
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesigned alpha-helical barrels for charge-selective peptide translocation†
Smrithi Krishnan
R.‡
ab,
Neethu
Puthumadathil‡
ab,
Amina H.
Shaji
a,
K.
Santhosh Kumar
a,
Gayathri
Mohan
a and
Kozhinjampara R.
Mahendran
 *a
*a
aMembrane Biology Laboratory, Transdisciplinary Research Program, Rajiv Gandhi Centre for Biotechnology, Thiruvananthapuram 695014, India. E-mail: mahendran@rgcb.res.in
bManipal Academy of Higher Education, Manipal, Karnataka, India-576104
First published on 4th November 2020
Abstract
Synthetic alpha-helix based pores for selective sensing of peptides have not been characterized previously. Here, we report large transmembrane pores, pPorA formed from short synthetic alpha-helical peptides of tunable conductance and selectivity for single-molecule sensing of peptides. We quantified the selective translocation kinetics of differently charged cationic and anionic peptides through these synthetic pores at single-molecule resolution. The charged peptides are electrophoretically pulled into the pores resulting in an increase in the dissociation rate with the voltage indicating successful translocation of peptides. More specifically, we elucidated the charge pattern lining the pore lumen and the orientation of the pores in the membrane based on the asymmetry in the peptide-binding kinetics. The salt and pH-dependent measurements confirm the electrostatic dominance and charge selectivity in controlling target peptide interaction with the pores. Remarkably, we tuned the selectivity of the pores to charged peptides by modifying the charge composition of the pores, thus establishing the molecular and electrostatic basis of peptide translocation. We suggest that these synthetic pores that selectively conduct specific ions and biomolecules are advantageous for nanopore proteomics analysis and synthetic nanobiotechnology applications.
Introduction
Protein pores are used as effective sensors for single-molecule detection of a wide variety of biological and chemical molecules in solution.1–7 Most previous studies have been focused on β-barrel pores such as α-hemolysin and MspA, as they are redesigned with atomic accuracy specifically for nucleic acid sequencing.7–9 Notably, several natural protein pores of sophisticated internal geometry such as α-hemolysin, Cytolysin A, Aerolysin, and Fragaceatoxin C were engineered for single-molecule sensing of peptides and proteins.10–21 The time-consuming nature of the production of natural protein pores and the demand for a superior single-molecule detection system have led to the development of synthetic pores.3,22 Also, ion channels and peptide-based biosensors were developed for the detection of various biomolecules.23–25 Recent advancements in the design and assembly of DNA-based nanopores are very remarkable.26–31 The majority of DNA nanopores are based on parallel-oriented DNA duplexes equipped with lipid anchors to facilitate their insertion into the lipid bilayers.32,33 However, the narrow DNA nanopores with the electrically leaky porous DNA walls of low membrane insertion efficiency have limited nanotechnology applications.22 The current scenario demands more advanced selective pores of desired geometry and charge pattern for chemical and biosensing effectively.34–37Notably, engineered β-barrel pores have produced limited changes in the charge selectivity of ion transport and high ion selectivity has not been produced, as seen with natural ion channels.3,38 While β-barrels and DNA nanopores have been engineered extensively, transmembrane alpha-helical pores remain relatively unexplored.22,39–41 Importantly, alpha-helical bundles are attractive targets because of similarities to natural membrane proteins.34,35,39–41 Therefore, engineering alpha-helical pores of new structural designs can play a significant role in nanobiotechnology and can be achieved with relatively simple sequences. In particular, modifications in the alpha-helix can alter its selectivity through conformational fine-tuning of the helical pattern due to numerous side-chain interactions dominating within the helix.35,42 This characteristic property of the helical bundles can be used to develop a selective sensor, which might be difficult to achieve with β-barrels.38,40,43 However, building membrane-spanning alpha-helical designs with balancing membrane solubility, structural stability and self-assembly is challenging.34,35,39–41 Despite this, membrane-spanning peptide and DNA–peptide hybrid pores of low conductance and selectivity were developed.22,27,39 More recently, we engineered and constructed large synthetic alpha-helical transmembrane pores, pPorA based on natural porin PorACj of Corynebacterium jeikeium.43,44 (Fig. 1a). Remarkably, they open possibilities for using designed alpha-helical barrels as components of membrane-based sensors. Here, we elucidated the selective translocation of cationic and anionic peptides through these alpha-helical pores and provide quantitative kinetics of peptide translocation. Further, we defined the molecular basis of peptide translocation controlled by electrostatics due to the charged residues in the pores. We also demonstrated the charge selectivity of the pores to the peptides, which will aid the design of pores for application in nanopore technology for peptide and protein characterization.
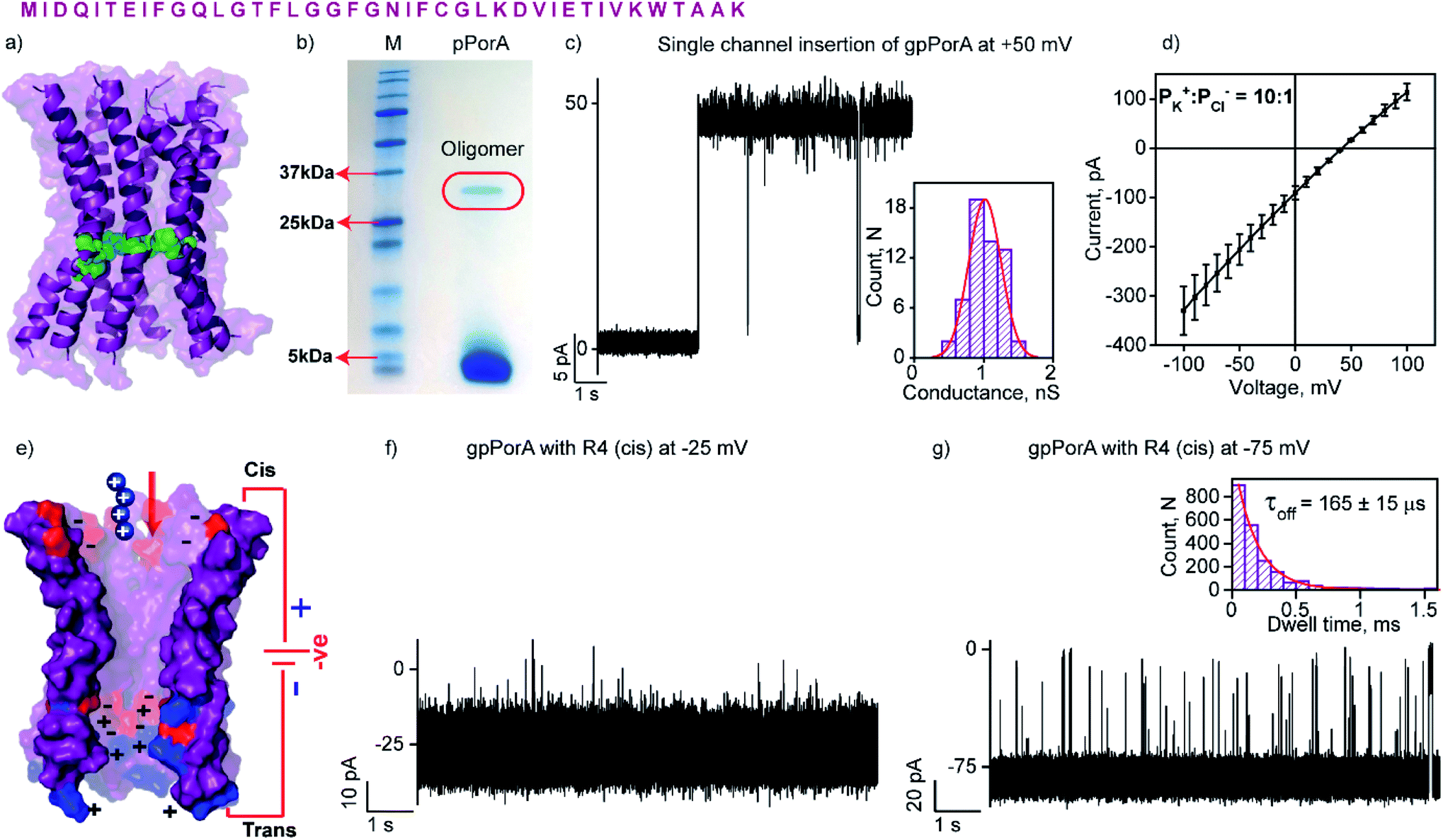 | ||
| Fig. 1 Structure and electrical properties of pPorA. (a) The sequence of pPorA and electrostatic distribution of modeled pPorA in deep purple colour. Cysteine residues are shown in green. (b) The pPorA peptides run on an SDS-PAGE showing the oligomers. (c) Electrical recording of a gel extracted single pPorA insertion (gpPorA) at +50 mV and unitary conductance histogram obtained by fitting the distribution to a single Gaussian (n = 70). (d) Reverse potential obtained from the I–V curve of a single gpPorA pore. (e) Model showing the interaction of tetraarginine (R4) with the cis side. Positively charged residues are shown in blue and negatively charged residues in red. (f) Electrical recordings showing R4 interaction with single gpPorA (10 μM, cis) at −25 mV and (g) −75 mV. Inset, recording at expanded time scale and the corresponding τoff dwell time histogram. Electrolyte: 0.15 M KCl, 10 mM HEPES, pH 7.4. The current signals (c and d) were filtered at 2 kHz and sampled at 10 kHz. The current signals (f and g) were filtered at 10 kHz and sampled at 50 kHz. | ||
Results and discussion
Biophysical and single-channel electrical properties of the pores
The natural 40 amino acid PorACj protein expressed in bacteria formed channels of variable conductance in lipid bilayers indicating heterogeneous structures.44 We synthesized the pPorA peptides corresponding to natural pores and characterized their biophysical and electrical properties (Fig. 1 and S1†). Remarkably, a 40-residue synthetic alpha-helical peptide containing cysteine at a specific position (pPorA-C24, cysteine at 24th position) in SDS-PAGE revealed a ∼36 kDa band corresponding to self-assembled stable octameric oligomers. Accordingly, we used SDS-PAGE to extract and purify pPorA oligomers to characterize single-channel properties (Fig. 1b).These gel extracted oligomers (gpPorA) rapidly inserted into DPhPC (1,2-diphytanoyl-sn-glycero-3-phosphocholine) lipid bilayers and formed stable pores (Fig. 1c). Remarkably, gpPorA formed unusually large conductance pores at +50 mV in 0.15 M KCl electrolyte buffer and a unitary conductance histogram was obtained based on the multiple pore insertions in the lipid bilayer (Fig. 1c). The pores exhibited a mean unitary conductance (G) of 1.1 ± 0.2 nS at ±50 mV (n = 70) that revealed the homogeneity and stability of the pores in the membrane. The pores existed in a fully stable open conductance state at voltages ≤ ±75 mV and showed a voltage-dependent gating at ±100 mV (Fig. S2†). The pores did not show significant asymmetry in the single-channel conductance with reference to the voltage polarity (n = 50) (Fig. S2†). Notably, ion selectivity measurements revealed that gpPorA formed cation-selective pores in the planar lipid bilayers (the permeability ratio PK+/PCl− = ∼10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) (Fig. 1d). The pores are presumed to be octameric alpha-helical barrels based on the modeled structure of the natural octameric porin PorACj of Corynebacterium jeikeium (Fig. 1a and S3†).43,44 We propose that the negatively charged aspartate and glutamate residues and the positively charged lysines aligned to the pore lumen could play a key role in selectivity. Further, we determined the number of side-chain charges exposed to the water exposed side of the pores based on 2.0 ns averaging via Gaussian accelerated molecular dynamics simulations (anions = −3.42; cations = +2.35) (Fig. S3†). Accordingly, we suggest that these self-assembled selective alpha-helical pores showing high conductance in the low salt buffer can be used for selective single-molecule sensing of a wide range of charged peptides.
:1) (Fig. 1d). The pores are presumed to be octameric alpha-helical barrels based on the modeled structure of the natural octameric porin PorACj of Corynebacterium jeikeium (Fig. 1a and S3†).43,44 We propose that the negatively charged aspartate and glutamate residues and the positively charged lysines aligned to the pore lumen could play a key role in selectivity. Further, we determined the number of side-chain charges exposed to the water exposed side of the pores based on 2.0 ns averaging via Gaussian accelerated molecular dynamics simulations (anions = −3.42; cations = +2.35) (Fig. S3†). Accordingly, we suggest that these self-assembled selective alpha-helical pores showing high conductance in the low salt buffer can be used for selective single-molecule sensing of a wide range of charged peptides.
Interaction of cationic peptides with the pores
The high cation-selectivity exhibited by gpPorA might be favourable for selective sensing. Therefore, we examined the binding and translocation of charged peptides through gpPorA at the single-molecule level in real-time.3,22 More specifically, we studied the interaction of cationic peptides differing in charge and length with gpPorA in low salt electrolyte buffer (0.15 M KCl). At first, we investigated the interaction of tetraarginine (R4) with gpPorA in a voltage-dependent manner (Fig. 1e, S4 and S5†). The addition of 10 μM R4 to the cis side produced occasional ion current blockages at −25 mV that are not accurately resolved (Fig. 1f). The frequency of the blockage events increased with the voltage and the mean dwell time of blocking (τoff) was calculated to be 165 ± 15 μs (n = 3) at −75 mV, demonstrating voltage-dependent peptide binding with the pores (Fig. 1g). No peptide-induced ion current blockages were detected at positive voltages suggesting electrophoretic repulsion of peptides from the pores (Fig. S4†). Subsequently, the trans side addition of R4 (10 μM) to the same pores after perfusion of peptides from the cis side resulted in short, less frequent blockage events exclusively at higher positive voltages (Fig. S5†). Here, the orientation of the pores remained the same that allowed us to study the asymmetrical translocation of peptides. Despite this, we cannot establish R4 translocation through the pores due to the weak electrostatic binding of these peptides. Next, we investigated the interaction of hexaarginine (R6) with the gpPorA in a voltage-dependent manner to confirm the peptide translocation (Fig. 2).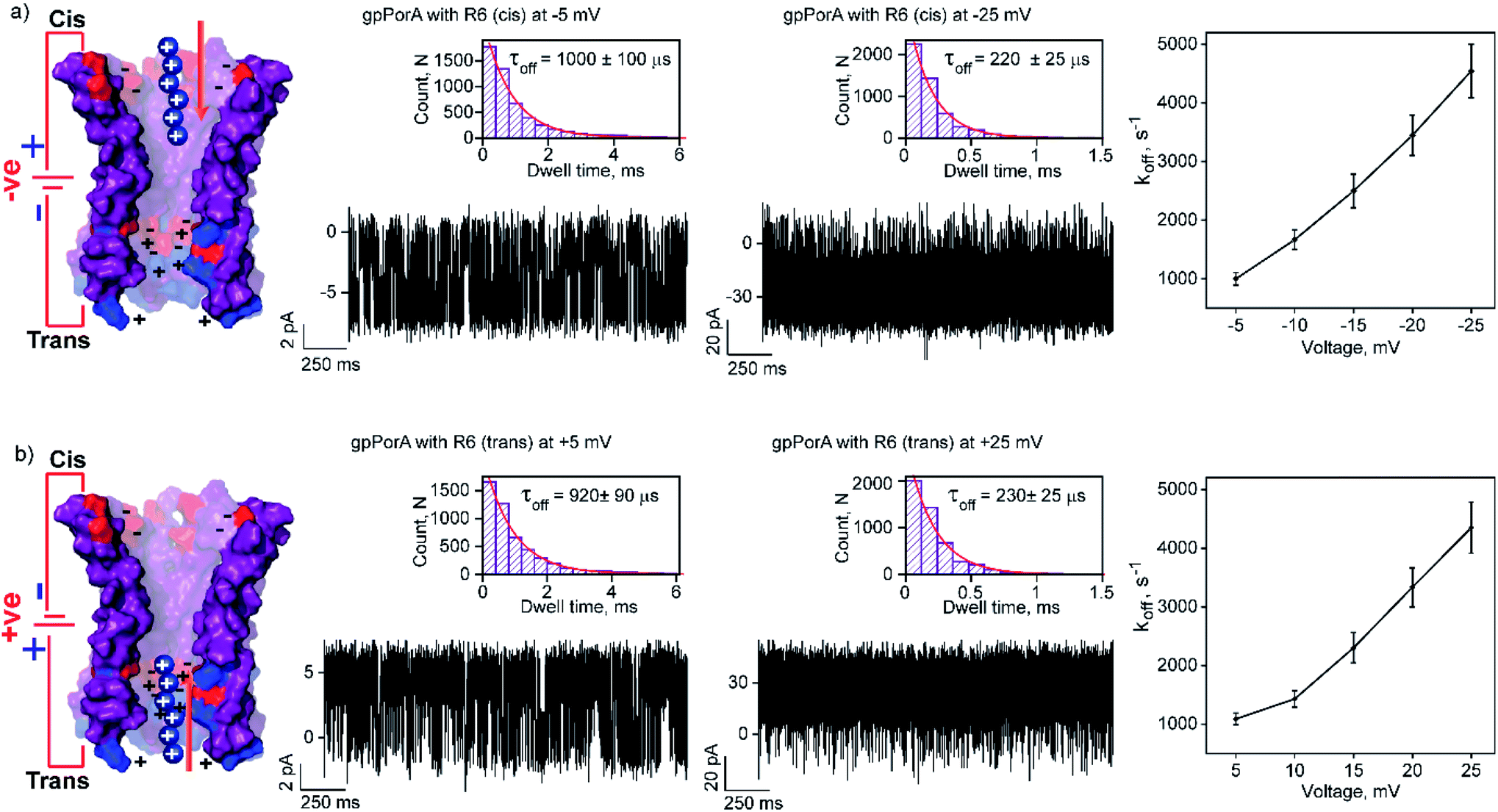 | ||
| Fig. 2 Interaction of hexaarginine with gpPorA. (a) Electrical recordings showing hexaarginine (R6) interaction with single gpPorA (0.5 μM, cis) at −5 mV, −25 mV and a plot of koffversus the applied voltage (mean values (±s.d.) from three independent experiments are shown). (b) Electrical recordings showing the interaction of R6 with single gpPorA (0.5 μM, trans) at +5 mV, +25 mV and a plot of koffversus the applied voltage (mean values (±s.d.) from three independent experiments are shown). Inset, recording at expanded time scale and the corresponding τoff dwell time histogram. Model showing the interaction of R6 with the cis and trans side of the pores. Electrolyte: 0.15 M KCl, 10 mM HEPES, pH 7.4. The current signals (a and b) ±5 mV and ±25 mV were digitally filtered at 1.5 kHz and 7 kHz using an 8-pole Bessel digital filter. | ||
Notably, the addition of 0.5 μM R6 to the cis side resulted in ion current blockages (100% block) even at low negative voltages, suggesting electrophoretic pulling and peptide binding with the pores (Fig. 2a and S6†). In particular, the τoff and frequency of blocking events were calculated for increasing voltages ranging from −5 mV to −25 mV to quantify the kinetics of R6 translocation. The τoff was estimated to be 1 ± 0.1 ms at −5 mV, which is substantially reduced to 0.23 ± 0.025 ms (n = 3) at −25 mV, suggesting an increase in the dissociation rate constant, koff (1/τoff), with increasing voltages (Fig. 2a, and Table S1†). Similarly, the frequency of blockage events decreased with an increase in the voltage and at higher negative voltages above −25 mV, the blockage events were not resolved accurately (Fig. S6†). We suggest that the R6 binds to the pores at lower voltages, whereas the binding is weakened at higher voltages resulting in rapid translocation consistent with the koff increase. The voltage-dependent blocking indicates electrostatic interaction of R6 with negatively charged residues in the pores that act as affinity sites facilitating target peptide translocation. Interestingly, ion current blockages were observed at lower positive voltages (+15 mV to +25 mV) on the cis side R6 addition, in contrast to the voltage-driven electrophoretic peptide binding with the pores (Fig. S6†). This could be due to packed negatively charged residues on the cis side of the pores promoting peptide binding against voltage induced electrostatic repulsion. Consistent with this at higher positive voltages, no blockage events were observed as the peptides are electrostatically pulled away from the pores. Similar to cis side addition, 0.5 μM R6 on the trans side produced time-resolved ion current blockages compatible with electrostatic binding and voltage-driven translocation of peptides (Fig. 2b, S7 and Table S1†). Notably, the peptide-binding kinetics revealed no significant asymmetry in the peptide translocation between the cis and trans sides of the pores.
Translocation kinetics: electrostatic peptide binding versus repulsion
To elucidate the effect of peptide charge on the pore binding, we investigated the interaction of highly charged nonaarginine (R9) with gpPorA and determined the binding affinity and translocation rate. The addition of 0.5 μM nonaarginine to the cis side resulted in prolonged ion current blockages (100% block) at negative voltages consistent with an effective peptide binding (Fig. 3). Surprisingly, at −5 mV, the pores predominantly remained closed, implying stable, tight binding of R9 (Fig. 3a). Further, an increase in the voltage resulted in the transition of the pores from the closed to the open state, producing clear-cut ion current blockages indicating relatively reduced peptide-binding compared to −5 mV (Fig. S8†). The kon and koff increased with the voltage from −10 mV to −35 mV, indicating the electrophoretic binding and voltage-driven peptide translocation (Fig. 3a, b, S8 and Table S2†). The frequency of ion current blockages considerably reduced with shorter blockage time close to the resolution limit at higher voltages above −40 mV, suggesting accelerated peptide translocation through the pores (Fig. S8†). Besides this, R9 induced well-resolved ion current blockages at positive voltages from +10 mV to +35 mV against the electrophoretic pulling (Fig. 3c and S8†).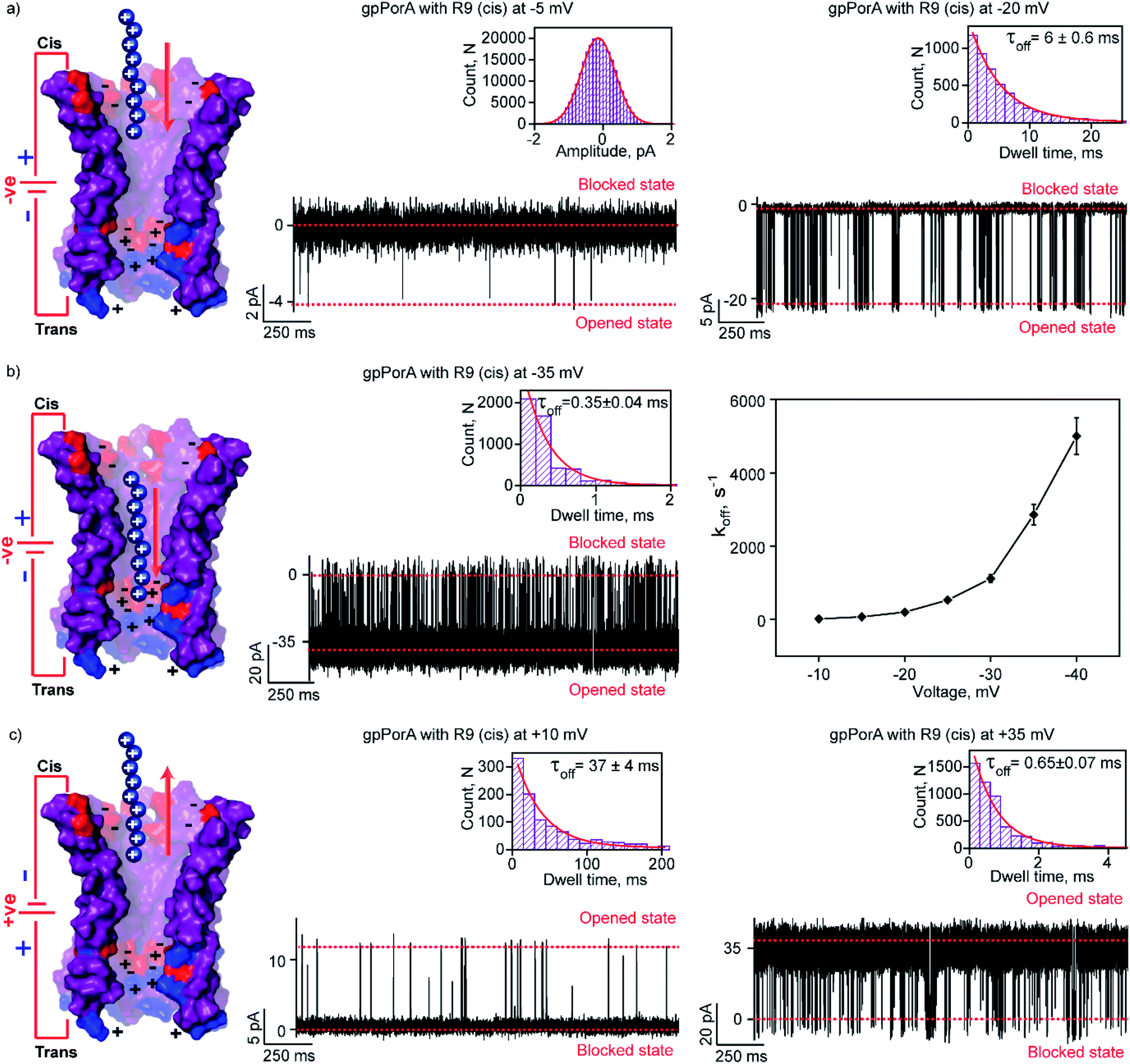 | ||
| Fig. 3 Interaction of nonaarginine with the cis side of the gpPorA. (a) Electrical recordings showing nonaarginine (R9) interaction with single gpPorA (0.5 μM, cis) at −5 mV and −20 mV. (b) Electrical recordings showing R9 interaction with single gpPorA (0.5 μM, cis) at −35 mV and a plot of koffversus the applied voltage (mean values (±s.d.) from three independent experiments are shown). (c) Electrical recordings showing R9 interaction with single gpPorA (0.5 μM, cis) at +10 mV, +35 mV against electrophoretic mobility (mean values (±s.d.) from three independent experiments are shown). Inset, recording at expanded time scale and the corresponding τoff dwell time histogram. Model showing the interaction of R9 with the cis side of the pores. Electrolyte: 0.15 M KCl, 10 mM HEPES, pH 7.4. The current signals (a) were digitally filtered at 1 kHz, (b) −35 mV at 5 kHz and (c) +10 mV and +35 mV at 1 kHz and 5 kHz using an 8-pole Bessel digital filter. | ||
We attribute this to densely packed negatively charged residues along the cis side of the pore lumen that acts as a predominant affinity site facilitating peptide-binding in spite of electrostatic repulsion. However, at higher voltages (above +40 mV), less frequent ion current blockages were observed with a rapid increase in koff as prominent electrostatic repulsive force inhibits R9 entry into the pores, suggesting disassociation of peptides (Fig. S8†). These data confirm the translocation of charged peptides guided by the voltage-controlled electrophoretic driving force that allowed us to distinguish binding from translocation. We conclude that R9 effectively binds to the pores with a high affinity (KDcis, 9.2 × 10−7 M at −10 mV) due to its higher charge than R6 (KDcis, 51.93 × 10−7 M at −10 mV) and R4. Following this, we examined the interaction of R9 with the trans side of the same pores (the same orientation in the membrane) after perfusion of peptides from the cis side to deduce the asymmetry in peptide binding kinetics (Fig. 4). The addition of 0.5 μM R9 to the trans side resulted in strong ion current blockages (100% block) with an increase in the kon and koff from +10 mV to +35 mV, establishing effective electrostatic peptide binding and translocation (Fig. 4a, b, S9 and Table S2†). Similar to the cis side, faster translocation of peptides at higher voltages and ion current blockages against the electrophoretic pulling was observed depending on the magnitude and polarity of the voltage (Fig. S9†). Here we show the bidirectional voltage-dependent translocation of peptides through the pores considering an increase in the koff with the voltage for both the cis and trans side R9 addition.
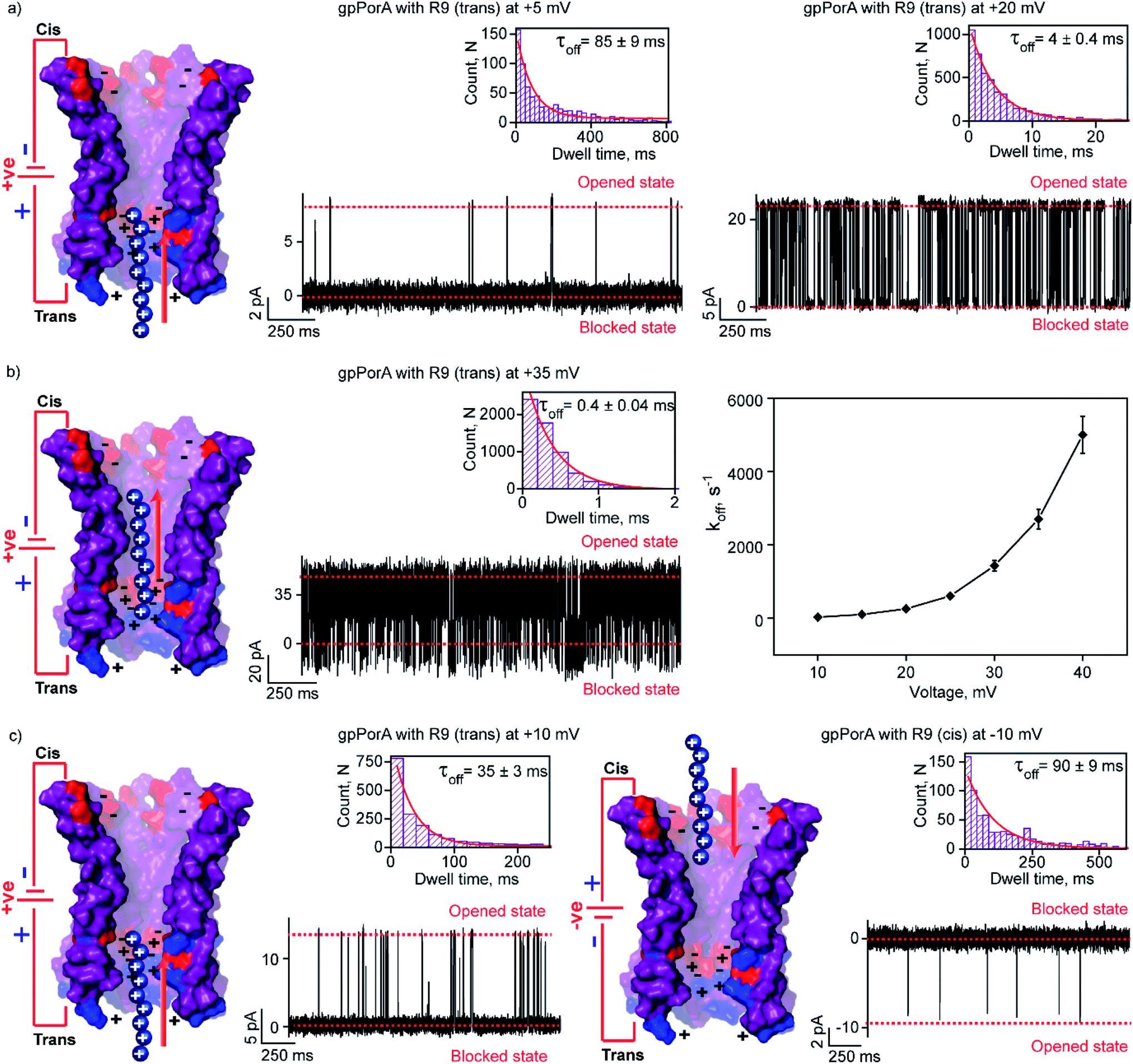 | ||
| Fig. 4 Interaction of nonaaarginine with the trans and cis side of the gpPorA. (a) Electrical recordings showing R9 interaction with single gpPorA (0.5 μM, trans) at +5 mV and +20 mV. (b) Electrical recordings showing R9 interaction with single pPorA (0.5 μM, trans) at +35 mV and a plot of koffversus the applied voltage (mean values (±s.d.) from three independent experiments are shown). (c) Electrical recordings showing R9 interaction with single gpPorA (0.5 μM, trans) at +10 mV and (0.5 μM, cis) at −10 mV inset, recording at expanded time scale and the corresponding τoff dwell time histogram. Model showing the interaction of R9 with the trans side and cis side of the pores is shown. Electrolyte: 0.15 M KCl, 10 mM HEPES, pH 7.4. The current signals (a and c) were digitally filtered at 1 kHz, (b) +35 mV at and 7 kHz using an 8-pole Bessel digital filter. | ||
Interestingly, R9 blocked the cis side of the pores more effectively than the trans side at specific voltages (Fig. 4c, S10 and Table S2†). For example, R9 blocked the cis side with τoff of 90 ± 9 ms (n = 3) at −10 mV, whereas it blocked the trans side with τoff of 35 ± 3 ms (n = 3) +10 mV confirming well-defined asymmetry in the peptide-binding kinetics (Fig. 4c, S10 and Table S2†). Subsequently, this asymmetry in R9 binding kinetics revealed an asymmetrical charge pattern in the pores. We propose that the cis side of the pores is associated explicitly with more densely packed negatively charged residues than the trans side resulting in the prominent cis side binding of R9. Accordingly, we deduced the orientation of the pores in the lipid membrane based on the asymmetry in the peptide-binding kinetics as peptides are added to either the cis or the trans side of the same pores (n = 25). We suggest that the gpPorA added to the cis side of the bilayer most likely results in the pore insertion with the densely packed negatively charged residues exposed towards the cis side resulting in effective R9 blocking. In competitive blocking measurements, the binding of R4 and R9 was discriminated based on τoff (Fig. S11†). This suggests the possibility of using these synthetic pores for complex peptide characterization and in related technologies.
Electrostatic basis of peptide translocation
Here, we examined the effect of salt and pH of electrolyte solution to define the electrostatic basis of charged peptide translocation through gpPorA pores. At first, we examined specifically the interaction of R9 with the gpPorA in 1 M KCl electrolyte buffer and compared the binding affinity with the one obtained in 0.15 M KCl (Fig. 5a and 3). The cis side addition of R9 resulted in ion current blockages with τoff of 0.14 ± 0.02 ms (n = 3) at −10 mV in 1 M KCl, indicating weak electrostatic peptide binding (Fig. 5a). An increase in the voltage to −20 mV, −30 mV and −50 mV resulted in less frequent to unresolved ion current blockages (Fig. 5a and S12†). A similar voltage-dependent peptide binding trend was observed for the trans side R9 addition (Fig. S12†). The nature of peptide blockages in 1 M KCl (∼70% at −10 mV) can be readily distinguished from that of blockages in 0.15 M KCl (100% block). These data indicate that R9 binds to the pores with low affinity in 1 M KCl compared to 0.15 M KCl, establishing strong electrostatic contribution in peptide binding with the pores due to charge screening.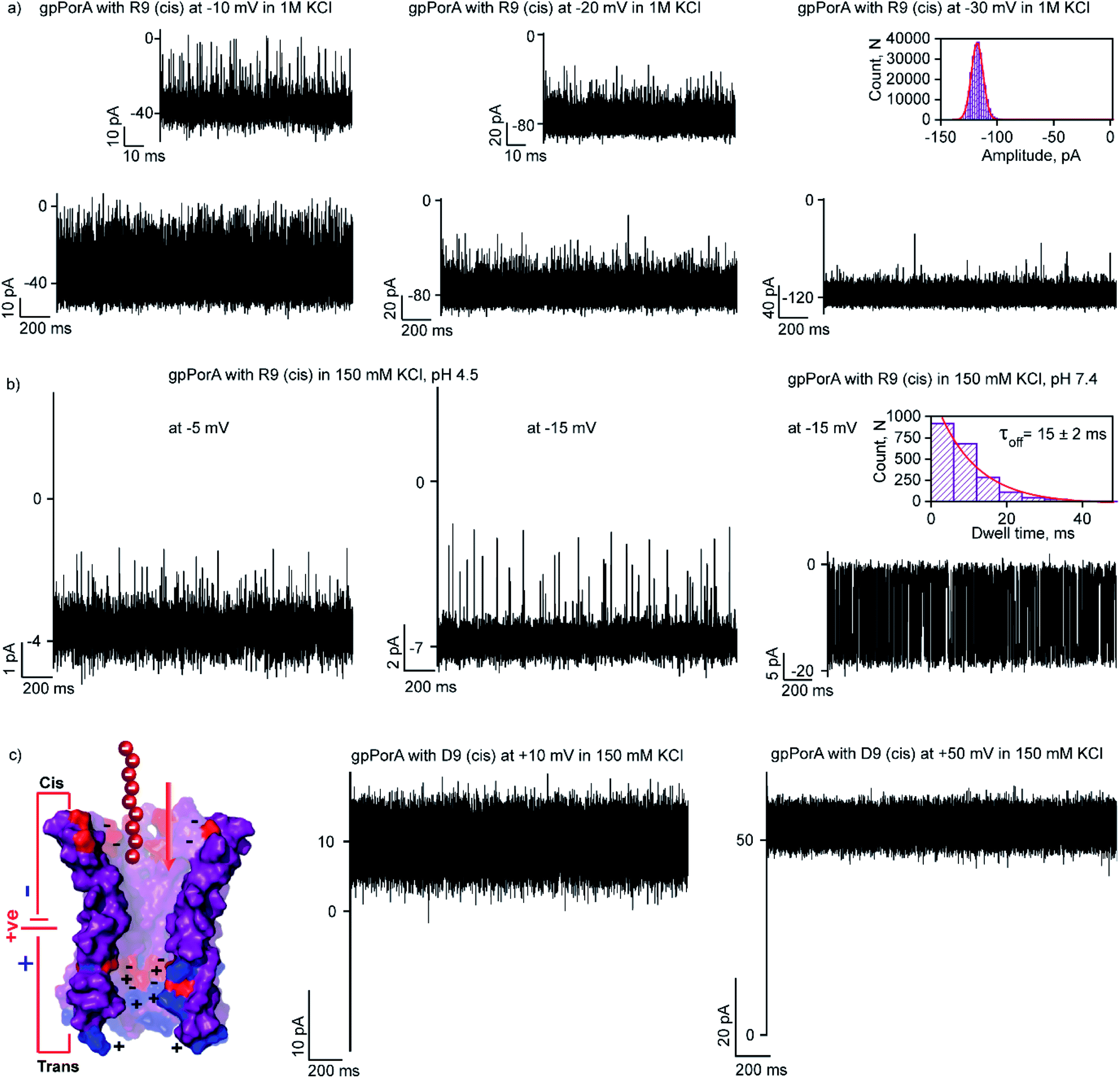 | ||
| Fig. 5 Electrostatic contribution in peptide binding and translocation through gpPorA. (a) Electrical recordings showing R9 interaction with single gpPorA (100 μM, cis) at −10 mV, −20 mV and −30 mV in 1 M KCl. (b) Electrical recordings showing R9 interaction with single gpPorA (0.5 μM, cis) at −5 mV and −15 mV (pH 4.5) and −15 mV (pH 7.4) in 0.15 M KCl. (c) Model showing the pPorA charge pattern with D9. Electrical recordings showing anionic D9 interaction with gpPorA (100 μM, cis) at +10 mV and +50 mV. Inset, recording at the expanded time scale and the corresponding τoff dwell time histogram. Electrolyte: 0.15 M KCl/1 M KCl, 10 mM HEPES, pH 7.4 and citrate buffer, pH 4.5. The current signals (a and c) were filtered at 10 kHz and sampled at 50 kHz and (b) were digitally filtered at 1 kHz and 2 kHz using an 8-pole Bessel digital filter. | ||
Next, we examined the effect of the pH of the electrolyte buffer on the interaction of R9 with the gpPorA. Specifically, we investigated R9 binding with the pores at pH 4.5 and 7.4 in 0.15 M KCl at different voltages. At lower pH (4.5) the pores exhibited a smaller unitary conductance of 0.6 ± 0.1 nS (n = 3) in 0.15 M KCl as acidic pH facilitated the decrease in the pore conductance (Fig. 5b). Interestingly, at pH 4.5, short-lived, less frequent ion current blockages were detected at −5 mV, −10 mV and −15 mV on the cis side addition of the peptide (0.5 μM) pores (Fig. 5b and S13†). Similarly, at higher voltages (−20 mV and −25 mV) no prominent blockage events were produced, indicating negligible interaction of R9 with the pores (Fig. S13†). This suggests weak peptide binding at lower pH irrespective of different voltages, most likely due to the reduced cation selectivity of the pores. Further, increasing the pH of the electrolyte solution to 7.4 resulted in an increased unitary conductance of 1.2 ± 0.2 nS (n = 25) and voltage-dependent well-defined ion current blockages (Fig. 5b and S13†). These data demonstrate pH-dependent R9 binding with the pores, indicating the charge selectivity for the cationic peptides. We propose that due to the presence of ionizable amino acids in the pore lumen, the overall charge varies with the pH of the electrolyte solution. For example, the pores are strongly cation-selective at pH 7.4 with an isoelectric point of pH ∼4.5, leading to strong R9 binding. In contrast, at pH 4.5, the pores tend to be less cation-selective as the acidic residues are protonated, resulting in weak interaction of R9 with the pores. Importantly, we tuned the selectivity of the pores to charged peptides by simply modulating the pH of the electrolyte solution. Altogether, we show how different pH and salt conditions modulate the charge distribution and subsequent electrostatic interaction of peptides with the pores. To validate the charge selectivity of the pores, we studied the interaction of negatively charged nonaaspartate (D9) with cation-selective gpPorA in low salt electrolyte buffer. The addition of D9 (100 μM) to the cis side of the gpPorA did not produce any ion current blockages at different peptide concentrations and voltages. Following this, we added 1 μM of positively charged R9 to the cis side of the same pores resulting in voltage-dependent intense blockage events. This is consistent with the negatively charged residues in the pore lumen hindering anionic D9 binding while facilitating cationic R9 interaction suggesting electric field-driven charge-selective peptide translocation (Fig. 5c and S14†). These data suggest high selectivity and specificity of synthetic alpha-helical pores for charged peptides compared to natural protein pores.11,12,45
Interaction of charged peptides with mutant pores and molecular basis of translocation
Here, we studied the interaction of charged peptides with a pPorA variant (pPorA-K24, lysine at 24th) that formed slightly anion-selective pores in the lipid membrane (PK+/PCl− as ∼1:3) (Fig. S15†).43 This particular peptide variant did not form self-assembled oligomers in SDS PAGE and the pores are formed by direct pPorA addition into the lipid bilayer (Fig. S15†). Notably, this pPorA variant exhibited a unitary conductance of ∼0.9 nS in 0.15 M KCl (n = 25), slightly lower than that of cation-selective gpPorA. The cationic R6 and R9 added to these pores did not produce any ion current blockages at different voltages, even at very high peptide concentrations (50 μM). This indicates that the additional positively charged residues in the mutant pores inhibit the electrostatic capture of cationic peptides resulting in no ion current blockage events (Fig. S15†). Therefore, we investigated the interaction of anionic D9 with this pPorA variant. The addition of 0.5 μM D9 to the cis side of the bilayer did not produce defined ion current blockages under different conditions. However, increasing the D9 concentration to 10 μM resulted in a very short, less frequent ion current blockage, specifically at higher positive voltages of +50 mV as peptides are electrophoretically pulled into the pores (Fig. 6a). These data indicate weak D9 peptide binding with the pores as blockage events were not correctly resolved to determine the kinetics. As expected, no ion current blockages were observed at negative voltages confirming the voltage-dependent selective translocation of peptides through the pPorA pores.
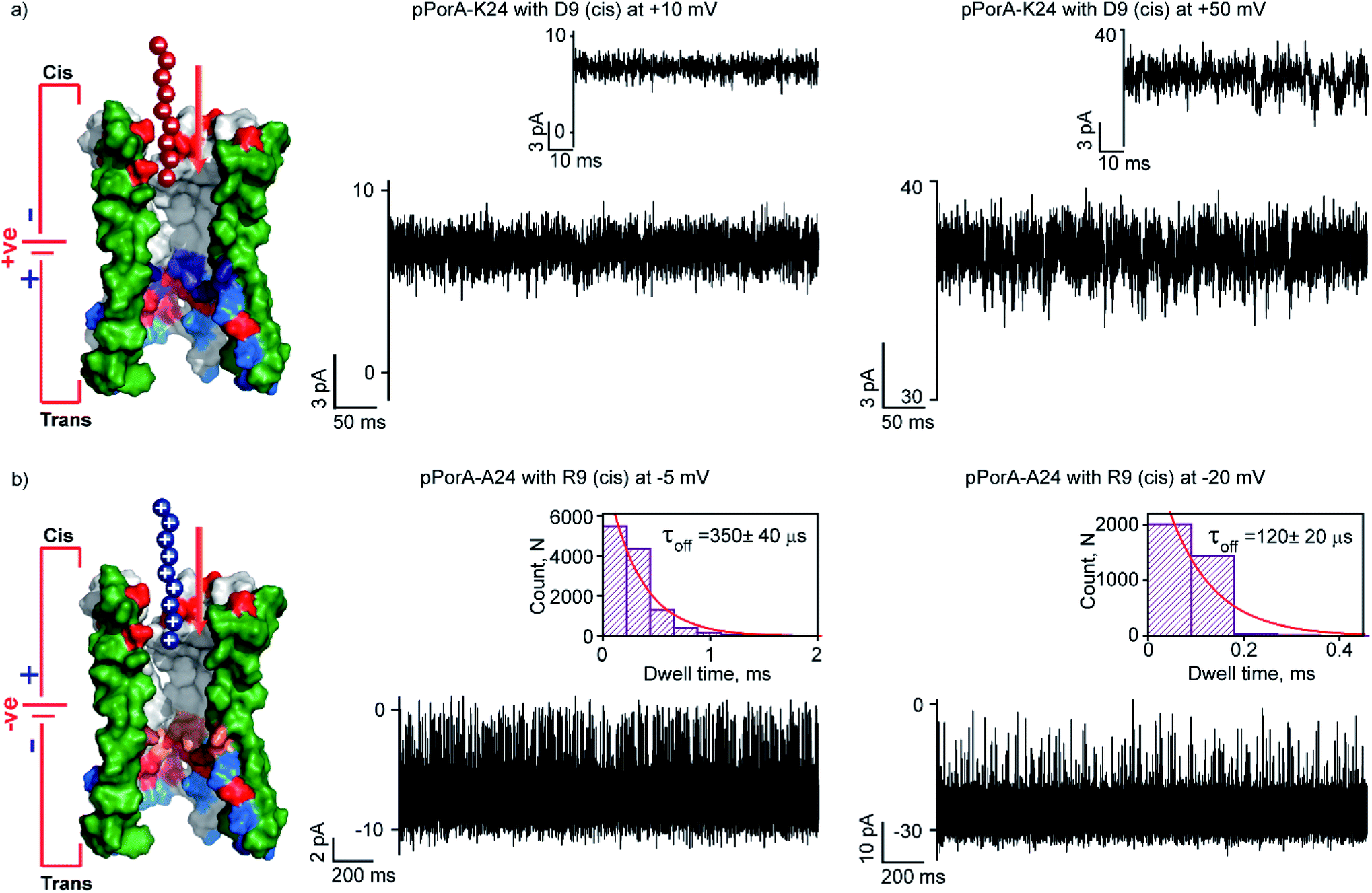 | ||
| Fig. 6 Charge selectivity regulating peptide translocation through pPorA variants. (a) Electrical recordings showing D9 interaction with single pPorA-K24 (10 μM, cis) at +10 mV and +50 mV. (b) Electrical recordings showing R9 interaction with single pPorA-A24 (0.5 μM, cis) at −5 mV and −20 mV. Electrolyte: 0.15 M KCl, 10 mM HEPES, pH 7.4. Inset, recording at expanded time scale and the corresponding τoff dwell time histogram. Model showing pPorA charge pattern. The current signals (a) were filtered at 2 kHz and sampled at 10 kHz and (b) −5 mV and −20 mV were digitally filtered at 4 kHz and 7 kHz using an 8-pole Bessel digital filter. | ||
Here, we demonstrate the molecular mechanism of peptide translocation and established charge selectivity of pores using peptide binding in cation-selective gpPorA and anion-selective pPorA-K24, respectively. Notably, the selectivity for cations or anions is observed depending on the surface charge pattern. We suggest that specific negatively charged residues and the overall electrostatic potential of the gpPorA promote effective cationic peptide binding and translocation (Fig. S3†). For example, we show that R9 binds to the cation-selective gpPorA (C24) with high affinity compared to the anionic D9 binding with slightly anion-selective pPorA (K24). We attribute this to abundant negatively charged residues in cationic-selective pores, compared to limited positively charges residues in the slightly anion-selective pores. This study elucidates the charge pattern of alpha-helical pores whose structure is unresolved based on the rate-limiting interactions of cationic and anionic peptides. Most recent peptide translocation studies focused on biological pores to identify individual amino acids as well as the mass and charge of the peptide.16,20,21 The X-ray crystal structure of these pores revealed the internal architecture and charge distribution that allowed precise engineering for sensing and to understand the molecular interactions.3,16,46 Finally, we designed and produced another pPorA variant (pPorA-A24, alanine at the 24th position) that formed slightly less cation-selective pores by the direct addition of pPorA peptides into the lipid bilayer in contrast to self-assembled gpPorA (Fig. 6b and S16†). The addition of 0.5 μM R9 to the cis side of the pores resulted in ion current blockages exclusively at negative voltages from −5 mV to −20 mV. The peptide-binding kinetics obtained at different voltages revealed the charge-dependent successful translocation of the peptides (Fig. 6b and S16†). Remarkably, R9 binds to directly formed pPorA with less affinity compared to self-assembled gpPorA. We propose that this difference in the kinetics of peptide binding could be most likely due to the diverse assembly pathway, less cation-selectivity and different subunit composition of the pPorA pores.
We have shown that a peptide containing cysteine pPorA-C24 self-assembled into stable octamers in SDS PAGE. This gel extracted oligomer (gpPorA) formed large uniform selective pores establishing high specificity in the peptide sequence, unique to alpha-helical structures. Interestingly, the direct addition of pPorA-C24 peptides to the bilayer chamber resulted in stable cation-selective pores identical to gpPorA. Notably, directly formed pores produced subconductance states with varying unitary conductance in the range of 1.0 nS to 1.4 nS in 0.15 M KCl, occasionally indicating the multimeric pore structures. As a result, R6 and R9 produced a combination of strong and weak ion current blockages through these subconductance pores (Fig. S17 and S18†). This suggests that gpPorA is more favorable to use as a selective single-molecule sensor as they are precisely made up of an equal number of subunits and produced consistent blocking with charged peptides.11,12,37,47–53 We show that this new class of synthetic self-assembled alpha-helical nanostructures are advantageous compared to biological and DNA pores in chemical synthesis, purification, unusually large conductance, selectivity and specificity for molecules.3,22,26 Additionally, they can be easily subjected to chemical and charge modification at specific residues. Hence, these pores of defined geometry and charge can be a promising tool for the stochastic sensing of complex peptides and protein fragments in a label-free manner.16,17,21,48,54,55 We emphasize that these nanopores are made by chemical synthesis and therefore, specialized functional pores of unique sequence and structure can be assembled for applications in nanopore technology for proteomics analysis.3,5,22
Conclusion
Here, we present synthetic monodisperse alpha-helical peptide pores for selective single-molecule sensing of differently charged cationic and anionic peptides. We quantified the peptide binding kinetics and demonstrated the voltage, pH and salt-dependent translocation of peptides through these pores at single-molecule resolution. Furthermore, we define the molecular mechanism of translocation dominated by the charge selectivity and electrostatics. We propose that alpha-helical peptide pores offer superior chemical tunability for selective sensing and sequencing. The ability to construct such pores of desirable geometry and charge composition would expand the scope of sensing complex biomacromolecules. We emphasize that understanding the molecular transport process across these pores will have important implications in synthetic chemical biology to design functionally versatile pores.Experimental section
Single-channel electrical recordings
Single-channel recordings were carried out using planar lipid bilayers of 1,2-diphytanoyl-sn-glycero-3-phosphocholine (DPhPC, Avanti Polar Lipids).56 Bilayers are formed across an aperture (∼70 μm in diameter) in a 25 μm thick polytetrafluoroethylene (Teflon) film (Goodfellow, Cambridge), which separated the apparatus into cis and trans compartments (500 μL each). Bilayers were formed by first prepainting the aperture with hexadecane in n-pentane (1 μL, 5 mg mL−1). Both compartments were filled with the low salt electrolyte buffer (0.15 M KCl, 10 mM HEPES, pH 7.4 and citrate buffer, pH citrate buffer 4.5). A few experiments were carried out using high salt electrolyte buffer on the requirement (1 M KCl, 10 mM HEPES, pH 7.4). Next, DPhPC dissolved in n-pentane (2 μL, 5 mg mL−1) was added to both sides of the compartments and a bilayer was formed when the electrolyte was raised, bringing the two lipid surface monolayers together at the aperture. The pPorA pores were formed by adding a solution of the gel extracted peptide (cis side) in (0.5 μL) under an applied potential of +100 mV. The cis compartment was connected to the grounded electrode and the trans compartment was attached to the working electrode. A potential difference was applied through a pair of Ag/AgCl electrodes, set in 3% agarose containing (3 M KCl). The current was amplified using an Axopatch 200B amplifier, digitized with a Digidata 1550B and recorded with the pClamp 11 acquisition software (Molecular Devices, CA) with a low-pass filter frequency of 10 kHz and a sampling frequency of 50 kHz. A few current signals were filtered using an 8-pole Bessel digital filter to resolve peptide blocking events accurately on the requirement. The data were analyzed with pClamp 11 (Molecular Devices, CA) and Origin 9.0. The average residence time of blocking (τoff) and the number of blockage events divided by peptide concentration (τon) is calculated in a single-channel analysis.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the ‘Innovative Young Biotechnologist Award’ of the Department of Biotechnology, Government of India (BT/010/IYBA/2016/06). KRM acknowledges the ‘Ramalingaswami Re-entry fellowship’ of Department of Biotechnology, Government of India (BT/RLF/Re-entry/49/2014) and the ‘Early Career Research Award’ of Science & Engineering Research Board (SERB), Department of Science and Technology, Government of India (ECR/2017/000533) for supporting this work. KRM thanks Prof. M. Radhakrishna Pillai, the Director, RGCB, for help and support to initiate peptide pores work. SKR is supported by the Senior Research Fellowship from the Indian Council of Medical Research (5/3/8/9/ITR-F/2020) and NP is supported by the University Grants Commission, Government of India.References
- J. J. Kasianowicz, E. Brandin, D. Branton and D. W. Deamer, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 13770–13773 CrossRef CAS.
- H. Bayley and P. S. Cremer, Nature, 2001, 413, 226–230 CrossRef CAS.
- M. Ayub and H. Bayley, Curr. Opin. Chem. Biol., 2016, 34, 117–126 CrossRef CAS.
- J. J. Kasianowicz, A. K. Balijepalli, J. Ettedgui, J. H. Forstater, H. Wang, H. Zhang and J. W. Robertson, Biochim. Biophys. Acta, 2016, 1858, 593–606 CrossRef CAS.
- S. Wang, Z. Zhao, F. Haque and P. Guo, Curr. Opin. Biotechnol., 2018, 51, 80–89 CrossRef CAS.
- G. Baaken, I. Halimeh, L. Bacri, J. Pelta, A. Oukhaled and J. C. Behrends, ACS Nano, 2015, 9, 6443–6449 CrossRef CAS.
- H. Bayley, Clin. Chem., 2015, 61, 25–31 CrossRef CAS.
- D. Deamer, M. Akeson and D. Branton, Nat. Biotechnol., 2016, 34, 518–524 CrossRef CAS.
- I. M. Derrington, T. Z. Butler, M. D. Collins, E. Manrao, M. Pavlenok, M. Niederweis and J. H. Gundlach, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 16060–16065 CrossRef.
- T. C. Sutherland, Y. T. Long, R. I. Stefureac, I. Bediako-Amoa, H. B. Kraatz and J. S. Lee, Nano Lett., 2004, 4, 1273–1277 CrossRef CAS.
- L. Movileanu, J. P. Schmittschmitt, J. M. Scholtz and H. Bayley, Biophys. J., 2005, 89, 1030–1045 CrossRef CAS.
- R. Stefureac, Y. T. Long, H. B. Kraatz, P. Howard and J. S. Lee, Biochemistry, 2006, 45, 9172–9179 CrossRef CAS.
- R. Stefureac, L. Waldner, P. Howard and J. S. Lee, Small, 2008, 4, 59–63 CrossRef CAS.
- M. Soskine, A. Biesemans, B. Moeyaert, S. Cheley, H. Bayley and G. Maglia, Nano Lett., 2012, 12, 4895–4900 CrossRef CAS.
- F. Piguet, H. Ouldali, M. Pastoriza-Gallego, P. Manivet, J. Pelta and A. Oukhaled, Nat. Commun., 2018, 9, 966 CrossRef.
- H. Ouldali, K. Sarthak, T. Ensslen, F. Piguet, P. Manivet, J. Pelta, J. C. Behrends, A. Aksimentiev and A. Oukhaled, Nat. Biotechnol., 2020, 38, 176–181 CrossRef CAS.
- Y. L. Ying and Y. T. Long, J. Am. Chem. Soc., 2019, 141, 15720–15729 CrossRef CAS.
- B. Cressiot, H. Ouldali, M. Pastoriza-Gallego, L. Bacri, F. G. Van der Goot and J. Pelta, ACS Sens., 2019, 4, 530–548 CrossRef CAS.
- M. Y. Li, Y. Q. Wang, Y. L. Ying and Y. T. Long, Chem. Sci., 2019, 10, 10400–10404 RSC.
- G. Huang, K. Willems, M. Soskine, C. Wloka and G. Maglia, Nat. Commun., 2017, 8, 935 CrossRef.
- G. Huang, A. Voet and G. Maglia, Nat. Commun., 2019, 10, 835 CrossRef.
- S. Howorka, Nat. Nanotechnol., 2017, 12, 619–630 CrossRef CAS.
- B. A. Cornell, V. L. Braach-Maksvytis, L. G. King, P. D. Osman, B. Raguse, L. Wieczorek and R. J. Pace, Nature, 1997, 387, 580–583 CrossRef CAS.
- M. Mayer, V. Semetey, I. Gitlin, J. Yang and G. M. Whitesides, J. Am. Chem. Soc., 2008, 130, 1453–1465 CrossRef CAS.
- M. X. Macrae, S. Blake, X. Jiang, R. Capone, D. J. Estes, M. Mayer and J. Yang, ACS Nano, 2009, 3, 3567–3580 CrossRef CAS.
- R. P. Thomsen, M. G. Malle, A. H. Okholm, S. Krishnan, S. S. Bohr, R. S. Sorensen, O. Ries, S. Vogel, F. C. Simmel, N. S. Hatzakis and J. Kjems, Nat. Commun., 2019, 10, 5655 CrossRef.
- E. Spruijt, S. E. Tusk and H. Bayley, Nat. Nanotechnol., 2018, 13, 739–745 CrossRef CAS.
- U. F. Keyser, Nat. Nanotechnol., 2016, 11, 106–108 CrossRef CAS.
- M. Langecker, V. Arnaut, T. G. Martin, J. List, S. Renner, M. Mayer, H. Dietz and F. C. Simmel, Science, 2012, 338, 932–936 CrossRef CAS.
- N. A. Bell, C. R. Engst, M. Ablay, G. Divitini, C. Ducati, T. Liedl and U. F. Keyser, Nano Lett., 2012, 12, 512–517 CrossRef CAS.
- R. Wei, T. G. Martin, U. Rant and H. Dietz, Angew. Chem., 2012, 51, 4864–4867 CrossRef CAS.
- J. R. Burns, E. Stulz and S. Howorka, Nano Lett., 2013, 13, 2351–2356 CrossRef CAS.
- K. Gopfrich, C. Y. Li, I. Mames, S. P. Bhamidimarri, M. Ricci, J. Yoo, A. Mames, A. Ohmann, M. Winterhalter, E. Stulz, A. Aksimentiev and U. F. Keyser, Nano Lett., 2016, 16, 4665–4669 CrossRef CAS.
- N. H. Joh, T. Wang, M. P. Bhate, R. Acharya, Y. Wu, M. Grabe, M. Hong, G. Grigoryan and W. F. DeGrado, Science, 2014, 346, 1520–1524 CrossRef CAS.
- D. N. Woolfson, G. J. Bartlett, A. J. Burton, J. W. Heal, A. Niitsu, A. R. Thomson and C. W. Wood, Curr. Opin. Struct. Biol., 2015, 33, 16–26 CrossRef CAS.
- Z. L. Hu, M. Y. Li, S. C. Liu, Y. L. Ying and Y. T. Long, Chem. Sci., 2019, 10, 354–358 RSC.
- X. Lin, A. P. Ivanov and J. B. Edel, Chem. Sci., 2017, 8, 3905–3912 RSC.
- L. Q. Gu, M. Dalla Serra, J. B. Vincent, G. Vigh, S. Cheley, O. Braha and H. Bayley, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 3959–3964 CrossRef CAS.
- K. R. Mahendran, A. Niitsu, L. Kong, A. R. Thomson, R. B. Sessions, D. N. Woolfson and H. Bayley, Nat. Chem., 2017, 9, 411–419 CrossRef CAS.
- C. Xu, P. Lu, T. M. Gamal El-Din, X. Y. Pei, M. C. Johnson, A. Uyeda, M. J. Bick, Q. Xu, D. Jiang, H. Bai, G. Reggiano, Y. Hsia, T. J. Brunette, J. Dou, D. Ma, E. M. Lynch, S. E. Boyken, P. S. Huang, L. Stewart, F. DiMaio, J. M. Kollman, B. F. Luisi, T. Matsuura, W. A. Catterall and D. Baker, Nature, 2020, 585, 129–134 CrossRef.
- D. N. Woolfson, Subcell. Biochem., 2017, 82, 35–61 CAS.
- K. T. O'Neil and W. F. DeGrado, Science, 1990, 250, 646–651 CrossRef.
- R. S. Krishnan, R. Satheesan, N. Puthumadathil, K. S. Kumar, P. Jayasree and K. R. Mahendran, J. Am. Chem. Soc., 2019, 141, 2949–2959 CrossRef.
- N. Abdali, E. Barth, A. Norouzy, R. Schulz, W. M. Nau, U. Kleinekathofer, A. Tauch and R. Benz, PLoS One, 2013, 8, e75651 CrossRef CAS.
- P. R. Singh, I. Barcena-Uribarri, N. Modi, U. Kleinekathofer, R. Benz, M. Winterhalter and K. R. Mahendran, ACS Nano, 2012, 6, 10699–10707 CrossRef CAS.
- H. Wang, J. Ettedgui, J. Forstater, J. W. F. Robertson, J. E. Reiner, H. Zhang, S. Chen and J. J. Kasianowicz, ACS Sens., 2018, 3, 251–263 CrossRef CAS.
- L. Restrepo-Perez, G. Huang, P. R. Bohlander, N. Worp, R. Eelkema, G. Maglia, C. Joo and C. Dekker, ACS Nano, 2019, 13, 13668–13676 CrossRef CAS.
- A. K. Thakur and L. Movileanu, Nat. Biotechnol., 2018, 37, 96–101 CrossRef.
- J. Houghtaling, C. Ying, O. M. Eggenberger, A. Fennouri, S. Nandivada, M. Acharjee, J. Li, A. R. Hall and M. Mayer, ACS Nano, 2019, 13, 5231–5242 CrossRef CAS.
- P. Waduge, R. Hu, P. Bandarkar, H. Yamazaki, B. Cressiot, Q. Zhao, P. C. Whitford and M. Wanunu, ACS Nano, 2017, 11, 5706–5716 CrossRef CAS.
- Z. Ji, X. Kang, S. Wang and P. Guo, Biomaterials, 2018, 182, 227–233 CrossRef CAS.
- J. Nivala, D. B. Marks and M. Akeson, Nat. Biotechnol., 2013, 31, 247–250 CrossRef CAS.
- S. Krishnan, D. Ziegler, V. Arnaut, T. G. Martin, K. Kapsner, K. Henneberg, A. R. Bausch, H. Dietz and F. C. Simmel, Nat. Commun., 2016, 7, 12787 CrossRef CAS.
- A. Asandei, I. Schiopu, M. Chinappi, C. H. Seo, Y. Park and T. Luchian, ACS Appl. Mater. Interfaces, 2016, 8, 13166–13179 CrossRef CAS.
- R. J. Yu, S. M. Lu, S. W. Xu, Y. J. Li, Q. Xu, Y. L. Ying and Y. T. Long, Chem. Sci., 2019, 10, 10728–10732 RSC.
- T. Gutsmann, T. Heimburg, U. Keyser, K. R. Mahendran and M. Winterhalter, Nat. Protoc., 2015, 10, 188–198 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Materials, text, table and Fig. S1–S18. See DOI: 10.1039/d0sc04856a |
| ‡ Contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |