
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Powering the next industrial revolution: transitioning from nonrenewable energy to solar fuels via CO2 reduction
Rami J. Batrice * and
John C. Gordon*
* and
John C. Gordon*
Chemistry Division, Inorganic, Isotope, and Actinide Chemistry, Los Alamos National Laboratory, Los Alamos, New Mexico 87545, USA. E-mail: batricer@lanl.gov; jgordon@lanl.gov
First published on 22nd December 2020
Abstract
Solar energy has been used for decades for the direct production of electricity in various industries and devices; however, harnessing and storing this energy in the form of chemical bonds has emerged as a promising alternative to fossil fuel combustion. The common feedstocks for producing such solar fuels are carbon dioxide and water, yet only the photoconversion of carbon dioxide presents the opportunity to generate liquid fuels capable of integrating into our existing infrastructure, while simultaneously removing atmospheric greenhouse gas pollution. This review presents recent advances in photochemical solar fuel production technology. Although efforts in this field have created an incredible number of methods to convert carbon dioxide into gaseous and liquid fuels, these can generally be classified under one of four categories based on how incident sunlight is utilised: solar concentration for thermoconversion (Category 1), transformation toward electroconversion (Category 2), natural photosynthesis for bioconversion (Category 3), and artificial photosynthesis for direct photoconversion (Category 4). Select examples of developments within each of these categories is presented, showing the state-of-the-art in the use of carbon dioxide as a suitable feedstock for solar fuel production.
Introduction
In the more than 300![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 years of human existence,1 no time period has been characterised by more rapid growth and technological advancement than the Industrial Revolution which began in the 18th century. This time period up to present day, commonly referred to as the Anthropocene,2 has been witness to profound advances in society which has allowed for: (1) greater affordability and accessibility to goods, (2) the development of labour-saving inventions, (3) rapid growth in medical understanding and technologies, (4) greater wealth and improved quality of life for the average person, and (5) increased supply and demand for specialized professions. These advances coupled with high levels of urban migration ultimately provided for an increased global carrying capacity, leading to unprecedented population growth from 791 million prior to the Industrial Revolution (1750) to 7.795 billion in 2020.3,4 While the quality of life for some, especially those in the legacy world, has increased dramatically in light of such innovations, several challenges have risen from the nearly ten-fold increase in population, not the least of which is the substantially greater energy demands required to power our modern society.
000 years of human existence,1 no time period has been characterised by more rapid growth and technological advancement than the Industrial Revolution which began in the 18th century. This time period up to present day, commonly referred to as the Anthropocene,2 has been witness to profound advances in society which has allowed for: (1) greater affordability and accessibility to goods, (2) the development of labour-saving inventions, (3) rapid growth in medical understanding and technologies, (4) greater wealth and improved quality of life for the average person, and (5) increased supply and demand for specialized professions. These advances coupled with high levels of urban migration ultimately provided for an increased global carrying capacity, leading to unprecedented population growth from 791 million prior to the Industrial Revolution (1750) to 7.795 billion in 2020.3,4 While the quality of life for some, especially those in the legacy world, has increased dramatically in light of such innovations, several challenges have risen from the nearly ten-fold increase in population, not the least of which is the substantially greater energy demands required to power our modern society.
Interestingly, the fuels used today to power transportation and industry vary little from those used nearly 300 years ago insomuch as we continue to rely on non-renewable fossil fuels in the form of coal, oil, and natural gas. This has led to two major problems; first, continued and increased use of fossil fuels has greatly diminished the global supply, and while their scarcity remains a point of debate, it is indisputable that they remain a finite and dwindling resource. Moreover, the geographical distribution of proved fossil fuel reserves (Fig. 1)5 raises questions about their long-term availability, especially when considering increased geopolitical tensions and mounting international sanctions. The second, and more substantial problem of this observed population growth and associated energy consumption is the proliferation of greenhouse gases (carbon dioxide (CO2), methane (CH4), nitrous oxide (N2O), etc.) in the environment and the resulting global climate change. Of these gases, CO2 poses the greatest threat, as it is by far the major greenhouse gas produced from fossil fuel combustion contributing to climate change, and also due to its persistence within the atmosphere.6,7 Historical records in fact show atmospheric CO2 levels rising from 277 ppm immediately prior to industrialisation in 1750, to 417 ppm by April of 2020, marking a nearly 51% increase.8,9 This corresponds to a 1.43 °C increase of the average global surface temperature from 13.42 °C to 14.85 °C within this time period,10,11 with the most dire projections showing a potential 4 °C increase beyond preindustrial levels by 2100.12,13 Even with the increase in temperature and atmospheric CO2 levels already observed, catastrophic effects have resulted including: oceans warming (0.11 °C per decade between 1971–2010), glaciers/ice sheets melting (∼4% recession between 1979–2012), sea level rise of 0.19 meters between 1901 and 2010, 26% increase in ocean acidity from the Industrial Revolution to 2014, increased coastal erosion, unpredictable precipitation patterns leading to regional drought/flooding, major disruption of terrestrial and marine ecosystems, increased frequency and intensity of meteorological natural disasters, compromised food supply and security, and forced migration of native populations.14 Presented with such data, it is immediately apparent that now, more than ever, the ability to mitigate these hazards is needed.
 | ||
| Fig. 1 2018 regional distribution of proved (a) oil reserves, (b) natural gas reserves, and (c) coal reserves. | ||
To address the current climate crisis and growing energy demands (Fig. 2),15,16 extensive work has been devoted toward developing renewable energy technologies to either augment or replace fossil fuel use globally. Nonetheless, while the Paris Climate Accords of 2015 showed an apparent willingness of nations to address the growing threats of climate change,17 progress since this treaty has been limited – the primary energy consumption generated from renewable sources increased from 9.3% in 2014 to only 10.9% in 2018.5,18 In order to meet current demands and prepare for the additional 380.7 exajoules (EJ) needed by 2050, all potential renewable energy sources must be measured against current standards; these sources include wind, hydropower, geothermal, ocean, biomass, and solar power. In determining the portfolio of viable alternative energy sources, three factors must initially be considered: (1) theoretical potential, (2) technical potential, and (3) energy return on investment (EROI). Theoretical potential refers to the upper limits of an energy source available without consideration of any potential restrictions. Technical potential is identified as the usable energy of a given source, limited by factors such as geographical distribution, terrain, land use rights, and others. Finally, EROI is an analytical metric useful for determining the efficiency and viability of an energy producing technology or industry, measured as the ratio of the energy obtained from a resource to the energy expended to produce that energy. Given that EROI is based upon current technologies, it is not necessarily a good indicator for the long-term potential viability of a technology, as future advancements can further increase EROI values. Analysis of these three factors has been studied extensively and given rise to theoretical and technical potentials for all major classes of renewable fuels (Table 1),19,20 as well as EROI models which reflect current technologies and economic analyses (Fig. 3).21,22 Even limited by the technical potentials, it is clear that solar energy is by far the most abundant and desirable renewable energy source,23 providing more than 60 times the 2050 projected annual global energy consumption.
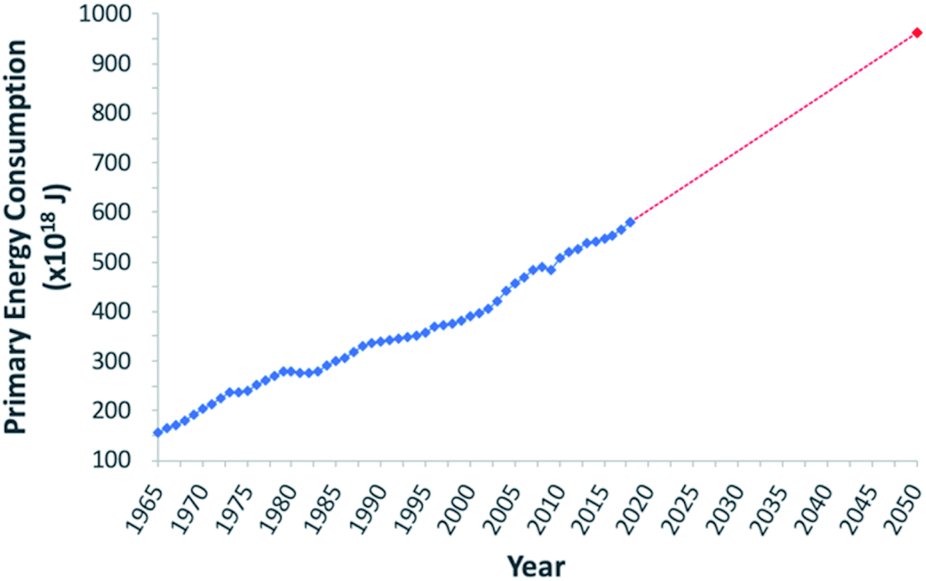 | ||
| Fig. 2 Graphical representation of global primary energy consumption. Blue trend line represents data from 1965–2018. Red dashed trend line shows projected increase between 2018 and 2050. | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) 000–280
000–280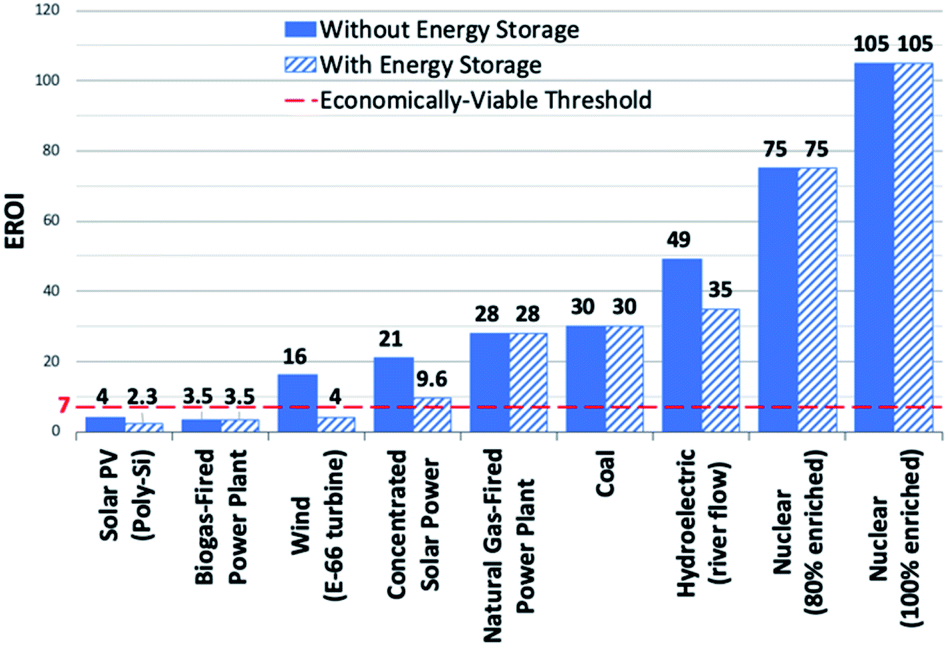 | ||
| Fig. 3 EROI's of electricity generating energy sources. Dashed red line denotes economically-viable threshold. Transportation energy sources not represented in figure. | ||
Towards mitigation of the more pressing issue of growing CO2 emissions, rising surface temperatures, and the ongoing detriment to the ecosystem caused by these factors, it is most prudent for efforts to be directed at the major source of greenhouse gases. According to 2017 energy statistics from the International Energy Agency, nearly 91% of CO2 emissions in this year were due to some form of fuel combustion across all industrial sectors.25 Unfortunately, the renewable energies discussed thus far address needs for electricity generation, though they remain invaluable as alternatives for power production in, for example, industrial and residential sectors. However, much of our global economy and livelihood relies heavily on both agriculture and transportation, economic sectors which require liquid fuels rather than direct electricity supply. The most promising emerging technology capable of filling this niche is solar fuels, serving as both a renewable energy source and combustible fuel with net-zero CO2 emissions. To date, the only industrial-scale renewable fuels in use are agro-biofuels, further illustrating the potential growth opportunities open in this field of research and industry.
Solar fuel processes and subsystems
As early as the turn of the 19th century, scientists of the day began uncovering the secrets of photosynthesis and the metabolic pathway by which autotrophs turn water, CO2, and sunlight into energy and biomass. Though being able to mimic these ubiquitous processes in a laboratory setting has been little more than a curiosity, recent years have witnessed a massive push to realize this objective,† with some of the earliest reports being published in the late 1970's.26,27 While nature's goal is generally to create glucose for plant growth, flower formation, and fruit development, our goal within the scientific community is rather to create solar fuels derived from these simple building blocks.Current research has led to the generation of solar fuels through two general pathways; either by splitting water to form hydrogen (H2) and oxygen (O2) gas, or by reducing CO2 in the presence of a sacrificial proton donor to generate carbon-based gas or liquid fuels. A cursory examination of the physical properties of some of the most common renewable fuels may point to H2 as an interesting option owing to its high energy density and the water which is formed as the sole product of combustion (Table 2); however, a major drawback globally is the lack of existing H2 based infrastructure and storage capacity, a shortcoming which would require major engineering overhauls worldwide. Moreover, observing the volumetric energy density of H2 compared to renewable liquid fuels, such as methanol, reveals that the H2 energy density per litre at STP is in fact 0.013 MJ, whereas the same volume of methanol would release 18.17 MJ upon combustion. The increased energy density of the fuels produced from CO2 in addition to the variety of liquid fuels which could be conveniently integrated to our existing infrastructure, point to the use of CO2 as the ideal feedstock for solar fuel production. An added advantage to this approach is the product diversity accessible, including gaseous fuels which can be used directly or converted to more useful products using known industrial methods (i.e. Fischer–Tropsch fuels), as well as other valuable liquid energy carriers. An initial concern is based upon the fact that creating and using these carbon derived energy sources would result in additional release of CO2 upon combustion, however, such an assumption fails to consider that the model for implementation of these technologies necessitates consumption of CO2 as a feedstock, making these carbon–neutral fuels (i.e. generated CO2 is ultimately recycled to create more solar fuels).32
| Fuel | Phase at STP | Composition | Mass density (kg L−1) | Energy density (HHV)a (MJ kg−1) |
|---|---|---|---|---|
| a Higher heating value: energy released as heat given complete combustion at STP.b HHV is highly variable due to different ratios of component gases and technology used to produce mixture.31 | ||||
| Hydrogen | Gas | H2 | 9.0 × 10−5 | 142 |
| Methane | Gas | CH4 | 6.6 × 10−4 | 55.6 |
| Biogas | Gas | CH4, CO2, etc. | >6.6 × 10−4 | <55.5 |
| Syngas | Gas | H2, CO, CH4, CO, etc. | 9.5 × 10−4 | 3–15b |
| Methanol | Liquid | CH3OH | 0.79 | 23.0 |
| Ethanol | Liquid | C2H5OH | 0.79 | 29.6 |
| Biodiesel | Liquid | Long-chain, mono-alkyl esters | 0.88 | 37.5 |
| Traditional biomass fuel | Solid | Woods, charcoal, agricultural residues, dung, ash | 0.6 | 18 |
| Dry green microalgal biomass | Solid | Proteins, carbohydrates, lipids, ash | 0.6 | 20 |
Despite this field of study having started in earnest merely a decade ago, extensive research has produced impressive results toward realizing this goal and mitigating the growing climate crisis.23,33–36 In assessing the feasible use of various solar fuel technologies, several studies have been conducted to determine factors such as system efficiencies or shipping/transportation costs.37,38 However, this review will focus on recent advances in the photochemical conversion of CO2 using natural or simulated solar radiation as the light source. In characterising the reactions to achieve this transformation, it is useful to identify the disparate activation pathways which facilitate the conversion of the feedstock to target chemicals. This was aptly done in a recent review by van der Zwaan and co-workers, wherein a taxonomical classification was developed for the characterisation of solar fuel generation according to the mode of solar energy harvesting.39 This was conveniently identified as Categories 1–4 (Fig. 4), where Category 1 is identified as concentration, Category 2 as transformation, Category 3 as natural photosynthesis, and Category 4 as artificial photosynthesis. Solar concentration (Cat1) relies upon harvesting solar energy and focusing the incident radiation to generate heat. While the most basic version of this can be demonstrated using a magnifying glass on a sunny day, modern solar concentrators are remarkably sophisticated and continue to be refined. In a further embodiment, transformation (Cat2) is characterised by the conversion of solar radiation into an electrical current; though this category is rather ubiquitous in the form of photovoltaic cells, in the context of solar fuels, this describes the generation of a voltage potential to power an electrochemical cell for the production of solar fuels. Category 3 encompasses natural photosynthetic processes by plants, cyanobacteria, algae, and other autotrophic organisms. This often produces glucose as the main carbon fixation product, useful for biomass generation, but biofuels may also directly or indirectly be generated from many such organisms. Modern advances in biotechnology have allowed for the creation of genetically modified and bioengineered organisms suitable for selective production of solar derived biofuels. While these transgenic species are not naturally occurring, their use for solar fuel production is typically considered to fall under the rubric of natural photosynthesis. Discussions of Cat3 within this review will therefore follow this convention and consider both wild-type and mutant species, as well as genetically modified organisms (GMO's). Artificial photosynthesis (Cat4) is inspired by the processes observed in Cat3, but has been tailored to harvest light energy and directly generate the desired solar fuels using either laboratory-prepared materials or bio-synthetic hybrids. Despite each of these categories being distinctly identified, several solar fuel systems have been developed which encompass more than one of these categories; these will not be discussed in detail in order to succinctly elaborate upon advances within each of the individual components.
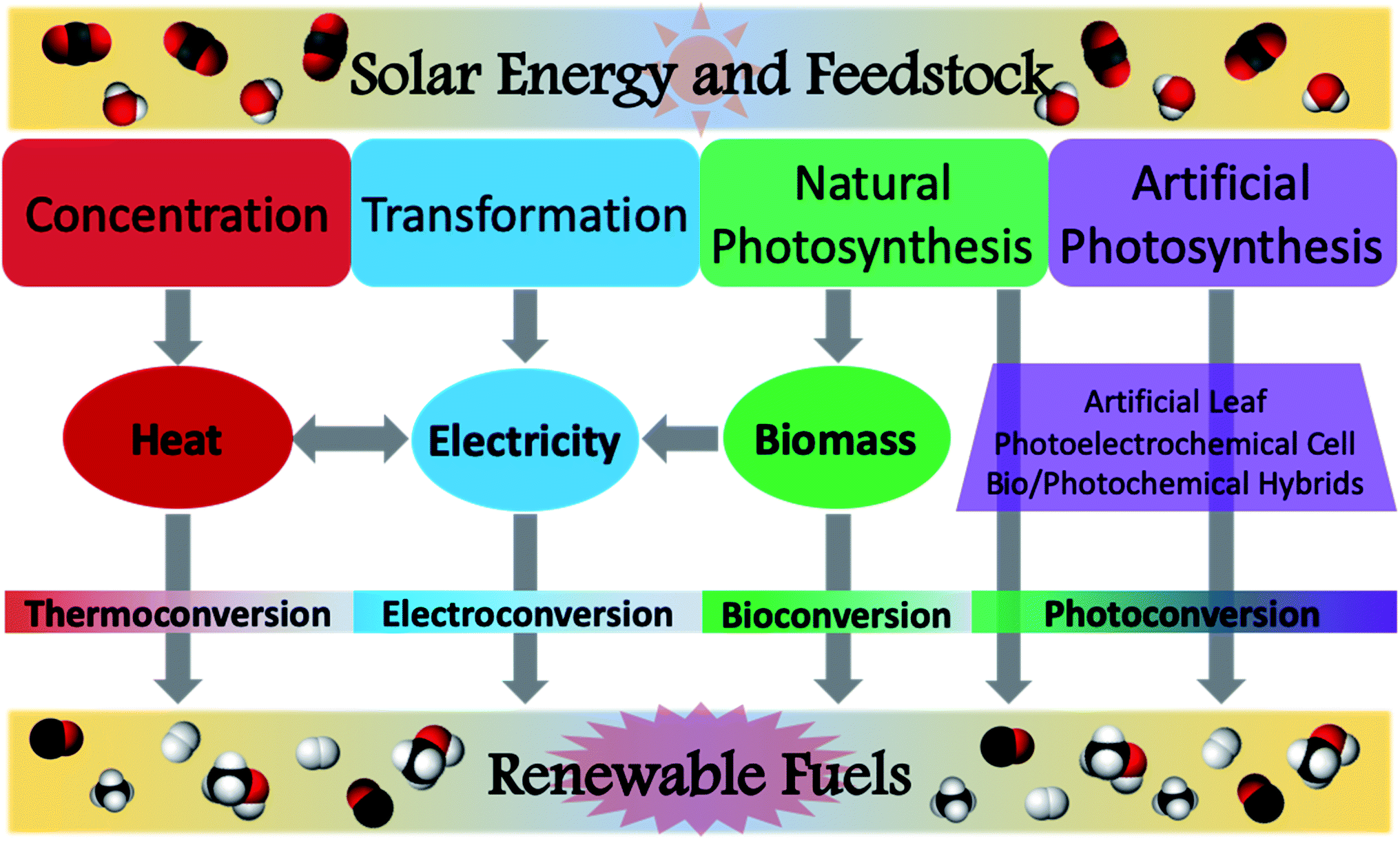 | ||
| Fig. 4 Modes of solar energy conversion of carbon dioxide and water into renewable fuels. Concentration (Category 1): solar radiation is concentrated for the generation of heat; generated heat can be used for thermochemical conversions or toward the generation of electricity. Transformation (Category 2): harnessed solar energy is converted into electricity, then used to power an electrochemical cell. Natural photosynthesis (Category 3): autotrophic conversion of H2O and CO2 into biomass, or direct conversion of feedstock into viable fuels. Artificial photosynthesis (Category 4): use of photoactive materials to harvest sunlight and achieve photoconversion of carbon dioxide and water to renewable fuels. Figure contents and layout adapted from previous literature.39 | ||
In examining the pathways to fuel generation from each of these solar conversion methods, water splitting toward H2 generation has arguably received the most attention and produced several impressive works.40–52 Nonetheless, as aforementioned, CO2 fixation and conversion better addresses current environmental needs while easily integrating into current infrastructures. Thus, the fuels of focus will generally be those presented in Table 2, as well as formate/formic acid. While the latter is not typically considered a fuel source directly, formate has been found to power novel designs of fuel cells,53–58 as well existing as an intermediate or byproduct of the catalytic conversion of CO2 to methanol (MeOH).59–62 Though the photochemical conversion of CO2 may be achieved by numerous synthetic routes, most work has been devoted to the hydrogenation of this substrate to oxygenates and/or hydrocarbons by pathways resembling a Sabatier (eqn (1)) or reverse water–gas shift reaction (RWGS) (eqn (2))
| CO2 + 4H2 → CH4 + 2H2O | (1) |
| CO2 + H2 → CO + H2O | (2) |
Alternatively, products of photochemical reactions may be further converted to higher value fuels by methods such as the Fischer–Tropsch (FT) conversion of syngas into pure hydrocarbons.
Category 1: thermoconversion by solar concentration
The warming effect of sunlight is a concept which is easily understood by all manner of species across the globe. As such, it is only logical that solar radiation can be harnessed to generate the thermal energy necessary to overcome enthalpic barriers for the chemical conversion of CO2 into combustible energy carriers. Currently, this may be achieved via one of two general pathways – either through indirect or direct thermoconversion, with several examples of each appearing regularly in scientific literature.63–68 Direct thermoconversion makes use of photonic thermomaterials which readily absorb incident light and convert this energy to heat, creating a localised temperature gradient at the site of reactivity. Conversely, indirect thermoconversion employs the use of solar concentrators which focus incident light energy (and where desired, filter the radiation to utilise a more narrow wavelength range) to a more concise area in order to heat the bulk reaction media. Developments in methods of direct photothermal reactions have relied heavily on research in pure and applied chemistry, whereas indirect photothermal reactions have advanced as a result of improvements in the engineering and design of solar concentrators. This section will focus more broadly on recent advances in direct thermoconversion, however developments in indirect thermoconversion processes will be discussed.:1 ratio of Fe:Ce at 300 °C (Fe2Ce1-300) showed the greatest activity, with a CO2 conversion of 43.63%, CO selectivity of 99.98%, and a reaction rate of 19.61 μmol g−1 h−1. When the reduction temperature is increased in the range of 300–500 °C, the reaction rate and product selectivity both decrease, generating greater amounts of CH4 owing to the formation of Fe0 during the catalyst synthesis.In order to optimise the amount of CH4 produced instead of carbon monoxide, other light-absorbing catalysts and reaction systems have also been investigated to these ends. Research from the Yu group sought to identify a phase of titania ideal for achieving thermal photoconversion of CO2 into viable solar fuels; using methods developed in their work, titania photonic crystals were formed by anodisation of titanium foil with alternating voltages of 20 and 30 V in thirty minute increments, followed by annealing at 550 °C for two hours, yielding macroporous photonic crystals with air cylinder diameters of 100 nm (Fig. 5).72 Systematic studies were performed using various phases of TiO2, revealing that the anatase titania photonic crystals prepared in this study significantly outperformed both P25 and titania nanotube arrays in the photomethanation of CO2 (35.0, 2.2, and 7.5 μmol h−1 m−2, respectively). The improved reactivity was attributed largely to the slow photon effect, wherein the incident light energy is slowed and propagated more efficiently due to interactions between the light and medium,73 in this case the photonic crystals. Improving even further upon this observed activity, Ye and co-workers presented the use of solid supports doped with various group VIII metals. Impregnation of ultrathin Mg–Al layered double hydroxides with ruthenium nanoparticles generated the highly stable catalyst Ru@FL-LDH.74 Introduction of this catalyst into a flow photoreactor in the presence of CO2, H2, and light irradiation resulted in photothermal methanation at rates as high as 277 mmol h−1 g−1, with sustained CO2 conversions of approximately 95%, and nearly 100% selectivity for CH4 production. Under sustained irradiation, Ru@FL-LDH maintained a surface temperature of roughly 350 °C and impressively retained its catalytic activity over several cycles of substrate introduction. Using an even more ubiquitous support, mesoporous Al2O3 was doped with numerous transition metal nanoparticles including Ru, Rh, Ni, Co, Pd, Pt, Ir, and Fe.75 The catalysts produced (M/Al2O3) were similarly placed in an atmosphere of CO2 and H2 under light irradiation, resulting in rapid photon-mediated heating of the catalyst nanoparticles (300–400 °C) and highly selective photomethanation of CO2. The most notable results were found with the alumina-supported ruthenium, rhodium, and cobalt nanoparticles, revealing a maximum CO2 reaction rate of 18.16 mol h−1 g−1 using Ru/Al2O3, CO2 conversion of 96.25% with Rh/Al2O3, and a CH4 selectivity of 99.51% when the reaction was performed with Co/Al2O3. Interestingly, it was found that these three catalysts in addition to the nickel analogue (Ni/Al2O3) significantly outperformed the palladium, platinum, and iridium nanocatalysts.
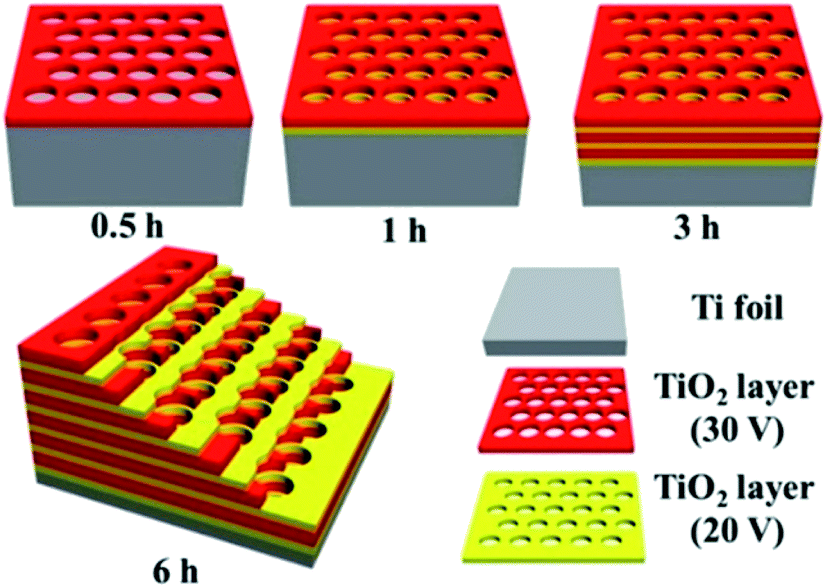 | ||
| Fig. 5 Schematic diagram of titania photonic crystals prepared by anodisation and calcination of titanium foil. Image adapted with permission from ACS Publications.72 | ||
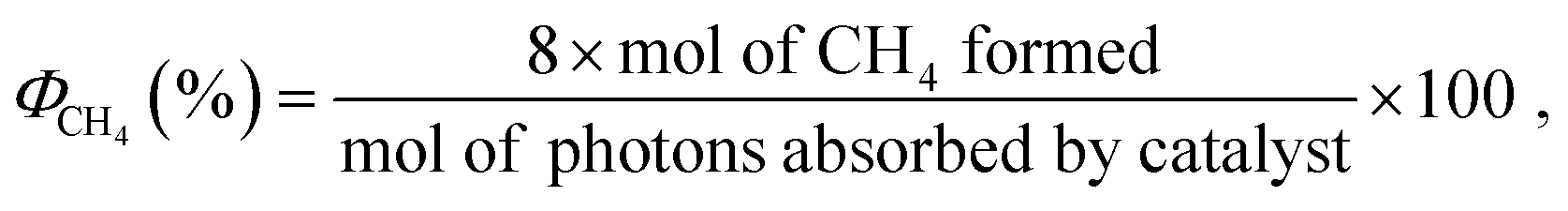 | (3) |
| Fe3O4 → 3FeO + ½O2 | (4a) |
| 3FeO + CO2 → Fe3O4 + CO | (4b) |
| CO2 → CO + ½O2. | (4c) |
 | ||
| Fig. 6 Aerial view of the Quarzazate Solar Power Station in Morocco. Image adapted from cited ref. 76. | ||
 | ||
| Fig. 7 Schematic diagram of solar reactor used in the ceria-mediated photothermal reduction of carbon dioxide. Image adapted with permission from ACS Publications.79 | ||
 | ||
| Fig. 8 Schematic diagram of CR5 used for CO2 photoconversion. *Reactive material is comprised of sets of counter-rotating rings. Current cross-sectional view shows single ring oriented coincident with the plane of this page.83,84 | ||
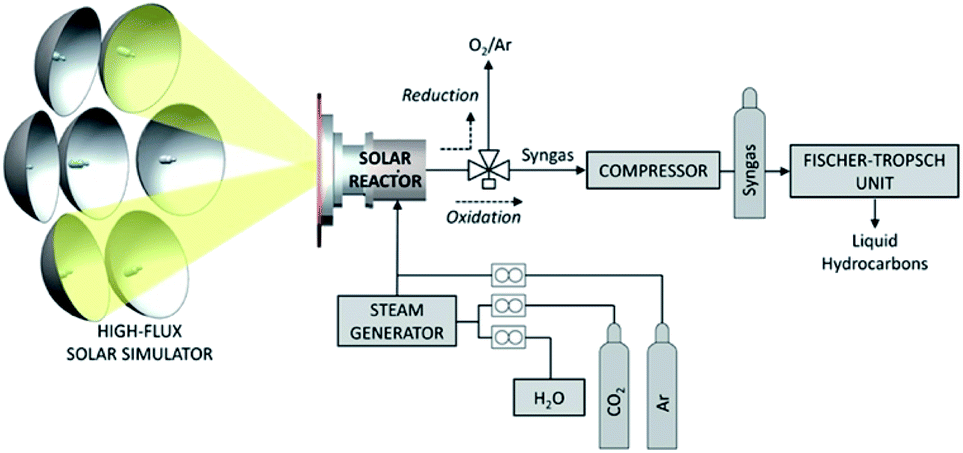
In this reaction, eqn (4a) is the high temperature thermal reduction of the iron catalyst, eqn (4b) is the comparatively lower temperature oxidation of ferrous oxide by CO2, and eqn (4c) represents the net CO2 splitting reaction. In order to generate the high temperatures needed to achieve the initial reduction step, the CR5 is integrated with an 88 m2 parabolic dish, giving rise to the Dish-CR5 system. The CO produced from the photothermal process described above is further processed in the subsequent subsystems which include: (1) a WGS reaction loop, (2) an amine-based CO2 absorption/separation system, (3) a methanol synthesis reactor, and (4) the methanol purification system (Fig. 9). The WGS reactor uses the CO produced from the Dish-CR5 in combination with a water feedstock to produce a H2/CO2/CO syngas mixture. Bubbling the gas mixture through aqueous monoethanolamine removes the CO2, a well-established industrial practice for removing CO2 in coal-fired power plants. The purified syngas mixture is reacted over a Cu/ZnO/Al2O3 catalyst in a fixed bed reactor to produce crude methanol, followed by purification of the methanol through a flash vessel to remove gaseous byproducts and starting material, then standard distillation to achieve the methanol–water separation, yielding 99% pure methanol. Using this setup, a 1050 hectare methanol production facility is proposed which would include a 17622 array of Dish-CR5 reactors including the four subsystems necessary to convert the syngas to methanol. The primary solar energy efficiency of this process was calculated to be 7.1%, significantly higher than other comparable thermoconverters, and the production capacity of this facility is expected to be 82700 Mg of methanol per year, the energetic equivalent of about 51.2 million litres of gasoline (13.5 million US gallons).
 | ||
| Fig. 9 Block flow diagram of photothermal conversion of CO2 to methanol by S2P process.84 | ||
Category 2: electroconversion by solar transformation
Electroconversion is a ubiquitous process owing to the widespread use of commercial and residential solar panels as well as other photovoltaic cells. Rather than using electroconversion toward large scale electricity production, however, the generated voltage may also be utilised downstream in the form of solar fuels. This approach thus combines the broader fields of photo- and electrochemistry and makes use of photoelectrochemical cells to store the electrical energy produced in the form of chemical bonds in combustible fuels.86,87 As with many solar fuel technologies, the splitting of water for the generation of dihydrogen has generally received the most attention and produced several notable works.40,42,44,45,49,88–90 Unlike the water splitting reaction achieved by PEC systems, CO2 reduction is considerably more complex and presents challenges not otherwise present when H2O alone is the substrate of interest.91–101 First, the generally low solubility and mass transfer of CO2 in aqueous and organic electrolytes limits the productivity of most PEC cells. Additionally, when performing the reduction in the liquid phase, only C1 products are detected.102 While we have seen that even C1 products of these reactions can serve as solar fuels or easily be converted into suitable energy carriers, decades of research have revealed that performing this class of reaction at the gas–solid interface (gas-phase reaction medium) is capable of generating ≥C2 products. The steps leading to this realisation have required discovery and extensive optimisation to determine the components necessary to create an efficient PEC cell, and current research continues to try and improve upon this technology. This has (generally) resulted in either sophisticated systems which incorporate light harvesting materials into electrode designs (integrated PEC cell), or wiring a photovoltaic cell to an electrolyser which performs the CO2 reduction reaction (modular PEC cell). Regardless of which approach is used, the constituents of a PEC cell at its most basic level include a light harvester, electrodes, and a catalyst, though each of these is not necessarily mutually exclusive and depends on the cell design. Within the construction of each PEC cell, these components operate in a highly synergistic manner wherein slight variations may result in drastic differences in properties such as efficiency, under/overpotential required, product distribution, and more. As such, the development of PEC cells for solar fuel generation has produced a diversity of works which have arduously investigated unique photoelectrolyser components and designs.While the previous study made use of gold only as a dopant, there remains considerable interest in developing a PEC cell which makes use of only earth-abundant metals. It is toward this goal that the Reisner lab has developed an integrated PEC cell driven by a cobalt-based photocathode.107 The photocathode was comprised of a p-type silicon electrode, mesoporous TiO2 as a scaffold, and an immobilised phosphonated cobalt bis(terpyridine) (CotpyP) catalyst interfaced to the titania layer (Fig. 10). Using the described photocathode in the photoelectrolysis of CO2 revealed strong solvent effects on the product distribution and reaction efficiency. Performing this experiment under anhydrous conditions in acetonitrile with tetrabutylammonium tetrafluoroborate (TBABF4) as electrolyte showed no catalytic turnover, however under aqueous conditions with KHCO3 electrolyte, dihydrogen and formate formed as the dominant reaction products. Systematically varying the H2O content in acetonitrile between 10 and 50% with TBABF4 altered the product distribution to differing ratios of CO, H2, and formate, with the optimum CO production being found at 40% water in acetonitrile. Under these conditions, the turnover number for CO2 conversion (TONCO2) was found to be 381 after 24 hours, with ca. 75% of gaseous product formed being CO. In additionally analysing the H2 and formate formed, it was found that the faradaic efficiency (FE) for CO, H2, and formate was 47.6, 16.7, and 12.8%, respectively, and the photocurrent onset potential under the conditions outlined was −0.44 V. Work from the same group soon elaborated upon this work by developing another photocathode composed of a cobalt porphyrin immobilised on buckypaper (CoMTPP@CNT).108 Incorporation of this cathode with a BiVO4–perovskite photovoltaic device demonstrated the construction of a tandem integrated PEC cell capable of achieving unassisted CO2 reduction to CO with simultaneous water oxidation and no detected production of formate. Under 1 sun irradiation, greatly improved turnover numbers for CO and H2 were achieved (5853 and 30725, respectively), and the total faradaic efficiency was calculated to be 91.9%; impressively, even at 0.1 sun irradiation, turnover numbers and efficiencies for this cell significantly outperformed the previously reported CotpyP photocathode (TONCO = 4546, TONH2 = 1359, FEH2 = 17.2%, FECO = 56.5%, and FEtotal = 73.7%), illustrating the ability for this cell to operate even under low irradiance conditions.
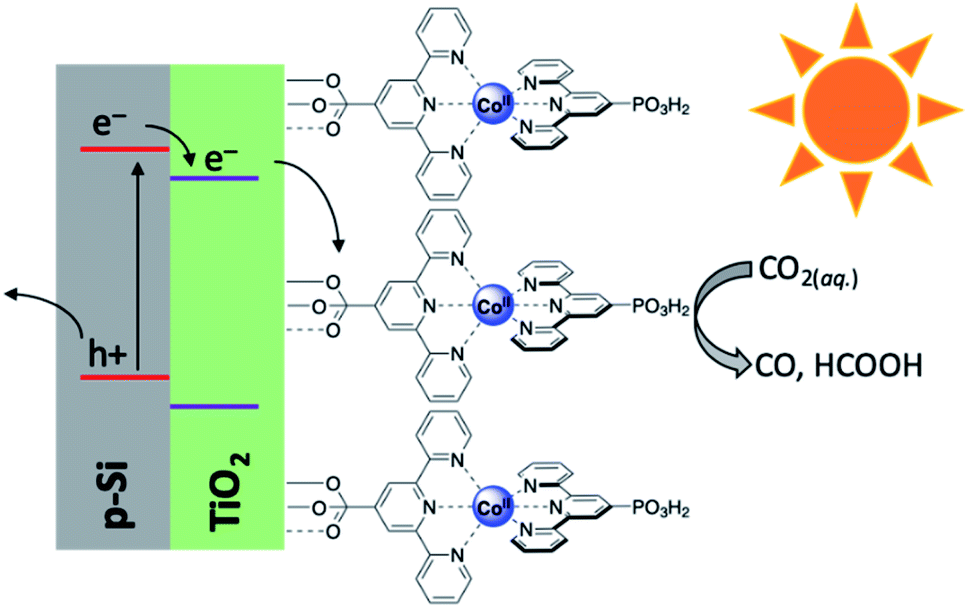 | ||
| Fig. 10 Schematic diagram of p-Si|meso-TiO2|CotpyP photocathode used in photoelectrolytic reduction of CO2.107 | ||
In a modular PEC reactor design, Grätzel and co-workers developed a bifunctional electrode composed of CuO nanowires coated with SnO2 by atomic layer deposition (ALD).109 This work was inspired by previous studies which have shown the utility of CuO as competent electrodes for both electrolytic CO2 reduction and water oxidation;110,111 a chief difference between the practical aspects of these two reactions is the medium in which the reactions optimally occur. For the aqueous reduction of CO2, the oxygen evolving reaction (OER) is most efficient under alkaline conditions, whereas CO2 reduction favours pH neutral conditions.112 In order to maintain ideal reaction conditions for the oxidation and reduction reaction independently, while minimising the risks of electrode degradation, a PEC reactor was designed consisting ALD–SnO2|CuO bifunctional electrodes, a bipolar membrane separating alkaline and neutral reaction media, and a triple junction PV cell (Fig. 11). When the performance of the PEC device was tested in the production of CO at a −0.9 V bias, it was quickly seen that ALD–SnO2 modification of the cathode (two ALD cycles) increased the CO turnover frequency drastically to 0.246 s−1 as compared to the TOFCO of 0.005 s−1 for bare CuO. The product selectivity was also enhanced in this process, evident by the ALD–SnO2|CuO TOFH2 being nearly ten times lower than the CuO TOFH2 (0.020 and 0.190 s−1, respectively). In addition, the number of SnO2 layers deposited on the CuO nanowires proved to be rather consequential; at −0.7 V, maximum CO selectivity was achieved when 2–5 layers of SnO2 were deposited by ALD (∼93% CO), and total current density reached its maximum at 5 layers of SnO2 with caesium electrolytes (∼1.6 mA cm−2). Beyond five ALD layers, the most notable change is the loss in CO selectivity and the increased production of H2 and formate. From calculations using the optimised reaction results, the FECO averaged 81%, the STFCO was equal to 13.4%, and the STFtotal was calculated to be 14.4%. Perhaps equally as notable as the calculated efficiency values is the observation that the prepared electrodes were not seen to degrade to any observable extent after 5 hours of operation, a problem which frequently plagues PEC reactors.
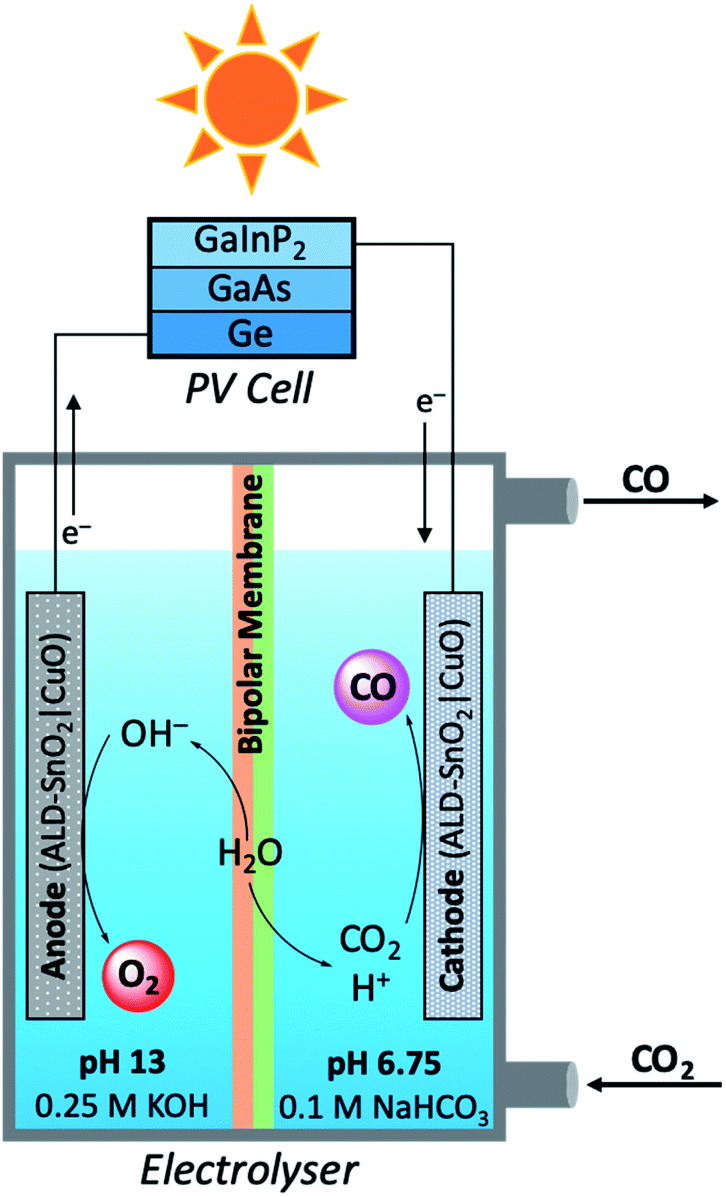 | ||
| Fig. 11 Schematic diagram of modular photoelectrolyser used in carbon dioxide reduction mediated by bifunctional ALD–SnO2|CuO electrodes.109 | ||
In 1994, work from the Bocarsly group discovered that under electrochemical conditions, the pyridinium cation was a surprisingly effective catalyst for the reduction of CO2 into methanol in the presence of a hydrogenated palladium electrode.113 It was theorised that under the described reaction conditions, pyridinium was readily reduced to a pyridinium radical; this intermediate would be expected to provide a source of both protons and electrons, thus allowing for shuttling of these species to effect the production of methanol from CO2. Moreover, the low overpotentials (∼200 mV) required to achieve this conversion made this a particularly appealing route for MeOH formation. It was more than a decade later that the same group integrated the electrochemical reaction described to a photovoltaic device.114 By incorporation of a p-GaP semiconducting electrode in the presence of a pyridinium catalyst, selective photoelectrochemical reduction of CO2 into methanol was achieved with faradaic and quantum efficiencies of nearly 100 and 44%, respectively, at an underpotential of 320 mV relative the thermodynamically predicted half-reaction. Later work by Lee et al. developed photocathodes used within an integrated PEC cell to similarly form methanol as a product of the photoreduction of CO2.115 The photocathodes were constructed of cuprous oxide nanowires (Cu2O NW) overcoated with a crystalline titania layer, (creating a p–n junction between Cu2O and TiO2, respectively), that is further decorated with Cu+ ions, abbreviated Cu2O|TiO2–Cu+ (Fig. 12). A CO2-saturated 0.3 M KHCO3 solution (pH 6.8) served as the reaction medium, and systematic studies of the ideal operating potential showed 0.3 V to be ideal for peak methanol production. The photocathode efficiency of bare Cu2O NW was compared to that of Cu2O|TiO2–Cu+ and showed two particular trends in reactivity; (1) bare Cu2O NW resulted in remarkably slow reactivity after 30 minutes due to photocorrosion, whereas the novel photocathode provided relatively constant reaction rates, and (2) at any point during the reaction timeline, RMeOH of Cu2O|TiO2–Cu+ was at least double that of bare Cu2O NW. Observing the faradaic efficiencies and methanol production for both of these systems over the course of a 2 hour reaction revealed that Cu2O NW showed a value of 23.6% and generated approximately 0.14 μmol of MeOH, whereas the titania overlayed nanowires more than doubled these values with a FEMeOH of 56.5% and produced ∼0.56 μmol of methanol.
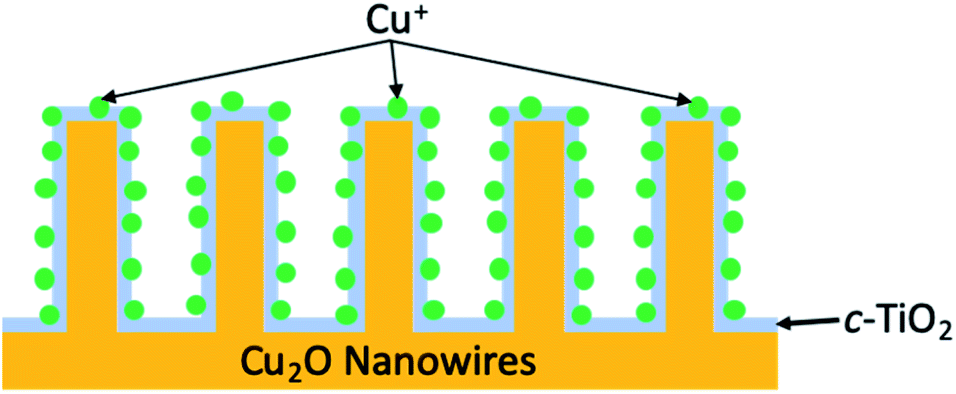 | ||
| Fig. 12 Schematic cross-sectional view of Cu2O|TiO2–Cu+ photocathode. | ||
Away from the classical structural motifs of powders, films, and nanostructures being used as scaffolds for photocathodes, Jing and co-workers utilised a nickel foam support to construct a photoactive cathode for methanol production, with a BiVO4 film used as the photoanode.116 Commercially available nickel foam (f-Ni) sheets served as a support for TiO2 which was later functionalised by amine or imine functionality. This was performed using a titania sol–gel followed by annealing to produce TiO2|f-Ni, which upon reaction with 3-aminopropyltriethoxysilane (APTES) generated the amine-modified cathode NH2–TiO2|f-Ni. Subsequent condensation in the presence of salicylaldehyde produced the imine-derived photocathode CHO–NH2–TiO2|f-Ni, thus providing three different nickel foam electrode motifs which were ultimately studied. When combined in a PEC cell with 0.1 M KHCO3, current densities up to −1.1 V were achieved, well beyond the required −0.6 V needed to achieve both water splitting and CO2 reduction. In this study, the reactivity of each of the aforementioned nickel foam electrodes was compared, and a screening of the bias voltage applied was performed to optimise reactivity and efficiency. It was found that the methanol production rate for the photocathodes increased in the order of: TiO2|f-Ni < NH2–TiO2|f-Ni ≪ CHO–NH2–TiO2|f-Ni. Seeing the latter of these photocathodes as the most productive, a screening of the applied bias voltage displayed remarkable results. At −1.0 V, the methanol production rate was 186.5 μM h−1 cm−2, corresponding to a FEMeOH of 27.3%. If the voltage intensity is decreased to −0.6 V, the RMeOH unsurprisingly is lowered to 153.4 μM h−1 cm−2, however the methanol faradaic efficiency increases drastically to 452.0%, with a FEtotal equal to 507.9%, and a cell quantum efficiency (Φcell) of 1.2%. In addition to methanol product formed in these PEC cells, trace amounts of formic acid, H2, CO, O2, and even ethanol (EtOH) were detected in the reaction mixtures at each voltage investigated. The significantly improved reactivity seen in CHO–NH2–TiO2|f-Ni is attributed to two separate factors. First, this photocathode is found to be a much better light harvester as compared to the other nickel foam electrodes, providing a greater impetus to drive the photocatalytic reactions. Second, the presence of the imine functionality serves as a superior scavenger for dissolved CO2, thus increasing the degree of CO2 adsorption to the photocathode surface.
While the methanol production seen thus far holds considerable promise for the production of useful solar fuels, the ability to directly generate ≥C2 liquid fuels would be even more advantageous owing to the greater energy density contained in such products, e.g. the energy density of ethanol versus methanol (Table 2). This goal is considerably more challenging, especially when beginning from CO2 feedstock, however work such as that presented by the Wang group has presented PEC cells capable of generating ethanol from CO2.117 Powdered samples of zinc telluride deposited on graphitic carbon nitride (g-C3N4/ZnTe) were dispersed on indium tin oxide (ITO) to create the photocathode of their PEC cell. When the photocathode described was irradiated while in a CO2-saturated 0.1 M KHCO3 aqueous solution at a −1.1 V bias potential, ethanol was produced as the major product at a rate of 17.1 μmol cm−2 h−1 (FEEtOH = 79.3%). Several factors were determined to facilitate this unusual, though highly valuable reactivity. The interface of the ZnTe and g-C3N4 creates a type-II heterojunction (staggered band gap), improving the charge separation, and thus the efficiency, of the catalysed reaction. The physical contact between the two materials resulting in the formation of a p–n semiconductor junction is further supported as only CO and H2 were formed when powders of zinc telluride and carbon nitride were utilised in the PEC cell without any chemical or hydrothermal treatment to create the binary cathode; this theory was also supported by the three fold increase in the standard electron transfer rate of g-C3N4/ZnTe as compared to ZnTe alone (26.68 × 10−3 cm s−1 and 7.75 × 10−3 cm s−1, respectively). Finally, a consideration of the binding affinities of the substrate and intermediates aid in providing a mechanistic explanation to the mode of ethanol production; in comparing the two components of the photocathode, it is found using DFT calculations that CO2 has a greater binding affinity to ZnTe, and CO preferentially binds to g-C3N4, more specifically to the nitrogen atoms which are electronically analogous to pyridine nitrogens. Under the PEC reaction conditions, this leads to a pipeline mechanism wherein, (1) CO2 is adsorbed to ZnTe and subsequently reduced to CO on this surface, (2) CO is transferred to g-C3N4, and (3) photo-induced electron transfer occurs from ZnTe to g-C3N4, resulting in a high degree of electron density on the conduction band of carbon nitride which drives the C–C coupling and proton-coupled electron transfer to generate ethanol. Of the byproducts formed in the reduction, propyl alcohol was the most abundant (ca. 3 μmol cm−2 h−1) with only trace amounts of CO and H2 being observed.
 | ||
| Fig. 13 General schematic diagram of hybrid bio-PEC cell.118 | ||
A broader diversity of products was formed by Yang et al. through the use of a nanowire-bacteria hybrid PEC reactor and subsequent reaction with engineered bacterial cells.121 Using a reactor setup similar to that used in the previous reference, titania (photoanode) and silica (photocathode) nanowire arrays were separated by an ion-conductive membrane with the anaerobic bacterium Sporomusa ovata being contained within the catholyte. Under visible light irradiation, the titania semiconductor effected water oxidation to yield protons which traversed the ion membrane; using these protons, electrons produced from the silicon nanowire array, and CO2 feedstock, the S. ovata converted CO2 into acetic acid. If the generated acetic acid was periodically removed from the cell and the lost volume replaced with bacterial growth media, the cell showed indefinite stability up to the maximum 120 hour reaction time. Depending on the culture media used, the hybrid S. ovata PEC cell could generate up to six grams acetic acid per litre of electrolyte volume with FEAcOH values up to 70%. Genetically engineered E. coli was used to convert the acetate to acetyl coenzyme A (acetyl-CoA), which may then be enzymatically converted to numerous value-added chemicals,122 most notable for this review being n-butanol. The n-butanol was produced up to a concentration of 198 mg per litre of growth medium and with a solar-to-fuel efficiency of 0.20%, however other products were produced from this study including amorphadiene, epi-aristolochene, cadinene, and polyhydroxybutyrate polymer. Though these four products reported are not viable solar fuels, they may still serve as suitable CO2-sinks to repurpose greenhouse gas emissions. While the production of acetic acid, acetyl-CoA, and the subsequent enzyme-derived products were all performed separately, it is conceivable that the full process may be streamlined either by possible incorporation into the hybrid PEC device, or by design of a modular reactor to perform these reactions stepwise.
Such a concept as designing a stepwise reaction system was demonstrated from the Schmid research group by incorporating a carbon dioxide photoelectrolyser system with a fermentation module.123 A modular photoelectrochemical cell was developed using an external photovoltaic device to create the charge potential needed to power the electrolyser. The cathode used was a commercially available silver-based gas diffusion electrode, and the anode was an iridium-mixed metal oxide (Ir-MMO) coated titania sheet. While the anolyte and catholyte were both aqueous solutions as is commonly employed in PEC reactors, the cell design used was unique in that a gas channel is present behind the cathode to allow direct introduction of CO2 into the reactor system without concern for the common limitations of CO2 solubility in aqueous media. When irradiated with simulated solar light, the photovoltaic generated 3.65 V which powered the electrolyser and succeeded in reducing CO2 to a syngas mixture. It was found that the faradaic efficiency for this process was nearly 100%, and equally as impressive is the consistent reaction rate observed even after running for more than 1200 hours. Owing to the reactor design, very low levels of oxygen flowed with the syngas product (≪100 ppm); the generated syngas was then flowed into a fermenter, and the products of this fermentation were found to be dependent on the bacterial cultures used in this system. If fermentation was performed with Clostridium autoethanogenum, acetate and ethanol were formed at a rate of 0.81 and 0.035 mmol h−1, respectively. When the bacterial cultures used were C. autoethanogenum and C. kluyveri, the products of syngas fermentation were acetate, ethanol, butyrate, butanol, hexanoate, and hexanol, each produced at rates of 1.45, 0.6, 0.21, 0.14, 0.05, and 0.04 mmol h−1, respectively. This work has also presented an industrial design for implementation of this technology which would potentially produce butanol and hexanol at a scale of 10000 tonnes per year, illustrating the viability of this solar fuel generation methodology. The researchers additionally pose questions regarding further optimisation of this process by the use of other bacteria or yeast species, perhaps foreshadowing elaboration of this technology toward greater selectivity for higher alcohols and other CO2-derived solar fuels.
Category 3: biotransformation by natural photosynthesis
Photosynthesis is undoubtedly the original solar fuel generating technology with plants representing the prime example, fixing CO2 and water to generate glucose as a basic energy source for metabolism. Humanity has long exploited this process as a food source, but additionally as a fuel source to achieve combustion. Primordial biomass combustion was likely performed using wood or other lignocellulosic material, however in the millennia to follow, society has sought to harness cleaner biomass sources as an energy source. By investigation of plants, cyanobacteria, algae, and similar autotrophic organisms, the scientific community has unveiled combustible biofuels created as photosynthetic products from such species; in addition, as the state of the art in bioengineering has improved exponentially over recent decades,124–126 our ability to tailor autotrophs toward production of specific photosynthetic products has reached an impressive level of sophistication.127–133 In developing the ability to harness natural photosynthesis to generate solar fuels, either using wild-type or genetically modified organisms, several challenges must be mitigated. Some of the most significant of these include blocking photosynthetic pathways which may favour cell growth and biomass production over solar fuel generation, and the selection or engineering of organisms which are not easily poisoned by the fuel generated in a given process. Despite the complexity involved in addressing these concerns, studies of recent years have proved ingenious in their ability to overcome such limitations, especially through the use of cyanobacteria.A 2012 report demonstrated that a recombinant cyanobacteria (Synechoscystis 6803) was capable of generating ethylene by overexpression of the Pseudomonas ethylene forming enzyme (EFE), albeit at modest rates.134 This work inspired the development of an engineered bacterium (JU547) possessing an enhanced ribosome binding site in the EFE toward forming an ethylene sink in the bacterial metabolism, resulting in significantly enhanced specific ethylene production rates.135 The mechanism of ethylene (C2H4) production was further investigated to determine the factors which improved C2H4 production in the in JU547 over Synechocystis 6803 and other wild-type cyanobacteria; it was found that the citric acid cycle (CAC) in JU547 operates in a closed cycle, whereas this system exists as a bifurcated cycle in wild-type cyanobacteria. The higher efficiency of the engineered bacterium is evident by the greater CO2 flux through the CAC, showing 37% total fixed carbon in the closed cycle, a nearly 3-fold increase over the 13% total fixed carbon in the bifurcated system. This was ultimately manifest in the in the ethylene production rate of 718 μL L−1 h−1 under 730 nm light irradiation.
Toward forming liquid fuels with more direct utility, Deng and Coleman integrated the photosynthetic utility of cyanobacteria with the ethanol production of a particular prokaryotic bacterium.136 The cyanobacteria used in this study was Synechococcus sp. strain PCC 7942, a unicellular organism well known to readily uptake foreign DNA, either through vector shuttling or homologous recombination;137 the prokaryote employed was Zymomonas mobilis, an especially useful bacterium for this purpose owing to its abundance of the enzymes pyruvate decarboxylase (PDC) and alcohol dehydrogenase II (ADH). In wild-type organisms these genes are necessary for regeneration of NAD+ for glycolysis under aerobic conditions in fungi, yeasts, and higher plants, though in organisms which produce high amounts of these enzymes, ethanol production has been seen. Using the E. coli plasmid vector pCB4, the PDC and ADH genes of Z. mobilis were cloned into this shuttle vector and incorporated into PCC 7942. In preparing the variants of PCC 7942, PDC and ADH gene expression could be controlled by either the cyanobacterial rbcLS promoter (the operon encoding for the subunits of ribulose-1,5-bisphosphate carboxylase/oxygenase) or the E. coli lac promotoer (the operon required for lactose transport and metabolism). The pCB4-Rpa cell line displayed PDC and ADH expression through the rbcLS promoter, whereas the pCB4-LRpa and pCB4-LR(TF)pa cell lines effected gene expression by combinations of control between rbcLS and E. coli lac promoters. These three cell lines, in addition to a pCB4 control group, were cultured in the presence of light for 21 days at 30 °C, after which cells were harvested to determine PDC and ADH activity from cell lysates, and the culture medium was analysed to determine the ethanol produced by PCC 7942 as a function of final ethanol concentration (Table 3). The highest production activity for the hybrid cyanobacteria was found for pCB4-LR(TF)pa, yielding a final ethanol concentration of 1710 μM (0.23 g L−1). Upon further investigation of the mechanism of selective ethanol production, it was determined that the presence of the PDC and ADH genes introduced a new reaction pathway not seen in the unmodified Synechococcus sp. PCC 7942 (Fig. 14). The metabolic activity of the cyanobacterium continues to rely on classical photosynthetic steps, including the Calvin cycle; this would generally produce 2-phosphoglyceric acid, which is subsequently transformed into phosphoenolpyruvic acid, then in to pyruvate, both of which may be transformed into other products which fuel the CAC. Rather, in the PDC/ADH-modified PCC 7942, the presence of PDC facilitates transformation of pyruvate into acetaldehyde, and the ADH enzyme converts this to ethanol. Perhaps most notable in this study are the conditions employed to achieve the fermentation to produce ethanol. While many natural photosynthetic processes only produce ethanol under dark, anaerobic conditions, the cyanobacterial–prokaryotic hybrid presented here produced this solar fuel during oxygenic photosynthesis without the need for any other special reaction conditions being applied.
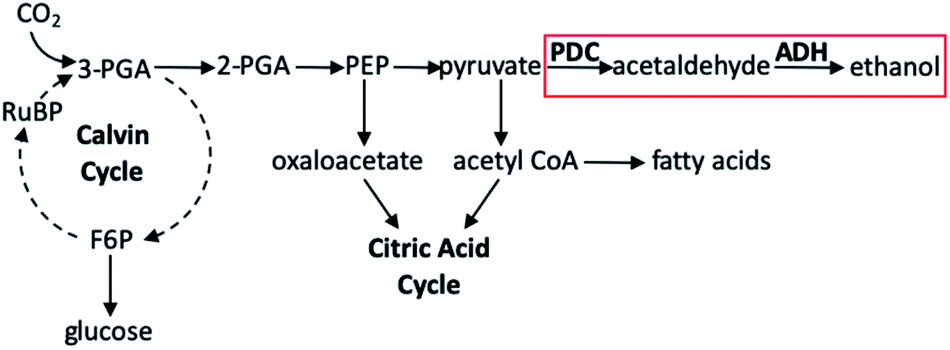 | ||
| Fig. 14 Partial diagram showing photosynthetic steps. Pathway in red box is added pathway from Synechococcus sp. strain PCC 7942 genetically modified with Z. mobilis PDC and ADH enzymes. Abbreviations: PGA, phosphoglyceric acid; F6P, fructose-6-phosphate; PEP, phosphoenolpyruvic acid; RuBP, ribulose-1,5-biphosphate.136 | ||
This work was further improved upon in the years to follow using Synechocystis sp. PCC 6803 by Dexter and Fu,138 as well as Duhring et al.,139 ultimately showing ethanol production as high as 0.46 and 3.60 g L−1, respectively. These studies exploited the endogenous ADH enzyme found in PCC 6803, however a 2012 study by Gao et al. successfully combined aspects of these works with the genetic modifications performed on PCC 7942 to produce a recombinant cyanobacterium with even greater ethanol productivity.140 A total of nine variants of PCC 6803 were produced in this study, but systematic modification revealed the most active mutant and elucidated the pathway by which the increased ethanol productivity was achieved. In a similar manner to the aforementioned research by Deng and Coleman,136 the alcohol producing pathway found in Z. mobilis was introduced to PCC 6803 by encoding the genes for PDC, ADH, and the promoter rbcLS into the slr0168 site141 of the cyanobacterial genome, producing the Syn-XT43 strain. Comparing the photosynthetic behaviour of this strain to a previously reported Syn-LY2 strain of Synechocystis142 provided valuable insight to the following iterations of PCC 6803 which were produced. Culturing the cells of both Syn-XT43 and Syn-LY2 under identical conditions in an atmosphere of 5% CO2 in air resulted in similar growth rates, however the cell density of the former strain was roughly half of that in Syn-LY2. Given that Synechocystis can tolerate ethanol concentrations up to 10.6 g L−1 without any notable impacts on cell growth,138 ethanol accumulation should have no direct effects on cell growth in this strain. The observed disparity of cell density between these strains was explained by one of two theories: (1) the carbon resources in Syn-XT43 are selectively utilised to produce ethanol rather than biomass, or (2) acetaldehyde accumulation may occur in the medium as a result of reverse catalysis of ADH,143 thus resulting in cell toxicity. Measuring the ethanol productivity of Syn-XT43 when sparged with 5% CO2 in air showed 0.4 g L−1 of EtOH generated, roughly four-fold greater than found when sparging with air alone. The second-generation cyanobacterial hybrid (Syn-ZG25) was formed by overexpressing the endogenous alcohol dehydrogenase gene of Synechocystis sp. PCC 6803 (slr1192) rather than transplanting the ADH gene of Z. mobilis. When cultured under the same conditions as Syn-XT43, the ethanol productivity of Syn-ZG25 was found to be roughly 50% higher (0.6 g L−1). Motivated by a previous study which diverted carbon flux toward poly-β-hydroxybutyrate (PHB) production and away from the glycogen pathway,144 Gao et al. further modified Syn-ZG25 to favour the ethanol-production pathway. The first attempt at this was performed by incorporating the PDC gene from Z. mobilis and the endogenous slr1192 gene into the regions coding for polyhydroxyalkanoate-specific β-ketothiolase (phaA or alr1993) and polyhydroxyalkanoate-specific acetoacetyl-CoA reductase (phaB or slr1994), forming the Syn-HZ23 strain of PCC 6803. These modifications were expected to block the PHB synthetic pathway, thus favouring ethanol production; surprisingly, blocking the PHB pathway did not show any appreciation in ethanol productivity as compared to Syn-ZG25. Conversely, when two copies of Z. mobilis PDC and endogenous slr1192 were placed at both the slr0168 site and the location of the phaAB gene, yielding the Syn-HZ24 strain of PCC 6803, the ethanol productivity increased drastically up to 5.50 g L−1. This result also served to further support the hypothesis that greater expression of pyruvate decarboxylase and alcohol dehydrogenase in cyanobacteria aids in generating greater amounts of ethanol by directing the carbon flux toward this metabolic pathway. When observing the ethanol production of the Syn-HZ24 mutant under the above described aerobic conditions (5% CO2 in air), it was found that the production dropped drastically after 15 days, however, if anoxic conditions were used by sparging with 5% CO2 in dinitrogen, ethanol productivity remained relatively constant over the course of 30 days. This finding is especially useful in understanding the parameters necessary for potential future industrialisation of this process using this or other genetically modified organisms.
Toward the production of more diverse polyols using GMO's, Atsumi and co-workers utilised Synechococcus elongatus PCC 7942 as the host organism as was previously demonstrated by Deng and Coleman.136 Rather than targeting ethanol production, however, 2,3-butanediol (23BD) was the product of choice in this study.145 Thorough review of precedent literature provided a feasible pathway to achieve this transformation starting from pyruvate (created as a product of the Calvin cycle), generating acetoin as an intermediate product, and ultimately yielding the desired diol (Fig. 15). The first step toward engineering PCC 7942 conversion of CO2 into 23BD required constructing a biosynthetic pathway to acetoin production; using E. coli as a vector shuttle, alsS and alsD genes possessing the expressed acetolactate synthase (ALS) were transferred to PCC 7942 to create the S. elongatus mutant 2-acetolactate decarboxylase (ALDC) proteins. In all PCC 7942 mutants created in this study, the source of the alsS gene was B. subtilis, however, alsD originated from one of six bacterium, allowing for a systematic investigation of the acetoin production as a function of the gene source. The control strain wherein only alsS was expressed produced only 0.2 g L−1 acetoin. The low level of acetoin yielded suggests that autodecarboxylation of (S)-2-acetolactate is the source of product rather than an enzyme-driven process.146 When alsD was coexpressed with alsS, the acetoin concentration increased up to ten-fold based on the gene carrier used. The highest activity was seen when the alsD gene originated from Aeromonas hydrophila (21.0 g L−1), followed by Gluconacetobacter xylinus (17.8 g L−1), Bacillus licheniformis (16.7 g L−1), Enterobacter aerogenes (16.0 g L−1), and Bacillus subtilis (6.6 g L−1). It was found that the Enterobacter cloacae alsD gene was not active in the E. coli vector and was not transferred to PCC 7942, resulting in no discernible increase in acetoin activity. From these results, the PCC 7942 mutant bearing the B. subtilis alsS and A. hydrophila alsD genes were used in subsequent modifications.
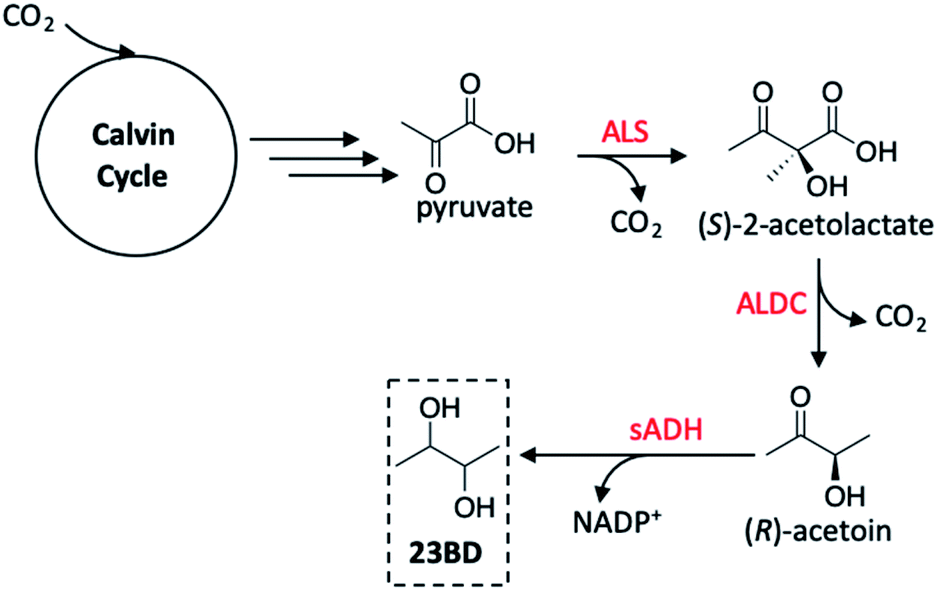 | ||
| Fig. 15 2,3-Butanediol (23BD) production pathway in Synechococcus elongatus PCC 7942. Red reagents are enzymes effecting pyruvate to 23BD conversion. sADH may exist as the (R) or (S) isomer, making the possible products (R,R), (meso), or (S,S)-23BD. The (S,S)-isomer is generated by spontaneous conversion of (S)-2-acetolactate to diacetyl, ALDC-mediated conversion to (S)-acetoin, then reaction with (S)-sADH to generate (S,S)-23BD. Abbreviations: ALS, acetolactate synthase; ALDC, alpha-acetolactate decarboxylase; sADH, secondary alcohol dehydrogenase.145 | ||
Although acetoin is a valuable chemical to be produced from CO2, its toxicity toward S. elongatus makes this an unattractive process as this would require continuous removal of product from the culture medium. To generate a more useful product from this system, the incorporation of a secondary alcohol dehydrogenase protein (sADH) was investigated to facilitate the rapid conversion of acetoin to 23BD. In considering the ideal candidates for the gene transfer, additional factors were considered including low oxygen sensitivity, NADPH-dependence (as this characteristic is expected to allow for greater bioavailability during photosynthesis), and the stereospecificity of the enzyme used depending on the desired product. When studying the 23BD productivity to determine the ideal strain for this reaction, it was important to consider both acetoin and 23BD production rates given that slower production of the diol as compared to acetoin would result in a bottleneck in the biosynthetic pathway and toxicity to the bacterial cultures. Four plasmids were tested to create sADH expression in PCC 7942, resulting in the greatest 23BD production in the alsS (B. subtilis)/alsD (A. hydrophila)/adh (T. brockii) strain at a concentration of 952 mg L−1 with limited acetoin accumulation (61 mg L−1). In long-term studies on the production of 23BD, this strain generated 1.97 g L−1 of the target product over three days at an average rate of 7.757 mg L−1 h−1, however the productivity of the alsS (B. subtilis)/alsD (A. hydrophila)/adh (C. beijerinckii) strain had an even higher 23BD productivity of 2.38 g L−1 after three days with an average rate of 9.847 mg L−1 h−1. This level of 23BD production was sustained for up to 21 days, after which the activity dropped off sharply. Replenishing the culture medium unfortunately did not accelerate 23BD production from the diminished rates, likely arising from spontaneous mutations which revert the cyanobacterial carbon flux toward the natural metabolic pathway of the organism. Though not yet scalable to an industrial process, these findings provide insight to routes of genetic engineering in autotrophs which may aid in improving productivity rates, diversifying products formed to a host of useful solar fuels, and increasing the longevity of the organisms used or created as solar fuel generators.
Category 4: photoconversion by artificial photosynthesis
Attempts to replicate natural photosynthesis in a man-made, laboratory-scale setting has been a long-standing endeavour of the scientific community. To realise this goal, several attempts have been made to create artificial photosynthesis wherein incident light energy facilitates direct photoconversion of CO2 into viable solar fuels.63,95,147–155 When considering the general mechanism of chemical reactivity of Cat4 on a quantum level, there are striking similarities to Cat2, especially when comparing to an integrated PEC device. While the following delineation could be qualified as a generality, we will consider such devices which do not require a bias potential or possess a dedicated photoelectrode as Cat4 systems for solar fuel generation. Within this category, researchers have subdivided developed technologies in several ways, e.g. suspended photocatalytic powders versus photocatalysts deposited on a surface. In this review, Cat4 systems will rather be considered based upon the nature of the catalyst used as either (1) homogeneous catalysts, (2) heterogeneous catalysts, or (3) hybrid bioinorganic catalyst. Each of these subcategories possess inherent advantages and weaknesses which will be discussed in further detail below.Even in relation to solar fuel generation, each of these factors continue to play a role, yet depending on the reaction, the shortcomings discussed can be easily mitigated. In the conversion of CO2, gaseous products can easily be separated from the liquid reaction mixtures. Naturally, when the products are liquid fuels, the challenges of separation/purification remain present, however it is most often seen that homogeneous photoconversion of CO2 yields either carbon monoxide or syngas mixtures, thus eliminating this challenge. Collaborative work from the labs of Neta and Fujita prepared a series of cobalt and iron porphyrins (MP) toward developing a homogeneous CO2 reduction catalyst (Fig. 16).156 Screening the activity of the CoTTP and FeTTP metalloporphyrins revealed that maximum conversion occurred in a CO2-saturated acetonitrile solution containing 5% triethylamine (TEA) as a reductive quencher, 3 mM para-terphenyl (TP) as a sensitizer, and 50 μM of the selected metalloporphyrin. Upon photoactivation, it was found that syngas was formed as the major product of the artificial photosynthesis; the Co3+ analogue showed an initial CO and H2 production rate of 0.45 and 0.2 mmol L−1 h−1, respectively, whereas the Fe3+ metalloporphyrin generated these gases at an initial rate of 0.84 and 0.1 mmol L−1 h−1, respectively. After 20 hours of photolysis, the final CO:H2 ratios for CoTTP and FeTTP were 3.1:1.6 and 2.1:3.4, respectively, and it was found that while CO production had generally ceased after this time, H2 production from FeTTP continued thereafter. Kinetic and mechanistic studies of this reaction showed that the precatalyst metalloporphyrins are activated by photolytic reduction, yielding M0L2− which subsequently reduces CO2. The TP introduced to the reaction mixtures serves to increase the quantum yields of MP and CO2 reduction by generating a radical anion (TP˙−) via TEA-mediated reduction. The abovementioned TP radical is found to effectively reduce the MP to its M0P2− state. The workers found that accumulation of CO in the reaction medium inhibits further reduction of CO2, likely due to CO binding to the metal centre in the MP complex. By screening the dozen MP complexes in this study under these conditions, the highest conversion was found using a 90 μM solution of CoT3FPP, yielding nearly 6 mmol L−1 of CO in the final reaction mixture. More than a decade later, Bonin et al. similarly showed the utility of metalloporphyrin complexes as homogeneous catalysts for the generation of solar fuels using modified iron(III) porphyrin complexes (Fig. 17). In a standard experiment, the turnover numbers (TON) and frequencies (TOF) of CO and H2 generation were studied as a function of the presence of a weak Brønsted acid, trifluoroethanol (TFE).157 The addition of this reagent was attempted to mimic the catalytic activity found for some works which apply a voltage bias. The results from this work showed that the addition of TFE did not generally improve catalytic activity or selectivity (Table 4), however, the iron porphyrins used were found to be comparable to other systems utilising electrochemical reductions of CO2 (ref. 159 and 161) without the need for rarer, more costly metal complexes.162–165 Unfortunately, the presence of the weak acid resulted in the addition of a catalyst degradation pathway in the proposed mechanism which greatly limited the lifetime of the active catalyst in the cycle. In a further improvement upon this work, the Brønsted acid was removed from the system and an organic photosensitiser, 9-cyanoanthracene (9-CNA), was introduced.158 The reaction was performed in CO2-saturated solutions of acetonitrile containing 2 μM FeCAT, 0.05 M of TEA, and 0.2 mM 9-CNA. After irradiation with visible light, CO production was consistently achieved over the course of 50 hours with no concomitant formation of H2; graphical analysis of the TON for carbon monoxide production revealed a linear plot, indicating that no degradation of the system occurred over the reaction time, a significant improvement over the system using TFE, and either comparable or better than previously reported systems which made use of Ru–Re dyads.166 Using 9-CNA as the photosensitiser, a TONCO of 60 was found, and a 100% product selectivity for carbon monoxide was found. Another common photosensitiser, fac-(tris-(2-phenylpyridine))iridium(III) (Fig. 18), was used in this study and ultimately showed superior TONCO of 140, however the product selectivity dropped to 93% in this case. Mechanistic studies of this process allowed for the proposal of a reaction mechanism using 9-CNA (Fig. 19).
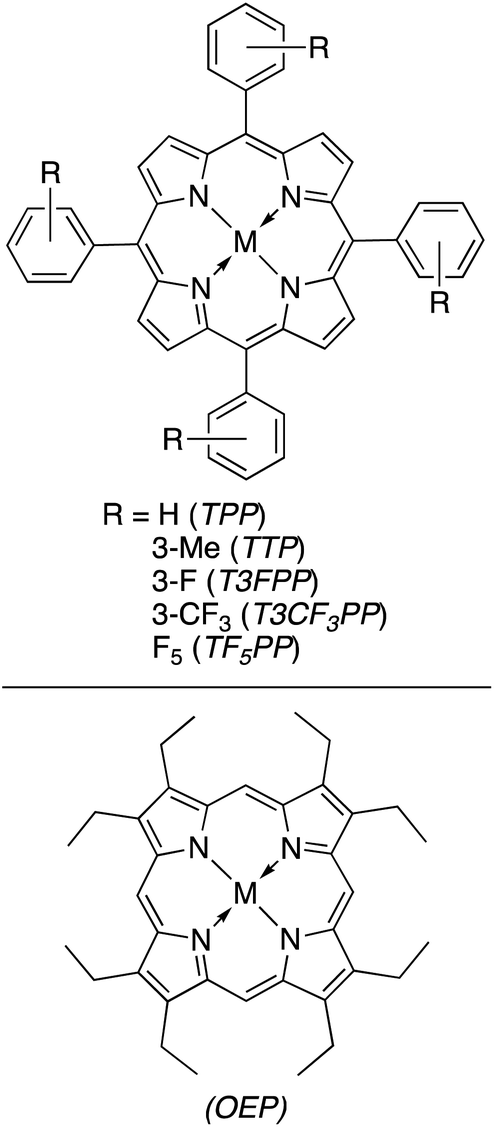 | ||
| Fig. 16 Metalloporphyrins used in the photoreduction of CO2.156 | ||
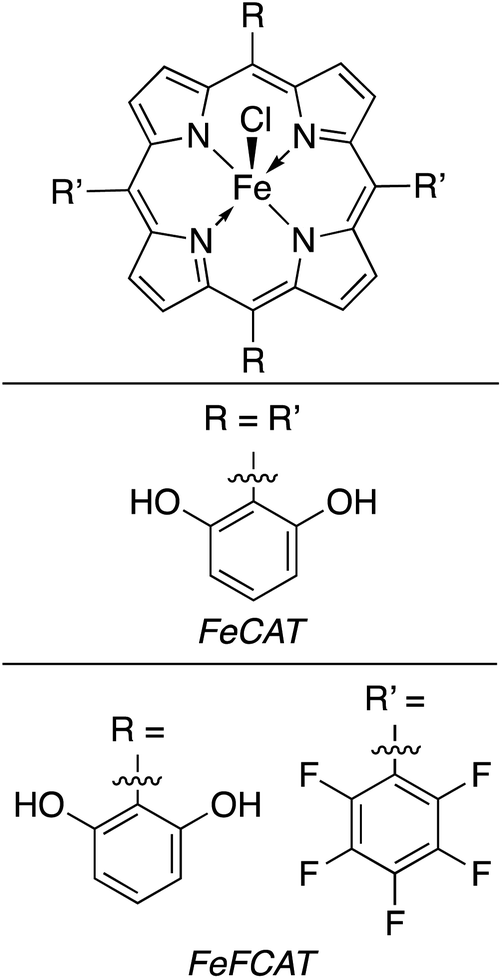 | ||
| Fig. 17 Iron(III) porphyrin complexes used by Bonin et al. in photocatalytic conversion of CO2.157,158 | ||
| Catalystc | TON (H2) | TON (CO) | ||
|---|---|---|---|---|
| No TFE | 50 mM TFE | No TFE | 50 mM TFE | |
| FeTPP | 37 | 23 | 17 | 7 |
| FeCAT | 10 | 10 | 28 | 30 |
| FeFCAT | 15 | 12 | 23 | 23 |
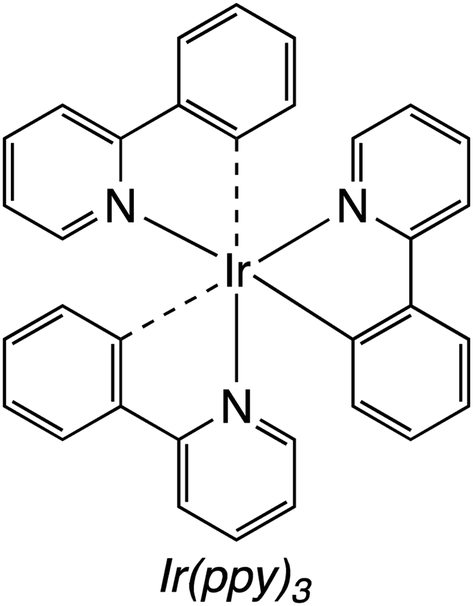 | ||
| Fig. 18 Iridium photosensitiser (Ir(ppy)3) studied in the FeCAT-mediated photoconversion of carbon dioxide to carbon monoxide.158 | ||
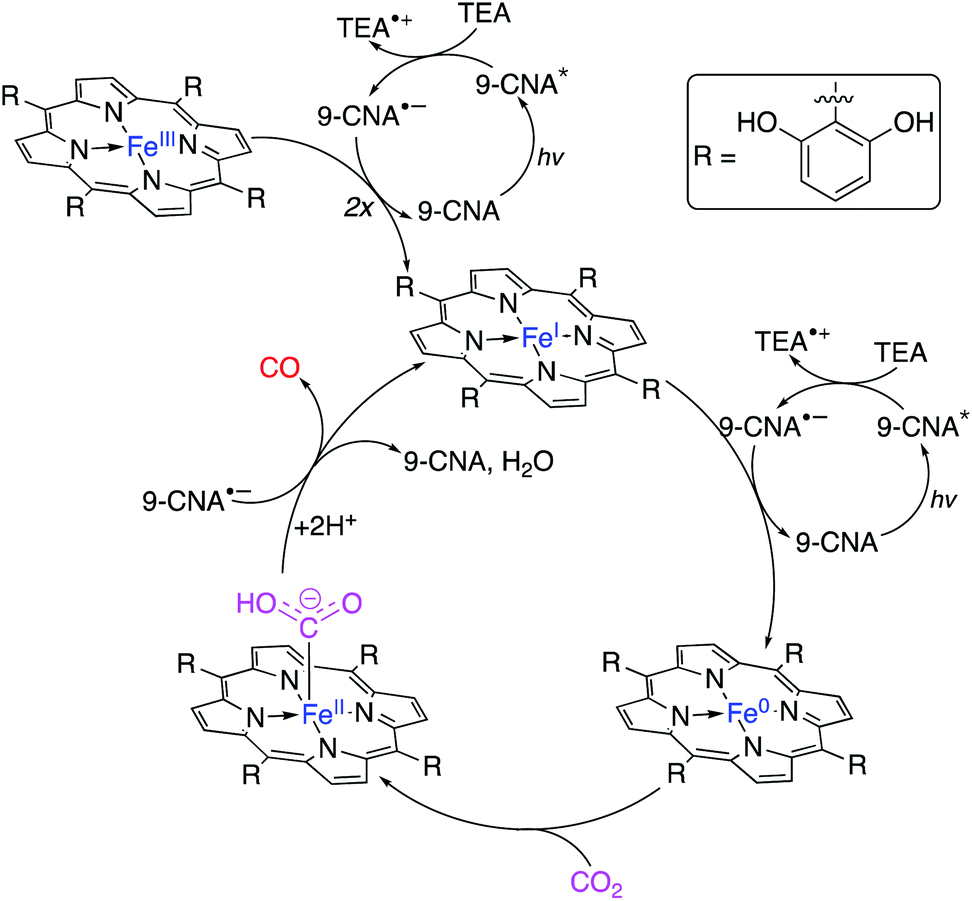 | ||
| Fig. 19 Proposed mechanism of CO2 photoconversion to carbon monoxide by FeCAT in the presence of 9-CNA photosensitiser.158 | ||
Photoexcitation of 9-CNA followed by reductive quenching by TEA yields the 9-CNA radical anion. Two equivalents of 9-CNA˙− reduce the FeCAT precatalyst to the iron(I) porphyrin active complex; upon reduction with an additional equivalent of 9-CNA˙−, Fe0CAT is formed which readily binds with CO2. Electron transfer from 9-CNA˙− and proton transfer from protonated TEA, both generated in situ from the photoactive steps, produces a FeIICAT–CO adduct which, upon additional electron transfer, releases the formed CO with concomitant formation of 9-CNA and water, and regeneration of the active catalyst.
In an attempt to create liquid fuels using homogeneous photoconversion, the MacDonnell research group developed an aqueous catalytic system to convert CO2. The catalytic mixture consisted of solvent water, ruthenium(II) trisphenanthroline (Ru(phen)3) as chromophore, pyridine (py) as the CO2 reduction catalyst, KCl as electrolyte, and ascorbic acid (AA) as the sacrificial reductant.167 The pH of the solution was adjusted using NaOH solution, and blue LED lights (470 ± 20 nm) were used to irradiate samples. The production of methanol and formate under these conditions was systematically investigated by varying concentrations of reagents, and by the addition of metal cocatalysts. Partial optimisation of the reaction showed that a minimum pyridine-to-ruthenium ratio of 100:1 is needed to achieve efficient conversion, and the optimum pH for the reaction is approximately 5.0. In addition, the use of potassium salts in the reaction was found to enhance the yield of methanol and formate considerably, however, using other alkali and alkali earth ions had no appreciable affect. After fully optimising this reaction, the ideal conditions were found to be 0.20 mM Ru(phen)3, 200 equivalents of pyridine (40 mM), 0.1 M ascorbic acid, and 0.1 M KCl. After adjusting the pH to 5.0 and irradiating with visible light, both methanol and formate were detected in the reaction mixture. After one hour under these conditions, formate was detected as the favoured product with a TON of 76 and a solution concentration of 18 mM; if irradiated for 6 hours, methanol was formed in significantly greater amounts with a TON of 0.33 and a final concentration of 66 μM. Interestingly, beyond six hours, little to no reaction was observed, likely owing to chromophore degradation. In a parallel set of experiments, small amounts of solid metal catalyst was added to the reaction mixture to determine if these would augment methanol production. In all cases (Pt, Pd, Ni and Au on carbon black), the addition of these metals proved inferior to the mixture without the precious metal catalysts, and the additional of colloidal platinum yielded no methanol from the reaction whatsoever.
In another example of selective formic acid formation from a photoactive homogeneous catalyst, Tamaki et al. prepared supramolecular complexes based upon the ruthenium(II) ion which contained both a photosensitiser subunit and a catalyst subunit.168 The photosensitiser base was comprised of RuII(bpy)3 units, the catalyst of RuII(bpy)2(CO)2, and the larger supramolecular structure was constructed by tethering different ratios of these two components together through alkyl bridges which joined adjacent ruthenium(II) centres by the bipyridine ligands. Using 1-benzyl-1,4-dihydronicotinamide (BNAH) and 1-(4-methoxybenzyl)-1,4-dihydronicotinamide (MeO–BNAH) as NADH model compounds to facilitate electron donation, various conditions were screened to optimise the CO2 reduction process. The most productive of these compounds possessed two photosensitiser units to each catalytic unit (2:1) and utilised MeO–BNAH as the electron donor (Fig. 20). In a 4:1 mixture of N,N-dimethylformamide/triethanolamine (TEOA), a CO2-saturated solution containing a concentration of 0.1 M reductant and 50 μM photocatalyst (2:1) was irradiated for 5 hours, yielding 36.8 μmol of formic acid, with small amounts of CO and H2 (2.4 and 1.9 μmol, respectively). By comparison, using BNAH as the electron donor produced 30.4, 1.8, and 1.8 μmol of formic acid, carbon monoxide, and dihydrogen, respectively. When the (1:2) supramolecular complex was used in the presence of BNAH, the activity dropped drastically, yielding only 8.4 μmol of formic acid with comparable amounts of CO and significantly smaller amounts of H2. In more closely observing the (2:1)/MeO–BNAH system, the researchers found that for the CO2 to formic acid conversion, an impressive turnover number of 671 was found in addition to a TOF of 11.6 min−1. It was ultimately theorised that the reaction proceeds by photoexcitation of the photosensitiser unit, followed by reductive quenching of this unit with BNAH, then intramolecular electron transfer from the reduced photosensitiser subunit to the catalyst subunit.
 | ||
| Fig. 20 (a) Select supramolecule used in photocatalytic conversion of CO2 to formic acid. Red subunits represent photosensitiser, and blue subunit shows catalyst unit. Supramolecular structures in this study are abbreviated according to the photosensitiser-to-catalyst ratio, in this case, (2:1). (b) NADH model compounds used as electron donors for the photocatalytic CO2 reduction.168 | ||
Niu and co-workers developed a spongy nickel–organic MOF to achieve photocatalytic conversion of CO2 into solar fuels using various ligands, metal dopants, and synthetic methodologies.169 The organic functionality supporting the MOF structure was terephthalic acid (TPA) and triethylene glycol (TEG), and a series of MOFs were prepared of the generic formula Ni(TPA), Ni(TEG), and Ni(TPA/TEG). In addition, the Ni–organic composites were synthesized using either laser irradiation (L) or traditional heating (H). These frameworks were prepared by combining solutions of nickel Ni(NO3)2, TEG, and/or TPA in DMF, stirring for 30 minutes, then heating at 110 °C for 48 hours, or irradiating with a nanosecond pulsed laser for three hours (1064 nm; 10 Hz; 7–8 ns pulse width; 0.9 cm beam diameter; 700 mJ per pulse). Examination of the catalytic activity of these materials revealed that Ni(TPA/TEG) (L) displayed the highest activity toward visible-light driven conversion of CO2 (Fig. 21). In a standard experiment, 3 mg of catalyst, 2.5 mmol of Ru(bpy)3Cl2·6H2O photosensitiser, and 2 mL of TEOA as sacrificial electron donor were added to an 8:2 solution of MeCN/H2O. After evacuation of the mixture, the system was pressurised with approximately 53 kPa of CO2 and irradiated with a solar simulator. Analysis of the CO2 reduction products revealed that carbon monoxide was produced as the major product with no detectable dihydrogen being formed, a remarkable finding as the HER which often accompanies CO2 reduction is entirely suppressed in this process. Irradiation for 2 hours generated 95.2 μmol of CO at a rate of 15.866 mmol h−1 g−1; while kinetic analysis of the evolved CO relative to amount of catalyst showed a linear relationship, suggesting first-order kinetics, decreasing the amount of catalyst to 1 mg of the catalyst under otherwise identical conditions revealed a CO production rate of 26.620 mmol h−1 g−1. This observation suggests that the relatively greater number of electrons generated from the photosensitiser were more effectively transferred to the catalyst in the reaction medium. Upon isolating and reusing the catalyst in subsequent reactions, the same activity and selectivity was observed, thus demonstrating the considerable durability of the catalyst. In trying to isolate liquid fuels, the nickel–organic composites were decorated with noble metal nanocrystals, namely Rh and Ag (Rh:Ni(TPA/TEG) and Ag:Ni(TPA/TEG), respectively). By comparison, the undecorated Ni(TPA/TEG) produced formic acid and acetic acid at final concentrations of 29.2 and 72.5 μM, respectively; when the noble metal nanocrystals were added, CO production decreased drastically while the liquid fuel generation increased. Rh:Ni(TPA/TEG) formed formic acid as the dominant product at a concentration of 313.5 μM after 2 hours of irradiation, and Ag:Ni(TPA/TEG) generated acetic acid (195.6 μM) as the major product within the same time period.
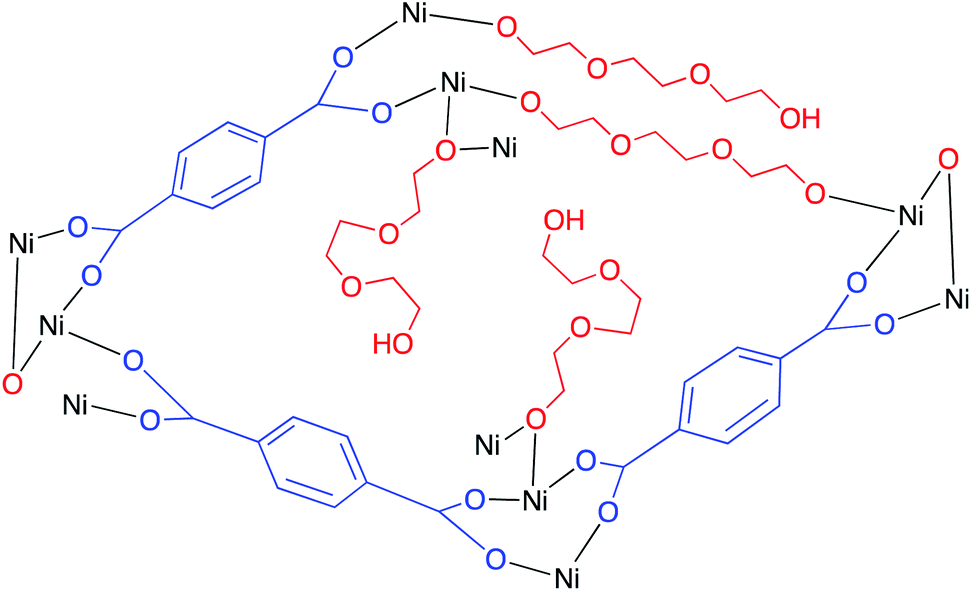 | ||
| Fig. 21 Ni(TPA/TEG) composite formed for the heterogeneous photoconversion of carbon dioxide. Red organic linkers are TEG, and blue organics are TPA.169 | ||
Using a ZnO-impregnated graphitic carbon nitride (ZnO/g-C3N4) catalyst, CO2 reduction was investigated while varying several factors including: ZnO:g-C3N4 ratios, reaction time, reaction temperature, and CO2 concentration.170 Samples containing 2, 4, 6, and 8 weight percent ZnO on g-C3N4 were prepared and labelled 2ZC, 4ZC, 6ZC, and 8ZC, respectively. Initial investigation of the CO2 conversion showed the dominant products of this process to be carbon monoxide, methanol, methane, and ethanol; further optimisation showed 6ZC to be the most active catalyst, which was subsequently used in further studies. In observing variations with reaction time, it was seen that longer reactions were marked by sharp increases in the rate of CO production, while the production rates for the other solar fuels was found to decrease over the course of the reaction. While the effects of temperature were negligible by comparison to other factors stated herein, the CO2 reduction rate increased gradually up to 60 °C, then decreased beyond this point. Finally, the CO2 concentration as a function of dilution in dinitrogen was investigated and showed that in the presence of water vapor, CO2 conversion occurred at a rate of 18 μmol g−1 h−1, even at a 1% CO2 concentration. As the reagent gas concentration rose, the reduction rate was found to continuously increase, even up to 100% CO2, thus demonstrating the robustness of the ZnO/g-C3N4 catalyst. Under these optimised conditions, the CO2 reduction was studied for one hour using simulated solar light, showing a reduction rate of 45.6 μmol g−1 h−1, with a solar fuel production rates for CO, MeOH, CH4, and EtOH of 38.7, 19.0, 5.4, and 2.5 μmol g−1 h−1, respectively. Using a single batch of catalyst for up to six reaction cycles showed a slight decrease in the reduction rate with no notable effect on product selectivity, indicating that the photocatalyst is relatively stable over several runs, but is likely undergoing some level of decomposition. Though not suitable for large scale expansion in its current form, this photoconversion system does display catalytic activity 4.9 and 8.2 times greater than g-C3N4 alone or P25 titania, respectively, providing guidance for future work to improve upon solar fuel generation from CO2.
Although P25 titania was outperformed as a CO2 reduction catalyst in the previous study, Sorcar and co-workers prepared and studied a highly active heterogeneous CO2 reduction catalyst, beginning from titanium dioxide as the precursor.171 P25 was ground with sodium borohydride and annealed under inert atmosphere at 350 °C, yielding reduced blue titania (RBT). Sonication of an RBT dispersion with various amounts of a 2 mg mL−1 graphene oxide solution, followed by annealing the recovered solids at 230 °C generated graphene-wrapped RBT (X-G/RBT), wherein X denotes the volume in millilitres of graphene oxide solution combined with 200 mL of the 10 mg mL−1 RBT suspension. Photodeposition of platinum nanoparticles was performed using hexachloroplatinic acid, yielding platinum sensitised samples of the general equation Pt%–X-G/RBT (% = 0.50, 1.00, 1.25, or 1.50 theoretical wt% platinum). Using the catalyst samples produced, photoreduction experiments were performed under ambient temperature and pressure under continuous gas flow of moist CO2, revealing that Pt1%–0.50-G/RBT was the most active of the catalysts synthesised. Irradiation of an aqueous suspension of this catalyst under flowing CO2 with simulated solar light for seven hours was found to produce a mixture of methane and ethane at rates of 37.0 and 11.0 μmol g−1 h−1, respectively. The roughly 3:1 ratio of methane and ethane produced varied according to the catalyst used, with this ratio being about 2.6 for Pt1.5%–0.50-G/RBT, and methane being the only product observed when using pure RBT. The reaction rate was found to decrease significantly after 7 hours, at which point thermo-vacuum treatment at 100 °C for two hours effectively regenerated the catalyst and restored the observed activity; this was theorised to be due to removal of gaseous products which were determined to be likely inhibitors of catalytic CO2 turnover, namely ethane. The prepared platinum sensitised graphene-wrapped RBT catalysts also showed excellent long-term stability, showing no loss in activity after 42 hours of operation given vacuum annealing was performed at seven hour increments.
In addition to gaseous fuels, heterogeneous photoconversion systems have shown excellent utility in generating liquid fuels from CO2 as demonstrated by Hsu and co-workers.172 In their work, graphene oxide (GO) was prepared by a variety methods to produce materials with different catalytic properties. GO-1 was prepared by an adaptation of Hummer's method by the combination of graphite, sodium nitrate, sulfuric acid, and potassium permanganate.173 Addition of different volumes of phosphoric acid followed by standard workup generated the modified graphene oxides, GO-2 and GO-3. The different synthetic methodologies used were found to affect the morphology, and thereby the optical properties, of the isolated material. GO-3 was found to have the roughest surface with the greatest topological variation, as well as the highest bandgap energy of the three materials (3.2–4.4). Consequently, GO-3 was also found to have the highest catalytic activity for photoconversion of CO2, generating methanol at a rate of 0.172 μmol g−1 h−1; while the RMeOH in GO-1 and GO-2 increased at comparable rates upon initiation of the reaction up to two hours, the methanol production rate of GO-3 continued to increase up to 4 hours and maintained greater stability over extended reaction times as compared to the other graphene oxide materials. Kumar et al. further utilised graphene oxide in the heterogeneous formation of methanol from CO2, however the GO used in this study served rather as a solid support for a cobalt(II) phthalocyanine complex (CoPc–GO, Fig. 22).174 Using the catalyst suspended in water with triethylamine as a sacrificial electron donor and 75 W m−2 irradiation for 48 hours, methanol was found to be produced as the major product at a rate of 78.79 μmol g−1 h−1, amounting to 3781.89 μmol MeOH per gram of catalyst. Analysis of the head space of the reaction showed 99.17% of the gas present to be CO2 with 0.82% being carbon monoxide as a minor product of the photoconversion. The potential to recycle the CoPc–GO was studied by isolating the catalyst at the conclusion of the 48 hour reaction and determining the identity and weight percent of CoPc remaining on the graphene oxide; after a single run, the cobalt content was diminished by 1.05 wt%, indicating that a small amount of leaching had occurred during the experiment. Though this points to limited recyclability, it also suggests that the heterogeneous catalyst could be used in additional reactions with limited loss of activity.
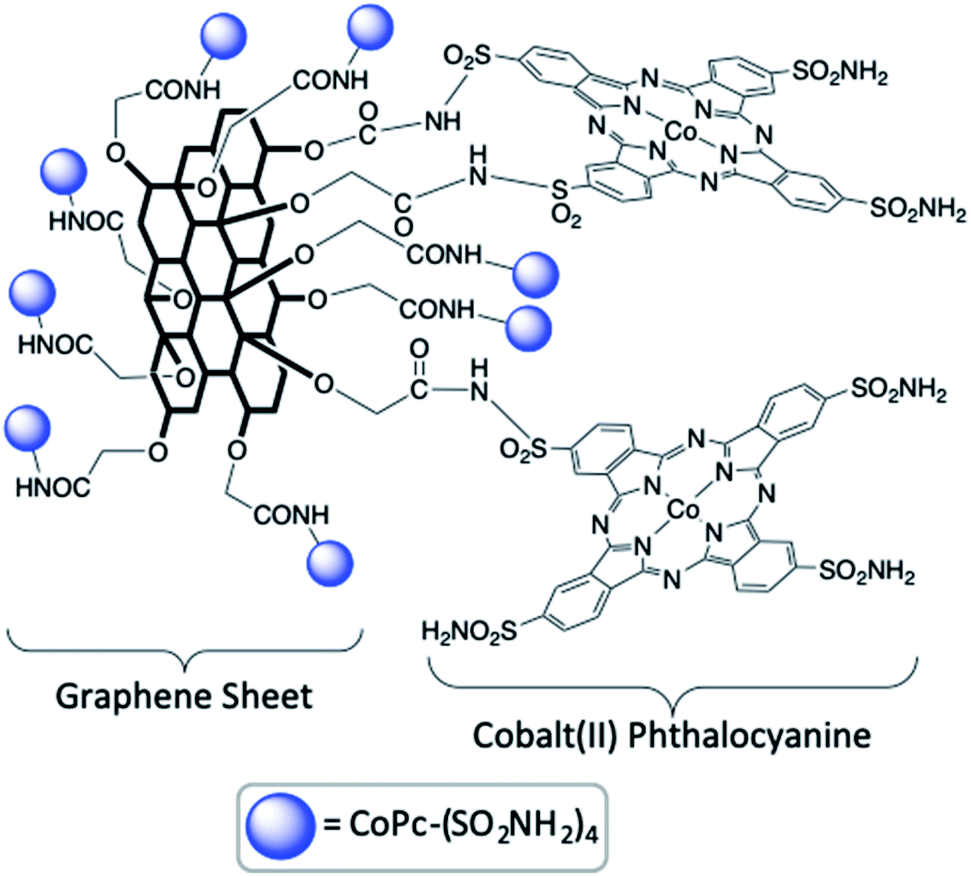 | ||
| Fig. 22 Graphene-tethered cobalt(II) phthalocyanine catalyst for heterogeneous photoconversion of CO2 to methanol.174 | ||
As the study of heterogeneous photoconverters and PEC devices has shown, seemingly trivial differences in the architecture of the catalyst can result in substantial changes to the chemical behaviour of the system, and research from the Xie lab has demonstrated this concept in their use of bismuth tungstate (Bi2WO6) layers to achieve conversion of CO2 to methanol.175 In this work, atomic layers of bismuth tungstate were prepared, and their catalytic activity in solar fuel generation compared to that of bulk and nanocrystalline Bi2WO6. Hydrothermal treatment of lamellar bismuth oleate with sodium tungstate (Na2WO4) yielded orthorhombic single-unit cell Bi2WO6 layers which were expected to display vastly improved CO2 conversion efficiencies. The underlying theory of this improved activity was based upon the single-unit cell thickness which would provide a higher specific surface area, an increased density of electronic states, and enhanced charge density on the catalyst surface. Of these factors, the surface area was able to be determined and showed significantly higher values in the atomic layer bismuth tungstate as compared to the bulk Bi2WO6 (27.6 and 0.7 m2 g−1, respectively). Performing the photocatalytic reduction using by the atomic layer Bi2WO6 catalyst with simulated solar radiation for 5 hours yielded 451.7 μmol g−1 of methanol as the dominant product, corresponding to an RMeOH of 75 μmol g−1 h−1; this production rate was found to be 3- and 125-times greater than that observed for bismuth tungstate nanocrystals (23 μmol g−1 h−1) and bulk Bi2WO6 (0.6 μmol g−1 h−1). In addition to the accelerated methanol production rate, use of the atomic layer Bi2WO6 heterogeneous catalyst presented other advantages in the enhanced photostability, demonstrated by the consistent RMeOH over the course of six reaction cycles, and the three-fold greater CO2 adsorption over bulk bismuth tungstate, likely due to the greater effective surface area discussed above. The activity seen for this bismuth tungstate notably outperformed previously reported atomic layer heterogeneous catalysts, including titania loaded zeolites (5.5 μmol g−1 h−1),176 and Ag/TiO2 (4.12 μmol g−1 h−1),177 however a 2017 report from Moriya prepared a titania/zirconia composite which revealed excellent photoconversion of CO2 to both methanol and formaldehyde using natural sunlight.178 The composite was comprised of nanometre-sized particles of TiO2 and micrometre-sized ZrO2, and the catalytic and weather conditions meticulously presented showing the impressive catalytic turnover. From combining different ratios of TiO2 and ZrO2, it was found that 1:1 and 6:4 ratios of the constituents were the most active in CO2 reduction, and mixing and pressing the two oxides produced greater catalytic activity than only mixing them. Water was introduced to the catalyst and resulting mixture by storing samples in a refrigerator to allow condensation to form upon the composite, thereby introducing a thin layer of water atop the oxide. To set a benchmark blank, the photoconversion reaction was attempted using the 1:1 composite in the absence of water, and in the dark; performing the reaction without water under solar irradiation showed no turnover of CO2, however running with water in the dark showed a low rate of methanol formation over the course of 30 minutes (134 μmol g−1 h−1), indicating that the catalyst, at minimum, is able to reduce carbon dioxide under classical catalytic conditions. When the wet samples of the 1:1 and 6:4 composites were irradiated with natural sunlight for five minutes, impressive generation of formaldehyde and methanol was observed, even given the short reaction time. Using the 6:4 composite, the sample was refrigerated for 48 hours then exposed to natural sunlight on a clear day (30.4 °C, 67% humidity, 1.16 mW cm−2 irradiation intensity); both formaldehyde and methanol were produced at rates of 1392 and 732 μmol g−1 h−1, respectively. The 1:1 composite performed even better under similar conditions (31.2 °C, 68% humidity, 1.33 mW cm−2 irradiation intensity), generating formaldehyde at a rate of 3084 μmol g−1 h−1 and methanol at a rate of 1008 μmol g−1 h−1. Impressively, even irradiation of the 1:1 composite on a cloudy day where the light intensity was 0.45 mW cm−2 was found to generate these fuels at notable rates (RHCOOH = 312 μmol g−1 h−1; RMeOH = 240 μmol g−1 h−1); the production of formaldehyde and increased productivity of methanol in this reaction indicate that even under low light, the photoconversion of CO2 readily occurs.
 | ||
| Fig. 23 (a) Amino acid residues replaced in sfYFP-BpA66 mutant protein. Tyrosine in 203 position and histidine in 148 position were replaced with aspartic acid and glutamic acid, respectively. (b) Incorporation of terpyridine ligand, nickel(II) centre, and BIH sacrificial reductant into PSP2 protein, forming PSP2-95C.179 | ||
In another work by Yadav et al., individual components of a larger CO2 reduction biocatalyst system were replaced with chemical derivatives rather than genetically engineering specific enzymes or proteins.183 The basis of this work revolved around the formate dehydrogenase (FDH)–formaldehyde dehydrogenase (FaldDH)–alcohol dehydrogenase (ADH) enzyme cascade which reduces CO2 to methanol. Electrons and protons are supplied to the enzymatic reaction via the NADH–NAD+ cycle, however in nature, stoichiometric amounts of NADH cofactors are necessary to regenerate NADH and support enzyme-mediated CO2 conversion. To avoid the use of such cofactors, a tandem photocatalyst cycle, inspired by previous studies on electrochemical regeneration of NADH,184,185 was developed to create an alternative catalytic cycle for methanol production from CO2 feedstocks. The proposed hybrid photocatalyst/biocatalyst (PC/BC) system requires an efficient light harvester capable of generating electrons and protons which are subsequently shuttled to NAD+, thus forming NADH. The researchers previously reported a chemically converted graphene coupled multi-anthraquinone-substituted porphyrin photocatalyst (CCGCMAQSP) which displayed photochemical conversion of CO2 to formic acid,186 however when used to generate methanol under the hybrid PC/BC conditions, only 5.62 μmol of methanol were formed after 90 minutes of irradiation. Testing chemically converted graphene (CCG) provided even poorer results, yielding only trace amounts of methanol after this reaction time. The results pointed to the need for porphyrin-based light harvesters to be covalently bound to the CCG, similar to what was seen in the CCGCMAQSP photocatalyst, albeit with substituents other than the anthraquinone previously used. To this end, isatin substitution on the porphyrin backbone was used, forming 1,1′,1′′-((20-(2-((7-amino-9,10-dioxo-9,10-dihydroanthracen-2-yl)amino)quinoline-3-yl)porphyrin-5,10,15-triyl) tris (quinolinne-3,2-diyl)) tris(indoline-2,3-dione), referred to as IP. Covalently linking IP to the chemically converted graphene formed the CCG-IP photocatalyst, an efficient visible light harvester (Fig. 24) which proved adept for the desired transformation. The CCG-IP photocatalyst and the FDH–FaldDH–ADH enzymatic biocatalyst served as the basis for the photoconversion, and triethanolamine (TEOA) and Cp*RhIII(bpy) were introduced as a sacrificial reductant and electron transfer shuttle, respectively. From electrochemical and mechanistic investigation, it was determined that photoexcitation of IP forms an excited state which transfers electrons to CCG, forming the graphene radical (CCG˙). The resulting IP+ is reduced by TEOA, forming the neutral IP and TEOA+. The rhodium catalyst abstracts a proton from solution, forming Cp*RhIII(bpy)(H) which then regenerates NADH from NAD+ by the photo-induced transfer of two electrons and a proton, with concomitant formation of Cp*RhIII(bpy)(OH2). The rhodium–aqua complex is reduced to its active form (Cp*RhI(bpy)) by CCG˙, and the regenerated NADH is used in the enzyme-mediated conversion of CO2 (Fig. 25). Bubbling CO2 at a rate of 0.5 mL min−1 through the aqueous reaction solution for 60 minutes under visible light irradiation resulted in the formation of 11.21 μmol of methanol (RMeOH = 14.95 mmol·gCCG-IP−1 h−1). From the experimental results, it was evident that isatin was superior as a photoabsorber and charge transfer reagent for the electron transfer to CCG as compared to CCGCMAQSP. In addition, the regeneration of NADH, which was one of the chief challenges of this reaction, was more efficiently accomplished by CCG-IP than CCGCMAQSP (38.99 and 28.46%, respectively). This study represents the first example of exclusive methanol formation from CO2 by a PC/BC hybrid system and informs of valuable methodologies by which bioinorganic photocatalysts may be developed to create solar fuels from carbon dioxide.
 | ||
| Fig. 24 CCG-IP photocatalyst used in photocatalytic/biocatalytic reduction of CO2 to methanol.183 | ||
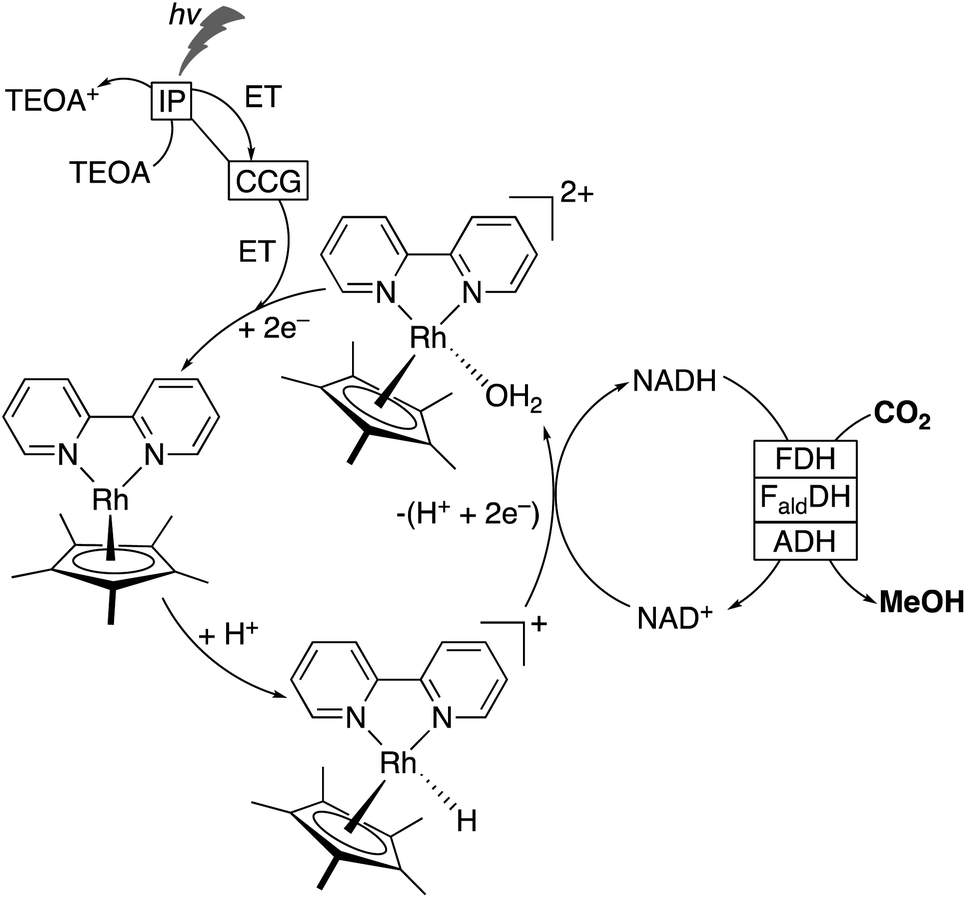 | ||
| Fig. 25 Proposed mechanism of photocatalytic/biocatalytic conversion of carbon dioxide to methanol.183 | ||
Outlook
Global climate change caused largely by CO2 emissions remains the most pressing environmental crisis facing society today. This threat in combination with the limited supply of fossil fuels prompts the necessity to find alternative energy sources to mitigate CO2 emissions and identify sustainable energy sources for long term use. To address both of these concerns, strategies of converting CO2 into viable solar fuels have emerged as promising, carbon–neutral options to sustainably generate energy carriers. Research in recent decades has resulted in impressive works which have created innovative routes to generate gas and liquid fuels from a CO2 feedstock, yet despite the progress made in this field, many challenges remain which must be overcome before a global implementation strategy can be pursued.Perhaps most pressing is the improvements needed in the technologies presented, including determination of the long-term stability of systems and further improvement/optimisation of solar fuel production. When aiming to store solar energy in chemical bonds as is proposed, the intermittent availability of sunlight limits the ability to harvest incident solar energy; in order to improve solar-to-fuel outputs, large areas are often required which incidentally creates other challenges in terms of land availability. In most instances, large scale pilot plants which have been constructed are located in isolated areas, thereby adding considerable transportation costs to the expenditures needed for wide-spread distribution. In order to avoid this and decrease the cost per unit of fuel, the efficiency and selectivity of solar fuel generation must be improved. Fortunately, as additional resources are dedicated to this work, it is increasingly likely that new technologies will be developed to achieve CO2 photoreduction to solar fuels on an industrial scale.
A further challenge to the implementation of solar fuel technology is the utilisation of CO2. In the vast majority of research dedicated to this process, concentrations of CO2 well beyond those found in the atmosphere are required to efficiently generate gaseous or liquid fuels. Alternatively, two general approaches have been investigated to maximise CO2 utilisation. The first includes direct use of CO2 in atmospheric or marine environments to create fuels,187 but the more widely investigated is direct carbon capture from power plants.188–190 The latter of these is the more promising design, providing a closed system wherein the combustion of solar fuels for energy production generates CO2 which is photoreduced to regenerate the solar fuel.
Several opportunities exist to utilise waste CO2 to produce energy carriers as viable replacements for conventional fossil fuels. Though several other alternative energy sources exist such as wind, hydropower, and others, harnessing solar energy in the form of chemical bonds in hydrocarbon fuels presents the only option which may conveniently be integrated into the current existing infrastructure. To realise this goal, additional resources are needed toward the fundamental research of creating new solar fuel technologies, as well as in capital investments which allow for expansion and industrialisation of discovered processes.
Conflicts of interest
The authors declare no competing interests.Acknowledgements
This work was supported by the Laboratory Directed Research and Development (LDRD) program of Los Alamos National Laboratory (LANL) under project numbers 20180705PRD1. LANL is operated by Triad National Security, LLC, for the National Nuclear Security Administration of U.S. Department of Energy (contract no. 89233218CNA000001).References
- D. Richter, R. Grün, R. Joannes-Boyau, T. E. Steele, F. Amani, M. Rué, P. Fernandes, J.-P. Raynal, D. Geraads, A. Ben-Ncer, J.-J. Hublin and S. P. McPherron, Nature, 2017, 546, 293–296 CrossRef CAS.
- C. N. Waters, J. Zalasiewicz, C. Summerhayes, A. D. Barnosky, C. Poirier, A. Gałuszka, A. Cearreta, M. Edgeworth, E. C. Ellis, M. Ellis, C. Jeandel, R. Leinfelder, J. R. McNeill, D. d. Richter, W. Steffen, J. Syvitski, D. Vidas, M. Wagreich, M. Williams, A. Zhisheng, J. Grinevald, E. Odada, N. Oreskes and A. P. Wolfe, Science, 2016, 351, aad2622 CrossRef.
- United Nations, Department of Economic and Social Affairs, The World at Six Billion, Population Division, 1999 Search PubMed.
- United Nations, Department of Economic and Social Affairs, World Population Prospects 2019, Volume I: Comprehensive Tables, ST/ESA/SER.A/426, Population Division, 2019 Search PubMed.
- BP Statistical Review of World Energy, British Petroleum, PLC, 68th edn, 2020 Search PubMed.
- U.S. Environmental Protection Agency, Office of Atmospheric Programs, Inventory of U.S. Greenhouse Gas Emissions and Sinks 1990–2018, EPA 430-R-20-002, 2020 Search PubMed.
- S. Solomon, G.-K. Plattner, R. Knutti and P. Friedlingstein, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 1704–1709 CrossRef CAS.
- E. Monnin, A. Indermühle, A. Dällenbach, J. Flückiger, B. Stauffer, T. F. Stocker, D. Raynaud and J.-M. Barnola, Science, 2001, 291, 112–114 CrossRef CAS.
- E. Dlugokencky and P. Tans, Trends in Atmospheric Carbon Dioxide, U.S. Department of Commerce, National Oceanic and Atmospheric Administration, Earth System Research Laboratory, http://www.esrl.noaa.gov/gmd/ccgg/trends/, accessed 1 May 2020 Search PubMed.
- S. A. Marcott, J. D. Shakun, P. U. Clark and A. C. Mix, Science, 2013, 339, 1198–1201 CrossRef CAS.
- State of the Climate: Global Climate Report for 2019, U.S. Department of Commerce, National Oceanic and Atmospheric Administration, National Centers for Environmental Information, https://www.ncdc.noaa.gov/sotc/global/201913/supplemental/page-1, accessed 1 May 2020 Search PubMed.
- M. Joshi, E. Hawkins, R. Sutton, J. Lowe and D. Frame, Nat. Clim. Change, 2011, 1, 407–412 CrossRef.
- P. T. Brown and K. Caldeira, Nature, 2017, 552, 45–50 CrossRef CAS.
- United Nations, Intergovernmental Panel on Climate Change, Climate Change 2014: Impacts, Adaptation, and Vulnerability. Part A: Global and Sectoral Aspects, Contribution to 5th Assessment Report, https://www.ipcc.ch/report/ar5/wg2/, 2014 Search PubMed.
- BP Statistical Review of World Energy – All Data (1965–2018), British Petroleum, PLC, https://www.bp.com/en/global/corporate/energy-economics/statistical-review-of-world-energy.html, accessed 2 May 2020 Search PubMed.
- U.S. Department of Energy, Energy Information Administration, Office of Energy Analysis, International Energy Outlook 2019 with Projection to 2050, IEO2019, 2019 Search PubMed.
- United Nations, Framework Convention on Climate Change, Adoption of the Paris Agreement, FCCC/CP/2015/L.9, https://unfccc.int/sites/default/files/english_paris_agreement.pdf, 2015 Search PubMed.
- BP Statistical Review of World Energy, British Petroleum, PLC, 64th edn, 2015 Search PubMed.
- P. Moriarty and D. Honnery, Renewable Sustainable Energy Rev., 2012, 16, 244–252 CrossRef.
- S. Teske, K. Nagrath, T. Morris and K. Dooley, in Achieving the Paris Climate Agreement Goals: Global and Regional 100% Renewable Energy Scenarios with Non-energy GHG Pathways for +1.5 °C and +2 °C, ed. S. Teske, Springer International Publishing, Cham, 2019, pp. 161–173, DOI:10.1007/978-3-030-05843-2_7.
- D. Weißbach, G. Ruprecht, A. Huke, K. Czerski, S. Gottlieb and A. Hussein, Energy, 2013, 52, 210–221 CrossRef.
- M. Carbajales-Dale, C. J. Barnhart and S. M. Benson, Energy Environ. Sci., 2014, 7, 1538–1544 RSC.
- N. Armaroli and V. Balzani, Chem.–Eur. J., 2016, 22, 32–57 CrossRef CAS.
- United Nations, Intergovernmental Panel on Climate Change, Renewable Energy Sources and Climate Change Mitigation, https://www.ipcc.ch/site/assets/uploads/2018/03/SRREN_Full_Report-1.pdf, 2011 Search PubMed.
- World Energy Balances 2019, International Energy Agency, Paris, 2019 Search PubMed.
- W. Keller, US Pat., 4149856A, 1977.
- J. R. Bolton, Science, 1978, 202, 705–711 CrossRef CAS.
- A. K. Ringsmuth, M. J. Landsberg and B. Hankamer, Renewable Sustainable Energy Rev., 2016, 62, 134–163 CrossRef CAS.
- S. A. Channiwala and P. P. Parikh, Fuel, 2002, 81, 1051–1063 CrossRef CAS.
- A. Mustafa, R. K. Calay and M. Y. Mustafa, Energy Procedia, 2017, 112, 529–536 CrossRef.
- J. C. Solarte-Toro, Y. Chacón-Pérez and C. A. Cardona-Alzate, Electron. J. Biotechnol., 2018, 33, 52–62 CrossRef CAS.
- S. J. Davis, N. S. Lewis, M. Shaner, S. Aggarwal, D. Arent, I. L. Azevedo, S. M. Benson, T. Bradley, J. Brouwer, Y.-M. Chiang, C. T. M. Clack, A. Cohen, S. Doig, J. Edmonds, P. Fennell, C. B. Field, B. Hannegan, B.-M. Hodge, M. I. Hoffert, E. Ingersoll, P. Jaramillo, K. S. Lackner, K. J. Mach, M. Mastrandrea, J. Ogden, P. F. Peterson, D. L. Sanchez, D. Sperling, J. Stagner, J. E. Trancik, C.-J. Yang and K. Caldeira, Science, 2018, 360, eaas9793 CrossRef.
- S. C. Roy, O. K. Varghese, M. Paulose and C. A. Grimes, ACS Nano, 2010, 4, 1259–1278 CrossRef CAS.
- G. Centi and S. Perathoner, ChemSusChem, 2010, 3, 195–208 CrossRef CAS.
- G. Centi and S. Perathoner, Green Carbon Dioxide: Advances in CO2 Utilization, John Wiley & Sons, Inc., Hoboken, New Jersey, 2014 Search PubMed.
- N. Z. Muradov and T. Veziroglu, Carbon–Neutral Fuels and Energy Carriers, CRC Press, Boca Raton, Florida, 1st edn, 2012 Search PubMed.
- R. H. Coridan, A. C. Nielander, S. A. Francis, M. T. McDowell, V. Dix, S. M. Chatman and N. S. Lewis, Energy Environ. Sci., 2015, 8, 2886–2901 RSC.
- F. H. Saadi, N. S. Lewis and E. W. McFarland, Energy Environ. Sci., 2018, 11, 469–475 RSC.
- R. J. Detz, J. N. H. Reek and B. C. C. van der Zwaan, Energy Environ. Sci., 2018, 11, 1653–1669 RSC.
- D. L. Ashford, M. K. Gish, A. K. Vannucci, M. K. Brennaman, J. L. Templeton, J. M. Papanikolas and T. J. Meyer, Chem. Rev., 2015, 115, 13006–13049 CrossRef CAS.
- D. G. Nocera, Acc. Chem. Res., 2012, 45, 767–776 CrossRef CAS.
- M. J. Katz, S. C. Riha, N. C. Jeong, A. B. F. Martinson, O. K. Farha and J. T. Hupp, Coord. Chem. Rev., 2012, 256, 2521–2529 CrossRef CAS.
- D. Gust, T. A. Moore and A. L. Moore, Acc. Chem. Res., 2009, 42, 1890–1898 CrossRef CAS.
- V. Artero and M. Fontecave, Chem. Soc. Rev., 2013, 42, 2338–2356 RSC.
- D. G. Nocera, Acc. Chem. Res., 2017, 50, 616–619 CrossRef CAS.
- B. M. Hunter, H. B. Gray and A. M. Müller, Chem. Rev., 2016, 116, 14120–14136 CrossRef CAS.
- K. Bourzac, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 4545–4548 CrossRef CAS.
- J. Z. Zhang and E. Reisner, Nat. Rev. Chem., 2020, 4, 6–21 CrossRef CAS.
- Y. Chen, S. Hu, C. Xiang and N. S. Lewis, Energy Environ. Sci., 2015, 8, 876–886 RSC.
- H. B. Gray, Nat. Chem., 2009, 1, 7 CrossRef CAS.
- S. Styring, Faraday Discuss., 2012, 155, 357–376 RSC.
- C. Liu, B. C. Colón, M. Ziesack, P. A. Silver and D. G. Nocera, Science, 2016, 352, 1210–1213 CrossRef CAS.
- L. An and R. Chen, J. Power Sources, 2016, 320, 127–139 CrossRef CAS.
- J. Jiang and A. Wieckowski, Electrochem. Commun., 2012, 18, 41–43 CrossRef CAS.
- X. Su, Z. Pan and L. An, Int. J. Energy Res., 2019, 43, 7433–7443 CrossRef CAS.
- H. Kawanami, Y. Himeda and G. Laurenczy, in Adv. Inorg. Chem., ed. R. van Eldik and C. D. Hubbard, Academic Press, 2017, vol. 70, pp. 395–427 Search PubMed.
- K. Sordakis, A. F. Dalebrook and G. Laurenczy, ChemCatChem, 2015, 7, 2332–2339 CrossRef CAS.
- E. Irtem, M. D. Hernández-Alonso, A. Parra, C. Fàbrega, G. Penelas-Pérez, J. R. Morante and T. Andreu, Electrochim. Acta, 2017, 240, 225–230 CrossRef CAS.
- E. Kalamaras, M. Belekoukia, J. Z. Y. Tan, J. Xuan, M. M. Maroto-Valer and J. M. Andresen, Faraday Discuss., 2019, 215, 329–344 RSC.
- S. Kar, A. Goeppert and G. K. S. Prakash, Acc. Chem. Res., 2019, 52, 2892–2903 CrossRef CAS.
- W.-H. Wang, Y. Himeda, J. T. Muckerman, G. F. Manbeck and E. Fujita, Chem. Rev., 2015, 115, 12936–12973 CrossRef CAS.
- A. Roldan and N. H. de Leeuw, Faraday Discuss., 2016, 188, 161–180 RSC.
- M. Tahir, B. Tahir, Z. Y. Zakaria and A. Muhammad, J. Cleaner Prod., 2019, 213, 451–461 CrossRef CAS.
- Y. Lu, L. Zhu, C. Agrafiotis, J. Vieten, M. Roeb and C. Sattler, Prog. Energy Combust. Sci., 2019, 75, 100785 CrossRef.
- A. Haeussler, S. Abanades, A. Julbe, J. Jouannaux and B. Cartoixa, Energy, 2020, 201, 117649 CrossRef CAS.
- S. Bhatta, D. Nagassou, S. Mohsenian and J. P. Trelles, Sol. Energy, 2019, 178, 201–214 CrossRef CAS.
- F. Piadehrouhi, B. Ghorbani, M. Miansari and M. Mehrpooya, Energy, 2019, 179, 938–959 CrossRef CAS.
- V. Kashyap, R. Medhi, P. Irajizad, P. Jafari, M. Nazari, A. Masoudi, M. D. Marquez, T. R. Lee and H. Ghasemi, Sustainable Energy Fuels, 2019, 3, 272–279 RSC.
- L. Wang, X. Liu, Y. Dang, H. Xie, Q. Zhao and L. Ye, Solid State Sci., 2019, 89, 67–73 CrossRef CAS.
- L. B. Hoch, P. G. O'Brien, A. Jelle, A. Sandhel, D. D. Perovic, C. A. Mims and G. A. Ozin, ACS Nano, 2016, 10, 9017–9025 CrossRef CAS.
- J. Zhao, Q. Yang, R. Shi, G. I. N. Waterhouse, X. Zhang, L.-Z. Wu, C.-H. Tung and T. Zhang, NPG Asia Mater., 2020, 12, 5 CrossRef CAS.
- J. Low, L. Zhang, B. Zhu, Z. Liu and J. Yu, ACS Sustainable Chem. Eng., 2018, 6, 15653–15661 CrossRef CAS.
- D. Giovannini, J. Romero, V. Potoček, G. Ferenczi, F. Speirits, S. M. Barnett, D. Faccio and M. J. Padgett, Science, 2015, 347, 857–860 CrossRef CAS.
- J. Ren, S. Ouyang, H. Xu, X. Meng, T. Wang, D. Wang and J. Ye, Adv. Energy Mater., 2017, 7, 1601657 CrossRef.
- X. Meng, T. Wang, L. Liu, S. Ouyang, P. Li, H. Hu, T. Kako, H. Iwai, A. Tanaka and J. Ye, Angew. Chem., Int. Ed., 2014, 53, 11478–11482 CrossRef CAS.
- C. L. Lormeus, Morocco's Noor III CSP Exceeds Performance Targets in Morocco World News, https://www.moroccoworldnews.com/2019/05/274576/noor-iii-morocco-performance-solar-energy/, 2019 Search PubMed.
- H. L. Zhang, J. Baeyens, J. Degrève and G. Cacères, Renewable Sustainable Energy Rev., 2013, 22, 466–481 CrossRef.
- S. Han, Y. Chen, S. Abanades and Z. Zhang, J. Energy Chem., 2017, 26, 743–749 CrossRef.
- D. Marxer, P. Furler, J. Scheffe, H. Geerlings, C. Falter, V. Batteiger, A. Sizmann and A. Steinfeld, Energy Fuels, 2015, 29, 3241–3250 CrossRef CAS.
- P. Furler, J. Scheffe, M. Gorbar, L. Moes, U. Vogt and A. Steinfeld, Energy Fuels, 2012, 26, 7051–7059 CrossRef CAS.
- P. Furler, J. R. Scheffe and A. Steinfeld, Energy Environ. Sci., 2012, 5, 6098–6103 RSC.
- W. C. Chueh, C. Falter, M. Abbott, D. Scipio, P. Furler, S. M. Haile and A. Steinfeld, Science, 2010, 330, 1797–1801 CrossRef CAS.
- J. Kim, T. A. Johnson, J. E. Miller, E. B. Stechel and C. T. Maravelias, Energy Environ. Sci., 2012, 5, 8417–8429 RSC.
- J. Kim, C. A. Henao, T. A. Johnson, D. E. Dedrick, J. E. Miller, E. B. Stechel and C. T. Maravelias, Energy Environ. Sci., 2011, 4, 3122–3132 RSC.
- T. Nakamura, Sol. Energy, 1977, 19, 467–475 CrossRef CAS.
- M. Grätzel, Nature, 2001, 414, 338–344 CrossRef.
- J. R. Galan-Mascaros, Catal. Sci. Technol., 2020, 10, 1967–1974 RSC.
- S. A. Bonke, M. Wiechen, D. R. MacFarlane and L. Spiccia, Energy Environ. Sci., 2015, 8, 2791–2796 RSC.
- M. K. Brennaman, R. J. Dillon, L. Alibabaei, M. K. Gish, C. J. Dares, D. L. Ashford, R. L. House, G. J. Meyer, J. M. Papanikolas and T. J. Meyer, J. Am. Chem. Soc., 2016, 138, 13085–13102 CrossRef CAS.
- C. R. Lhermitte and K. Sivula, ACS Catal., 2019, 9, 2007–2017 CrossRef CAS.
- V. Kumaravel, J. Bartlett and S. C. Pillai, ACS Energy Lett., 2020, 5, 486–519 CrossRef CAS.
- Z. J. Mast, T. H. T. Myren, C. G. Huntzinger, T. A. Stinson, R. M. Kharbouch, E. M. Almanza, S. E. Zygmont, J. R. Miecznikowski and O. R. Luca, Inorg. Chem. Front., 2020, 7, 1012–1015 RSC.
- B. Zhou, P. Ou, N. Pant, S. Cheng, S. Vanka, S. Chu, R. T. Rashid, G. Botton, J. Song and Z. Mi, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 1330–1338 CrossRef CAS.
- T. Kunene, L. Xiong and J. Rosenthal, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 9693–9695 CrossRef CAS.
- H. Jun, S. Choi, M. Y. Yang and Y. S. Nam, J. Mater. Chem. A, 2019, 7, 17254–17260 RSC.
- C. M. Gabardo, C. P. O'Brien, J. P. Edwards, C. McCallum, Y. Xu, C.-T. Dinh, J. Li, E. H. Sargent and D. Sinton, Joule, 2019, 3, 2777–2791 CrossRef CAS.
- Gurudayal, D. Perone, S. Malani, Y. Lum, S. Haussener and J. W. Ager, ACS Appl. Energy Mater., 2019, 2, 4551–4559 CrossRef CAS.
- D. Ren, J. Gao, L. Pan, Z. Wang, J. Luo, S. M. Zakeeruddin, A. Hagfeldt and M. Grätzel, Angew. Chem., Int. Ed., 2019, 58, 15036–15040 CrossRef CAS.
- J. F. d. Brito, J. A. L. Perini, S. Perathoner and M. V. B. Zanoni, Electrochim. Acta, 2019, 306, 277–284 CrossRef CAS.
- Q. Gong, P. Ding, M. Xu, X. Zhu, M. Wang, J. Deng, Q. Ma, N. Han, Y. Zhu, J. Lu, Z. Feng, Y. Li, W. Zhou and Y. Li, Nat. Commun., 2019, 10, 2807 CrossRef.
- W.-H. Cheng, M. H. Richter, I. Sullivan, D. M. Larson, C. Xiang, B. S. Brunschwig and H. A. Atwater, ACS Energy Lett., 2020, 5, 470–476 CrossRef CAS.
- W. A. Smith, in Photoelectrochemical Solar Fuel Production: From Basic Principles to Advanced Devices, ed. S. Giménez and J. Bisquert, Springer International Publishing, Cham, 2016, pp. 163–197, DOI:10.1007/978-3-319-29641-8_4.
- L. Ye, X. Jin, X. Ji, C. Liu, Y. Su, H. Xie and C. Liu, Chem. Eng. J., 2016, 291, 39–46 CrossRef CAS.
- L. Ye, H. Wang, X. Jin, Y. Su, D. Wang, H. Xie, X. Liu and X. Liu, Sol. Energy Mater. Sol. Cells, 2016, 144, 732–739 CrossRef CAS.
- C. Ding, L. Ye, Q. Zhao, Z. Zhong, K. Liu, H. Xie, K. Bao, X. Zhang and Z. Huang, J. CO2 Util., 2016, 14, 135–142 CrossRef CAS.
- Y. Bai, L. Ye, L. Wang, X. Shi, P. Wang and W. Bai, Environ. Sci.: Nano, 2016, 3, 902–909 RSC.
- J. J. Leung, J. Warnan, K. H. Ly, N. Heidary, D. H. Nam, M. F. Kuehnel and E. Reisner, Nat. Catal., 2019, 2, 354–365 CrossRef CAS.
- V. Andrei, B. Reuillard and E. Reisner, Nat. Mater., 2020, 19, 189–194 CrossRef CAS.
- M. Schreier, F. Héroguel, L. Steier, S. Ahmad, J. S. Luterbacher, M. T. Mayer, J. Luo and M. Grätzel, Nat. Energy, 2017, 2, 17087 CrossRef CAS.
- X. Liu, S. Cui, M. Qian, Z. Sun and P. Du, Chem. Commun., 2016, 52, 5546–5549 RSC.
- X. Liu, S. Cui, Z. Sun, Y. Ren, X. Zhang and P. Du, J. Phys. Chem. C, 2016, 120, 831–840 CrossRef CAS.
- Y. Hori, Handbook of Fuel Cells, 2010, DOI:10.1002/9780470974001.f207055.
- G. Seshadri, C. Lin and A. B. Bocarsly, J. Electroanal. Chem., 1994, 372, 145–150 CrossRef CAS.
- E. E. Barton, D. M. Rampulla and A. B. Bocarsly, J. Am. Chem. Soc., 2008, 130, 6342–6344 CrossRef CAS.
- K. Lee, S. Lee, H. Cho, S. Jeong, W. D. Kim, S. Lee and D. C. Lee, J. Energy Chem., 2018, 27, 264–270 CrossRef.
- L. Wang, Y. Jia, R. Nie, Y. Zhang, F. Chen, Z. Zhu, J. Wang and H. Jing, J. Catal., 2017, 349, 1–7 CrossRef CAS.
- Q. Wang, X. Wang, Z. Yu, X. Jiang, J. Chen, L. Tao, M. Wang and Y. Shen, Nano Energy, 2019, 60, 827–835 CrossRef CAS.
- E. M. Nichols, J. J. Gallagher, C. Liu, Y. Su, J. Resasco, Y. Yu, Y. Sun, P. Yang, M. C. Y. Chang and C. J. Chang, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 11461–11466 CrossRef CAS.
- R. A. Mah, M. R. Smith and L. Baresi, Appl. Environ. Microbiol., 1978, 35, 1174–1184 CrossRef CAS.
- K. D. Olson, C. W. McMahon and R. S. Wolfe, Appl. Environ. Microbiol., 1991, 57, 2683–2686 CrossRef CAS.
- C. Liu, J. J. Gallagher, K. K. Sakimoto, E. M. Nichols, C. J. Chang, M. C. Y. Chang and P. Yang, Nano Lett., 2015, 15, 3634–3639 CrossRef CAS.
- B. B. Bond-Watts, R. J. Bellerose and M. C. Y. Chang, Nat. Chem. Biol., 2011, 7, 222–227 CrossRef CAS.
- T. Haas, R. Krause, R. Weber, M. Demler and G. Schmid, Nat. Catal., 2018, 1, 32–39 CrossRef CAS.
- From Cellulose to Cellulase: Strategies to Improve Biofuel Production, ed. N. Srivastave, M. Strivastava, P. K. Mishra, P. W. Ramteke and R. L. Singh, Elsevier, Cambridge, 2019 Search PubMed.
- Microbial Biotechnology in Agro-Environmental Sustainability, ed. J. S. Singh and D. P. Singh, Elsevier, Cambridge, 2019 Search PubMed.
- Microbes in Soil, Crop and Environmental Sustainability, ed. J. S. Singh, Elsevier, Cambridge, 2019 Search PubMed.
- L. Yuan, S.-F. Hung, Z.-R. Tang, H. M. Chen, Y. Xiong and Y.-J. Xu, ACS Catal., 2019, 9, 4824–4833 CrossRef CAS.
- C.-Y. Lee, J. Zou, J. Bullock and G. G. Wallace, J. Photochem. Photobiol., C, 2019, 39, 142–160 CrossRef CAS.
- M. Flores-Flores, E. Luévano-Hipólito, L. M. Torres-Martínez and T.-O. Do, Mater. Chem. Phys., 2019, 227, 90–97 CrossRef CAS.
- S. Cestellos-Blanco, H. Zhang and P. Yang, Faraday Discuss., 2019, 215, 54–65 RSC.
- X. Chen, Y. Liu and J. Wu, Mol. Catal., 2020, 483, 110716 CrossRef CAS.
- M. Kumar, P. C. Sahoo, S. Srikanth, R. Bagai, S. K. Puri and S. S. V. Ramakumar, Bioresour. Technol., 2019, 272, 300–307 CrossRef CAS.
- P.-L. Tremblay, M. Xu, Y. Chen and T. Zhang, iScience, 2020, 23, 100784 CrossRef CAS.
- J. Ungerer, L. Tao, M. Davis, M. Ghirardi, P.-C. Maness and J. Yu, Energy Environ. Sci., 2012, 5, 8998–9006 RSC.
- W. Xiong, J. A. Morgan, J. Ungerer, B. Wang, P.-C. Maness and J. Yu, Nat. Plants, 2015, 1, 15053 CrossRef CAS.
- M.-D. Deng and J. R. Coleman, Appl. Environ. Microbiol., 1999, 65, 523–528 CrossRef CAS.
- S. S. Golden, J. Brusslan and R. Haselkorn, in Methods Enzymol., Academic Press, 1987, vol. 153, pp. 215–231 Search PubMed.
- J. Dexter and P. Fu, Energy Environ. Sci., 2009, 2, 857–864 RSC.
- U. Duhring, K. Ziegler and D. Kramer, US Pat. US2010/0003739 A1, 2010 Search PubMed.
- Z. Gao, H. Zhao, Z. Li, X. Tan and X. Lu, Energy Environ. Sci., 2012, 5, 9857–9865 RSC.
- J. G. K. Williams, in Methods Enzymol., Academic Press, 1988, vol. 167, pp. 766–778 Search PubMed.
- X. Tan, L. Yao, Q. Gao, W. Wang, F. Qi and X. Lu, Metab. Eng., 2011, 13, 169–176 CrossRef CAS.
- S. Kinoshita, T. Kakizono, K. Kadota, K. Das and H. Taguchi, Appl. Microbiol. Biotechnol., 1985, 22, 249–254 CrossRef CAS.
- G. F. Wu, Z. Y. Shen and Q. Y. Wu, Enzyme Microb. Technol., 2002, 30, 710–715 CrossRef CAS.
- J. W. K. Oliver, I. M. P. Machado, H. Yoneda and S. Atsumi, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 1249–1254 CrossRef CAS.
- S. H. Park, R. Xing and W. B. Whitman, Biochim. Biophys. Acta, Gen. Subj., 1995, 1245, 366–370 CrossRef.
- M. Miller, W. E. Robinson, A. R. Oliveira, N. Heidary, N. Kornienko, J. Warnan, I. A. C. Pereira and E. Reisner, Angew. Chem., Int. Ed., 2019, 58, 4601–4605 CrossRef CAS.
- S. Zhang, J. Shi, Y. Sun, Y. Wu, Y. Zhang, Z. Cai, Y. Chen, C. You, P. Han and Z. Jiang, ACS Catal., 2019, 9, 3913–3925 CrossRef CAS.
- P. B. Pati, R. Wang, E. Boutin, S. Diring, S. Jobic, N. Barreau, F. Odobel and M. Robert, Nat. Commun., 2020, 11, 3499 CrossRef CAS.
- A. H. Chowdhury, A. Das, S. Riyajuddin, K. Ghosh and S. M. Islam, Catal. Sci. Technol., 2019, 9, 6566–6569 RSC.
- L. Wan, Q. Zhou, X. Wang, T. E. Wood, L. Wang, P. N. Duchesne, J. Guo, X. Yan, M. Xia, Y. F. Li, A. A. Jelle, U. Ulmer, J. Jia, T. Li, W. Sun and G. A. Ozin, Nat. Catal., 2019, 2, 889–898 CrossRef CAS.
- N. U. M. Nor and N. A. S. Amin, J. CO2 Util., 2019, 33, 372–383 CrossRef CAS.
- A. Kumar, P. K. Prajapati, M. S. Aathira, A. Bansiwal, R. Boukherroub and S. L. Jain, J. Colloid Interface Sci., 2019, 543, 201–213 CrossRef CAS.
- R. Kim, J. Kim, J. Y. Do, M. W. Seo and M. Kang, Catalysts, 2019, 9, 998 CrossRef CAS.
- S. E. Lee, A. Nasirian, Y. E. Kim, P. T. Fard, Y. Kim, B. Jeong, S.-J. Kim, J.-O. Baeg and J. Kim, J. Am. Chem. Soc., 2020, 142, 19142–19149 CrossRef CAS.
- T. Dhanasekaran, J. Grodkowski, P. Neta, P. Hambright and E. Fujita, J. Phys. Chem. A, 1999, 103, 7742–7748 CrossRef CAS.
- J. Bonin, M. Chaussemier, M. Robert and M. Routier, ChemCatChem, 2014, 6, 3200–3207 CrossRef CAS.
- J. Bonin, M. Robert and M. Routier, J. Am. Chem. Soc., 2014, 136, 16768–16771 CrossRef CAS.
- C. Costentin, S. Drouet, M. Robert and J.-M. Savéant, Science, 2012, 338, 90–94 CrossRef CAS.
- C. Costentin, G. Passard, M. Robert and J.-M. Savéant, J. Am. Chem. Soc., 2014, 136, 11821–11829 CrossRef CAS.
- C. Costentin, G. Passard, M. Robert and J.-M. Savéant, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 14990–14994 CrossRef CAS.
- H. Takeda and O. Ishitani, Coord. Chem. Rev., 2010, 254, 346–354 CrossRef CAS.
- Z. Chen, J. J. Concepcion, M. K. Brennaman, P. Kang, M. R. Norris, P. G. Hoertz and T. J. Meyer, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 15606–15611 CrossRef CAS.
- G. A. Andrade, A. J. Pistner, G. P. A. Yap, D. A. Lutterman and J. Rosenthal, ACS Catal., 2013, 3, 1685–1692 CrossRef CAS.
- J. Chauvin, F. Lafolet, S. Chardon-Noblat, A. Deronzier, M. Jakonen and M. Haukka, Chem.–Eur. J., 2011, 17, 4313–4322 CrossRef CAS.
- S. Sato, K. Koike, H. Inoue and O. Ishitani, Photochem. Photobiol. Sci., 2007, 6, 454–461 RSC.
- D. J. Boston, C. Xu, D. W. Armstrong and F. M. MacDonnell, J. Am. Chem. Soc., 2013, 135, 16252–16255 CrossRef CAS.
- Y. Tamaki, T. Morimoto, K. Koike and O. Ishitani, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 15673–15678 CrossRef CAS.
- K. Niu, Y. Xu, H. Wang, R. Ye, H. L. Xin, F. Lin, C. Tian, Y. Lum, K. C. Bustillo, M. M. Doeff, M. T. M. Koper, J. Ager, R. Xu and H. Zheng, Sci. Adv., 2017, 3, e1700921 CrossRef.
- Y. He, Y. Wang, L. Zhang, B. Teng and M. Fan, Appl. Catal., B, 2015, 168–169, 1–8 CAS.
- S. Sorcar, J. Thompson, Y. Hwang, Y. H. Park, T. Majima, C. A. Grimes, J. R. Durrant and S.-I. In, Energy Environ. Sci., 2018, 11, 3183–3193 RSC.
- H.-C. Hsu, I. Shown, H.-Y. Wei, Y.-C. Chang, H.-Y. Du, Y.-G. Lin, C.-A. Tseng, C.-H. Wang, L.-C. Chen, Y.-C. Lin and K.-H. Chen, Nanoscale, 2013, 5, 262–268 RSC.
- M. Hirata, T. Gotou, S. Horiuchi, M. Fujiwara and M. Ohba, Carbon, 2004, 42, 2929–2937 CAS.
- P. Kumar, A. Kumar, B. Sreedhar, B. Sain, S. S. Ray and S. L. Jain, Chem.–Eur. J., 2014, 20, 6154–6161 CrossRef CAS.
- L. Liang, F. Lei, S. Gao, Y. Sun, X. Jiao, J. Wu, S. Qamar and Y. Xie, Angew. Chem., Int. Ed., 2015, 54, 13971–13974 CrossRef CAS.
- M. Anpo, H. Yamashita, Y. Ichihashi, Y. Fujii and M. Honda, J. Phys. Chem. B, 1997, 101, 2632–2636 CrossRef CAS.
- J. C. S. Wu, T.-H. Wu, T. Chu, H. Huang and D. Tsai, Top. Catal., 2008, 47, 131–136 CrossRef CAS.
- I. Moriya, Sci. Rep., 2017, 7, 14446 CrossRef.
- X. Liu, F. Kang, C. Hu, L. Wang, Z. Xu, D. Zheng, W. Gong, Y. Lu, Y. Ma and J. Wang, Nat. Chem., 2018, 10, 1201–1206 CrossRef CAS.
- R. Breslow, S. Baldwin, T. Flechtner, P. Kalicky, S. Liu and W. Washburn, J. Am. Chem. Soc., 1973, 95, 3251–3262 CrossRef CAS.
- R. Breslow, S. Kitabatake and J. Rothbard, J. Am. Chem. Soc., 1978, 100, 8156–8160 CrossRef CAS.
- J. C. Kauer, S. Erickson-Viitanen, H. R. Wolfe and W. F. DeGrado, J. Biol. Chem., 1986, 261, 10695–10700 CAS.
- R. K. Yadav, G. H. Oh, N.-J. Park, A. Kumar, K.-j. Kong and J.-O. Baeg, J. Am. Chem. Soc., 2014, 136, 16728–16731 CrossRef CAS.
- F. Hollmann, A. Schmid and E. Steckhan, Angew. Chem., Int. Ed., 2001, 40, 169–171 CrossRef CAS.
- H. Wu, C. Tian, X. Song, C. Liu, D. Yang and Z. Jiang, Green Chem., 2013, 15, 1773–1789 RSC.
- R. K. Yadav, J.-O. Baeg, G. H. Oh, N.-J. Park, K.-j. Kong, J. Kim, D. W. Hwang and S. K. Biswas, J. Am. Chem. Soc., 2012, 134, 11455–11461 CrossRef CAS.
- B. D. Patterson, F. Mo, A. Borgschulte, M. Hillestad, F. Joos, T. Kristiansen, S. Sunde and J. A. van Bokhoven, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 12212–12219 CrossRef CAS.
- R. Steeneveldt, B. Berger and T. A. Torp, Chem. Eng. Res. Des., 2006, 84, 739–763 CrossRef CAS.
- E. S. Rubin, J. E. Davison and H. J. Herzog, Int. J. Greenhouse Gas Control, 2015, 40, 378–400 CrossRef CAS.
- K. Möllersten, J. Yan and J. R. Moreira, Biomass Bioenergy, 2003, 25, 273–285 CrossRef.
Footnote |
| † A database search of the term “solar fuels” showed no more than six published works per year from 1977–2008. Beginning in 2009, the average number of publications per year was more than 134. |
| This journal is © The Royal Society of Chemistry 2021 |