
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRetrosynthetic accessibility score (RAscore) – rapid machine learned synthesizability classification from AI driven retrosynthetic planning†
Amol
Thakkar
 *ab,
Veronika
Chadimová
a,
Esben Jannik
Bjerrum
a,
Ola
Engkvist
a and
Jean-Louis
Reymond
*b
*ab,
Veronika
Chadimová
a,
Esben Jannik
Bjerrum
a,
Ola
Engkvist
a and
Jean-Louis
Reymond
*b
aHit Discovery, Discovery Sciences, R&D, AstraZeneca, Gothenburg, 431 50, Sweden. E-mail: amol.thakkar@dcb.unibe.ch
bDepartment of Chemistry and Biochemistry, University of Bern, Bern, CH-3012, Switzerland. E-mail: jean-louis.reymond@dcb.unibe.ch
First published on 22nd January 2021
Abstract
Computer aided synthesis planning (CASP) is part of a suite of artificial intelligence (AI) based tools that are able to propose synthesis routes to a wide range of compounds. However, at present they are too slow to be used to screen the synthetic feasibility of millions of generated or enumerated compounds before identification of potential bioactivity by virtual screening (VS) workflows. Herein we report a machine learning (ML) based method capable of classifying whether a synthetic route can be identified for a particular compound or not by the CASP tool AiZynthFinder. The resulting ML models return a retrosynthetic accessibility score (RAscore) of any molecule of interest, and computes at least 4500 times faster than retrosynthetic analysis performed by the underlying CASP tool. The RAscore should be useful for pre-screening millions of virtual molecules from enumerated databases or generative models for synthetic accessibility and produce higher quality databases for virtual screening of biological activity.
Introduction
Artificial intelligence (AI) in chemical discovery has been driving improvements in the tools available to the chemical community. This has occurred primarily in the areas of de novo generation of new chemical entities (NCE),1,2 toxicology/bioactivity,3 and computer aided synthesis planning (CASP).4,5 The question as to which molecule to make and how to make it, is at the center of chemical discovery programs across academia and a range of industries, ranging from agrochemical to pharmaceutical.6 Typically virtual screening (VS) workflows have been used to decide which compounds to make, starting from generated, enumerated, commercial, or public datasets which are then filtered using a variety of statistical and physics based modelling techniques until the search space is refined (Fig. 1).7–10 The question and decision of which and how to make a given set of compounds is left to a team of chemists at the end of the VS workflow, prior to synthesis in the laboratory. To aid this filtering process a variety of computational tools which take synthesizability considerations into account have been employed over the last two decades.11–13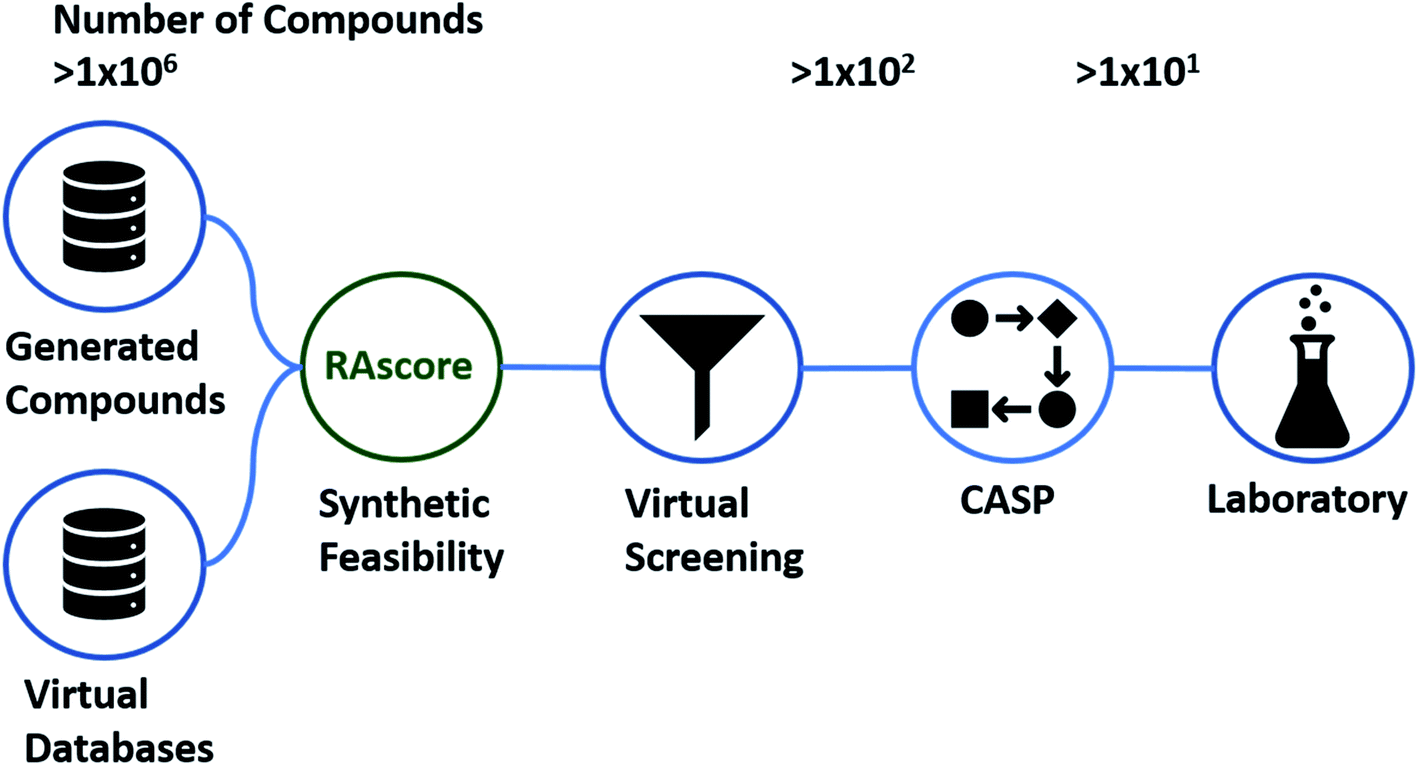 | ||
| Fig. 1 Example of a virtual screening (VS) workflow. The synthesis of compounds is typically considered at the end of the workflow as a final selection criteria, and it is at this point CASP is also used to filter compound libraries to synthesizable compounds. RAscore allows for pre-screening of compounds that may be synthetically accessible by CASP enabling use earlier in the VS workflow (green). | ||
CASP has emerged as a method by which compounds can be filtered in the VS workflow, and during optimization cycles throughout the generative modelling process. Several recent CASP tools have been developed which may be used for these purposes, including but not limited to: Synthia (formerly Chematica),14 ICSYNTH,15 ASKCOS,5 AiZynthFinder,16 and IBM RXN.17 These can be used at two potential stages of the generation process, either to bias the generation process or as a post hoc filter after the molecules have been generated.18 Given a target compound, CASP can predict each step of the synthesis pathway towards commercially available building blocks. This makes it suitable for the in silico filtering of large compound libraries, and has been demonstrated by Gao and Coley for the case of generated compounds.18 However, despite the vast amount of progress that has contributed to making the prediction of full synthetic routes computationally tractable,6,12,19,20 to the extent that some predictions may be made within a minute.5,6 The scale at which predictions must be conducted for large compound libraries consisting of several million or even billions of compounds can still be limiting.
To tackle the challenge of screening large compound libraries with synthesizability considerations, existing scores include the synthetic accessibility score (SAscore), synthetic complexity score (SCscore), and synthetic Bayesian accessibility (SYBA).16,21–23 The SAscore and SYBA are estimations of synthetic feasibility based on the occurrence of molecular fragments in public databases, whereas SCscore is learned from a reaction corpus, with the underlying assumption that products are more complex than their constituent reactants.
Herein, we propose the retrosynthetic accessibility score (RAscore) that enables rapid estimation of synthetic feasibility as determined from the predictions of CASP, in this case AiZynthFinder.16 We investigate a machine learning classifier for retrosynthetic accessibility (RA) assessment called RAscore, trained on the outcomes generated from AiZynthFinder, which we have shown can increase the speed at which synthetic accessibility can be estimated, and separate compounds for which retrosynthetic routes can be found by AiZynthFinder. This is an improvement that adds value to existing synthesis scores, and when used in combination with the previous scores, the RAscore should enable pre-screening of compounds that can be later subjected to full retrosynthetic analysis. Thus, this enables CASP to be used at earlier stages of a VS workflow or during the generative modelling process.
We further emphasize that the RAscore may be retrained on data generated from any CASP tool. Therefore, the score will serve to reflect improvements in the continuously changing synthesis planning technology landscape, thereby overcoming current limitations, and can be customized to the specific needs of a project or user. The models and training protocols have therefore been made available for public use: https://github.com/reymond-group/RAscore.
Methods
AiZynthFinder – a tool for computer aided synthesis planning
AiZynthFinder is a template-based retrosynthetic planning tool based on the methodology of Segler and Waller.4,16 It consists of a neural network policy, which determines which reaction to use at a given retrosynthetic step, with Monte-Carlo tree search, as reported in our previous studies.24 The code, data, and models are open source and available to the public: https://github.com/MolecularAI/AiZynthFinder. The reaction transforms have been extracted from the US patent office extracts (USPTO) and used to train the model by which retrosynthetic expansion was conducted.25 Models on Reaxys and proprietary datasets have been examined in our previous studies, but have been omitted in this study due to their proprietary nature.24 These can equally be used in place of the USPTO policy for those who have access to the data, and the extraction and training protocols can be found in the repository linked above.AiZynthFinder considers retrosynthetic routes to be solved if the precursors or building blocks are commercially available. Therefore, as stopping criteria we use the ACD catalogue,26 Enamine building block set,27 and AstraZeneca internal database. These are available from the respective vendors with the exception of the AstraZeneca internal catalogue. In place of the vendors mentioned here, the AiZynthFinder GitHub repository contains a set of compounds extracted from the ZINC database,28 as highlighted in our previous work.16
The score is inherently limited by the underlying CASP tool, however retraining of the RAscore is possible following the procedures outlined herein. Thus, the score can be customized for individual projects and users, as well as kept up to date with developments in synthesis planning technology. We emphasize that any synthesis planning tool should be able to be used for these purposes.
Retrosynthesis prediction for training set generation
Training and test datasets were generated by randomly sampling 200![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 compounds from ChEMBL,29 as a reference set, and 100000 compounds each from GDBChEMBL and GDBMedChem, to resemble compounds that would usually be out with the applicability domain of CASP.30,31 The compounds were subsequently subjected to retrosynthetic analysis using AiZynthFinder, and labelled as solved or unsolved. The time limit to search for retrosynthetic routes was set as 3 minutes per target compound, with a maximum of seven steps, a maximum of two hundred iterations, and expansion of fifty actions at each stage of the search as determined by the policy network up to a cumulative cutoff threshold of 0.995.
000 compounds from ChEMBL,29 as a reference set, and 100000 compounds each from GDBChEMBL and GDBMedChem, to resemble compounds that would usually be out with the applicability domain of CASP.30,31 The compounds were subsequently subjected to retrosynthetic analysis using AiZynthFinder, and labelled as solved or unsolved. The time limit to search for retrosynthetic routes was set as 3 minutes per target compound, with a maximum of seven steps, a maximum of two hundred iterations, and expansion of fifty actions at each stage of the search as determined by the policy network up to a cumulative cutoff threshold of 0.995.
Machine learning classifiers for estimation of retrosynthetic accessibility
Estimation of retrosynthetic accessibility (RA) was framed as a binary classification problem, as the goal of the study was not to score complexity but rather identify with rapid approximation whether a compound could be synthesized or not by CASP, for which we use AiZynthFinder in this study. We trained a series of classifiers on the retrosynthetic predictions of AiZynthFinder using the label generation method stated previously. The trained classifier predicts whether or not a given compound is synthetically accessible as found by AiZynthFinder.We examined the following classification algorithms: (a) a feed forward neural network classifier, (b) XGBoost classifier, and (c) random forest classifier. For each algorithm 2048 dimensional counted extended connectivity fingerprints were used with a radius set to 3 (ECFP6), and ECFP6 counts with features as generated by RDKit.32,33 In total six different models were trained for each dataset, ChEMBL, GDBChEMBL, and GDBMedChem. SAscore, SCscore, and SYBA are continuous scores for complexity, thus we trained a classifier for each score for comparative purposes, where the score was used as the sole descriptor. For the score-based classifiers we used a feed forward neural network and logistic regression. The scores used as descriptors were calculated using RDKit and the models published by the authors of the corresponding publications.21–23,33
Scikit-Learn was used to train the random forest model,34 XGBoost for the XGB classifier, and Keras with Tensorflow for the feed forward neural networks.35,36 In each case the models where wrapped within an objective function using the Optuna framework for hyperparameter optimisation.37 All models with the exception of the feed neural network were optimized using a five-fold cross validation. The framework used to train the classifiers and models are available at https://github.com/reymond-group/RAscore, and can be used for any binary classification problem.
Each model was optimized with the Optuna hyperparameter optimization framework to find the optimal parameter set.37 In the case of the feed forward neural network, we treated the number of layers, the size of the layers, the activation function, the dropout rate, and the learning rate as hyperparameters, to find the optimal architecture within the bounds of the starting criterion as given in the ESI.†
There was no overlap of compounds between training, validation, and test sets. This was determined by computing the InChI-keys of the compounds in the two sets and using the Python built-in set methods to find the intersection.38 We did not check whether a compound was present in the training data used to train AiZynthFinder, however this is likely not to influence the performance of multi-step retrosynthesis, as the reaction datasets only consider single steps and is supported by our previous studies.24
Average linkage as a method for evaluating machine learning based classifiers
We assessed model performance by computing how well solved and unsolved routes are separated using the concept of average linkage. Average linkage is a statistical method by which the distance between two clusters are treated as the average distance between all pairs of items, where each member of the pair belongs to one of the two clusters. In this instance, the two clusters are solved and unsolved compounds as determined by AiZynthFinder (other CASP tools may be used in place). The average linkage or separation between solved and unsolved compounds was determined by min–max scaling the values of each score such that they were normalized between 1 and 0 using the Scikit-Learn MinMaxScaler. The absolute pairwise distances where computed, and the average of the distances taken to yield a value that corresponds to the separation of the clusters as shown in (Fig. 2).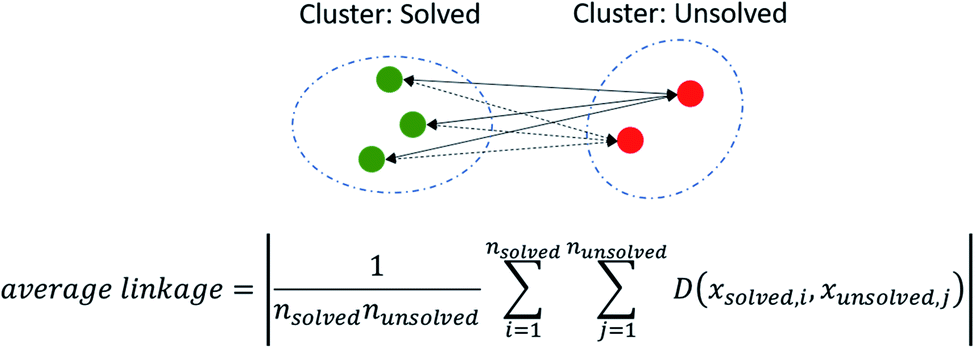 | ||
| Fig. 2 Illustrates the computation of the average linkage. The average linkage is a method by which the distance between two clusters are treated as the average distance between all pairs of items, where one member of the pair belongs to each cluster. | ||
Results and discussion
Route statistics from the generation of labels for machine learning classifiers
Initially training and test datasets were generated by randomly sampling 200000 compounds from ChEMBL,29 as a reference set, and 100000 compounds each from GDBChEMBL and GDBMedChem.30,31 The two are subsets of the GDB17 database.39 ChEMBL was chosen to represent a selection of bioactive molecules and the GDB subsets chosen to be more challenging owing to their differing structural and physiochemical property distribution.30 The compounds were subsequently subjected to retrosynthetic analysis using AiZynthFinder, and labelled as solved or unsolved.
Fig. 3 shows statistics gathered for the predicted retrosynthetic routes during the label generation process. The percentage of solved routes increases monotonically, and the rate at which routes are solved decreases with the number of steps for each dataset. This is most noticeable for compounds requiring synthetic routes between 5 and 7 steps, where we observe a significant increase in the dataset coverage (Fig. 3b), but no corresponding increase in the percentage of solved compounds (Fig. 3a). ChEMBL has the highest percentage of solved compounds, whereas GDBMedChem and GDBChEMBL are consistently lower.
 | ||
| Fig. 3 Statistics gathered for the retrosynthesis predicted during the label generation process for each dataset ChEMBL, GDBChEMBL, and GDBMedChem. The statistics are shown for all compounds sampled: 200000 from ChEMBL, and 100000 from each of the GDB subsets. (a) The percentage of solved compounds as a function of the number of steps, (b) the dataset coverage as a function of the number of steps. (c–f) Histograms depicting the distribution of the compounds in each dataset for each of the currently used scores. For SAscore and SCscore the lower the score the less complex and easier to synthesise a given compounds, whereas for SYBA positive values indicate easy to synthesise compounds and negative values hard to synthesise. (g–j) The percentage of solved compounds as a function of each of the currently used scores as computed for each bin in the histogram. | ||
We observed a correlation between the percentage of solved compounds and the SAscore,23 SCscore,21 and SYBA,22 as well as SMILES length (Fig. 3g–j), which are in agreement with the results obtained by Coley and Gao.18 In the case of SAscore and SCscore, the lower the score the more likely it is that a synthetic route can be obtained for a compound, as found for all datasets. The ChEMBL sample exhibits a lower range of SAscore than the GDBMedChem and GDBChEMBL samples (Fig. 3c), which may explain the higher percentage of solved compounds in ChEMBL as compared to GDBMedChem and GDBChEMBL (Fig. 3a).
However, for SCscore (Fig. 3d) the GDB subsets exhibit a lower range of scores in comparison to the ChEMBL sample. Thus, the inverse of the distribution we obtain for SAscore and can be rationalized by considering the assumptions made in the SCscore model. The SCscore is based on reactions rather than molecular fragments and assumes that the products of a reaction are more complex than the reactants. In most cases the products are also larger than the reactants, thus the assumption for SCscore falters for the GDB subsets because of their restricted size as shown by the difference in SMILES length between the ChEMBL and GDB subsets (Fig. 3f and j). This is further supported by the lower percentage of solved routes for the GDB subsets (Fig. 3f and j).
In the case of SYBA, the higher the score the more likely it is that a route can be found, negative values indicate hard to synthesize compounds. The distribution shown for SMILES length reflects the fact that the GDB subsets are skewed towards smaller molecules, and with lower heavy atom counts than those found in ChEMBL by virtue of the rules used in their enumeration.30,31 The Fig. 3j reveals that the rate at which compounds can be solved falls off much more rapidly with SMILES length for the GDB subsets than for ChEMBL.
Attempts at using SAscore, SCscore, and SYBA
We assessed the existing scores SAscore, SCscore, and SYBA for their ability to distinguish between compounds that could be solved by AiZynthFinder and those that could not (Fig. 4). These scores have often been used to filter or estimate the synthetic accessibility of large datasets of virtual compounds.39–41 However, we have found that there is no threshold value at which the SA, SC, and SYBA scores can be set that clearly separates compounds that can and cannot be solved by AiZynthFinder, as shown by the overlapping histograms. This was observed for all datasets examined in this study (refer to ESI†). Thus, there is potential for them to be misused when filtering large virtual libraries. To resolve this issue, we propose that the existing scores be used alongside the classifiers trained in this study to determine whether a synthetic route can be found, and how difficult it may be to realize the route in the wet lab.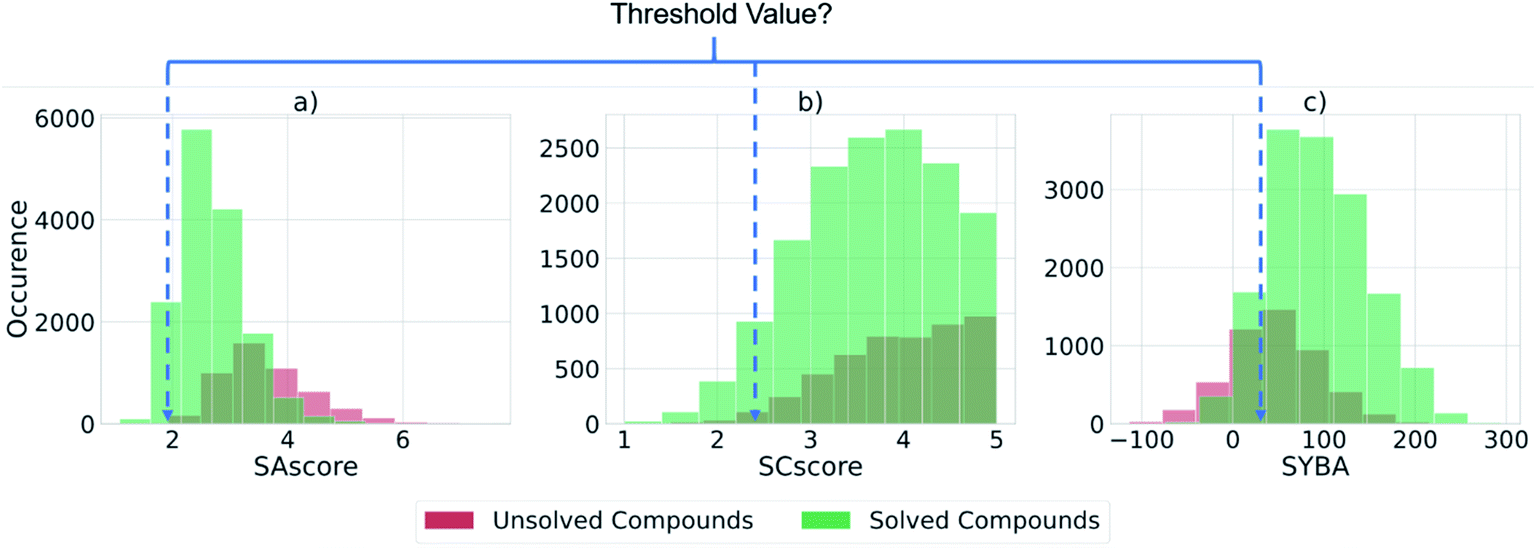 | ||
| Fig. 4 Histograms computed for the test set of ca. 20000 ChEMBL compounds showing whether a retrosynthetic route could be found by AiZynthFinder for a given compound (green) or not (red), and their distributions across each of the scores in current use. There is no threshold value at which the current scores are able to separate compounds that can be solved by AiZynthFinder (green) from those that cannot (red). This highlights how the scores have potential for misuse in generative modelling and filtering sets of compounds. | ||
Machine learning classifiers for estimation of retrosynthetic accessibility
The overlaps shown in Fig. 4, demonstrate the need to be able to differentiate between compounds that can and cannot be synthesized by AiZynthFinder. Therefore, we trained a series of ML based classifiers to determine whether a given compound could be solved by AiZynthFinder. A selection of the results obtained for the trained classifiers are shown in Table 1 (refer to the ESI† for all trained models). In each case the classifiers outperform the existing scores which were used as a baseline (SAscore, SCscore, and SYBA) both in terms of the AUC (area under the curve) and average linkage with respect to their ability to classify compounds as solved or unsolved. When using the existing scores as descriptors to train the classifiers, we observed a marginal improvement in comparison to the score itself. This is because the existing scores are complexity based scores, thus have not been developed with the separation of compounds found synthetically accessible by CASP in mind. A more significant improvement in classifier performance was obtained when using ECFP6 counted vectors as molecular descriptors, both with and without features. The feed forward neural network (NN) based models consistently outperformed random forest and showed comparable performance to gradient boosting methods (XGB).| Dataset | Model | Descriptor | AUC | Accuracy | Precision | Recall | Average linkage |
|---|---|---|---|---|---|---|---|
| a For comparative purposes a baseline has been included which are the SAscore, SCscore, and SYBA. The metrics for these have been computed using Scikit-Learn and the average linkage computed as described in the methods. Classifiers were trained using each of the respective scores as descriptors to enable a direct comparison of classifier performance. These marginally outperform the baseline models in terms of AUC and average linkage. The top 3 classifiers for each dataset using ECFP6 variants consistently outperform the baseline models and their classifiers. For RAscore the top performing classifier on the ChEMBL dataset was chosen, and a separate GDB specific model chosen termed GDBscore which was the top performing classifier on the GDBChEMBL dataset. | |||||||
| ChEMBL | NN (RAscore) | ECFP6 counts with features | 0.93 | 0.90 | 0.92 | 0.95 | 0.69 |
| NN | ECFP6 counts | 0.94 | 0.90 | 0.92 | 0.95 | 0.68 | |
| XGB | ECFP6 counts | 0.95 | 0.91 | 0.92 | 0.96 | 0.65 | |
| NN | SAscore | 0.85 | 0.81 | 0.84 | 0.92 | 0.37 | |
| NN | SCscore | 0.61 | 0.75 | 0.61 | 1.00 | 0.27 | |
| NN | SYBA score | 0.74 | 0.78 | 0.78 | 0.97 | 0.21 | |
| Baseline | SAscore | 0.15 | — | — | — | 0.17 | |
| Baseline | SCscore | 0.39 | — | — | — | 0.22 | |
| Baseline | SYBA | 0.74 | — | — | — | 0.17 | |
| GDBChEMBL | NN (GDBscore) | ECFP6 counts | 0.93 | 0.87 | 0.76 | 0.73 | 0.64 |
| NN | ECFP6 counts with features | 0.94 | 0.88 | 0.78 | 0.74 | 0.63 | |
| XGB | ECFP6 counts | 0.94 | 0.89 | 0.81 | 0.73 | 0.61 | |
| Baseline | SAscore | 0.11 | — | — | — | 0.26 | |
| Baseline | SCscore | 0.38 | — | — | — | 0.14 | |
| Baseline | SYBA | 0.72 | — | — | — | 0.17 | |
| GDBMedChem | NN | ECFP6 counts | 0.93 | 0.88 | 0.75 | 0.64 | 0.64 |
| NN | ECFP6 counts with features | 0.94 | 0.89 | 0.77 | 0.66 | 0.63 | |
| XGB | ECFP6 counts | 0.94 | 0.89 | 0.78 | 0.64 | 0.61 | |
| Baseline | SAscore | 0.13 | — | — | — | 0.22 | |
| Baseline | SCscore | 0.39 | — | — | — | 0.14 | |
| Baseline | SYBA | 0.70 | — | — | — | 0.17 | |
We identified that the following classifiers were consistently the top three models across each of the datasets: feed forward neural networks using ECFP6 counts, feed forward neural networks using ECFP6 counts with features, and XGBoost using ECFP6 counts. For the RAscore we chose the top performing classifier for separating the compounds as determined by the average linkage. We also identified a GDB specific classifier which we term GDBscore in the same manner. The GDBscore classifier was trained on the GDBChEMBL dataset, the classifier trained on GDBMedChem was found to have equivalent performance.
Prediction time
The importance of training ML based classifiers rather than simply predicting the full retrosynthetic pathway becomes clear when examining Table 2. Full retrosynthetic route prediction of the ChEMBL sample of 200000 compounds to a set of commercially available building blocks took approximately 239 CPU days on a single machine with 8 CPUs and 64 GB of RAM, using AiZynthFinder. Parallelization of full synthetic route prediction is not possible on a single machine under the current implementation of AiZynthFinder, however, it is possible to split the compounds over several cores and distribute the workload over several machines as has been done in this study. In comparison 77 minutes were required for classifying retrosynthetic accessibility using RAscore. The increase in prediction speed by ca. 4500 times opens up the possibility of estimating the retrosynthetic accessibility of virtual compounds, for instance in drug discovery projects, and for the scoring of compounds resulting from generative models earlier in the virtual screening workflow. Similar increases in prediction time are also observed for the GDB subsets (Table 1).
| ChEMBL | GDBChEMBL | GDBMedChem | |
|---|---|---|---|
| a Expressed in days taken on a single machine with 8 CPUs and 64 GB of RAM, rounded to the nearest day. The time taken in minutes for the neural network classifier with ECFP6 counted fingerprints is also given for comparative purposes. The neural network classifier, RAscore, is able to reproduce the results obtained from AiZynthFinder in a fraction of the time taken to predict full retrosynthetic routes. b RAscore. c GDBscore. | |||
| Percentage solved | 75.21 | 25.54 | 20.79 |
| Size | 200000 |
100000 |
100000 |
| AiZynthFinder run time (days) | 239 | 149 | 151 |
| Score run time (min) | 79b | 30c | 30c |
Applicability domain
Gao and Coley previously published the results of running retrosynthetic analysis with ASKCOS for a series of datasets consisting of both published and generated compounds.18 We tested our trained classifier on the dataset used by Gao and Coley to determine the applicability domain of the classifier and gauge how well the ASKCOS predictions could be reproduced. We also used AiZynthFinder to predict retrosynthetic routes to the same set of compounds to establish whether the classifier could reproduce the underlying CASP tool.For each dataset AiZynthFinder marginally outperforms ASKCOS, and is most striking for the GDB17 sample (Fig. 5).39 This is because AiZynthFinder only considers retrosynthetic analysis, whereas ASKCOS additionally factors in reaction prediction which enables pruning of unfeasible or low probability retrosynthetic pathways. Furthermore, as the reaction prediction models are trained on published chemistry, and the majority of GDB17 compounds are unpublished or dissimilar to published compounds,39 the pathways suggested are likely to be pruned resulting in the lower percentage of solved compounds for ASKCOS. Another difference that should be considered when comparing the two models is the building blocks available to each respective model. This can affect the ability of the CASP tool to find retrosynthetic routes and influences whether or not a compound is labelled as solved.
 | ||
| Fig. 5 Applicability domain as determined by application to a set of compounds published by Gao and Coley in a previous study, full details of each dataset can be found in the referenced manuscript.18 | ||
We found that the feed forward neural network classifier trained on ChEMBL that we term RAscore, overestimates the synthetic accessibility of GDB17 in comparison to ASKCOS and AiZynthFinder. This is also observed for the other datasets examined, however the extent to which RAscore overpredicts is less striking. To replicate the GDB17 dataset, we use GDBscore, which is a classifier trained on GDBChEMBL and find we can better reproduce the underlying AiZynthFinder synthesis planning tool. The MOSES dataset is based on the ZINC Clean Leads collection and GaucaMol is based on the ChEMBL database, both are used for evaluating distribution learning algorithms for drug discovery.42,43 The overprediction on both ZINC and the prediction in line with the MOSES dataset is surprising considering the compounds originate from the same database. However, this may be rationalized considering the samples differ in their distribution, and have been obtained from different collections within the ZINC database.40,43,44
The overprediction on the Sheridan et al. dataset can be seen as positive as all compounds in the dataset were previously synthesized at Merck.45 In addition, the prediction in line with the GuacaMol set, implies that the classifier performs well on ChEMBL like compounds by virtue of the underlying training data.
Examples – limitations of RAscore arising from CASP
We examined the test set from our ChEMBL sample for compounds within a Tanimoto similarity of 0.8 or greater. Some examples of pairs of compounds are shown in Fig. 6. In the pairs shown one compound was unsolved by our retrosynthetic tool and the other labelled as solved. For each example we show that the topology is largely unchanged and only small edits have been made to the functionality of the molecule. The change in outcome with minor changes in functionality highlight a limitation of AiZynthFinder and likely other template based CASP tools. This can originate from: the representation of the input molecules, the way the templates are specified, and the distribution of similar samples in the dataset from which the reactions originate. The templates suggested for disconnections are unable to account for subtle changes in the reaction center, thus the appropriate precursors were not able to be enumerated. This arises because the molecular graph underlying the template does not match that of the substrate, thus there is no substructure match. These examples are not ‘true’ negatives in the sense that they cannot be experimentally realised in the wet-lab and are only negative in relation to the ability of the AiZynthFinder to conduct a retrosynthetic analysis. Some examples of such compounds which have led to poor separation of solved/unsolved compounds are shown in Fig. 6.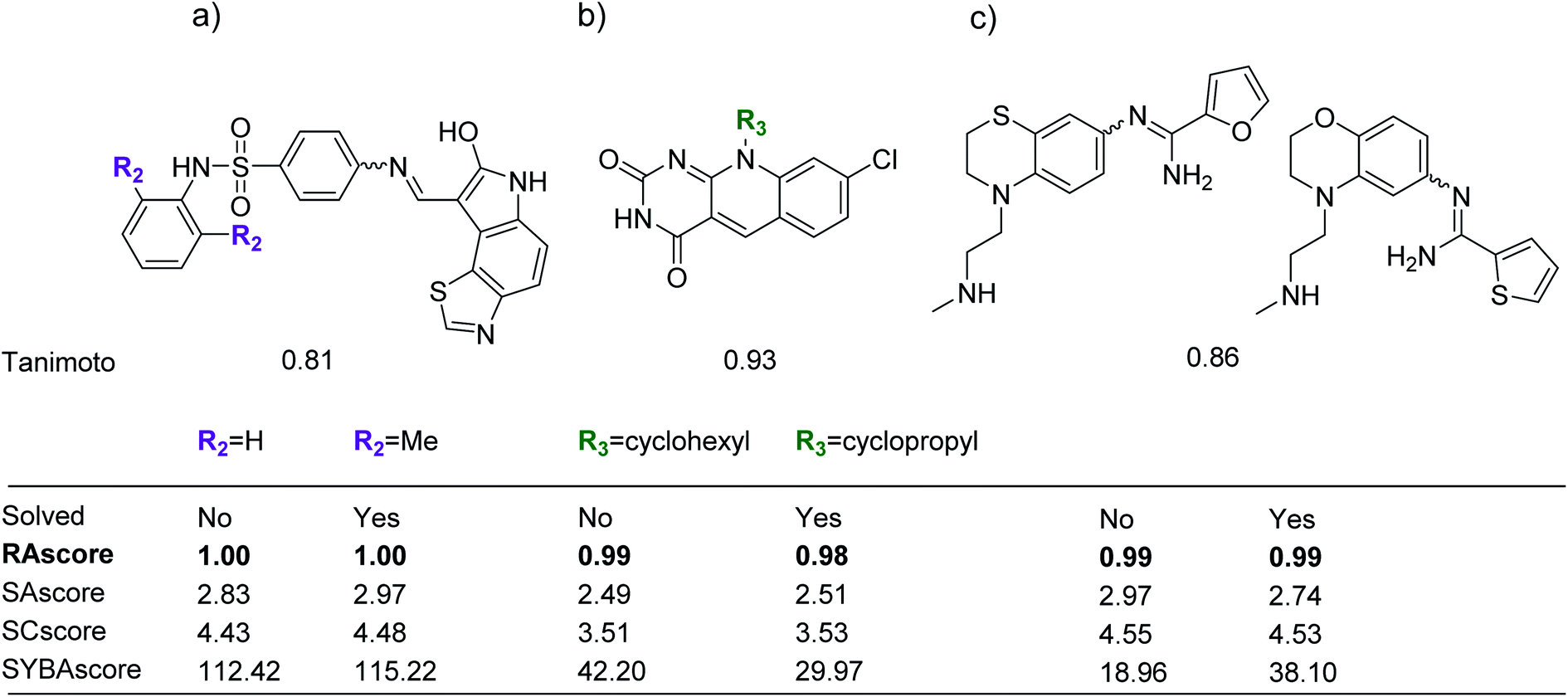 | ||
| Fig. 6 Examples of pairs of compounds from the test set that are similar to each other (Tanimoto > 0.8), where a retrosynthetic route could be found for one example in the pair but not the other. In each case only a slight modification of the compound leads to a change in the outcome from the CASP tool, consider (a) addition of two ortho-methyl groups on the terminal phenyl ring, (b) substitution of a cyclohexane moiety for a cyclopropane, and (c) a change in substitution pattern and ring morphology, leads to a change in outcome from solved to unsolved. | ||
To understand why the solved/unsolved test cases were not easily separable, consider the examples in Fig. 6. In the case of similar compounds, both solved and unsolved compounds are scored as synthetically feasible with values tending towards 1.0, despite AiZynthFinder not having found a synthetic route. The example in Fig. 6a, is a case for which RAscore predicts the compounds as synthetically accessible by AiZynthFinder despite a synthetic route having been found for only the compound with two ortho-methyl groups on the terminal phenyl ring. The RAscore learns that such minor changes to functionality are feasible by virtue of the machine learning approach, which does not take into consideration the inner workings of AiZynthFinder, but rather learns a mapping between inputs (compounds) and outputs (synthesisable by AiZynthFinder/unsynthesisable by AiZynthFinder). This behavior is an artefact of the subset of compounds from ChEMBL the model was trained on, and examples in which the model misclassifies compounds as synthesizable can also be found. Similar substitutions are shown in Fig. 6b and c, whereby AiZynthFinder failed to suggest retrosynthetic disconnections leading to commercially building blocks.
In most cases the most similar molecule in the training set was below a Tanimoto value of 0.8 (ESI†), and potentially requires a different synthetic strategy as compared to the compounds shown for the test set (Fig. 6). This raises another limitation of AiZynthFinder and potentially other CASP tools, which can be overcome by RAscore. The performance of a CASP tool is limited by the number and type of building blocks available. In some cases it may be that the building blocks necessary are not included in the database underlying the CASP tool, but are in fact available from other vendors. Furthermore, it can also be the case that similar building blocks are available that a medicinal chemist may consider for functionalization. In these cases the RAscore is able to learn that it is likely that two analogues are synthetically accessible despite a retrosynthetic route having not been found. This is because RAscore is not based on a library of building blocks, and has been trained with the compound as input and label (synthesisable by AiZynthFinder/unsynthesisable by AiZynthFinder) as output, thus has no knowledge of building blocks explicitly. The RAscore model learns similarity between compounds internally, and by doing so learns where to place a decision boundary between datapoints belonging to each cluster. This is the basis on which most machine learning techniques enable the models to extrapolate to similar compounds.
To exemplify the aforementioned arguments consider the routes predicted by AiZynthFinder shown in Fig. 7. If we again take the case of the phenyl moiety both with and without the ortho-methyl groups of the compound shown in Fig. 6a, and examine the routes predicted for each, Fig. 7a and b respectively, we observe differences in the predicted route in terms of the synthetic strategy used, thus step count. Therefore, similar compounds with largely unchanged topology can have considerably different synthetic routes predicted for them. One of the reasons this occurs is because each step in the route prediction is treated independently from the others. Thus, the neural network used in AiZynthFinder does not learn that similar compounds have the potential to be synthesized via similar routes as it has not been fed information about the route. Whilst a chemist may consider first synthesizing the scaffold, and subsequently functionalising it to yield the desired analogues, AiZynthFinder is currently unable to take into account such considerations. This is further exemplified in Fig. 7a and b, whereby different synthetic routes necessitate different starting materials. The synthetic route proposed in Fig. 7a can be used to synthesize the compound in Fig. 7b with only a slight variation in how the building blocks are initially synthesized.
 | ||
| Fig. 7 (a and b) Synthetic routes predicted by AiZynthFinder. We show that small variations in the queried compound lead to considerably different synthetic routes, thus require different building blocks. This is a limitation of CASP tools that models such as RAscore may be able to overcome as they do not explicitly take into account the inner workings of the CASP tool but are rather based on learned similarity and how this maps to a given output. The fragments that are modified at each step are highlighted in blue in the synthetic scheme. | ||
The RAscore has potential to overcome some of these limitations as it does not take into account route information explicitly. Rather the RAscore is based on the predictions of AiZynthFinder, and equally the predictions of any CASP tool should be able to be used in their place. This has the advantage that the RAscore is then able to approximate whether a synthetic route can be found using CASP for any given molecule, without having to compute the synthetic route each time.
Conclusions
Herein we have built on the improvements in AI driven CASP in recent years by combining the predictions made with our CASP tool, AiZynthFinder, with ML, to train a classifier returning a retrosynthetic accessibility score (RAscore). RAscore addresses the challenge of classifying compounds as synthetically feasible and is orders of magnitude faster than full retrosynthetic analysis by CASP, and with comparable performance. The RAscore demonstrates potential for rapid pre-screening of compounds for synthetic accessibility, enabling enrichment of synthetically feasible chemical space. Whereas previous synthetic accessibility and complexity based scores have potential for misuse when filtering large virtual libraries, as a result of being unable to determine a threshold value (Fig. 4), we resolve this issue by proposing that the existing scores be used alongside the RAscore to determine whether a synthetic route can be found, and how difficult it may be to realize the route in the wet lab.In addition, we highlight inherent limitations to be aware of in the RAscore arising from the performance and applicability of the underlying CASP tool, namely: (1) availability of building blocks, (2) different synthetic strategies toward the same scaffold, and (3) route predictions are treated independently to each other. The concept presented herein can be extended to any CASP tool and the predictions it generates, and the score retrained. The score will be made available under an MIT license at: https://github.com/reymond-group/RAscore.
Author contributions
A. Thakkar designed, conducted the research, and wrote the manuscript. V. Chadimová performed preliminary modelling studies. E. J. Bjerrum contributed ideas to the project. O. Engkvist, and J. L. Reymond supervised the project and assisted in writing the manuscript. All authors read and approved the final manuscript.Availability of data and materials
The score will be made available under an MIT license at, as well as instructions on how to access the datasets and the framework for training the classifiers: https://github.com/reymond-group/RAscore.AiZynthFinder is open source and is available under an MIT license at: https://github.com/MolecularAI/AiZynthFinder.
The dataset used to assess the applicability domain can be found at: https://github.com/wenhao-gao/askcos_synthesizability/tree/master/results/dataset.csv.
Funding
A. Thakkar received support from AstraZeneca, the Swiss National Science Foundation (SNF), and the European Union's Horizon 2020 research and innovation program under the Marie Skłodowska-Curie grant agreement no. 676434, “Big Data in Chemistry” (“BIGCHEM,” http://bigchem.eu). V. Chadimová was sponsored by the AstraZeneca R&D graduate programme.Conflicts of interest
The authors declare that they have no competing interests.Acknowledgements
The authors would like to thank NextMove software for providing access to NameRxn for atom-mapping. Samuel Genheden for valuable feedback and discussions regarding the manuscript and analysis. The Molecular AI group at AstraZeneca and Reymond group at the University of Bern for their support.References
- M. H. S. Segler, T. Kogej, C. Tyrchan and M. P. Waller, Generating Focused Molecule Libraries for Drug Discovery with Recurrent Neural Networks, ACS Cent. Sci., 2018, 4(1), 120–131 CrossRef CAS.
- T. Blaschke, J. Arús-Pous, H. Chen, C. Margreitter, C. Tyrchan, O. Engkvist, K. Papadopoulos and A. Patronov, Reinvent 2.0 – an Ai Tool for De Novo Drug Design, ChemRxiv, 2020 Search PubMed.
- A. Mayr, G. Klambauer, T. Unterthiner and S. Hochreiter, Deeptox: Toxicity Prediction Using Deep Learning, Front. Environ. Sci., 2016, 3, 80 CrossRef.
- M. H. S. Segler, M. Preuss and M. P. Waller, Planning Chemical Syntheses with Deep Neural Networks and Symbolic Ai, Nature, 2018, 555, 604 CrossRef CAS.
- C. W. Coley, D. A. Thomas, J. A. M. Lummiss, J. N. Jaworski, C. P. Breen, V. Schultz, T. Hart, J. S. Fishman, L. Rogers, H. Gao, R. W. Hicklin, P. P. Plehiers, J. Byington, J. S. Piotti, W. H. Green, A. J. Hart, T. F. Jamison and K. F. Jensen, A Robotic Platform for Flow Synthesis of Organic Compounds Informed by Ai Planning, Science, 2019, 365(6453), eaax1566 CrossRef CAS.
- T. J. Struble, J. C. Alvarez, S. P. Brown, M. Chytil, J. Cisar, R. L. DesJarlais, O. Engkvist, S. A. Frank, D. R. Greve, D. J. Griffin, X. Hou, J. W. Johannes, C. Kreatsoulas, B. Lahue, M. Mathea, G. Mogk, C. A. Nicolaou, A. D. Palmer, D. J. Price, R. I. Robinson, S. Salentin, L. Xing, T. Jaakkola, W. H. Green, R. Barzilay, C. W. Coley and K. F. Jensen, Current and Future Roles of Artificial Intelligence in Medicinal Chemistry Synthesis, J. Med. Chem., 2020, 63(16), 8667–8682 CrossRef CAS.
- W. P. Walters, Virtual Chemical Libraries, J. Med. Chem., 2019, 62(3), 1116–1124 CrossRef CAS.
- P. Schneider, W. P. Walters, A. T. Plowright, N. Sieroka, J. Listgarten, R. A. Goodnow, J. Fisher, J. M. Jansen, J. S. Duca, T. S. Rush, M. Zentgraf, J. E. Hill, E. Krutoholow, M. Kohler, J. Blaney, K. Funatsu, C. Luebkemann and G. Schneider, Rethinking Drug Design in the Artificial Intelligence Era, Nat. Rev. Drug Discovery, 2020, 19(5), 353–364 CrossRef CAS.
- F. Chevillard and P. Kolb, A Large yet Screenable and Easily Searchable Database of Computationally Created Chemical Compounds Optimized toward High Likelihood of Synthetic Tractability, J. Chem. Inf. Model., 2015, 55(9), 1824–1835 CrossRef CAS.
- A. Zhavoronkov, Y. A. Ivanenkov, A. Aliper, M. S. Veselov, V. A. Aladinskiy, A. V. Aladinskaya, V. A. Terentiev, D. A. Polykovskiy, M. D. Kuznetsov, A. Asadulaev, Y. Volkov, A. Zholus, R. R. Shayakhmetov, A. Zhebrak, L. I. Minaeva, B. A. Zagribelnyy, L. H. Lee, R. Soll, D. Madge, L. Xing, T. Guo and A. Aspuru-Guzik, Deep Learning Enables Rapid Identification of Potent Ddr1 Kinase Inhibitors, Nat. Biotechnol., 2019, 37(9), 1038–1040 CrossRef CAS.
- J. C. Baber and M. Feher, Predicting Synthetic Accessibility: Application in Drug Discovery and Development, Mini-Rev. Med. Chem., 2004, 4(6), 681–692 CrossRef CAS.
- W.-D. Ihlenfeldt and J. Gasteiger, Computer-Assisted Planning of Organic Syntheses: The Second Generation of Programs, Angew. Chem., Int. Ed. Engl., 1996, 34(23–24), 2613–2633 CrossRef.
- V. J. Gillet, G. Myatt, Z. Zsoldos and A. P. Johnson, Sprout, Hippo and Caesa: Tools for De Novo Structure Generation and Estimation of Synthetic Accessibility, Perspect. Drug Discovery Des., 1995, 3(1), 34–50 CrossRef CAS.
- B. Mikulak-Klucznik, P. Gołębiowska, A. A. Bayly, O. Popik, T. Klucznik, S. Szymkuć, E. P. Gajewska, P. Dittwald, O. Staszewska-Krajewska, W. Beker, T. Badowski, K. A. Scheidt, K. Molga, J. Mlynarski, M. Mrksich and B. A. Grzybowski, Computational Planning of the Synthesis of Complex Natural Products, Nature, 2020, 588, 83–88 CrossRef CAS.
- A. Bøgevig, H.-J. Federsel, F. Huerta, M. G. Hutchings, H. Kraut, T. Langer, P. Löw, C. Oppawsky, T. Rein and H. Saller, Route Design in the 21st Century: The Icsynth Software Tool as an Idea Generator for Synthesis Prediction, Org. Process Res. Dev., 2015, 19(2), 357–368 CrossRef.
- S. Genheden, A. Thakkar, V. Chadimová, J.-L. Reymond, O. Engkvist and E. Bjerrum, Aizynthfinder: A Fast, Robust and Flexible Open-Source Software for Retrosynthetic Planning, J. Cheminf., 2020, 12(1), 70 Search PubMed.
- P. Schwaller, T. Laino, T. Gaudin, P. Bolgar, C. A. Hunter, C. Bekas and A. A. Lee, Molecular Transformer: A Model for Uncertainty-Calibrated Chemical Reaction Prediction, ACS Cent. Sci., 2019, 5(9), 1572–1583 CrossRef CAS.
- W. Gao and C. W. Coley, The Synthesizability of Molecules Proposed by Generative Models, J. Chem. Inf. Model., 2020, 60(12), 5714–5723 CrossRef CAS.
- E. Corey, A. Long and S. Rubenstein, Computer-Assisted Analysis in Organic Synthesis, Science, 1985, 228(4698), 408–418 CrossRef CAS.
- A. Cook, A. P. Johnson, J. Law, M. Mirzazadeh, O. Ravitz and A. Simon, Computer-Aided Synthesis Design: 40 Years On, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2(1), 79–107 CAS.
- C. W. Coley, L. Rogers, W. H. Green and K. F. Jensen, Scscore: Synthetic Complexity Learned from a Reaction Corpus, J. Chem. Inf. Model., 2018, 58(2), 252–261 CrossRef CAS.
- M. Voršilák, M. Kolář, I. Čmelo and D. Svozil, Syba: Bayesian Estimation of Synthetic Accessibility of Organic Compounds, J. Cheminf., 2020, 12(1), 35 Search PubMed.
- P. Ertl and A. Schuffenhauer, Estimation of Synthetic Accessibility Score of Drug-Like Molecules Based on Molecular Complexity and Fragment Contributions, J. Cheminf., 2009, 1(1), 8 Search PubMed.
- A. Thakkar, T. Kogej, J.-L. Reymond, O. Engkvist and E. J. Bjerrum, Datasets and Their Influence on the Development of Computer Assisted Synthesis Planning Tools in the Pharmaceutical Domain, Chem. Sci., 2020, 11(1), 154–168 RSC.
- D. Lowe, Chemical Reactions from Us Patents, 1976–Sep2016, https://figshare.com/articles/Chemical_reactions_from_US_patents_1976-Sep2016_/5104873, accessed Apr 31, 2018 Search PubMed.
- https://www.acdlabs.com/index.php .
- Enamine, https://enamine.net/building-blocks Search PubMed.
- Zinc, http://zinc.docking.org/rings/subsets/, accessed Aug 27, 2019 Search PubMed.
- A. Gaulton, A. Hersey, M. Nowotka, A. P. Bento, J. Chambers, D. Mendez, P. Mutowo, F. Atkinson, L. J. Bellis, E. Cibrián-Uhalte, M. Davies, N. Dedman, A. Karlsson, M. P. Magariños, J. P. Overington, G. Papadatos, I. Smit and A. R. Leach, The Chembl Database in 2017, Nucleic Acids Res., 2017, 45(D1), D945–D954 CrossRef CAS.
- S. Bühlmann and J.-L. Reymond, ChEMBL-Likeness Score and Database GDBChEMBL, Front. Chem., 2020, 8(46) DOI:10.3389/fchem.2020.00046.
- M. Awale, F. Sirockin, N. Stiefl and J.-L. Reymond, Medicinal Chemistry Aware Database Gdbmedchem, Mol. Inf., 2019, 38, 1900031 CrossRef CAS.
- D. Rogers and M. Hahn, Extended-Connectivity Fingerprints, J. Chem. Inf. Model., 2010, 50(5), 742–754 CrossRef CAS.
- Rdkit: Open-Source Cheminformatics, http://www.Rdkit.org Search PubMed.
- F. Pedregosa, G. Varoquaux, A. Gramfort, V. Michel, B. Thirion, O. Grisel, M. Blondel, P. Prettenhofer, R. Weiss, V. Dubourg, J. Vanderplas, A. Passos, D. Cournapeau, M. Brucher, M. Perrot and E. Duchesnay, Scikit-Learn: Machine Learning in {P}Ython, J. Mach. Learn. Res., 2011, 12, 2825–2830 Search PubMed.
- M. Abadi, A. Agarwal, P. Barham, E. Brevdo, Z. Chen, C. Citro, G. S. Corrado, A. Davis, J. Dean, M. Devin, S. Ghemawat, I. Goodfellow, A. Harp, G. Irving, M. Isard, R. Jozefowicz, Y. Jia, L. Kaiser, M. Kudlur, J. Levenberg, D. Mané, M. Schuster, R. Monga, S. Moore, D. Murray, C. Olah, J. Shlens, B. Steiner, I. Sutskever, K. Talwar, P. Tucker, V. Vanhoucke, V. Vasudevan, F. Viégas, O. Vinyals, P. Warden, M. Wattenberg, M. Wicke, Y. Yu and X. Zheng, Tensorflow: Large-Scale Machine Learning on Heterogeneous Systems, 2015, software available from, https://www.tensorflow.org Search PubMed.
- F. Chollet, Keras, Github, 2015, https://Github.Com/Fchollet/Keras Search PubMed.
- T. Akiba, S. Sano, T. Yanase, T. Ohta and M. Koyama, Optuna: A Next-Generation Hyperparameter Optimization Framework, in Proceedings of the 25th ACM SIGKDD International Conference on Knowledge Discovery & Data Mining, Association for Computing Machinery: Anchorage, AK, USA, 2019, pp. 2623–2631 Search PubMed.
- S. R. Heller, A. McNaught, I. Pletnev, S. Stein and D. Tchekhovskoi, Inchi, the Iupac International Chemical Identifier, J. Cheminf., 2015, 7(1), 23 Search PubMed.
- L. Ruddigkeit, R. van Deursen, L. C. Blum and J.-L. Reymond, Enumeration of 166 Billion Organic Small Molecules in the Chemical Universe Database Gdb-17, J. Chem. Inf. Model., 2012, 52(11), 2864–2875 CrossRef CAS.
- R. Gómez-Bombarelli, J. N. Wei, D. Duvenaud, J. M. Hernández-Lobato, B. Sánchez-Lengeling, D. Sheberla, J. Aguilera-Iparraguirre, T. D. Hirzel, R. P. Adams and A. Aspuru-Guzik, Automatic Chemical Design Using a Data-Driven Continuous Representation of Molecules, ACS Cent. Sci., 2018, 4(2), 268–276 CrossRef.
- O. Méndez-Lucio, B. Baillif, D.-A. Clevert, D. Rouquié and J. Wichard, De Novo Generation of Hit-Like Molecules from Gene Expression Signatures Using Artificial Intelligence, Nat. Commun., 2020, 11(1), 10 CrossRef.
- N. Brown, M. Fiscato, M. H. S. Segler and A. C. Vaucher, Guacamol: Benchmarking Models for De Novo Molecular Design, J. Chem. Inf. Model., 2019, 59(3), 1096–1108 CrossRef CAS.
- D. Polykovskiy, A. Zhebrak, B. Sanchez-Lengeling, S. Golovanov, O. Tatanov, S. Belyaev, R. Kurbanov, A. Artamonov, V. Aladinskiy, M. Veselov, A. Kadurin, S. Johansson, H. Chen, S. Nikolenko, A. Aspuru-Guzik and A. J. a. e.-p. Zhavoronkov, Molecular Sets (Moses): A Benchmarking Platform for Molecular Generation Models, 2018, p. arXiv:1811.12823, https://ui.adsabs.harvard.edu/abs/2018arXiv181112823P, accessed November 01, 2018 Search PubMed.
- J. J. Irwin, T. Sterling, M. M. Mysinger, E. S. Bolstad and R. G. Coleman, Zinc: A Free Tool to Discover Chemistry for Biology, J. Chem. Inf. Model., 2012, 52(7), 1757–1768 CrossRef CAS.
- R. P. Sheridan, N. Zorn, E. C. Sherer, L.-C. Campeau, C. Chang, J. Cumming, M. L. Maddess, P. G. Nantermet, C. J. Sinz and P. D. O'Shea, Modeling a Crowdsourced Definition of Molecular Complexity, J. Chem. Inf. Model., 2014, 54(6), 1604–1616 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc05401a |
| This journal is © The Royal Society of Chemistry 2021 |