
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceControllable DNA strand displacement by independent metal–ligand complexation†
Liang-Liang
Wang
,
Qiu-Long
Zhang
,
Yang
Wang
,
Yan
Liu
,
Jiao
Lin
,
Fan
Xie
and
Liang
Xu
 *
*
MOE Key Laboratory of Bioinorganic and Synthetic Chemistry, School of Chemistry, Sun Yat-Sen University, Guangzhou, 510275, China. E-mail: xuliang33@mail.sysu.edu.cn
First published on 18th May 2021
Abstract
Introduction of artificial metal–ligand base pairs can enrich the structural diversity and functional controllability of nucleic acids. In this work, we revealed a novel approach by placing a ligand-type nucleoside as an independent toehold to control DNA strand-displacement reactions based on metal–ligand complexation. This metal-mediated artificial base pair could initiate strand invasion similar to the natural toehold DNA, but exhibited flexible controllability to manipulate the dynamics of strand displacement that was only governed by its intrinsic coordination properties. External factors that influence the intrinsic properties of metal–ligand complexation, including metal species, metal concentrations and pH conditions, could be utilized to regulate the strand dynamics. Reversible control of DNA strand-displacement reactions was also achieved through combination of the metal-mediated artificial base pair with the conventional toehold-mediated strand exchange by cyclical treatments of the metal ion and the chelating reagent. Unlike previous studies of embedded metal-mediated base pairs within natural base pairs, this metal–ligand complexation is not integrated into the nucleic acid structure, but functions as an independent toehold to regulate strand displacement, which would open a new door for the development of versatile dynamic DNA nanotechnologies.
Introduction
Ligand-modified oligonucleotides are a type of artificial nucleic acid that can bind and position metal ions into the specific sites of the nucleic acid structures.1–4 Metal-mediated base pairs, formed by metal–ligand complexation between two ligand-type nucleoside analogues, have been widely integrated into the nucleic acid structures as substitutes for natural base pairs.4–6 Compared with a canonical Watson–Crick base pair, due to the formation of coordinate bonds, this artificial base pair is typically more thermostable.7–12 Besides, this coordination is mostly reversible by adding or chelating specific metal ions to regulate the formation of metal-mediated base pairs. Given these unique properties, the metal–ligand complexation between two ligand-modified sites can be treated as a strengthened and tunable base pair with metal functionality. Previous studies using these metal-mediated artificial base pairs have generated a variety of metal-based DNA assemblies,13–17 achieved diverse regulations of DNA structures,10–12,18,19 and also expanded the penitential genetic codes.20 These applications generally focus on the formation and conversion of DNA nanostructures, but less attention has been paid to utilization of ligand-modified nucleic acids in the dynamic DNA nanotechnology, particularly in strand-displacement reactions.The application of nucleic acid strand-displacement reactions is the essential concept in the field of dynamic DNA nanotechnology.21,22 In the enzyme-free system, the toehold-mediated strand displacement is the basic foundation to construct diverse DNA reaction networks23 that can exhibit sophisticated dynamic behaviors, such as acting as logic gates, making decisions, and transducing and amplifying signals.24–28 Toeholds are generally single-stranded segments of DNA with versatile designs including but not limited to toehold exchange,29 hidden toehold,25,30 remote toehold,31 combinatory toehold32 and allosteric toehold,33 to which an invader strand can bind to initiate strand-displacement reactions. In the past two decades, with the employment of toehold-mediated strand-displacement reactions, dynamic nucleic acid technology has been widely applied in information technology (DNA computation),26,27 biomedical analysis,34–36 drug delivery and release37–40 and synthetic biology.41–48
Unlike direct strand displacement, the toehold-mediated reaction is both thermodynamically and kinetically favored. In order to achieve full potential for the displacement kinetics, the minimal binding ability between the invader strand and the toehold is needed, with a typical length of single-stranded DNA over 6 nt for the conventional toehold (Fig. 1a).29 As aforementioned that the metal-mediated base pair can be much more thermostable than the natural base pairs, can the toehold be replaced by a strong artificial base pair? Would a single metal-mediated base pair be able to initiate a strand-displacement reaction (Fig. 1b)? This concept is fundamentally different from previous investigations of embedded metal-mediated base pairs within natural base pairs to regulate the strand displacement,49–51 as those studies still basically rely on the conformational switch of nucleic acid structures induced by metal ions. In this new scenario, however, the metal–ligand complexation is not integrated into the nucleic acid structure, but functions as an independent toehold. Compared with the metal-induced duplex formation of nucleic acids, the great advantage of this design is that the strand dynamics can be governed solely based on the intrinsic coordination properties between the metal and the ligand without the necessity for consideration and involvement of nucleic acid sequences and structures. If feasible, it sets up a great opportunity to introduce all kinds of artificial metal–ligand complexation as versatile triggers for dynamic DNA nanotechnologies.
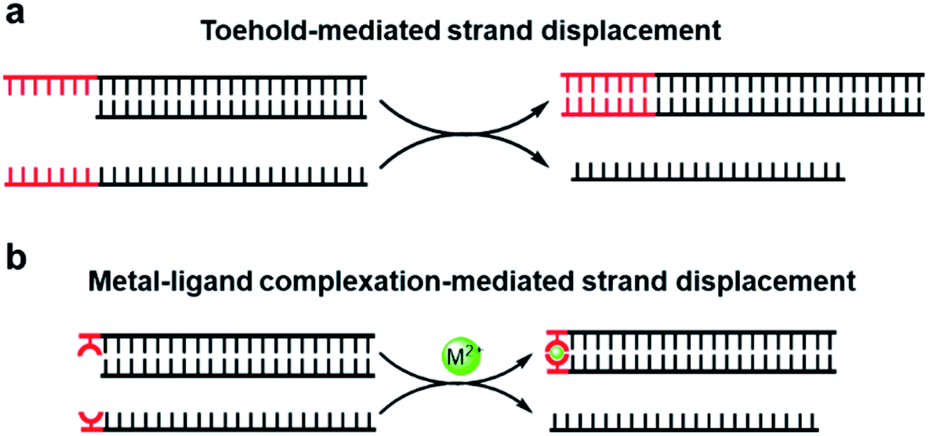 | ||
| Fig. 1 DNA strand-displacement mediated by the conventional toehold (a) and the metal–ligand complexation (b). | ||
Herein, we selected 8-hydroxyquinoline (8-HQ) as the ligand in the modified DNA to examine this concept. 8-Hydroxyquinoline is a potent ligand that can chelate a variety of metal ions and has also been previously applied in metal-mediated base pairs.7,52 Taking this ligandoside as an independent toehold, we investigated how the metal-mediated base pair regulated the dynamics of the strand invasion and displacement process. Our systematical evaluation of metal–ligand complexation in the strand-displacement reactions suggested a highly efficient but controllable dynamics, which would not only expand the applicable scenarios of metal-mediated base pairs, but also provide alternative approaches for the development of versatile dynamic DNA nanotechnologies.
Results and discussion
In our design, the 8-hydroxyquinoline ligand was conjugated with DNA through an amide group in a simplified three carbon backbone, glycidol, instead of deoxyribose, as previous studies7,53–55 suggested that this acyclic and relatively flexible linker can also form a highly stable metallo-base pair with greatly reduced complexity of synthesis (Fig. 2a). This glycidol skeleton contains a flexible methylene group, allowing the conjugated 8-HQ to adjust for an appropriate orientation during the coordination process to form a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 complex between the ligand and the metal ion. Synthesis of this artificial nucleoside was started from the construction of the carboxyl substituted 8-hydroxyquinoline, followed by coupling the ligand with 3-aminopropane-1,2-diol via the formation of amide as described in Scheme S1.† The ligand modified DNA was prepared through solid phase synthesis by the conventional phosphoramidite method as described in the ESI.† The incorporated ligand was located in either the 3′ or 5′ terminal of the DNA strand. All the synthesized DNA strands were purified by HPLC and characterized by mass spec.
:1 complex between the ligand and the metal ion. Synthesis of this artificial nucleoside was started from the construction of the carboxyl substituted 8-hydroxyquinoline, followed by coupling the ligand with 3-aminopropane-1,2-diol via the formation of amide as described in Scheme S1.† The ligand modified DNA was prepared through solid phase synthesis by the conventional phosphoramidite method as described in the ESI.† The incorporated ligand was located in either the 3′ or 5′ terminal of the DNA strand. All the synthesized DNA strands were purified by HPLC and characterized by mass spec.
 | ||
Fig. 2 DNA strand invasion by coordination between the copper ion and the 8-hydroxyquinoline ligand. (a) Scheme for the formation of the metal-mediated base pair between the 8-hydroxyquinoline ligand and the divalent metal ions. The star indicates the chiral center. The ligand investigated in this figure was the R-configuration. (b) Observation of strand displacement by gel electrophoresis. The fluorophore labeled strand was denoted by ![[S with combining macron]](https://www.rsc.org/images/entities/char_0053_0304.gif) F; SL indicated the 8-HQ modified strand that preformed a duplex structure (duplex 1) with the F strand; the competing strand (the input strand) during strand invasion was denoted by L. Concentrations for these strands were 0.5 μM (F), 0.75 μM (SL) and 1 μM (L). The reaction buffer was 20 mM Tris–HCl (pH = 7.5) with 100 mM NaCl. Effective concentrations of divalent metal ions were 1 μM; for the absence of metal ions, 1 mM EDTA was added into the reaction buffer. Native PAGE was performed after 1 hour incubation. Only the FAM fluorophore labeled strand was visualized in the gel shift. The upper bands indicated the original duplex DNA without strand invasion (duplex 1). The lower bands indicated the output single stranded DNA that was displaced from the duplex 1. (c) Quantitative comparison of the percentages of strand invasion. The control sample indicated the direct invasion without ligand modification. Error bars were derived from at least three independent replicates. (d) Measurements of melting temperatures (Tm) of ligand modified duplex DNA (SL-Q: L-F) in the presence of different metal ions. Tm was calculated from the FRET-based assay. The concentration of the ligand-modified duplex was 0.5 μM. Effective concentrations of divalent metal ions were 1 μM; for the absence of metal ions, 1 mM EDTA was added into the reaction buffer before the measurements. F; SL indicated the 8-HQ modified strand that preformed a duplex structure (duplex 1) with the F strand; the competing strand (the input strand) during strand invasion was denoted by L. Concentrations for these strands were 0.5 μM (F), 0.75 μM (SL) and 1 μM (L). The reaction buffer was 20 mM Tris–HCl (pH = 7.5) with 100 mM NaCl. Effective concentrations of divalent metal ions were 1 μM; for the absence of metal ions, 1 mM EDTA was added into the reaction buffer. Native PAGE was performed after 1 hour incubation. Only the FAM fluorophore labeled strand was visualized in the gel shift. The upper bands indicated the original duplex DNA without strand invasion (duplex 1). The lower bands indicated the output single stranded DNA that was displaced from the duplex 1. (c) Quantitative comparison of the percentages of strand invasion. The control sample indicated the direct invasion without ligand modification. Error bars were derived from at least three independent replicates. (d) Measurements of melting temperatures (Tm) of ligand modified duplex DNA (SL-Q: L-F) in the presence of different metal ions. Tm was calculated from the FRET-based assay. The concentration of the ligand-modified duplex was 0.5 μM. Effective concentrations of divalent metal ions were 1 μM; for the absence of metal ions, 1 mM EDTA was added into the reaction buffer before the measurements. | ||
To investigate whether the metal–ligand complexation can initiate the strand-displacement reaction, we first annealed the ligand-modified strand with an unmodified strand to form a stable duplex DNA with a length of 21 nt. The complementary ligand-modified strand was then incubated with the duplex DNA as the competitor (Fig. 2b). The unmodified DNA strand was labeled by a FAM fluorophore, and when the displacement occurred, this FAM labeled sequence was displaced to become a single stranded DNA, which would be observed in the gel electrophoresis at different migration rates. Given that 8-HQ is a good divalent metal chelator but does not bind to alkali metals, we utilized 100 mM NaCl as the necessary salt in all the displacement reaction buffers. To make sure the strand displacement can proceed extensively, we incubated the reaction mixture at room temperature for 1 hour before gel electrophoresis. As shown in Fig. 2b, with the addition of selected metal ions, the strand-displacement reactions occurred as observed from the appearance of the single-strand DNA band. Interestingly, different divalent metals presented dramatically distinct behaviors during strand-displacement reactions. The Cu2+ ion possessed the strongest displacement ability, whereas other metals promoted much weaker displacement reactions. Quantitative analysis indicated that the 8-HQ was highly selective to the copper ion rather than other divalent metal ions (Fig. 2c). Notably, although metal ions, particularly Cu2+, could greatly boost the strand invasion, in the absence of any divalent metal ions, the ligand-modified competitor could also slightly induce the strand-displacement reaction compared with the direct invasion with unmodified DNA strands (left two columns in Fig. 2c). This might be caused by the formation of a weak HQ–HQ base pair as an initiating toehold with potential interactions of hydrogen bonding and base stacking.52 In fact, the duplex thermostability investigations suggested that this HQ–HQ base pair was fully comparable to an A:T pair at the end of a DNA duplex (Fig. S1†). To exclude that the Cu2+ ion itself might benefit the strand displacement, we performed the direct strand displacement of unmodified DNA in the presence and absence of Cu2+ as comparison. The results suggested that addition of Cu2+ could not obviously enhance the performance as observed in Fig. S2.† Measurements of melting temperatures in the presence and absence of these divalent metal ions indicated that the copper ion can form the strongest metallo-base pair (ΔTm = 16 °C with a 21-bp duplex) (Fig. 2d and Table S1†). Other metal ions, however, exhibited much weaker impacts on the thermostabilities of this ligand-modified base pair, which were highly consistent with the gel analysis on the strand-displacement reactions. As a control, the thermostabilities of the unmodified duplex with the same sequence could be hardly affected by these divalent metal ions (Fig. S3†). Clearly, the formation of the potent metal–ligand coordination as the base pair would drive the efficient displacement of strand invasion. In this system, the Cu2+ ion mediated complexation with 8-HQ was highly selective for triggering the strand-displacement reactions.
Since the Cu2+ mediated 8-HQ base pair was the most potent one to facilitate the strand displacement, we then performed kinetic analysis to check how efficiently the strand displacement could proceed. Both fluorescence and gel-based methods were utilized to measure the kinetic behaviors (Fig. 3a and b). Gel-based analysis could directly analyze the appearance of the displaced strand to show the invasion process (Fig. 3a), whereas the fluorescence change could monitor the real-time process (Fig. 3b). Both methods showed similar kinetic behaviors for 1 μM Cu2+ triggered strand-displacement reactions (∼66 s calculated from the gel-based measurements and ∼39 s calculated from the fluorescence change to reach the 50% conversion of strand displacement). This was a highly efficient process, which was in sharp contrast to the observation in the absence of the copper ion (Fig. 3b, black curve), suggesting a highly controllable strand-displacement behavior mediated by the metal–ligand coordination. Besides, given that a chiral center exists in the three-carbon backbone, the ligand with the other configuration was also generated and tested in the above experiments. Due to the flexibility in the terminal of the DNA strand, the modified DNA with the other chiral configuration presented similar behaviors in terms of both thermodynamic stability of metal-mediated base pairs (Fig. S4–S6†) and kinetic efficiency of strand-displacement reactions (Fig. S7†). Considering the chiral center has limited effect in this terminal metallo-base pair, we only investigated one of the configurations in the following experiments.
 | ||
| Fig. 3 Kinetic behaviors of metal–ligand complexation-mediated strand-displacement reactions. (a) Measurements of kinetic behaviors for the strand invasion by gel electrophoresis. The gel-based results were determined by the generation of the displaced single stranded DNA (the output strand F) as described in Fig. 2b. (b) Measurements of kinetic behaviors by the increasing fluorescence of the FAM fluorophore. Before invasion, the FAM-labeled strand (F) was quenched by the BHQ-labeled strand (SL-Q). After the invasion of the competing strand (L), the fluorophore was released with increasing signals. The concentration of the copper ion was 1 μM. The same strands without any divalent metal ions (with the presence of 1 mM EDTA) (black curve) and the unmodified DNA strands with the copper ion (pink curve) was also monitored as the control. Concentrations for these strands in (a) and (b) were 0.5 μM (F), 0.75 μM (SL or SL-Q) and 1 μM (L). The reaction buffer was 20 mM Tris–HCl (pH = 7.5) with 100 mM NaCl. (c) Concentration-dependent alterations of the HQ–Cu2+–HQ complexation triggered strand-displacement reactions. The concentrations of copper ions were 1, 3, 10, 30 and 100 μM, respectively. (d) pH-dependent alterations of HQ–Cu2+–HQ complexation triggered strand-displacement reactions. The pH values of the reaction buffer (20 mM Tris–HCl) were 5, 6, 7, 7.5 and 8, respectively. The values of t0.5 represented the time needed for 50% conversion of strand displacement, which were calculated based on the results from gel-based analysis. | ||
Given that the formation of the metal-mediated base pair is the key driving force to initiate the strand-displacement process, any factors that can impact the metal–ligand complexation would potentially regulate the dynamics. First, the concentration of the metal ion could impact the coordination mode between the metal and the ligand. We observed that high concentrations of copper ion could reduce the efficiency of the strand-displacement reactions (Fig. S8†) as increasing the concentration of the metal ion would move the metal–ligand binding equilibrium towards the mono-chelating complex instead of the metal-mediated base pairs. Quantitative kinetic analysis from the gel shift experiments indicated that with the concentrations of the copper ion varying between 1 μM and 100 μM, the time needed for 50% conversion could be significantly changed with a more than two-order of magnitude from ∼66 s to more than 7200 s (Fig. 3c and S9†). When the copper ion concentration was reduced to a sub-stoichiometric ratio level, the plateau signal obviously descended (Fig. S10†). This was mainly attributed to the limited metal ions for the complex formation. These data showed a regulatory approach to control displacement kinetics through different concentrations of the metal ion. Besides, deprotonation of the hydroxyl group in the 8-HQ ligand is a necessary step during complexation with metal ions, and the basic pH would promote the formation of coordinate bonds. Indeed, pH variations significantly changed the displacement kinetics as described in Fig. 3d and S11† from the gel-based analysis (the time to reach the 50% conversion could be greatly altered from ∼51 s to ∼1200 s between pH 8 and 5). These variations provided another effective pathway to regulate the dynamic strand invasion by altering the ligand properties. Collectively, these results suggested that the metal–ligand complexation triggered strand-displacement reactions can be easily manipulated by external conditions to control the dynamics of strand invasion.
We then next asked whether this metal-mediated base pair can function similarly to the conventional toehold. First, we compared the kinetic behaviors of this metal–ligand complexation triggered strand displacement with the conventional toehold-mediated reaction (a 7 nt toehold DNA that can achieve the maximum kinetic performance). Intriguingly, the kinetic process initiated by this single metal-mediated base pair stayed at a close level compared to the natural toehold DNA (Fig. 4a, ∼30 s for the metal-mediated displacement vs. ∼12 s for the conventional toehold to reach the 50% conversion at pH 8 monitored by the fluorescence). Second, the formation of the metal-mediated base pair was highly specific to the ligand modified DNA (Fig. 4b), as the competing experiment suggested that the same DNA sequence without the conjugated ligand hardly influenced the displacement kinetics even in a highly excessive amount (10-fold). Third, similarly to the conventional toehold, once the invading strand was constrained into a duplex structure, the strand displacement was greatly inhibited even in the presence of metal–ligand complexation (Fig. 4c). Fourth, this metal–ligand complexation mediated strand displacement was also highly sensitive to the mismatched nucleotide in the invaded duplex structure through an asymmetric way as previously observed in the conventional toehold mediated displacement (Fig. 4d).56 Taken together, we revealed that the strand invasion by the metal–ligand complexation also possessed the efficiency, specificity, tunability and sensitivity similarly to the Watson–Crick base-paired toehold DNA, which could potentially achieve all kinds of dynamic strand-displacement processes like a conventional toehold.
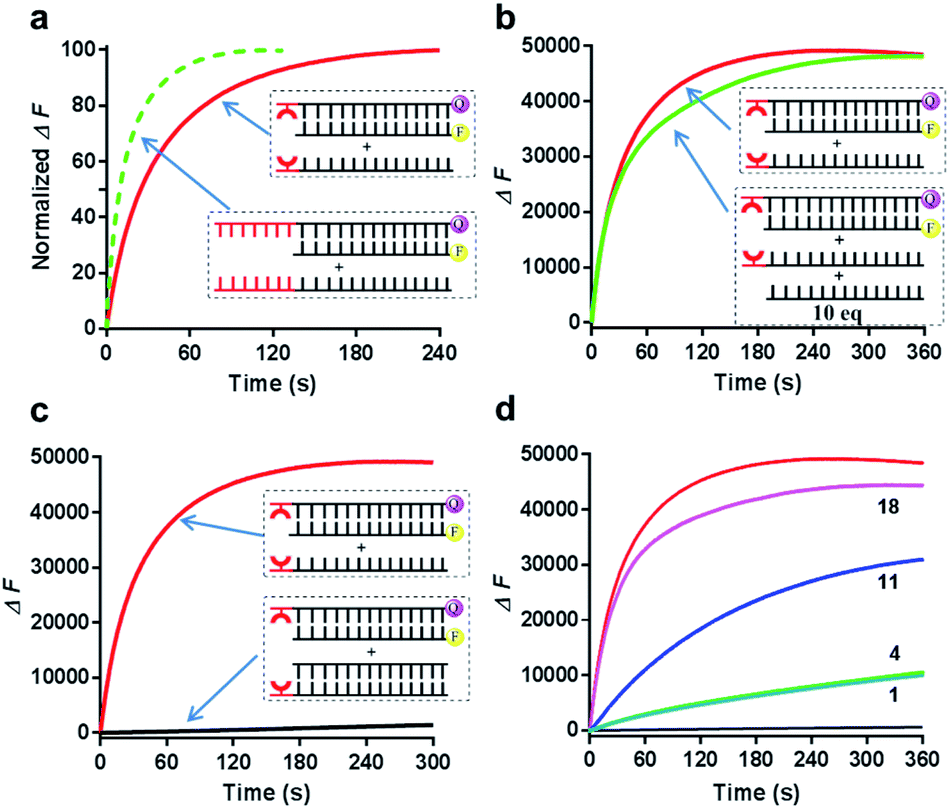 | ||
| Fig. 4 Comparison between the conventional toehold-mediated and the metal–ligand complexation-mediated strand-displacement reactions. (a) Direct comparison of the kinetic behaviors for the strand invasion. The t0.5 value for the conventional toehold was ∼12 s and it was ∼30 s for the metal–ligand complexation. (b) Kinetic comparison in the presence of a high concentration (10×) of competing strand without ligand modification (the unmodified strand 1). (c) Kinetic comparison when the ligand-modified competing strand (L) was masked by the complementary sequence (S1). (d) Kinetic comparison when the ligand-modified competing strand (L) was not fully matched with the strand SL. The numbers indicated the mismatched position in the sequence counted from the ligand modified site. All these results were measured based on the fluorescence analysis. The reaction buffer was 20 mM Tris–HCl (pH = 8.0) with 100 mM NaCl in the presence or absence of 1 μM copper ion. Concentrations for all these strands related to this figure were 0.5 μM (F), 0.75 μM (SL-Q), 1 μM (L), 10 μM (1) and 1.2 μM (S1). | ||
Notably, compared with the Watson–Crick base-paired toehold, the metal–ligand paired toehold can present a unique feature of controllability during the dynamic strand displacement. This controllability originated from the controllable metal–ligand complexation. Herein, through introducing this controllability with the combination of the Watson–Crick base-paired toehold into the dynamic strand-displacement reactions, we examined a controllable and reversible DNA dynamic process based on toehold exchange as a proof of principle. As described in Fig. 5a, once the input DNA strand was added, the strand displacement was uncontrollable with the natural base-paired toehold, and eventually could achieve the equilibrium during the seesaw system. In the metal–ligand complexation mediated strand displacement, the absence of Cu2+ would not initiate the strand exchange; addition of Cu2+ would trigger the strand invasion towards one direction; however, if EDTA was added into the system, the displaced strand would bind back to reset the toehold exchange system (Fig. 5a).
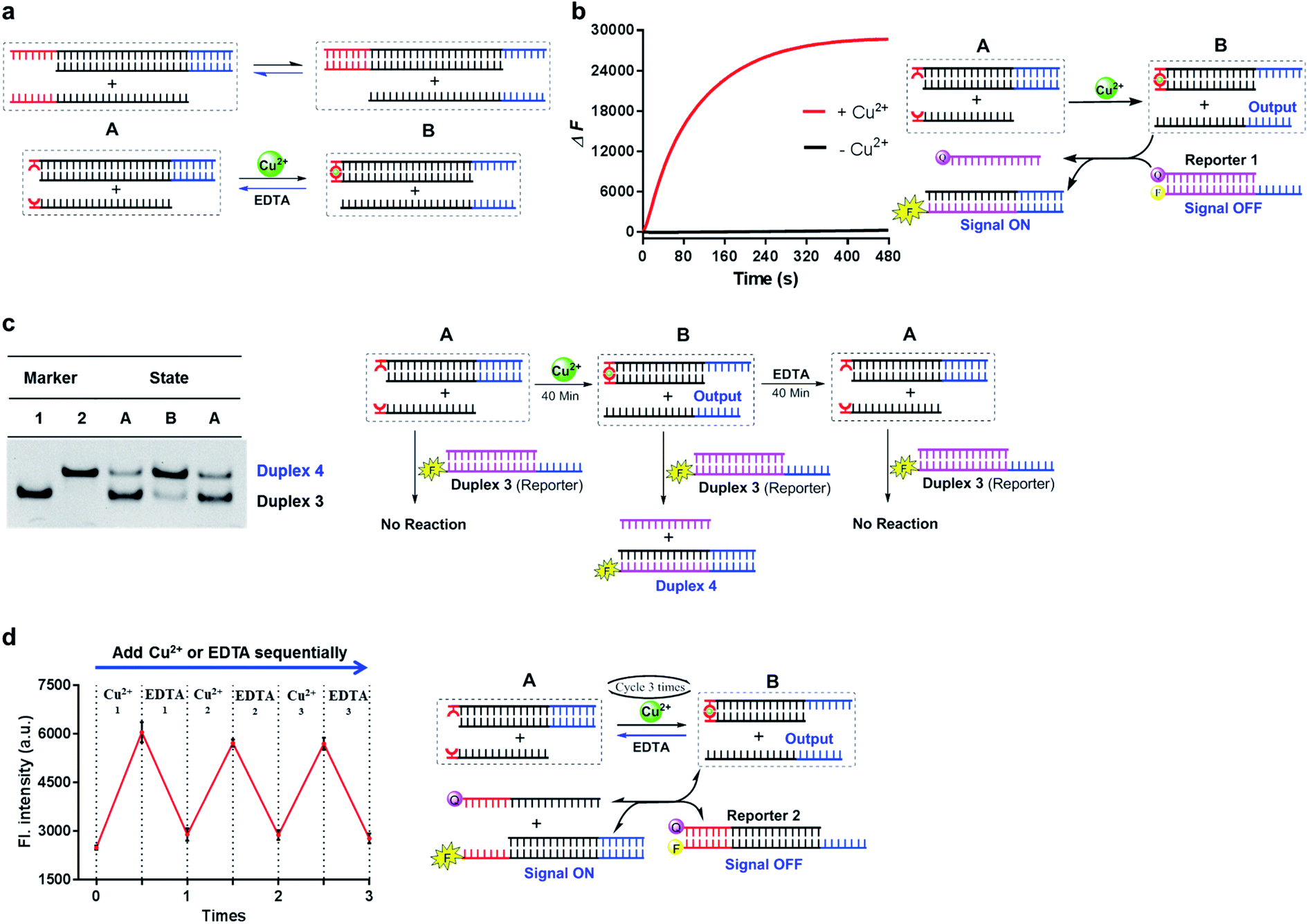 | ||
Fig. 5 Combination of the metal–ligand complexation-mediated and the conventional toehold-mediated strand displacement to achieve controllable and reversible toehold exchange. (a) A comparison between the conventional toehold exchange (upper panel) and the model with both the metal–ligand complexation and the natural base pairs as toeholds (lower panel). In the typical toehold exchange, States A and B are uncontrollable once the input strand has been added. However, for the combinatory model, the coordination between the HQ ligand and the copper would shift the strand displacement towards State B, resulting in a release of a natural toehold in the presence of the copper ion; when the copper ion was removed by a chelator, such as EDTA, the natural toehold would dominate the displacement and push the displacement reaction backward, which would release the ligand-modified strand (State A). (b) Monitoring the toehold exchange using the unidirectional reporter system from the fluorescence measurements. In the absence of copper ion, State A was the stable form (the duplex formed by SLT and T1, and the single strand L) without the release of the output strand (T1), and as a result, the unidirectional reporter system (R1Q and  ) could not report any strand-displacement reaction (black curve). After the addition of the copper ion, the State B could be generated with the release of the output strand, and therefore, the reporting signal could be monitored from fluorescence. (c) Sequential detection of the output strand by the unidirectional reporter system from the gel-based assays. In the State A, the reporter duplex 3 (R1 and ) could not report any strand-displacement reaction (black curve). After the addition of the copper ion, the State B could be generated with the release of the output strand, and therefore, the reporting signal could be monitored from fluorescence. (c) Sequential detection of the output strand by the unidirectional reporter system from the gel-based assays. In the State A, the reporter duplex 3 (R1 and  ) was unaffected, whereas the reporter duplex 4 (T1 and ) was unaffected, whereas the reporter duplex 4 (T1 and 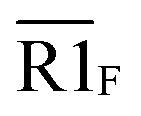 ) was only generated in the presence of State B with the release of the output strand. (d) Reversible control of toehold exchange by the bidirectional reporter system. In the bidirectional reporter system (R2Q and ) was only generated in the presence of State B with the release of the output strand. (d) Reversible control of toehold exchange by the bidirectional reporter system. In the bidirectional reporter system (R2Q and 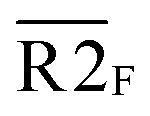 ), all the conventional toehold-mediated strand displacement was reversible without any control. The only controllable reversibility was governed by the metal–ligand complexation between States A and B. In the first cycle, the addition of copper ion was 1 μM and the addition of EDTA was 25 μM; in the second cycle, the addition of copper ion was 25 μM and the addition of EDTA was 50 μM; in the third cycle, the addition of copper ion was 50 μM and the addition of EDTA was 50 μM. After each treatment, the reaction mixture was incubated for 40-minute to reach the equilibrium before the measurements of fluorescence. Concentrations for all these strands were described in the ESI.† The reaction buffer was 20 mM Tris–HCl (pH = 8.0) with 100 mM NaCl. Error bars were derived from at least three independent replicates. ), all the conventional toehold-mediated strand displacement was reversible without any control. The only controllable reversibility was governed by the metal–ligand complexation between States A and B. In the first cycle, the addition of copper ion was 1 μM and the addition of EDTA was 25 μM; in the second cycle, the addition of copper ion was 25 μM and the addition of EDTA was 50 μM; in the third cycle, the addition of copper ion was 50 μM and the addition of EDTA was 50 μM. After each treatment, the reaction mixture was incubated for 40-minute to reach the equilibrium before the measurements of fluorescence. Concentrations for all these strands were described in the ESI.† The reaction buffer was 20 mM Tris–HCl (pH = 8.0) with 100 mM NaCl. Error bars were derived from at least three independent replicates. | ||
To monitor this process, we introduced two types of reporter systems. One was a unidirectional reporter, in which the equilibrium would be shifted towards the forward direction (described in Fig. 5b and c); the other one was a bidirectional reporter, in which the reporting signals were altered based on the shift of the seesaw system (described in Fig. 5d). When mixed with the unidirectional reporter system, the strand-displacement process was directly controlled by the metal–ligand complexation as real-time observed in Fig. 5b. To check the individual step during the toehold exchange, we prepared three sequential samples, and examined the strand information using the unidirectional reporter through gel shift as shown in Fig. 5c. In the absence of metal ions, the toehold exchange could not be initiated (State A); the following incubation with the reporter duplex could not induce any structural change, and therefore, the reporter duplex stayed unchanged. 40-Min after addition of Cu2+, the output strand was almost fully displaced by the metal–ligand complexation (State B), which was then reported by the fluorescence labeled long duplex. Once EDTA was mixed together after incubation of Cu2+, the previously displaced output strand then bound back through the Watson-Crick base-paired toehold (back to State A); therefore, the reporter duplex was unable to detect the significant release of the output strand. In addition to the gel-based analysis, we also observed the change of the reporter duplex from the fluorescence measurements (Fig. S12†). This multistep monitoring process explicitly indicated the controllable and reversible strand displacement by the metal-mediated base pair. Furthermore, with utilization of the bidirectional reporter system, this reversible operation could be directly monitored by fluorescence measurements as shown in Fig. 5d. Addition of Cu2+ initiated the toehold exchange, and subsequently the output strand was reported as the equilibrium moved to the forward direction (fluorescence increase); with EDTA treatment in this same system, loss of metal–ligand complexation shifted the equilibrium backward (fluorescence decrease). Multiple cycles were achieved to control the directions of strand dynamics (Fig. 5d and S13†). Notably, compared with the first cycle, the real-time fluorescence monitoring revealed that the reaction rate in the following cycle was significantly slower, and it took a longer time to reach equilibrium towards either the forward or backward direction (Fig. S14†). The reduced kinetic efficiency might be attributed to the competitive chelation between 8-HQ and EDTA in this cycling system. As a negative control, the conventional toehold exchange system could not induce any significant fluorescence response through an identical procedure (Fig. S15†). With the combination of metal–ligand complexation and toehold-mediated strand exchange, these results together confirmed the controllability and reversibility of the strand-displacement dynamics in the metal-mediated seesaw system.
Conclusions
This study demonstrated a highly efficient and regulatory DNA strand-displacement process through metal–ligand complexation as a controllable toehold. This toehold consisted of an unnatural chelating ligand, 8-hydroxyquinoline, as the artificial nucleoside and functioned based on the selective HQ-Cu2+-HQ complexation. Given the potent thermostability of the metal–ligand coordination, a single metal-mediated artificial base pair could initiate the highly dynamic DNA strand invasion, similarly to the natural base paired toehold. Notably, the DNA strand-displacement reactions could be diversely controlled by a variety of regulators related to the metal–ligand complexation, such as metal species, metal concentrations, and pH conditions, which would provide different effectors to manipulate the strand dynamics. Moreover, with the introduction of the metal–ligand complexation into the toehold exchange system, controllable and reversible strand release could also be achieved. Since this design was only based on the intrinsic properties of metal–ligand coordination without involvement of the nucleic acid context, it presented a highly expandable opportunity to manipulate all kinds of DNA nanomachines.Previous investigations on metal-mediated base pairs generally focused on how the coordinate bonds could be integrated into DNA structures to form new metallo-nanostructures or to achieve conformational alterations. Unlike any reported function and application of metal-mediated base pairs, we revealed that the coordination between the artificial ligand and the metal ion can independently initiate a highly efficient DNA strand-displacement process without the consideration of nucleic acid sequences and structures. In this work, this metal-mediated base pair was treated as an artificial toehold that could regulate the dynamic DNA nanomachine. The complexation between the metal and ligand would be the driving force to initiate strand dynamics. Herein, we revealed that the highly selective coordination between Cu2+ and 8-HQ was an efficient toehold to control the DNA strand-displacement reactions. Given that so many types of metal–ligand interactions have been reported as artificial base pairs, such as Ni2+,57 Cu2+,11,57–61 Cu+,10 Zn2+,62 Hg2+,14 Ag+,8,63–65 and Pd2+ based66 coordination, as well as some natural bases mediated metallo-base pairs,67–69 one would expect that it could be feasible to introduce these ligands as toeholds to control the strand-displacement dynamics by different metal ions. More importantly, different ligands generally exhibited a high selectivity towards some specific metal ions; hence, multiplex control and logical manipulation of DNA strand-displacement reactions through different metal–ligand base pairs as specific toeholds could be also foreseeable. Since DNA strand displacement has been widely utilized as a signal transduction and amplification strategy for biochemical analysis,70 our design of versatile controllability of strand dynamics mediated by metal–ligand complexation may have established a general basis for potential applications in sensing metal ions.
In the dynamic DNA technology, controllable toeholds are essential to manipulate the processes and outcomes of DNA strand-displacement reactions. Great efforts have been devoted to design and construct different controllable toeholds in the past few years, including alteration of DNA secondary structures,49,50,71,72 ligand-induced aptamer interactions73,74 and photo-released single-strand toeholds.75 The general idea of these conditional toeholds is that they functioned based on the conformational switch of nucleic acid structures. However, our ligand-modified DNA, separating the metal–ligand complexation out of the nucleic acid structures, can control the strand dynamics solely based on the properties of the ligand itself. In this concept, toehold-mediated strand-displacement reactions employ the metal–ligand complexation as the trigger to achieve controllable dynamics that is only governed by its intrinsic coordination properties. Therefore, the controllability of strand displacement can be more easily manipulated with a great dynamic switch. In addition to the controllable behaviors reported in this work, other conditions that could regulate the metal–ligand interaction, such as redox signals10 and photo-irradiation,76 might be also applicable to mediate the strand-displacement reactions. Metal–ligand complexation can provide alternative approaches for the development of versatile dynamic DNA nanotechnologies related to dynamic nanostructures, nucleic acid computation and synthetic biology, and this work opens a new door towards these possibilities.
Author contributions
L. Xu designed the project. L.-L. Wang performed the experiments and analyzed the data with assistance from Q.-L. Zhang, Y. Wang, Y. Liu, J. Lin, and F. Xie. L.-L. Wang and L. Xu wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the National Natural Science Foundation of China (No. 21708054 and No. 21977122), the Innovative Research Team in University of Ministry of Education of China (IRT_17R111) and the National Key R&D Program of China (2020YFA0211200).Notes and references
- H. Yang, K. L. Metera and H. F. Sleiman, Coord. Chem. Rev., 2010, 254, 2403–2415 CrossRef CAS.
- S. Mandal and J. Muller, Curr. Opin. Chem. Biol., 2017, 37, 71–79 CrossRef CAS PubMed.
- S. Naskar, R. Guha and J. Muller, Angew. Chem., Int. Ed., 2020, 59, 1397–1406 CrossRef CAS PubMed.
- Y. Takezawa and M. Shionoya, Acc. Chem. Res., 2012, 45, 2066–2076 CrossRef CAS PubMed.
- G. H. Clever, C. Kaul and T. Carell, Angew. Chem., Int. Ed., 2007, 46, 6226–6236 CrossRef CAS PubMed.
- J. Muller, Coord. Chem. Rev., 2019, 393, 37–47 CrossRef.
- L. L. Zhang and E. Meggers, J. Am. Chem. Soc., 2005, 127, 74–75 CrossRef CAS PubMed.
- I. Sinha, C. Fonseca Guerra and J. Muller, Angew. Chem., Int. Ed., 2015, 54, 3603–3606 CrossRef CAS PubMed.
- N. Santamaria-Diaz, J. M. Mendez-Arriaga, J. M. Salas and M. A. Galindo, Angew. Chem., Int. Ed., 2016, 55, 6170–6174 CrossRef CAS PubMed.
- B. Jash and J. Muller, Angew. Chem., Int. Ed., 2018, 57, 9524–9527 CrossRef CAS PubMed.
- T. Nakama, Y. Takezawa, D. Sasaki and M. Shionoya, J. Am. Chem. Soc., 2020, 142, 10153–10162 CrossRef CAS PubMed.
- Y. Takezawa, L. Y. Hu, T. Nakama and M. Shionoya, Angew. Chem., Int. Ed., 2020, 59, 21488–21492 CrossRef CAS PubMed.
- H. Yang, C. K. McLaughlin, F. A. Aldaye, G. D. Hamblin, A. Z. Rys, I. Rouiller and H. F. Sleiman, Nat. Chem., 2009, 1, 390–396 CrossRef CAS PubMed.
- S. Mandal, M. Hebenbrock and J. Muller, Angew. Chem., Int. Ed., 2016, 55, 15520–15523 CrossRef CAS PubMed.
- J. Kondo, Y. Tada, T. Dairaku, Y. Hattori, H. Saneyoshi, A. Ono and Y. Tanaka, Nat. Chem., 2017, 9, 956–960 CrossRef CAS PubMed.
- A. Ono, H. Kanazawa, H. Ito, M. Goto, K. Nakamura, H. Saneyoshi and J. Kondo, Angew. Chem., Int. Ed., 2019, 58, 16835–16838 CrossRef CAS PubMed.
- S. Johannsen, N. Megger, D. Bohme, R. K. O. Sigel and J. Muller, Nat. Chem., 2010, 2, 229–234 CrossRef CAS PubMed.
- Y. Takezawa, S. Yoneda, J. L. H. A. Duprey, T. Nakama and M. Shionoya, Chem. Sci., 2016, 7, 3006–3010 RSC.
- O. P. Schmidt, S. Jurt, S. Johannsen, A. Karimi, R. K. O. Sigel and N. W. Luedtke, Nat. Commun., 2019, 10, 4818 CrossRef PubMed.
- C. Kaul, M. Muller, M. Wagner, S. Schneider and T. Carell, Nat. Chem., 2011, 3, 794–800 CrossRef CAS PubMed.
- D. Y. Zhang and G. Seelig, Nat. Chem., 2011, 3, 103–113 CrossRef CAS PubMed.
- F. C. Simmel, B. Yurke and H. R. Singh, Chem. Rev., 2019, 119, 6326–6369 CAS.
- D. Soloveichik, G. Seelig and E. Winfree, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 5393–5398 CrossRef CAS PubMed.
- G. Seelig, D. Soloveichik, D. Y. Zhang and E. Winfree, Science, 2006, 314, 1585–1588 CrossRef CAS PubMed.
- D. Y. Zhang, A. J. Turberfield, B. Yurke and E. Winfree, Science, 2007, 318, 1121–1125 CrossRef CAS PubMed.
- L. Qian and E. Winfree, Science, 2011, 332, 1196–1201 CrossRef CAS PubMed.
- L. Qian, E. Winfree and J. Bruck, Nature, 2011, 475, 368–372 CrossRef CAS PubMed.
- N. Srinivas, J. Parkin, G. Seelig, E. Winfree and D. Soloveichik, Science, 2017, 358, eaal2052 CrossRef PubMed.
- D. Y. Zhang and E. Winfree, J. Am. Chem. Soc., 2009, 131, 17303–17314 CrossRef CAS PubMed.
- B. L. Li, A. D. Ellington and X. Chen, Nucleic Acids Res., 2011, 39, e110 CrossRef CAS PubMed.
- A. J. Genot, D. Y. Zhang, J. Bath and A. J. Turberfield, J. Am. Chem. Soc., 2011, 133, 2177–2182 CrossRef CAS PubMed.
- A. J. Genot, J. Bath and A. J. Turberfield, Angew. Chem., Int. Ed., 2013, 52, 1189–1192 CrossRef CAS PubMed.
- X. L. Yang, Y. N. Tang, S. M. Traynor and F. Li, J. Am. Chem. Soc., 2016, 138, 14076–14082 CrossRef CAS PubMed.
- C. Jung and A. D. Ellington, Acc. Chem. Res., 2014, 47, 1825–1835 CrossRef CAS PubMed.
- Y. F. Dai, A. Furst and C. C. Liu, Trends Biotechnol., 2019, 37, 1367–1382 CrossRef CAS PubMed.
- Y. X. Zhao, X. L. Zuo, Q. Li, F. Chen, Y. R. Chen, J. Q. Deng, D. Han, C. L. Hao, F. J. Huang, Y. Y. Huang, G. L. Ke, H. Kuang, F. Li, J. Li, M. Li, N. Li, Z. Y. Lin, D. B. Liu, J. W. Liu, L. B. Liu, X. G. Liu, C. H. Lu, F. Luo, X. H. Mao, J. S. Sun, B. Tang, F. Wang, J. B. Wang, L. H. Wang, S. Wang, L. L. Wu, Z. S. Wu, F. Xia, C. L. Xu, Y. Yang, B. F. Yuan, Q. Yuan, C. Zhang, Z. Zhu, C. Y. Yang, X. B. Zhang, H. H. Yang, W. H. Tan and C. H. Fan, Sci. China Chem., 2021, 64, 171–203 CrossRef CAS PubMed.
- Y. Krishnan and F. C. Simmel, Angew. Chem., Int. Ed., 2011, 50, 3124–3156 CrossRef CAS PubMed.
- Y. J. Chen, B. Groves, R. A. Muscat and G. Seelig, Nat. Nanotechnol., 2015, 10, 748–760 CrossRef CAS PubMed.
- J. Kim, D. Jang, H. Park, S. Jung, D. H. Kim and W. J. Kim, Adv. Mater., 2018, 30, e1707351 CrossRef PubMed.
- F. Wang, X. Q. Liu and I. Willner, Angew. Chem., Int. Ed., 2015, 54, 1098–1129 CrossRef CAS PubMed.
- K. Rinaudo, L. Bleris, R. Maddamsetti, S. Subramanian, R. Weiss and Y. Benenson, Nat. Biotechnol., 2007, 25, 795–801 CrossRef CAS PubMed.
- A. A. Green, P. A. Silver, J. J. Collins and P. Yin, Cell, 2014, 159, 925–939 CrossRef CAS PubMed.
- A. A. Green, J. Kim, D. Ma, P. A. Silver, J. J. Collins and P. Yin, Nature, 2017, 548, 117–121 CrossRef CAS PubMed.
- M. H. Hanewich-Hollatz, Z. Chen, L. M. Hochrein, J. Huang and N. A. Pierce, ACS Cent. Sci., 2019, 5, 1241–1249 CrossRef CAS PubMed.
- J. Kim, Y. Zhou, P. D. Carlson, M. Teichmann, S. Chaudhary, F. C. Simmel, P. A. Silver, J. J. Collins, J. B. Lucks, P. Yin and A. A. Green, Nat. Chem. Biol., 2019, 15, 1173–1182 CrossRef CAS PubMed.
- L. Oesinghaus and F. C. Simmel, Nat. Commun., 2019, 10, 2092 CrossRef PubMed.
- K. H. Siu and W. Chen, Nat. Chem. Biol., 2019, 15, 217–220 CrossRef CAS PubMed.
- J. Lin, Y. Liu, P. Lai, H. Ye and L. Xu, Nucleic Acids Res., 2020, 48, 11773–11784 CrossRef CAS PubMed.
- A. Porchetta, A. Vallee-Belisle, K. W. Plaxco and F. Ricci, J. Am. Chem. Soc., 2013, 135, 13238–13241 CrossRef CAS PubMed.
- W. Ding, W. Deng, H. Zhu and H. J. Liang, Chem. Commun., 2013, 49, 9953–9955 RSC.
- W. Deng, H. Xu, W. Ding and H. Liang, PLoS One, 2014, 9, e111650 CrossRef PubMed.
- T. Ehrenschwender, W. Schmucker, C. Wellner, T. Augenstein, P. Carl, J. Harmer, F. Breher and H. A. Wagenknecht, Chem.–Eur. J., 2013, 19, 12547–12552 CrossRef CAS PubMed.
- M. K. Schlegel, X. Xie, L. Zhang and E. Meggers, Angew. Chem., Int. Ed., 2009, 48, 960–963 CrossRef CAS PubMed.
- P. Scharf, B. Jash, J. A. Kuriappan, M. P. Waller and J. Muller, Chem.–Eur J., 2016, 22, 295–301 CrossRef CAS PubMed.
- M. K. Schlegel, L. L. Zhang, N. Pagano and E. Meggers, Org. Biomol. Chem., 2009, 7, 476–482 RSC.
- P. Irmisch, T. E. Ouldridge and R. Seidel, J. Am. Chem. Soc., 2020, 142, 11451–11463 CrossRef CAS PubMed.
- C. Switzer and D. Shin, Chem. Commun., 2005, 10, 1342–1344 RSC.
- K. Tanaka, A. Tengeiji, T. Kato, N. Toyama, M. Shiro and M. Shionoya, J. Am. Chem. Soc., 2002, 124, 12494–12498 CrossRef CAS PubMed.
- N. Sandmann, J. Bachmann, A. Hepp, N. L. Doltsinis and J. Muller, Dalton Trans., 2019, 48, 10505–10515 RSC.
- Y. Takezawa, T. Nakama and M. Shionoya, J. Am. Chem. Soc., 2019, 141, 19342–19350 CrossRef CAS PubMed.
- M. Su, M. Tomas-Gamasa and T. Carell, Chem. Sci., 2015, 6, 632–638 RSC.
- B. Jash and J. Muller, J. Inorg. Biochem., 2018, 186, 301–306 CrossRef CAS PubMed.
- K. Tanaka, Y. Yamada and M. Shionoya, J. Am. Chem. Soc., 2002, 124, 8802–8803 CrossRef CAS PubMed.
- N. Zimmermann, E. Meggers and P. G. Schultz, J. Am. Chem. Soc., 2002, 124, 13684–13685 CrossRef CAS PubMed.
- D. W. Shin and C. Switzer, Chem. Commun., 2007, 42, 4401–4403 RSC.
- K. Tanaka and M. Shionoya, J. Org. Chem., 1999, 64, 5002–5003 CrossRef CAS.
- Y. Miyake, H. Togashi, M. Tashiro, H. Yamaguchi, S. Oda, M. Kudo, Y. Tanaka, Y. Kondo, R. Sawa, T. Fujimoto, T. Machinami and A. Ono, J. Am. Chem. Soc., 2006, 128, 2172–2173 CrossRef CAS PubMed.
- A. Ono, S. Cao, H. Togashi, M. Tashiro, T. Fujimoto, T. Machinami, S. Oda, Y. Miyake, I. Okamoto and Y. Tanaka, Chem. Commun., 2008, 39, 4825–4827 RSC.
- Y. Takezawa, A. Suzuki, M. Nakaya, K. Nishiyama and M. Shionoya, J. Am. Chem. Soc., 2020, 142, 21640–21644 CrossRef CAS PubMed.
- Y. Dai, A. Furst and C. C. Liu, Trends Biotechnol., 2019, 37, 1367–1382 CrossRef CAS PubMed.
- A. Amodio, B. Zhao, A. Porchetta, A. Idili, M. Castronovo, C. H. Fan and F. Ricci, J. Am. Chem. Soc., 2014, 136, 16469–16472 CrossRef CAS PubMed.
- W. Tang, H. Wang, D. Wang, Y. Zhao, N. Li and F. Liu, J. Am. Chem. Soc., 2013, 135, 13628–13631 CrossRef CAS PubMed.
- J. H. Monserud, K. M. Macri and D. K. Schwartz, Angew. Chem., Int. Ed., 2016, 55, 13710–13713 CrossRef CAS PubMed.
- L. Li, X. G. Chen, C. Cui, X. S. Pan, X. W. Li, H. S. Yazd, Q. Wu, L. P. Qiu, J. Li and W. H. Tan, J. Am. Chem. Soc., 2019, 141, 17174–17179 CrossRef CAS PubMed.
- F. J. Huang, M. X. You, D. Han, X. L. Xiong, H. J. Liang and W. H. Tan, J. Am. Chem. Soc., 2013, 135, 7967–7973 CrossRef CAS PubMed.
- B. Tylkowski, A. Trojanowska, V. Marturano, M. Nowak, L. Marciniak, M. Giamberini, V. Ambrogi and P. Cerruti, Coord. Chem. Rev., 2017, 351, 205–217 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc01041g |
| This journal is © The Royal Society of Chemistry 2021 |