
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAlloying a single and a double perovskite: a Cu+/2+ mixed-valence layered halide perovskite with strong optical absorption†
Bridget A.
Connor
a,
Rebecca W.
Smaha
 ab,
Jiayi
Li
a,
Aryeh
Gold-Parker
ac,
Alexander J.
Heyer
a,
Michael F.
Toney
cd,
Young S.
Lee
be and
Hemamala I.
Karunadasa
*ab
ab,
Jiayi
Li
a,
Aryeh
Gold-Parker
ac,
Alexander J.
Heyer
a,
Michael F.
Toney
cd,
Young S.
Lee
be and
Hemamala I.
Karunadasa
*ab
aDepartment of Chemistry, Stanford University, Stanford, California 94305, USA. E-mail: hemamala@stanford.edu
bStanford Institute for Materials and Energy Sciences, SLAC National Accelerator Laboratory, Menlo Park, California 94025, USA
cStanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory, Menlo Park, California 94025, USA
dDepartment of Chemical and Biological Engineering, University of Colorado Boulder, Boulder, CO 80309, USA
eDepartment of Applied Physics, Stanford University, Stanford, California 94305, USA
First published on 14th May 2021
Abstract
Introducing heterovalent cations at the octahedral sites of halide perovskites can substantially change their optoelectronic properties. Yet, in most cases, only small amounts of such metals can be incorporated as impurities into the three-dimensional lattice. Here, we exploit the greater structural flexibility of the two-dimensional (2D) perovskite framework to place three distinct stoichiometric cations in the octahedral sites. The new layered perovskites AI4[CuII(CuIInIII)0.5Cl8] (1, A = organic cation) may be derived from a CuI–InIII double perovskite by replacing half of the octahedral metal sites with Cu2+. Electron paramagnetic resonance and X-ray absorption spectroscopy confirm the presence of Cu2+ in 1. Crystallographic studies demonstrate that 1 represents an averaging of the CuI–InIII double perovskite and CuII single perovskite structures. However, whereas the highly insulating CuI–InIII and CuII perovskites are colorless and yellow, respectively, 1 is black, with substantially higher electronic conductivity than that of either endmember. We trace these emergent properties in 1 to intervalence charge transfer between the mixed-valence Cu centers. We further propose a tiling model to describe how the Cu+, Cu2+, and In3+ coordination spheres can pack most favorably into a 2D perovskite lattice, which explains the unusual 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:1 ratio of these cations found in 1. Magnetic susceptibility data of 1 further corroborate this packing model. The emergence of enhanced visible light absorption and electronic conductivity in 1 demonstrates the importance of devising strategies for increasing the compositional complexity of halide perovskites.
:2:1 ratio of these cations found in 1. Magnetic susceptibility data of 1 further corroborate this packing model. The emergence of enhanced visible light absorption and electronic conductivity in 1 demonstrates the importance of devising strategies for increasing the compositional complexity of halide perovskites.
1. Introduction
The double perovskite lattice considerably increases the versatility of halide perovskites by enabling the incorporation of a much wider range of elements compared to single perovskites. Here, the divalent octahedral B-site cation of the three-dimensional (3D) AIBIIX3 (X = halide) single perovskite structure is replaced with an ordered mixture of two different B-site occupants, yielding the general formula AI2BB′X6. Nevertheless, the double perovskite framework is still restrictive in that it requires exactly two distinct B-sites that satisfy charge balance requirements.1 Thus, double perovskites most commonly feature B-site combinations of 1+ and 3+ (e.g., Cs2AgIBiIIIBr6)2,3 or 4+ and 0 (vacancy) (e.g., Cs2SnIVCl6).4 Recent work has explored B-site alloying as a means of further expanding the compositional phase space of double perovskites.5–7 Isovalent alloying, in which the impurity cation carries the same charge as one of the two B-site cations, is a highly effective method for tuning the optical and electronic properties of these materials. Often, such alloys exist as solid solutions where the composition and materials properties can be varied continuously between two pure endmember phases.5,8–11 Aliovalent alloying, in which the charge of the impurity cation does not match the charge of either B-site cation, can have a similarly large impact on the properties of a given double perovskite composition.7,12–14 However, compositional flexibility approaching that seen in isovalent alloys remains rare.15,16 Likely due to lattice mismatch, local charge imbalances, and formation of charge-compensating defects,7 most examples of aliovalent alloys exist only in a narrow composition window with low (ca. 1 atom%) impurity concentrations. Nevertheless, complex perovskite compositions, which incorporate heterovalent cations in high concentrations, may exhibit unique optoelectronic properties and strategies for their syntheses are, therefore, highly desirable.The close structural analogy between halide perovskites and their oxide analogues points to one exciting avenue for such work. Layered CuII-oxide perovskite derivatives feature S = 1/2 spins in a square lattice. Famously, members of this family display a high-temperature superconducting transition induced by electron17 or hole18 doping. A similar electronic structure to that of electron-doped oxide cuprates may be accessible in the halide perovskites by incorporating both Cu+ and Cu2+ as B-site cations, which, to our knowledge, has not yet been accomplished. Here, we achieve this mixed-valence Cu state in an unusual layered perovskite containing three distinct metal cations disordered over the octahedral B sites with the formula: AI4[CuII(CuIInIII)0.5Cl8] (1; A = organic cation). Perovskite 1 may be derived from the layered CuI–InIII double perovskite by replacing half of the octahedral metal sites with Cu2+, or equivalently from the layered CuII perovskites by replacing half of the Cu2+ sites with an equal mixture of Cu+ and In3+. Importantly, spectroscopic characterization of 1 demonstrates that incorporation of three heterovalent B-site cations affords unique optical and electronic properties including enhanced optical absorption in the visible region (Fig. 1) and substantially increased electronic conductivity. We uncover the origins of the new optical, transport, and magnetic properties in 1 and propose a packing model that explains why the specific 1:2:1 Cu+:Cu2+:In3+ metal ratio is stabilized in the layered perovskite lattice.
 | ||
| Fig. 1 Photographs of crystals of (A) the colorless double perovskite, (PEA)4CuIInIIICl8, (B) the yellow single perovskite, (PCA)2CuIICl4, and (C) the black perovskite, (BA)4[CuII(CuIInIII)0.5Cl8] (1BA), featuring a 1:1 blend of the inorganic lattices in A and B. The black color arises from Cu+/2+ mixed valency. PEA = phenethylammonium, PCA = p-chloroanilinium, and BA = butylammonium. | ||
2. Results and discussion
2.1. Perovskite structure
The 3D AIBIIX3 single perovskite structure consists of a lattice of corner-sharing BX6 octahedra (B = divalent cation, X = halide anion) where small monovalent A-site cations fill the voids between octahedra. In the double perovskite structure, the B site is split into two different occupants that form an ordered arrangement yielding the general formula AI2BB′X6. The B and B′ sites can be vacant or filled by cations with charges of 1+ through 5+,19–21 provided that the B sites maintain an average charge of 2+. Employing large alkylammonium A-site cations in the synthesis of halide perovskites affords lower-dimensional derivatives where the inorganic perovskite lattice is partitioned by the organic cations, forming structures with two-dimensional (2D), one-dimensional (1D), or zero-dimensional (0D) connectivity.22 Recently, dimensional reduction of the double perovskite lattice to 2D layered structures with perovskite layers of mono- and bilayer thickness (n = 1 and 2, respectively) has emerged as a handle for diversifying double perovskites.23,24 In addition to exhibiting unique optoelectronic properties,24,25 layered double perovskites have greatly expanded the compositional phase space of this materials family due to the greater structural flexibility of the 2D lattice.25–27 In the 2D structure, coordination spheres have greater freedom to distort and tilt with respect to one another, stabilizing compositions predicted to be unstable in 3D form.28,29 Here, we exploit the greater structural freedom of the 2D framework to bring three distinct octahedral metal sites into the perovskite lattice.2.2. Tracking the black phase in the synthesis of a CuI–InIII layered double perovskite
The layered double perovskite (PEA)4CuInCl8 (PEA = phenethylammonium; Fig. 2A),30 consists of sheets of corner-sharing [CuICl6]5− and [InIIICl6]3− octahedra partitioned by bilayers of PEA cations. Crystals of (PEA)4CuInCl8 grow as colorless plates under an inert N2 atmosphere (Fig. 1), but we observed the formation of thin, plate-like, black crystals (1PEA) when droplets of the mother liquor in Paratone-N® oil were exposed to air for ca. 15 minutes. We hypothesized that formation of this black phase is related to the oxidation of Cu+ to Cu2+ and therefore attempted to prepare it directly. (A recent study on a layered CuI–InIII perovskite also speculated that oxidation of Cu+ led to formation of black material, although this product was not characterized further.)31 Indeed, quickly cooling a 100 °C solution of CuCl, CuCl2, In2O3, and phenethylamine in HCl under N2 affords black plate-like crystals (see ESI†). Unfortunately, these crystals diffracted poorly, yet slowing the cooling rate or allowing the black crystals to remain in the mother liquor afforded colorless crystals, hindering characterization and suggesting that the black phase was a kinetic product. Given the importance of the organoammonium A-site cation in dictating both the stability and crystallinity of perovskite phases,22,25,32,33 we sought to stabilize the black phase using different organic cations. In Sections 2.3 and 2.4, we prepare this black phase using two different organic cations. With butylammonium (BA) cations, we obtain a stable, phase-pure perovskite (1BA) enabling accurate compositional characterization. However, we obtained higher-quality single-crystal X-ray diffraction (SCXRD) data using p-chloroanilinium (PCA) as the A-site cation, and thus, we used this analogue (1PCA) to study structural details.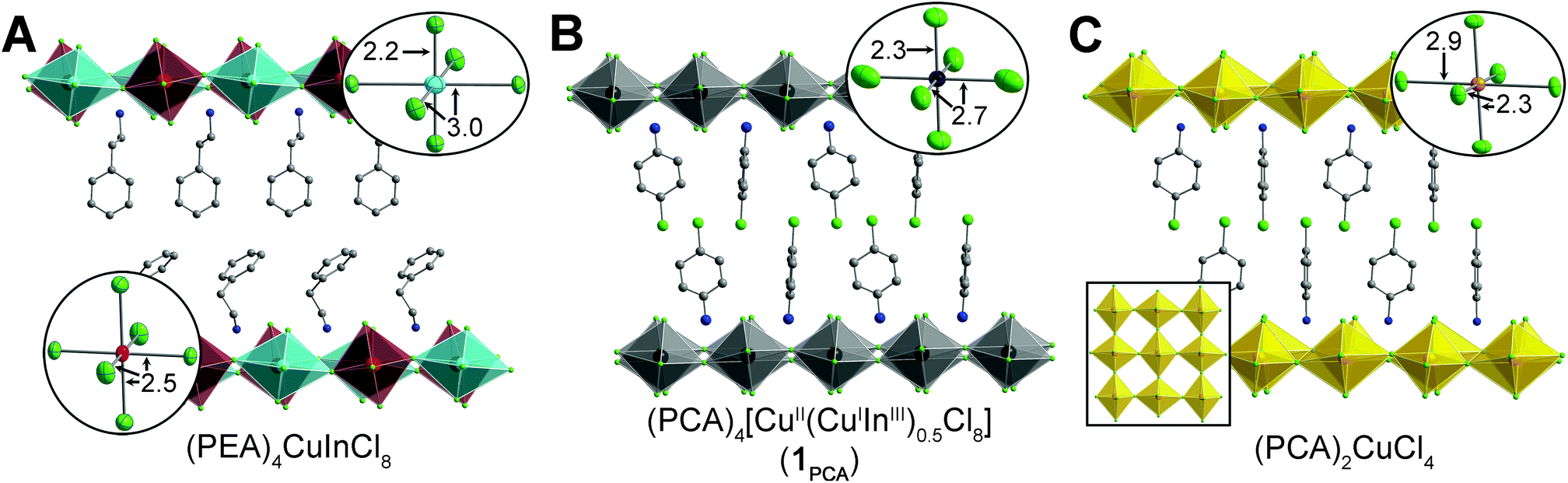 | ||
| Fig. 2 SCXRD structures of (A) (PEA)4CuInCl8,30 (B) (PCA)4[CuII(CuIInIII)0.5Cl8] (1PCA), and (C) (PCA)2CuCl4 (ref. 36) at 300 K with insets showing the local coordination environments of the B sites in each structure and the antiferrodistortive arrangement of the elongated axes of the Cu2+ octahedra in (PCA)2CuCl4 (C). Turquoise, red, yellow, green, gray, and blue spheres represent Cu+, In3+, Cu2+, Cl, C, and N atoms, respectively. Black spheres represent mixed Cu/In sites. H and disordered atoms are omitted for clarity. | ||
2.3. Composition of 1
Single-crystal structure determination of 1BA was complicated by weak diffraction, disorder, and a low-temperature phase transition (see ESI†). Nevertheless, our data reveal that 1BA has an n = 1 layered perovskite structure consisting of 2D sheets of corner sharing metal-halide octahedra partitioned by bilayers of BA cations (Fig. S3†). Interestingly, the structure is best modeled when the B sites are treated as a disordered mixture of Cu and In cations. Although too much emphasis should not be placed on structural parameters extracted from this solution due to the relatively low data quality, we were nevertheless intrigued by the ca. 78:22 Cu:In site-occupancy ratio found for the B sites (Table S1†). This ratio is very different than the 50:50 ratio expected for a disordered n = 1 perovskite with only Cu+ and In3+ at the B sites. Because 1BA forms only in the presence of Cu2+, we posited that Cu2+ cations could replace equal amounts of In3+ and Cu+ in the perovskite lattice, thus altering the Cu:In occupancy ratio of the B site (see Section 2.4 for an in-depth structural analysis of 1 performed on a higher-quality SCXRD data set).
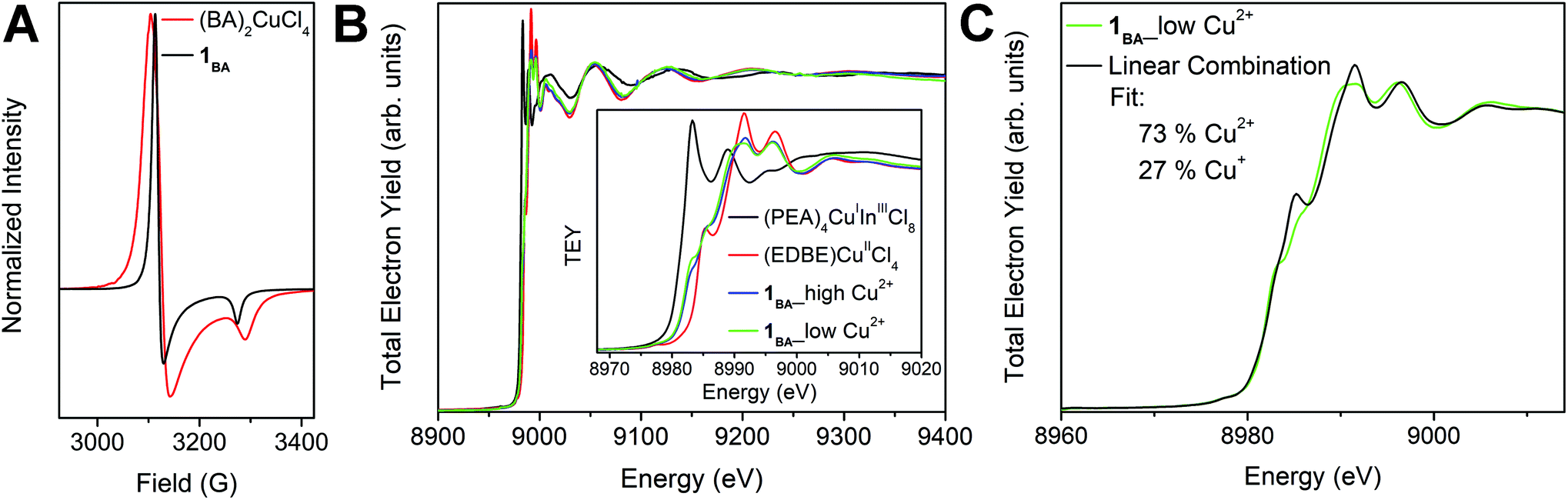 | ||
| Fig. 3 (A) EPR spectra of 1BA (black) and (BA)2CuCl4 (red). (B) XAS spectra of (PEA)4CuInCl8 (black), (EDBE)CuCl4 (red), and 1BA prepared using high (blue) and low (green) solution concentrations of Cu2+ (EDBE = 2,2′-(ethylenedioxy)bis(ethylammonium)). (C) A linear combination of the (PEA)4CuInCl8 and (EDBE)CuCl4 XAS spectra (black) provides an approximation to the XAS spectrum of 1BA prepared using a low solution concentration of Cu2+ (green), giving a rough estimate of the Cu2+:Cu+ ratio (73:27) for this sample (see Section 2.3.3 for a more accurate value). | ||
X-ray absorption spectroscopy (XAS) was also used to confirm the presence of both Cu+ and Cu2+ in 1BA and to estimate the Cu+:Cu2+ ratio. Measurements were collected at the Cu K-edge on finely ground powders of 1BA, (EDBE)CuIICl4 (EDBE = 2,2′-(ethylenedioxy)bis(ethylammonium)), and (PEA)4CuIInIIICl8, with the latter two materials serving as Cu2+ and Cu+ standards, respectively (see ESI†). Here, we report data obtained using the total electron yield (TEY) method; similar results were obtained using the fluorescence method (Fig. S5 and Table S3†). We found that samples of 1BA, which were phase-pure by PXRD, could be prepared using a range of precursor stoichiometries (see ESI†), and we tested these for compositional variability using XAS. Phase-pure samples of 1BA prepared using high (Cu2+:Cu+:In3+ ratio of ca. 1:2:3) and low (Cu2+:Cu+:In3+ ratio of ca. 1:10:10) concentrations of Cu2+ yield very similar XAS spectra (Fig. 3B). Both feature absorption onsets intermediate between those of the Cu+ and Cu2+ standards, indicating the presence of both oxidation states in 1BA. We fit these spectra using a linear combination of the Cu+ and Cu2+ standard spectra (see ESI†), obtaining estimated Cu+:Cu2+ ratios of 22:78 and 27:73 for high and low Cu2+ solution concentrations, respectively (Fig. 3C and S6†). These ratios are only estimates as they assume that the Cu+ and Cu2+ environments in 1BA are identical to those in the Cu+ and Cu2+ standards. Notably, these results indicate relatively little flexibility in the Cu+:Cu2+ ratio of 1BA despite the widely ranging precursor ratios employed in the different syntheses.
:In ratio of 1BA, we used inductively coupled plasma mass spectrometry (ICP-MS). We measured the same samples made with high and low Cu2+ concentrations used for XAS, finding Cu:In weight % ratios of 1.73 and 1.59, respectively (Table S4†). Assuming that all Cu+ B-site cations are charge compensated by In3+ B-site cations, these ratios yield extremely similar formulas of (BA)4[(CuII)1.04(CuIInIII)0.48Cl8] and (BA)4[(CuII)0.97(CuIInIII)0.515Cl8], respectively (see ESI†). Formulas extracted from our SCXRD and XAS data (see ESI†) are consistent with these results, though the ICP-MS formulas are the most reliable given the greater accuracy of this method for such analyses.
Importantly, both our XAS and ICP-MS data indicate little variability in the B-site composition of 1BA and we propose the approximate formula of (BA)4[CuII(CuIInIII)0.5Cl8]. In addition, EPR spin quantification (Section 2.3.2) and magnetic susceptibility measurements (Section 2.6.3) provide two independent estimates of the amount of Cu2+ in 1BA, both of which agree with this empirical formula. This validates our assumption that charge balance of the perovskite lattice is maintained by incorporating equal amounts of In3+ and Cu+ at the B sites. Furthermore, C, H, and N analyses of 1BA agree well with our proposed formula (see ESI†). Therefore, our analysis indicates that, in terms of composition, 1BA is a 50:50 mixture of a CuII single perovskite and a CuI–InIII double perovskite.
2.4. Structure of 1
Further characterization of 1PCA using the techniques employed above for 1BA was hindered by its instability in solution (similar to the case of 1PEA, see ESI†). Nevertheless, our structural data for 1PCA are fully consistent with the presence of both Cu and In cations at the B sites, and the Cu:In ratio of 78:22 (100 K structure) is in good agreement with our ICP-MS data for 1BA.
We can glean further insights into the black phase from the SCXRD structure of 1PCA. At both 300 and 100 K, the structure of 1PCA has only one unique B site, indicating that the Cu2+ cations disrupt the checkerboard-like ordering of Cu+ and In3+ cations found in the double-perovskite endmember. As seen in the SCXRD structures of the CuII and CuI–InIII endmember perovskites, these three B-site cations exhibit very different coordination environments. The Cu+ cations of (PEA)4CuInCl8 adopt a linear [2 + 4] coordination, with two short axial bonds and four long equatorial bonds (Fig. 2A).30 Meanwhile, the elongated axis of the Jahn–Teller distorted Cu2+ cations of (PCA)2CuCl4 (ref. 36) (and other CuII perovskites)37 lies in the plane of the 2D perovskite sheets with the long axes of adjacent [CuCl6]4− units positioned orthogonally to one another (an antiferrodistortive arrangement; Fig. 2C). In contrast, the In3+ cations of (PEA)4CuInCl8 (ref. 30) (and other known n = 1 perovskite structures)25,26 display relatively little deviation from octahedral coordination, with similar axial and equatorial bond lengths (Fig. 2A). Disordering of Cu+, Cu2+, and In3+ is expected to yield a B-site coordination environment that is an average of the three very different coordination spheres of these cations, with two axial B–Clax bonds that are substantially shorter than the four equatorial B–Cleq bonds. This is, in fact, what we observe in both the 300 and 100 K SCXRD structures of 1PCA. Also consistent with this picture, we find that the thermal ellipsoids of the equatorial Cl atoms are elongated along the directions of B–Cl–B connectivity (Fig. 2B, inset and Fig. S7†) due to the presence of both very long (CuII–Cleq = 2.9041(7) Å for (PCA)2CuCl4 at 300 K, CuI–Cleq = 3.0340(2) Å for (PEA)4CuInCl8 at 300 K) and very short (CuII–Cleq = 2.3067(6) Å for (PCA)2CuCl4 at 300 K) B–Cleq bond lengths. The thermal ellipsoids of the axial Cl atoms are less elongated, as expected based on the smaller variation in B–Clax bond lengths between the Cu+, Cu2+, and In3+ coordination spheres of the CuI–InIII and CuII endmember perovskites (CuI–Clax = 2.1779(1) Å, CuII–Clax = 2.2674(7) Å, InIII–Clax = 2.5483(1) Å at 300 K).
Although we performed this in-depth structural analysis for 1PCA rather than for 1BA due to the higher-quality SCXRD data obtained for the former, it is important to note that the structural parameters obtained from the SCXRD structure of 1BA are extremely similar (see ESI†), and thus, the above analysis can be extended to the case of 1BA.
2.5. Impact of tiling three coordination spheres on the composition of 1
It is interesting to consider how the very differently shaped Cu+, Cu2+, and In3+ coordination spheres (Fig. 4A) can be arranged into an n = 1 perovskite lattice. The metal-halide octahedra pack efficiently in (PEA)4CuInCl8 and (PCA)2CuCl4, with the large [CuICl6]5− and small [InIIICl6]3− units adopting an alternating checkerboard pattern in the former and the elongated [CuIICl6]4− units forming an antiferrodistortive arrangement in the latter. However, mixing these three units in a single 2D lattice disturbs both of these well-defined arrangements. The elongated [CuIICl6]4− units do not fit the CuI–InIII perovskite lattice, introducing large packing mismatch (Fig. 4B). Likewise, [CuICl6]5− and [InIIICl6]3− units disturb the arrangement of Jahn–Teller distorted [CuIICl6]4− octahedra (Fig. 4C). However, a new efficiently packed arrangement can be generated from the cell outlined in Fig. 4D that contains one [InIIICl6]3−, one [CuICl6]5−, and two [CuIICl6]4− units. This cell can be tessellated in two dimensions to generate an n = 1 perovskite lattice with no significant packing mismatch. Notably, the resulting lattice has a 1:2:1 ratio of Cu+:Cu2+:In3+ cations, matching the results of our ICP-MS data for 1BA (Section 2.3.3). Thus, we propose that the problem of packing the differently shaped Cu+, Cu2+, and In3+ coordination spheres into an n = 1 perovskite lattice explains the robust composition adopted by 1BA: a composition in which 50% of the B sites of the CuI–InIII perovskite are replaced by Cu2+ cations represents a sweet spot that enables the most efficient packing arrangement of the different coordination spheres. Deviations from this composition will introduce packing mismatch (see ESI and Fig. S8†), which may explain the lack of compositional flexibility observed for 1BA.
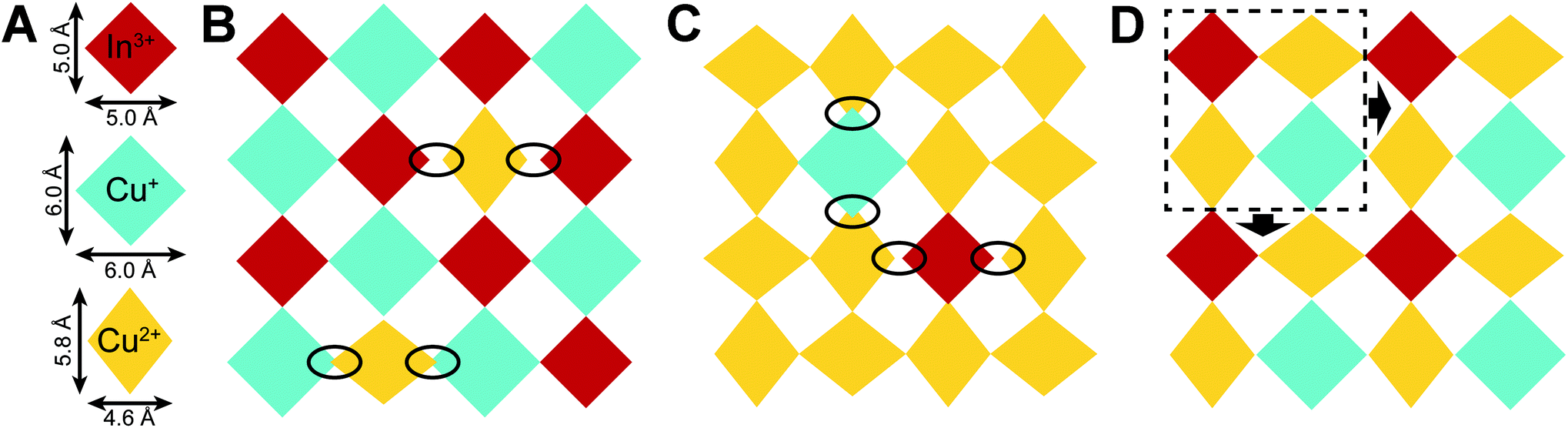 | ||
| Fig. 4 Schematic top-down view of n = 1 perovskite sheets where [InIIICl6]3−, [CuICl6]5−, and [CuIICl6]4− units are represented by red, blue, and yellow diamonds, respectively, with dimensions proportional to the coordination spheres found in the 300 K SCXRD structures of (PEA)4CuInCl8 and (PCA)2CuCl4 (A). (B) Packing arrangements for Cu2+ doped into the CuI–InIII perovskite lattice and (C) Cu+ and In3+ doped into the CuII perovskite lattice. Large packing mismatches are circled. (D) Proposed packing arrangement for 1: One [InIIICl6]3−, one [CuICl6]5−, and two [CuIICl6]4− units can be arranged into a cell and tessellated to yield a 2D lattice without large packing mismatch. See Fig. S9† for alternative tessellations of the same cell, which may explain the B-site disordering of 1. | ||
Slight variations on the tessellation pattern illustrated in Fig. 4D are also possible, generating 2D lattices with different packing arrangements (Fig. S9†). Some of these packing arrangements are expected to be similarly favorable (Fig. S9B and C†) which may explain the B-site disordering of 1. It is also important to point out that 1, (PCA)2CuCl4, and (PEA)4CuInCl8 all exhibit similar degrees of octahedral tilting, making it unlikely that large packing mismatches are present in 1 as they would likely need to be compensated by severe octahedral tilting.
2.6. Emergent properties in 1
Our characterization of 1BA and 1PCA has shown that the composition and structure of these materials are well-described as an average of the CuII and CuI–InIII endmember perovskites. However, 1 displays new optical, electronic, and magnetic properties not seen in the endmembers.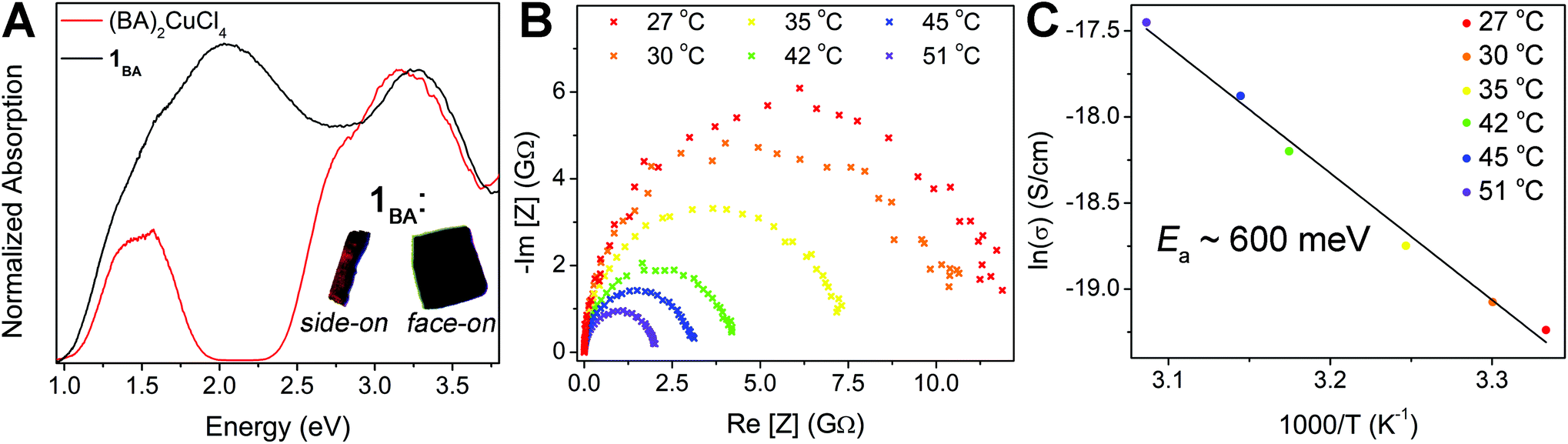 | ||
| Fig. 5 (A) Diffuse reflectance spectra of 1BA (black) and (BA)2CuCl4 (red) with insets showing the dichroism of a plate-like crystal of 1BA in side-on and face-on orientations under linearly polarized light. (B) Nyquist plots for a single crystal of 1BA measured at a range of temperatures demonstrating the increase in conductivity with temperature (Z = electrochemical impedance). (C) Arrhenius relationship for the data plotted in B showing an activation energy (Ea) for electronic conductivity (σ) of ca. 600 meV. | ||
Materials containing the same element in multiple valence states that are in electronic contact often exhibit intense colors because electron transfer between the different ions results in strong optical absorption in the visible region.40 Thus, we propose that the feature at 2 eV in 1BA arises due to the presence of both Cu+ and Cu2+ cations in 1BA and assign it as a Cu+ → Cu2+ intervalence charge transfer (IVCT) transition.41 Intriguingly, this IVCT transition endows crystals of 1BA with clear dichroic effects. Under linearly polarized light, thick plate-like single crystals of 1BA appear black when viewed face-on but translucent red-orange when viewed in the proper side-on orientation (Fig. 5A, inset). This indicates that the 2 eV transition can only be excited by light polarized in the plane of the perovskite sheet and thus corresponds to an IVCT transition between Cu ions in the same layer.
According to the Robin and Day classification,401 is best described as a class II mixed-valence material. The spectral features of the component ions (most notably the d–d and LMCT transitions of the [CuCl6]4− unit) are preserved in the mixed-valence material, while a new IVCT absorption appears in the visible region at a location close to that observed for similar class II mixed-valent Cu+/2+ materials.41–43 Assignment of 1 as a class II species is consistent with our structural characterization, which suggests that Cu+ and Cu2+ cations retain distinct coordination environments (excluding assignment as a class III material) but are both six-fold coordinated making structural rearrangement of the coordination spheres relatively easy (unlike in a class I material). Thus, 1 joins the small number of class II mixed-valence halide perovskites, including lattices with Au+/3+ and Sb3+/5+.15,40,44
Perovskite 1 allows us to test the impact of magnetically diluting this Cu2+ square lattice, with nonmagnetic Cu+ and In3+, on the long-range FM order. Further, 1BA presents an intriguing system for study since the magnetic occupancy of the metal square lattice (approximately 50% Cu2+ at the B sites) is close to the site percolation threshold of a 2D simple square lattice with four nearest neighbors (59.2%).53,54 At and above this value, enough magnetic ions should exist to support long-range magnetic order. While the overall stoichiometry of 1BA is below this threshold, if some areas of the square lattice have a high enough concentration of Cu2+ ions, local puddles of long-range order may arise.
Our measurements reveal weak interactions between local Cu2+ moments with no long-range order down to T = 2 K, as shown by the lack of a peak in plots of the low-temperature zero-field AC susceptibility (Fig. 6A, insets) and low-field DC susceptibility (Fig. 6A). A spin glass ground state, which may also be expected for dilute magnetic ions, is ruled out by the lack of splitting between the zero-field cooled and field cooled susceptibility data at low temperature (Fig. 6A), the lack of a peak in the variable-temperature AC susceptibility (χ′ and χ′′; Fig. 6A, insets), and the frequency-independence of the AC susceptibility (Fig. S17†). High-temperature DC susceptibility measured in an applied field of μ0H = 5 T (μ0 = permeability of free space, H = magnetic field strength) is shown in Fig. 6B. After subtraction of a large temperature-independent diamagnetic contribution (likely from BA cations, χ0 = −0.00108 emu mol−1) a Curie–Weiss fit of the inverse susceptibility data from T = 100–300 K yields a Curie constant C = 0.362(4) K emu mol−1 and a Weiss temperature Θ = 10.9(2) K. Note that these calculations assume a formula weight given by the formula of 1BA determined above ((BA)4[CuII(CuIInIII)0.5Cl8]). The slightly positive value of Θ may indicate that the weak interactions between moments, or the interactions in somewhat more concentrated puddles, are FM. Our fit results are in line with those reported for (BA)2CuCl4, which has a higher value of Θ (18.3(3) K), consistent with its long-range FM order at TC = 7.3 K.47,52 The effective moment (μeff) of 1BA is 1.702(1) μB (μB = Bohr magneton), consistent with magnetically isolated Cu2+ moments, and the Landé g-factor is 1.965(1), consistent with the g-factors extracted from EPR measurements (Section 2.3.2). The overall paramagnetic behavior and the absence of magnetic long-range order are consistent with our ICP-MS analysis, which yields a Cu2+ content below the site percolation threshold. In addition, as noted in Section 2.3.3, our magnetic susceptibility measurements allow us to estimate the amount of Cu2+ in 1BA as 0.97 mol Cu2+ per formula unit (see ESI†), in agreement with the values obtained from ICP-MS (Section 2.3.3) and EPR data (Section 2.3.2).
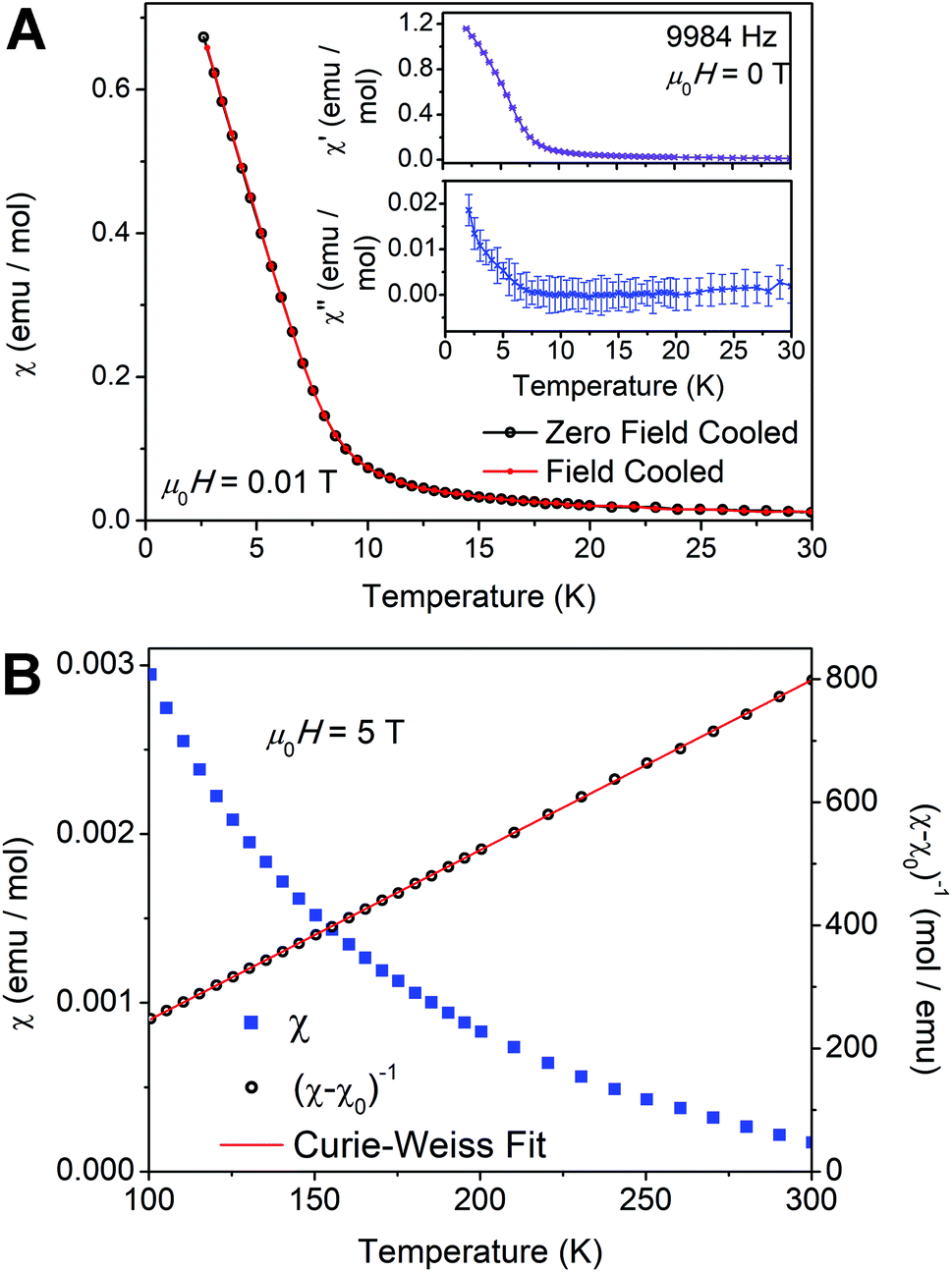 | ||
| Fig. 6 (A) Zero-field cooled and field cooled DC susceptibility (χ) of 1BA measured in a small applied field (μ0H = 0.01 T) from T = 2–30 K. The top and bottom insets show the real (χ′) and imaginary (χ′′) parts of the AC susceptibility, respectively, measured in zero applied field. (B) High-temperature DC susceptibility (χ) and inverse susceptibility ((χ − χ0)−1) of 1BA measured in an applied field μ0H = 5 T (χ0 is the diamagnetic contribution to χ). | ||
These findings present an interesting contrast with a report of an n = 1 ZnII fluoride perovskite where 40% of the B sites are replaced with Cu2+. Here, the Cu2+ content is well below the site percolation threshold and also lower than that of 1BA, yet EPR data provide evidence for long-range magnetic order at 4.2 K.55 This suggests that Cu2+ cations may form clusters within the Zn2+ lattice, leading to the stronger magnetic interactions. The absence of long-range order in 1BA despite the higher Cu2+ content suggests that here, Cu2+ cluster formation may be disfavored. This further validates our proposed packing scheme discussed in Section 2.5 and the ESI,† which shows that arrangements containing adjacent Cu2+ centers generally have larger packing mismatch (Fig. S9D and F†), potentially disfavoring formation of Cu2+ clusters.
3. Conclusion
We report the new layered perovskite (A)4[CuII(CuIInIII)0.5Cl8] (1, A = BA, PCA, PEA), which contains a 1:2:1 mixture of Cu+, Cu2+, and In3+ cations at the B sites. The composition and structure of 1 are well-described as an average of a CuII single perovskite and a CuI–InIII double perovskite. Simple packing arguments suggest that this specific composition is enforced by the differently shaped Cu+, Cu2+, and In3+ coordination spheres that must pack efficiently into an n = 1 perovskite lattice. Magnetic studies of 1BA demonstrate the loss of long-range ferromagnetic order observed in the CuII perovskite, corroborating our proposed packing model, which disfavors segregated domains of CuII in the lattice of 1.
The presence of mixed-valent Cu in 1 affords new properties not seen in the endmember lattices. In particular, 1 is black, displaying strong optical absorption across the visible region, which is highly unusual for an n = 1 chloride perovskite. We further measure a large increase in conductivity between 1BA and its CuII and CuI–InIII endmembers. Thus, alloying two different 2D perovskites affords a new perovskite with properties not seen in either endmember.
The novel optoelectronic properties of 1 evidence the significant advantages of incorporating B-site cations with a range of valence states into the perovskite structure and set forth the tantalizing possibility of using this strategy to access exotic physical properties, similar to the discovery of high-temperature superconductivity in analogous electron-doped copper-oxide perovskites. We expect that the simple tiling arguments made here, based on using a mixture of long and short metal-halide bond lengths to construct the 2D footprint of the inorganic sheets, may be extended to other compositions to incorporate multiple metal ions in the perovskite lattice. Thus, increasing the complexity of the inorganic sheets, beyond the simple rock-salt ordering of most double perovskites,1 promises to enhance the optoelectronic diversity of halide perovskites with emergent properties arising from new electronic transitions.
Author contributions
B. A. C. performed all experimental work and analysis, except as noted below. J. L. synthesized materials for elemental analysis and EPR studies. B. A. C. and J. L. were advised by H. I. K. R. W. S. conducted the magnetic susceptibility measurements and data analysis, advised by Y. S. L. A. J. H. conducted EPR spin quantification studies. A. G.-P. conducted the XAS measurements and data analysis, advised by M. F. T. The project was conceived by B. A. C. with guidance from H. I. K.; they wrote the manuscript with contributions from all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the U.S. Department of Energy, Office of Science, Basic Energy Sciences (DOE, BES), Division of Materials Sciences and Engineering under Contract DE-AC02-76SF00515. SC-XRD studies were performed at beamline 12.2.1 at the Advanced Light Source (ALS) at the Lawrence Berkeley National Laboratory and at the Stanford Nano Shared Facilities, supported by the National Science Foundation (NSF; award ECCS-1542152). Use of the Stanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory, is supported by the DOE, BES under Contract No. DE-AC02-76SF00515. B. A. C. was supported by an NSF Graduate Research Fellowship (DGE-114747) and the Evelyn McBain award from Stanford Chemistry. R. W. S. was supported by an NSF Graduate Research Fellowship (DGE-1656518). We thank Dr M. A. Manumpil and N. R. Wolf for experimental assistance and Prof. E. I. Solomon for access to equipment and helpful discussions.Notes and references
- N. R. Wolf, B. A. Connor, A. H. Slavney and H. I. Karunadasa, Angew. Chem., Int. Ed., 2021, 60, 2–17 CrossRef.
- A. H. Slavney, T. Hu, A. M. Lindenberg and H. I. Karunadasa, J. Am. Chem. Soc., 2016, 138, 2138–2141 CrossRef CAS PubMed.
- E. T. McClure, M. R. Ball, W. Windl and P. M. Woodward, Chem. Mater., 2016, 28, 1348–1354 CrossRef CAS.
- G. Engel, Naturwissenschaften, 1933, 21, 704 CrossRef CAS.
- A. H. Slavney, L. Leppert, D. Bartesaghi, A. Gold-Parker, M. F. Toney, T. J. Savenije, J. B. Neaton and H. I. Karunadasa, J. Am. Chem. Soc., 2017, 139, 5015–5018 CrossRef CAS PubMed.
- J. Luo, X. Wang, S. Li, J. Liu, Y. Guo, G. Niu, L. Yao, Y. Fu, L. Gao, Q. Dong, C. Zhao, M. Leng, F. Ma, W. Liang, L. Wang, S. Jin, J. Han, L. Zhang, J. Etheridge, J. Wang, Y. Yan, E. H. Sargent and J. Tang, Nature, 2018, 563, 541–545 CrossRef CAS PubMed.
- K. P. Lindquist, S. A. Mack, A. H. Slavney, L. Leppert, A. Gold-Parker, J. F. Stebbins, A. Salleo, M. F. Toney, J. B. Neaton and H. I. Karunadasa, Chem. Sci., 2019, 10, 10620–10628 RSC.
- A. H. Slavney, L. Leppert, A. Saldivar Valdes, D. Bartesaghi, T. J. Savenije, J. B. Neaton and H. I. Karunadasa, Angew. Chem., Int. Ed., 2018, 57, 12765–12770 CrossRef CAS PubMed.
- A. E. Maughan, A. M. Ganose, M. M. Bordelon, E. M. Miller, D. O. Scanlon and J. R. Neilson, J. Am. Chem. Soc., 2016, 138, 8453–8464 CrossRef CAS PubMed.
- T. T. Tran, J. R. Panella, J. R. Chamorro, J. R. Morey and T. M. McQueen, Mater. Horiz., 2017, 4, 688–693 RSC.
- K.-z. Du, W. Meng, X. Wang, Y. Yan and D. B. Mitzi, Angew. Chem., Int. Ed., 2017, 56, 8158–8162 CrossRef CAS PubMed.
- A. Karmakar, M. S. Dodd, S. Agnihotri, E. Ravera and V. K. Michaelis, Chem. Mater., 2018, 30, 8280–8290 CrossRef CAS.
- N. Nandha K and A. Nag, Chem. Commun., 2018, 54, 5205–5208 RSC.
- J. D. Majher, M. B. Gray, T. A. Strom and P. M. Woodward, Chem. Mater., 2019, 31, 1738–1744 CrossRef CAS.
- L. Atkinson and P. Day, J. Chem. Soc. A, 1969, 2423–2431 RSC.
- M. Retuerto, Z. Yin, T. J. Emge, P. W. Stephens, M.-R. Li, T. Sarkar, M. C. Croft, A. Ignatov, Z. Yuan, S. J. Zhang, C. Jin, R. P. Sena, J. Hadermann, G. Kotliar and M. Greenblatt, Inorg. Chem., 2015, 54, 1066–1075 CrossRef CAS PubMed.
- N. P. Armitage, P. Fournier and R. L. Greene, Rev. Mod. Phys., 2010, 82, 2421–2487 CrossRef CAS.
- C. W. Chu, L. Z. Deng and B. Lv, Phys. C, 2015, 514, 290–313 CrossRef CAS.
- N. Elliott and L. Pauling, J. Am. Chem. Soc., 1938, 60, 1846–1851 CrossRef CAS.
- R. W. G. Wyckoff and E. Posnjak, J. Am. Chem. Soc., 1921, 43, 2292–2309 CrossRef CAS.
- K. Prassides, P. Day and A. K. Cheetham, Inorg. Chem., 1985, 24, 545–552 CrossRef CAS.
- D. B. Mitzi, J. Chem. Soc., Dalton Trans., 2001, 1–12 RSC.
- L. M. Castro-Castro and A. M. Guloy, Angew. Chem., Int. Ed., 2003, 42, 2771–2774 CrossRef CAS PubMed.
- B. A. Connor, L. Leppert, M. D. Smith, J. B. Neaton and H. I. Karunadasa, J. Am. Chem. Soc., 2018, 140, 5235–5240 CrossRef CAS PubMed.
- B. A. Connor, R.-I. Biega, L. Leppert and H. I. Karunadasa, Chem. Sci., 2020, 11, 7708–7715 RSC.
- L. Mao, S. M. L. Teicher, C. C. Stoumpos, R. M. Kennard, R. A. DeCrescent, G. Wu, J. A. Schuller, M. L. Chabinyc, A. K. Cheetham and R. Seshadri, J. Am. Chem. Soc., 2019, 141, 19099–19109 CrossRef CAS PubMed.
- L.-Y. Bi, Y.-Q. Hu, M.-Q. Li, T.-L. Hu, H.-L. Zhang, X.-T. Yin, W.-X. Que, M. S. Lassoued and Y.-Z. Zheng, J. Mater. Chem. A, 2019, 7, 19662–19667 RSC.
- G. Volonakis, A. A. Haghighirad, R. L. Milot, W. H. Sio, M. R. Filip, B. Wenger, M. B. Johnston, L. M. Herz, H. J. Snaith and F. Giustino, J. Phys. Chem. Lett., 2017, 8, 772–778 CrossRef CAS PubMed.
- Z. Xiao, K.-Z. Du, W. Meng, D. B. Mitzi and Y. Yan, Angew. Chem., Int. Ed., 2017, 56, 12107–12111 CrossRef CAS PubMed.
- M. L. Aubrey, A. Saldivar Valdes, M. R. Filip, B. A. Connor, K. P. Lindquist, J. B. Neaton and H. I. Karunadasa, Directed assembly of layered halide perovskite heterostructures as single crystals, in review.
- E. T. McClure, A. P. McCormick and P. M. Woodward, Inorg. Chem., 2020, 59, 6010–6017 CrossRef CAS PubMed.
- L.-Y. Bi, T.-L. Hu, M.-Q. Li, B.-K. Ling, M. S. Lassoued, Y.-Q. Hu, Z. Wu, G. Zhou and Y.-Z. Zheng, J. Mater. Chem. A, 2020, 8, 7288–7296 RSC.
- E. R. Dohner, E. T. Hoke and H. I. Karunadasa, J. Am. Chem. Soc., 2014, 136, 1718–1721 CrossRef CAS PubMed.
- C.-C. Chao, J. Magn. Reson., 1973, 10, 1–6 CAS.
- C. P. Landee, K. E. Halvorson and R. D. Willett, J. Appl. Phys., 1987, 61, 3295–3297 CrossRef CAS.
- A. K. Vishwakarma, P. S. Ghalsasi, A. Navamoney, Y. Lan and A. K. Powell, Polyhedron, 2011, 30, 1565–1570 CrossRef CAS.
- R. D. Willett, J. Chem. Phys., 1964, 41, 2243–2244 CrossRef CAS.
- R. D. Willett, O. L. Liles Jr and C. Michelson, Inorg. Chem., 1967, 6, 1885–1889 CrossRef CAS.
- R. Valiente and F. Rodríguez, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 60, 9423–9429 CrossRef CAS.
- M. B. Robin and P. Day, in Advances in Inorganic Chemistry and Radiochemistry, ed. H. J. Emeléus and A. G. Sharpe, Academic Press, 1968, vol. 10, pp. 247–422 Search PubMed.
- N. S. Hush, in Prog. Inorg. Chem., ed. F. A. Cotton, John Wiley & Sons, Inc., 1967, ch. 7, vol. 8, pp. 391–444 Search PubMed.
- M. Mori, Bull. Chem. Soc. Jpn., 1961, 34, 1249–1253 CrossRef CAS.
- D. Culpin, P. Day, P. R. Edwards and R. J. P. Williams, Chem. Commun., 1965, 450–451 RSC.
- N. Kojima and H. Kitagawa, J. Chem. Soc., Dalton Trans., 1994, 327–331 RSC.
- C. P. Tigges, J. E. Drumheller and L. O. Snively, J. Appl. Phys., 1983, 54, 5473–5475 CrossRef CAS.
- D. M. D'Alessandro and F. R. Keene, Chem. Soc. Rev., 2006, 35, 424–440 Search PubMed.
- L. J. de Jongh, A. C. Botterman, F. R. de Boer and A. R. Miedema, J. Appl. Phys., 1969, 40, 1363–1365 CrossRef CAS.
- L. J. de Jongh and A. R. Miedema, Adv. Phys., 1974, 23, 1–260 CrossRef CAS.
- J. B. Goodenough, Phys. Rev., 1955, 100, 564–573 CrossRef CAS.
- J. B. Goodenough, J. Phys. Chem. Solids, 1958, 6, 287–297 CrossRef CAS.
- J. Kanamori, J. Phys. Chem. Solids, 1959, 10, 87–98 CrossRef CAS.
- C. Aruta, F. Licci, A. Zappettini, F. Bolzoni, F. Rastelli, P. Ferro and T. Besagni, Appl. Phys. A, 2005, 81, 963–968 CrossRef CAS.
- S. Galam and A. Mauger, Phys. Rev. E: Stat. Phys., Plasmas, Fluids, Relat. Interdiscip. Top., 1996, 53, 2177–2181 CrossRef CAS PubMed.
- K. Malarz and S. Galam, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2005, 71, 016125 CrossRef PubMed.
- D. Reinen and S. Krause, Inorg. Chem., 1981, 20, 2750–2759 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: The CIFs for (BA)4[Cu0.5InCl7][Cu0.5Cl][(BA)Cl] (300 K), 1BA (300 K), 1PCA (300 K), and 1PCA (100 K) have been deposited in the CCDC under deposition numbers 2049961–2049964, respectively. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc01159f |
| This journal is © The Royal Society of Chemistry 2021 |