
Multicatalysis from renewable resources: a direct route to furan-based polyesters†
Lucie
Guillaume
,
Adam
Marshall
,
Nicolas
Niessen
,
Pingping
Ni
,
Régis M.
Gauvin
 * and
Christophe M.
Thomas
*
* and
Christophe M.
Thomas
*
Chimie ParisTech, PSL University, CNRS, Institut de Recherche de Chimie Paris, 75005 Paris, France. E-mail: regis.gauvin@chimieparistech.psl.eu; christophe.thomas@chimieparistech.psl.eu; Web: http://www.ircp.cnrs.fr/la-recherche/equipe-cocp/
First published on 20th August 2021
Abstract
Here we present a multicatalytic route to produce 2,5-bis(hydroxymethyl)furan and the corresponding (co)polymers from stable 2,5-furanedicarboxylic acid. This approach combines the use of several commercial catalysts and in particular allows to quantitatively obtain two key furan intermediates, not contaminated by humins.
Renewable materials have recently gained much attention as an alternative to petroleum-based polymers.1–8 However, the main challenge is still to design efficient and selective transformations of abundant, renewable, low-cost raw materials into innovative polymeric products.9,10 Among the new biobased polymers that have emerged over the past decade, furanic materials are of particular interest.11–15 The availability of monomer feedstocks from renewable resources has given these polymers an increasing prominence in the market. As such, 2-hydroxymethyl-5-furfuraldehyde, also known as 5-(hydroxymethyl)furfural (HMF), which is obtained by dehydrating hexoses from biomass, has been viewed as having great potential, as it can be used to synthesize many valuable products. HMF can be converted to various 2,5-disubstituted furans, including 2,5-bis(hydroxymethyl)furan (BHMF), which has a rigid symmetric structure, allowing to obtain polymers with higher glass transition temperatures and better mechanical properties.16 Surprisingly, the potential of BHMF is largely underestimated, although it can be used as a monomer for the synthesis of polyesters, polycarbonates or polyurethanes. BHMF can be obtained by a variety of synthetic routes, but selective hydrogenation of HMF is the most widely employed.17,18 However, HMF from renewable resources is often contaminated with highly coloured polymeric impurities called humins.19 Since the purity and especially the colour of the monomer are important to obtain high quality colourless polymers (required for example by the packaging industry), the removal of humins from HMF production processes remains a problem. DuPont and ADM have recently developed effective processes to produce stable 2,5-furandicarboxylic acid (FDCA), that is virtually free of humins, from contaminated HMF.20 Therefore, there is an interest in investigating the hydrogenation of 2,5-furandicarboxylic acid esters in order to produce BHMF that would be of high quality for polymer synthesis. This would complement the scope of reported furan-based polyesters, in particular those based on FDCA and on various aliphatic diols.11
To synthesize BHMF-derived polyesters, we chose to use one-pot catalytic transformations, which have significant advantages over conventional multi-step syntheses, such as time and cost savings, reduced waste and energy consumption.21–23 These synthetic schemes, which proceed through two or more consecutive catalytic steps, can serve as a versatile method in polymerization reactions.24–28 However, the different catalytic systems used must be compatible with each other but also with the solvent, the substrate and the by-products of the reaction in order to achieve high activity and selectivity.29–32 Multicatalytic synthesis is therefore not only a useful methodology to follow for the production of (macro)molecules, but also a promising green approach for polymer synthesis.33 In this regard, oligomeric furan-based diols are also interesting synthetic targets due to the reactivity of the hydroxyl end groups with different functional groups.34 However, oligomeric diols are mainly synthesized industrially by depolymerization routes that are carried out under severe/drastic conditions (e.g., high temperature and pressure).35 Since furans are temperature sensitive, there is a real interest in developing alternative synthetic routes for the production of oligomeric furan diols. A multicatalytic approach can overcome these pitfalls with a combination of catalysts performing the desired reactions.
Herein we present a practical route to biobased furan-incorporating polyesters by way of a multicatalytic reaction using simple commercial catalysts. This process provides direct access to BHMF and the corresponding copolymers in high yields.
Monomer formation sequence
To carry out a one-pot procedure, it is necessary to have a monomer synthesis process that can produce BHMF with a high yield and selectivity. We chose to start from FDCA, first forming its dimethyl ester (MeFDCA), then reducing it by hydrogenation, which avoids salt formation. As the first step, we thus studied the esterification of FDCA using dimethyl dicarbonate (Moc2O) as a coupling agent (Scheme 1). On the basis of the mechanism proposed by Bartoli,36 we assumed that the presence of a Lewis acid (MgCl2) can cause a double addition of FDCA to the dicarbonate, affording a carboxylic anhydride and an alcohol. Then, the resulting symmetrical anhydride can react with the in situ released methanol from Moc2O. Thus, reaction conditions were identified, leading to complete, selective conversion of FDCA into MeFDCA in THF (Table S2†). The reaction also proceeds efficiently in the more environmentally friendly MeTHF,37 though with longer reaction times due to FDCA solubility issues.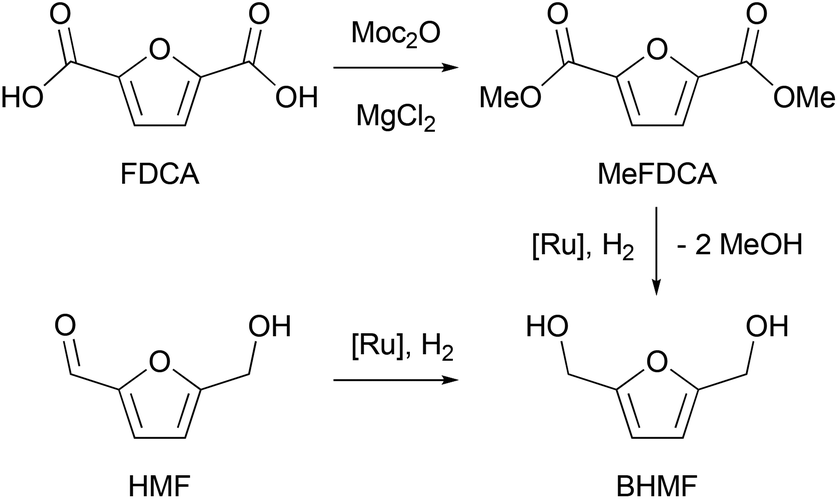 | ||
| Scheme 1 Catalytic access to BHMF from FDCA or HMF. | ||
Then, we investigated the next step of our multicatalytic sequence: the hydrogenation of 2,5-furandicarboxylic acid dimethyl ester (MeFDCA) in the presence of a molecular catalyst. Production of BHMF from HMF is a proven route with both homogeneous and heterogeneous catalytic processes.17 However, the above-mentioned issues with HMF intrinsic stability and storage motivate the search for alternative pathways. Along these lines, FDCA, and its ester derivatives could be efficiently harnessed as starting materials for polyester synthesis, especially when bearing in mind their use in implemented commercial solutions (Dupont-ADM process). Significant progress with the reduction of esters using hydrogenation38 motivated us to probe this route when looking for efficient entry into the production of BHMF. Interestingly, the dimethylester MeFDCA has been very scarcely used as a substrate. More specifically, the single report was from Beller and coworkers, who reported unselective reduction, with a manganese pincer catalyst.39 In spite of extended reaction time and use of a robust catalyst, the conversion of the aromatic diester into a mixture of the targeted diol and the monoreduced derivative evidenced that reduction of such a substrate may prove challenging. In the view of the breakthroughs in the field stemming from use of non-innocent pincer ligands, we probed two archetypical commercially available systems, namely Ru-PNP from Takasago40 and Ru-SNS (Gusev catalyst)41 given their unique robustness and versatility, as well as their recognized potential in the hydrogenation of esters (Fig. 1).42 The latter is most particularly interesting when aiming at developing large scale processes, as it relies on a phosphine-free pincer ligand scaffold. Such an air-stable compound is thus easier and cheaper to produce and handle compared to phosphine-derived systems.
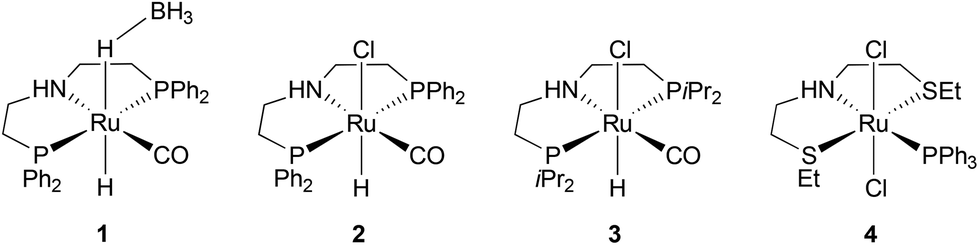 | ||
| Fig. 1 Ester hydrogenation precatalysts used for this study. | ||
Representative results are summarized on Table 1. Under moderately demanding conditions (100 °C, 50 bar H2) and in the presence of excess alkoxide, full conversion of 100 equivalents of diester is achieved within 2 hours in toluene. In the case of the PNP systems, the selectivity towards diol remains moderate, with marginal influence from either phosphine substituent (Ph vs. iPr) or pre-catalyst coordination sphere (BH4vs. Cl) (Table 1, entries 1–3). The Gusev catalyst displayed superior performances, which could even be improved by raising H2 pressure up to 80 bar (Table 1, entries 4 and 5). In the search for a less harmful solvent,37 we probed anisole and 2-methyltetrahydrofuran (MeTHF) as solvent for the hydrogenation step (Table 1, entries 6 and 7). Rewardingly, use of MeTHF resulted in improved selectivity, as full conversion and quasi-total selectivity were achieved under a 50 bar H2 pressure. Lowering the reaction temperature does only marginally impact selectivity, contrary to use of a shorter reaction time (Table 1, entries 8 and 9). Finally, use of a lesser amount of alkoxide base (5% vs. 10%) resulted in comparable results from our best conditions (entry 10 vs. entry 7). The effectiveness of the bench-top stable SNS system in achieving full and selective conversion of MeFDCA into BHMF demonstrates its applicative potential, as previously underlined.43 Under these conditions, no reduction of the aromatic furanic system was observed.44 Thus, using our multicatalytic methodology, the combination of MgCl2 and Gusev's catalyst provides BHMF from 2,5-furandicarboxylic acid with excellent selectivity. As a side note, we checked that HMF can also be reduced into BHMF with full conversion and selectivity using identical reaction conditions than with MeFDCA (as in entry 10 from Table 1).
| Entry | [Ru] | Temperature (°C) | P(H2) (bar) | Time (h) | Solvent | Sel.b (%) |
|---|---|---|---|---|---|---|
| a Unless otherwise noted, the reaction was carried out on MeFDCA (0.81 mmol, 100 eq.) with catalyst (1 eq.) and KOtBu (10 eq.) in solvent (2.5 mL) in a 25 mL Parr reactor. Conversion of MeFDCA, determined by 1H NMR, is quantitative for each entry. b Selectivity towards BHMF, determined by 1H NMR. c No base additive. d 5 mol% KOtBu. | ||||||
| 1c | 1 | 100 | 50 | 2 | PhMe | 42 |
| 2 | 2 | 100 | 50 | 2 | PhMe | 54 |
| 3 | 3 | 100 | 50 | 2 | PhMe | 39 |
| 4 | 4 | 100 | 50 | 2 | PhMe | 83 |
| 5 | 4 | 100 | 80 | 2 | PhMe | 96 |
| 6 | 4 | 100 | 50 | 2 | PhOMe | 80 |
| 7 | 4 | 100 | 50 | 2 | MeTHF | 99 |
| 8 | 4 | 80 | 50 | 2 | MeTHF | 96 |
| 9 | 4 | 80 | 50 | 1 | MeTHF | 88 |
| 10d | 4 | 100 | 50 | 2 | MeTHF | 99 |
Polymer formation step
With an efficient and quantitative synthesis of BHMF in hand, we then explored the step-growth polymerisation of the resulting mixture using a dicarboxylic acid as a comonomer. Industrially, polyesters are mainly produced by reacting a diester (i.e., an activated acid function) with a diol. This reaction in equilibrium requires the energy-intensive use of high temperatures, which favors certain secondary reactions. In particular, etherification reactions can take place, especially in the case of BHMF, where the OH groups are very reactive, and can dehydrate to form BHMF ethers.16 In order to conduct our polymerization under mild conditions, we therefore investigated the use of di-tert-butyl dicarbonate (Boc2O) as a coupling agent for the reaction of BHMF with different dicarboxylic acids (Scheme 2). Indeed, as already observed in the first step of the sequence, the reaction of carboxylic acids in the presence of dialkyl dicarbonates and a Lewis acid catalyst allows the formation of anhydrides.45–47 These intermediates can then react with the in situ released methanol in the case of Moc2O or with an alcohol more nucleophilic than tBuOH that is formed when using Boc2O.48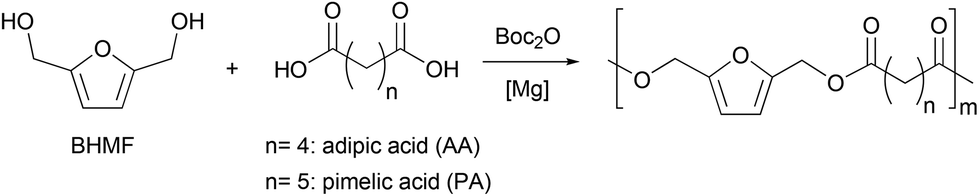 | ||
| Scheme 2 Copolymerisation of BHMF and linear aliphatic diacids. | ||
To evaluate the feasibility of the overall process, we conducted preliminary experiments with the magnesium catalyst able to perform the first step with a [catalyst]/[BHMF] ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :100 (Table 2, entry 1). We first tested the copolymerization of BHMF with adipic acid. The choice of adipic acid is justified by the fact that copolymers formed from this diacid should have a higher degradability than other furan derivatives, as shown by the Bikiaris group for adipate-based copolymers.49 By reacting one equivalent of adipic acid and BHMF with di-tert-butyl dicarbonate and a suitable catalyst, it is indeed possible to obtain quantitatively the corresponding polymer. More precisely, the mild Lewis acid MgCl2 selectively catalyses the formation of the polyester within 115 hours at 30 °C (Table 2, entry 1). As expected, the reactivity of the catalytic system is improved by increasing the reaction temperature to 60 °C which allows to reach a complete conversion in 20 h (Table 2, entry 2). We then thought that the poor solubility of MgCl2 made it difficult to control the polymerization properly. Therefore, we decided to investigate the reactivity of a well-defined magnesium chloride derivative, namely the sterically demanding amido (TMP)MgCl·LiCl (TMP = 2,2,6,6-tetramethylpiperidyl) for the polycondensation step.50 Although several bimetallic combinations are known for organic applications,51–54 magnesiate complexes, such as (TMP)MgCl·LiCl, have been scarcely reported for polyester synthesis.55 However, the presence of the halide salt in (TMP)MgCl·LiCl is argued to increase the solubility and the polarity of the magnesium complex,56 therefore enhancing its reactivity thanks to increased basicity compared to the lithium chloride free compound.57 In our case, (TMP)MgCl·LiCl should therefore readily deprotonate the acid to a carboxylate that would make it more reactive for nucleophilic attack on the dicarbonate. Gratifyingly, polyesterification of adipic acid by BHMF is efficiently carried out in the presence of (TMP)MgCl·LiCl to give a polymer with a Mw value of 16000 (Table 2, entry 3). Again, by increasing the reaction temperature to 80 °C with either MgCl2 or (TMP)MgCl·LiCl, we were able to obtain copolymers more rapidly, albeit with lower masses (Table 2, entries 4 and 5). For the copolymerization of BHMF with pimelic acid, non-sequential addition of Boc2O did not lead to a high molar mass polymer with MgCl2 as a catalyst (Table 2, entry 6). Therefore, we attempted a sequential addition (i.e., of two portions of 100 eq. of Boc2O, the second one being added upon complete diacid conversion) to avoid decomposition of the dicarbonate involved. Under these conditions, MgCl2 was able to convert both substrates within 30 h while allowing for an increase in molar mass (Table 2, entry 7). For (TMP)MgCl·LiCl, the same reactivity was observed (Table 2, entry 8). It should be noted that the copolymer obtained from adipic acid and BHMF has a Tg of −9 °C (Table 2, entry 3), while the copolymer of pimelic acid and BHMF has a glass transition of −23 °C, due to the more flexible nature of the pimelate backbone (Table 2, entry 8). In agreement with literature data, the two copolymers have very close melting temperatures (47 °C vs. 42 °C).16 Furthermore, in order to gather qualitative mechanistic information, in situ IR follow-up of the copolymerization of BHMF and PA was performed in THF at 40 °C in the presence of MgCl2. It showed in a first stage the consumption of the acid with transient formation of pimelic anhydride. Then, the anhydride species reacted with the dialcohol monomer, resulting in polyester formation (Fig. S17†).
:100 (Table 2, entry 1). We first tested the copolymerization of BHMF with adipic acid. The choice of adipic acid is justified by the fact that copolymers formed from this diacid should have a higher degradability than other furan derivatives, as shown by the Bikiaris group for adipate-based copolymers.49 By reacting one equivalent of adipic acid and BHMF with di-tert-butyl dicarbonate and a suitable catalyst, it is indeed possible to obtain quantitatively the corresponding polymer. More precisely, the mild Lewis acid MgCl2 selectively catalyses the formation of the polyester within 115 hours at 30 °C (Table 2, entry 1). As expected, the reactivity of the catalytic system is improved by increasing the reaction temperature to 60 °C which allows to reach a complete conversion in 20 h (Table 2, entry 2). We then thought that the poor solubility of MgCl2 made it difficult to control the polymerization properly. Therefore, we decided to investigate the reactivity of a well-defined magnesium chloride derivative, namely the sterically demanding amido (TMP)MgCl·LiCl (TMP = 2,2,6,6-tetramethylpiperidyl) for the polycondensation step.50 Although several bimetallic combinations are known for organic applications,51–54 magnesiate complexes, such as (TMP)MgCl·LiCl, have been scarcely reported for polyester synthesis.55 However, the presence of the halide salt in (TMP)MgCl·LiCl is argued to increase the solubility and the polarity of the magnesium complex,56 therefore enhancing its reactivity thanks to increased basicity compared to the lithium chloride free compound.57 In our case, (TMP)MgCl·LiCl should therefore readily deprotonate the acid to a carboxylate that would make it more reactive for nucleophilic attack on the dicarbonate. Gratifyingly, polyesterification of adipic acid by BHMF is efficiently carried out in the presence of (TMP)MgCl·LiCl to give a polymer with a Mw value of 16000 (Table 2, entry 3). Again, by increasing the reaction temperature to 80 °C with either MgCl2 or (TMP)MgCl·LiCl, we were able to obtain copolymers more rapidly, albeit with lower masses (Table 2, entries 4 and 5). For the copolymerization of BHMF with pimelic acid, non-sequential addition of Boc2O did not lead to a high molar mass polymer with MgCl2 as a catalyst (Table 2, entry 6). Therefore, we attempted a sequential addition (i.e., of two portions of 100 eq. of Boc2O, the second one being added upon complete diacid conversion) to avoid decomposition of the dicarbonate involved. Under these conditions, MgCl2 was able to convert both substrates within 30 h while allowing for an increase in molar mass (Table 2, entry 7). For (TMP)MgCl·LiCl, the same reactivity was observed (Table 2, entry 8). It should be noted that the copolymer obtained from adipic acid and BHMF has a Tg of −9 °C (Table 2, entry 3), while the copolymer of pimelic acid and BHMF has a glass transition of −23 °C, due to the more flexible nature of the pimelate backbone (Table 2, entry 8). In agreement with literature data, the two copolymers have very close melting temperatures (47 °C vs. 42 °C).16 Furthermore, in order to gather qualitative mechanistic information, in situ IR follow-up of the copolymerization of BHMF and PA was performed in THF at 40 °C in the presence of MgCl2. It showed in a first stage the consumption of the acid with transient formation of pimelic anhydride. Then, the anhydride species reacted with the dialcohol monomer, resulting in polyester formation (Fig. S17†).
| Entry | Catalyst | Temperature (°C) | Time (h) | Conv. BHMFb (%) | M2c | M expn (g mol−1) | M expw (g mol−1) | Đ | T g (°C) | T m (°C) |
|---|---|---|---|---|---|---|---|---|---|---|
| a Conditions: BHMF (100 eq.), M2 (100 eq.), catalyst (1 eq.), Boc2O (220 eq.), MeTHF ([M2] = 1.2 M). b Determined by 1H NMR spectroscopy. c Conversion of M2 is quantitative for each entry. d Determined by size-exclusion chromatography (SEC-RI) at 35 °C. e Determined by differential scanning calorimetry (DSC; second scan). f Reaction was initiated with 100 eq. of Boc2O until complete consumption of PA, then an additional 100 eq. of Boc2O was added. | ||||||||||
| 1 | MgCl2 | 30 | 115 | 94 | AA | 2970 | 11950 |
4.0 | ||
| 2 | MgCl2 | 60 | 20 | 100 | AA | 3890 | 9600 | 2.5 | ||
| 3 | (TMP)MgCl·LiCl | 60 | 24 | 100 | AA | 6410 | 16080 |
2.5 | −9 | 47 |
| 4 | MgCl2 | 80 | 15 | 100 | AA | 1820 | 4520 | 2.5 | ||
| 5 | (TMP)MgCl·LiCl | 80 | 15 | 100 | AA | 6240 | 11940 |
1.9 | ||
| 6 | MgCl2 | 60 | 17 | 100 | PA | 2800 | 6640 | 2.4 | ||
| 7f | MgCl2 | 60 | 30 | 100 | PA | 4150 | 10230 |
2.5 | ||
| 8f | (TMP)MgCl·LiCl | 60 | 22 | 100 | PA | 4340 | 10320 |
2.3 | −23 | 42 |
Finally, we focused on the one-pot process (Scheme 3). We hypothesized that the Lewis acidity of the magnesium complex involved in the first step would be sufficient to catalyze the final polymerization reaction. Therefore, an esterification was performed with 100 eq. of furandicarboxylic acid in MeTHF. After complete conversion of FDCA with MgCl2 as catalyst, the solvent was removed in vacuo, Gusev catalyst and KOtBu were added in MeTHF. After 2 hours at 100 °C under 50 bar H2 pressure, the volatiles were removed under vacuum and the polymerization was conducted at 60 °C in the presence of adipic acid and Boc2O. Polymers with a Mw of 2400 g mol−1 and a dispersity of 1.8 were obtained.
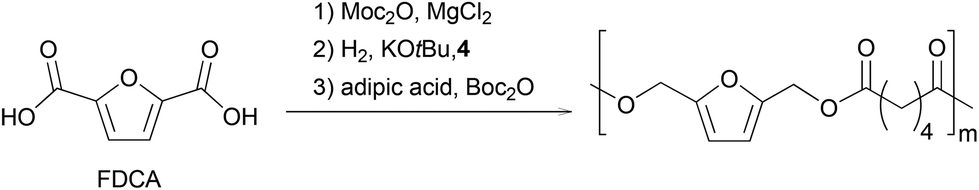 | ||
| Scheme 3 Sequence from FDCA to furan-based polyesters. | ||
Even if the full one-pot sequence did not deliver at this stage a polymer with characteristics up to those obtained via dedicated synthesis, adjustment of the third catalytic system should improve the outcome of the process in terms of copolymer molecular weight.
Conclusions
A new multicatalytic synthetic route to produce 2,5-bis(hydroxymethyl)furan and the corresponding (co)polymers has been developed from stable FDCA. This approach makes it possible to directly obtain bio-based materials in the form of copolymers, without needing to isolate and purify intermediates. Our future efforts are oriented towards further study of the reaction mechanism, as well as development of catalytic systems that exhibit higher reactivities for the whole process.Author contributions
L. G., N. N. and P. N. performed the experiments. L. G., N. N., P. N., R. M. G. and C. M. T. analysed the data. A. M., R. M. G. and C. M. T. wrote the paper. All authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
CNRS and ENSCP are thanked for financial support. Île-de-France Region is gratefully acknowledged for financial support of 500 MHz NMR spectrometer of Chimie ParisTech in the framework of the SESAME equipment project. CMT is grateful to the Institut Universitaire de France.Notes and references
- E. MacArthur, Science, 2017, 358, 843–843 CrossRef PubMed.
- M. A. Hillmyer, Science, 2017, 358, 868–870 CrossRef CAS PubMed.
- M. Poliakoff and P. Licence, Nature, 2007, 450, 810–812 CrossRef CAS PubMed.
- A. J. Ragauskas, C. K. Williams, B. H. Davison, G. Britovsek, J. Cairney, C. A. Eckert, W. J. Frederick Jr., J. P. Hallett, D. J. Leak, C. L. Liotta, J. R. Mielenz, R. Murphy, R. Templer and T. Tschaplinski, Science, 2006, 311, 484–489 CrossRef CAS.
- F. S. Güner, Y. Yağcı and A. T. Erciyes, Prog. Polym. Sci., 2006, 31, 633–670 CrossRef.
- J. F. Jenck, F. Agterberg and M. J. Droescher, Green Chem., 2004, 6, 544 RSC.
- A. Corma, S. Iborra and A. Velty, Chem. Rev., 2007, 107, 2411–2502 CrossRef CAS PubMed.
- U. Biermann, U. T. Bornscheuer, I. Feussner, M. A. R. Meier and J. O. Metzger, Angew. Chem., Int. Ed., 2021 DOI:10.1002/anie.202100778.
- M. J.-L. Tschan, E. Brulé, P. Haquette and C. M. Thomas, Polym. Chem., 2012, 3, 836–851 RSC.
- H. Fouilloux and C. M. Thomas, Macromol. Rapid Commun., 2021, 42, 2000530 CrossRef CAS PubMed.
- A. F. Sousa, C. Vilela, A. C. Fonseca, M. Matos, C. S. R. Freire, G.-J. M. Gruter, J. F. J. Coelho and A. J. D. Silvestre, Polym. Chem., 2015, 6, 5961–5983 RSC.
- A. Gandini, A. J. D. Silvestre, C. P. Neto, A. F. Sousa and M. Gomes, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 295–298 CrossRef CAS.
- G. Guidotti, M. Soccio, M. C. García-Gutiérrez, T. Ezquerra, V. Siracusa, E. Gutiérrez-Fernández, A. Munari and N. Lotti, ACS Sustainable Chem. Eng., 2020, 8, 9558–9568 CrossRef CAS PubMed.
- K. Loos, R. Zhang, I. Pereira, B. Agostinho, H. Hu, D. Maniar, N. Sbirrazzuoli, A. J. D. Silvestre, N. Guigo and A. F. Sousa, Front. Chem., 2020, 8, 585 CrossRef CAS.
- J. G. Rosenboom, D. K. Hohl, P. Fleckenstein, G. Storti and M. Morbidelli, Nat. Commun., 2018, 9, 2701 CrossRef PubMed.
- Y. Jiang, A. J. J. Woortman, G. O. R. Alberda van Ekenstein, D. M. Petrović and K. Loos, Biomacromolecules, 2014, 15, 2482–2493 CrossRef CAS PubMed.
- S. Chen, R. Wojcieszak, F. Dumeignil, E. Marceau and S. Royer, Chem. Rev., 2018, 118, 11023–11117 CrossRef CAS PubMed.
- S. Chen, C. Ciotonea, K. De Oliveira Vigier, F. Jérôme, R. Wojcieszak, F. Dumeignil, E. Marceau and S. Royer, ChemCatChem, 2020, 12, 2050–2059 CrossRef CAS.
- A. Sangregorio, A. Muralidhara, N. Guigo, L. G. Thygesen, G. Marlair, C. Angelici, E. de Jong and N. Sbirrazzuoli, Green Chem., 2020, 22, 2786–2798 RSC.
- P. S. Metkar, R. Ozer and B. Rajagopalan, Processes for preparing 2,5-furandicarboxylic acid and esters thereof, WO-2017019441-A1.
- D. E. Fogg and E. N. dos Santos, Coord. Chem. Rev., 2004, 248, 2365–2379 CrossRef CAS.
- A. Ajamian and J. L. Gleason, Angew. Chem., Int. Ed., 2004, 43, 3754–3760 CrossRef CAS PubMed.
- C. Robert and C. M. Thomas, Chem. Soc. Rev., 2013, 42, 9392–9402 RSC.
- H. Lu, J. Wang, Y. Lin and J. Cheng, J. Am. Chem. Soc., 2009, 131, 13582–13583 CrossRef CAS PubMed.
- M. Eriksson, A. Boyer, L. Sinigoi, M. Johansson, E. Malmström, K. Hult, S. Trey and M. Martinelle, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 5289–5297 CrossRef CAS.
- C. Cheng, K. Qi, E. Khoshdel and K. L. Wooley, J. Am. Chem. Soc., 2006, 128, 6808–6809 CrossRef CAS PubMed.
- G. Chen, D. Huynh, P. L. Felgner and Z. Guan, J. Am. Chem. Soc., 2006, 128, 4298–4302 CrossRef CAS PubMed.
- R. B. Grubbs, C. J. Hawker, J. Dao and J.-M. J. Fréchet, Angew. Chem., Int. Ed. Engl., 1997, 36, 270–272 CrossRef CAS.
- Z. J. A. Komon and G. C. Bazan, Macromol. Rapid Commun., 2001, 22, 467–478 CrossRef CAS.
- J.-C. Wasilke, S. J. Obrey, R. T. Baker and G. C. Bazan, Chem. Rev., 2005, 105, 1001–1020 CrossRef CAS PubMed.
- S. L. Poe, M. Kobašlija and D. T. McQuade, J. Am. Chem. Soc., 2007, 129, 9216–9221 CrossRef CAS.
- J. Zhou, Chem. – Asian J, 2010, 5, 422–434 CrossRef CAS.
- Y. Hayashi, Chem. Sci., 2016, 7, 866–880 RSC.
- C. Oh, E. H. Choi, E. J. Choi, T. Premkumar and C. Song, ACS Sustainable Chem. Eng., 2020, 8, 4400–4406 CrossRef CAS.
- P. Skoczinski, M. K. Espinoza Cangahuala, D. Maniar, R. W. Albach, N. Bittner and K. Loos, ACS Sustainable Chem. Eng., 2020, 8, 1068–1086 CrossRef CAS.
- G. Bartoli, M. Bosco, A. Carlone, R. Dalpozzo, E. Marcantoni, P. Melchiorre and L. Sambri, Synthesis, 2007, 3489–3496 CrossRef CAS.
- F. P. Byrne, S. Jin, G. Paggiola, T. H. M. Petchey, J. H. Clark, T. J. Farmer, A. J. Hunt, C. R. McElroy and J. Sherwood, Sustainable Chem. Processes, 2016, 4, 7 CrossRef.
- J. Pritchard, G. A. Filonenko, R. van Putten, E. J. M. Hensen and E. A. Pidko, Chem. Soc. Rev., 2015, 44, 3808–3833 RSC.
- S. Elangovan, M. Garbe, H. Jiao, A. Spannenberg, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2016, 55, 15364–15368 CrossRef CAS PubMed.
- W. Kuriyama, T. Matsumoto, Y. Ino, O. Ogata and N. Saeki, WO 2011048727, 2011, (Takasago Int. Co.).
- D. Spasyuk, S. Smith and D. G. Gusev, Angew. Chem., Int. Ed., 2013, 52, 2538–2542 CrossRef CAS PubMed.
- D. H. Nguyen, G. Raffa, Y. Morin, S. Desset, F. Capet, V. Nardello-Rataj, F. Dumeignil and R. M. Gauvin, Green Chem., 2017, 19, 5665–5673 RSC.
- J. Schörgenhumer, A. Zimmermann and M. Wase, Org. Process Res. Dev., 2018, 22, 862–870 CrossRef.
- A. Cadu, K. Sekine, J. Mormul, D. M. Ohlmann, T. Schaub and A. S. K. Hashmi, Green Chem., 2018, 20, 3386–3393 RSC.
- C. Robert, F. de Montigny and C. M. Thomas, Nat. Commun., 2011, 2, 586 CrossRef PubMed.
- C. Robert, F. de Montigny and C. M. Thomas, ACS Catal., 2014, 4, 3586–3589 CrossRef CAS.
- L. Fournier, C. Robert, S. Pourchet, A. Gonzalez, L. Williams, J. Prunet and C. M. Thomas, Polym. Chem., 2016, 7, 3700–3704 RSC.
- H. Fouilloux, W. Qiang, C. Robert, V. Placet and C. M. Thomas, Angew. Chem., Int. Ed., 2021, 60, 19374–19382 CrossRef CAS PubMed.
- L. Papadopoulos, A. Magaziotis, M. Nerantzaki, Z. Terzopoulou, G. Z. Papageorgiou and D. N. Bikiaris, Polym. Degrad. Stab., 2018, 156, 32–42 CrossRef CAS.
- A. Krasovskiy, V. Krasovskaya and P. Knochel, Angew. Chem., Int. Ed., 2006, 45, 2958–2961 CrossRef CAS.
- S. Dumouchel, F. Mongin, F. Trécourt and G. Quéguiner, Tetrahedron, 2003, 59, 8629–8640 CrossRef CAS.
- M. Uchiyama, T. Furuyama, M. Kobayashi, Y. Matsumoto and K. Tanaka, J. Am. Chem. Soc., 2006, 128, 8404–8405 CrossRef CAS PubMed.
- T. Furuyama, M. Yonehara, S. Arimoto, M. Kobayashi, Y. Matsumoto and M. Uchiyama, Chem. – Eur. J., 2008, 14, 10348–10356 CrossRef CAS.
- N. T. T. Chau, M. Meyer, S. Komagawa, F. Chevallier, Y. Fort, M. Uchiyama, F. Mongin and P. C. Gros, Chem. – Eur. J., 2010, 16, 12425–12433 CrossRef CAS PubMed.
- J. Char, E. Brulé, P. C. Gros, M.-N. Rager, V. Guérineau and C. M. Thomas, J. Organomet. Chem., 2015, 796, 47–52 CrossRef CAS.
- W. Schlecker, A. Huth, E. Ottow and J. Mulzer, J. Org. Chem., 1995, 60, 8414–8416 CrossRef CAS.
- For a recent review, see R. E. Mulvey and S. D. Robertson, Top. Organomet. Chem., 2014, 47, 129–158 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and characterization data. See DOI: 10.1039/d1gc01889b |
| This journal is © The Royal Society of Chemistry 2021 |