 | ||
| Scheme 2 Asymmetric synthesis of cyclobutanes and their derivatives. (a) Enantioselective functionalization of four-membered ring substrates. (b) Synthesis of chiral benzocyclobutenone via memory of chirality. (c) This work: sequential enantioselective reduction/C–H functionalization. | ||
In line with our continued interest in precision modification of four-membered ring skeletons,9d,10c,12a we initiated our studies on the synthesis of chiral benzocyclobutenes via enantioselective functionalization of highly strained benzocyclobutenones. It is well known that benzocyclobutene derivatives are labile to undergo a ring-opening reaction to release their inherent ring strains.17 Therefore, it is a challenging task to modify the benzocyclobutenone and preserve the four-membered ring skeleton at the same time. We envisioned that a carbonyl group directed C–H functionalization18 of the gem-dimethyl group could furnish enantioenriched α-quaternary benzocyclobutenones (Scheme 2c). This could be viewed as an alternative approach to achieve the alkylation of benzocyclobutenone, which was otherwise directly inaccessible using enolate chemistry through the unstable anti-aromatic intermediate.19 In addition, a highly regioselective C–H activation would be required to functionalize the methyl group instead of the aryl ring. Here we report our work on sequential enantioselective reduction and intramolecular C–H silylation to provide enantioenriched benzocyclobutenols and cyclobutanols with all-carbon quaternary centers. The excellent diastereoselectivity and regioselectivity of silylation were attributed to rigid structural organization of the 4/5 fused ring. Furthermore, this strategy has been utilized to accomplish the total synthesis of natural products phyllostoxin (proposed structure), grandisol and fragranol.
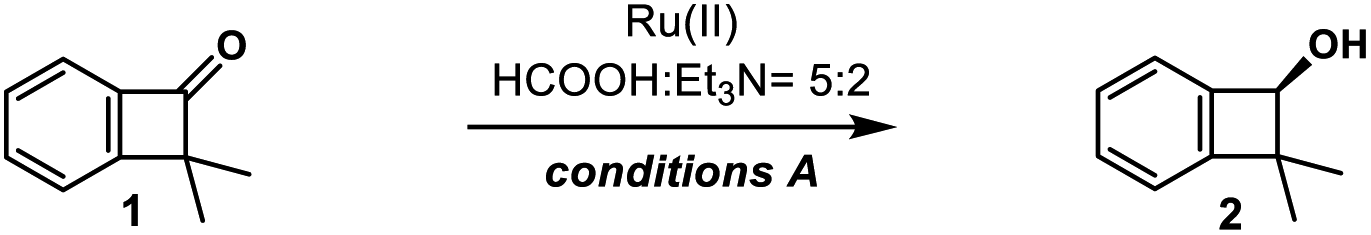
We commenced our studies with enantioselective reduction of readily prepared dimethylbenzocyclobutenone 1a (Scheme 3).15,20 Surprisingly, enantioselective reduction of the carbonyl group of cyclobutanone derivatives received little attention. The first reduction of parent benzocyclobutenone was studied in 1996 by Kündig using chlorodiisopinocamphenylborane21 or chiral oxazaborolidines (CBS reduction),22 and only moderate enantioselectivity (44–68% ee) was obtained.23 Although copper-catalyzed asymmetric hydrosilylation of benzocyclobutenone 1a using CuCl/(R)-BINAP gave the benzocyclobutenol ent-2a in 88% ee, optimization of ligands gave no further improvement (Scheme 3a, see Tables S1–S4† for details).24 Gladly, excellent enantioselective reduction could be achieved in 94% yield and 97% ee under Noyori's asymmetric transfer hydrogenation conditions (Scheme 3b, conditions A, RuCl[(S,S)-Tsdpen](p-cymene)).25 The product 2a showed remarkable stability and no ring-opening byproduct 2a′ was observed. The reduction of parent benzocyclobutenone was examined under conditions A, and benzocyclobutenol was obtained in 90% yield and 81% ee. Apparently, the steric influence imposed by the α-dimethyl group enhanced the enantioselectivity of the reduction. Similarly, the CBS reduction ((S)-B–Me) of benzocyclobutenone 1a gave better results compared with parent benzocyclobutenone, affording the product 2a in 86% yield and 92% ee (Scheme 3c).
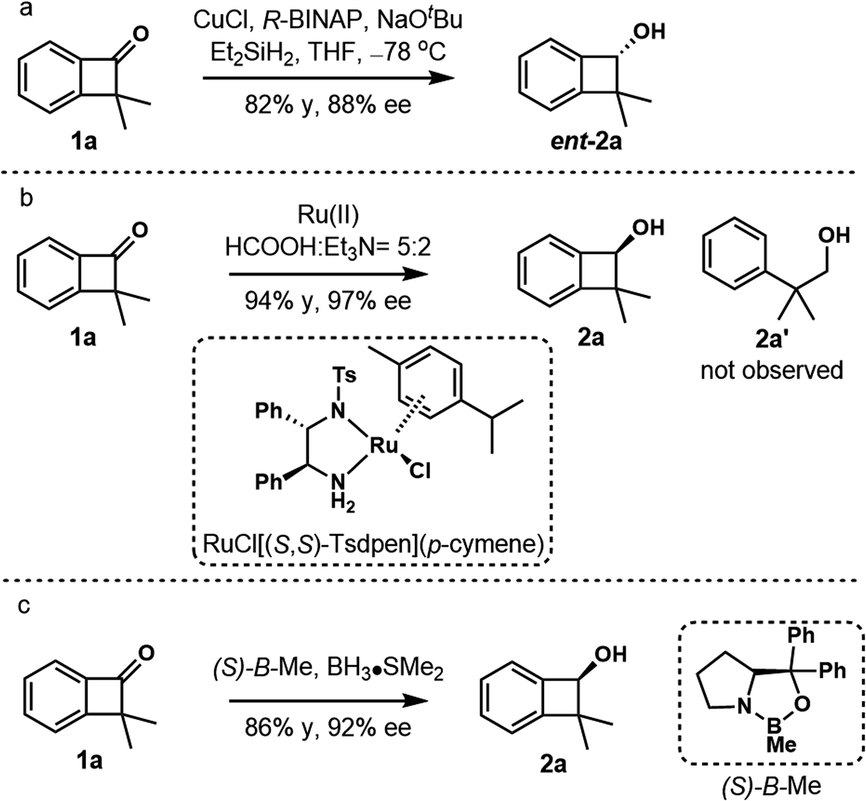 | ||
| Scheme 3 Enantioselective reduction of benzocyclobutenone 1a. (a) Copper hydride reduction. (b) Ru-catalyzed asymmetric transfer hydrogenation. (c) CBS reduction. | ||
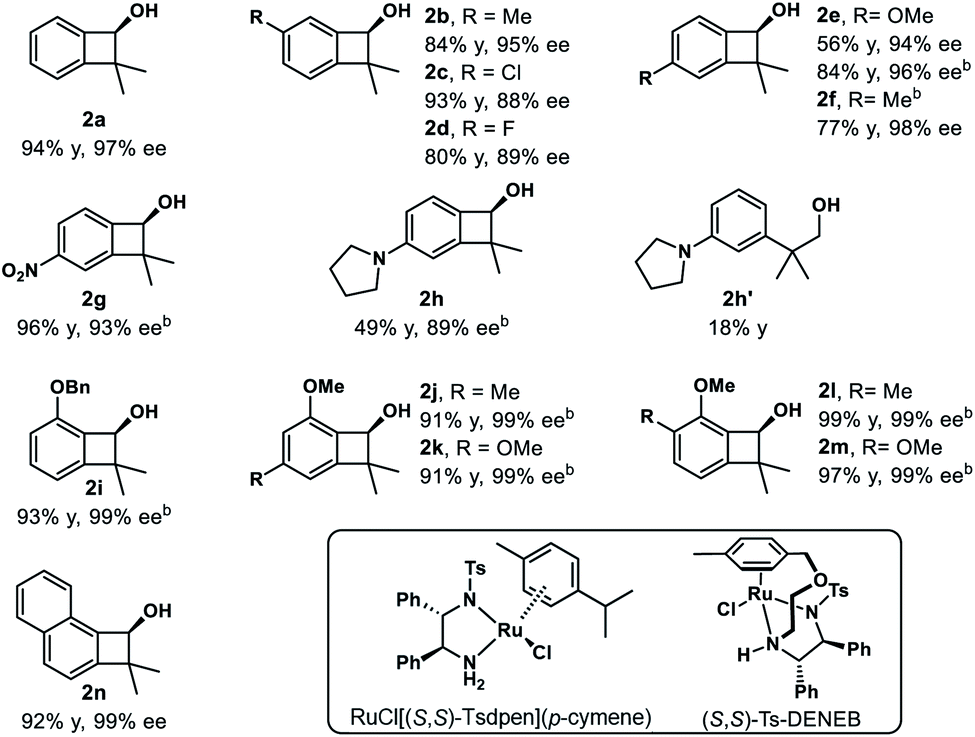
We then examined the substrate scope of the reduction reaction (Table 1). A variety of substituted benzocyclobutenones were tolerated, giving the corresponding benzocyclobutenols 2b–2n in 56–99% yield and 88–99% ee. Notably, benzocyclobutenones with electron-rich substitutions (2e, 2f and 2i–2n) showed much lower reactivity towards reduction, and thus a more reactive catalyst (S,S)-Ts-DENEB26 was chosen to improve the yield and enantioselectivity. Besides, benzocyclobutenol 2g with nitro substitution could be obtained in 96% yield and 93% ee. Treatment of pyrrolidinyl substituted benzocycobutenone 1h with catalyst (S,S)-Ts-DENEB afforded desired product 2h in 49% yield and 89% ee, together with ring-opening product 2h′ (18%).
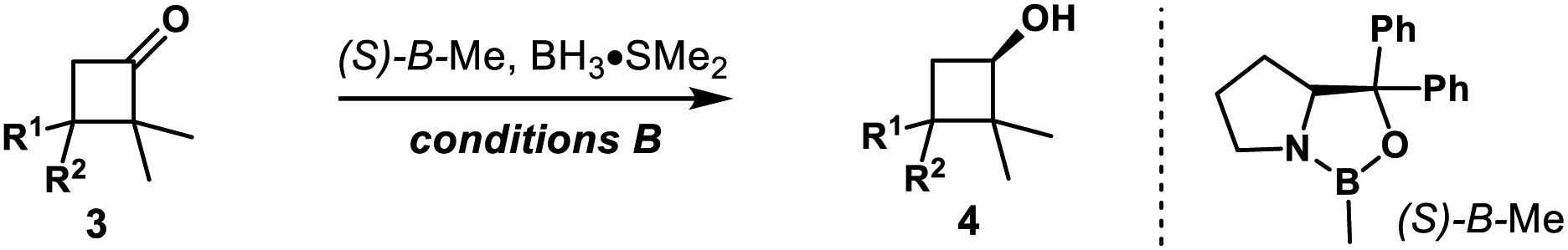
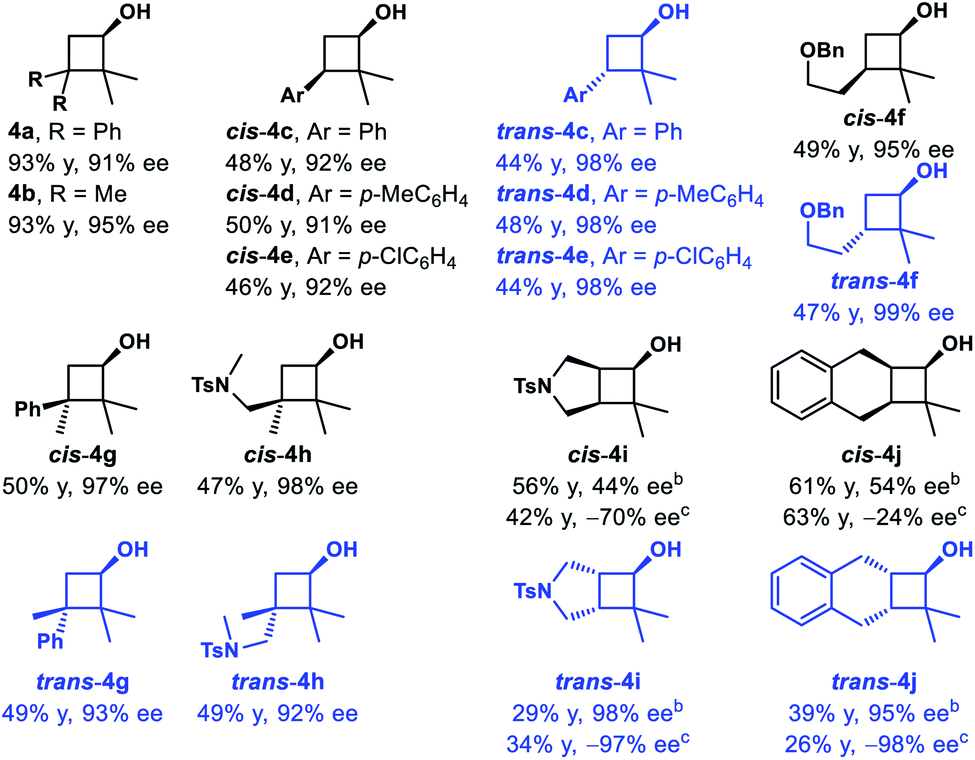
3,3-Disubstituted cyclobutanones were also explored (Table 2). Using catalyst (S,S)-Ts-DENEB, the reaction of 2,2-dimethyl-3,3-diphenylcyclobutanone 3a gave cyclobutanol 4a only in 44% ee. After optimization, we were glad to find that oxazaborolidine (S)-B–Me turned out to be the best catalyst, and cyclobutanol 4a could be obtained in 93% yield and 91% ee (conditions B). The reduction of cyclobutanone 3b gave alcohol 4b in excellent yield and enantioselectivity as well. Interestingly, racemic cyclobutanones underwent efficient optical resolution to give the corresponding two diastereomers, both with high enantiomeric purity. Treatment of cyclobutanones 3c–3h with (S)-B–Me and BH3·Me2S provided the corresponding cis- and trans-cyclobutanols 4c–4h in a nearly 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 diastereomeric ratio and 91–99% ee. In addition, CBS reduction of bicyclic cyclobutanones was also examined. The reaction of 3i afforded trans-4i in 29% yield and 98% ee, along with cis-4i in 56% yield and 44% ee. And reduction with (−)-Ipc2BCl afforded ent-trans-4i in 34% yield and 97% ee, along with ent-cis-4i in 42% yield and 70% ee. Dess–Martin periodinane oxidation of ent-trans-4i (97% ee), followed by selective reduction with L-selectride gave cis-4i as a single product in 99% yield and 96% ee. The reaction of 3j gave similar results, and enantioenriched cyclobutanols cis-4j could be furnished in 78% yield and 97% ee from ent-trans-4j (98% ee) following the above oxidation–reduction procedure. The absolute configurations of 2a, ent-2j and trans-4i were unambiguously determined by single-crystal X-ray diffraction analysis of their corresponding nitrobenzoate derivative.27
:1 diastereomeric ratio and 91–99% ee. In addition, CBS reduction of bicyclic cyclobutanones was also examined. The reaction of 3i afforded trans-4i in 29% yield and 98% ee, along with cis-4i in 56% yield and 44% ee. And reduction with (−)-Ipc2BCl afforded ent-trans-4i in 34% yield and 97% ee, along with ent-cis-4i in 42% yield and 70% ee. Dess–Martin periodinane oxidation of ent-trans-4i (97% ee), followed by selective reduction with L-selectride gave cis-4i as a single product in 99% yield and 96% ee. The reaction of 3j gave similar results, and enantioenriched cyclobutanols cis-4j could be furnished in 78% yield and 97% ee from ent-trans-4j (98% ee) following the above oxidation–reduction procedure. The absolute configurations of 2a, ent-2j and trans-4i were unambiguously determined by single-crystal X-ray diffraction analysis of their corresponding nitrobenzoate derivative.27
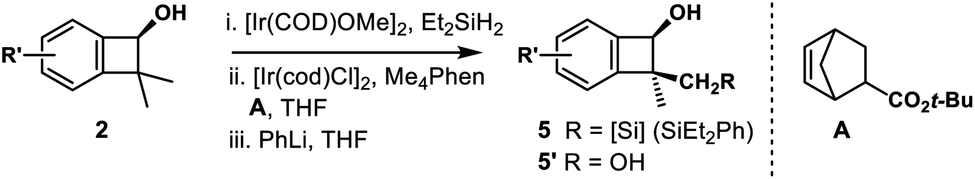
Inspired by powerful and reliable directed C–H silylation chemistry pioneered by Hartwig,28 we envisioned that the transition-metal catalyzed intramolecular C–H silylations of the above alcohols would provide a single diastereomer owing to rigid structural organization. The challenges here are the control of regioselectivity in the cyclization step and inhibition of the ring-opening pathway. Benzocyclobutenol 2a was chosen as a model substrate to study this intramolecular C–H silylation. The transition-metal catalyst system and alkene acceptors were screened (Scheme 4, see Tables S5–S9† for details). Acceptor norbornene (nbe) derivative A gave the optimal yield in the cyclization step (63% NMR yield), and other phenanthroline ligands gave inferior results. The reaction showed remarkable regio- and diastereoselectivity; no silylation of the arene was detected.With optimal intramolecular silylation conditions in hand, sequential hydroxysilylation/C–H silylation/phenyllithium addition reaction of 2a provided desired product 5a in 56% overall yield without any obvious erosion of enantiomeric purity (Table 3, conditions C). Then the reactions of the above enantioenriched benzocyclobutenols 2b–2m were examined. The corresponding products 5b–5f and 5h–5m could be obtained in 30–83% overall yield without obvious enantiomeric purity erosion (96–99% es). However, the reaction of 2g gave no expected product 5g. Notably, we did observe ring-opening byproduct 5e′′ (14% yield) when using [Ir(COD)OMe]2 as a catalyst and NBE as a hydrogen acceptor in the reaction of 4e. This byproduct was efficiently suppressed under optimal conditions, giving product 5e in 52% yield. Besides the nucleophilic addition approach, treatment of the cyclization product with H2O2 provided diol 5l′ in 84% yield and 99% ee. Surprisingly, the reaction of substrate 2n (99% ee) gave the product 5n in only 77% ee. We assumed the partial racemization took place during the cyclization step, since acidic treatment of the hydroxysilylation product gave recovered alcohol 2n in 99% ee. Gladly, we were able to improve the selectivity by lowering the reaction temperature (60 °C, 49% yield, 93% ee, 94% es).
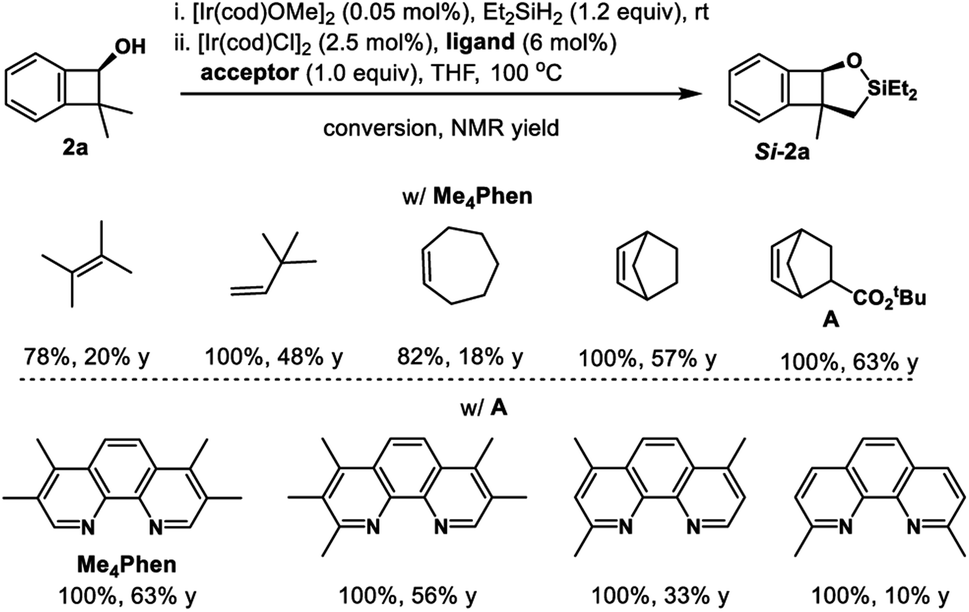 | ||
| Scheme 4 Optimization of intramolecular C–H silylation of benzocyclobutenol 1a. | ||
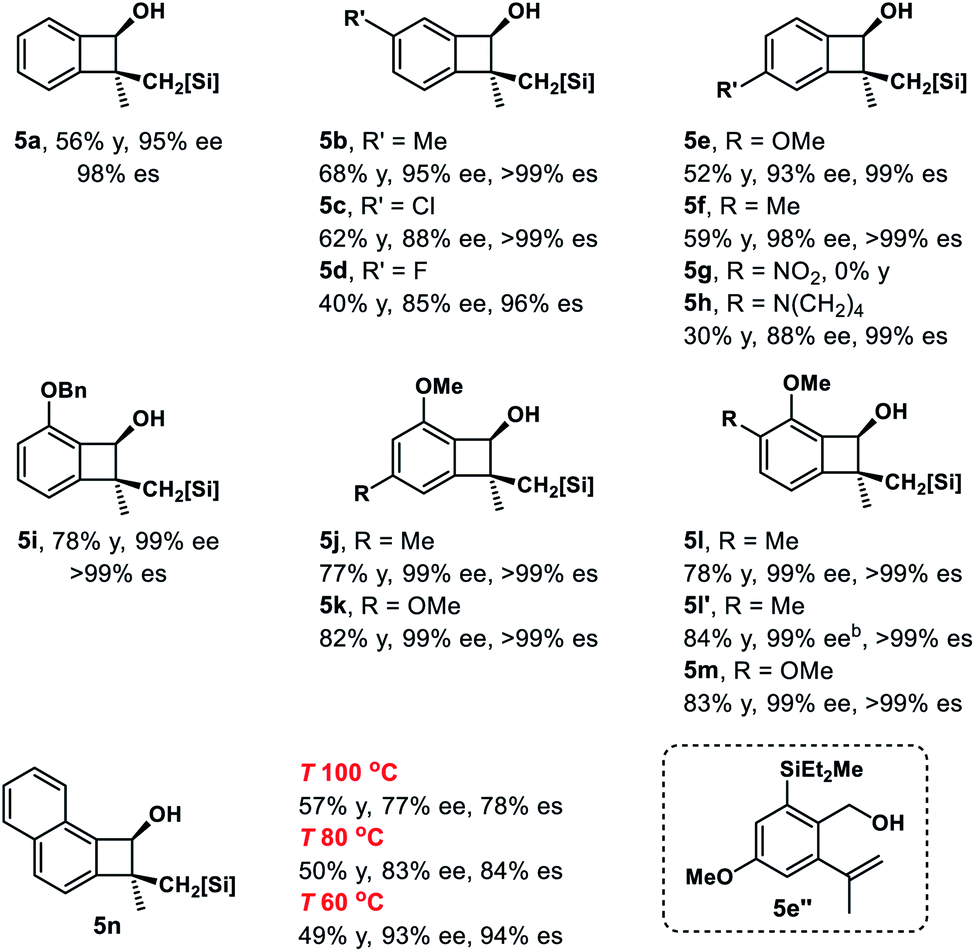
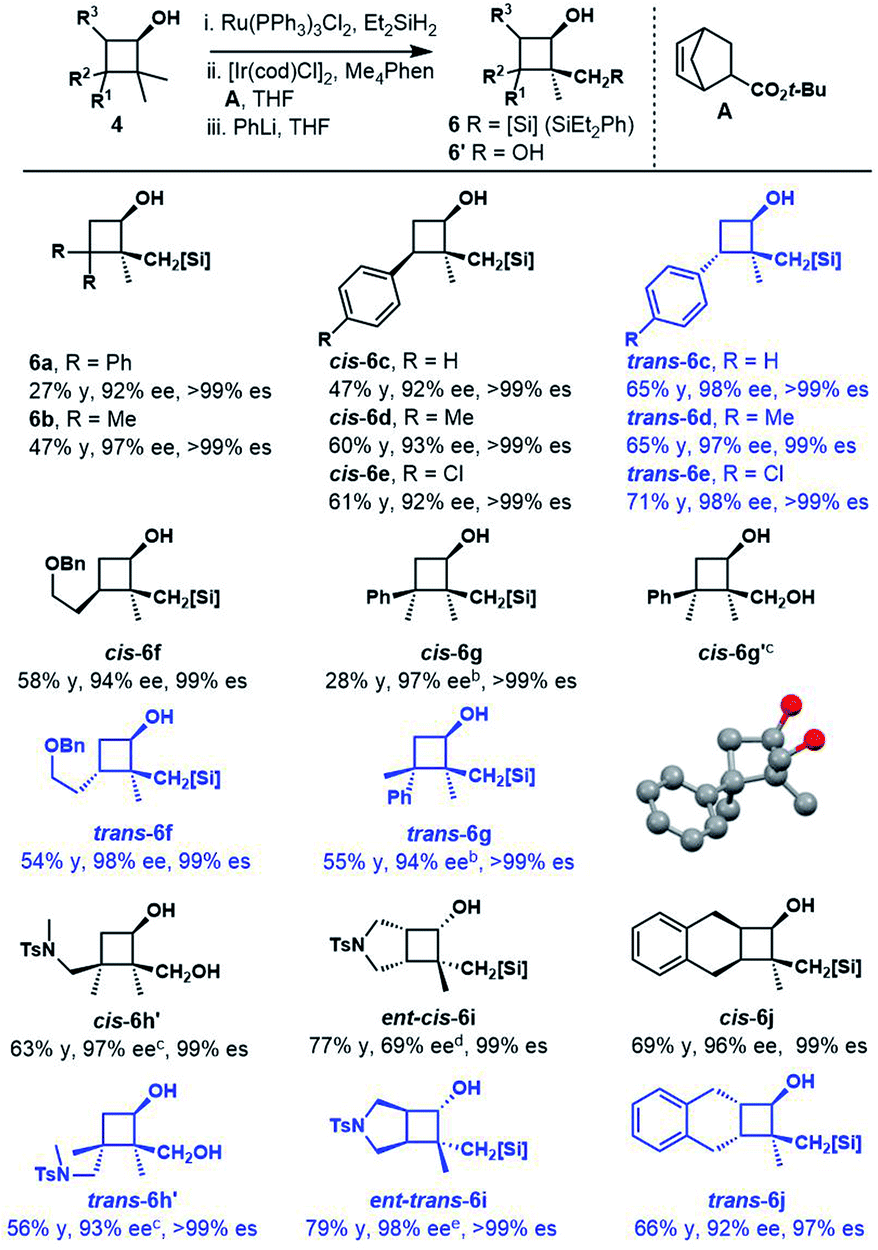
Cyclobutanols were examined under optimal conditions as well (Table 4). Products 6a and 6b could be obtained in 27–48% yield. We assumed that the low yield of 6a was due to the steric interaction of the phenyl and trialkylsilylhydroxy group, both were in a cis relationship. The reaction of 3-monosubstituted cyclobutanols 4b–4g afforded the corresponding products cis-6b–6g and trans-6b–6g in good yields with the retention of ee. In the above cases, three contiguous chiral centers, even two quaternary centers were installed efficiently. The absolute configuration of cis-6g′ was unambiguously determined by single-crystal X-ray diffraction analysis of its corresponding diol.27 The diols cis-6h′ and trans-6h′ could be achieved upon treatment of cyclization products with H2O2 instead of phenyllithium. In addition, bicyclic substrates 4i, 4j smoothly furnished the corresponding enantioenriched products cis-6i, 6j and trans-6i, 6j with four contiguous carbon centers in good yields.
|
a Reaction conditions: 4 (0.5 mmol), Ru(PPh3)3Cl (0.2 mol%), Et2SiH2 (1.5 equiv.), THF, 35 °C; ii. [Ir(COD)Cl]2 (2.5 mol%), Me4Phen (6 mol%), A (1.0 equiv.), THF, 100 °C; iii. PhLi, THF, −78 °C; see the ESI for more details.
b ii. [Ir(COD)Cl]2 (5 mol%), Me4Phen (12 mol%).
c iii. KHCO3 (2.5 equiv.), KF (2.5 equiv.), H2O2 (10 equiv.), THF/MeOH (1:1), 50 °C.
d
ent-cis-4i (70% ee) was used.
e
ent-trans-4i (97% ee) was used.
|
|---|
|
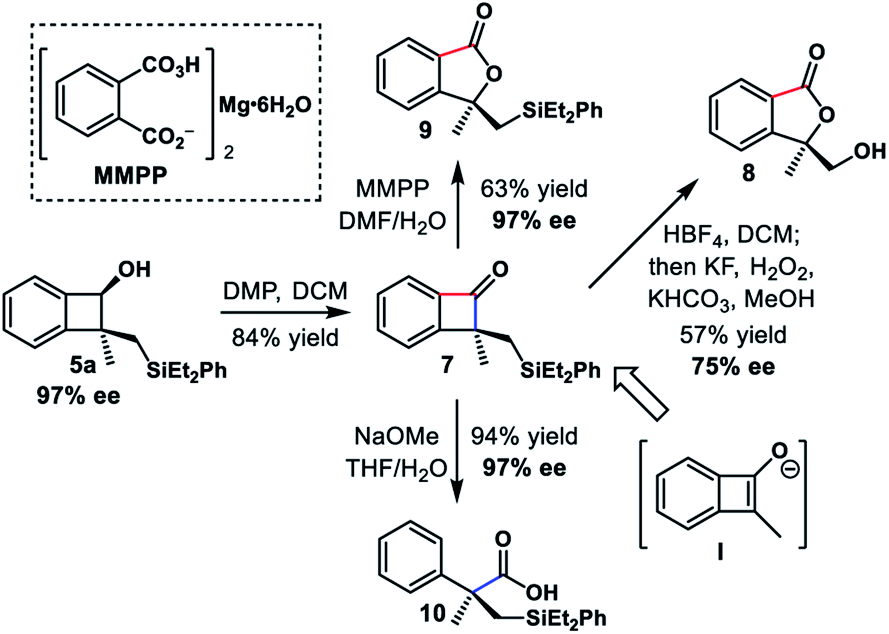
At this point, we conducted further transformations to explore the utilities of the chiral benzocyclobutene derivatives (Scheme 5). The oxidation of benzocyclobutenol 5a afforded benzocyclobutenone 7 smoothly using Dess–Martin periodinane. This product could be viewed as the result of the alkylation of α-substituted benzocyclobutenone via elusive enolate intermediate I.
 | ||
| Scheme 5 Further transformations of benzocyclobutenol 5a. | ||
Subsequent Tamao–Fleming oxidation29 with a concomitant cyclobutanone oxidation provided alcohol 8 in 57% yield, albeit with partial loss of enantiopurity. Furthermore, the regioselective Bayer–Villiger oxidation of 7 was achieved using MMPP,30 giving phthalide 9 in 63% yield and 97% ee. Poor regioselectivity was observed when parent benzocyclobutenone was treated with a base.31 In contrast, exposure of 7 to sodium methoxide afforded phenylacetic acid derivative 10 as a single product in 94% yield and 97% ee via proximal bond cleavage.
Phyllostoxin (11) was isolated from fungal pathogen Phyllosticta cirsii, and it could represent a potential natural herbicide (Scheme 6).32 The structure was proposed to contain chiral α-quaternary benzocyclobutenone moiety. We envisioned that our strategy would provide a straightforward way to assemble the quaternary center of benzocyclobutenone, thereby confirming the proposed structure and determining the absolute configuration. Our synthesis commenced with enantioselective transfer hydrogenation of substrate 1o. Enantioenriched benzocyclobutenol 2o could be obtained in 93% yield and 99% ee using catalyst (R,R)-Ts-DENEB. Standard procedure, including hydrosilylation/C–H silylation/oxidation, provided diol 5o′ in 89% overall yield and 99% ee. Various oxidation conditions were examined to oxidize diol 5o′, including Swern oxidation, Dess–Martin periodinane and PCC; unfortunately, the reaction only gave messy mixtures. Thus we turned to selective protection of the diol. Selective benzoylation could be achieved via three-step manipulation, giving primary alcohol 12 in 82% overall yield. Swern oxidation and nucleophilic addition of EtMgBr, followed by global deprotection, provided triol 13 in 54% yield over 3 steps. Of mention, benzoyl migration was observed in the EtMgBr addition step. Finally, selective acylation of the phenol and subsequent oxidation furnished benzocyclobutenone 11 in 39% overall yield. However, the optical rotation and NMR spectral data did not match those reported for the natural product.
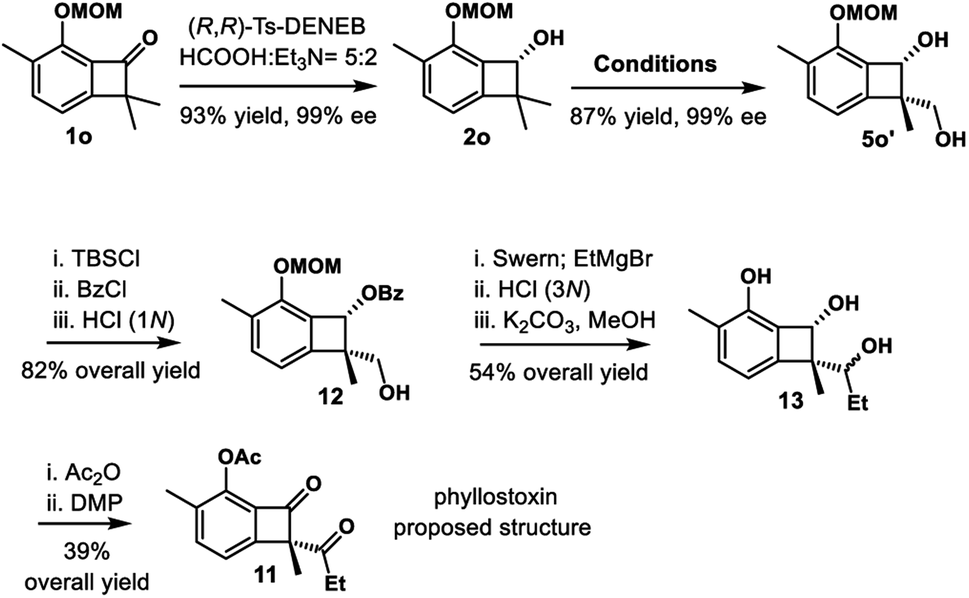 | ||
| Scheme 6 Total synthesis of the proposed structure of phyllostoxin. Conditions: [Ir(COD)OMe]2, Et2SiH2, THF, rt; ii. [Ir(COD)Cl]2, Me4Phen, A, THF, 100 °C; iii. KHCO3, H2O2, THF/MeOH (1:1), 50 °C. | ||
The monoterpene grandisol (14) was known as a main component of the sex pheromone of the cotton boll weevil, Anthonomous grandis Boheman, and other insects.33,34 The diastereomer fragranol (15) was isolated in many essential oil aerial parts of plant species such as Achillea fragrantissima, A. falcata and Geranium tuberosum.33 Surprisingly, in comparison to grandisol, there is only one report on enantioselective synthesis of fragranol yet.35 We postulated that our strategy would enable a divergent synthesis of these two diastereomers, starting from an optical resolution of cyclobutanone 3k (Scheme 7). As expected, the CBS reduction of 1x provided cyclobutanols cis-4k and trans-4k (90% yield, 1:1.1 dr, 90–99% ee). Subsequent C–H functionalization and oxidation gave diastereomers cis-6k′ and trans-6k′ in good yield. And both diastereomers could be easily separated by column chromatography. Debenzylation, selective silylation of the primary alcohol and Barton–McCombie deoxygenation provided cyclobutanes 17 and 20 uneventfully. Starting from cyclobutane 17, deprotection and subsequent oxidation afforded lactone 18 in 56% overall yield, which led to formal total synthesis of (−)-grandisol 14. Starting from cyclobutane 20, regioselective dehydration with Martin sulfurane and removal of the TBS group furnished alkene 21 in 70% overall yield. Finally, (−)-fragranol 15 was obtained in three additional steps, which included oxidation to an aldehyde, olefination/hydrolysis and reduction.
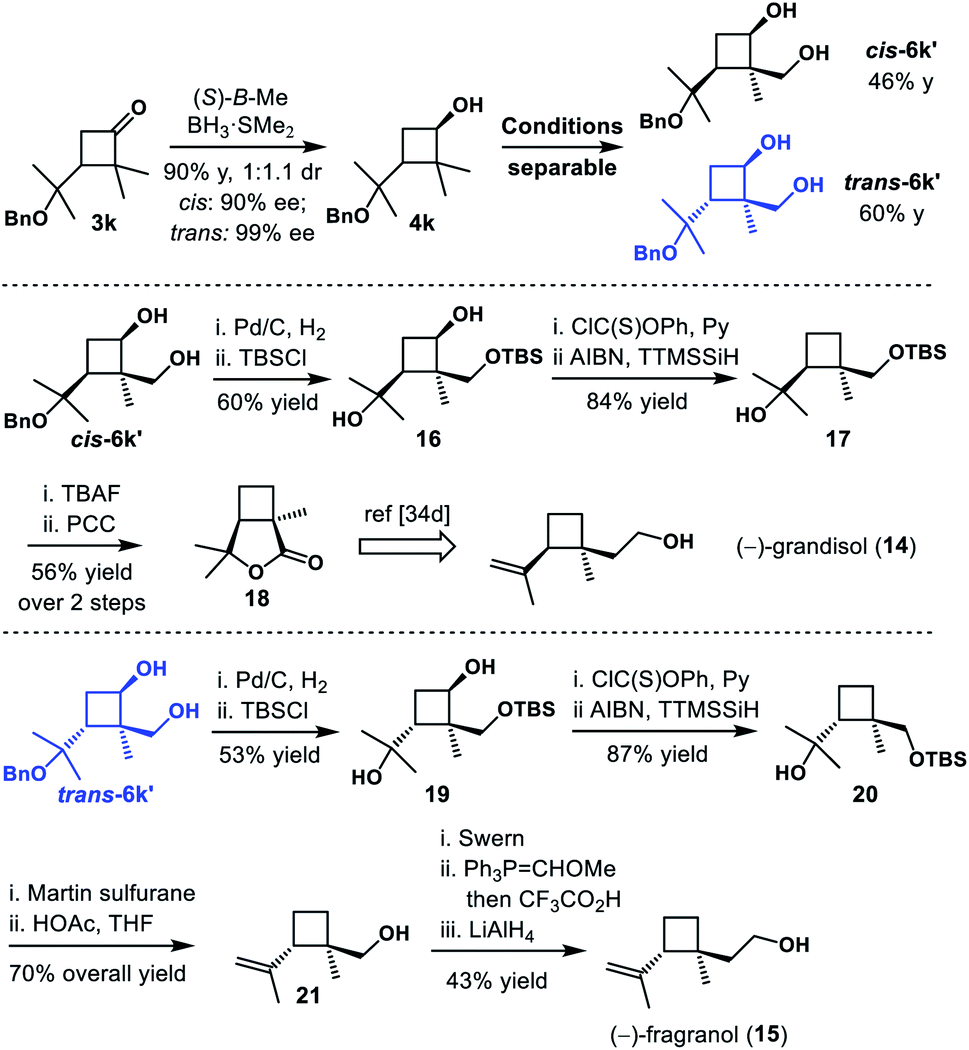 | ||
| Scheme 7 Divergent synthesis of grandisol and fragranol. Conditions: Ru(PPh3)3Cl, Et2SiH2, THF, 35 °C; ii. [Ir(COD)Cl]2, Me4Phen, A, THF, 100 °C; iii. KHCO3, H2O2, THF/MeOH (1:1), 50 °C. | ||
Conclusions
In conclusion, we developed a practical and robust approach to accessing enantioenriched cyclobutanols and benzocyclobutenols with all-carbon quaternary centers from readily available cyclobutanones and benzocyclobutenones. This strategy provided an alternative way to synthesize chiral α-quaternary cyclobutanones and benzocyclobutenones, which are otherwise directly inaccessible using enolate chemistry. Further transformations, including regioselective ring expansion and ring opening reactions were explored. This strategy was also applied to the synthesis of phyllostoxin (proposed structure), grandisol and fragranol. Finally, this sequential enantioselective reduction/C–H functionalization strategy could be utilized as a general method to synthesize α-quaternary cyclic carbonyl compounds, for example, 1-indanone, 2-indanone and cyclopentanone derivatives. These results will be reported in due course.Data availability
Detailed condition optimization, experimental procedure, characterization data are available in the ESI.Author contributions
J. C. designed the approach and performed the experiments, analyzed the experimental data and prepared the Supplementary Information. Z. S. and C. L. expanded the scope of the substrates. P. L. directed the investigations and prepared the manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was supported by the National Natural Science Foundation of China (22071028, 21772024, and 21921003) and Zhuhai Fudan Innovation Institute.Notes and references
- K. B. Wiberg, Angew. Chem., Int. Ed. Engl., 1986, 25, 312–322 CrossRef.
- (a) J. C. Namyslo and D. E. Kaufmann, Chem. Rev., 2003, 103, 1485–1538 CrossRef CAS PubMed; (b) A. K. Sadana, R. K. Saini and W. E. Billups, Chem. Rev., 2003, 103, 1539–1602 CrossRef CAS PubMed; (c) E. M. Carreira and T. C. Fessard, Chem. Rev., 2014, 114, 8257–8322 CrossRef CAS PubMed.
- (a) M. Murakami and N. Ishida, Chem. Rev., 2021, 121, 264–299 CrossRef CAS PubMed; (b) G. Fumagalli, S. Stanton and J. F. Bower, Chem. Rev., 2017, 117, 9404–9432 CrossRef CAS PubMed; (c) T. Seiser, T. Saget, D. N. Tran and N. Cramer, Angew. Chem., Int. Ed., 2011, 50, 7740–7752 CrossRef CAS; (d) P.-H. Chen, B. A. Billett, T. Tsukamoto and G. Dong, ACS Catal., 2017, 7, 1340–1360 CrossRef CAS PubMed.
- (a) Y.-Y. Fan, X.-H. Gao and J.-M. Yue, Sci. China: Chem., 2016, 59, 1126–1141 CrossRef CAS; (b) M. A. Beniddir, L. Evanno, D. Joseph, A. Skiredj and E. Poupon, Nat. Prod. Rep., 2016, 33, 820–842 RSC; (c) C. Zhang, F. Li, Y. Yu, A. Huang, P. He, M. Lei, J. Wang, L. Huang, Z. Liu, J. Liu and Y. Wei, J. Med. Chem., 2017, 60, 3618–3625 CrossRef CAS PubMed.
- (a) M. Wang and P. Lu, Org. Chem. Front., 2018, 5, 254–259 RSC; (b) K.-G. Wen, Y.-Y. Peng and X.-P. Zeng, Org. Chem. Front., 2020, 7, 2576–2597 RSC; (c) F. Secci, A. Frongia and P. P. Piras, Molecules, 2013, 18, 15541–15572 CrossRef PubMed.
- For selected examples, see: (a) S. Poplata, A. Tröster, Y.-Q. Zou and T. Bach, Chem. Rev., 2016, 116, 9748–9815 CrossRef CAS PubMed; (b) Y. Xu, M. L. Conner and M. K. Brown, Angew. Chem., Int. Ed., 2015, 54, 11918–11928 CrossRef CAS PubMed; (c) V. V. Pagar and T. V. RajanBabu, Science, 2018, 361, 68–72 CrossRef CAS PubMed; (d) J. Zheng, W. B. Swords, H. Jung, K. L. Skubi, J. B. Kidd, G. J. Meyer, M.-H. Baik and T. P. Yoon, J. Am. Chem. Soc., 2019, 141, 13625–13634 CrossRef CAS PubMed; (e) L. Zeng, J. Xu, D. Zhang, Z. Yan, G. Cheng, W. Rao and L. Gao, Angew. Chem., Int. Ed., 2020, 59, 21890–21894 CrossRef CAS PubMed; (f) X. Huang, T. R. Quinn, K. Harms, R. D. Webster, L. Zhang, O. Wiest and E. Meggers, J. Am. Chem. Soc., 2017, 139, 9120–9123 CrossRef CAS; (g) R. Brimioulle and T. Bach, Science, 2013, 342, 840–843 CrossRef CAS PubMed; (h) M. L. Conner, Y. Xu and M. K. Brown, J. Am. Chem. Soc., 2015, 137, 3482–3485 CrossRef CAS PubMed; (i) Ł. Albrecht, G. Dickmeiss, F. C. Acosta, C. Rodríguez-Escrich, R. L. Davis and K. A. Jørgensen, J. Am. Chem. Soc., 2012, 134, 2543–2546 CrossRef PubMed; (j) J.-L. Hu, L.-W. Feng, L. Wang, Z. Xie, Y. Tang and X. Li, J. Am. Chem. Soc., 2016, 138, 13151–13154 CrossRef CAS PubMed; (k) J. N. Du, K. L. Skubi, D. M. Schultz and T. P. Yoon, Science, 2014, 344, 392–396 CrossRef CAS PubMed.
- (a) F. Kleinbeck and F. D. Toste, J. Am. Chem. Soc., 2009, 131, 9178–9179 CrossRef CAS PubMed; (b) S. Y. Shim, Y. Choi and D. H. Ryu, J. Am. Chem. Soc., 2018, 140, 11184–11188 CrossRef CAS PubMed; (c) B. M. Trost and T. A. Yasukata, J. Am. Chem. Soc., 2001, 123, 7162–7163 CrossRef CAS PubMed.
- (a) C. M. Reeves, C. Eidamshaus, J. Kim and B. M. Stoltz, Angew. Chem., Int. Ed., 2013, 52, 6718–6721 CrossRef CAS PubMed; (b) A. Misale, S. Niyomchon, M. Luparia and N. Maulide, Angew. Chem., Int. Ed., 2014, 53, 7068–7073 CrossRef CAS PubMed; (c) A. Misale, S. Niyomchon and N. Maulide, Acc. Chem. Res., 2016, 49, 2444–2458 CrossRef CAS PubMed.
- (a) D. Mailhol, M. d. M. Sanchez Duque, W. Raimondi, D. Bonne, T. Constantieux, Y. Coquerel and J. Rodriguez, Adv. Synth. Catal., 2012, 354, 3523–3532 CrossRef CAS; (b) D. J. Aitken, P. Caboni, H. Eijsberg, A. Frongia, R. Guillot, J. Ollivier, P. P. Piras and F. Secci, Adv. Synth. Catal., 2014, 356, 941–945 CrossRef CAS; (c) Y. Zhou, Y.-L. Wei, J. Rodriguez and Y. Coquerel, Angew. Chem., Int. Ed., 2018, 58, 456–460 CrossRef PubMed; (d) M. Wang, J. Chen, Z. Chen, C. Zhong and P. Lu, Angew. Chem., Int. Ed., 2018, 57, 2707–2711 CrossRef CAS PubMed; (e) S. A. Moteki, J. Han, S. Arimitsu, M. Akakura, K. Nakaya-ma and K. Maruoka, Angew. Chem., Int. Ed., 2012, 51, 1187–1190 CrossRef CAS PubMed.
- (a) Y.-J. Chen, T.-J. Hu, C.-G. Feng and G.-Q. Lin, Chem. Commun., 2015, 51, 8773–8776 RSC; (b) H. A. Clement, M. Boghi, R. M. Mcdonald, L. Bernier, J. W. Coe, W. Farrell, C. J. Helal, M. R. Reese, N. W. Sach, J. C. Lee and D. G. Hall, Angew. Chem., Int. Ed., 2019, 58, 18405–18409 CrossRef CAS PubMed; (c) C. Zhong, Y. Huang, H. Zhang, Q. Zhou, Y. Liu and P. Lu, Angew. Chem., Int. Ed., 2020, 59, 2750–2754 CrossRef CAS; (d) K. Nguyen, H. A. Clement, L. Bernier, J. W. Coe, W. Farrell, C. J. Helal, M. R. Reese, N. W. Sach, J. C. Lee and D. G. Hall, ACS Catal., 2021, 11, 404–413 CrossRef CAS.
- (a) K.-J. Xiao, D. W. Lin, M. Miura, R.-Y. Zhu, W. Gong, M. Wasa and J.-Q. Yu, J. Am. Chem. Soc., 2014, 136, 8138–8142 CrossRef CAS PubMed; (b) J. He, Q. Shao, Q. Wu and J.-Q. Yu, J. Am. Chem. Soc., 2017, 139, 3344–3347 CrossRef CAS PubMed; (c) X. Chen, L. Chen, H. Zhao, Q. Gao, Z. Shen and S. Xu, Chin. J. Chem., 2020, 38, 1533–1537 CrossRef CAS.
- For a recent review, see: (a) M. Wang, C. Zhong and P. Lu, Synlett, 2021 DOI:10.1055/a-1493-9489. For other examples, see: (b) M. Guisán-Ceinos, A. Parra, V. Martín-Heras and M. Tortosa, Angew. Chem., Int. Ed., 2016, 55, 6969–6972 CrossRef PubMed; (c) J. Xia, Y. Nie, G. Yang, Y. Liu, I. D. Gridnev and W. Zhang, Chin. J. Chem., 2018, 36, 612–618 CrossRef CAS; (d) L. Deng, Y. Fu, S. Y. Lee, C. Wang, P. Liu and G. Dong, J. Am. Chem. Soc., 2019, 141, 16260–16265 CrossRef CAS PubMed.
- R. Martin and A. Flores-Gaspar, Synthesis, 2013, 45, 563–580 CrossRef.
- M. Chaumontet, R. Piccardi, N. Audic, J. Hitce, J.-J. Peglion, E. Clot and O. Baudoin, J. Am. Chem. Soc., 2008, 130, 15157–15166 CrossRef CAS PubMed.
- P. Álvarez-Bercedo, A. Flores-Gaspar, A. Correa and R. Martin, J. Am. Chem. Soc., 2010, 132, 466–467 CrossRef PubMed.
- K. Kasamatsu, T. Yoshimura, A. Mandi, T. Taniguchi, K. Monde, T. Furuta and T. Kawabata, Org. Lett., 2017, 19, 352–355 CrossRef CAS PubMed.
- (a) P.-H. Chen and G. Dong, Chem.–Eur. J., 2016, 22, 18290–18315 CrossRef CAS PubMed; (b) J. G. Allen and S. J. Danishefsky, J. Am. Chem. Soc., 2001, 123, 351–352 CrossRef CAS PubMed; (c) M. Chaumontet, R. Piccardi and O. Baudoin, Angew. Chem., Int. Ed., 2009, 48, 179–182 CrossRef CAS PubMed; (d) K. Ohmori, K. Mori, Y. Ishikawa, H. Tsuruta, S. Kuwahara, N. Harada and K. Suzuki, Angew. Chem., Int. Ed., 2004, 43, 3167–3171 CrossRef CAS PubMed; (e) Y. Matsuya, N. Ohsawa and H. Nemoto, J. Am. Chem. Soc., 2006, 128, 13072–13073 CrossRef CAS PubMed.
- (a) J. Loup, U. Dhawa, F. Pesciaioli, J. Wencel-Delord and L. Ackermann, Angew. Chem., Int. Ed., 2019, 58, 12803–12818 CrossRef CAS PubMed; (b) T. G. Saint-Denis, R.-Y. Zhu, G. Chen, Q.-F. Wu and J.-Q. Yu, Science, 2018, 359, eaao4798 CrossRef PubMed; (c) G. Liao, T. Zhang, Z.-K. Lin and B.-F. Shi, Angew. Chem., Int. Ed., 2020, 59, 19773–19786 CrossRef CAS PubMed.
- D. J. Bertelli and P. Crews, J. Am. Chem. Soc., 1986, 90, 3889–3890 CrossRef.
- P.-H. Chen, N. A. Savage and G. Dong, Tetrahedron, 2014, 70, 4135–4146 CrossRef CAS PubMed.
- H. C. Brown, J. Chandrasekharan and P. V. Ramachandran, J. Am. Chem. Soc., 1988, 110, 1539–1546 CrossRef CAS.
- E. J. Corey, R. K. Bakshi and S. Shibata, J. Am. Chem. Soc., 1987, 109, 5551–5553 CrossRef CAS.
- E. P. Kündig, J. Leresche, L. Saudan and G. Bernardinelli, Tetrahedron, 1996, 52, 7363–7378 CrossRef.
- C. Deutsch, N. Krause and B. H. Lipshutz, Chem. Rev., 2008, 108, 2916–2927 CrossRef CAS PubMed.
- A. Fujii, S. Hashiguchi, N. Uematsu, T. Ikariya and R. Noyori, J. Am. Chem. Soc., 1996, 118, 2521–2522 CrossRef CAS.
- T. Touge, T. Hakamata, H. Nara, T. Kobayashi, N. Sayo, T. Saito, Y. Kayaki and T. Ikariya, J. Am. Chem. Soc., 2011, 133, 14960–14963 CrossRef CAS PubMed.
- Deposition numbers 2058586 (2a), 2058587 (ent-2j), 2058588 (trans-4i) and 2058693 (cis-6g′) contain the supplementary crystallographic data for this paper. These data are provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service.
- (a) E. M. Simmons and J. F. Hartwig, Nature, 2012, 483, 70–73 CrossRef CAS PubMed; (b) J. F. Hartwig and E. A. Romero, Tetrahedron, 2019, 75, 4059–4070 CrossRef CAS PubMed.
- (a) I. Fleming, R. Henning and H. Plaut, J. Chem. Soc., Chem. Commun., 1984, 29–31 RSC; (b) K. Tamao, T. Nakajima, R. Sumiya, H. Arai, N. Higuchi and Y. Ito, J. Am. Chem. Soc., 1986, 108, 6090–6093 CrossRef CAS PubMed.
- T. Hosoya, Y. Kuriyama and K. Suzuki, Synlett, 1995, 635–638 CrossRef CAS.
- (a) M. P. Cava and K. Muth, J. Am. Chem. Soc., 1960, 82, 652–654 CrossRef CAS; (b) A. Gokhale and P. Schiess, Helv. Chim. Acta, 1998, 81, 251–267 CrossRef CAS.
- A. Evidente, A. Cimmino, A. Andolfi, M. Vurro, M. C. Zonno and A. Motta, J. Agric. Food Chem., 2008, 56, 884–888 CrossRef CAS PubMed.
- (a) J. H. Tumlinson, D. D. Hardee, R. C. Gueldner, A. C. Thompson, P. A. Hedin and J. P. Minyard, Science, 1969, 166, 1010–1012 CrossRef CAS PubMed; (b) F. Bohlmann, C. Zdero and U. Faas, Chem. Ber., 1973, 106, 2904–2909 CrossRef CAS.
- (a) S. Antonsen, R. B. Østby and Y. Stenstrøm, Stud. Nat. Prod. Chem., 2018, 57, 1–40 CAS; (b) D. P. Hari, J. C. Abell, V. Fasano and V. K. Aggarwal, J. Am. Chem. Soc., 2020, 142, 5515–5520 CrossRef CAS PubMed; (c) S. Poplata and T. Bach, J. Am. Chem. Soc., 2018, 140, 3228–3231 CrossRef CAS PubMed; (d) V. N. Wakchaure and B. List, Angew. Chem., Int. Ed., 2010, 49, 4136–4139 CrossRef CAS PubMed.
- T. Martin, C. M. Rodriguez and V. S. Martin, Tetrahedron: Asymmetry, 1995, 6, 1151–1164 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2058586–2058588 and 2058693. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc02119b |
| ‡ Current address: Department of Chemistry, The Scripps Research Institute, 10550 North Torrey Pines Road, La Jolla, California 92037, USA. |
| This journal is © The Royal Society of Chemistry 2021 |