
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Defunctionalisation catalysed by boron Lewis acids
Huaquan
Fang
 and
Martin
Oestreich
*
and
Martin
Oestreich
*
Institut für Chemie, Technische Universität Berlin, Strasse des 17. Juni 115, 10623 Berlin, Germany. E-mail: martin.oestreich@tu-berlin.de
First published on 28th July 2020
Abstract
Selective defunctionalisation of organic molecules to valuable intermediates is a fundamentally important transformation in organic synthesis. Despite the advances made in efficient and selective defunctionalisation using transition-metal catalysis, the cost, toxicity, and non-renewable properties limit its application in industrial manufacturing processes. In this regard, boron Lewis acid catalysis has emerged as a powerful tool for the cleavage of carbon–heteroatom bonds. The ground-breaking finding is that the strong boron Lewis acid B(C6F5)3 can activate Si–H bonds through η1 coordination, and this Lewis adduct is a key intermediate that enables various reduction processes. This system can be tuned by variation of the electronic and structural properties of the borane catalyst, and together with different hydride sources high chemoselectivity can be achieved. This Perspective provides a comprehensive summary of various defunctionalisation reactions such as deoxygenation, decarbonylation, desulfurisation, deamination, and dehalogenation, all of which catalysed by boron Lewis acids.
 Huaquan Fang | Huaquan Fang (born in 1992 in Jiangxi/China) studied chemistry at Beijing Normal University (2009–2013), completing his final year research project with Professor Guohua Hou. He then pursued his doctoral studies with Professor Zheng Huang at the Shanghai Institute of Organic Chemistry, CAS (2013–2018), where he received his Ph.D. degree developing new pincer ruthenium complexes for catalytic activation and conversion of inert chemical bonds. He is currently funded by the Humboldt Foundation to support postdoctoral studies with Professor Martin Oestreich at the Technische Universität Berlin, where he is developing new applications of the B(C6F5)3/hydrosilane combinations. |
 Martin Oestreich | Martin Oestreich (born in 1971 in Pforzheim/Germany) is Professor of Organic Chemistry at the Technische Universität Berlin. He received his diploma degree with Paul Knochel (Marburg, 1996) and his doctoral degree with Dieter Hoppe (Münster, 1999). After a two-year postdoctoral stint with Larry E. Overman (Irvine, 1999–2001), he completed his habilitation with Reinhard Brückner (Freiburg, 2001–2005) and was appointed as Professor of Organic Chemistry at the Westfälische Wilhelms-Universität Münster (2006–2011). He also held visiting positions at Cardiff University in Wales (2005), at The Australian National University in Canberra (2010), and at Kyoto University in Japan (2018). Martin recently edited a monograph entitled Organosilicon Chemistry: Novel Approaches and Reactions together with Tamejiro Hiyama. |
Introduction
The conversion of organic functional groups into hydrogen atoms, namely defunctionalisation, is an important transformation in synthetic chemistry. Although it turns more functionalised raw materials into less functionalised products, the latter are considered to be more valuable than their precursors for diverse aspects such as high-value feedstocks produced by degradation of biomass sources1 and environmentally friendly fuels prepared by desulfurisation and deamination of crude liquid fuels.2 In addition, this process has also found application in environmental remediation, for example, dechlorination of toxic persistent polychlorinated biphenyls (PCBs).3Numerous methods have been developed for the defunctionalisation of a variety of functional groups. Traditional approaches generally utilise stoichiometric amounts of pyrophoric metallic hydrides as reductant. Although widely used in laboratories, this approach is only applicable to certain leaving groups and suffers from the formation of inorganic salts as stoichiometric waste as well as poor selectivity and functional-group tolerance. Catalytic approaches are highly demanded and can serve as convenient, efficient, and economic alternatives for established methods. Three common catalytic strategies for the defunctionalisation of a variety of functional groups, including radical catalysis, transition-metal catalysis, and Lewis acid/frustrated Lewis pair catalysis, have been developed (Scheme 1). Reductive radical chain defunctionalisation of I with tin hydrides as the hydrogen source in the presence of a radical starter had been developed over the last 60 years.4 However, this method suffers from the use of toxic organotin compounds and the difficulty to completely remove the corresponding tin by-products. Several improvements including the use of a catalytic amount of tin hydrides and the use of other hydrogen sources have also been developed. Transition metal-catalysed defunctionalisation of I with a hydrogen source has provided an efficient and selective protocol for the cleavage of carbon–heteroatom bonds.5 However, the use of rare, expensive, and toxic transition metal catalysts limit their applications in industrial manufacturing processes. Recently, Lewis acid/frustrated Lewis pair-catalysed defunctionalisation of I with various hydride sources has emerged as a promising tool to this end.6
 | ||
| Scheme 1 Ways of catalytic defunctionalisation. FG = functional group. | ||
Among various Lewis acids investigated, boron Lewis acids are particularly attractive due to their high Lewis acid strength, low cost, and benign environmental impact and have been developed rapidly in the last two decades. What's more, the Lewis acidity and reactivity of boron Lewis acid can be easily tuned by changing or modifying substituents attached to the boron atom.7 A variety of defunctionalisation reactions catalysed by boron Lewis acids, such as deoxygenation, decarbonylation, desulfurisation, deamination, and dehalogenation, have been reported (Scheme 2). This review provides a comprehensive summary of defunctionalisation catalysed by boron Lewis acids. The condensation of alkoxysilanes and hydrosilanes to synthesize structurally complex functional silicones and an alkane as a by-product catalysed by a B(C6F5)3 catalyst, known as the Piers–Rubinsztajn reaction,8 is not covered.
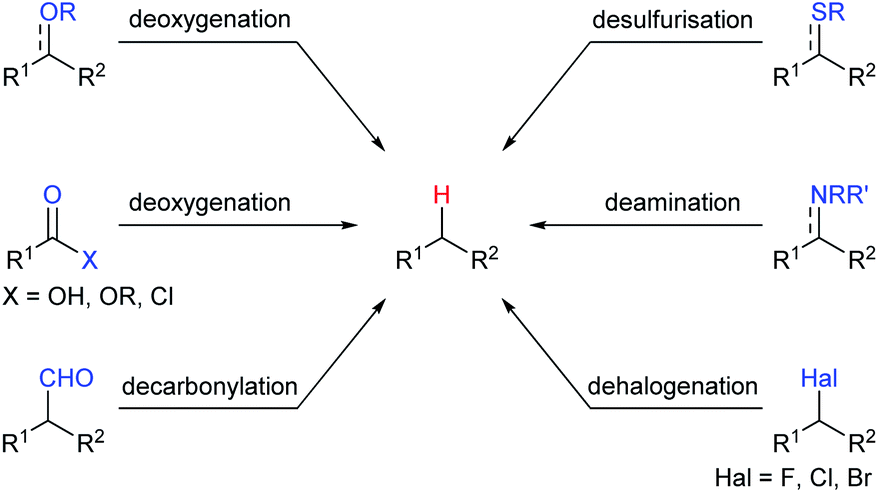 | ||
| Scheme 2 Scope of boron Lewis acids-catalysed defunctionalisation. | ||
Boron Lewis acids-catalysed deoxygenation
Deoxygenation of organic molecules to hydrocarbons is a step frequently encountered in organic synthesis. In fact, boron Lewis acid-mediated deoxygenation of alcohols and their derivatives with a hydrosilane as the reductant is known since the 1970s.9 However, the application of boron Lewis acid in catalysis remained undeveloped until 1999. Inspired by the pioneering work of Piers et al. on B(C6F5)3-catalysed hydrosilylation of carbonyl functions,10 Gevorgyan, Yamamoto, and co-workers found that alcohols and ethers were effectively reduced by excess Et3SiH in the presence of catalytic amounts of B(C6F5)3 to give the corresponding hydrocarbons at room temperature (Scheme 3).11 This catalytic system is efficient for the deoxygenation of primary alcohols, however, the deoxygenation of secondary and tertiary alcohols, except for those alcohols possessing strong carbocation-stabilising groups, was failed. Hence, the relative reactivity order of different types of alcohols was found to be 1° ≫ 2° > 3°. | ||
| Scheme 3 Deoxygenation of alcohols and ethers catalysed by B(C6F5)3 with Et3SiH as the reductant. | ||
A proposed mechanism for B(C6F5)3-catalysed deoxygenation of alcohols and ethers is depicted in Scheme 4.11b,12 The association of B(C6F5)3 with hydrosilane generates an η1-adduct IV, either represented as Si–H⋯B(C6F5)3 or Si⋯H–B(C6F5)3. The adduct is subsequently attacked by the substrate to form ion pairs V or VI; hydride transfer from the borohydride to the electrophilic carbon atom of the oxonium ion produces the silyl ether and hydrocarbon, respectively, with regeneration of III. It is worth noting that Lewis adduct IV is usually spectroscopically undetectable when mixing B(C6F5)3 and a hydrosilane. However, an isolable borane–hydrosilane adduct formed by mixing 1,2,3-tris(pentafluorophenyl)-4,5,6,7-tetrafluoro-1-boraindene and Et3SiH was reported by Piers, Tuononen, and co-workers.13
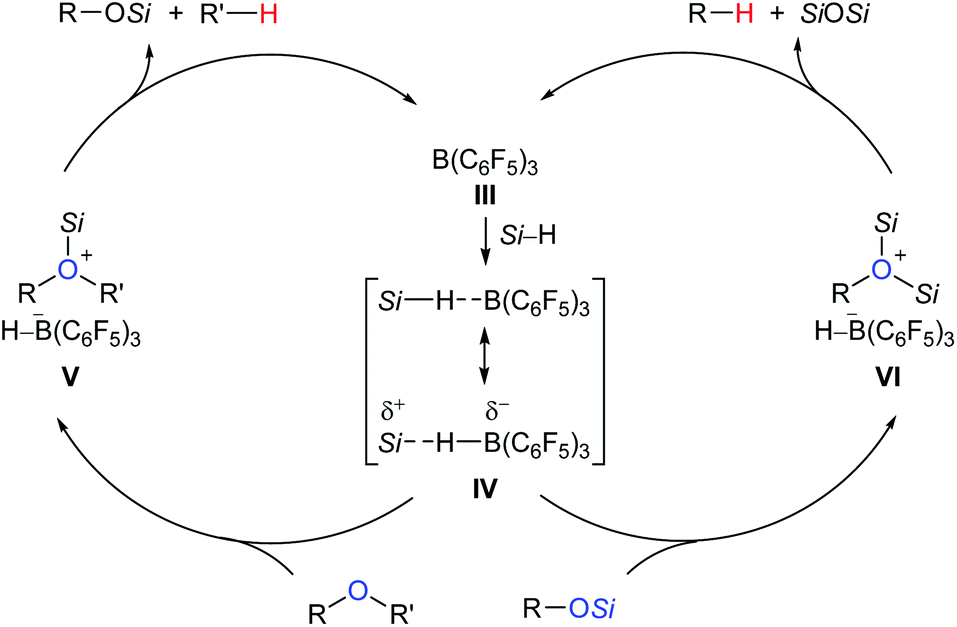 | ||
| Scheme 4 Proposed mechanism for B(C6F5)3-catalysed deoxygenation of alcohols and ethers. | ||
By using that catalytic system, Gevorgyan, Yamamoto, and co-workers described the exhaustive deoxygenation of a variety of carbonyl functions such as carboxylic acids, aldehydes, acyl chlorides, and esters to give the corresponding hydrocarbons at room temperature (Scheme 5).14
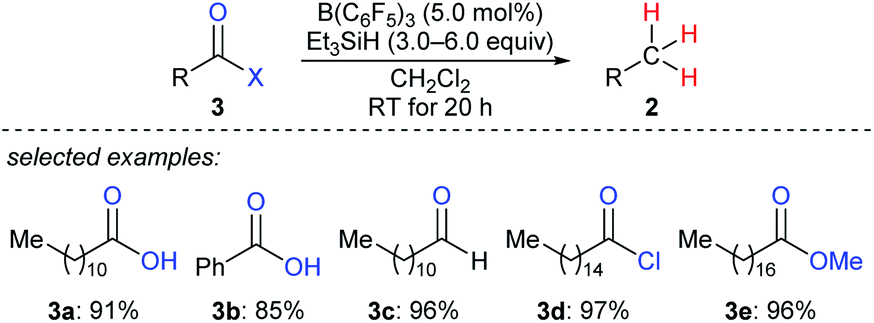 | ||
| Scheme 5 Deoxygenation of carbonyl and carboxy compounds catalysed by B(C6F5)3 with Et3SiH as the reductant. | ||
To produce long-chain hydrocarbons (carbon number > 10), Fu and co-workers developed a B(C6F5)3-catalysed deoxygenation of biomass-derived fatty acids and derivatives thereof with the silicone industry byproduct polymethylhydrosiloxane (PMHS) as the reductant (Scheme 6).15 The successful conversion of commercially available plant oils to hydrocarbons demonstrated the value of this method and also provided a useful strategy for the production of liquid hydrocarbon fuels by upgrading of biodiesel. Later, B(C6F5)3-catalysed deoxygenation of triglycerides to give a mixture of alkanes and alkenes with hydrosiloxanes as reductants was further explored by Gale and Brook (not shown).16
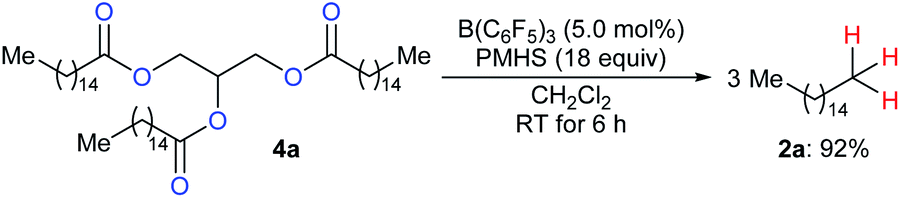 | ||
| Scheme 6 Deoxygenation of biomass-derived fatty acid esters catalysed by B(C6F5)3 with PMHS as the reductant. | ||
A mild and rapid B(C6F5)3-catalysed deoxygenation of a variety of ketones 5a–c to afford the corresponding hydrocarbons with PMHS as the reductant was disclosed by Chandrasekhar and co-workers (Scheme 7).17 This system exhibits good efficiency and selectivity and is compatible with various functional groups such as chloride, silyl ether, ester, and alkenyl groups. Later, these authors employed this catalytic system for the deoxygenation of Baylis–Hillman adducts to form (Z)- or (E)-trisubstituted alkenes (not shown);18 the process involves the elimination of the hydroxy group followed by double bond migration. In addition, Cantat and co-workers developed B(C6F5)3-catalysed deoxygenation of oxalic acid with tetramethyldisiloxane (TMDS) or PMHS as reductants (not shown).19 Nimmagadda and McRae found the combined use of B(C6F5)3 with the less sterically hindered Et2SiH2 or nBuSiH3 proved to be a more reactive catalytic system for the deoxygenation of polycarboxylic acids, aldehydes, ketones, and alcohols (not shown).20 The B(C6F5)3/Ph2SiH2 catalytic system demonstrated by Tan and Zhang was shown to be efficient for the reduction of an alcohol, a ketone, an α,β-unsaturated carboxylic acid, and an enol ether to their corresponding hydrocarbons (not shown).21
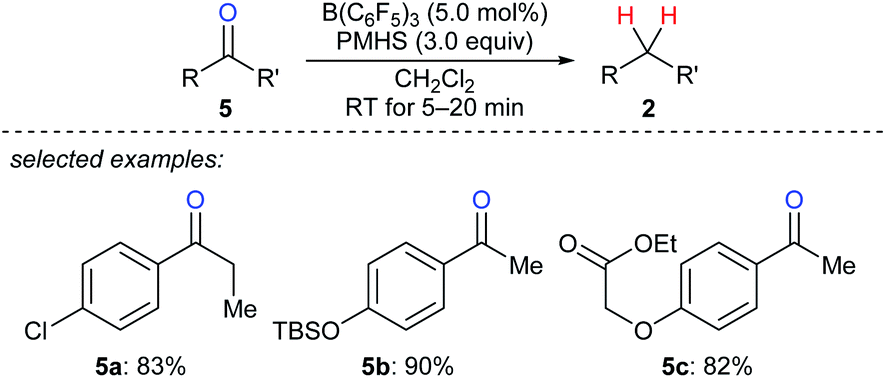 | ||
| Scheme 7 Deoxygenation of ketones catalysed by B(C6F5)3 with PMHS as the reductant. | ||
The use of H2 as a reductant in boron Lewis acid-catalysed deoxygenation is challenging yet highly attractive, and only water is formed as a by-product. Repo and co-workers found that Lewis pair of B(C6F5)3 and aromatic carbonyl compounds can heterolytically activate H2 at elevated temperature (110 °C).22 By this, the stoichiometric reduction of benzophenone with H2 as the reductant became feasible. However, the catalytic deoxygenation of ketones with H2 by B(C6F5)3 still is a difficult task due to the hydrolysis of B(C6F5)3 with the by-product H2O. By employing molecular sieves as a heterogeneous Lewis base and a desiccant to adsorb water, Mahdi and Stephan developed a B(C6F5)3-catalysed deoxygenation of diaryl ketones 5d–g with H2 as the reductant at 70 °C (Scheme 8).23
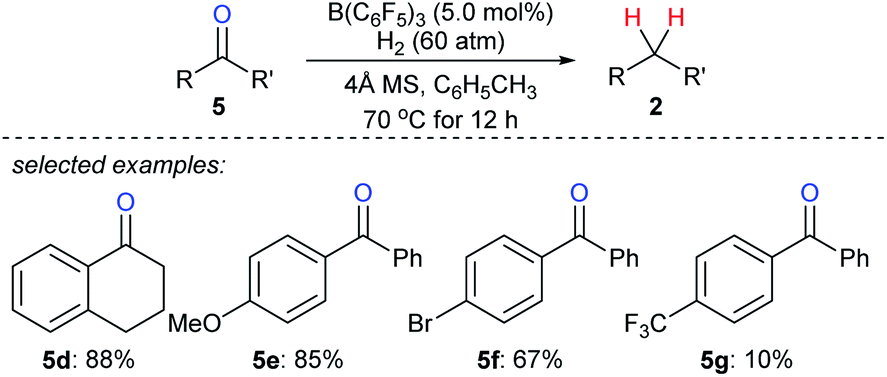 | ||
| Scheme 8 Deoxygenation of ketones catalysed by B(C6F5)3 with H2 as the reductant. | ||
The degradation of readily available carbohydrates to value-added feedstocks and fuels is an attractive yet challenging endeavour which requires the activation of several nonactivated C–O bonds. In 2014, Gagné and co-workers reported the B(C6F5)3-catalysed deoxygenation of carbohydrates 6 to afford mixtures of hexanes and hexenes with Et2SiH2 as the reductant (Scheme 9).24 The degree of deoxygenation was influenced by the choice of hydrosilane. Secondary hydrosilanes as reductants led to exhaustive reduction while tertiary hydrosilanes generated partially deoxygenated products. Later, B(C6F5)3-catalysed deoxygenation of lignin25 and various polymeric materials based on polyethers, polyesters, polycarbonates,26 and poly(methyl acrylate)27 with hydrosilanes as reductants were demonstrated by the groups of Cantat, Chang, and Seo (not shown).
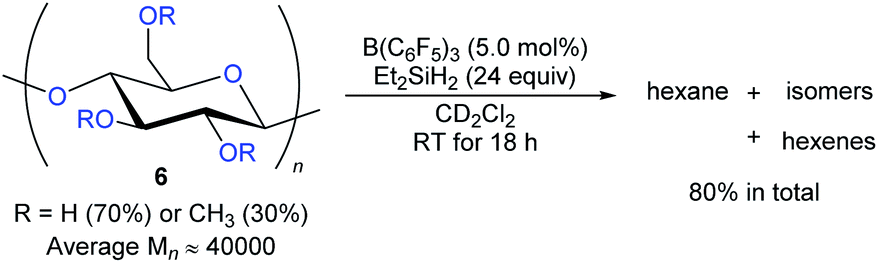 | ||
| Scheme 9 Deoxygenation of carbohydrates catalysed by B(C6F5)3 with Et2SiH2 as the reductant. | ||
The chemoselective partial deoxygenation of a variety of biologically sourced polyols to produce various oxygen-functionalised chiral synthons using a combination of B(C6F5)3 and tertiary hydrosilanes was described by Gagné and co-workers (Scheme 10).28 For example, the deoxygenation of galactitol 7 with Me2EtSiH (7.0 equiv.) in the presence of B(C6F5)3 (10 mol%) generated a C2-reduced triol 9 with inversion at C5. The deoxygenation of 7 with 2.5 equivalents of Me2EtSiH gave 1,6-deoxygenated tetraol 8, which underwent intramolecular nucleophilic attack from the C2–OSi group to the activated C5–OSi2+ in VII to generate the cyclic oxonium ion intermediate VIII. Subsequent hydride transfer from borohydride to the C2 position of VIII formed the observed triol 9 with inversion at C5. The formation of cyclic intermediates caused by neighbouring group participation is crucial for achieving high site- and chemoselectivity. Later, these authors reported B(C6F5)3-catalysed chemoselective deoxygenation of unsaturated polyols to produce highly enriched (Z)-triols and partial deoxygenation of disaccharides to yield 1,6-deoxygenated tetraols and 1-deoxyglucose with a tertiary hydrosilane as the reductant (not shown).29 Moreover, site- and chemoselective deoxygenations of carbohydrates and its derivatives by a combination of B(C6F5)3/catecholborane (HBcat) and B(3,5-(CF3)2C6H3)3/tertiary hydrosilane combinations were also developed (not shown).30 More recently, the Gagné group found the polarity of solvent to exert a profound influence on reactivity and regioselectivity of the deoxygenation of sugars using the B(C6F5)3/hydrosilane catalytic system (not shown).31 Mechanistic investigations indicated low-dielectric solvents can shorten inter-ion bond lengths of the key ion-pair intermediates due to electrostatic compressive forces.
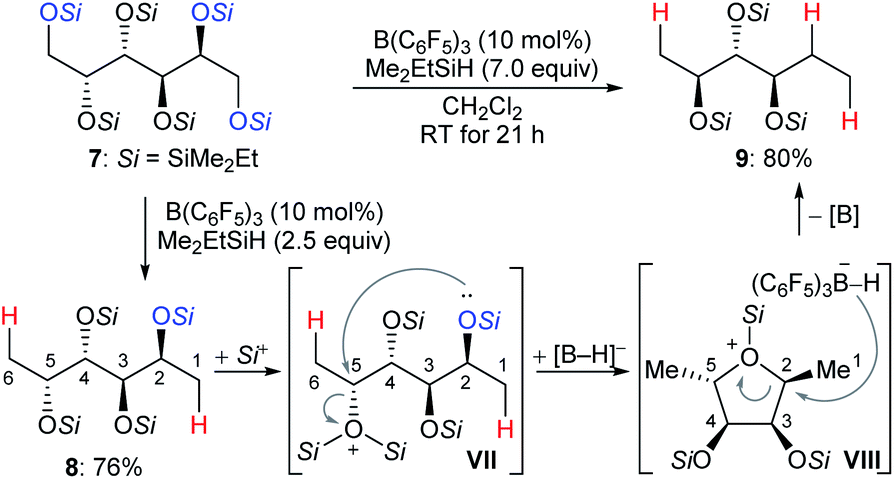 | ||
| Scheme 10 Selective deoxygenation of biologically sourced polyols catalysed by B(C6F5)3 with Me2EtSiH as the reductant. | ||
By tuning the electronic properties of fluoroaryl borane catalysts and utilising different reductants, chemo- and site-selective modifications of various complex natural products to yield divergent products were achieved by Gagné and co-workers (Scheme 11).32 For example, the reaction of gibberellic acid (10a) with excess Et3SiH in the presence of a catalytic amount of B(C6F5)3 generated the known diacid 11a in 93% yield. This process involves a sequence of dehydrosilylation and ring-opening of the lactone group accompanied by an allylic transposition. In addition, 11a can also be obtained in excellent yields by the deoxygenation of pre-silylated gibberellic acid 10b with a combination of B(C6F5)3/HBcat or B(2,4,6-F3C6H2)3/Me2EtSiH (not shown). By employing a combination of B(C6F5)3/HBcat, full isomerisation of 10a to 11b was observed after deprotection. The deoxygenation of 10b with excess Et3SiH catalysed by B(3,5-(CF3)2C6H3)3 provided a conjugated diene derivative of 11c in 51% yield after deprotection.
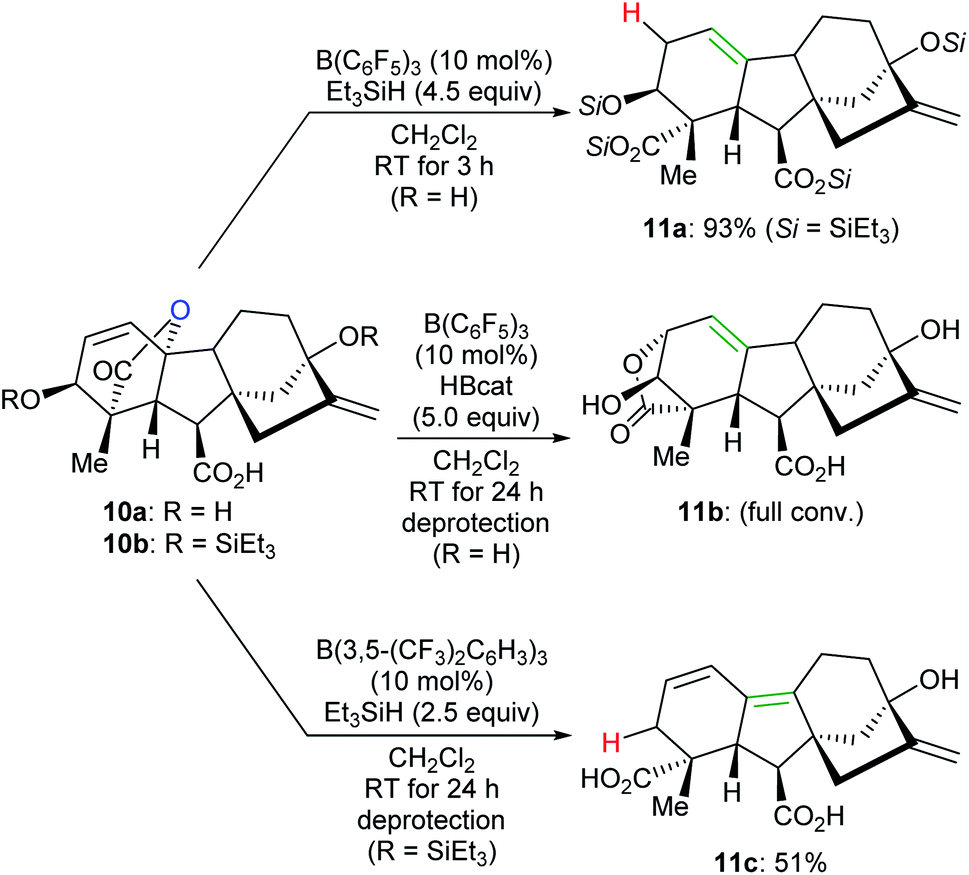 | ||
| Scheme 11 Chemoselective deoxygenation of gibberellic acids catalysed by B(C6F5)3 or B(3,5-(CF3)2C6H3)3 with Et3SiH or HBcat as reductants. | ||
The beautiful work of Gagné prompted chemists to develop new approaches to selective deoxygenation. In 2015, Drosos and Morandi introduced a highly selective B(C6F5)3-catalysed monodeoxygenation of terminal 1,2- and 1,3-diols 12a–d to give 2-alkanols by using a combination of Ph2SiH2 and Et3SiH (Scheme 12).33 The overall reaction is a sequence of protection to form cyclic siloxane intermediates and selective reduction at their primary position to afford 2-alkanols. Computational studies reveals that the formation of cyclic siloxane intermediates, which facilitates the deoxygenation by minimizing the steric repulsions between cyclic siloxane and borane–hydrosilane complex and hinders the further deoxygenation due to the bulky disiloxane moiety, plays a significant role.34
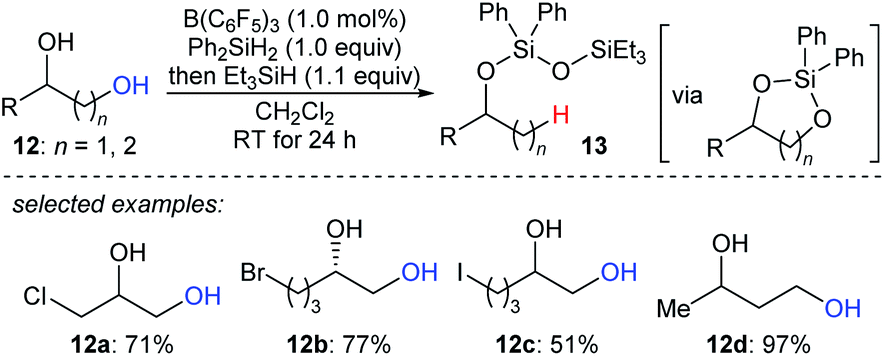 | ||
| Scheme 12 Selective deoxygenation of terminal 1,2- and 1,3-diols catalysed by B(C6F5)3 with Et3SiH as the reductant. | ||
A two-step strategy for the B(C6F5)3-catalysed chemoselective deoxygenation of 1,n-diols and the hydroxymethyl group of an orthogonally protected carbohydrate with Et3SiH as the reductant was disclosed by Oestreich and co-workers (Scheme 13).35 The cleavage of C–O bonds of primary tosylates 14a–c proceeds preferentially over that of bromide, silyl ethers, and aryl ethers at room temperature. Later, Song and co-workers used (HMe2SiCH2)2 as a new reductant for the chemoselective deoxygenation of ether-substituted alcohols and carbonyl compounds (not shown), and the authors proposed that (HMe2SiCH2)2 promotes an intramolecular Si–O activation pathway.36
 | ||
| Scheme 13 Selective deoxygenation of primary alkyl tosylates catalysed by B(C6F5)3 with Et3SiH as the reductant. | ||
Selective deoxygenation of enol ethers 15a–e with Et3SiH as the reductant catalysed by B(C6F5)3 was achieved by Chulsky and Dobrovetsky (Scheme 14).37 This process involves the selective “indirect” cleavage of alkenyl–oxygen bonds in the presence of alkyl–oxygen bonds; the mechanism is believed to be a sequence of hydrosilylation followed by silicon-assisted β-elimination.
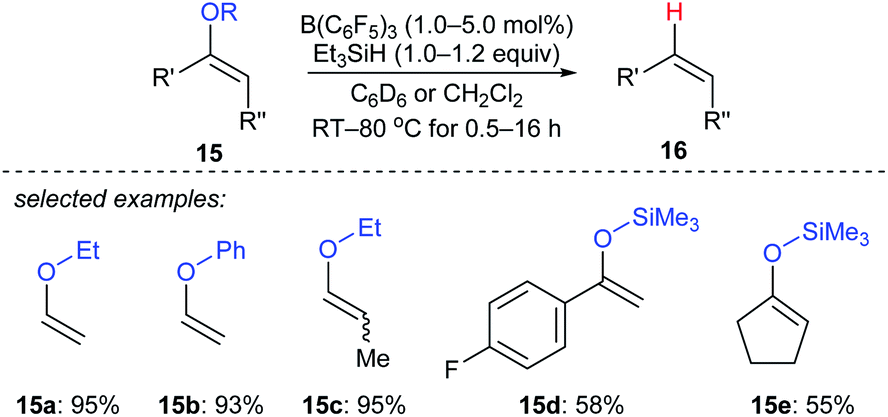 | ||
| Scheme 14 Selective deoxygenation of enol ethers catalysed by B(C6F5)3 with Et3SiH as the reductant. | ||
The reduction of amides to the corresponding amines, which is another synthetically useful transformation in organic synthesis,38 with hydrosilanes as reductants catalysed by a boron Lewis acid was first described by Tan and Zhang (Scheme 15).21 Various N-phenylamides 17a–c were successfully reduced to the corresponding amines at 75 °C. However, the reduction of the parent benzamide using this catalytic system was unsuccessful, even at 120 °C. Later, McGrath and co-workers found that various functional groups such as ether, ketone, and ester groups were tolerated in the B(C6F5)3-catalysed reduction of acetanilides to secondary amines with Et3SiH as the reductant (not shown).39 The reactivity of hydrosilanes with different steric demand in this reaction was also examined.
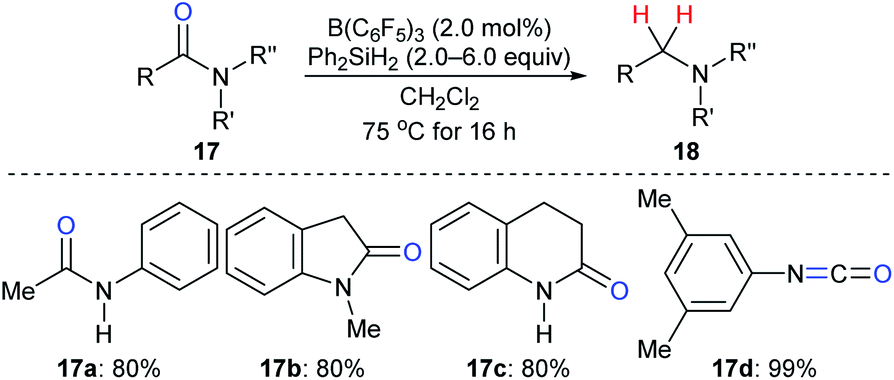 | ||
| Scheme 15 Deoxygenation of amides and isocyanate catalysed by B(C6F5)3 with Ph2SiH2 as the reductant. | ||
By utilising TMDS or PMHS as reductants, reduction of various secondary and tertiary amides 17e–h to the corresponding amines catalysed by B(C6F5)3 were independently described by the groups of Cantat40 and Adronov41 (Scheme 16). The reduction of benzamide with TMDS (2.0 equiv.) in the presence of B(C6F5)3 (10 mol%) gave mixtures of dibenzylamine, N-benzylbenzamide, and (E)-N-benzyl-1-phenylmethanimine at 100 °C after 18 h. To prevent the formation of benzonitrile, which was formed by slow dehydrogenative silylation of the N–H bonds of benzamide and subsequent elimination of a siloxane, the protection of benzamide using Me3SiCl prior to the reduction was performed. Using this strategy, primary amides were successfully converted into the corresponding primary amines in excellent yields.
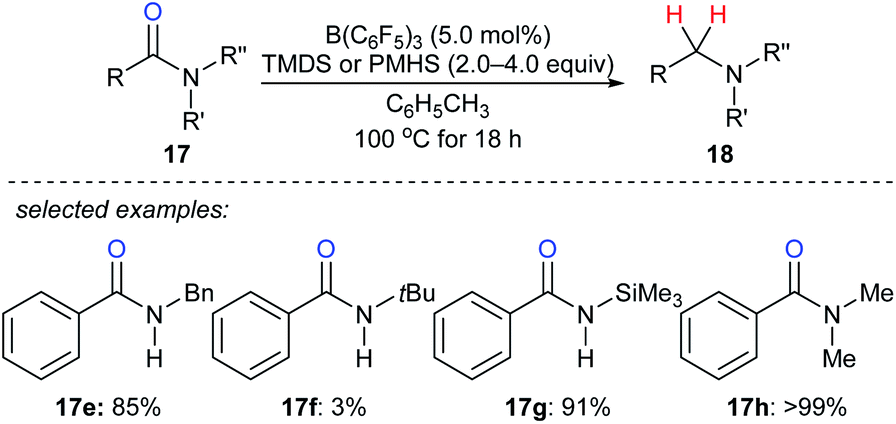 | ||
| Scheme 16 Deoxygenation of amides catalysed by B(C6F5)3 with TMDS or PMHS as reductants. | ||
By merging Tf2O/2-F-pyridine activation and B(C6F5)3/TMDS reduction, Huang and co-workers found that various N-alkyl secondary amides, which had been difficult to reduce previously, were efficiently reduced to secondary amines at room temperature (not shown).42 A variety of functional groups such as methoxy, trifluoromethyl, bromo, nitro, ester, cyano, alkenyl, alkynyl, cyclopropyl, and silyl ether was compatible.
In 2018, Sohma, Kanai, and co-workers described a B(C6F5)3-catalysed chemo- and regioselective reduction of various hydroxy amides 17i–k with MePhSiH2 as the reductant to synthesize 1,n-aminoalcohols under mild conditions with high functional group tolerance (Scheme 17).43 This process undergoes a sequence of B(C6F5)3-catalysed dehydrogenative silylation of the hydroxy group and selective deoxygenation through intramolecular Lewis acid/base type interaction between the silicon atom and oxygen atom of the amide carbonyl group. The application of this catalytic system to chemo- and site-selective reduction of a specific amide bond in cyclosporin A, which contains four secondary and seven tertiary amide bonds, demonstrated the power of this catalytic system when applied to complex molecules.
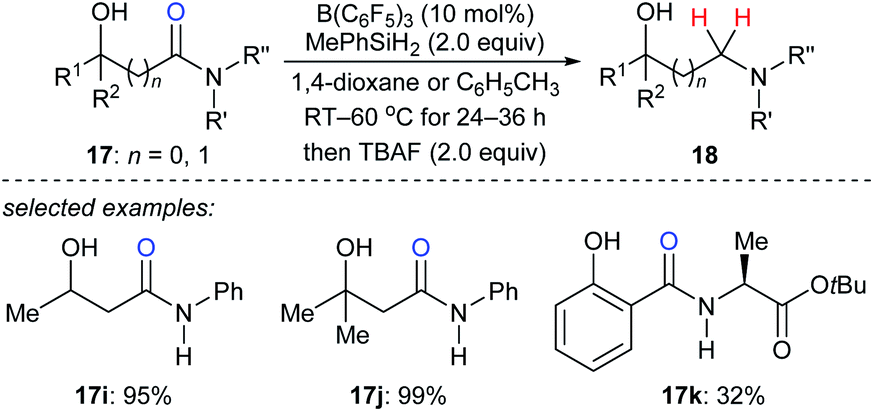 | ||
| Scheme 17 Hydroxy-directed deoxygenation of amides catalysed by B(C6F5)3 with MePhSiH2 as the reductant. | ||
As described above, the strong boron Lewis acid B(C6F5)3 proved to be a potent catalyst in the reduction of amides. In 2013, Beller and co-workers introduced benzothiophene-functionalised boronic acids 19 for the reduction of tertiary, secondary, and primary amides with PhSiH3 as the reductant. At 110–130 °C, the corresponding amines were obtained and the functional-group tolerance was good (Scheme 18).44 Later, Blanchet and co-workers reported bis(2-chlorophenyl)borinic acid as an efficient catalyst for the reduction of tertiary amides with PhSiH3 under mild reaction conditions (not shown).45 Mechanistic investigations indicated that this process involves the formation of borane and an amine–borane complex.
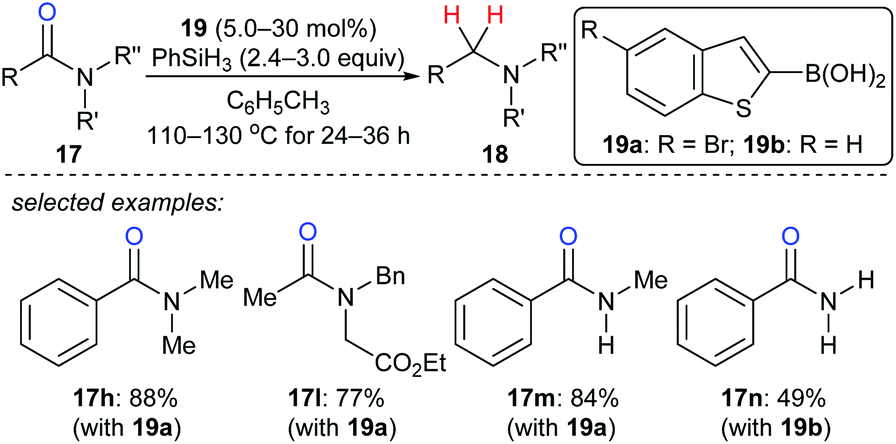 | ||
| Scheme 18 Deoxygenation of amides catalysed by boronic acids 19 with PhSiH3 as the reductant. | ||
In 2016, Okuda and co-workers described the reduction of tertiary amides 17o–q with MePhSiH2 as the reductant catalysed by moderately Lewis acidic BPh3 to give amines under mild conditions with high chemoselectivity in the presence of halide, nitro, ether, ketone, ester, imine, and isocyanate functions (Scheme 19).46 The authors proposed a carbonyl activation pathway for this catalytic system instead of the known Piers–Oestreich-type hydrosilane activation mechanism.
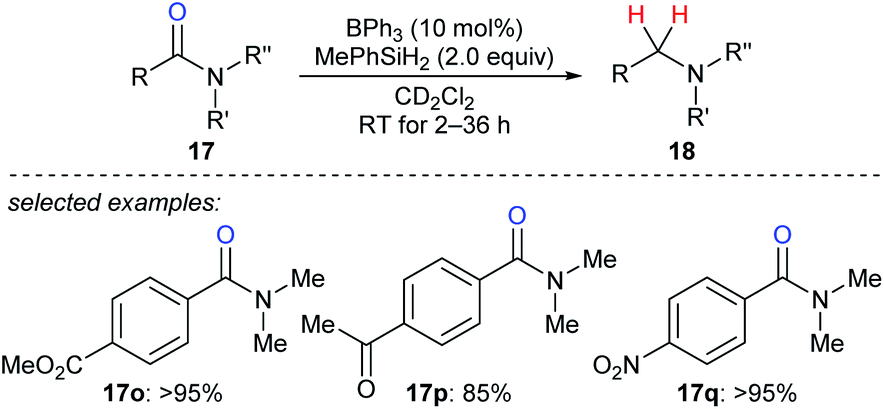 | ||
| Scheme 19 Deoxygenation of amides catalysed by BPh3 with MePhSiH2 as the reductant. | ||
Based on the successful application of three boron catalysts with modified steric and electronic profiles for the selective modifications of natamycin, Gagné and co-workers developed the mixed alkyl(fluoroaryl)borane catalyst B(C6F5)2(hex-3-yl) (20), which is generated in situ by the hydroboration of hex-3-ene with Piers' borane HB(C6F5)2, for the chemoselective reduction of mycosamine acetamides (Scheme 20).32a The reaction of acetamide derivatives of natamycin 17r and 17s with Et2SiH2 (4.5 equiv.) in the presence of 20 (10 mol%) led to the selective reduction of the N-acetamide to the N-ethyl mycosamine derivatives of natamycin 18r and 18s in useful yields without the competing reduction of other sites. Later, these authors developed a heteroleptic borane catalyst B(C6F5)2(CH2CH2CH2Bpin) for the mild reduction of tertiary alkyl amides, N-acetyl proline dipeptides, and even cyclosporine A with Me2EtSiH or Et2SiH2 as reductants with good functional-group tolerance (not shown).47
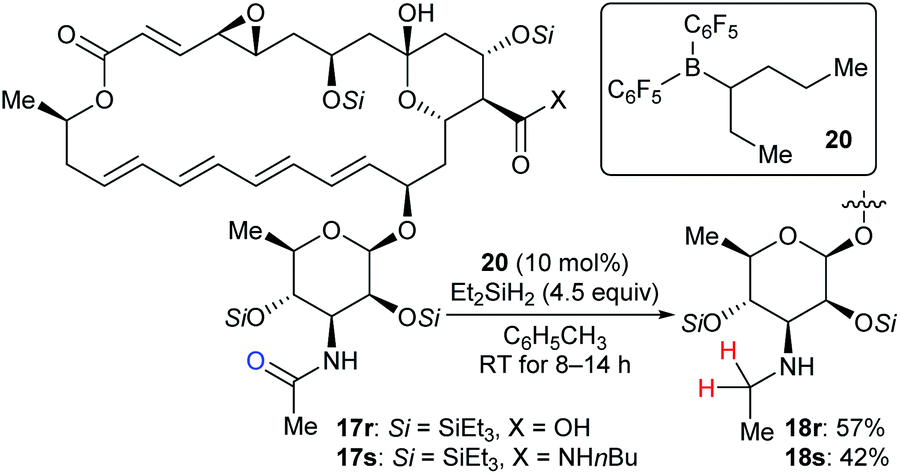 | ||
| Scheme 20 Selective deoxygenation of mycosamine acetamides catalysed by B(C6F5)2(hex-3-yl) (20) with Et2SiH2 as the reductant. | ||
The reduction of tertiary amides with H2 as the reductant with the aid of oxalyl chloride as an activating agent catalysed by B(2,6-F2C6H3)3 was disclosed by Paradies, Grimme, and co-workers (Scheme 21).48 The process involves the in situ formation of a chloroiminium chloride intermediate by the reaction of the amide with oxalyl chloride and exhibits high functional-group tolerance towards ester, ether, nitro, cyano, or thiophenyl groups. The corresponding amines were isolated as their HCl salts. The reduction of acetylamide 17v resulted in a low yield, and the authors attributed this to the polymerisation of the corresponding chloroiminium chloride intermediate. Mechanistic investigations indicated the key role of chloride as an active Lewis base in borane-mediated H2 activation.
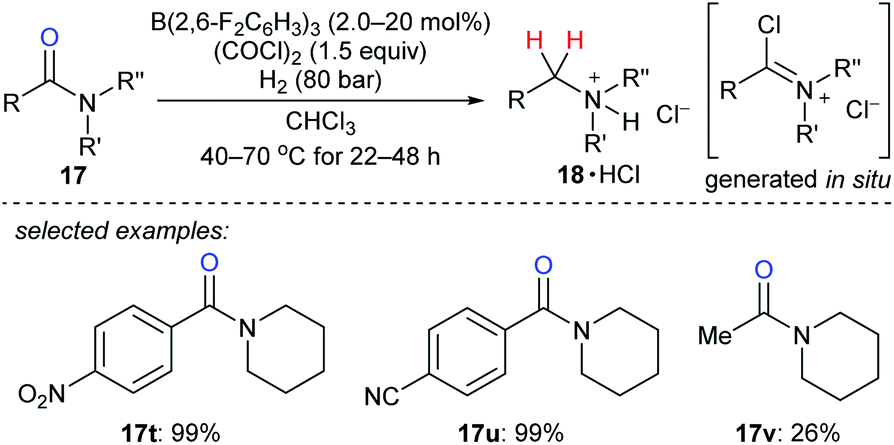 | ||
| Scheme 21 Deoxygenation of tertiary amides catalysed by B(2,6-F2C6H3)3 with H2 as the reductant. | ||
Ammonia borane is an ideal H2 storage material49 owing to its high storage capacity (19.6 weight% H), low molecular weight (30.87 g mol−1), good stability against air and moisture, easy availability, and simple handling. It has been intensively investigated as a reductant for the reduction of unsaturated C–C and carbon–heteroatom bonds.50 Xu, Fan, Xiao, and co-workers reported the reduction of various amides with ammonia borane as the reductant in the presence of catalytic amounts of B(C6F5)3 and BF3·OEt2 to provide a wide range of structurally diverse amines in good to excellent yields under mild reaction conditions with high functional-group tolerance (Scheme 22).51 The role of the BF3·OEt2 co-catalyst is to activate the carbonyl group of amide by Lewis adduct formation.
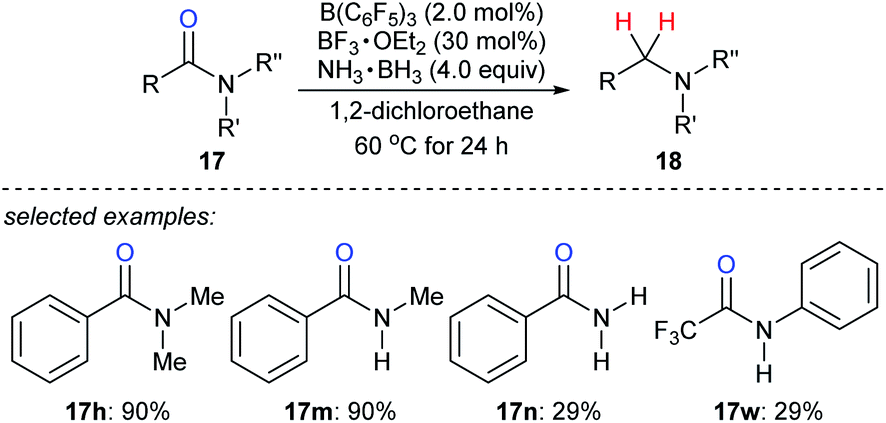 | ||
| Scheme 22 Deoxygenation of amides catalysed by B(C6F5)3 with NH3·BH3 as the reductant. | ||
Some deoxygenation reactions accompanied by ring closure and rearrangement processes have been explored. This further highlights the synthetic applicability of defunctionalisation with B(C6F5)3/hydrosilane combinations. In 2016, Gagné and co-workers disclosed a B(C6F5)3-catalysed reductive carbocyclisation of unsaturated carbohydrates with hydrosilanes to give cyclopropanes and cyclopentanes with high efficiency and excellent diastereoselectivity (Scheme 23).52 The reaction of gluco-derived 21a with Ph3SiH (1.1 equiv.) in the presence of B(C6F5)3 (10 mol%) generated a single diastereomer of cyclopropane 22a in 95% yield (Scheme 23, top). This process involves B(C6F5)3-catalysed intramolecular nucleophilic substitution of the activated primary C7 position by C4–OSi of 21a to form 23a, which captures a “silylium ion” by the more basic cyclic oxygen atom to generate silyloxonium IX. Borohydride reduction of alkene moiety in IX induces a cyclisation event to yield cyclopropane 22a after deprotection. Conversely, the allylic polyol derivative 21b provided a single cyclopentane diastereomer 22b in 82% yield under similar conditions (Scheme 23, bottom). After the formation of silyloxonium X, a 1,2-migration of the styryl group with inversion at C4 produces an silyloxycarbenium/silylcarboxonium ion intermediate XI, which is reduced by borohydride to the observed intermediate XII. Subsequent silylation of the primary silyl ether group of XII is followed by cyclisation through nucleophilic attack of the alkene to the activated C7 carbon atom. This generates a benzylic cation which is further reduced by borohydride to give cyclopentane 22b after deprotection.
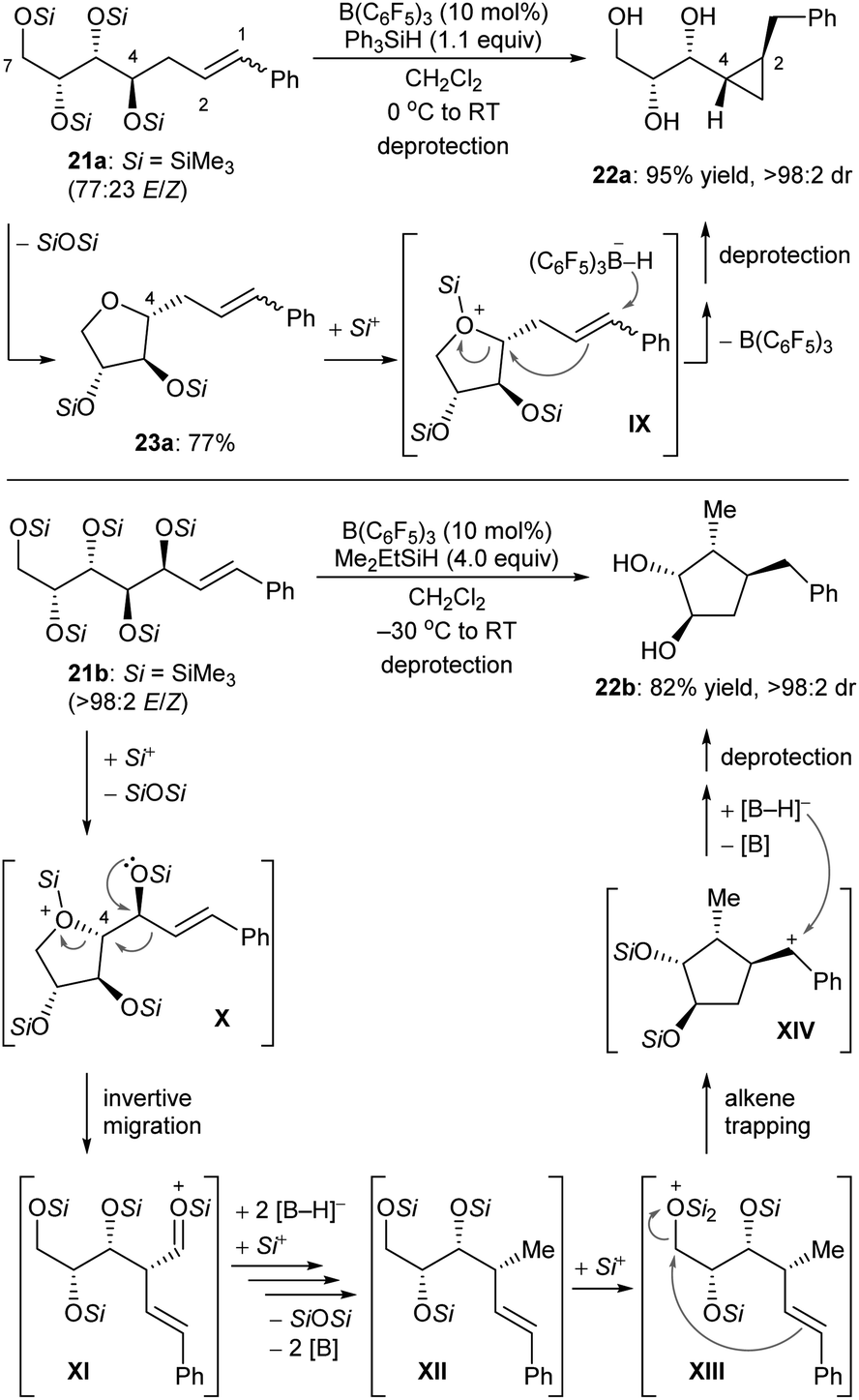 | ||
| Scheme 23 Carbocyclisation of unsaturated carbohydrates catalysed by B(C6F5)3 with Ph3SiH or Me2EtSiH as the reductant. | ||
Chang and co-workers reported the stereocontrolled conversion of furans into Z-configured homoallylic silanes 25 and anti-substituted cyclopropyl silanes 26 through selective ring-opening and subsequent ring-closing processes (Scheme 24).53 The B(C6F5)3-catalysed double hydrosilylation of furans with Me2PhSiH (2.05 equiv.) afforded 25 in excellent yields and with high stereoselectivity (Scheme 24, top). The subsequent cyclopropanation can be simply achieved by the addition of further equivalents of the hydrosilane to furnish 26 with exclusive trans-selectivity. Isolation of ring-opened intermediates is not required. The authors proposed a cascade of B(C6F5)3-catalysed ring-opening (by two-fold hydrosilylation) and ring-closing reactions (by intramolecular cyclopropanation) (Scheme 24, bottom). The selective borohydride attack at the α-carbon of intermediate XVII and at the C4 of silyloxonium species XVIII leads to trans-(2-alkyl)cyclopropyl silanes exclusively. Later, these authors further reported the B(C6F5)3-catalysed reductive carbocyclisation of homoallylic alcohols and dihydro-2H-pyrans with Me2EtSiH or PhSiH3 as reductants to construct a range of 1,2-disubstituted (hetero)arylcyclobutanes under mild reaction conditions with high efficiency and excellent cis-selectivity.54 Mechanistic studies suggested a stepwise, dual ring-closing pathway (not shown).
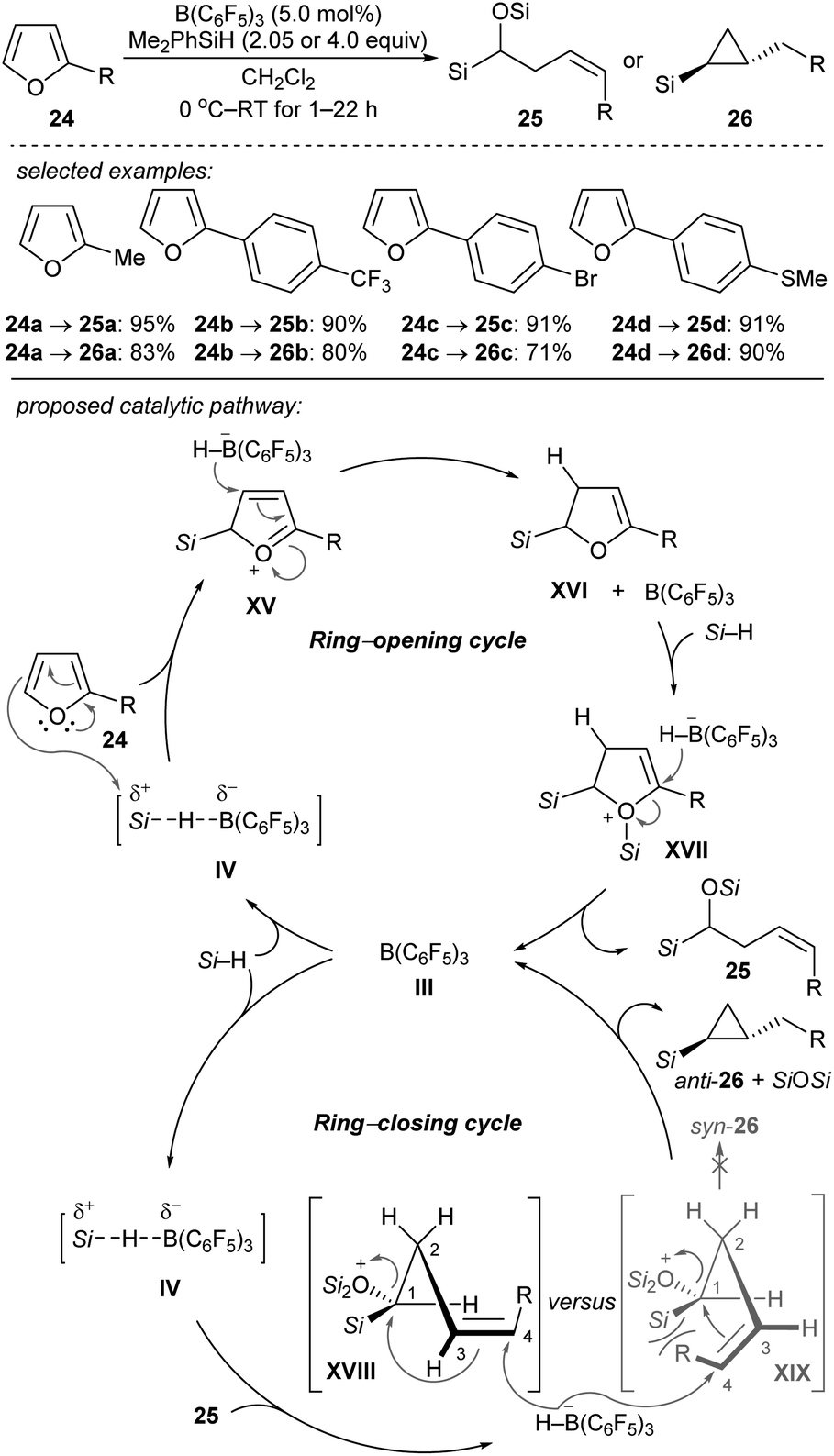 | ||
| Scheme 24 Ring-opening and -closing cascades of furans catalysed by B(C6F5)3 with Me2PhSiH as the reductant. | ||
During their investigation of chemoselective deoxygenation of protected 1,n-diols, Oestreich and co-workers found that diols 14d–f were partially or fully transformed into the rearranged products (Scheme 25).35 The authors proposed that these processes involve phenonium ion intermediates XX for substrates 14d and 14e with anchimeric assistance by an adjacent aryl group or a three-membered silyloxonium ion intermediate XXI for aliphatic 14f. A similar rearrangement was also observed by Song and co-workers (not shown).36
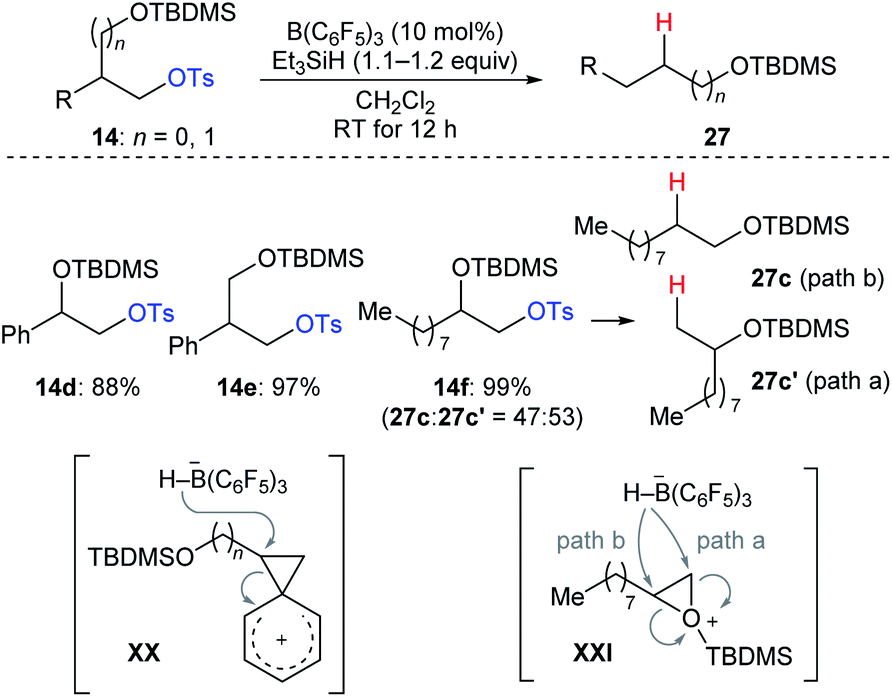 | ||
| Scheme 25 Rearrangement of primary alkyl tosylates catalysed by B(C6F5)3 with Et3SiH as the reductant. | ||
A reductive pinacol-type rearrangement of internal 1,2-diols was described by Morandi and co-workers (Scheme 26).55 By employing a combination of B(C6F5)3 and two hydrosilanes, a broad range of structurally diverse 1,2-diols 12e–h underwent reductive rearrangement with inversion to give primary and secondary alcohols. This process involves the formation of a cyclic siloxane, and mechanistic investigations indicated that alkyl migration occurs prior to deoxygenation in internal diols due to the hyperconjugative and steric effects of the alkyl substituent.
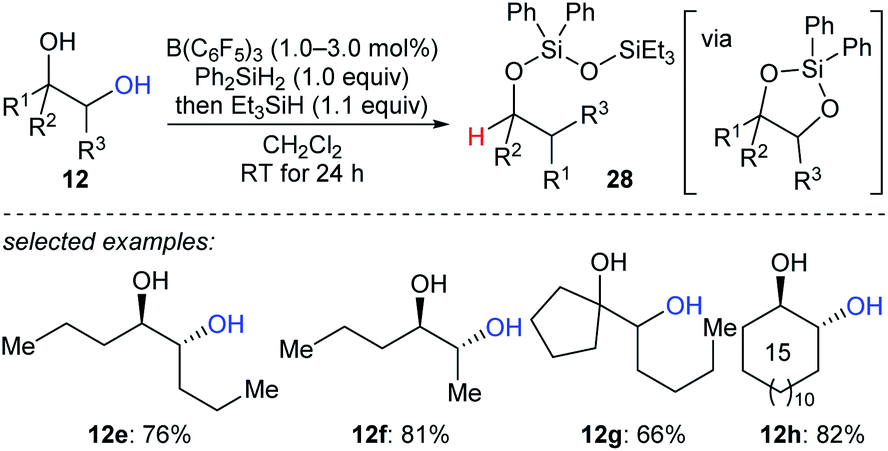 | ||
| Scheme 26 Pinacol-type rearrangement of 1,2-diols catalysed by B(C6F5)3 with Et3SiH as the reductant. | ||
Boron Lewis acids-catalysed decarbonylation
A formal decarbonylation of aliphatic aldehydes 29a–dvia a sequence of Baeyer–Villiger oxidation and B(C6F5)3- or BF3·OEt2-catalysed deoxygenation of the resulting formate with Et3SiH as the reductant was developed by Richter and Oestreich (Scheme 27).56 Mechanistic investigations suggested that an SN1 mechanism is involved for the deoxygenation process.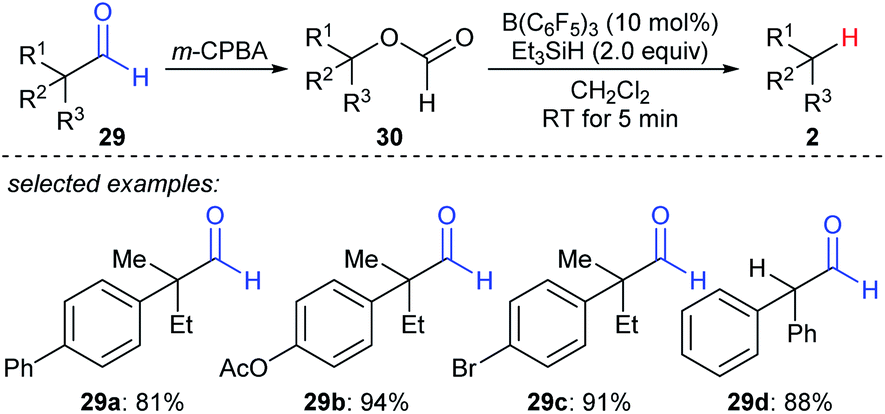 | ||
| Scheme 27 Formal decarbonylation of α-branched aliphatic aldehydes catalysed by B(C6F5)3 with Et3SiH as the reductant. | ||
Boron Lewis acids-catalysed desulfurisation
The combination of a Lewis acid catalyst and a hydrosilane has been widely used for the activation of C–O bonds in the above deoxygenation processes. However, application of this catalytic system to the cleavage of other carbon–heteroatom bonds is far less explored. During their investigation of B(C6F5)3-catalysed chemoselective postpolymerisation modification of poly(phenylsilane), Rosenberg and co-workers observed the formation of diphenylmethane as a result of overreduction of thiobenzophenone (31a).57 The authors demonstrated that the desulfurisation of 31a with PhSiH3 or Ph2SiH2 as reductants occurred rapidly to furnish diphenylmethane (2c) in quantitative yield (Scheme 28).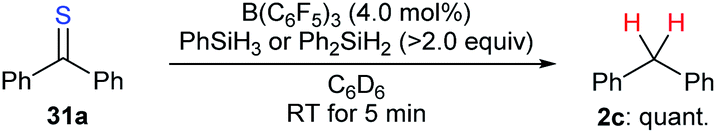 | ||
| Scheme 28 Desulfurisation of thiobenzophenone catalysed by B(C6F5)3 with PhSiH3 or Ph2SiH2 as reductants. | ||
A detailed investigation of B(C6F5)3-catalysed desulfurisation of various sulfides 32a–d was disclosed by Akiyama and co-workers (Scheme 29).58 The desulfurisation of various benzyl and alkyl sulfides and dithianes with Et3SiH as the reductant in the presence of a catalytic amount of B(C6F5)3 generated the corresponding hydrocarbons in good yields under mild reaction conditions with high chemoselectivity. This process could be applied to the deprotection of dithioacetals.
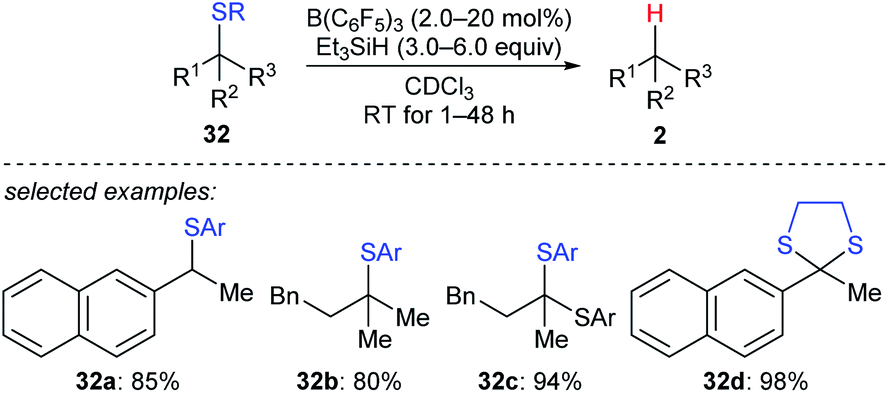 | ||
| Scheme 29 Desulfurisation of sulfides catalysed by B(C6F5)3 with Et3SiH as the reductant. Ar = p-ClC6H4. | ||
Boron Lewis acids-catalysed deamination
More recently, the utility of boron Lewis acid/hydrosilane combinations in the cleavage of C–N bonds to effect catalytic deamination was described by Fang and Oestreich (Scheme 30).59 With B(C6F5)3 as the catalyst and PhSiH3 as the reductant, a broad range of 1°, 2°, and 3° amines 33a–d as well as heterocumulenes (not shown) was converted into the corresponding hydrocarbons at 120 °C. Yields were moderate to good. The relative reactivity of 1°, 2°, and 3° benzylic amines under catalytic conditions was investigated and was opposite to the order of reactivity seen in the deoxygenation of C–O bonds. This process involves the formation of bissilylammonium borohydride intermediates. These dissociate into the corresponding benzylic carbocations which could be further captured by the borohydride to generate the defunctionalised products.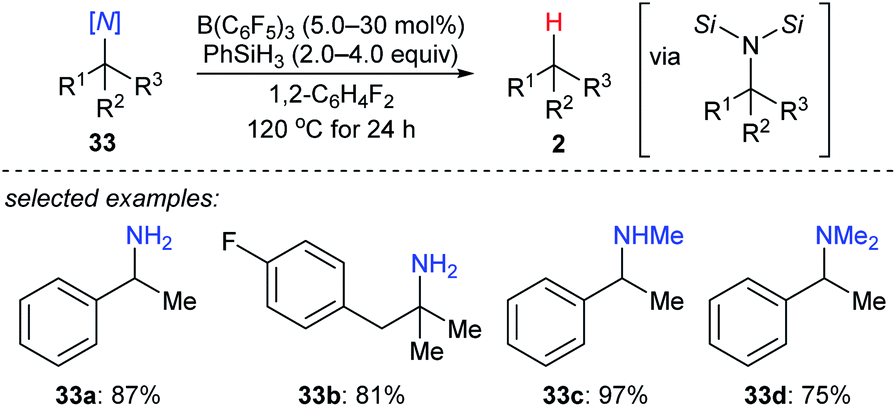 | ||
| Scheme 30 Deamination of amines catalysed by B(C6F5)3 with PhSiH3 as the reductant. | ||
Boron Lewis acids-catalysed dehalogenation
The combined use of boron Lewis acid and hydrosilanes can also be employed to the cleavage of carbon–halogen bonds. In 2012, Caputo and Stephan reported a mild B(C6F5)3-catalysed hydrodefluorination of 1°, 2°, and 3° alkyl fluorides with Et3SiH as the reductant to afford the corresponding hydrocarbons in good to excellent yields (Scheme 31).60 The hydrodefluorination of 1,1,1,3,3,3-hexafluoro-2-(fluoromethoxy)propane (34d) was more sluggish, and a temperature of 60 °C was required at which the trifluoromethyl groups remained intact. The authors attributed the slower reaction to the presence of the ethereal oxygen rendering the C–F bond less polar, thus leading to a weak donor–acceptor interaction between the substrate and the boron Lewis acid. Later, the selective hydrodefluorination of a C1-fluorinated glucose derivative with TMDS as the reductant catalysed by Piers' borane, (C6F5)2BH, generated in situ from (C6F5)2BOH was described by Zhang, Park, and Chang (not shown).61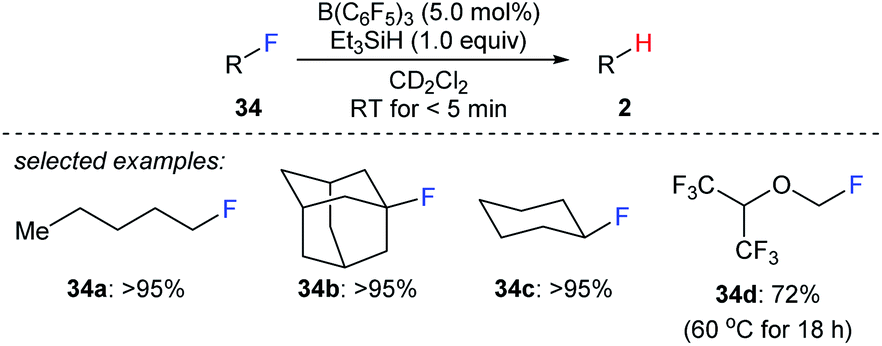 | ||
| Scheme 31 Hydrodefluorination of alkyl fluorides catalysed by B(C6F5)3 with Et3SiH as the reductant. | ||
The hydrodefluorination of trifluorotoluenes with Et3SiH as the reductant catalysed by B(C6F5)3 alone was unsuccessful. By adding an extra group 4 metal complex as a co-catalyst, Lamač and co-workers realised this transformation.62 Among the metallocene co-catalysts screened, 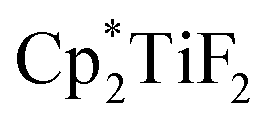 was the most active co-catalyst and also promoted the hydrodechlorination of the aliphatic halogenated solvent, CHCl3. A quantitative yield of toluene was obtained for the hydrodefluorination of trifluorotoluene (35a) with Et3SiH catalysed by the combination of B(C6F5)3 and
was the most active co-catalyst and also promoted the hydrodechlorination of the aliphatic halogenated solvent, CHCl3. A quantitative yield of toluene was obtained for the hydrodefluorination of trifluorotoluene (35a) with Et3SiH catalysed by the combination of B(C6F5)3 and 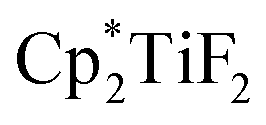 in PhCl (Scheme 32). Et3SiF was formed as a by-product, and a higher selectivity was achieved compared to previously reported catalytic systems by suppressing Friedel–Crafts side products generated by the alkylation of toluene with substrate.
in PhCl (Scheme 32). Et3SiF was formed as a by-product, and a higher selectivity was achieved compared to previously reported catalytic systems by suppressing Friedel–Crafts side products generated by the alkylation of toluene with substrate.
 | ||
Scheme 32 Hydrodefluorination of trifluorotoluenes catalysed by the combination of B(C6F5)3 and 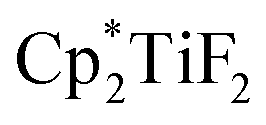 with Et3SiH as the reductant. with Et3SiH as the reductant. | ||
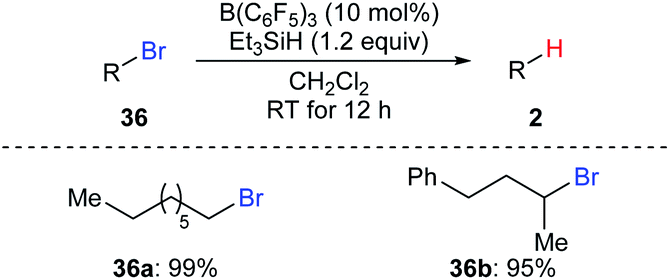
Oestreich and co-workers also demonstrated B(C6F5)3-catalysed hydrodebromination of primary and secondary alkyl bromides 36a and 36b with Et3SiH as the reductant at room temperature (Scheme 33).35 However, hydrodechlorination of the corresponding alkyl chloride using this catalytic system was unsuccessful.
 | ||
| Scheme 33 Hydrodebromination of alkyl bromides catalysed by B(C6F5)3 with Et3SiH as the reductant. | ||
Summary and outlook
During the past two decades, significant achievements have been made in the field of defunctionalisation on the basis of boron Lewis acid catalysis, which exhibits comparable or even superior catalytic activity and selectivity to transition metal catalysis. Starting from Piers's seminal discovery of B(C6F5)3-catalysed hydrosilylation and Gevorgyan's early works on B(C6F5)3-catalysed deoxygenation, numerous reductive alcohol deoxygenations by combinations of boron Lewis acids and hydride sources have been developed. Especially Gagné showcased the impressive chemo-, regio-, and stereoselectivity that can be achieved with this tool. In addition, this boron Lewis acid catalysis has been successfully extended to the cleavage of C–S, C–N, and carbon–halogen bonds. However, the efficiency and selectivity of boron Lewis acid catalysis still needs to be further improved. Functional-group tolerance remains an issue. Thus, the development of air- and moisture-stable and easy-to-prepare and -handle catalysts with high activity and selectivity is desirable. Next to the significant advances made in the area of deoxygenation, selective decarbonylation, desulfurisation, deamination, and dehalogenation have just begun to flourish.Conflicts of interest
There are no conflicts to declare.Acknowledgements
H. F. gratefully acknowledges the Humboldt Foundation for a postdoctoral fellowship (2018–2020). M. O. is indebted to the Einstein Foundation (Berlin) for an endowed professorship.References
- For reviews, see: (a) A. M. Robinson, J. E. Hensley and J. W. Medlin, ACS Catal., 2016, 6, 5026–5043 CrossRef CAS; (b) C. Chatterjee, F. Pong and A. Sen, Green Chem., 2015, 17, 40–71 RSC.
- J. J. Eisch, L. E. Hallenbeck and M. A. Lucarelli, Fuel, 1985, 64, 440–442 CrossRef CAS.
- For selected examples, see: (a) R. DeVor, K. Carvalho-Knighton, B. Aitken, P. Maloney, E. Holland, L. Talalaj, S. Elsheimer, C. A. Clausen and C. L. Geiger, Chemosphere, 2009, 76, 761–766 CrossRef CAS PubMed; (b) B.-Z. Wu, H.-Y. Chen, S. J. Wang, C. M. Wai, W. Liao and K. Chiu, Chemosphere, 2012, 88, 757–768 CrossRef CAS PubMed; (c) A. Ido, S. Ishihara, A. Kume, T. Nakanishi, Y. Monguchi, H. Sajiki and H. Nagase, Chemosphere, 2013, 90, 57–64 CrossRef PubMed.
- For a review, see: A. Studer and S. Amrein, Synthesis, 2002, 835–849 CrossRef CAS.
- For a review, see: A. Modak and D. Maiti, Org. Biomol. Chem., 2016, 14, 21–35 RSC.
- For reviews of boron Lewis acids/frustrated Lewis pair-catalysed H–H and Si–H bond activation, see: (a) D. Weber and M. R. Gagné, in Organosilicon Chemistry: Novel Approaches and Reactions, ed. T. Hiyama and M. Oestreich, Wiley-VCH, Weinheim, 2019, pp. 33–85 Search PubMed; (b) T. Hackel and N. A. McGrath, Molecules, 2019, 24, 432–461 CrossRef PubMed; (c) M. Oestreich, J. Hermeke and J. Mohr, Chem. Soc. Rev., 2015, 44, 2202–2220 RSC; (d) D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2015, 54, 6400–6441 CrossRef CAS PubMed; (e) D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2010, 49, 46–76 CrossRef CAS PubMed.
- For a review, see: J. L. Carden, A. Dasgupta and R. L. Melen, Chem. Soc. Rev., 2020, 49, 1706–1725 RSC.
- For reviews, see: (a) X. J. Chen, M. H. Yi, S. F. Wu, L. W. Tan, X. Ge, M. He and G. Q. Yin, Materials, 2019, 12, 304–316 CrossRef CAS PubMed; (b) M. A. Brook, Chem.–Eur. J., 2018, 24, 8458–8469 CrossRef CAS PubMed; (c) R. Wakabayashi and K. Kuroda, ChemPlusChem, 2013, 78, 764–774 CrossRef CAS PubMed; for selected examples of Piers–Rubinsztajn reaction, see: (d) S. Rubinsztajn and J. A. Cella, Macromolecules, 2005, 38, 1061–1063 CrossRef CAS; (e) J. Chojnowski, S. Rubinsztajn, J. A. Cella, W. Fortuniak, M. Cypryk, J. Kurjata and K. Kazmierski, Organometallics, 2005, 24, 6077–6084 CrossRef CAS; (f) J. Cella and S. Rubinsztajn, Macromolecules, 2008, 41, 6965–6971 CrossRef CAS; (g) J. Chojnowski, S. Rubinsztajn, W. Fortuniak and J. Kurjata, Macromolecules, 2008, 41, 7352–7358 CrossRef CAS; (h) J. B. Grande, D. B. Thompson, F. Gonzaga and M. A. Brook, Chem. Commun., 2010, 46, 4988–4990 RSC; (i) B. A. Kamino, J. B. Grande, M. A. Brook and T. P. Bender, Org. Lett., 2011, 13, 154–157 CrossRef CAS PubMed; (j) M. J. Gretton, B. A. Kamino, M. A. Brook and T. P. Bender, Macromolecules, 2012, 45, 723–728 CrossRef CAS; (k) M. J. Gretton, B. A. Kamino and T. P. Bender, Macromol. Symp., 2013, 324, 82–94 CrossRef CAS; (l) J. F. Zhang, Y. Chen and M. A. Brook, ACS Sustainable Chem. Eng., 2014, 2, 1983–1991 CrossRef CAS; (m) J. F. Zhang, E. Fleury and M. A. Brook, Green Chem., 2015, 17, 4647–4656 RSC; (n) J. F. Zhang, S. Liang, L. Y. Yu, A. L. Skov, H. M. Etmimi, P. E. Mallon, A. Adronov and M. A. Brook, J. Polym. Sci., Part A: Polym. Chem., 2016, 54, 2379–2385 CrossRef CAS; (o) M. C. Liao, A. F. Schneider, S. E. Laengert, C. B. Gale, Y. Chen and M. A. Brook, Eur. Polym. J., 2018, 107, 287–293 CrossRef CAS.
- For selected examples, see: (a) M. G. Adlington, M. Orfanopoulos and J. L. Fry, Tetrahedron Lett., 1976, 17, 2955–2958 CrossRef; (b) J. L. Fry, M. Orfanopoulos, M. G. Adlington, W. P. Dittman and S. B. Silverman, J. Org. Chem., 1978, 43, 374–375 CrossRef CAS; (c) J. W. Larsen and L. W. Chang, J. Org. Chem., 1979, 44, 1168–1170 CrossRef CAS; (d) M. Orfanopoulos and I. Smonou, Synth. Commun., 1988, 18, 833–839 CrossRef CAS; (e) I. Smonou, Synth. Commun., 1994, 24, 1999–2002 CrossRef CAS.
- D. J. Parks and W. E. Piers, J. Am. Chem. Soc., 1996, 118, 9440–9441 CrossRef CAS.
- (a) V. Gevorgyan, J.-X. Liu, M. Rubin, S. Benson and Y. Yamamoto, Tetrahedron Lett., 1999, 40, 8919–8922 CrossRef CAS; (b) V. Gevorgyan, M. Rubin, S. Benson, J.-X. Liu and Y. Yamamoto, J. Org. Chem., 2000, 65, 6179–6186 CrossRef CAS PubMed.
- For some mechanistic investigations on B(C6F5)3-catalysed hydrosilylation of ketones and imines, see: (a) D. J. Parks, J. M. Blackwell and W. E. Piers, J. Org. Chem., 2000, 65, 3090–3098 CrossRef CAS PubMed; (b) J. M. Blackwell, E. R. Sonmor, T. Scoccitti and W. E. Piers, Org. Lett., 2000, 2, 3921–3923 CrossRef CAS PubMed; (c) S. Shinke, T. Tsuchimoto and Y. Kawakami, Silicon Chem., 2007, 3, 243–249 CrossRef CAS; (d) S. Rendler and M. Oestreich, Angew. Chem., Int. Ed., 2008, 47, 5997–6000 CrossRef CAS PubMed; (e) D. T. Hog and M. Oestreich, Eur. J. Org. Chem., 2009, 5047–5056 CrossRef CAS; (f) J. Hermeke, M. Mewald and M. Oestreich, J. Am. Chem. Soc., 2013, 135, 17537–17546 CrossRef CAS PubMed; (g) K. Sakata and H. Fujimoto, J. Org. Chem., 2013, 78, 12505–12512 CrossRef CAS PubMed; (h) T. Fallon and M. Oestreich, Angew. Chem., Int. Ed., 2015, 54, 12488–12491 CrossRef CAS PubMed.
- A. Y. Houghton, J. Hurmalainen, A. Mansikkamaki, W. E. Piers and H. M. Tuononen, Nat. Chem., 2014, 6, 983–988 CrossRef CAS PubMed.
- (a) V. Gevorgyan, M. Rubin, J.-X. Liu and Y. Yamamoto, J. Org. Chem., 2001, 66, 1672–1675 CrossRef CAS PubMed; (b) G. B. Bajracharya, T. Nogami, T. Jin, K. Matsuda, V. Gevorgyan and Y. Yamamoto, Synthesis, 2004, 308–311 CAS.
- X. Y. Li, R. Shang, M. C. Fu and Y. Fu, Green Chem., 2015, 17, 2790–2793 RSC.
- C. B. Gale and M. A. Brook, Green Chem., 2018, 20, 3717–3721 RSC.
- S. Chandrasekhar, C. R. Reddy and B. N. Babu, J. Org. Chem., 2002, 67, 9080–9082 CrossRef CAS PubMed.
- S. Chandrasekhar, G. Chandrashekar, K. Vijeender and M. S. Reddy, Tetrahedron Lett., 2006, 47, 3475–3478 CrossRef CAS.
- E. Feghali, O. Jacquet, P. Thuery and T. Cantat, Catal. Sci. Technol., 2014, 4, 2230–2234 RSC.
- (a) R. D. Nimmagadda and C. McRae, Tetrahedron Lett., 2006, 47, 3505–3508 CrossRef CAS; (b) R. D. Nimmagadda and C. McRae, Tetrahedron Lett., 2006, 47, 5755–5758 CrossRef CAS; (c) R. D. Nimmagadda and C. McRae, Org. Geochem., 2007, 38, 1061–1072 CrossRef CAS.
- M. X. Tan and Y. G. Zhang, Tetrahedron Lett., 2009, 50, 4912–4915 CrossRef CAS.
- M. Lindqvist, N. Sarnela, V. Sumerin, K. Chernichenko, M. Leskela and T. Repo, Dalton Trans., 2012, 41, 4310–4312 RSC.
- T. Mahdi and D. W. Stephan, Angew. Chem., Int. Ed., 2015, 54, 8511–8514 CrossRef CAS PubMed.
- L. L. Adduci, M. P. McLaughlin, T. A. Bender, J. J. Becker and M. R. Gagné, Angew. Chem., Int. Ed., 2014, 53, 1646–1649 CrossRef CAS PubMed.
- (a) E. Feghali and T. Cantat, Chem. Commun., 2014, 50, 862–865 RSC; (b) E. Feghali, G. Carrot, P. Thuery, C. Genre and T. Cantat, Energy Environ. Sci., 2015, 8, 2734–2743 RSC.
- E. Feghali and T. Cantat, ChemSusChem, 2015, 8, 980–984 CrossRef CAS PubMed.
- C. Jeon, D. W. Kim, S. Chang, J. G. Kim and M. Seo, ACS Macro Lett., 2019, 8, 1172–1178 CrossRef CAS.
- L. L. Adduci, T. A. Bender, J. A. Dabrowski and M. R. Gagné, Nat. Chem., 2015, 7, 576–581 CrossRef CAS PubMed.
- T. A. Bender, J. A. Dabrowski and M. R. Gagné, ACS Catal., 2016, 6, 8399–8403 CrossRef CAS.
- (a) Y. Seo and M. R. Gagné, ACS Catal., 2018, 8, 81–85 CrossRef CAS; (b) J. M. Lowe, Y. Seo and M. R. Gagné, ACS Catal., 2018, 8, 8192–8198 CrossRef CAS; (c) Y. Seo, J. M. Lowe and M. R. Gagné, ACS Catal., 2019, 9, 6648–6652 CrossRef CAS.
- J. Lowe, B. Bowers, Y. Seo and M. R. Gagné, Angew. Chem., Int. Ed., 2020, 59 DOI:10.1002/anie.202007415.
- (a) T. A. Bender, P. R. Payne and M. R. Gagné, Nat. Chem., 2018, 10, 85–90 CrossRef CAS PubMed; (b) Y. Seo, A. Gudz, J. M. Lowe and M. R. Gagné, Tetrahedron, 2019, 75, 130712 CrossRef CAS PubMed.
- N. Drosos and B. Morandi, Angew. Chem., Int. Ed., 2015, 54, 8814–8818 CrossRef CAS PubMed.
- G. J. Cheng, N. Drosos, B. Morandi and W. Thiel, ACS Catal., 2018, 8, 1697–1702 CrossRef CAS.
- I. Chatterjee, D. Porwal and M. Oestreich, Angew. Chem., Int. Ed., 2017, 56, 3389–3391 CrossRef CAS PubMed.
- W. Y. Yang, L. Gao, J. Lu and Z. L. Song, Chem. Commun., 2018, 54, 4834–4837 RSC.
- K. Chulsky and R. Dobrovetsky, Org. Lett., 2018, 20, 6804–6807 CrossRef CAS PubMed.
- For reviews, see: (a) A. Volkov, F. Tinnis, T. Slagbrand, P. Trillo and H. Adolfsson, Chem. Soc. Rev., 2016, 45, 6685–6697 RSC; (b) A. M. Smith and R. Whyman, Chem. Rev., 2014, 114, 5477–5510 CrossRef CAS PubMed.
- K. M. Lucas, A. F. Kleman, L. R. Sadergaski, C. L. Jolly, B. S. Bollinger, B. L. Mackesey and N. A. McGrath, Org. Biomol. Chem., 2016, 14, 5774–5778 RSC.
- E. Blondiaux and T. Cantat, Chem. Commun., 2014, 50, 9349–9352 RSC.
- R. C. Chadwick, V. Kardelis, P. Lim and A. Adronov, J. Org. Chem., 2014, 79, 7728–7733 CrossRef CAS PubMed.
- P. Q. Huang, Q. W. Lang and Y. R. Wang, J. Org. Chem., 2016, 81, 4235–4243 CrossRef CAS PubMed.
- J. Z. Ni, T. Oguro, T. Sawazaki, Y. Sohma and M. Kanai, Org. Lett., 2018, 20, 7371–7374 CrossRef CAS PubMed.
- Y. H. Li, J. A. M. de la Torre, K. Grabow, U. Bentrup, K. Junge, S. L. Zhou, A. Brückner and M. Beller, Angew. Chem., Int. Ed., 2013, 52, 11577–11580 CrossRef CAS PubMed.
- A. Chardon, T. M. El Dine, R. Legay, M. De Paolis, J. Rouden and J. Blanchet, Chem.–Eur. J., 2017, 23, 2005–2009 CrossRef CAS PubMed.
- D. Mukherjee, S. Shirase, K. Mashima and J. Okuda, Angew. Chem., Int. Ed., 2016, 55, 13326–13329 CrossRef CAS PubMed.
- (a) M. T. Peruzzi, Q. Q. Mei, S. J. Lee and M. R. Gagné, Chem. Commun., 2018, 54, 5855–5858 RSC; (b) M. T. Peruzzi, F. Gallon, S. J. Lee and M. R. Gagné, Org. Lett., 2019, 21, 3451–3455 CrossRef CAS PubMed.
- N. A. Sitte, M. Bursch, S. Grimme and J. Paradies, J. Am. Chem. Soc., 2019, 141, 159–162 CrossRef CAS PubMed.
- For reviews, see: (a) A. Rossin and M. Peruzzini, Chem. Rev., 2016, 116, 8848–8872 CrossRef CAS PubMed; (b) Z. Huang and T. Autrey, Energy Environ. Sci., 2012, 5, 9257–9268 RSC; (c) A. Staubitz, A. P. M. Robertson and I. Manners, Chem. Rev., 2010, 110, 4079–4124 CrossRef CAS PubMed; (d) C. W. Hamilton, R. T. Baker, A. Staubitz and I. Manners, Chem. Soc. Rev., 2009, 38, 279–293 RSC; (e) T. B. Marder, Angew. Chem., Int. Ed., 2007, 46, 8116–8118 CrossRef CAS PubMed.
- For selected examples, see: (a) S. Li, G. Li, W. Meng and H. Du, J. Am. Chem. Soc., 2016, 138, 12956–12962 CrossRef CAS PubMed; (b) Q. Zhou, L. Zhang, W. Meng, X. Feng, J. Yang and H. Du, Org. Lett., 2016, 18, 5189–5191 CrossRef CAS PubMed; (c) S. Li, W. Meng and H. Du, Org. Lett., 2017, 19, 2604–2606 CrossRef CAS PubMed; (d) F. Ding, Y. Zhang, R. Zhao, Y. Jiang, R. L.-Y. Bao, K. Lin and L. Shi, Chem. Commun., 2017, 53, 9262–9264 RSC.
- Y. Pan, Z. Luo, J. Han, X. Xu, C. Chen, H. Zhao, L. Xu, Q. Fan and J. Xiao, Adv. Synth. Catal., 2019, 361, 2301–2308 CrossRef CAS.
- T. A. Bender, J. A. Dabrowski, H. Y. Zhong and M. R. Gagné, Org. Lett., 2016, 18, 4120–4123 CrossRef CAS PubMed.
- C. K. Hazra, N. Gandhamsetty, S. Park and S. Chang, Nat. Commun., 2016, 7, 13431–13439 CrossRef CAS PubMed.
- C. K. Hazra, J. Jeong, H. Kim, M. H. Baik, S. Park and S. Chang, Angew. Chem., Int. Ed., 2018, 57, 2692–2696 CrossRef CAS PubMed.
- N. Drosos, G. J. Cheng, E. Ozkal, B. Cacherat, W. Thiel and B. Morandi, Angew. Chem., Int. Ed., 2017, 56, 13377–13381 CrossRef CAS PubMed.
- S. C. Richter and M. Oestreich, Chem.–Eur. J., 2019, 25, 8508–8512 CrossRef CAS PubMed.
- P. T. K. Lee, M. K. Skjel and L. Rosenberg, Organometallics, 2013, 32, 1575–1578 CrossRef CAS.
- K. Saito, K. Kondo and T. Akiyama, Org. Lett., 2015, 17, 3366–3369 CrossRef CAS PubMed.
- H. Fang and M. Oestreich, Angew. Chem., Int. Ed., 2020, 59, 11394–11398 CrossRef CAS PubMed.
- C. B. Caputo and D. W. Stephan, Organometallics, 2012, 31, 27–30 CrossRef CAS.
- J. Zhang, S. Park and S. Chang, Angew. Chem., Int. Ed., 2017, 56, 13757–13761 CrossRef CAS PubMed.
- D. Dunlop, J. Pinkas, M. Horáček, N. Žilková and M. Lamač, Dalton Trans., 2020, 49, 2771–2775 RSC.
| This journal is © The Royal Society of Chemistry 2020 |