
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDendritic fibrous nanosilica-supported dendritic IL/Ru(II) as photocatalysts for the dicarbofunctionalization of styrenes with carbon dioxide and amines
Can Liu*a and
Jalal Rouhi *b
*b
aSchool of Electronic Engineering, Xi'an Shiyou University, Xi'an, 710065, China. E-mail: liucanxk@126.com
bFaculty of Physics, University of Tabriz, Tabriz 51566, Iran. E-mail: jalalrouhi@gmail.com
First published on 9th March 2021
Abstract
The effectual utilization of heterogeneous catalysts from nano sources through chemical moderation for the α-aminomethylcarboxylation of alkenes with carbon dioxide and amines is an attractive area to study. Dendritic fibrous nanosilica (DFNS) is a cost-effective, resistant, plenteous, and reproducible source with dandelion-like fibrous anatomy. The present paper is a report on an easy method to provide a family of new DFNS-supported dendritic imidazolium IL/Ru(II) heterogeneous catalysts DFNS/IL/Ru (1–3) with high ionic density from DFNS. A positive dendritic effect was perceived in the chemical stabilization performance of CO2. DFNS/IL/Ru(II) was appropriately identified by UV-vis spectroscopy, XPS, SEM, TEM, FT-IR spectroscopy, and TGA. It was discovered that DFNS/IL/Ru(II) has high catalytic activity for the synthesis of quinoline-2-one through the annulation of ortho-heteroaryl anilines and CO2. DFNS/IL/Ru (3) could be reutilized ten continuous times with no notable reduction in the catalytic activity. Notably, the coveted quinoline-2-one was prepared on a multi-gram scale by deploying DFNS/IL/Ru (3) as a green heterogeneous catalyst. Owing to the attendance of the zwitterionic liquid functional groups on the exterior layer of the bio-based DFNS/IL/Ru (3) catalyst, DFNS/IL/Ru (3) expressed the highest catalytic activity. This approach provides highly functional γ-amino acids in proper yields with great selective power. This paper announces the first nanocatalyst for this transformation, comprising the DFNS-supported Ru N-heterocyclic carbine complex.
Introduction
γ-Amino acids are worthwhile composites and used in pharmacology with broad biological applications, including antagonists and agonists of mammalian central nervous system.1,2 Therefore, considerable efforts have been made to synthesize γ-amino acids,3,4 including Michael addition of carbonyl compounds to nitroethylenes,5–9 or conjugate addition of nitroalkanes to α,β-unsaturated carbonyl compounds10,11 to provide γ-amino acids. Lately, Ye et al. announced the γ-amination of α,β-unsaturated acyl chlorides with azodicarboxylates as well as the opening of the dihydropyridazinone reducing ring to prepare γ-amino acids.12,13 However, these synthetic approaches are associated with disadvantages such as restricted substrate area, numerous stages, and/or severe reaction circumstances. It was hypothesized that the concurrent combination of CO2, α-aminoalkyl, and alkenes through the dicarbofunctionalization of alkenes is a perfect pathway for the delivery of γ-amino acids.The visible-light-mediated photoredox catalysis provides an operational method for achieving open-shell radical species,14–17 resulting in innovative approaches for the performance of alkenes.18–20 Lately, the photoredox-promoted single-electron oxidation and subsequent deprotonation of amines to produce α-aminoalkyl radicals was explained.21–25 Adding α-aminoalkyl radicals to alkenes led to the production of an alkyl radical family that endures a single electron drop to form an electrophile.26–29 It was predicted that the concatenation of CO2 as an electrophile in the reaction blend may lead to the emergence of γ-amino acids.
CO2, as a recyclable and nontoxic carbon source, is used in the synthesis of various compounds.30–36 Among them, the catalytic carboxylation of unsaturated compounds with CO2 has received more interest.37–40 However, compared to the broadly focused hydrocarboxylation of CO2 with alkenes,41–46 the catalytic functional carboxylation of CO2 with alkenes is not widely reported since it is not easy to perform. Lately, Martin et al. demonstrated the photocatalytic process under visible light between carbon dioxide and alkylcarboxylic acid.47–51 Considering the possibility of these alterations and our continuing desire for the carboxylation of CO2 with alkenes for the effectual dicarbofunctionalization reaction, herein, the photoredox-catalyzed α-aminomethylcarboxylation of alkenes with CO2 and amines is reported. This durable, common, and practical method demonstrates an uncommon sample of the redox-neutral dicarbofunctionalization of alkenes to produce highly efficient γ-amino acids under mild reaction circumstances.
Ru complexes are currently being investigated regarding their attractive anatomical, biological, catalytic, and electrochemical characteristics.52–56 These studies include research on ruthenium complexes possessing diimino tetradentate Schiff bases, such as salophen and salen ligands.57,58 It has recently been discovered that several Ru/salen ligands are active catalysts in various chemical alterations.59,60 In particular, Ru macrocyclic complexes, which are resistant to side demetallation, illustrate reversible electrochemistry and prepare an appropriate framework for mechanistic studies of proton-coupled multielectron transfer reactions.61,62 For arranging rational conversions catalyzed by Ru/salen compounds, knowledge of the anatomical and redox properties is required.63
Dendrimers have been extensively analyzed due to their great fork 3D anatomies, specific dendritic effects, and numerous circumferential groups.64,65 The immobilization of dendrimer catalysts on support materials is a new field that has attracted the attention of many chemists due to the facile recoverability.66–68 ILs have a special place in green chemistry because of their unique attributes such as light vapor pressure and slight ignition. The adaptability of the anatomy and characteristics is a significant feature of ILs. By adding various functional groups to anions or cations with various anatomies, it is possible to modify the physical and chemical characteristics of ILs. Dendritic ILs (DILs), with a combination of dendrimer and IL characteristics, have been broadly utilized in catalysis, as transporters, and in the adsorption of heavy metal cations.69,70
This study reports the production of DFNS-supported dendritic IL/Ru(II) catalysts DFNS/IL/Ru (1), DFNS/IL/Ru (2), and DFNS/IL/Ru (3) with high ionic density and various generations. Results show that the dendritic IL/Ru catalysts DFNS/IL/Ru (1), DFNS/IL/Ru (2), and DFNS/IL/Ru (3) positively influence the efficiency of the α-aminomethylcarboxylation of alkenes with amines and CO2.
Experimental section
The universal approach to provide DFNS NPs
First, 5.0 g of tetraethyl orthosilicate was dissolved in a blend of 3.0 mL of 1-pentanol and 60 mL of cyclohexane. Then, a blend of urea (1.2 g), cetylpyridinium bromide (2 g), and H2O (60 mL) was released into it. The obtained blend was constantly mixed at room temperature for 45 minutes. Next, it was put in a Teflon-sealed hydrothermal reactor and heated at 130 °C (4 hours). The generated silica was centrifuged, cleansed with a blend of acetone and deionized water, and oven-dried. Then, it was calcined in air at 600 °C for 4 hours.The universal approach to provide DFNS/APTS NPs
DFNS (3.0 g) was released into a solution of NaOH (50 mL, 0.5 M), and the blend was refluxed for 3 hours to enhance the hydrophilicity. Then, resulting nanoparticles were cleansed several times with H2O to neutralize the pH of the deionized H2O and then vacuum-dried at 70 °C for 6 hours. Next, 3-aminopropyltrimethoxysilane (APTS) was supported on the DFNS exterior layer to synthesize amino-modified DFNS/APTS. Then, the blend of activated DFNS (3 g) and 3-aminopropyltrimethoxysilane (APTS) (6 mL) in 100 mL ethanol was mixed at 100 °C for 17 hours. In the last step, DFNS/APTS was filtered, cleansed twice with water, and dried at 90 °C for 12 hours.The universal approach to provide DFNS/IL (1)
DFNS/APTS (2.0 g), diisopropylethylamine (DIPEA) (3.0 mL, 16.4 mmol) and cyanuric chloride (CC) (2.0 g) in 50.0 mL of tetrahydrofuran were mixed at 30 °C for 10 hours. DFNS/APTS/CC-1 fibers were cleansed with ethyl acetate (5 mL × 4) and dried for 10 hours. Then, 3.0 g of DFNS/APTS/CC-1 was combined with 1-imidazole (3.0 g), tetrahydrofuran (50.0 mL) was released into it, and the blend was mixed for 20 hours at 70 °C. DFNS/APTS/imidazole (80 mg) was released into deionized water:EtOH blend (5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 v/v, 80 mL) containing 1,4-butanesultone (3.0 g), and the reaction was performed for 2.5 hours at r.t. Upon separating the solvent, the final blend was filtered. Then, the filtrate was vacuum-dried at 50 °C.
:5 v/v, 80 mL) containing 1,4-butanesultone (3.0 g), and the reaction was performed for 2.5 hours at r.t. Upon separating the solvent, the final blend was filtered. Then, the filtrate was vacuum-dried at 50 °C.
The universal approach to provide DFNS/IL (2) and DFNS/IL (3)
DFNS/APTS/CC-1 (2.0 g), ethylenediamine (3.5 mL), tetrahydrofuran (50.0 mL), and N,N-diisopropylethylamine (9.0 mL) were combined. The blend was mixed at 90 °C for 20 hours. The obtained product was filtered, cleansed with ethyl acetate (15 mL × 4), and vacuum-dried at 60 °C for 10 hours. The obtained nanoparticles (2.0 g), DIPEA (9.0 mL), cyanuric chloride (CC) (5.0 g), and tetrahydrofuran (50 mL) were mixed at 30 °C for 10 hours. In the next step, DFNS/APTS/CC-2 was easily filtered, DFNS/APTS/CC-2 was cleansed with ethyl acetate (15 mL × 4), and vacuum-dried for 10 hours. Then, 2.0 g of DFNS/APTS/CC-2 was suspended in tetrahydrofuran (50 mL), imidazole (3.0 g) and diisopropylethylamine (17.0 mL) were released into it, and the blend was mixed for 20 hours at 100 °C. Then, it was cleansed with ethyl acetate (15 mL × 4) and vacuum-dried at 50 °C for 10 hours. DFNS/APTS/imidazole (3) was synthesized by the same method as above. DFNS/APTS/imidazole (2) or DFNS/APTS/imidazole (3) (110 mg) was released into H2O:EtOH mixture (1:1 v/v, 50 mL) containing 1,4-butanesultone (3.0 g), and the reaction was performed at r.t. for 3 hours. Upon separating the solvent, the final blend was filtered. Then, the filtrate was vacuum-dried at 50 °C.
The universal approach to provide DFNS/IL/Ru(II)
The ruthenium(II) dimer [{RuCl(μ-Cl)(η6-p-cymene)}2] (0.15 g) was released into a blend of DFNS/IL (0.25 g) in 25 mL of ethanol and then mixed for 5 hours at r.t. Next, the ethanol was removed under decreased pressure and the remnant was cleansed first with Et2O, then with CH2Cl2:Et2O (4:1 v/v), and finally with diethylether:acetone (1:1 v/v) to give an orange solid.
Photocatalytic reactions
DFNS/IL/Ru(II) was cleansed with dimethylformamide to eliminate the unreacted ligands and metal salts and then soaked in MeCN for 5 hours. Then, the specimens were purified and evaporated with CO2. DFNS/IL/Ru(II) (25.0 mg) was released into a blend of TEOA/CH3CN (v/v = 20/1, 40 mL) which has been degassed with CO2 for 5 hours. Catalytic reactions were performed under a CO2 gas at atmospheric pressure, excluding the control experiment under nitrogen. A 22 W compact fluorescent lamp (CFL) was used to irradiate the reaction. The light and the bottle were 10 cm apart. Twenty milliliters of DFNS/IL/Ru(II) (25 mg) was released into a Schlenk tube with a magnetic stir bar. The tube was emptied and filled with CO2 thrice. Then, anhydrous DMSO (1 mL), methyl 4-vinylbenzoate (15 mg), and N,N-dimethylaniline (70 mg) were released into the tube under a positive CO2 environment. The reaction tube was sealed and mixed at r.t. under a 22 W compact fluorescent lamp (CFL) for 24 hours. When the reaction was over, it was watchfully extinguished with 2 M HCl. The blend was drawn out with EtOAc (4 × 5 mL). The composite organic layers were dried over anhydrous Na2SO4. Then, ether:methanol (1:1) (1.0 mL) and TMSCHN2 (200 μL, 1 M in hexane) were released into the reaction blend and stirred at 0 °C for 30 minutes. Upon esterification, the efficiency was distinguished by GC method utilizing n-dodecane (20 μL) as an internal standard.
2-(4-(Methoxycarbonyl)phenyl)-4-(methyl(phenyl)amino)butanoic acid: colorless oil, 1H NMR (CDCl3, 600 MHz) δ 7.99 (d, J = 8.0 Hz, 2H), 7.36 (d, J = 7.9 Hz, 2H), 7.21 (t, J = 7.8 Hz, 2H), 6.68 (t, J = 7.2 Hz, 1H), 6.60 (d, J = 8.1 Hz, 2H), 3.93 (s, 3H), 3.69 (s, 1H), 3.28 (m, 1H), 3.25–3.15 (m, 1H), 2.82 (s, 3H), 2.38 (m, 1H), 2.14–1.94 (m, 1H); 13C NMR (CDCl3, 600 MHz) δ 178.46, 166.92, 149.07, 143.19, 130.24, 129.71, 129.37, 128.29, 117.13, 112.95, 52.30, 50.90, 49.24, 38.71, 29.93.
Results and discussion
In this study, a set of new DFNS-supported dendritic zwitterionic IL/Ru heterogeneous catalysts with high-density active sites DFNS/IL/Ru (1), DFNS/IL/Ru (2), and DFNS/IL/Ru (3) from DFNS were provided and deployed as highly effective and reusable heterogeneous catalysts for the α-aminomethylcarboxylation of alkene with CO2 and amines (Scheme 1). This sustainable, common, and functional approach illustrates an unusual example of the redox-neutral dicarbofunctionalization of alkenes for the synthesis of highly efficient important γ-amino acids under mild reaction conditions (Scheme 2).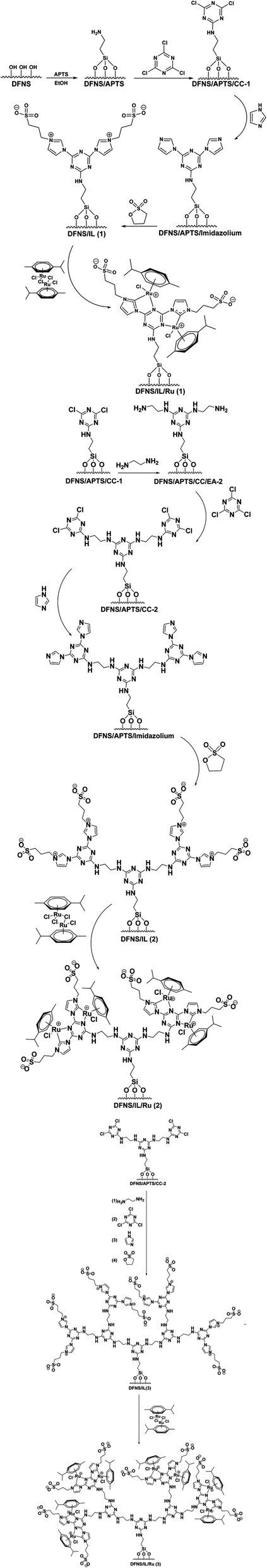 | ||
| Scheme 1 Synthesis of DFNS/IL/Ru (1), DFNS/IL/Ru (2), and DFNS/IL/Ru (3). | ||
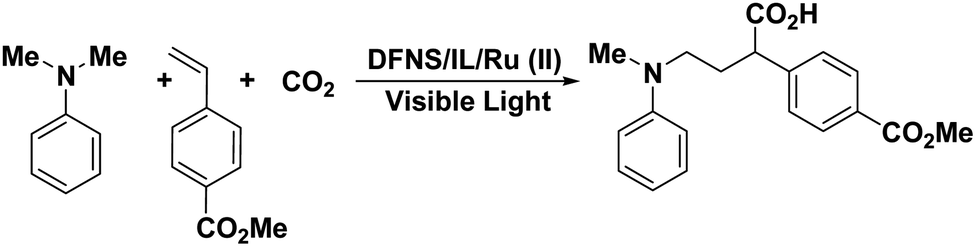 | ||
| Scheme 2 Synthesis of γ-amino acid from CO2 in the attendance of DFNS/IL/Ru NPs. | ||
The morphology and anatomy of the DFNS and DFNS/IL/Ru NPs were identified by TEM and SEM. Fig. 1a and e illustrate the TEM and SEM images of highly textured spherical DFNS specimens with a diameter of ∼300 nm and corrugated radial anatomy. As the pictures illustrate, the corrugated fibers (∼10 nm) protrude from the core of the spheres and are organized radially in 3 dimensions. The overlap of the corrugated radial anatomy creates open conical holes. The SEM picture illustrates the solidity of the sphere and its fibrous structure. Moreover, the radial anatomy and fibers facilitate the transport of chemicals and improve the access to active sites. According to the TEM and SEM pictures, the morphology of DFNS does not change after rectification (Fig. 1b–d and f–h).
 | ||
| Fig. 1 SEM pictures of dendritic fibrous nanosilica (DFNS) (a); DFNS/IL/Ru (1) NPs (b); DFNS/IL/Ru (2) NPs (c); DFNS/IL/Ru (3) NPs (d); and TEM pictures of dendritic fibrous nanosilica (DFNS) (e); DFNS/IL/Ru (1) NPs (f); DFNS/IL/Ru (2) NPs (g); DFNS/IL/Ru (3) NPs (h). | ||
The decomposition manner of the DFNS fibers and dendritic IL/Ru(II) catalysts DFNS/IL/Ru (1), DFNS/IL/Ru (2), and DFNS/IL/Ru (3) were compared to perceive the influences of the grafted dendritic IL molecules. Fig. 2 illustrates the thermogravimetric analysis (TGA) of DFNS/IL/Ru (1), DFNS/IL/Ru (2), and DFNS/IL/Ru (3). As can be seen, the specimens show the same thermal stability. The TGA curves experience two trends of weight reduction. The initial trend (28–120 °C) shows a low weight reduction of 5.0% that is ascribed to the vaporization of the absorbed water by specimens. The second trend (300–500 °C) shows a significant weight reduction of around 40–50%, which may occurr due to the degradation of lignin.
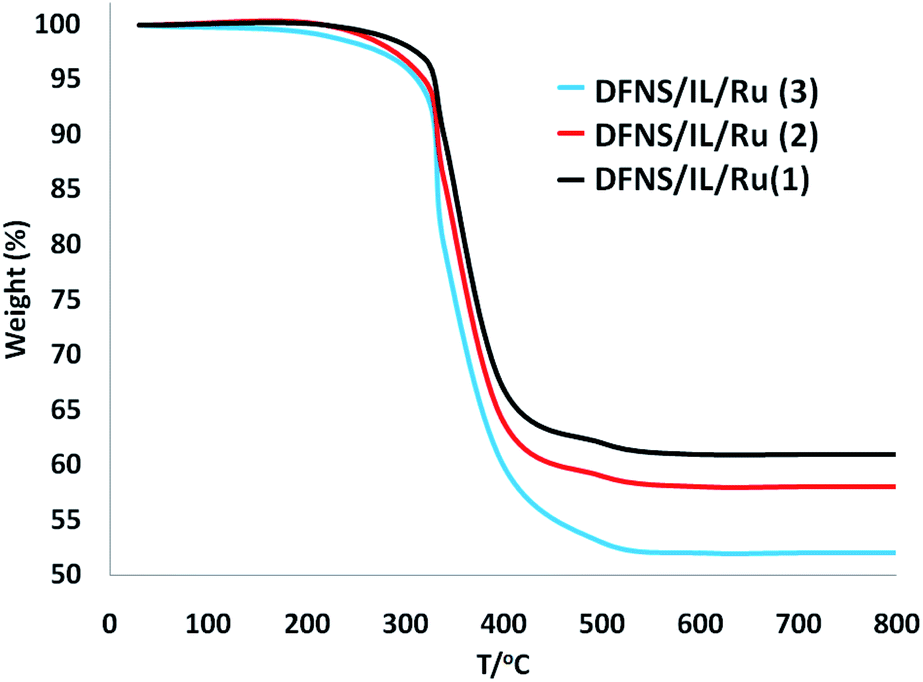 | ||
| Fig. 2 TGA of DFNS/IL/Ru (1), DFNS/IL/Ru (2), and DFNS/IL/Ru (3). | ||
FT-IR spectroscopy was used to identify the changes in the exterior layer of the synthesized DFNS/IL/Ru (3) (Fig. 3). The bands at 1631, 1106, and 803 cm−1 in the FT-IR spectrum of both DFNS and DFNS/IL/Ru (3) correspond to the tensile state of the water absorbed on the solid exterior layer, Si–O–Si vibrations, and Si–OH, respectively (Fig. 3a).71 In comparison with the FT-IR spectrum of luffa sponge (Fig. 3a), several novel absorption peaks are revealed at 1691, 1047, and 852 cm−1 (Fig. 3b), which appertain to the tensile vibration of Si–O bond and the flexural vibrations of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond on the exterior layer of DFNS/IL. Furthermore, the wide peak at 1042 cm−1 appertains to SO tensile vibrations in the sulphonate functional groups. Bands at 1497 cm−1 are related to N–H bending vibrations in the ammonium groups. The findings showed that IL was immobilized on DFNS NPs with success.72
N bond on the exterior layer of DFNS/IL. Furthermore, the wide peak at 1042 cm−1 appertains to SO tensile vibrations in the sulphonate functional groups. Bands at 1497 cm−1 are related to N–H bending vibrations in the ammonium groups. The findings showed that IL was immobilized on DFNS NPs with success.72
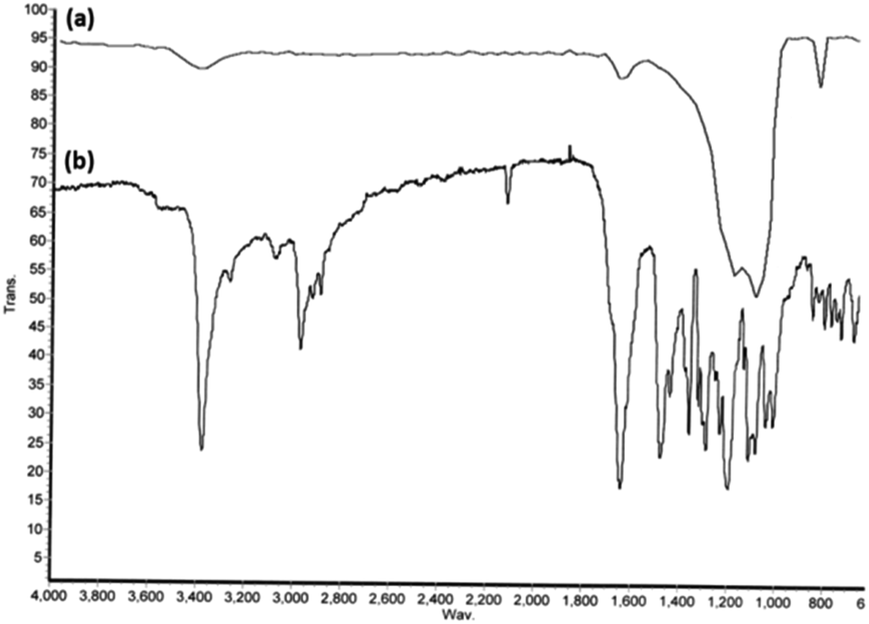 | ||
| Fig. 3 FTIR spectra of DFNS (a) and DFNS/IL/Ru (3) (b). | ||
The UV/vis spectra of the Ru(II) complex are shown in Fig. 4. The spectra of the supported Ru(II) illustrate properties alike the neat compound. The bands at 210 nm and 325 nm can be ascribed to the ligand charge transmission. The existence of the band at 475 nm is because of the ligand-to-metal charge transmission of the ruthenium complex. In the immobilization of ruthenium(II), all the bands existed in its spectra. The UV-vis spectra proved the immobilization of ruthenium compound on DFNS.73
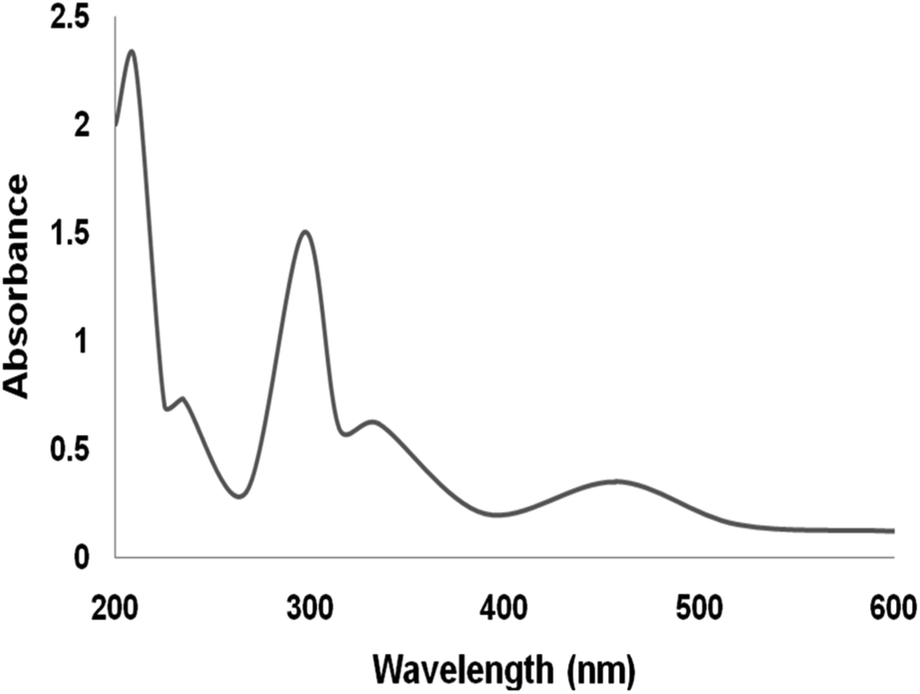 | ||
| Fig. 4 UV-vis spectra of DFNS/IL/Ru (3) NPs. | ||
XPS was utilized for examining the chemical parts on the DFNS/IL/Ru (3) NPs level. Fig. 5 depicts an XPS design for the assembled catalyst. Peaks of Ru, O, Cl, N, C, Si, and S are perceived and the attendance of N (1s) confirms that DFNS is functionalized by the imidazolium. Furthermore, the attendance of S, which is indicated with a sharp peak, illustrates the presence of sulfonate moiety in the catalyst. Furthermore, the XPS scheme of Ru 3p represents a doublet indicative of the metallic complex.
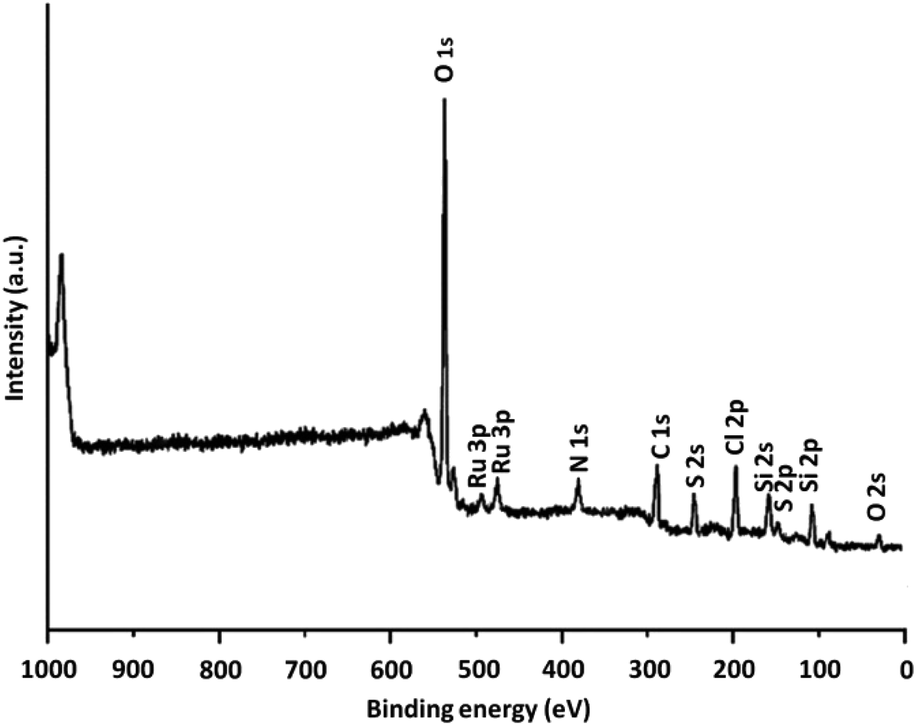 | ||
| Fig. 5 XPS spectra of DFNS/IL/Ru (3). | ||
The N2 adsorption–desorption isotherms of DFNS/IL/Ru (1), DFNS/IL/Ru (2), and DFNS/IL/Ru (3) illustrate the characteristic type IV curve that is in accordance with the previous studies on standard fibrous silica spheres (Fig. 6). For DFNS/IL/Ru (1), DFNS/IL/Ru (2), and DFNS/IL/Ru (3), the BET exterior layer areas were 257, 243, and 211 m2 g−1; pore diameters were 11.61, 11.38, and 11.09 nm; and pore volumes were 1.09, 1.01, and 0.92 cm3 g−1, respectively. Besides, the nitrogen sorption analysis of DFNS/IL/Ru proved an even and unvaried structure with a reduction in the exterior layer area, pore diameter, and pore volume, compared to pristine DFNS. Functionalization by X–Si (X = IL/Ru (1), IL/Ru (2), and IL/Ru (3)) reduced the corresponding pore volume. This might be due to overloading with the excavator that engrosses a vast volume in silica fields (Table 1).
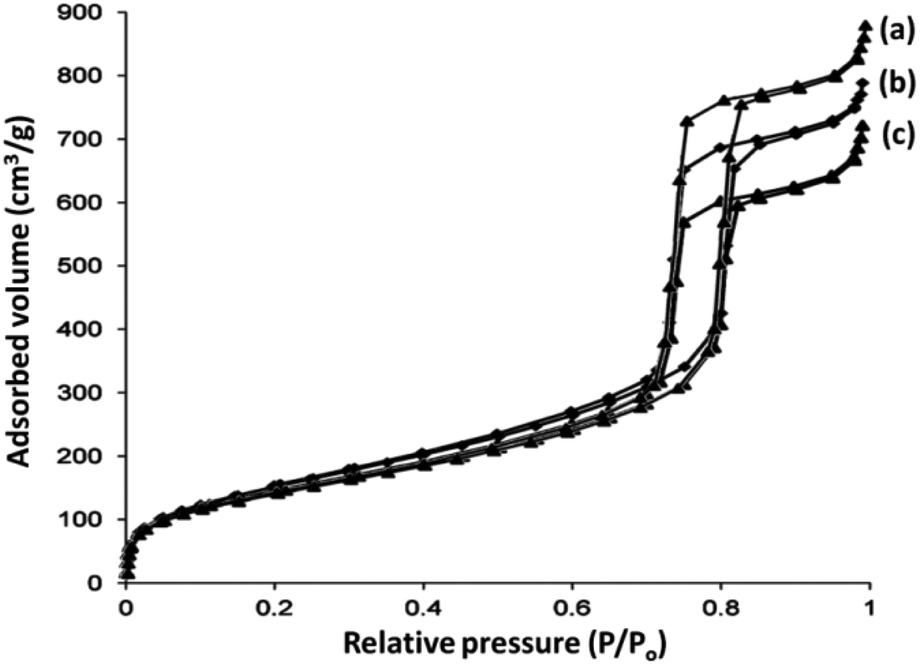 | ||
| Fig. 6 Adsorption–desorption isotherms of (a) DFNS/IL/Ru (1), (b) DFNS/IL/Ru (2), and (c) DFNS/IL/Ru (3). | ||
| Catalysts | SBET (m2 g−1) | Vt (cm3 g−1) | DBJH (nm) |
|---|---|---|---|
| DFNS | 439 | 1.49 | 14.78 |
| DFNS/IL/Ru (1) | 257 | 1.09 | 11.61 |
| DFNS/IL/Ru (2) | 243 | 1.01 | 11.38 |
| DFNS/IL/Ru (3) | 211 | 0.92 | 11.09 |
We tested the catalytic activity of the different generations of IL/Ru catalysts DFNS/IL/Ru (1), DFNS/IL/Ru (2), and DFNS/IL/Ru (3) by the carbocarboxylation of styrenes employing CO2 and amines (Table 2). As shown in Table 2, we first employed carbon dioxide (1.0 MPa) in the presence of IL/Ru catalyst DFNS/IL/Ru (1), DFNS/IL/Ru (2), and DFNS/IL/Ru (3) (50 mg). Product yields of 85–97% were obtained by DFNS/IL/Ru (1), DFNS/IL/Ru (2), and DFNS/IL/Ru (3) (50 mg) after 7 hours using TEOA as co-catalyst. According to the results, enhancing IL/Ru improved the catalytic activity. Moreover, a positive dendritic influence on the efficiency of the α-aminomethylcarboxylation of alkenes with amines and CO2 was perceived. Besides, by reducing the amount of the catalyst from 50 mg to 25 mg, the product yield was increased to 97% due to the high catalytic activity of DFNS/IL/Ru (3). Regarding the better performance of the ionic liquid, it was selected as a catalyst in this study, and further research was done on it.
There are various parameters such as light with various intensities to figure out the effects on the product constitution. The catalyst was screened at different intensities of light with other constant indicators to achieve the corresponding efficiencies of 6%, 28%, 89%, 97%, and 97% (Fig. 7). The generation of γ-amino acids was enhanced from 8–22 W and remained stable at 22 W. The deployment of higher CFL watts (32 W) had no significant influence on the efficiency or reaction time.
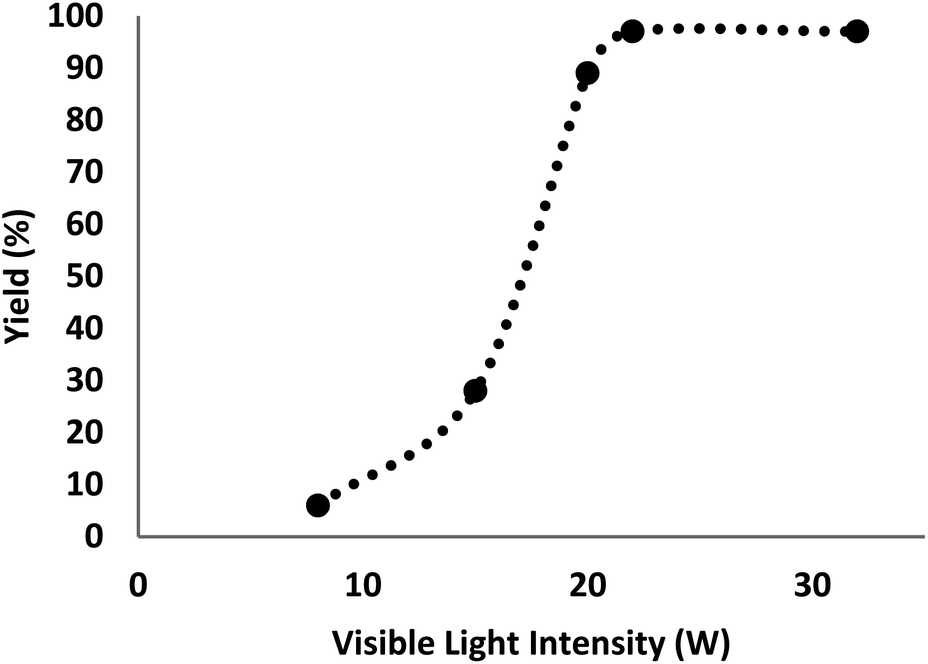 | ||
| Fig. 7 Effects of the light source on the reaction process in the presence of DFNS/IL/Ru (3). | ||
The quantity of the catalyst had a critical influence on the carbocarboxylation of styrenes employing CO2 and amines since the reaction did not occur in the absence of the catalyst. It was perceived that the production of γ-amino acids began with at least 5 mg of DFNS/IL/Ru (3) as catalyst under optimal reaction circumstances, with an efficiency of 11.25 mg catalyst showing the highest efficiency (97%) for formate, and further enhancement in catalyst quantity showed no improvement in product formation (Fig. 8).
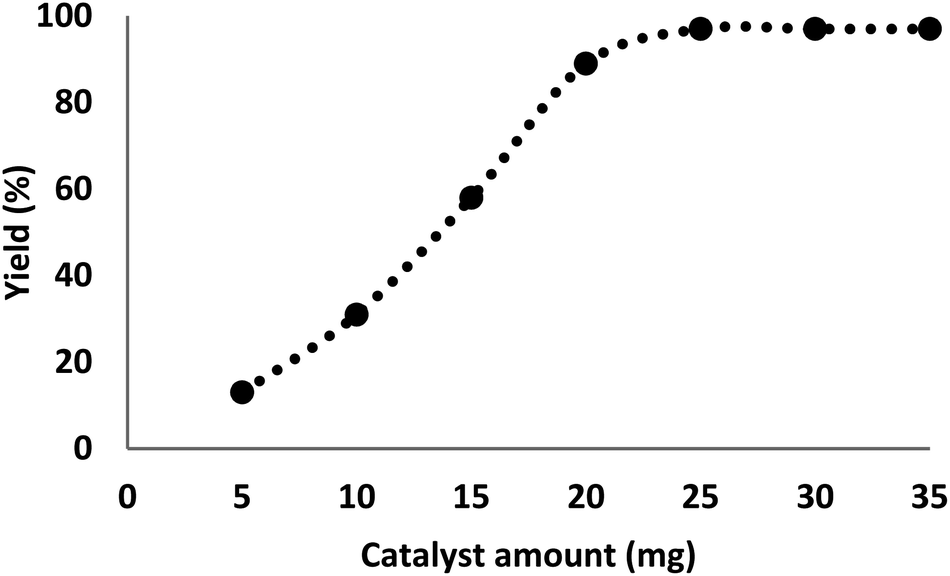 | ||
| Fig. 8 The effect of DFNS/IL/Ru (3) quantity. | ||
Furthermore, a set of comparative investigations were done. The reaction was identified as photocatalytic since under dark condition no γ-amino acid was produced. In the absence of DFNS/IL/Ru (3) NPs or TEOA, the reaction did not illustrate any γ-amino acid formation under visible light irradiation. With the aim of investigating the source of the generated γ-amino acids, we employed N2 instead of CO2 for the processing of the DFNS/IL/Ru (3) NPs and the soluble system. There was merely a trace of γ-amino acids under 5 hours of irradiation with DFNS/IL/Ru (3) NPs as the photocatalyst, which showed that the generated γ-amino acids were obtained from CO2 and not from the decomposition of ligands (Fig. 9).
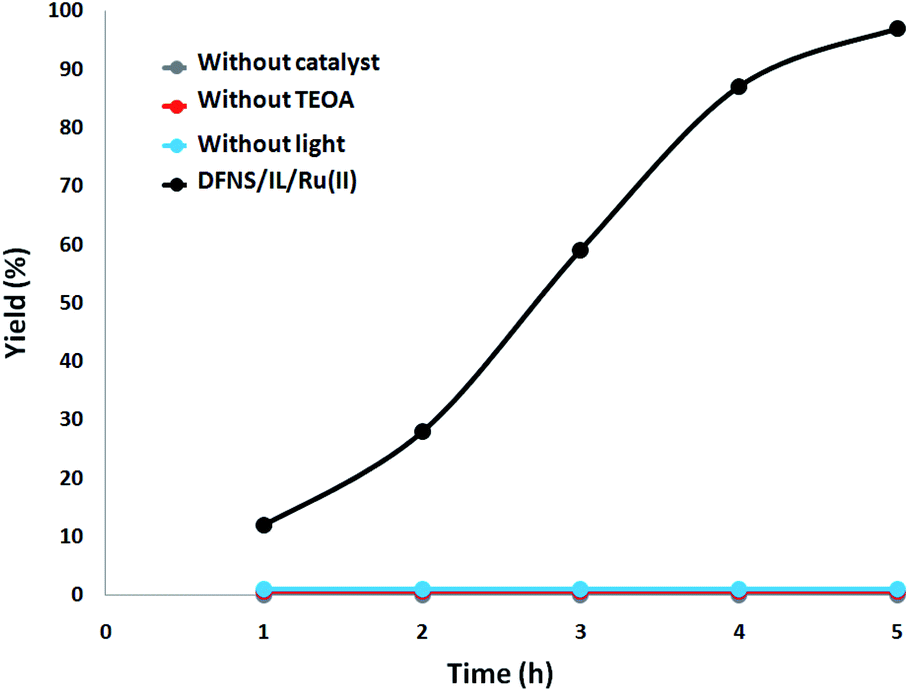 | ||
| Fig. 9 The amount of γ-amino acids generated at different times of irradiation under various circumstances. | ||
For a deeper evaluation of the efficiency of the catalyst, various control experiments were conducted and the results are shown in Table 3. The reaction performed deploying DFNS showed that no amount of γ-amino acids was formed after 5 hours (Table 3, row 1). Moreover, no reaction was perceived when DFNS/IL (3) was employed as the catalyst (Table 3, row 2). IL was not able to show good catalytic activity in standard reactions. We compared the results with many other similar catalysts. Due to the unfavorable results, we continued research to increase the efficiency by adding Ru(II) (Table 3, row 3). Our results showed that the reaction cycle is primarily catalyzed using Ru(II) complexes in the DFNS/IL (3) nanostructure. The nanoparticles increase the activity of the catalyst due to the increase in surface area to volume, so they significantly increase the sensitivity between the reactants and the catalyst and act as homogeneous catalysts (Table 3, rows 3 and 4).
Based on previous research,74 an acceptable mechanism is presented and shown in Scheme 3. Photo-excited DFNS/IL/Ru is reductively quenched by aniline leading to (A) and a radical cation intermediate (1), which then deprotonates and gives out α-aminoalkyl radical (2) in the presence of a base. The carbon radical B undergoes addition to the CC bond of styrene (B) to selectively generate the γ-amino benzylic radical (3). Further, the nucleophilic addition to CO2 and protonation complete this reaction and yield the expected γ-amino acids.
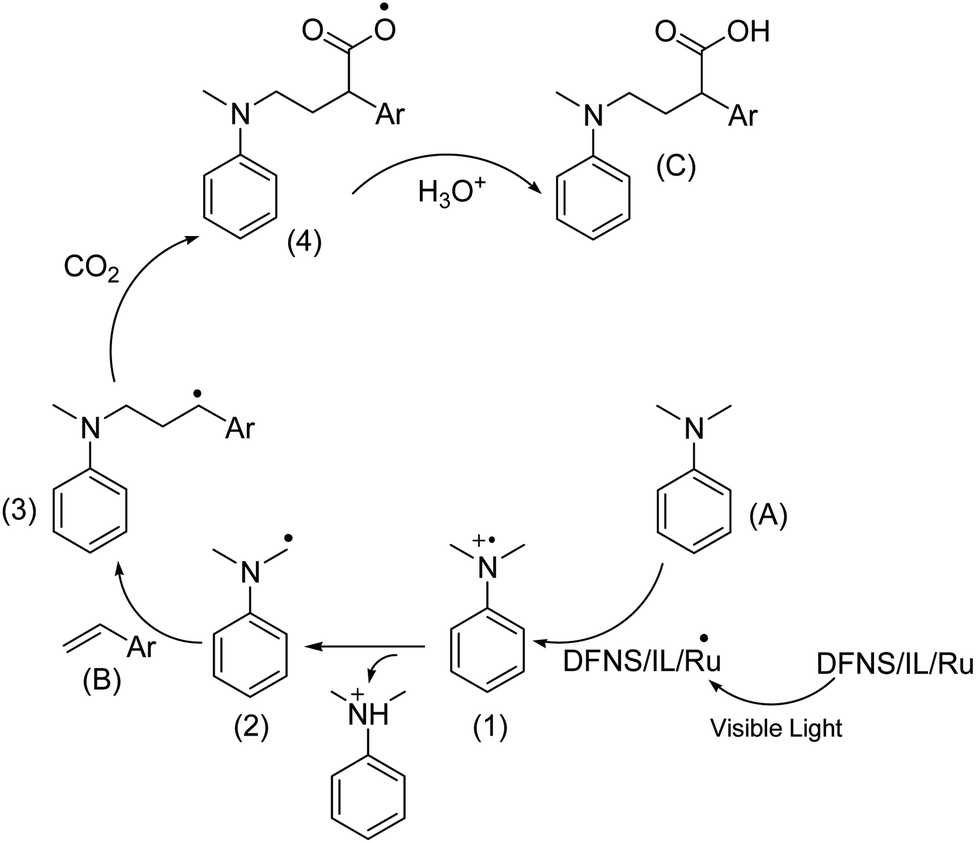 | ||
| Scheme 3 The proposed reaction mechanism. | ||
The recyclability of a catalyst is considered as an important characteristic in green chemistry. Therefore, the recyclability of the NPs of DFNS/IL/Ru (3) was optimally examined for the carbocarboxylation of styrenes employing CO2 and amines. Upon completion, the solid NPs of DFNS/IL/Ru (3) were easily removed from the liquid reaction area in seconds. After cleaning the solvent, the catalyst could be recycled immediately. Fig. 10 illustrates the reuse of the catalyst ten times in a row. The efficiency of the product in the tenth run was 97%, which shows merely a 4% reduction (93%).
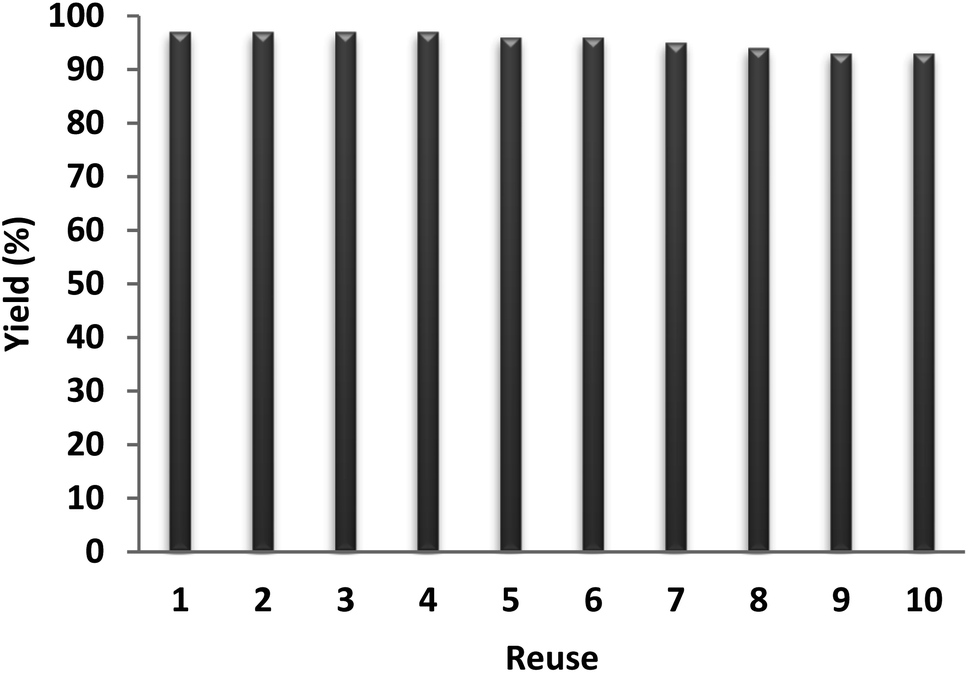 | ||
| Fig. 10 Recyclability of the catalyst. | ||
Besides, a comprehensive study was conducted on the heterogeneous essence of the catalyst. First, the hot filtration test for generating γ-amino acids was done under superior circumstances, and it showed that the catalyst was eliminated at a yield of 43% (about 2.5 hours). After the removal of the heterogeneous catalyst, it was found that the free catalyst residues were relatively active, and a conversion of 74% was achieved after ten hours of cyclic carbonate production. This showed the heterogeneity of the catalyst during the reaction, with partial leaching. Subsequently, a mercury-poisoning test was done to prove the heterogeneity of the catalyst.
Mercury(0) significantly impoverished the metal catalyst on the active exterior layer, calming the activity of the catalyst. This investigation illustrated the heterogeneity of the catalyst. This examination was done on the mentioned model of reaction at optimal circumstances. About 300 molar mercury was added to the reaction composition after 2.5 hours. After 5 hours, no conversion was observed from the poisoned catalyst. Fig. 11 illustrates a kinetic design of the reaction at the attendance of Hg(0). The negative findings of the tests indicated that the catalyst is heterogeneous and no significant ruthenium leaching happened during cyclic carbonate formation.
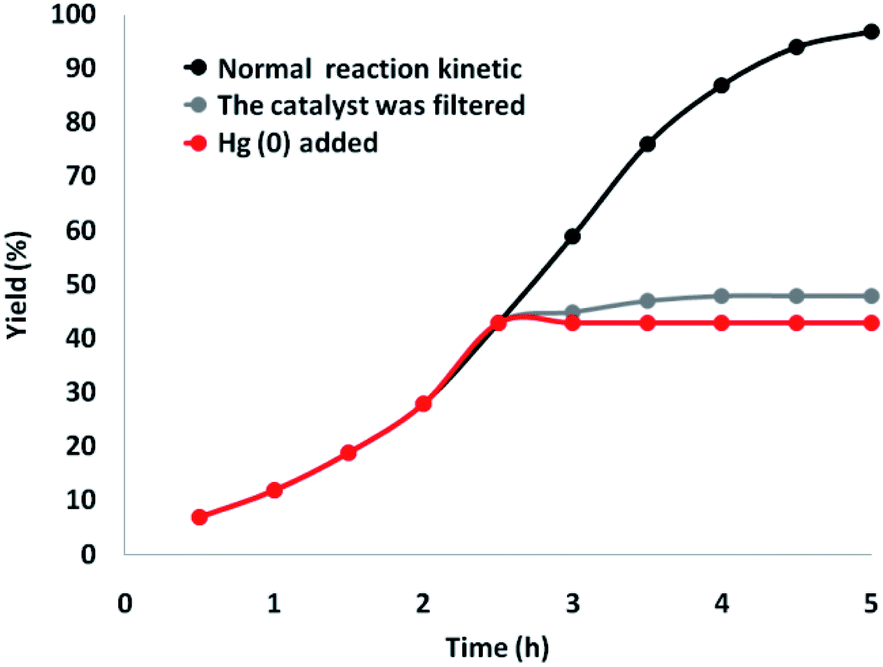 | ||
| Fig. 11 Reaction kinetics, Hg(0) poisoning, and hot filtration studies for synthesis of γ-amino acids. | ||
The SEM and TEM picture analyses illustrated the more fibrous nanoparticles of DFNS/IL/Ru (3) than ever before. Fig. 12a and b show the SEM and TEM pictures of the fibrous nanoparticles of DFNS/IL/Ru (3) that were reused ten times. The catalyst preserved its fibrous anatomy after being used 10 times. A similar anatomy was perceived for fresh fibrous nanoparticles of DFNS/IL/Ru (3) and DFNS/IL/Ru (3) after 10 times of recycling. Furthermore, the thermal stability of the recycled DFNS/IL/Ru (3) catalyst after ten times of recycling was weaker than that of the fresh catalyst. This may happen due to the disappearance of IL in DFNS during the course of recycling (Fig. 13). The operation was not influenced at 100 °C.
 | ||
| Fig. 12 (a) FE-SEM and (b) TEM pictures of the recycled DFNS/IL/Ru (3) NPs after the 10th run for the synthesis of quinoline-2-one. | ||
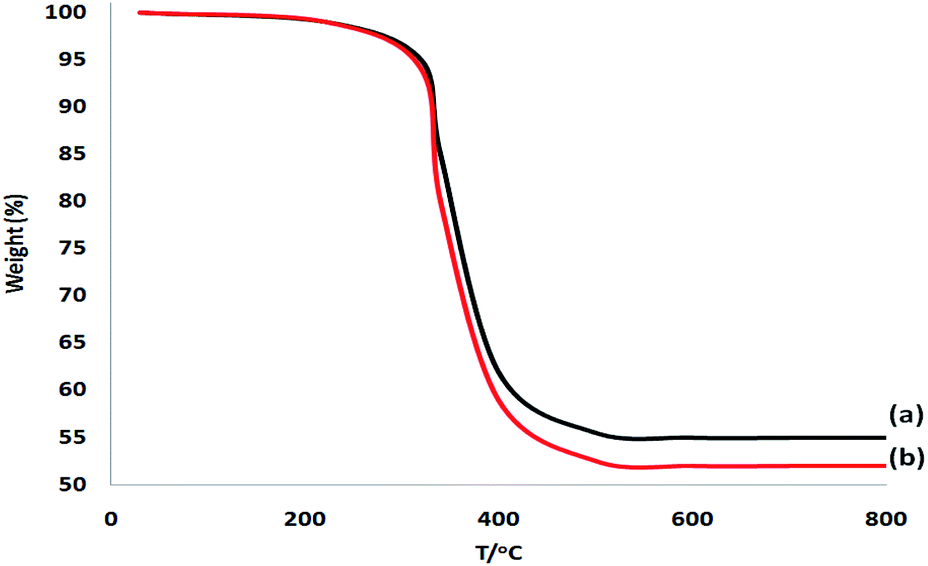 | ||
| Fig. 13 TGA of the reused DFNS/IL/Ru (3) catalyst after 10 times of recycling (a) and that of the fresh DFNS/IL/Ru (3) catalyst (b). | ||
Conclusions
For the first time, this study reports the production of DFNS-supported dendritic IL/Ru(II) catalysts DFNS/IL/Ru (1), DFNS/IL/Ru (2), and DFNS/IL/Ru (3) with high ionic density and various generations. Results showed that the dendritic IL/Ru catalysts DFNS/IL/Ru (1), DFNS/IL/Ru (2), and DFNS/IL/Ru (3) positively influence the efficiency of the α-aminomethylcarboxylation of alkenes with amines and CO2. DFNS/IL/Ru (3) was the optimal system with superb catalytic activity for the chemical stabilization of CO2 in quinoline-2-one. DFNS/IL/Ru (3) could be reused to up to ten continuous runs without any significant reduction in catalytic activity. Compared to the reported heterogeneous catalysts, DFNS/IL/Ru(II) signaled a number of advantages, including inexpensiveness, recyclable essence, facile production, and high catalytic power to fix CO2 reaction. These properties make it a suitable candidate for industrial operations.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Young Talent Innovation Project of the Education Department of Heilongjiang Province of China (Grants No. UNPYSCT-2018114), and the Key Project of College Student Innovation and Entrepreneurship Training Program of Heilongjiang Province of China (Grants No. 2020102254).Notes and references
- P. Conti, L. Tamborini, A. Pinto, A. Blondel, P. Minoprio, A. Mozzarelli and C. De Micheli, Chem. Rev., 2011, 111, 6919–6946 CrossRef CAS PubMed.
- R. B. Silverman, Angew. Chem., Int. Ed., 2008, 47, 3500–3504 CrossRef CAS PubMed.
- M. Ordóñez and C. Cativiela, Tetrahedron: Asymmetry, 2007, 18, 3–99 CrossRef.
- K. Maruoka and T. Ooi, Chem. Rev., 2003, 103, 3013–3028 CrossRef CAS PubMed.
- R. Kastl and H. Wennemers, Angew. Chem., Int. Ed., 2013, 52, 7228–7232 CrossRef CAS PubMed.
- J. H. Sim and C. E. Song, Angew. Chem., Int. Ed., 2017, 56, 1835–1839 CrossRef CAS PubMed.
- Y. Chi, L. Guo, N. A. Kopf and S. H. Gellman, J. Am. Chem. Soc., 2008, 130, 5608–5609 CrossRef CAS PubMed.
- T. Okino, Y. Hoashi, T. Furukawa, X. Xu and Y. Takemoto, J. Am. Chem. Soc., 2005, 127, 119–125 CrossRef CAS PubMed.
- M. Wiesner, J. D. Revell, S. Tonazzi and H. Wennemers, J. Am. Chem. Soc., 2008, 130, 5610–5611 CrossRef CAS PubMed.
- K. Akagawa and K. Kudo, Angew. Chem., Int. Ed., 2012, 51, 12786–12789 CrossRef CAS PubMed.
- A. Baschieri, L. Bernardi, A. Ricci, S. Suresh and M. F. Adamo, Angew. Chem., Int. Ed., 2009, 48, 9342–9345 CrossRef CAS PubMed.
- X. Y. Chen, F. Xia, J. T. Cheng and S. Ye, Angew. Chem., Int. Ed., 2013, 52, 10644–10647 CrossRef CAS PubMed.
- L. T. Shen, L. H. Sun and S. Ye, J. Am. Chem. Soc., 2011, 133, 15894–15897 CrossRef CAS PubMed.
- D. Ravelli, S. Protti and M. Fagnoni, Chem. Rev., 2016, 116, 9850–9913 CrossRef CAS PubMed.
- D. Stanveness, I. Bosque and C. R. Stephenson, Acc. Chem. Res., 2016, 49, 2295–2306 CrossRef PubMed.
- J.-P. Goddard, C. Ollivier and L. Fensterbank, J. Am. Chem. Soc., 2016, 49, 1924–1936 CAS.
- N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS PubMed.
- M. Y. Cao, X. Ren and Z. Lu, Tetrahedron Lett., 2015, 56, 3732–3742 CrossRef CAS.
- T. Koike and M. Akita, Org. Chem. Front., 2016, 3, 1345–1348 RSC.
- T. Koike and M. Akita, Chem, 2018, 4, 1–29 Search PubMed.
- A. McNally, C. K. Prier and D. W. MacMillan, Science, 2016, 352, 1304–1308 CrossRef PubMed.
- Y. Cai, Y. Tang, L. Fan, Q. Lefebvre, H. Hou and M. Rueping, ACS Catal., 2018, 8, 9471–9476 CrossRef CAS.
- L. Shi and W. Xia, Chem. Soc. Rev., 2012, 41, 7687–7697 RSC.
- K. Nakajima, Y. Miyake and Y. Nishibayashi, Acc. Chem. Res., 2016, 49, 1946–1956 CrossRef CAS PubMed.
- S. A. Morris, J. Wang and N. Zheng, Acc. Chem. Res., 2016, 49, 1957–1968 CrossRef CAS PubMed.
- Y. Miyake, K. Nakajima and Y. Nishibayashi, J. Am. Chem. Soc., 2012, 134, 3338–3341 CrossRef CAS PubMed.
- P. Kohls, D. Jadhav, G. Pandey and O. Reiser, Org. Lett., 2012, 14, 672–675 CrossRef CAS PubMed.
- Y. Yin, Y. Dai, H. Jia, J. Li, L. Bu, B. Qiao, X. Zhao and Z. Jiang, J. Am. Chem. Soc., 2018, 140, 6083–6087 CrossRef CAS PubMed.
- M. A. Ashley, C. Yamauchi, J. C. K. Chu, S. Otsuka, H. Yorimitsu and T. Rovis, Angew. Chem., Int. Ed., 2019, 58, 4002–4006 CrossRef CAS PubMed.
- D. Yu, S. P. Teong and Y. Zhang, Coord. Chem. Rev., 2015, 293–294, 279–291 CrossRef CAS.
- S. Wang, G. Du and C. Xi, Org. Biomol. Chem., 2016, 14, 3666–3676 RSC.
- S. Wang and C. Xi, Chem. Soc. Rev., 2019, 48, 382–404 RSC.
- Y. Cao, X. He, N. Wang, H.-R. Li and L.-N. He, Chin. J. Chem., 2018, 36, 644–659 CrossRef CAS.
- Q. Liu, L. Wu, R. Jackstell and M. Beller, Nat. Commun., 2015, 6, 5933 CrossRef PubMed.
- B. Su, Z.-C. Cao and Z.-J. Shi, Acc. Chem. Res., 2015, 48, 886–896 CrossRef CAS PubMed.
- F. Tan and G. Yin, Chin. J. Chem., 2018, 36, 545–554 CrossRef CAS.
- A. Tortajada, F. Julia-Hernandez, M. Borjesson, T. Moragas and R. Martin, Angew. Chem., Int. Ed., 2018, 57, 15948–15982 CrossRef CAS PubMed.
- M. Bçrjesson, T. Moragas, D. Gallego and R. Martin, ACS Catal., 2016, 6, 6739–6749 CrossRef PubMed.
- F. Julia-Hernandez, M. Gaydou, E. Serrano, M. Gemmeren and R. Martin, Top. Curr. Chem., 2016, 374, 45 CrossRef PubMed.
- Y. Tsuji and T. Fujihara, Chem. Commun., 2012, 48, 9956–9964 RSC.
- B. Yu, Z.-F. Diao, C.-X. Guo and L.-N. He, J. CO2 Util., 2013, 1, 60–68 CrossRef CAS.
- S. M. Saadati and S. M. Sadeghzadeh, Catal. Lett., 2018, 148, 1692–1702 CrossRef CAS.
- S. M. Sadeghzadeh, R. Zhiani and S. Emrani, Catal. Lett., 2018, 148, 119–124 CrossRef CAS.
- S. M. Sadeghzadeh, R. Zhiani and M. Moradi, ChemistrySelect, 2018, 3, 3516–3522 CrossRef CAS.
- S. Esfandiari Baghbamidi, A. Hassankhani, E. Sanchooli and S. M. Sadeghzadeh, Appl. Organomet. Chem., 2018, 32, e4251 CrossRef.
- P. Shao, S. Wang, C. Chen and C. Xi, Org. Lett., 2016, 18, 2050–2053 CrossRef CAS PubMed.
- V. R. Yatham, Y. Shen and R. Martin, Angew. Chem., Int. Ed., 2017, 56, 10915–10919 CrossRef CAS PubMed.
- J. H. Ye, M. Miao, H. Huang, S. S. Yan, Z. B. Yin, W. J. Zhou and D. G. Yu, Angew. Chem., Int. Ed., 2017, 56, 15416–15420 CrossRef CAS PubMed.
- Q. Fu, Z.-Y. Bo, J.-H. Ye, T. Ju, H. Huang, L.-L. Liao and D.-G. Yu, Nat. Commun., 2019, 10, 3592 CrossRef PubMed.
- J. Hou, A. Ee, H. Cao, H.-W. Ong, J. Xu and J. Wu, Angew. Chem., Int. Ed., 2018, 57, 17220–17224 CrossRef CAS PubMed.
- H. Wang, Y. Gao, C. Zhou and G. Li, J. Am. Chem. Soc., 2020, 142, 8122–8129 CrossRef CAS PubMed.
- R. J. Crutchky, Adv. Inorg. Chem., 1994, 41, 273 CrossRef.
- S. Nag, P. Gupta, R. J. Butcher and S. Bhattacharya, Inorg. Chem., 2004, 43, 4814–4816 CrossRef CAS PubMed.
- R. Acharyya, S. M. Peng, G. H. Lee and S. Bhattacharya, Inorg. Chem., 2003, 42, 7378 CrossRef CAS PubMed.
- T. Koiwa, Y. Masuda, J. Shono, Y. Kawmoto, Y. Hashino, T. Hashimoto, K. Natarajan and K. Shimizu, Inorg. Chem., 2004, 43, 6215–6223 CrossRef CAS PubMed.
- O. Novakova, J. Kasparkova, O. Vrana, P. M. Vanvliet, J. Reedijik and V. Brabec, Biochemistry, 1995, 34, 12369–12378 CrossRef CAS PubMed.
- G. D. Frey, Z. R. Bell, J. C. Jeffery and M. D. Ward, Polyhedron, 2001, 20, 3231–3237 CrossRef CAS.
- P. A. Vigato and S. Tamburini, Coord. Chem. Rev., 2004, 248, 1717–2128 CrossRef CAS.
- J. A. Miller, B. A. Gross, M. A. Zhuravel, W. Jin and S. T. Nguyen, Angew. Chem., 2005, 117, 3953–3957 CrossRef.
- W. L. Man, H. K. Kwong, W. W. Y. Lam, J. Xiang, T. W. Wong, W. H. Lam, W. T. Wong, S. M. Peng and T. C. Lau, Inorg. Chem., 2008, 47, 5936–5944 CrossRef CAS PubMed.
- C. M. Che and C. K. Poon, Pure Appl. Chem., 1998, 60, 1201–1207 Search PubMed.
- C. M. Che, W. T. Tang, W. T. Wong and T. F. Lai, J. Am. Chem. Soc., 1989, 111, 9048 CrossRef CAS.
- W. H. Leung, E. Y. Y. Chan, E. K. F. Chow, I. D. Williams and S. M. Peng, J. Chem. Soc., Dalton Trans., 1996, 1229 RSC.
- D. Astruc, E. Boisselier and C. Ornelas, Chem. Rev., 2010, 110, 1857–1959 CrossRef CAS PubMed.
- D. A. Tomalia, New J. Chem., 2012, 36, 264–281 RSC.
- F. Giacalone, V. Campisciano, C. Calabrese, V. L. Parola, Z. Syrgiannis and M. Prato, ACS Nano, 2016, 10, 4627–4636 CrossRef CAS PubMed.
- H. Karimi-Maleh, M. Alizadeh, Y. Orooji, F. Karimi, M. Baghayeri, J. Rouhi, S. Tajik, H. Beitollahi, S. Agarwal, V. K. Gupta, S. Rajendran, S. Rostamnia, L. Fu, F. Saberi-Movahed and S. Malekmohammadi, Ind. Eng. Chem. Res., 2021, 60, 816–823 CrossRef CAS.
- E. Murugan, J. N. Jebaranjitham, K. J. Raman, A. Mandal, D. Geethalakshmi, M. D. Kumarc and A. Saravanakumarc, New J. Chem., 2017, 41, 10860–10871 RSC.
- T. Y. Qin, X. Y. Li, J. P. Chen, Y. Zeng, T. J. Yu, G. Q. Yang and Y. Li, Chem.–Asian J., 2014, 9, 3641–3649 CrossRef CAS PubMed.
- S. Hayouni, A. Robert, C. Maes, A. Conreux, B. Marin and A. Mohamadou, New J. Chem., 2018, 42, 18010–18020 RSC.
- R. Singh, R. Belgamwar, M. Dhimana and V. Polshettiwar, J. Mater. Chem. B, 2018, 6, 1600–1604 RSC.
- S. Lai, J. Gao, H. Zhang, L. Cheng and X. Xiong, J. CO2 Util., 2020, 38, 148–157 CrossRef CAS.
- R. Zhiani, M. Khoobi and S. M. Sadeghzadeh, Catal. Today, 2020, 340, 197–203 CrossRef CAS.
- B. Zhang, Y. Yi, Z. Q. Wu, C. Chen and C. Xi, Green Chem., 2020, 22, 5961–5965 RSC.
| This journal is © The Royal Society of Chemistry 2021 |