
Mechanical bonding activation in rotaxane-based organocatalysts†
Jesus de Maria
Perez
 a,
Julio
Puigcerver
a,
Tainara
Orlando
b,
Aurelia
Pastor
a,
Marcos A. P.
Martins
b,
Mateo
Alajarin
a,
Alberto
Martinez-Cuezva
*a and
Jose
Berna
*a
a,
Julio
Puigcerver
a,
Tainara
Orlando
b,
Aurelia
Pastor
a,
Marcos A. P.
Martins
b,
Mateo
Alajarin
a,
Alberto
Martinez-Cuezva
*a and
Jose
Berna
*a
aDepartamento de Química Orgánica, Facultad de Química, Regional Campus of International Excellence “Campus Mare Nostrum”, Universidad de Murcia, E-30100, Murcia, Spain. E-mail: amcuezva@um.es; ppberna@um.es
bNúcleo de Química de Heterociclos (NUQUIMHE), Departamento de Química, Universidade Federal de Santa Maria, 97105-900 Santa Maria-RS, Brazil
First published on 1st June 2021
Abstract
We report herein the enhanced efficiency as organocatalysts of a series of succinamide-based hydrogen-bonded [2]rotaxanes functionalized with an acyclic secondary amine as the catalytically active site. We also evaluated their catalytic activity, compareds with that of their non-interlocked threads, in an iminium-type process between crotonaldehyde and acetylacetone. The presence of an interlocked polyamide macrocycle notably increased the catalytic activity of the entwined organocatalysts. The mechanized catalysts rapidly form a reactive iminium intermediate with the aldehyde, increasing its population. The hydrogen-bonding interaction established between the macrocycle and the electrophile has been proposed as one of the reasons for the rapid formation and stabilization of this key intermediate.
Introduction
The synthesis and study of mechanically interlocked molecules (MIMs) have experienced a huge evolution during the last few decades.1 The mechanical bond provides stability and unique internal dynamics to the entwined molecules, making them ideal candidates to be applied in numerous fields, highlighting that of the molecular machinery.2 Amongst the different uses of the MIMs, different research groups have recently focused on the applicability of these systems as ligands in metal-catalysed processes3 and organocatalysts,4 including their use in selective asymmetric transformations.5 In catalysts with the rotaxane structure,6 the mechanical bond offers singular features7 that can be advantageous for the control of the reaction outcomes, such as their ability to create dynamic enzyme-like 3D cavities due to the orthogonal disposition of the entwined components, a feature difficult to build in conventional covalent catalysts.8 In addition, the well-known protecting effect of the mechanical bond,9 kinetically stabilizing the functional groups located inside the macrocyclic cavity, has been successfully exploited for the development of an extensive number of rotaxane-based switchable catalysts.10 The precise control of the position of the bulky macrocycle along the thread allows concealing or exposing the active site placed on the thread, altering the catalytic capability of the systems in terms of activity (ON/OFF),11 enantio- or diastereoselectivity switching,12 or selection between diverse activation modes.13 As a consequence, non-interlocked threads are generally more reactive than the hampered interlocked systems.Herein we evaluate the behaviour as organocatalysts of a series of succinamide-based hydrogen-bonded [2]rotaxanes, bearing a catalytically active acyclic secondary amine at the thread (Fig. 1). These interlocked organocatalysts contain either one single or two binding-sites embedded into the axis, which offer different internal dynamics. The comparison of the catalytic activity of the non-interlocked threads versus the rotaxanes will allow us to determine the effect of the mechanical bond on their catalytic performance (Fig. 1, inset I). Aiming to explore the consequences of the ring translational motion on the catalysis, the comparison between the one single and two binding-site rotaxanes is also examined. The translational ring dynamics at the two-binding-site rotaxane largely contrasts with that of a single succinamide-based surrogate. The ring is located most of the time over the succinamide station in the one binding-site system, leading to a nearly full exposure of the active site (Fig. 1a). In the case of the two identical binding-site rotaxane (a so-called degenerate rotaxane),14 the ring continuously moves back and forward from one station to the other (Fig. 1, inset II). Thus, two possible scenarios may be envisioned when the system is used as a catalyst: the bulky polyamide macrocycle (blue ring in Fig. 1) momentarily conceals the active site, restricting the approaching of the substrates and decreasing the catalytic activity of the catalyst (Fig. 1b) or the polyamide macrocycle15 activates the process by establishing hydrogen-bonding (HB) interactions with the substrates (Fig. 1c). The weakly acidic protons of the –CONH– groups present on the interlocked macrocycle might be enabled to orient and/or activate the substrates, stabilizing the transition states and cooperatively increasing the overall catalytic rate. The Michael addition between acetylacetone and crotonaldehyde was selected as the model iminium-type catalytic process.
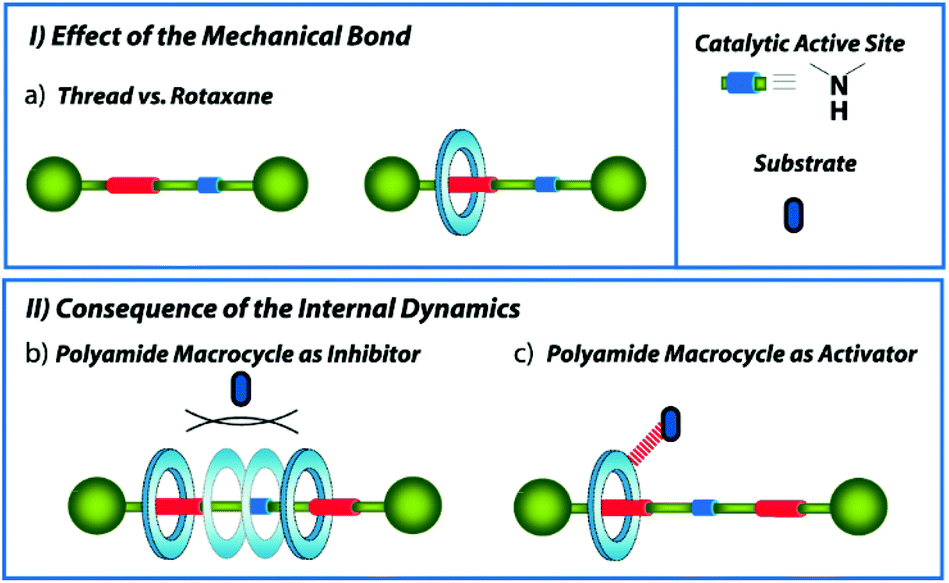 | ||
| Fig. 1 Different scenarios studied in this work: (I.a) Effect of the mechanical bond on the catalysis: thread versus rotaxane-based organocatalysts; (II.b) catalytic activity decrease due to the steric hindrance of the polyamide macrocycle; (II.c) catalytic activity enhancement by hydrogen-bonding activation of the substrates triggered by the polyamide macrocycle. | ||
Results and discussion
Aiming to study the different scenarios proposed in Fig. 1, we assembled rotaxane 2a, having one single succinamide as the binding-site, and rotaxane 2b, with two identical succinamide stations (Fig. 2). An acyclic secondary amine was placed in the systems as the active site. The five-component reaction between p-xylylenediamine, isophthaloyl chloride, and the corresponding Boc protected threads Boc1 afforded Leigh-type [2]rotaxanes Boc2,16 which after acid deprotection, yielded organocatalysts 2 (see the ESI† for the complete synthetic protocols). The corresponding [3]rotaxane 3b was also isolated in a minor amount during the assembly reaction of 2b (Fig. 2).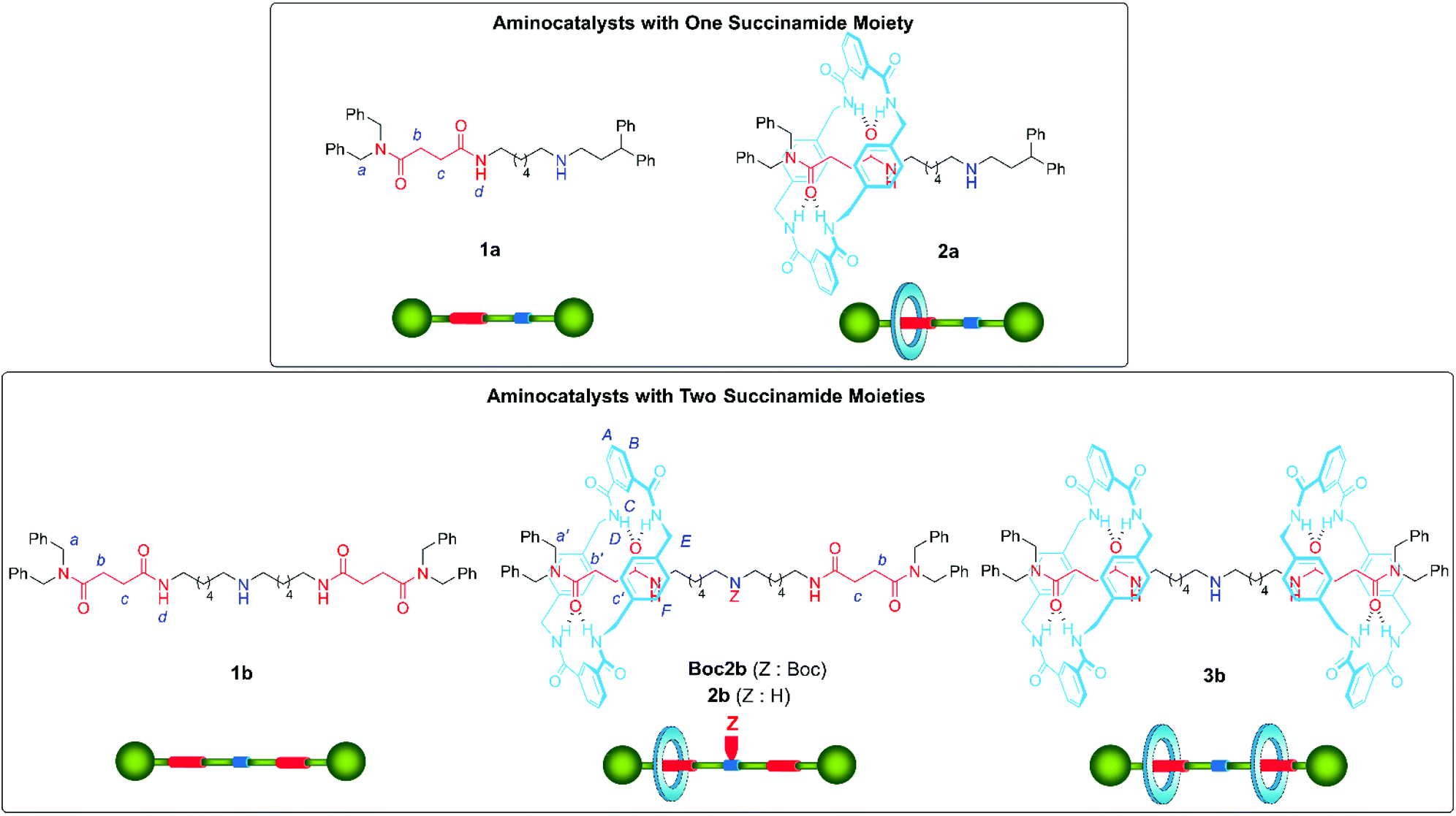 | ||
| Fig. 2 Synthesized systems evaluated as organocatalysts. | ||
The comparison of the 1H NMR spectra of the single succinamide-based thread 1a and its rotaxane 2a indicates that the macrocycle predominantly settles over the succinamide station (the signals related to succinamides Hb and Hc experienced a significant shifting due to the shielding by the macrocycle, Δδ = 1.48 ppm; see Fig. S2†). Similarly, the comparison of the 1H NMR spectra of thread 1b and rotaxanes Boc2b and 2b, recorded in CDCl3, allowed us to trace in each case the relative position of the macrocycle along the thread (Fig. 3). The methylene protons of the succinamide binding site (Hb+c, in red) in thread 1b appeared at 2.78 and 2.58 ppm (Fig. 3a). The bulky Boc group in rotaxane Boc2b, which acts as a steric barrier, prevents the free translational movement of the macrocycle along the embedded thread, leading to the compartmentalization of the interlocked system (Fig. 3b).17
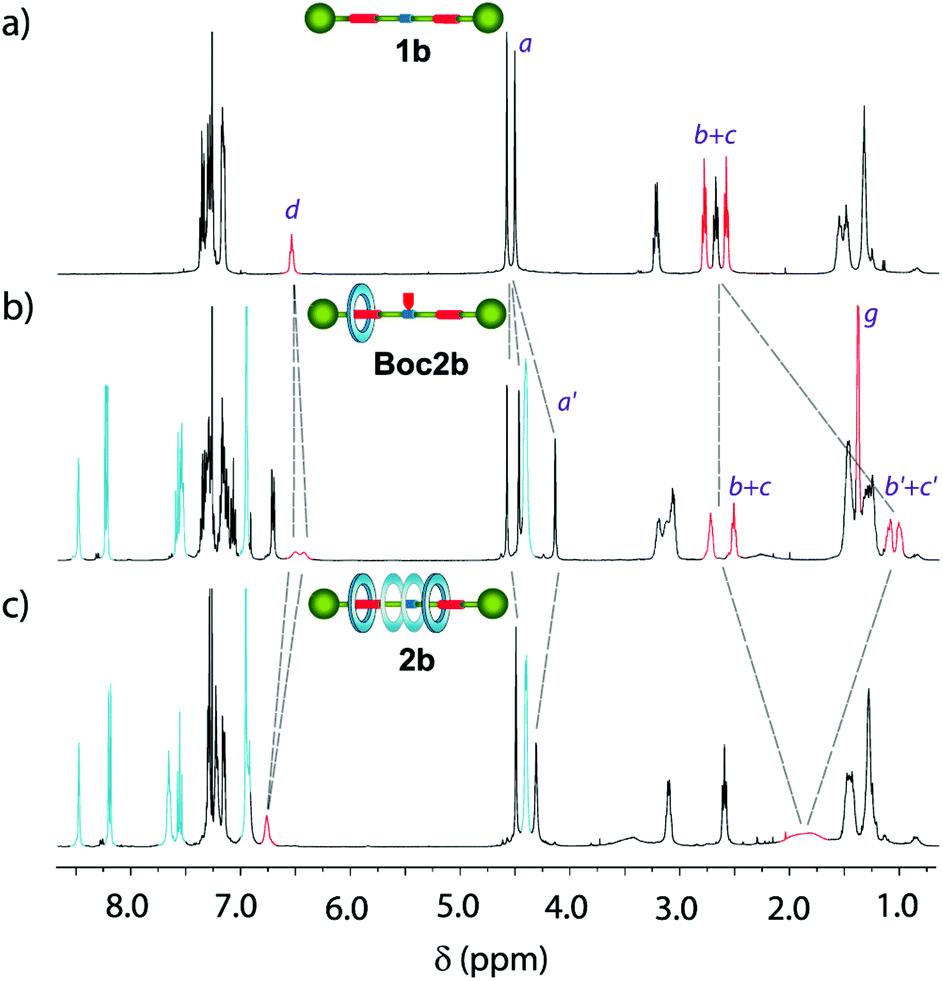 | ||
| Fig. 3 Stacked 1H NMR spectra (400 MHz, CDCl3, 298 K) of (a) thread 1b; (b) compartmentalised [2]rotaxane Boc2b; and (c) degenerate [2]rotaxane 2b. See lettering in Figure 2. | ||
Two succinamide stations with different magnetic environments are then noticeable: one occupied by the macrocycle, whose Hb′+c′ nuclei are shielded (δ = 1.08 and 1.00 ppm), and an unoccupied one in which nuclei Hb+c appeared at similar chemical shifts to those of thread 1b (δ = 2.72 and 2.51 ppm). Moreover, the signals related to the methylene protons of the transoid benzyl groups (Ha′) also experienced important changes, upshifting Δδ = 0.37 ppm. After the Boc removal, the macrocycle can move back and forward between the two stations of 2b (Fig. 3c). The signals related to the two equivalent succinamides appeared between 2.00 and 1.60 ppm as a broad multiplet. Note that its chemical shift is approximately the average of the corresponding shifts of the occupied and unoccupied binding sites in Boc2b.
The kinetics of the shuttling movement of the macrocyclic tetralactam along the thread in 2b was analysed by 1H VT-NMR spectroscopy (see the ESI for further details, Fig. S1 and Table S1†).18 Above room temperature the macrocycle is uninterruptedly moving between both succinamide stations, and an averaged co-conformation is observed by 1H NMR. When the temperature is gradually decreased, the shuttling motion slowed down until the splitting of the resonances for the succinamide moieties is observed below 278 K (Tc). At this temperature, a co-conformational “freezing” occurred, observing one succinamide shielded by the macrocycle and the second one naked. From the separation of the signals corresponding to the two succinamides in the slow dynamic regime, an energy barrier of 12.17 kcal mol−1 for macrocycle shuttling was calculated, which is in the range of other amide-based degenerate rotaxanes,19 with a shuttling rate of 1571 s−1.
Next, we explored the catalytic activity of the synthesized systems 1 and 2. Dibutylamine was also tested as a simplified catalytic model. The secondary amine as the active site allows these systems to participate as aminocatalysts in iminium-type transformations.20,21 We chose the conjugate addition of acetylacetone 5a to crotonaldehyde 4a as the model iminium-type catalysed process, monitoring the formation of Michael adduct 6aa over time by 1H NMR spectroscopy (Fig. 4, see the ESI for further experimental details, Fig. S3–S21†). The reactions were conducted with freshly distilled crotonaldehyde (4a, 2 equiv.), acetylacetone (5a, 1 equiv., 0.125 M) and 5 mol% of the corresponding catalyst in CDCl3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 22 at 298 K without the need for additives23 (see Tables S2 and S3† for the optimization of the reaction conditions). CDCl3 is a non-competitive solvent in which the intercomponent hydrogen-bonding network between the thread and the macrocycle in the interlocked systems survives.24 Thread 1b (with 2 succinamides,
22 at 298 K without the need for additives23 (see Tables S2 and S3† for the optimization of the reaction conditions). CDCl3 is a non-competitive solvent in which the intercomponent hydrogen-bonding network between the thread and the macrocycle in the interlocked systems survives.24 Thread 1b (with 2 succinamides,  ) was more active than thread 1a (one succinamide,
) was more active than thread 1a (one succinamide,  ) in catalysing the Michael addition.25 Note that the plot with dibutylamine (
) in catalysing the Michael addition.25 Note that the plot with dibutylamine ( ) is almost overlapped with that catalysed by thread 1a. Rotaxanes 2 are more effective catalysts than threads 1, pointing to an active role of the threaded polyamide macrocycle. Rotaxane 2a (t1/2 ∼ 2 h,
) is almost overlapped with that catalysed by thread 1a. Rotaxanes 2 are more effective catalysts than threads 1, pointing to an active role of the threaded polyamide macrocycle. Rotaxane 2a (t1/2 ∼ 2 h,  ) showed a faster reaction rate than non-interlocked thread 1a (reaction too slow to measure the t1/2,
) showed a faster reaction rate than non-interlocked thread 1a (reaction too slow to measure the t1/2,  ). The same trend was found when comparing the activities of rotaxane 2b (t1/2 ∼ 1.8 h,
). The same trend was found when comparing the activities of rotaxane 2b (t1/2 ∼ 1.8 h,  ) and that of thread 1b (t1/2 ∼ 7 h,
) and that of thread 1b (t1/2 ∼ 7 h,  ). Rotaxane 2b (
). Rotaxane 2b ( ) (Fig. S15† shows the monitoring of the reaction over time followed by 1H NMR) was the most active catalytic species, obtaining almost 90% conversion of the adduct 6aa after 12 h, versus the 70% conversion achieved with rotaxane 1a. Having in mind that the macrocycle in two-station rotaxane 2b is continuously translocating between the two succinamide moieties, and thus temporally encapsulating the amine active site, this activity enhancement is an intriguing fact. As we envisioned, the reaction catalysed by the highly hindered [3]rotaxane 3b was slower than that catalysed by [2]rotaxane 2b (less than 50% conversion after 12 h, not shown in Fig. 4; see Fig. S22†). The presence of two macrocycles mechanically linked to the thread hampers the access of the substrates to the active site, drastically reducing the catalytic performance of the system.
) (Fig. S15† shows the monitoring of the reaction over time followed by 1H NMR) was the most active catalytic species, obtaining almost 90% conversion of the adduct 6aa after 12 h, versus the 70% conversion achieved with rotaxane 1a. Having in mind that the macrocycle in two-station rotaxane 2b is continuously translocating between the two succinamide moieties, and thus temporally encapsulating the amine active site, this activity enhancement is an intriguing fact. As we envisioned, the reaction catalysed by the highly hindered [3]rotaxane 3b was slower than that catalysed by [2]rotaxane 2b (less than 50% conversion after 12 h, not shown in Fig. 4; see Fig. S22†). The presence of two macrocycles mechanically linked to the thread hampers the access of the substrates to the active site, drastically reducing the catalytic performance of the system.
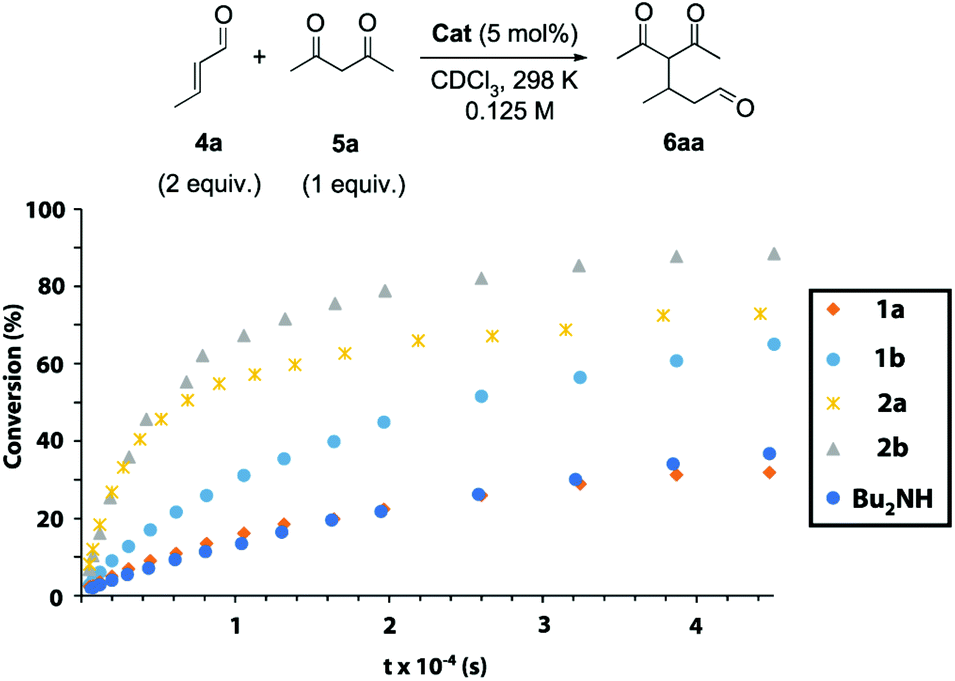 | ||
Fig. 4 Plot of the conversion (%) versus time of the Michael addition between crotonaldehyde and acetylacetone catalyzed by: thread 1a ( ), thread 1b ( ), thread 1b ( ), rotaxane 2a ( ), rotaxane 2a ( ), rotaxane 2b ( ), rotaxane 2b ( ) and dibutylamine ( ) and dibutylamine ( ). The conversion was determined by 1H NMR using CH2Br2 as an internal standard. ). The conversion was determined by 1H NMR using CH2Br2 as an internal standard. | ||
Our interest in uncovering the role of the mechanical bond, keeping both components (macrocycle and thread) linked, in the improved catalytic performance led us to carry out an additional experiment by employing a mixture composed of thread 1b and an acyclic tetraamide surrogate, N1,N3-bis(4-(benzamidomethyl)benzyl)isophthalamide,15a as a catalytic system (see Fig. S23 and S24†). The activation effect when this surrogate is present is negligible, observing a behaviour similar to that obtained for thread 1b itself. Note that the same experiment using the free macrocycle was unfeasible due to the high insolubility of the polyamide ring in CDCl3.26
Aiming to gain insight into the enhanced catalytic activity showed by the interlocked systems, we designed further experiments. The addition of acetylacetone 5a to crotonaldehyde 4a catalysed by secondary amines proceeds via the formation of the corresponding iminium intermediates.21 Thus, we mixed the corresponding catalyst (dibutylamine, thread 1b or rotaxane 2b) in the presence of variable amounts of crotonaldehyde 4a in CDCl3 (0.01 M) at room temperature. After this time, the 1H NMR spectra were registered. In the case of dibutylamine, no change in the spectra was observed after 48 h when 4 equiv. of crotonaldehyde 4a were added. It is known that the presence of catalytic amounts of Brønsted acid helps in the stabilization of the iminium derivatives.27 Thus, the addition of TFA allowed us to observe a considerable amount of the corresponding iminium salt intermediate (verified by 1D and 2D NMR experiments, Fig. S25–S27†). In contrast, tiny signals appeared in the spectra of the equimolecular mixture of thread 1b and crotonaldehyde 4a after 48 hours, which can be attributable to an iminium intermediate (Fig. S28†). Remarkably, in the case of rotaxane 2b (for clarity, Fig. 5 also shows the spectra of rotaxanes Boc2b and 2b), the appearance of new signals, which increased upon addition of more equivalents of aldehyde and time, rapidly arose (4 hours) (Fig. 5c and d). The desymmetrization of the signals related to the rotaxane, increasing the complexity of the spectra, points to the compartmentalization of the system, as in rotaxane Boc2b (Fig. 5a). Thus, the formation of the corresponding iminium salt 7b by the reaction of crotonaldehyde with the secondary amine in 2b precludes the free motion of the macrocycle.28 Accordingly, a signal emerged at 4.14 ppm, attributable to a shielded benzyl group of the stopper (Ha′), at a similar chemical shift to that of rotaxane Boc2b. Other signals also arose in the macrocycle region (8.5–8.00 ppm) and the aliphatic one (two succinamides are clearly observable, highlighted in red in Fig. 5c and d), indicative of the formation of a desymmetrized rotaxane derivative. Moreover, several peaks emerged between 5.50 and 6.00 ppm (H2+3, see numbering in Fig. 7), attributable to the olefinic protons of the iminium salt 7b, at 9.44 ppm (iminium hydrogen, H1) and 1.00 ppm (CH3, H4). The complex 13C NMR spectra of this mixture also point to the compartmentalization of rotaxane 2b upon reaction with the aldehyde (see ESI, Fig. S29†). Additionally, when acetylacetone 5a was added to this mixture, signals attributable to Michael adduct 6aa arose after a short time (in yellow, Fig. 5e), indicating that the formed intermediate is an active species in the process.
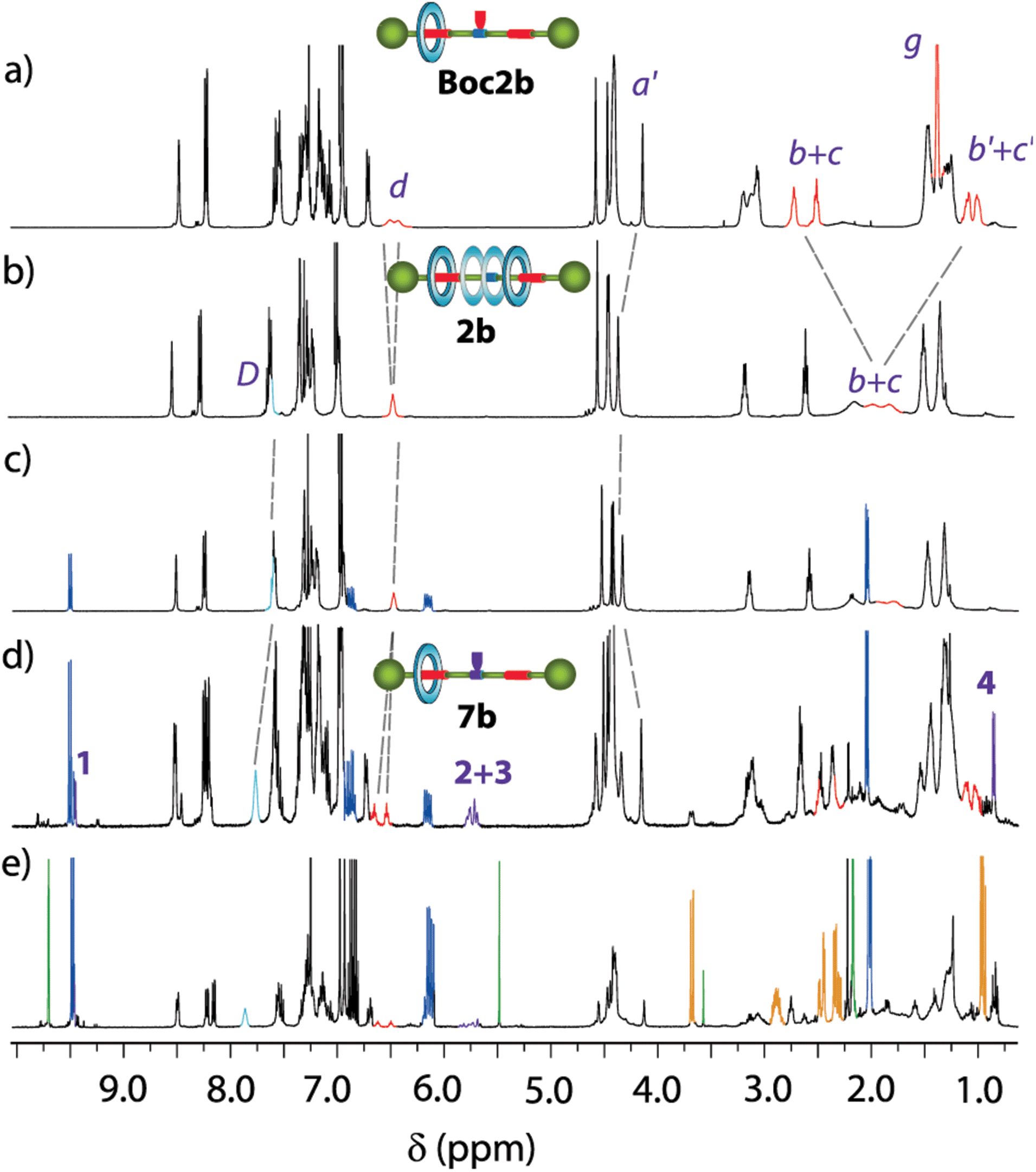 | ||
| Fig. 5 Stacked 1H NMR spectra (400 MHz, CDCl3, 298 K) of (a) Boc-protected rotaxane Boc2b; (b) rotaxane 2b; (c) a mixture of rotaxane 2b and 1 equivalent of crotonaldehyde 4a (dark blue) after 3 min; (d) a mixture of rotaxane 2b and 2 equivalents of crotonaldehyde 4a (dark blue) after 24 h; (e) addition of acetylacetone 5a (green) to (d) after 6 hours. Signals corresponding to the Michael adduct 6aa are highlighted in orange. Signals assigned to the new species 7b are highlighted in purple and marked with numbers. The assignments correspond to the lettering shown in Scheme 1 and numbering in Fig. 7. | ||
Aiming to gain further evidence of the formation of the iminium salt intermediate 7b, we performed 1H PGSE (Pulsed Gradient Spin Echo) diffusion measurements with a mixture of rotaxane 2b and crotonaldehyde 4a (600 MHz, CDCl3, 298 K). The obtained diffusion coefficients (D) could help to distinguish among the species present in solution, having in mind the large difference in these values expected for crotonaldehyde 4a, rotaxane 2b and the putative intermediate 7b. Thus, we determined a D value of 2.20 × 10−9 m2 s−1 for crotonaldehyde 4a, and a value of 4.7 × 10−10 m2 s−1 for rotaxane 2b. The intensity decays were followed on the resonances marked with asterisks in Fig. 6 (see the ESI for experimental details, Fig. S31–S34†). The new signals that emerged in the mixture of rotaxane 2b and crotonaldehyde (mixture 2b:4a, 1:10), tentatively attributable to the hydrogens of iminium intermediate 7b, showed a D value of 4.2 × 10−10 m2 s−1, slightly smaller than that obtained for rotaxane 2b. This data strongly supports the formation of iminium salt 7b.
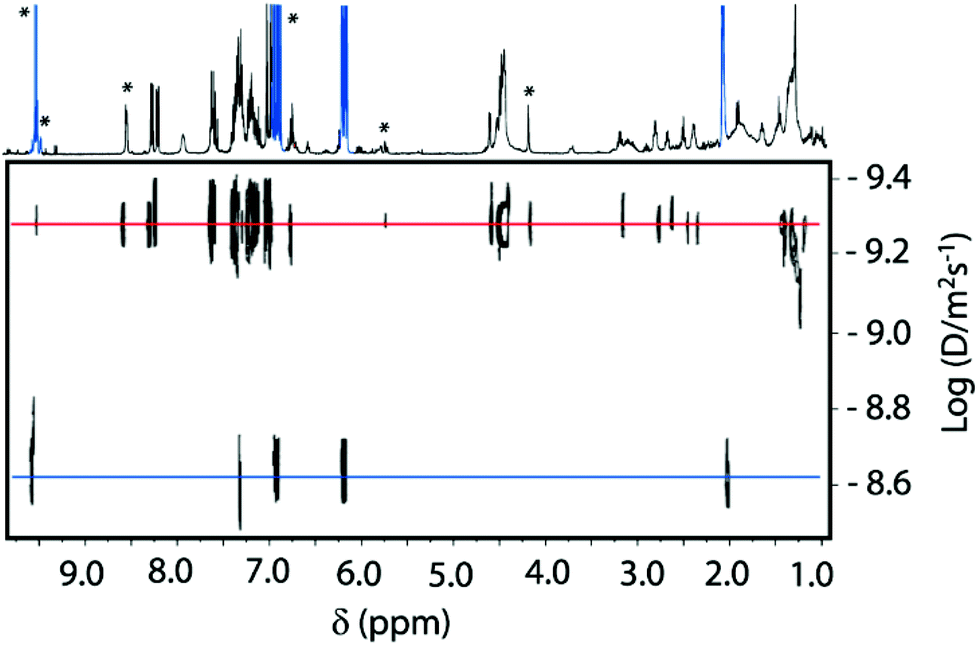 | ||
| Fig. 6 DOSY NMR spectra (600 MHz, CDCl3, 298 K) of a mixture of rotaxane 2b (2 mM) and crotonaldehyde 4a (10 equivalents) after 24 hours. | ||
After analysis of the data, we propose that the higher catalytic performance showed by rotaxanes 2 when compared with their non-interlocked threads 1 (and with dibutylamine) is predominantly a consequence of the amide functions present at the macrocycle. As an initial point, the establishment of hydrogen-bonding (HB) interactions between the amide groups29 with crotonaldehyde 4a might induce an increase of its electrophilic character towards the ulterior nucleophilic attack of the amine group (Fig. 7, step 1). Indeed, the addition of crotonaldehyde 4a to rotaxane 2b instantly triggers a small downfield displacement of the signals related to the NH amide groups of the [2]rotaxane as a result of the association between the catalyst and the aldehyde (Fig. 5c). In addition, the formation of the intermediate iminium salt 7b+·OH− becomes favoured by intramolecular assistance of the macrocycle to bind the hydroxyl anion (see Fig. 7, step 2–3). It is known that thioureas and squaramides neutralise the hydroxyl anion formed during iminium type processes, promoting the stabilization of the active iminium salts in the absence of an additional acid as a co-catalyst.30 Considering this precedent, we propose that the acidic hydrogens of the aromatic amide groups in rotaxanes 2 can play a similar role to those reported systems, by binding the hydroxyl group of intermediate 7b+·OH− (see Fig. 7b and c). The deprotonation of one of the amide groups of the ring (or thread) by the hydroxyl group will form compartmentalised zwitterion 7b (see Fig. 5d and Fig. S29†).31 Indeed, we experimentally proved that an organic base such as tetrabutylammonium hydroxide (TBAH) is able to deprotonate the hydrogens of the NH amides of the ring in rotaxane 2b (see ESI, Fig. S30†). The addition of 1 equiv. of lyophilized TBAH to rotaxane 2b triggers the decrease of the intensity and shift of the 1H NMR signals related to the amides.
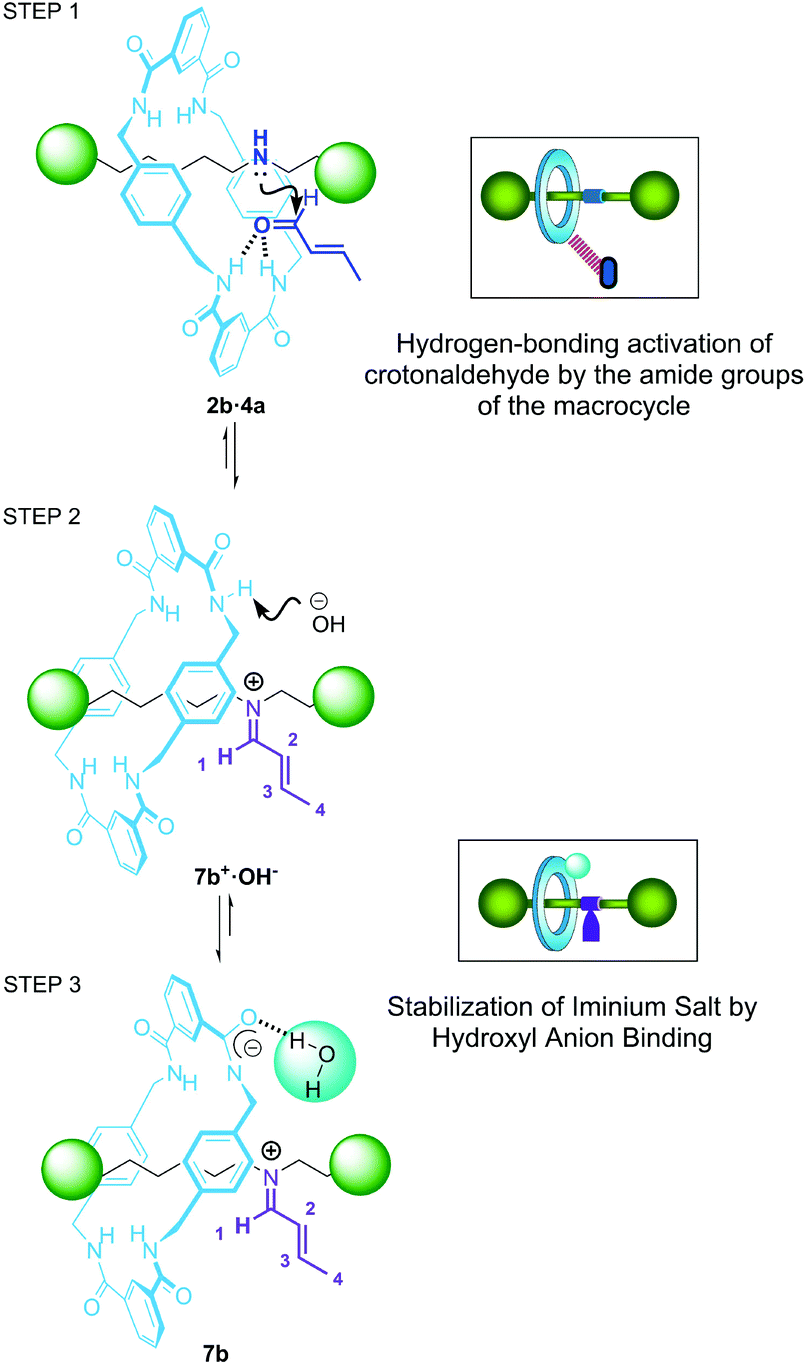 | ||
| Fig. 7 Proposed pathways for the formation of iminium intermediate 7b: (step 1) hydrogen-bonding activation of crotonaldehyde 4a; (step 2) formation of iminium salt 7b+·OH−; (step 3) macrocycle-assisted hydroxyl anion binding for the stabilization of zwitterion 7b. | ||
It is worth noting that mechanical shuttling in two-station rotaxane 2b is also important for the enhancement of the catalytic activity of the system,32 enabling the amide groups of the ring to easily interact with the crotonaldehyde 4a in a suitable orientation. In contrast, these amide–aldehyde interactions may be less noticeable in the case of one binding-site rotaxane 2a, in which the macrocycle is predominantly fixed over the succinamide binding-site by establishing stable bifurcated hydrogen bonds (see Fig. S2†) and decreasing its ability to form stabilizing HB with the electrophile. This causes a slight decrease of the catalytic activity of 2a compared with the two-binding site system 2b.
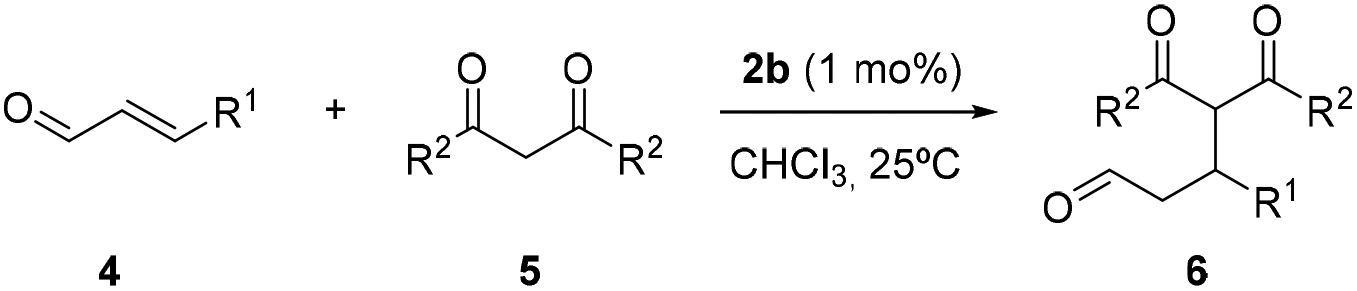
By using the most active catalyst 2b, we explore the scope of this process, decreasing the catalyst loading down to 1 mol% (Table 1, see ESI, Fig. S35–S37†). The reaction of diketones 5a,b as nucleophiles with crotonaldehyde 4a smoothly afforded adducts 6aa and 6ab in high conversions in 24 hours (Table 1, entries 1 and 2), showing that catalyst 2b remains active at the low catalyst loading employed. The variation of the aldehyde was also tested. The reaction of cinnamaldehyde 4b with acetylacetone 5a provided the corresponding adduct 6ba in a moderate conversion (Table 1, entry 3). The reaction between crotonaldehyde 4a and acetylacetone 5a, without a catalyst, was also tested, not observing the formation of the corresponding adduct (Table S2,† entry 1). It is worth mentioning that this iminium-type transformation has been used for the evaluation of various switchable mechanically interlocked catalysts.11b,13a,b In these reports, the employment of sodium acetate as an additive was required in order to activate the nucleophile. Moreover, high catalyst loadings (15–20 mol%) and long reaction times (2–5 days) were necessary for obtaining good conversions of the adducts. Remarkably, additives, such as acids, were not required by using our interlocked catalyst, obtaining high conversions in hours at low catalyst loadings (1 mol%).
a
|
|
|||
|---|---|---|---|
| Entry | 4 (R1) | 5 (R2) |
6b (%) |
| a Reaction conditions: aldehyde (2 equiv.), nucleophile (1 equiv., 0.5 M), 2b (1 mol%), CHCl3, 25 °C, 24 h. b Conversion calculated by 1H NMR after 24 h of reaction. c The reaction without the catalyst was fully unproductive. | |||
| 1 | 4a (Me) | 5a (Me) | 6aa (92)c |
| 2 | 4a (Me) | 5b (Ph) | 6ab (80) |
| 3 | 4b (Ph) | 5a (Me) | 6ba (57) |
Conclusions
In conclusion, we have synthesized a series of mechanically interlocked systems having one or two succinamides as binding-sites. The presence of an acyclic secondary amine in the thread allows these systems to be evaluated as organocatalysts in amino-catalysed transformations. In the case of a two binding-site system, the shuttling dynamics was studied, and the shuttling rate was calculated. We found that the mechanical bond has a deep influence on the catalytic performance of the interlocked systems, importantly increasing the reaction rates when compared with their non-interlocked threads. Moreover, bis(succinamide)-based [2]rotaxane showed the highest activity when compared with the rest of the systems, probably due to its ring dynamics enabling the activation of the electrophile. Moreover, the generation of the iminium ions without the addition of an external acid highlights the assistance of the mechanical bond (polyamide macrocycle) with hydroxyl anion binding, contributing to the enhanced catalytic performance. This activation effect strongly contrasts with the well-established hindering consequence of the mechanical bond reported in switchable rotaxane-based catalysts, opening the door to the development of new activation modes in catalysis via mechanical bonding. Research on this line is ongoing in our laboratories, focused on the optimization of the interlocked catalysts for their use in other catalytic processes, including their asymmetric versions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the MINECO and MICINN (CTQ2017-87231-P, PID2020-113686GB-I00 and RYC-2017-22700) with joint financing by FEDER Funds and Fundacion Seneca-CARM (Project 20811/PI/18). T. O. is grateful for the fellowship from CAPES/Print (Proc. 8881.310412/2018-01). J. P. thanks the Ministerio de Ciencia, Innovación y Universidades, for his predoctoral contract (FPU19/05419).Notes and references
- (a) J.-P. Sauvage and P. Garpard, From Non-Covalent Assemblies to Molecular Machines, Wiley, Weinheim, 2011 Search PubMed; (b) C. J. Bruns and J. F. Stoddart, The Nature of the Mechanical Bond: From Molecules to Machines, Wiley, New York, 2016 Search PubMed.
- (a) J. M. Abendroth, O. S. Bushuyev, P. S. Weiss and C. J. Barrett, Controlling Motion at the Nanoscale: Rise of the Molecular Machines, ACS Nano, 2015, 9, 7746–7768 Search PubMed; (b) S. Erbas-Cakmak, D. A. Leigh, C. T. McTernan and A. L. Nussbaumer, Artificial Molecular Machines, Chem. Rev., 2015, 115, 10081–10206 Search PubMed; (c) L. van Dijk, M. J. Tilby, R. Szpera, O. A. Smith, H. A. P. Bunce and S. P. Fletcher, Molecular Machines for Catalysis, Nat. Rev. Chem., 2018, 2, 0117 Search PubMed.
- (a) P. Thordarson, E. J. A. Bijsterveld, A. E. Rowan and R. J. M. Nolte, Epoxidation of Polybutadiene by a Topologically Linked Catalyst, Nature, 2003, 424, 915–918 Search PubMed; (b) G. Hattori, T. Hori, Y. Miyake and Y. Nishibayashi, Design and Preparation of a Chiral Ligand Based on a Pseudorotaxane Skeleton: Application to Rhodium-Catalyzed Enantioselective Hydrogenation of Enamides, J. Am. Chem. Soc., 2007, 129, 12930–12931 Search PubMed; (c) Y. Li, Y. Feng, Y.-M. He, F. Chen, J. Pan and Q.-H. Fan, Supramolecular Chiral Phosphorous Ligands Based on a [2]Pseudorotaxane Complex for Asymmetric Hydrogenation, Tetrahedron Lett., 2008, 49, 2878–2881 Search PubMed; (d) N. Miyagawa, M. Watanabe, T. Matsuyama, Y. Koyama, T. Moriuchi, T. Hirao, Y. Furusho and T. Takata, Successive Catalytic Reactions Specific to Pd-Based Rotaxane Complexes as a Result of Wheel Translation along the Axle, Chem. Commun., 2010, 46, 1920–1922 Search PubMed; (e) Y. Suzaki, K. Shimada, E. Chihara, T. Saito, Y. Tsuchido and K. Osakada, [3]Rotaxane-Based Dinuclear Palladium Catalysts for Ring-closure Mizoroki–Heck Reaction, Org. Lett., 2011, 13, 3774–3777 Search PubMed; (f) S. Hoekman, M. O. Kitching, D. A. Leigh, M. Papmeyer and D. Roke, Goldberg Active Template Synthesis of a [2]Rotaxane Ligand for Asymmetric Transition-Metal Catalysis, J. Am. Chem. Soc., 2015, 137, 7656–7659 Search PubMed; (g) A. W. Heard and S. M. Goldup, Synthesis of a Mechanically Planar Chiral Rotaxane Ligand for Enantioselective Catalysis, Chem, 2020, 6, 1–13 Search PubMed; (h) F.-C. Hsueh, C.-Y. Tsai, C.-C. Lai, Y.-H. Liu, S.-M. Peng and S.-H. Chiu, N–Heterocyclic Carbene Copper(I) Rotaxanes Mediate Sequential Click Ligations with All Reagents Premixed, Angew. Chem., Int. Ed., 2020, 132, 11374–11378 Search PubMed; (i) L. Zhu, J. Li, J. Yang and H. Y. Au-Yeung, Cross Dehydrogenative C–O Coupling Catalysed by a Catenane-Coordinated Copper(I), Chem. Sci., 2020, 11, 13008–13014 Search PubMed.
- (a) B. Lewandowski, G. De Bo, J. W. Ward, M. Papmeyer, S. Kuschel, M. J. Aldegunde, P. M. E. Gramlich, D. Heckmann, S. M. Goldup, D. M. D'Souza, A. E. Fernandes and D. A. Leigh, Sequence-Specific Peptide Synthesis by an Artificial Small-Molecule Machine, Science, 2013, 339, 189–193 Search PubMed; (b) J. Berna, M. Alajarin and R.-A. Orenes, Azodicarboxamides as Template Binding Motifs for the Building of Hydrogen-Bonded Molecular Shuttles, J. Am. Chem. Soc., 2010, 132, 10741–10747 Search PubMed; (c) J. Y. C. Lim, N. Yuntawattana, P. D. Beer and C. K. Williams, Isoselective Lactide Ring Opening Polymerisation using [2]Rotaxane Catalysts, Angew. Chem., Int. Ed., 2019, 58, 6007–6011 Search PubMed; (d) C. McTernan, G. De Bo and D. A. Leigh, A Track-Based Molecular Synthesizer that Builds a Single-Sequence Oligomer through Iterative Carbon-Carbon Bond Formation, Chem, 2020, 6, 2964–2973 Search PubMed.
- (a) Y. Tachibana, N. Kihara and T. Takata, Asymmetric Benzoin Condensation Catalyzed by Chiral Rotaxanes Tethering a Thiazolium Salt Moiety via the Cooperation of the Component: Can Rotaxane Be an Effective Reaction Field?, J. Am. Chem. Soc., 2004, 126, 3438–3439 Search PubMed; (b) K. Xu, K. Nakazono and T. Takata, Design of Rotaxane Catalyst for O-Acylative Asymmetric Desymmetrization of meso-1,2-Diol Utilizing the Cooperative Effect of the Components, Chem. Lett., 2016, 45, 1274–1276 Search PubMed; (c) Y. Cakmak, S. Erbas-Cakmak and D. A. Leigh, Asymmetric Catalysis with a Mechanically Point-Chiral Rotaxane, J. Am. Chem. Soc., 2016, 138, 1749–1751 Search PubMed; (d) R. Mitra, H. Zhu, S. Grimme and J. Niemeyer, Functional Mechanically Interlocked Molecules: Asymmetric Organocatalysis with a Catenated Bifunctional Brønsted Acid, Angew. Chem., Int. Ed., 2017, 56, 11456–11459 Search PubMed; (e) A. Martinez-Cuezva, M. Marin-Luna, D. A. Alonso, D. Ros-Ñiguez, M. Alajarin and J. Berna, Interlocking the Catalyst: Thread versus Rotaxane-Mediated Enantiodivergent Michael Addition of Ketones to β-Nitrostyrene, Org. Lett., 2019, 21, 5192–5196 Search PubMed; (f) M. Calles, J. Puigcerver, D. A. Alonso, M. Alajarin, A. Martinez-Cuezva and J. Berna, Enhancing the Selectivity of Prolinamide Organocatalysts Using the Mechanical Bond in [2]Rotaxanes, Chem. Sci., 2020, 11, 3629–3635 Search PubMed; (g) N. Pairault, H. Zhu, D. Jansen, A. Huber, C. G. Daniliuc, S. Grimme and J. Niemeyer, Heterobifunctional Rotaxanes for Asymmetric Catalysis, Angew. Chem., Int. Ed., 2020, 59, 5102–5107 Search PubMed; (h) D. Jansen, J. Gramüller, F. Niemeyer, T. Schaller, M. C. Letzel, S. Grimme, H. Zhu, R. M. Gschwind and J. Niemeyer, What is the Role of Acid–Acid Interactions in Asymmetric Phosphoric Acid Organocatalysis? A Detailed Mechanistic Study using Interlocked and Non-interlocked Catalysts, Chem. Sci., 2020, 11, 4381–4390 Search PubMed.
- D. A. Leigh, V. Marcos and M. R. Wilson, Rotaxane Catalysts, ACS Catal., 2014, 4, 4490–4497 Search PubMed.
- (a) E. A. Neal and S. M. Goldup, Chemical Consequences of Mechanical Bonding in Catenanes and Rotaxanes: Isomerism, Modification, Catalysis and Molecular Machines for Synthesis, Chem. Commun., 2014, 50, 5128–5142 Search PubMed; (b) S. F. M. van Dongen, S. Cantekin, J. A. A. W. Elemans, A. E. Rowan and R. J. M. Nolte, Functional Interlocked Systems, Chem. Soc. Rev., 2014, 43, 99–122 Search PubMed; (c) M. Xue, Y. Yang, X. Chi, X. Yan and F. Huang, Development of Pseudorotaxanes and Rotaxanes: From Synthesis to Stimuli-Responsive Motions to Applications, Chem. Rev., 2015, 115, 7398–7501 Search PubMed.
- (a) N. H. Evans, Chiral Catenanes and Rotaxanes: Fundamentals and Emerging Applications, Chem. – Eur. J., 2018, 24, 3101–3112 Search PubMed; (b) E. M. G. Jamieson, F. Modicom and S. M. Goldup, Chirality in Rotaxanes and Catenanes, Chem. Soc. Rev., 2018, 47, 5266–5311 Search PubMed; (c) A. Martinez-Cuezva, A. Saura-Sanmartin, M. Alajarin and J. Berna, Mechanically Interlocked Catalysts for Asymmetric Synthesis, ACS Catal., 2020, 10, 7719–7733 Search PubMed; (d) C. Kwamen and J. Niemeyer, Functional Rotaxanes in Catalysis, Chem. – Eur. J., 2021, 27, 175–186 Search PubMed.
- (a) A. H. Parham, B. B. Windisch and F. Vögtle, Chemical Reactions in the Axle of Rotaxanes–Steric Hindrance by the Wheel, Eur. J. Org. Chem., 1999, 1233–1238 Search PubMed; (b) P. Ghosh, O. Mermagen and C. A. Schalley, Novel Template Effect for the Preparation of [2]Rotaxanes with Functionalised Centre Pieces, Chem. Commun., 2002, 2628–2629 Search PubMed; (c) T. Oku, Y. Furusho and T. Takata, Rotaxane-Stabilized Thiophosphonium Salt from Disulfide and Phosphine, Org. Lett., 2003, 5, 4923–4925 Search PubMed; (d) D. M. D'Souza, D. A. Leigh, L. Mottier, K. M. Mullen, F. Paolucci, S. J. Teat and S. Zhang, Nitrone [2]Rotaxanes: Simultaneous Chemical Protection and Electrochemical Activation of a Functional Group, J. Am. Chem. Soc., 2010, 132, 9465–9470 Search PubMed; (e) J. Winn, A. Pinczewska and S. M. Goldup, Synthesis of a Rotaxane CuI Triazolide under Aqueous Conditions, J. Am. Chem. Soc., 2013, 135, 13318–13321 Search PubMed.
- V. Blanco, D. A. Leigh and V. Marcos, Artificial Switchable Catalysts, Chem. Soc. Rev., 2015, 44, 5341–5370 Search PubMed.
- (a) V. Blanco, A. Carlone, K. D. Hänni, D. A. Leigh and B. Lewandowski, A Rotaxane-Based Switchable Organocatalyst, Angew. Chem., Int. Ed., 2012, 51, 5166–5169 Search PubMed; (b) V. Blanco, D. A. Leigh, U. Lewandowska, B. Lewandowski and V. Marcos, Exploring the Activation Modes of a Rotaxane-Based Switchable Organocatalyst, J. Am. Chem. Soc., 2014, 136, 15775–15780 Search PubMed; (c) V. Blanco, D. A. Leigh, V. Marcos, J. A. Morales-Serna and A. L. Nussbaumer, A Switchable [2]Rotaxane Asymmetric Organocatalyst That Utilizes an Acyclic Chiral Secondary Amine, J. Am. Chem. Soc., 2014, 136, 4905–4908 Search PubMed; (d) C. M. Álvarez, H. Barbero and D. Miguel, Multivalent Molecular Shuttles–Effect of Increasing the Number of Centers in Switchable Catalysts, Eur. J. Org. Chem., 2015, 6631–6640 Search PubMed; (e) Y.-J. Lee, K.-S. Liu, C.-C. Lai, Y.-H. Liu, S.-M. Peng, R. P. Cheng and S.-H. Chiu, Na+ Ions Induce the Pirouetting Motion and Catalytic Activity of [2]Rotaxanes, Chem. – Eur. J., 2017, 23, 9756–9760 Search PubMed; (f) C. Biagini, S. D. P. Fielden, D. A. Leigh, F. Schaufelberger, S. Di Stefano and D. Thomas, Dissipative Catalysis with a Molecular Machine, Angew. Chem., Int. Ed., 2019, 58, 9876–9880 Search PubMed.
- (a) M. Galli, J. E. M. Lewis and S. M. Goldup, A Stimuli-Responsive Rotaxane–Gold Catalyst: Regulation of Activity and Diastereoselectivity, Angew. Chem., Int. Ed., 2015, 54, 13545–13549 Search PubMed; (b) A. Martinez-Cuezva, A. Saura-Sanmartin, T. Nicolas-Garcia, C. Navarro, R.-A. Orenes, M. Alajarin and J. Berna, Photoswitchable Interlocked Thiodiglycolamide as a Cocatalyst of a Chalcogeno-Baylis–Hillman Reaction, Chem. Sci., 2017, 8, 3775–3780 Search PubMed; (c) M. Dommaschk, J. Echavarren, D. A. Leigh, V. Marcos and T. A. Singleton, Dynamic Control of Chiral Space Through Local Symmetry Breaking in a Rotaxane Organocatalyst, Angew. Chem., Int. Ed., 2019, 58, 14955–14958 Search PubMed.
- (a) J. Beswick, V. Blanco, G. De Bo, D. A. Leigh, U. Lewandowska, B. Lewandowski and K. Mishiro, Selecting Reactions and Reactants using a Switchable Rotaxane Organocatalyst with two Different Active Sites, Chem. Sci., 2015, 6, 140–143 Search PubMed; (b) C.-S. Kwan, A. S. C. Chan and K. C.-F. Leung, A Fluorescent and Switchable Rotaxane Dual Organocatalyst, Org. Lett., 2016, 18, 976–979 Search PubMed; (c) K. Eichstaedt, J. Jaramillo-Garcia, D. A. Leigh, V. Marcos, S. Pisano and T. A. Singleton, Switching between Anion-Binding Catalysis and Aminocatalysis with a Rotaxane Dual-Function Catalyst, J. Am. Chem. Soc., 2017, 139, 9376–9381 Search PubMed.
- A degenerate [2]rotaxane consists in a thread functionalized by two identical recognition sites, separated by a spacer, surrounded by a macrocycle. The shuttling process of the macrocycle back and forth between the two binding sites is isoenergetic.
- For previous studies reporting the activation of the reactivity due to the presence of an polyamide macrocycle around a functionality, see: (a) A. Martinez-Cuezva, C. Lopez-Leonardo, D. Bautista, M. Alajarin and J. Berna, Stereocontrolled Synthesis of β-Lactams within [2]Rotaxanes: Showcasing the Chemical Consequences of the Mechanical Bond, J. Am. Chem. Soc., 2016, 138, 8726–8729 Search PubMed; (b) A. Martinez-Cuezva, D. Bautista, M. Alajarin and J. Berna, Enantioselective Formation of 2-Azetidinones by Ring-Assisted Cyclization of Interlocked N-(α-Methyl)benzyl Fumaramides, Angew. Chem., Int. Ed., 2018, 57, 6563–6567 Search PubMed; (c) A. Martinez-Cuezva, C. Lopez-Leonardo, M. Alajarin and J. Berna, Stereocontrol in the Synthesis of β-lactams arising from the Interlocked Structure of Benzylfumaramide-based Hydrogen-bonded [2]Rotaxanes, Synlett, 2019, 893–902 Search PubMed; (d) A. Martinez-Cuezva, A. Pastor, M. Marin-Luna, C. Diaz-Marin, D. Bautista, M. Alajarin and J. Berna, Cyclization of Interlocked Fumaramides into β-lactams: Experimental and Computational Mechanistic Assessment of the Key Intercomponent Proton Transfer and the Stereocontrolling Active Pocket, Chem. Sci., 2021, 12, 747–756 Search PubMed.
- J. Berna, G. Bottari, D. A. Leigh and E. M. Perez, Amide-based Molecular Shuttles (2001–2006), Pure Appl. Chem., 2007, 79, 39–54 Search PubMed.
- (a) L. Jiang, J. Okano, A. Orita and J. Otera, Intermittent Molecular Shuttle as a Binary Switch, Angew. Chem., Int. Ed., 2004, 43, 2121–2124 Search PubMed; (b) M. N. Chatterjee, E. R. Kay and D. A. Leigh, Beyond Switches: Ratcheting a Particle Energetically Uphill with a Compartmentalized Molecular Machine, J. Am. Chem. Soc., 2006, 128, 4058–4073 Search PubMed; (c) A. Coskun, D. C. Friedman, H. Li, K. Patel, H. A. Khatib and J. F. Stoddart, A Light-Gated STOP–GO Molecular Shuttle, J. Am. Chem. Soc., 2009, 131, 2493–2495 Search PubMed; (d) A. Carlone, S. M. Goldup, N. Lebrasseur, D. A. Leigh and A. Wilson, A Three-Compartment Chemically-Driven Molecular Information Ratchet, J. Am. Chem. Soc., 2012, 134, 8321–8323 Search PubMed.
- (a) P.-L. Anelli, M. Asakawa, P. R. Ashton, R. A. Bissell, G. Clavier, R. Gorski, A. E. Kaifer, S. J. Langford, G. Mattersteig, S. Menzer, D. Philp, A. M. Z. Slawin, N. Spencer, J. F. Stoddart, M. S. Tolley and D. J. Williams, Toward Controllable Molecular Shuttles, Chem. – Eur. J., 1997, 3, 1113–1135 Search PubMed; (b) S. J. Rowan and J. F. Stoddart, Precision Molecular Grafting: Exchanging Surrogate Stoppers in [2]Rotaxanes, J. Am. Chem. Soc., 2000, 122, 164–165 Search PubMed; (c) T. Iijima, S. A. Vignon, H.-R. Tseng, T. Jarrosson, J. K. M. Sanders, F. Marchioni, M. Venturi, E. Apostoli, V. Balzani and J. F. Stoddart, Controllable Donor–Acceptor Neutral [2]Rotaxanes, Chem. – Eur. J., 2004, 10, 6375–6392 Search PubMed; (d) S. Nygaard, K. C. F. Leung, I. Aprahamian, T. Ikeda, S. Saha, B. W. Laursen, S.-Y. Kim, S. W. Hansen, P. C. Stein, A. H. Flood, J. F. Stoddart and J. O. Jeppesen, Functionally Rigid Bistable [2]Rotaxanes, J. Am. Chem. Soc., 2007, 129, 960–970 Search PubMed; (e) M. Hmadeh, A. C. Fahrenbach, S. Basu, A. Trabolsi, D. Benitez, H. Li, A.-M. Albrecht-Gary, M. Elhabiri and J. F. Stoddart, Electrostatic Barriers in Rotaxanes and Pseudorotaxanes, Chem. – Eur. J., 2011, 17, 6076–6087 Search PubMed.
- (a) P. Ghosh, G. Federwisch, M. Kogej, C. A. Schalley, D. Haase, W. Saak, A. Luetzen and R. M. Gschwind, Controlling the Rate of Shuttling Motions in [2]Rotaxanes by Electrostatic Interactions: a Cation as Solvent-Tunable Brake, Org. Biomol. Chem., 2005, 3, 2691–2700 Search PubMed; (b) J. Berna, M. Alajarin, C. Marin-Rodriguez and C. Franco-Pujante, Redox Divergent Conversion of a [2]Rotaxane into two Distinct Degenerate Partners with Different Shuttling Dynamics, Chem. Sci., 2012, 3, 2314–2320 Search PubMed; (c) D. Günbaş and A. M. Brouwer, Degenerate Molecular Shuttles with Flexible and Rigid Spacers, J. Org. Chem., 2012, 77, 5724–5735 Search PubMed; (d) M. Douarre, V. Martí-Centelles, C. Rossy, I. Pianet and N. D. McClenaghan, Regulation of Macrocycle Shuttling Rates in [2]Rotaxanes by Amino–Acid Speed Bumps in Organic–Aqueous Solvent Mixtures, Eur. J. Org. Chem., 2020, 5820–5827 Search PubMed.
- (a) B. List, The Ying and Yang of Asymmetric Aminocatalysis, Chem. Commun., 2006, 819–824 Search PubMed; (b) A. Erkkilä, I. Majander and P. M. Pihko, Iminium Catalysis, Chem. Rev., 2007, 107, 5416–5470 Search PubMed.
- Reports in which acyclic secondary amines are employed as organocatalysts in iminium type processes are scarce due to their lower reactivity when compared with cyclic secondary amines or primary amines. For some selected examples where the corresponding iminium salts are isolated or employed as catalysts, see: (a) N. J. Leonard and J. V. Paukstelis, Direct Synthesis of Ternary Iminium Salts by Combination of Aldehydes or Ketones with Secondary Amine Salts, J. Org. Chem., 1963, 28, 3021–3024 Search PubMed; (b) J. W. Yang, M. T. Hechavarria Fonseca and B. List, A Metal-Free Transfer Hydrogenation: Organocatalytic Conjugate Reduction of α,β-Unsaturated Aldehydes, Angew. Chem., Int. Ed., 2004, 43, 6660–6662 Search PubMed; (c) S. Lakhdar, T. Tokuyasu and H. Mayr, Electrophilic Reactivities of α,β-Unsaturated Iminium Ions, Angew. Chem., Int. Ed., 2008, 47, 8723–8726 Search PubMed.
- CDCl3 was distilled over CaCl2 and kept with molecular sieves prior use.
- X. Companyó and J. Bures, Distribution of Catalytic Species as an Indicator to Overcome Reproducibility Problems, J. Am. Chem. Soc., 2017, 139, 8432–8435 Search PubMed.
- A. Saura-Sanmartin, A. Martinez-Cuezva, A. Pastor, D. Bautista and J. Berna, Light-Driven Exchange between Extended and Contracted Lasso-Like Isomers of a Bistable [1]Rotaxane, Org. Biomol. Chem., 2018, 16, 6980–6987 Search PubMed.
- Each experiment has been run in duplicate, showing a consistent reproducibility, with an estimated error less than 10% in all the cases. See ESI for further details.†.
- The solubility of the polyamide macrocycle in chloroform is less than 1 mg L−1: A. G. Johnston, D. A. Leigh, A. Murphy, J. P. Smart and M. D. Deegan, The Synthesis and Solubilization of Amide Macrocycles via Rotaxane Formation, J. Am. Chem. Soc., 1996, 118, 10662–10663 Search PubMed.
- H. Gotoh, T. Uchimaru and Y. Hayashi, Two Reaction Mechanisms via Iminium Ion Intermediates: The Different Reactivities of Diphenylprolinol Silyl Ether and Trifluoromethyl–Substituted Diarylprolinol Silyl Ether, Chem. – Eur. J., 2015, 21, 12337–12346 Search PubMed.
- A. S. Lane, D. A. Leigh and A. Murphy, Peptide-Based Molecular Shuttles, J. Am. Chem. Soc., 1997, 119, 11092–11093 Search PubMed.
- (a) X. Liu, L. Lin and X. Feng, Amide-based Bifunctional Organocatalysts in Asymmetric Reactions, Chem. Commun., 2009, 6145–6158 Search PubMed; (b) Ł. Albrecht, H. Jiang and K. A. Jørgensen, Hydrogen-Bonding in Aminocatalysis: From Proline and Beyond, Chem. – Eur. J., 2014, 20, 358–368 Search PubMed.
- V. Juste-Navarro, L. Prieto, I. Delso, R. Manzano, T. Tejero, E. Reyes, J. L. Vicario and P. Merino, A Case Study of Thiourea-Assisted Iminium Formation by Hydroxyl Anion Binding: Kinetic, Spectroscopic and Computational Evidences, Adv. Synth. Catal., 2017, 359, 4122–4128 Search PubMed.
- Note this amidate can be further stabilized by the establishment of intramolecular hydrogen-bonding interactions with the nearest amide group of the macrocycle.
- For other studies relating the internal dynamics of the catalysts with their catalytic activity, see: (a) P. Kumar Biswas, S. Saha, S. Gaikwad and M. Schmittel, Reversible Multicomponent AND Gate Triggered by Stoichiometric Chemical Pulses Commands the Self-Assembly and Actuation of Catalytic Machinery, J. Am. Chem. Soc., 2020, 142, 7889–7897 Search PubMed; (b) A. Goswami and M. Schmittel, Double Rotors with Fluxional Axles: Domino Rotation and Azide–Alkyne Huisgen Cycloaddition Catalysis, Angew. Chem., Int. Ed., 2020, 59, 12362–12366 Search PubMed; (c) M. Schmittel and P. Howlader, Toward Molecular Cybernetics – the Art of Communicating Chemical Systems, Chem. Rec., 2021, 21, 523–543 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental, characterization, and other additional information. See DOI: 10.1039/d1qo00789k |
| This journal is © the Partner Organisations 2021 |