
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePalladium-catalyzed direct asymmetric C–H bond functionalization enabled by the directing group strategy
Ke
Yang
 a,
Mengjie
Song
a,
Hao
Liu
b and
Haibo
Ge
*b
a,
Mengjie
Song
a,
Hao
Liu
b and
Haibo
Ge
*b
aJiangsu Key Laboratory of Advanced Catalytic Materials & Technology, School of Petrochemical Engineering, Changzhou University, Changzhou, Jiangsu 213164, China
bDepartment of Chemistry and Biochemistry, Texas Tech University, Lubbock, Texas 79409, USA. E-mail: haibo.ge@ttu.edu
First published on 9th July 2020
Abstract
In the past decade, selective C–C and C-heteroatom bond construction through palladium-catalyzed direct C–H bond functionalization has been extensively studied by employing a variety of directing groups. Within this category, direct asymmetric C(sp2)–H and C(sp3)–H activation for the construction of highly enantiomerically enriched skeletons still progressed at a slow pace. This minireview briefly introduces the major advances in the field for palladium-catalyzed direct asymmetric C–H bond functionalization via the directing group strategy.
 Ke Yang | Ke Yang obtained his PhD degree from Nanjing University in 2014. Next, he became a postdoctoral fellow at Nanjing University with Professor Guigen Li. In 2015, he joined the group of Professor Haibo Ge at the Department of Chemistry and Chemical Biology, Indiana University – Purdue University Indianapolis (IUPUI) in the USA. After that, he moved to Changzhou University and started his independent academic career. His research interests involve novel transition metal-catalyzed C–H bond functionalization reactions and green organic synthesis methodology. |
 Mengjie Song | Mengjie Song completed her B.E. degree in Applied Chemistry at Nanyang Institute of Technology. From 2019, she began her master's degree study in the School of Petrochemical Engineering at Changzhou University and joined Yang's group. Now, her research interests involve green organic synthetic methodology and transition metal-catalyzed C–H bond functionalization. |
 Hao Liu | Hao Liu obtained his bachelor's degree of science in chemical engineering from the University of New Brunswick, Canada. He is currently pursuing his PhD at Texas Tech University and has joined Prof. Ge Haibo's research team to study the activation of C–H bonds. |
 Haibo Ge | Haibo Ge received his PhD degree in Medicinal Chemistry from The University of Kansas in 2006, and then moved to The Scripps Research Institute for postdoctoral study. In 2009, he began his independent academic career at the Department of Chemistry and Chemical Biology at Indiana University – Purdue University Indianapolis and relocated to the Department of Chemistry and Biochemistry at Texas Tech University in 2020. Research by his group is mainly focused on the development of novel methods for carbon–carbon and carbon–heteroatom bond formation through transition metal catalyzed C–H functionalization. |
1. Introduction
Transition metal-catalyzed C–H bond functionalization is one of most efficient approaches for selective C–C and C-heteroatom bond construction in organic synthesis.1 Among various transition metal catalysts, palladium species have been recognized as the most important ones in the direct C–H bond functionalization reactions.2In the past decade, palladium-catalyzed selective C(sp2)–H and C(sp3)–H bond functionalization has been demonstrated well by covalently attaching a directing group to a substrate.3 In spite of being a powerful approach, additional reaction steps are required for the pre-installation and subsequent removal of the directing group, which decreases the overall efficiency of the process. To overcome this drawback, the transient directing group strategy has recently been developed and successfully applied in the field of C–H bond functionalization. In this process, an external ligand is added to the reaction system to bind to the substrate in reversible mode and subsequently coordinate with the metal centre.4 As a result, no additional synthetic steps are needed for pre-functionalization of the substrate and removal of the directing group, which greatly improves the compatibility and efficiency of the reaction.
Meanwhile, direct asymmetric C–H bond functionalization has also attracted considerable attention due to its potential as the most efficient method to access highly enantiomerically enriched skeletons,5–11 and significant progress has been made in directing group strategy enabled palladium catalysis. Remarkably, the asymmetric version of the process has also been achieved with the assistance of a catalytic amount of the chiral ligand or chiral directing group. It was demonstrated by Yu and co-workers in 2008 that a catalytic amount of mono-N-protected amino acids (MPAAs) could serve as chiral ligands for the Pd-catalyzed asymmetric C–H bond functionalization.6 Furthermore, a breakthrough in the transient directing group strategy was achieved by the same group7 using L-tert-leucine as a transient ligand for the Pd-catalyzed enantioselective benzylic C(sp3)–H functionalization.
Recently, several related reviews on or accounts of transition metal-catalyzed direct asymmetric C–H functionalization have been reported.5a,8–11 Ackermann's minireview summarized the remarkable recent advances in direct asymmetric C–H functionalization catalyzed by earth-abundant 3d transition metals.5a Yu's account highlighted their development of bifunctional MPAA ligands for the diverse C–H bond functionalization reactions,8a and the review mainly illustrated recent literature about transition-metal (Pd-, Ir- and Rh-) catalyzed asymmetric C(sp3)–H bond functionalization by using a specific chiral ligand scaffold.8b The You group summarized the recent progress in the construction of planar chiral ferrocenes through transition-metal (Cu-, Pd-, Ir-, Rh-, Au- and Pt-) catalyzed asymmetric C–H bond functionalization.9 Shi's review provided an overview principally for the synthesis of axially chiral biaryls.10 Cramer's review focused mainly on the Rh- and Ir-catalyzed asymmetric transformations.11
In this mini-review, we will introduce and discuss the significant progress made in the field of palladium-catalyzed asymmetric C–H functionalization using a monodentate, bidentate, or transient directing group (Scheme 1). We will also provide informative summarization of the directing group strategy and outlook for transient directing groups in the transition metal-catalyzed asymmetric C–H bond functionalization.
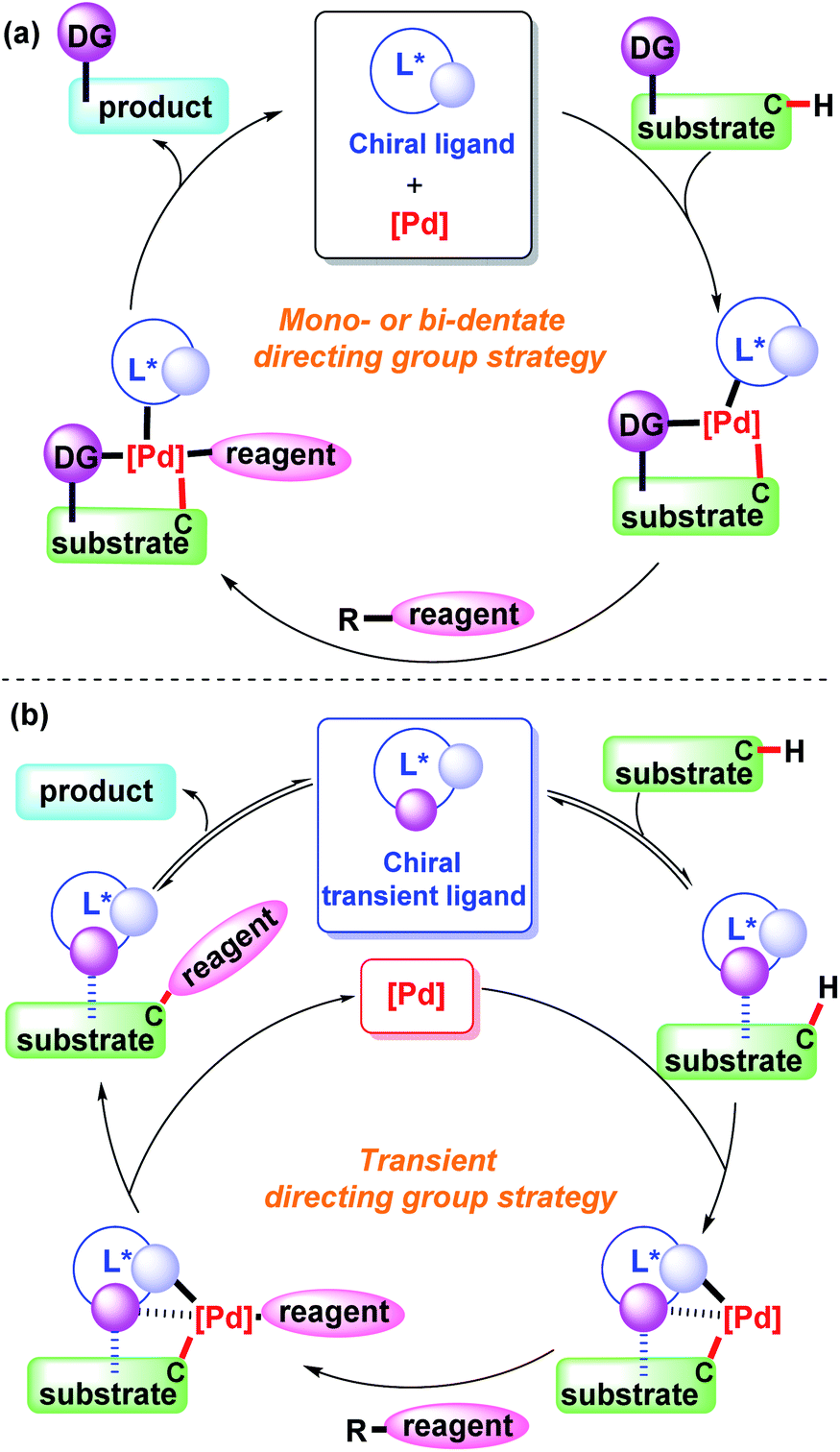 | ||
| Scheme 1 Concept of palladium-catalyzed direct asymmetric C–H bond functionalization enabled by the directing group strategy. | ||
2. Monodentate directing group-enabled asymmetric C–H functionalization
Monodentate directing groups, such as amide, ester, carboxylic acid, ketone, heterocyclic, amine, imine, hydroxyl and so on, have been well studied for the Pd-catalyzed C–H functionalization reactions. Moreover, sulfur and phosphorus-containing monodentate directing groups have also been developed. Despite these great achievements, realization of Pd-catalyzed asymmetric C–H functionalization in the presence of a catalytic amount of a chiral ligand is still a challenging process.3Asymmetric desymmetrization is a unique and important asymmetric synthesis strategy in organic chemistry, and its targets are structurally symmetric compounds with prochirality or meso-substrates.12 In 2005, the Yu group utilized a chiral tert-leucine derived oxazoline directing group to achieve Pd(II)-catalyzed diastereoselective iodination and acetoxylation of C(sp3)–H bonds.13a,b Encouraged by this important work, efficient Pd(II)-catalyzed asymmetric desymmetrization of prochiral diaryl-2-pyridylmethanes with alkyl boronic acids was then established by employing MPAA ligands (Scheme 2a).6 After evaluating various commercially available chiral carboxylates and chiral phosphates, (−)-Men-Leu-OH (L1) was proved to be the optimal ligand and provided the desired products in excellent yields and enantioselectivities.
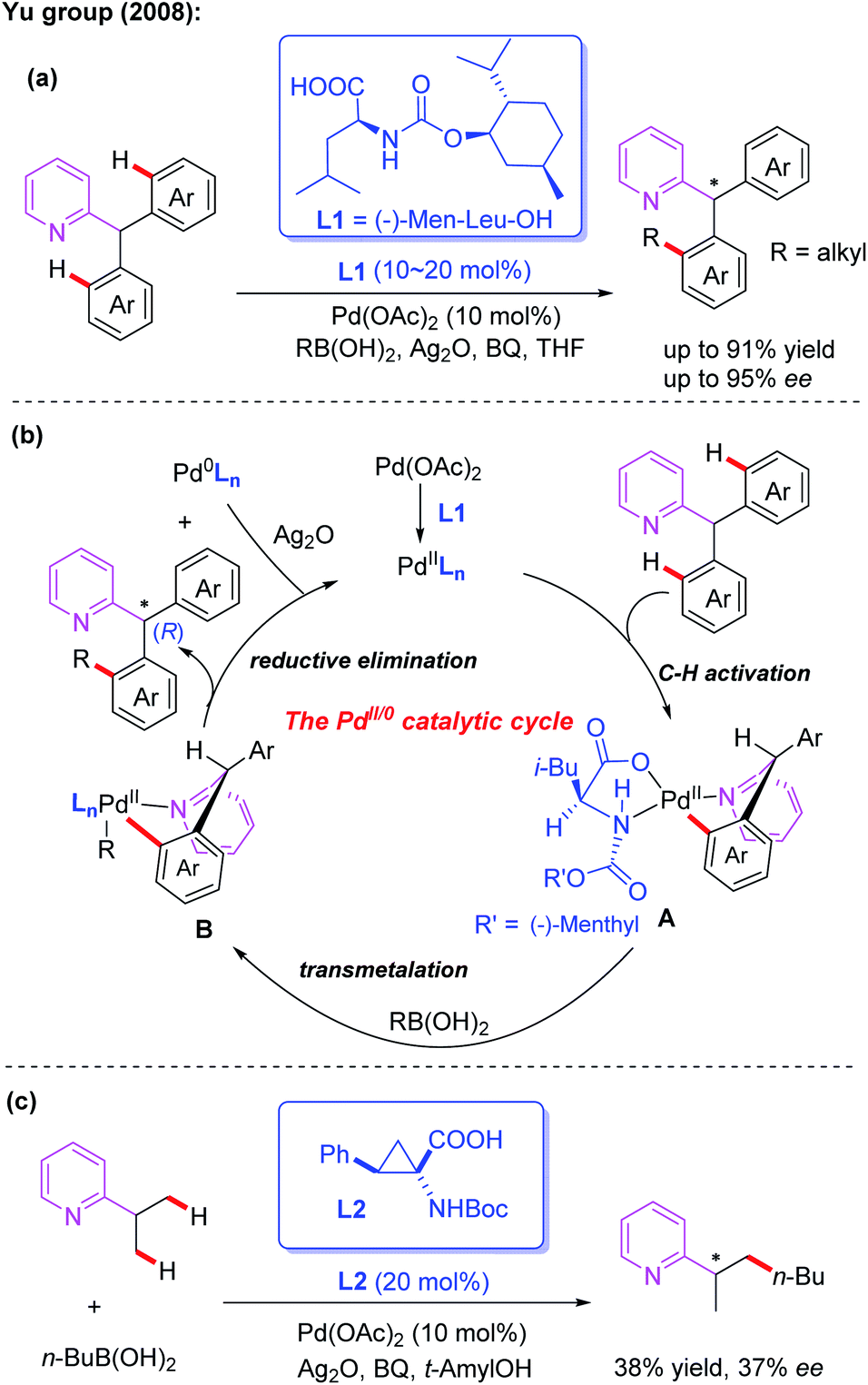 | ||
| Scheme 2 External MPAA ligands for the Pd(II)-catalyzed C–H asymmetric desymmetrization of diaryl-2-pyridylmethanes and 2-isopropylpyridine with alkyl boronic acids. | ||
A plausible catalytic cycle involving Pd(II)/Pd(0) catalysis was proposed (Scheme 2b). The selective C–H bond cleavage of diaryl-2-pyridylmethane with a Pd(II) catalyst in the presence of (−)-Men-Leu-OH generates the corresponding cyclic Pd(II) intermediate A. Next, transmetalation between this intermediate and an alkyl boronic acid provides the intermediate B. Finally, the desired chiral product is formed through a reductive elimination process along with a Pd(0) complex.
In the structure of the proposed cyclic Pd(II) intermediate A, the i-Bu group is above the palladium plane, and the (−)-menthyloxycarbonyl group is on the palladium plane. Due to the conformational requirements, the non-participating aryl group prefers a less space-crowded axial position, which ultimately results in an (R)-configured alkyl product.
In this reaction, BQ was found to be very critical for both the C–H bond activation and reductive elimination. The silver salt was proposed to act as an oxidizing reagent in the process to re-oxidize the resulting Pd(0) complex to the corresponding Pd(II) complex. Notably, a very recent DFT theoretical calculation study indicates that an important molecular interaction between the Pd(II) catalyst and the silver salt additive may exist in the critical transition states of the aryl C(sp2)–H bond activation process. Silver salt is likely to manifest in the form of a Pd–Ag heterobimetallic species in the whole catalytic cycle, not just as a terminal oxidant.13c Moreover, in the presence of a cyclopropane amino acid ligand, L2, the enantioselective C(sp3)–H alkylation of 2-isopropylpyridine with butyl boronic acid was also developed and the desired product was obtained in 38% yield and 37% ee (Scheme 2c).
Carboxylic acids, as cheap and readily available reagents, are widely used in organic chemistry.13d In 2010, the Yu group demonstrated desymmetric C–H olefination of diarylacetic acids using Boc-L-isoleucine (L3) as the chiral ligand (Scheme 3a).14 In order to obtain high yield and enantioselectivity, the unique combination of sodium diphenylacetate and KHCO3 is applied in this reaction. Moreover, the structure of the (R)-configured olefination product was confirmed by X-ray analysis and was consistent with the proposed cyclic Pd(II) intermediate (Scheme 3b). These results demonstrated that MPAA ligands could effectively promote stereoinduction in the Pd(II)-catalyzed asymmetric C–H bond functionalization.
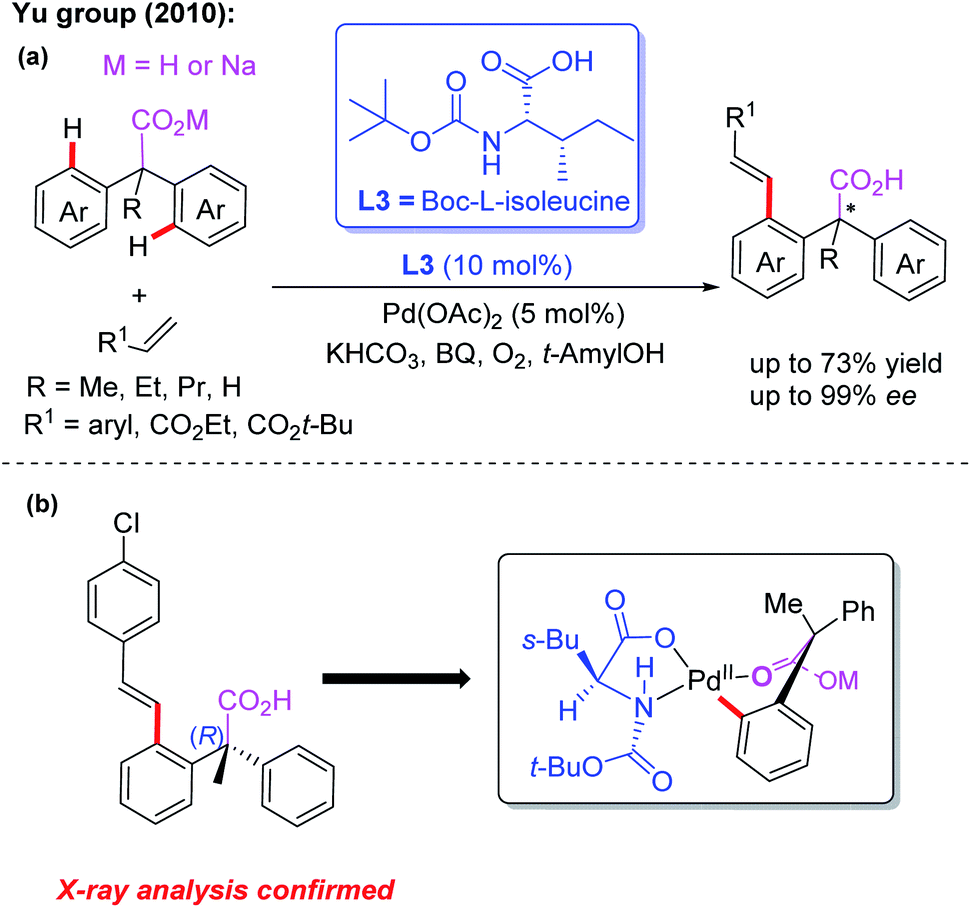 | ||
| Scheme 3 Pd(II)-catalyzed desymmetric C–H olefination of diphenylacetic acids using Boc-L-isoleucine as the chiral ligand. | ||
In 2011, the Yu group reported the first example of asymmetric C–H bond activation of cyclopropane carboxylic acid derived 4-cyanotetrafluorophenyl amides in the presence of a novel mono-N-protected amino acid ligand, L4 (Scheme 4).15 A range of organoboron reagents, such as aryl, vinyl, and alkylboron compounds, were used to afford cis-substituted chiral products in good to excellent enantioselectivities (up to 81% yield and 99% ee). It was proposed that use of the strongly electron-withdrawing 4-cyanotetrafluorophenyl-substituted amide as a weakly coordinating group could increase the acidity of the N–H bond, enabling deprotonation of substrates under weakly basic conditions, and thus facilitating subsequent C–H bond cleavage with a Pd(II) catalyst. Furthermore, this novel ligand L4 presents two specific features: (1) the withdrawing trichloromethyl group increases steric size while also adjusting the electronic properties of the nitrogen atom and (2) the 2,6-difluorophenyl group protects the arene from metalation due to its rigid steric environment.
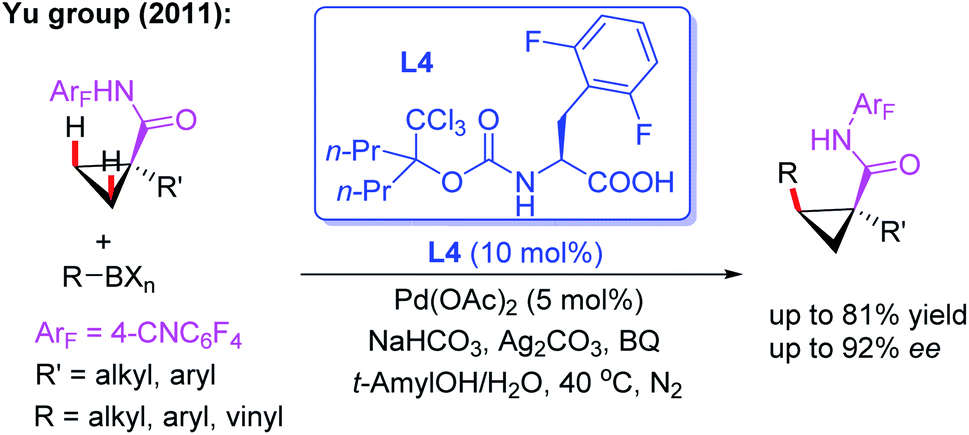 | ||
| Scheme 4 Pd(II)-catalyzed asymmetric C–H bond activation of cyclopropane acid derived amides in the presence of a chiral ligand, L4. | ||
After a short while, the first example of Pd(II)-catalyzed asymmetric methylene β-C(sp3)–H functionalization of cyclobutane carboxylic acid derived 4-cyanotetrafluorophenyl amides with arylboron reagents was also reported by the Yu group (Scheme 5).16 In this study, it was found that with MPAAs as chiral ligands, only low yield and poor ee could be achieved. However, mono N-protected α-amino-O-methylhydroxamic acid (MPAHA) ligand L5 afforded high enantioselectivities. A possible reason for the improved performance is presumably the much tighter binding of N-methoxyamide to the palladium catalyst. However, cyclobutane substrates bearing α-hydrogen atoms only afford poor yields.
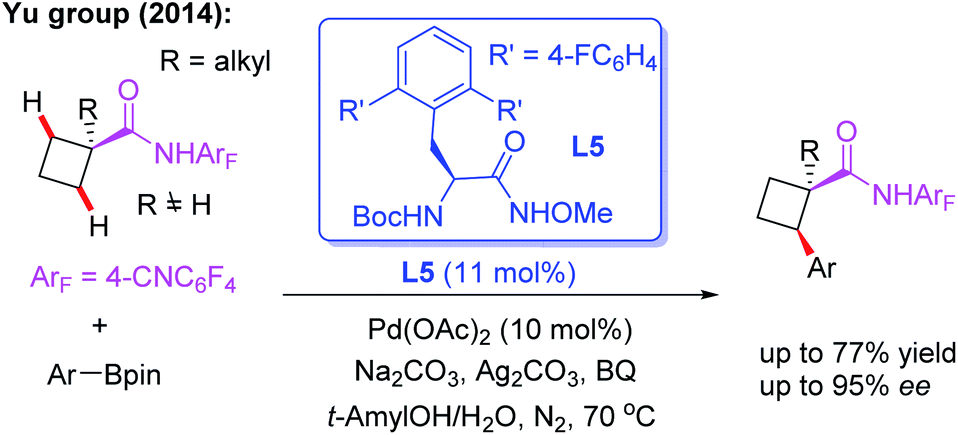 | ||
| Scheme 5 Pd(II)-catalyzed asymmetric β-C(sp3)–H arylation of cyclobutyl carboxylic acid derived amide with arylboron reagents. | ||
As discussed, asymmetric C(sp3)–H activation reactions could be achieved by employing MPAA or MPAHA ligands. However, the substrate scope was limited in early studies. In 2017, the Yu group reported the first example of Pd(II)-catalyzed enantioselective β-C(sp3)–H borylation of carboxylic acid derived 4-trifluoromethyltetrafluorophenyl amides with (Bpin)2 in the presence of a chiral acetyl-protected aminomethyloxazoline (APAO) ligand, L6. In this reaction, various substrates, including cyclopropyl, cyclobutyl and cyclohexyl acid derived amides, were coupled with (Bpin)2 to afford the desired products in good yields and enantioselectivities (Scheme 6).17 Notably, this process could provide a complementary approach to achieve enantioselective borylation of amide substrates containing α-tertiary as well as α-quaternary carbon centers.
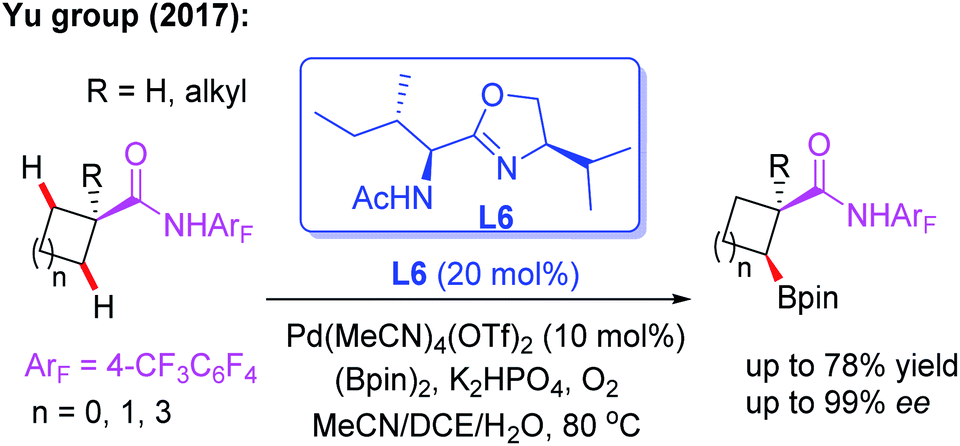 | ||
| Scheme 6 Pd(II)-catalyzed enantioselective β-C(sp3)–H borylation of carboxylic acid derived amides. | ||
Subsequently, the same group demonstrated Pd-catalyzed enantioselective arylation of the cyclobutyl carboxylic acid derived 4-trifluoromethyltetrafluorophenyl amides using the same chiral ligand L6 (Scheme 7a).18 Moreover, the first example of enantioselective C(sp3)–H vinylation of cyclobutyl carboxylic acid derived 4-trifluoromethyltetrafluorophenyl amides was developed (Scheme 7b).18 It was also found that only amide substrates containing an α-hydrogen atom were compatible in this process. Notably, compared with MPAA or MPAHA ligand promoted asymmetric C(sp3)–H activation of carboxylic acid derived amides, the use of the acidic 4-trifluoromethyltetrafluorophenyl amide as a directing group gave better results in the presence of chiral aminomethyl oxazoline ligands.
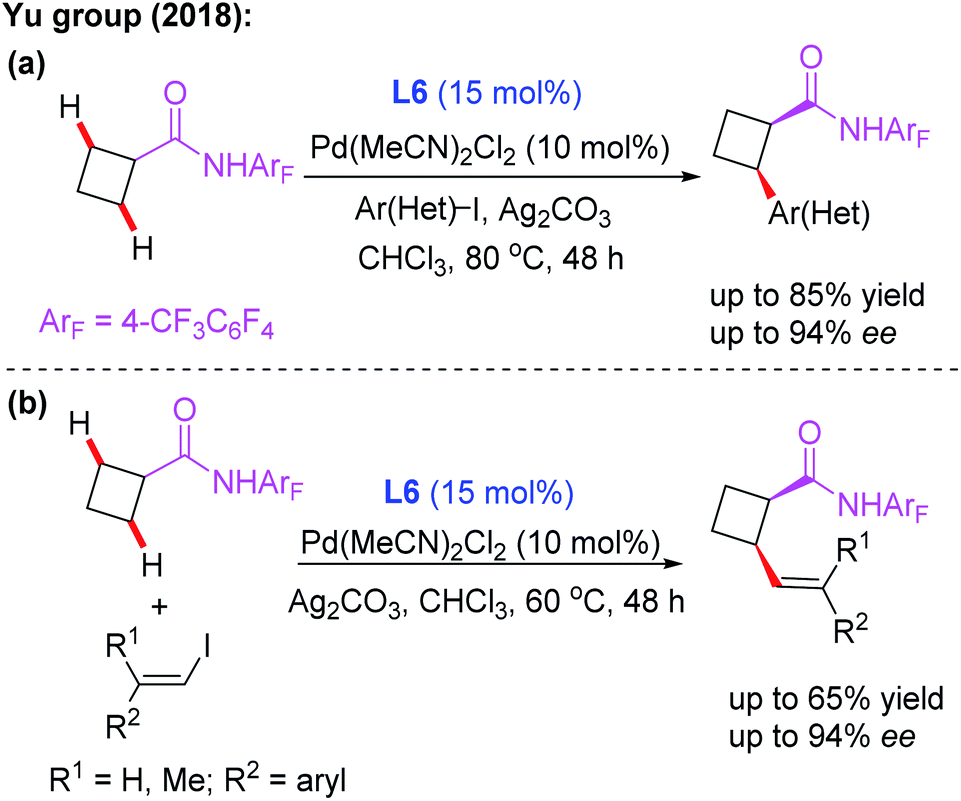 | ||
| Scheme 7 Pd(II)-catalyzed asymmetric arylation and vinylation of the cyclobutyl carboxylic acid derived amide. | ||
A plausible catalytic cycle involving APAO ligand L6-enabled Pd(II)/Pd(IV) catalysis is depicted in Scheme 8, which is different from the previously reported enantioselective Pd(II)/Pd(0) catalysis. First, the pre-coordination of MPAO ligand L6 with Pd(MeCN)2Cl2 provides the chiral Pd(II) species A. Subsequent coordination of the substrate with the chiral Pd(II) species A followed by a site-selective C–H bond activation step produces the chiral Pd(II) intermediate B. Next, oxidative addition of the intermediate B with an aryl or vinyl iodide generates the Pd(IV) complex C which undergoes reductive elimination to provide the desired product and release the chiral Pd(II) species A.
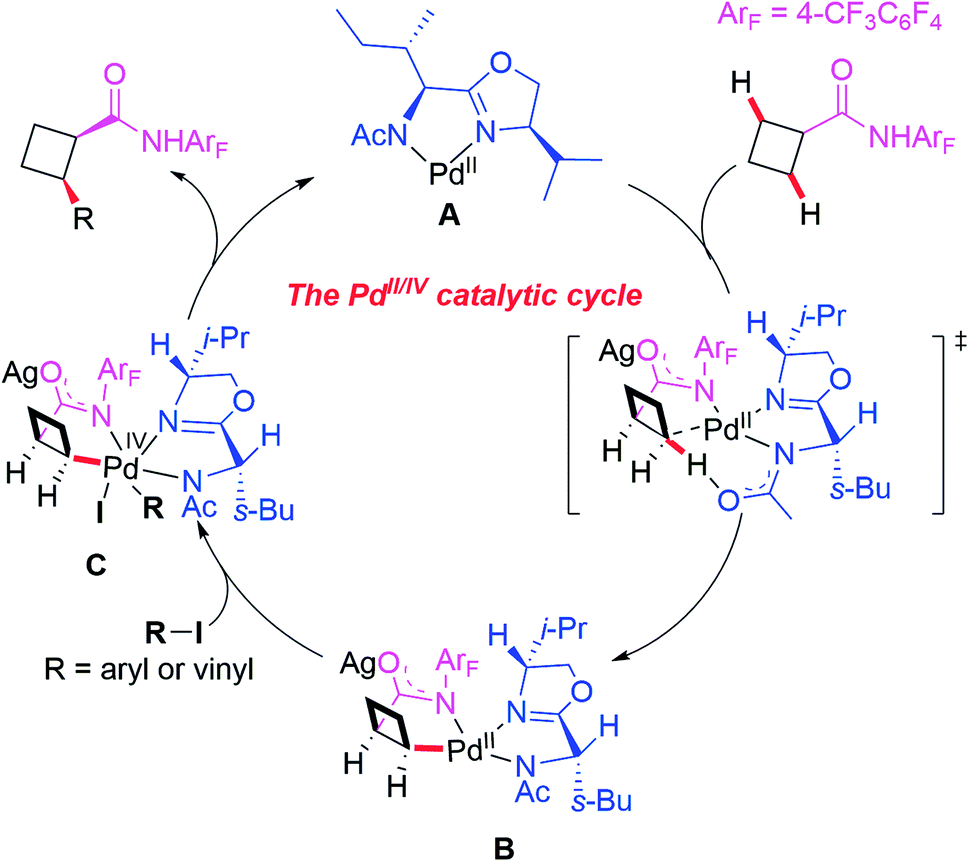 | ||
| Scheme 8 The proposed mechanism. | ||
The novel chiral ligand Boc-Leu-NHOMe L7 was found to be effective for the desymmetric C(sp3)–H activation of prochiral gem-dimethyl groups. However, only the large sterically hindered substrate (R = t-Bu) afforded the desired product in 61% yield and 80% ee (Scheme 9).16 Later, Yu and co-workers designed a chiral acetyl-protected aminomethyl oxazoline (Ac-PAO) ligand, L8, for the Pd(II)-catalyzed enantioselective β-arylation, alkenylation and alkynylation of isobutyric acid derived 4-trifluoromethyltetrafluorophenyl amides (Scheme 10a). Furthermore, in the presence of a chiral benzoyl-protected aminomethyl oxazoline (Bz-PAO) ligand, L9, desymmetrization of 2-aminoisobutyric acid derived 4-trifluoromethyltetrafluorophenyl amides has also been achieved (Scheme 10b).19 The desired α,α-dialkyl α-amino acid derivatives were isolated in good yields and enantioselectivities. Moreover, these derivatives could be used as basic fragments for the construction of peptide-based drugs. The control experiments indicated that coordination of the chiral center on the oxazoline ring with the substrate is crucial for the above reactions.
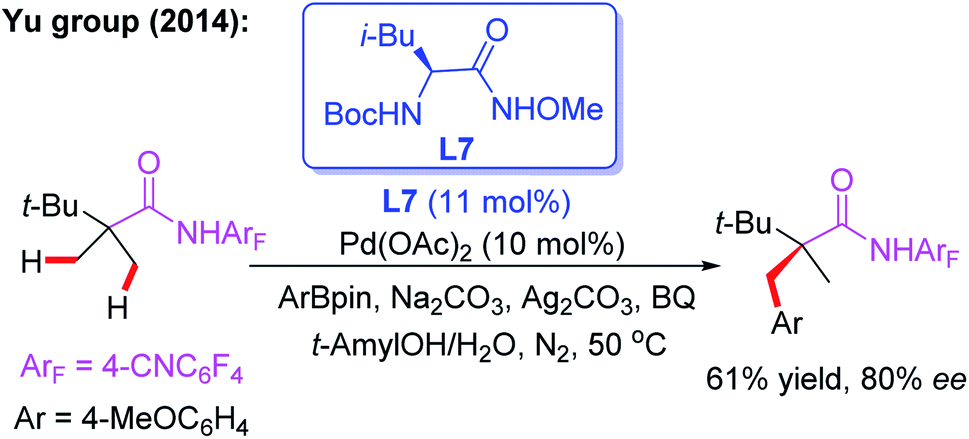 | ||
| Scheme 9 The external chiral ligand L7 for the Pd(II)-catalyzed desymmetric C(sp3)–H activation of prochiral gem-dimethyl groups. | ||
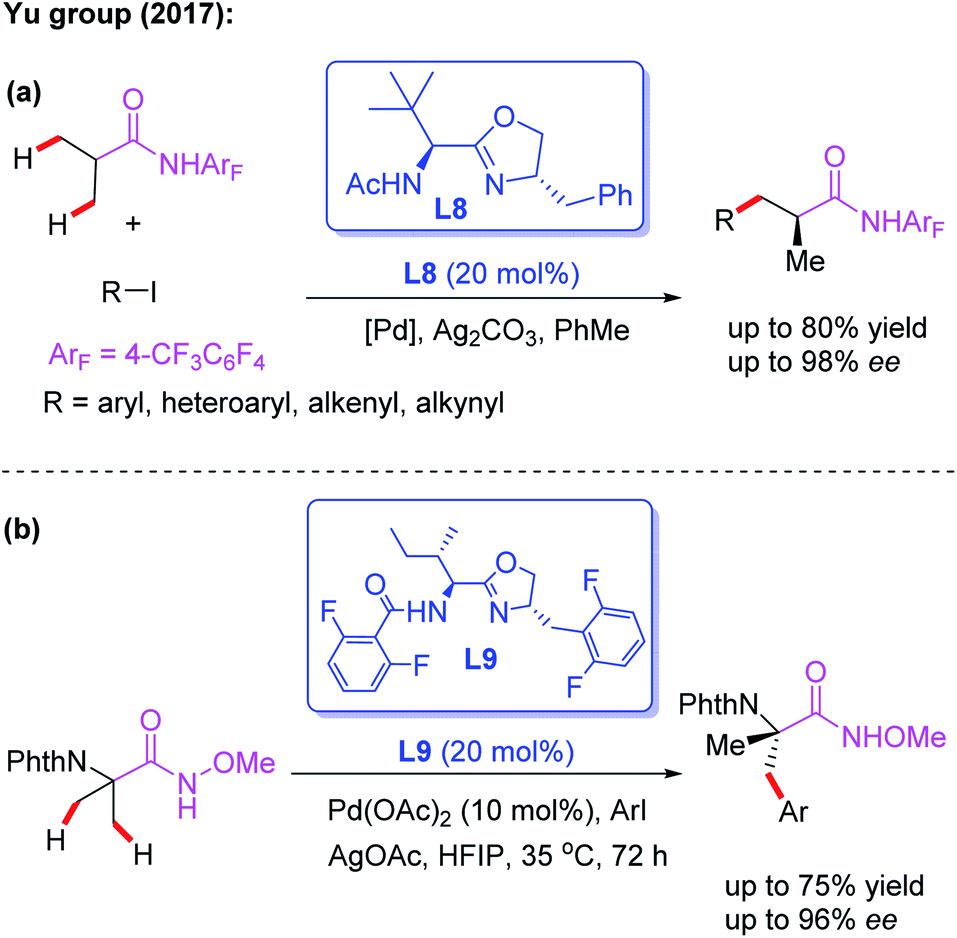 | ||
| Scheme 10 Chiral acetyl- and Bz-protected aminomethyl oxazoline ligands for the Pd(II)-catalyzed asymmetric C–H functionalization. | ||
P-Stereogenic phosphorus derivatives exhibit a prominent chiral induction. However, due to the lack of effective synthetic methods, the applications are greatly limited.20 In 2015, the Han group presented the first example of Pd(II)-catalyzed desymmetric C–H arylation of diaryl phosphinamide with boronic ester by using a chiral ligand, L10. This novel desymmetric strategy afforded a wide array of P-stereogenic phosphinamides in up to 74% yield and 98% ee. Furthermore, this process could be used to synthesize various P-stereogenic phosphorus derivatives form P-stereogenic phosphinamides (Scheme 11).21
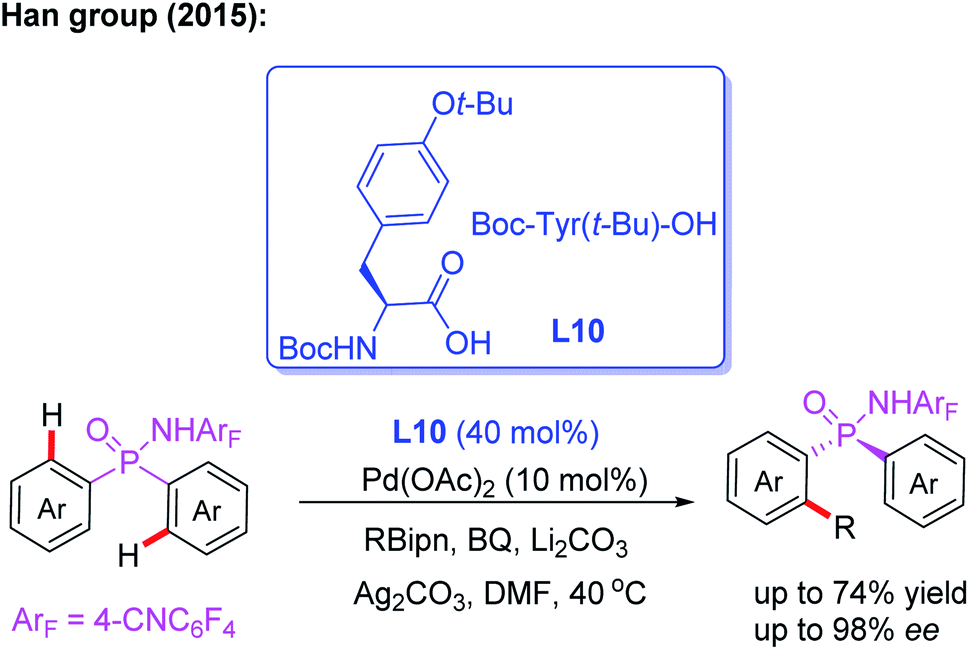 | ||
| Scheme 11 Pd(II)-catalyzed desymmetric C–H arylation of diaryl phosphinamide with boronic ester. | ||
The direct asymmetric desymmetrization of free carboxylic acids provides excellent atom and step economies, because this reaction strategy does not require additional steps to install and remove the directing groups. In 2018, Yu and co-workers designed a novel mono-protected aminoethyl amine (MPAAM) chiral ligand, L11a, and achieved the Pd-catalyzed asymmetric arylation of free cyclopropane carboxylic and 2-aminoisobutyric acids (Scheme 12a).22 It was observed that only arylation through a PdII/PdIV catalytic cycle is compatible with this chiral catalyst. Furthermore, this reaction was not compatible with cyclobutane substrates.
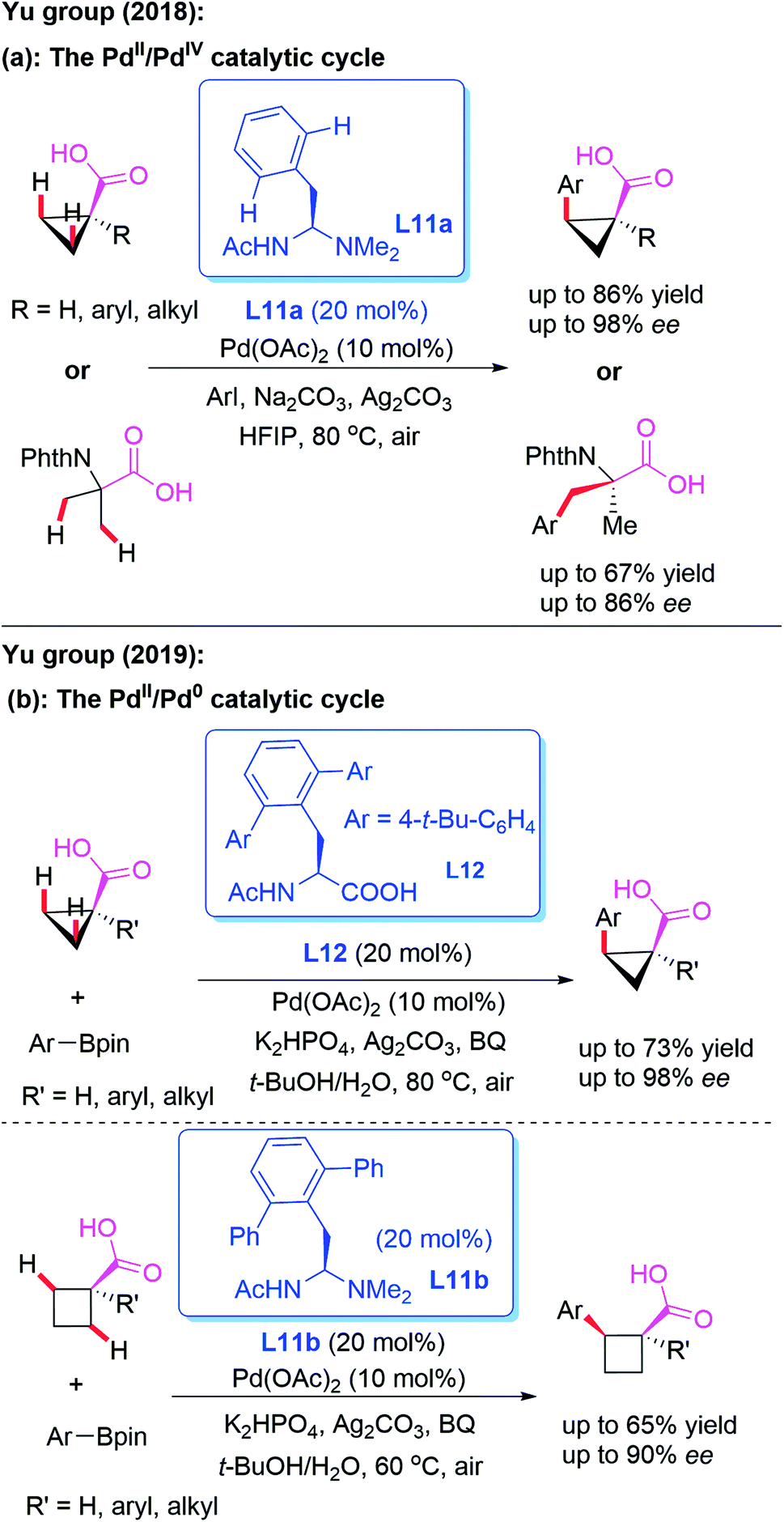 | ||
| Scheme 12 The direct asymmetric desymmetrization of free carboxylic acids catalyzed by Pd(OAc)2. | ||
Very recently, the same group revealed a novel method for the asymmetric desymmetrization of free cyclopropane and cyclobutene carboxylic acids with aryl and vinyl organoboron reagents in the presence of either mono-protected amino acid (MPAA) ligand L12 or mono-protected aminoethyl amine (MPAAM) ligand L11b through a Pd-catalyzed β-C(sp3)–H activation process (Scheme 12b).23 Notably, these reactions could also access the desired chiral arylated acids containing α-tertiary and α-quaternary carbon centers. It is proposed that a PdII/Pd0 catalytic cycle might be involved in this process.
Amine derivatives are an important class of synthetically useful compounds in organic chemistry.24 The first example of asymmetric C–H iodination of Tf-protected diarylmethylamines by using a chiral MPAA ligand, L13, at ambient temperatures was reported by Yu and co-workers in 2013 (Scheme 13).25 In this process, inexpensive I2 was used both as the sole oxidant and reaction reagent. In addition, they developed the Pd-catalyzed asymmetric C–H arylation of Tf-protected cyclopropyl methylamines with aryl iodides in the presence of a chiral ligand, L14 (Boc-L-Val-OH), via a PdII/PdIV catalytic cycle (Scheme 14). Chiral cis-aryl-products were obtained in excellent yields (up to 99%) and enantiomeric excesses (up to 99.5% ee).26
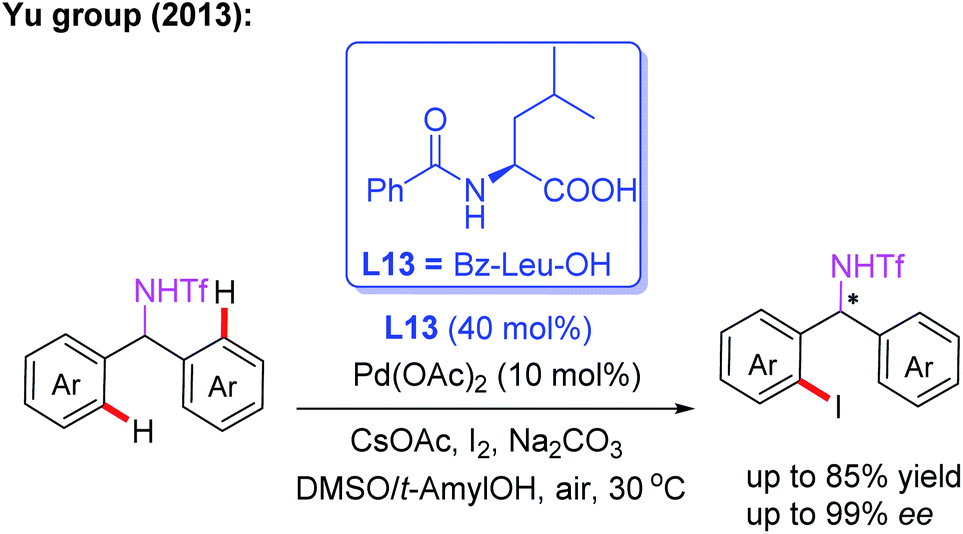 | ||
| Scheme 13 MPAA-enabled Pd(II)-catalyzed enantioselective C(sp2)–H iodination of Tf-protected diarylmethylamines. | ||
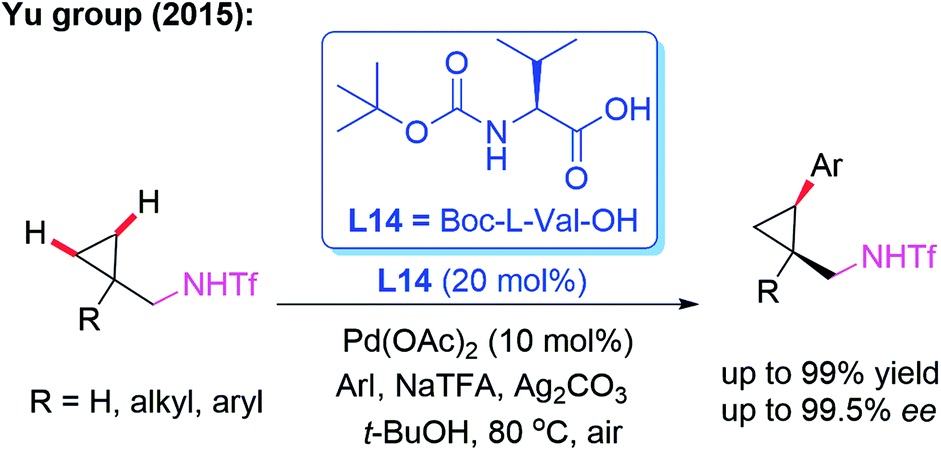 | ||
| Scheme 14 Pd(II)-catalyzed highly enantioselective C–H arylation of Tf-protected cyclopropylmethylamines. | ||
In 2016, Yu and co-workers reported the asymmetric desymmetrization of amines through a Pd(II)-catalyzed α-C(sp3)–H arylation process in the presence of a chiral phosphoric acid ligand, L15 (Scheme 15).27 In this reaction, various amines, including ethyl amines, pyrrolidines, azetidines, piperidines, azepanes, indolines, and tetrahydroisoquinolines, were well coupled with aryl boronic acids to construct chiral aryl-amines in excellent yields and enantioselectivities. It is worth noting that chiral phosphoric acids were demonstrated to be effective anionic ligands for this asymmetric C–H bond activation. Very recently, the same group reported the asymmetric desymmetrization of Tf-protected alkyl amines through the Pd(II)-catalyzed enantioselective γ-C–(sp3)–H arylation and vinylation by employing the chiral acetyl-protected aminomethyl oxazoline (APAO) ligands L8 and L16 (Scheme 16).26
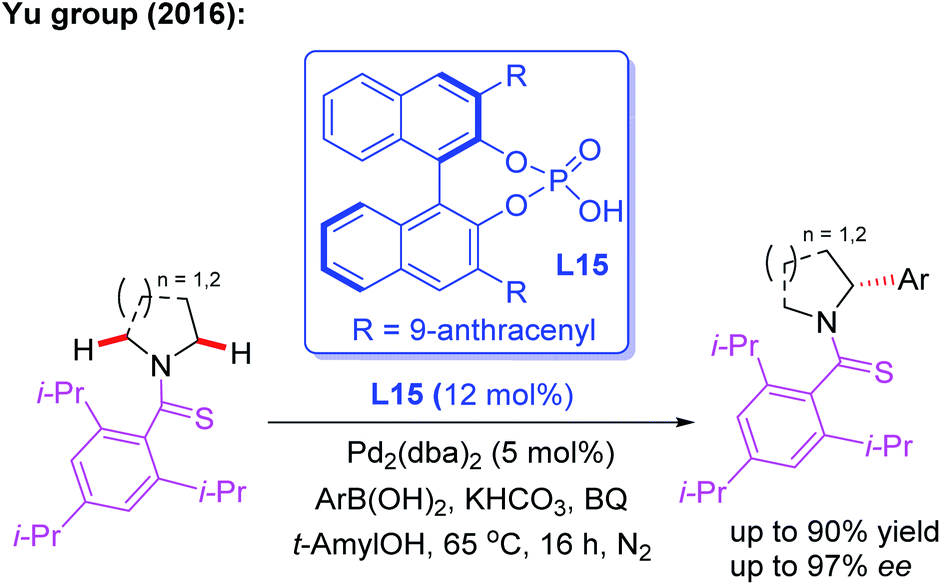 | ||
| Scheme 15 The asymmetric desymmetrization of amines through a Pd(II)-catalyzed α-C(sp3)–H arylation reaction. | ||
 | ||
| Scheme 16 The asymmetric desymmetrization of Tf-protected alkyl amines through the Pd(II)-catalyzed enantioselective γ–C–(sp3)–H arylation and vinylation. | ||
While the asymmetric desymmetrization approach is limited to substrates with two prochiral C–H bonds, the kinetic resolution approach is conceptually different, requiring a different reaction rate between the chiral catalyst and each enantiomer in the racemic mixture.11
In 2016, the Yu group developed a kinetic resolution strategy for the Pd(II)-catalyzed asymmetric C–H olefination of racemic α-hydroxy and amino phenylacetic acids. Employing (S)-MPAA (L17) as the chiral ligand, the enantio-enriched products were obtained in up to 92% ee. Moreover, the recovered starting material could be converted to the other enantiomer in excellent yield and enantioselectivity through Pd-catalyzed C–H olefination using an (R)-MPAA ligand L18 (Scheme 17).29
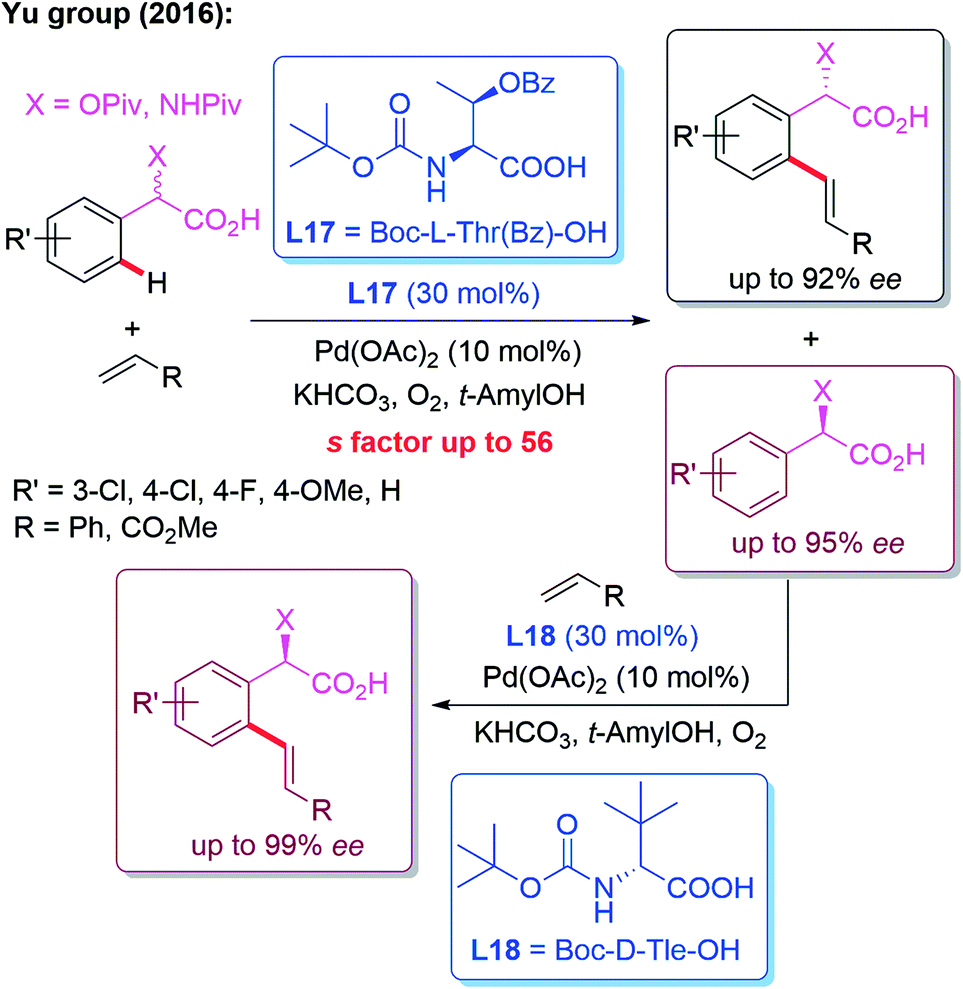 | ||
| Scheme 17 A kinetic resolution strategy for the Pd(II)-catalyzed asymmetric C–H olefination of racemic α-hydroxy and amino phenylacetic acids. | ||
Meanwhile, the Pd(II)-catalyzed asymmetric C–H arylation of Ns-protected benzylamines in the presence of a chiral mono-N-protected α-amino-O-methylhydroxamic acid ligand, L19, has also been achieved via a kinetic resolution process. In this reaction, it is essential to use the easy-to-remove nosyl (Ns) protected amino group as the directing group, and both chiral benzylamines and ortho-arylated benzylamines were isolated in excellent enantioselectivities (Scheme 18).30 Very recently, they developed a chiral ligand, L20, for asymmetric γ-C(sp3)–H activation of alkyl amines with ArBpin through a kinetic resolution process (Scheme 19).28
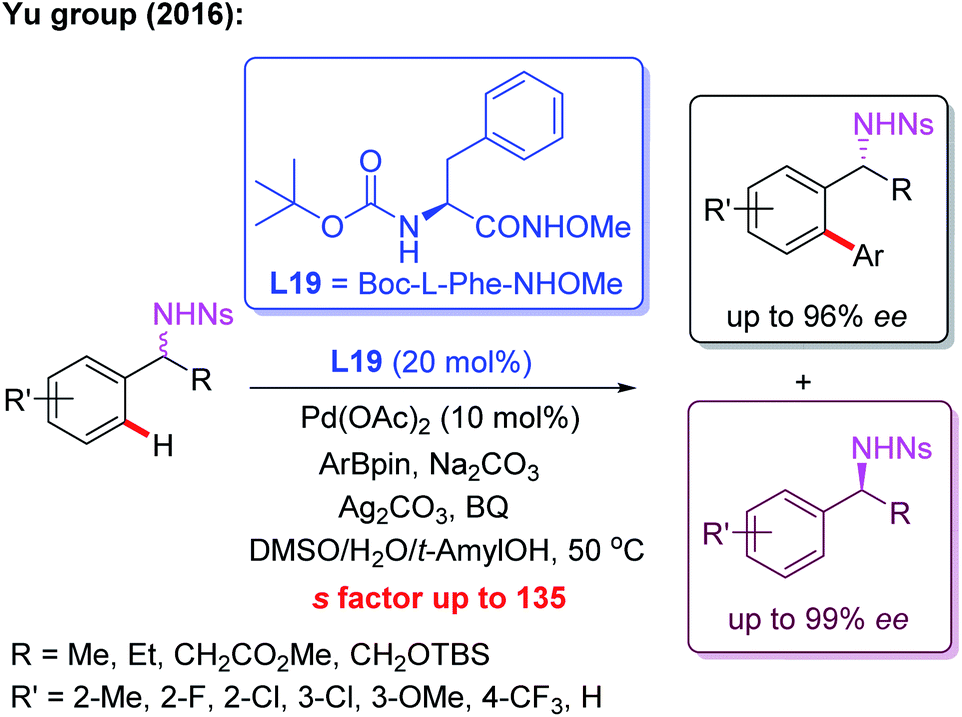 | ||
| Scheme 18 Kinetic Resolution of Ns-protected benzylamines via a Pd(II)-catalyzed asymmetric C–H arylation. | ||
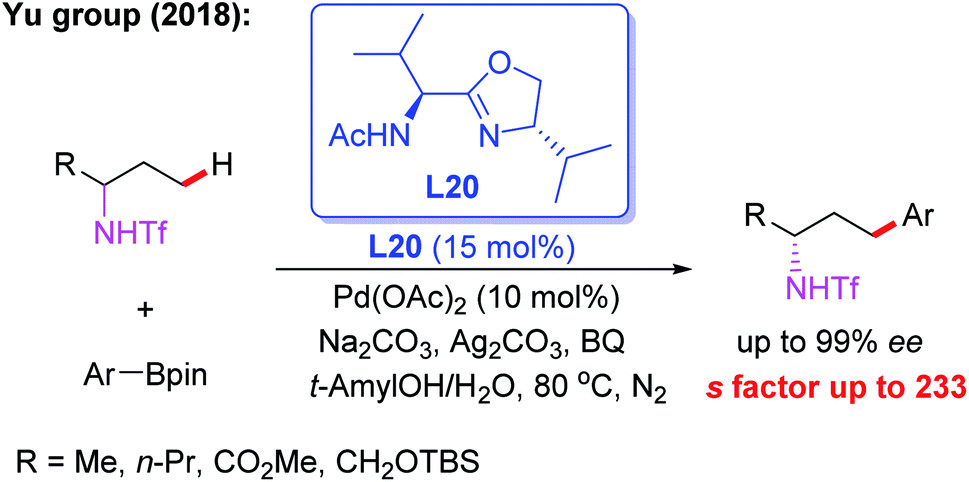 | ||
| Scheme 19 Pd(II)-catalyzed asymmetric γ-C(sp3)–H activation of alkyl amines with ArBpin through a kinetic resolution process. | ||
Axially chiral biaryls are widely present in natural products as important skeleton.31 Moreover, they can also be used as chiral organic catalysts or chiral ligands in asymmetric synthesis.32 The Murai group reported the first example of asymmetric C(sp2)–H alkylation of naphthylpyridine derivatives employing a chiral ferrocenyl phosphine ligand through the Rh(I)-catalyzed dynamic kinetic resolution process; however, only low yields and enantioselectivities were obtained.33
The first example of the Pd-catalyzed atroposelective C–H iodination reaction through a kinetic resolution process was developed by the You group in 2014. In this reaction, N-monoprotected phenylalanine (L21), as the most effective chiral ligand, was used to construct axially chiral biaryls in good yields and enantioselectivities (Scheme 20).34
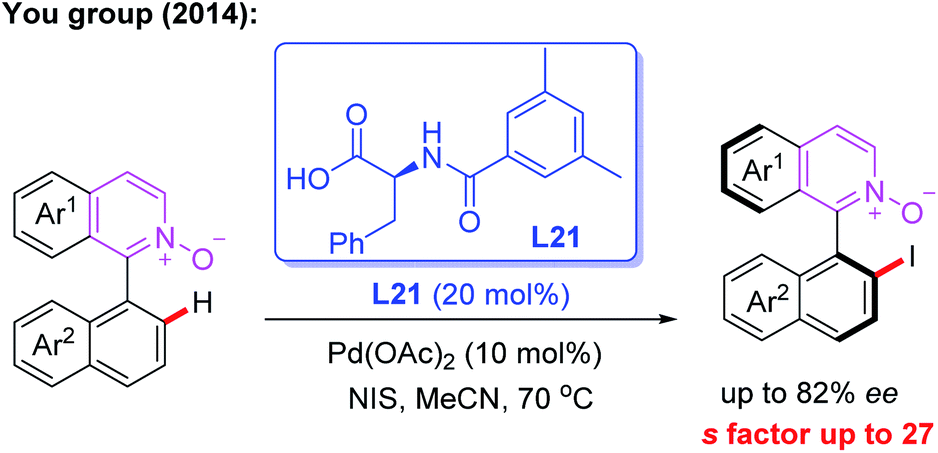 | ||
| Scheme 20 Pd(II)-catalyzed atroposelective C–H iodination reaction through a kinetic resolution process. | ||
Moreover, the Yang group reported a Pd-catalyzed atroposelective C–H olefination for the construction of axially chiral biaryls in 2017 (Scheme 21). Using P(O)Ph2 as the directing group and Boc-L-Val-OH (L14) as the chiral ligand, the racemic biaryl phosphine oxides were transformed into the desired chiral biaryl phosphine–olefin products in excellent yields (up to 99%) and enantioselectivities (up to 96% ee). In addition, both electron-donating and electron-withdrawing substituents on the aromatic rings were well tolerated in this catalytic system.35
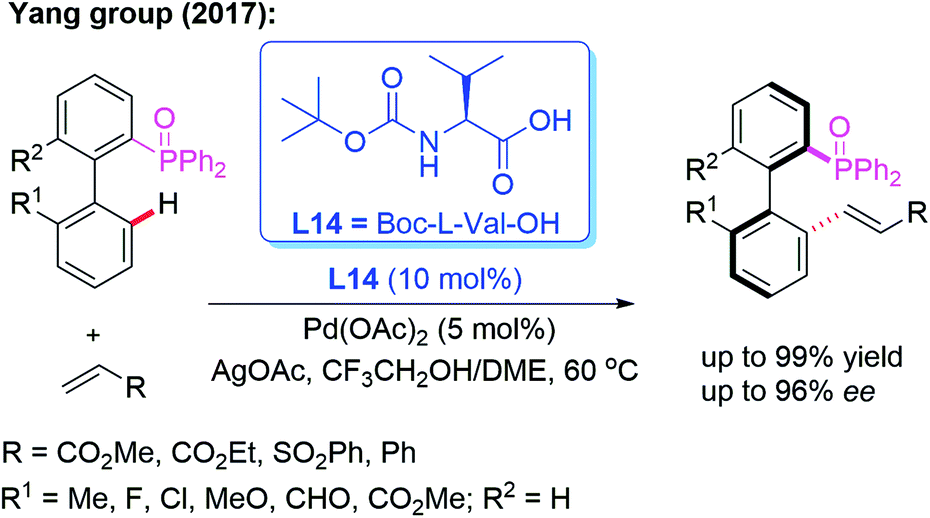 | ||
| Scheme 21 Pd(II)-catalyzed atroposelective C–H olefination for the construction of axially chiral biaryls. | ||
Very recently, Pd-catalyzed atroposelective C–H olefination for the synthesis of axially chiral biaryls by using a novel chiral spiro phosphoric acid ligand, L22, was developed by Shi and co-workers (Scheme 22a).36 Various axially chiral quinolines were isolated in excellent yields (up to 99% yields) and enantioselectivities (up to 98% ee). Subsequently, they reported an NH2-directed Pd-catalyzed atroposelective C–H olefination for the preparation of axially chiral biaryl-2-amines (Scheme 22b).37 In this strategy, chiral spiro phosphoric acid L23 was used as an efficient ligand to access a broad range of axially chiral biaryl-2-amines in excellent yields (up to 91%) and enantioselectivities (up to 97% ee). During the study, they also developed a novel approach to construct axially chiral styrenes via Pd(II)-catalyzed atroposelective C–H alkenylation and alkynylation by employing L-pyroglutamic acid L24 as a chiral ligand (Scheme 23).38
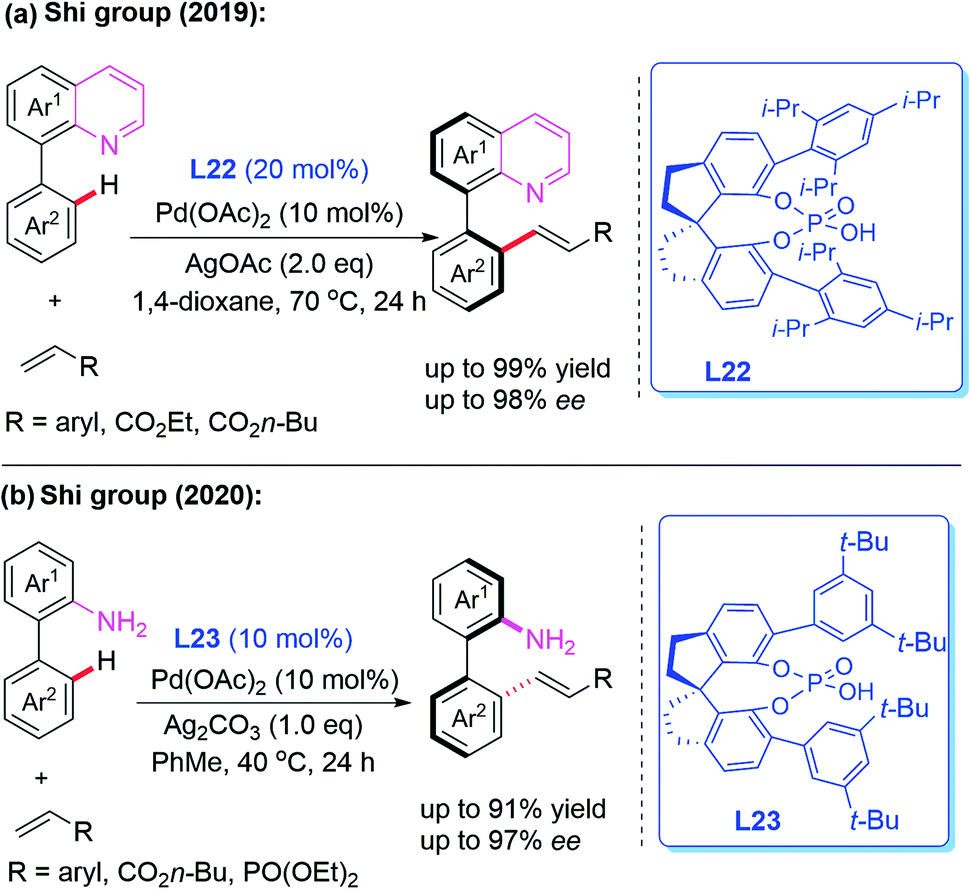 | ||
| Scheme 22 Pd(II)-catalyzed atroposelective C–H olefination for the synthesis of axially chiral biaryls. | ||
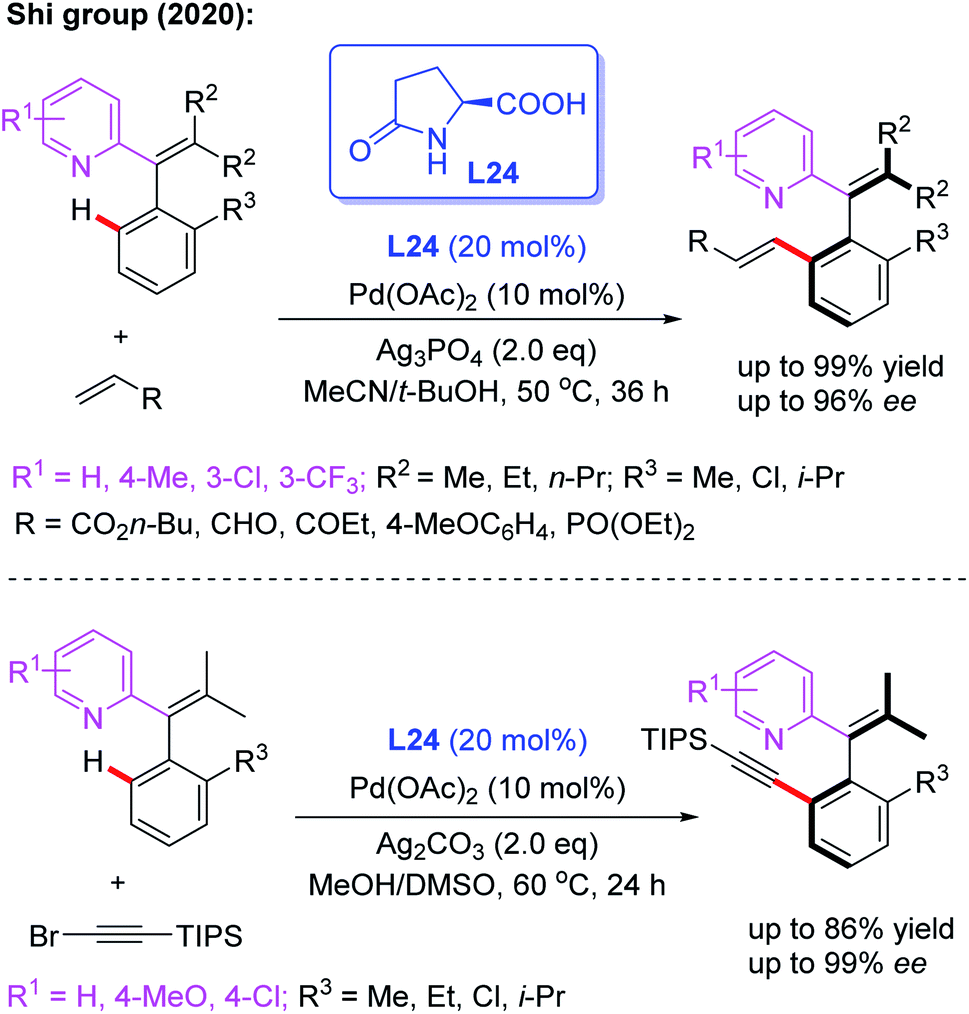 | ||
| Scheme 23 Pd(II)-catalyzed atroposelective C–H olefination for the synthesis of axially chiral biaryls. | ||
Ferrocenes bearing planar chirality have received much attention due to their application in asymmetric catalysis as efficient ligands or catalysts.39 In the field of Pd-catalyzed asymmetric C–H bond functionalization, the Yu group developed a series of asymmetric reactions by using a catalytic amount of chiral monoprotected amino acids.8 Inspired by Yu's work on asymmetric C–H bond functionalization reactions, the You group realized the first example of Pd-catalyzed asymmetric C–H arylation of N,N-disubstituted dialkylaminomethyl ferrocenes in 2013 by using Boc-L-Val-OH (L14) as the chiral ligand and arylboronic acids as coupling reagents (Scheme 24a).40 The desired chiral products were obtained in good yields and excellent enantioselectivities under mild reaction conditions. Notably, with methylboronic acid as the coupling partner, the yield of the desired product was dramatically decreased (14%) under even a higher temperature and prolonged reaction time.
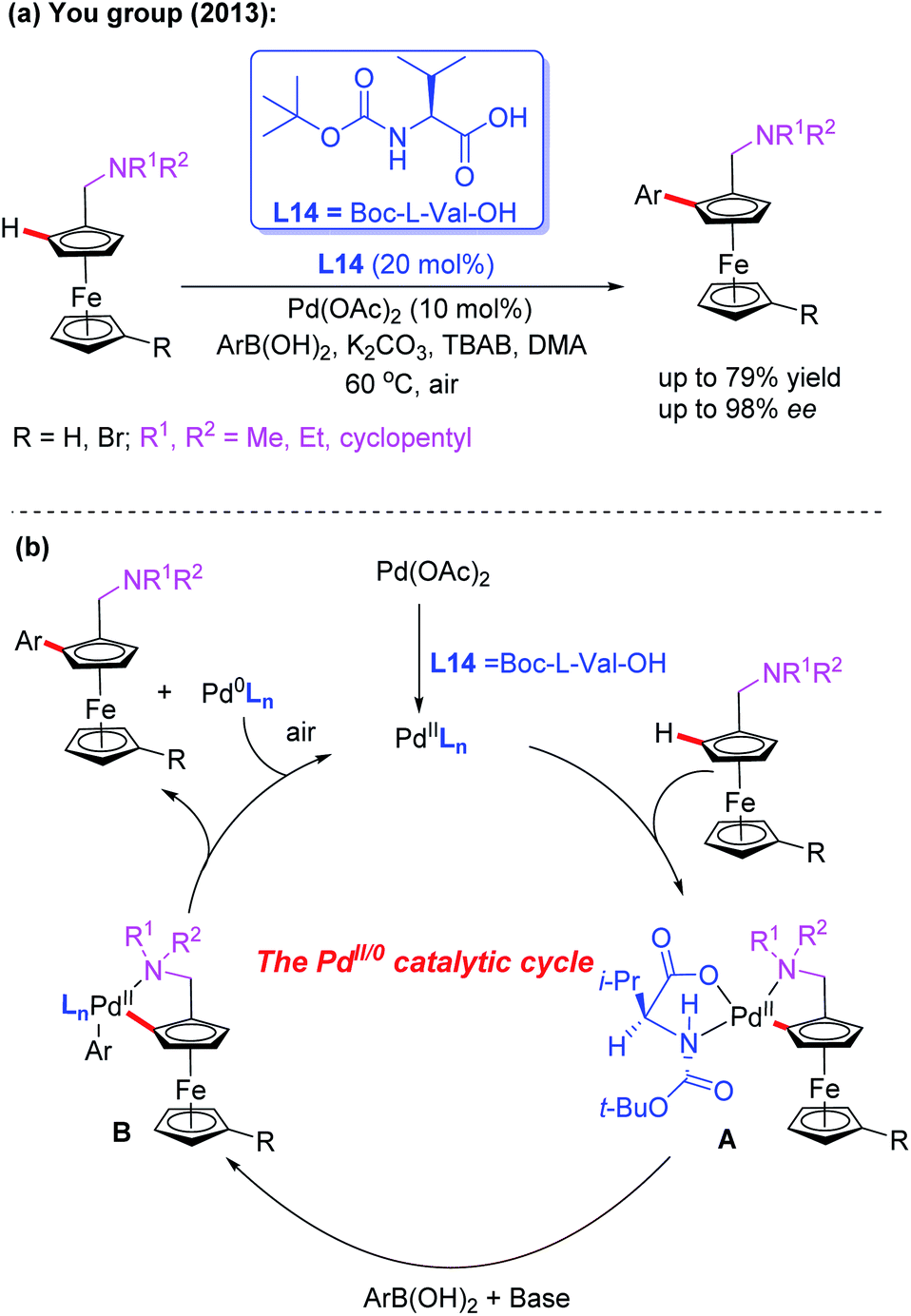 | ||
| Scheme 24 Pd(II)-catalyzed asymmetric C–H arylation of N,N-disubstituted dialkylaminomethyl ferrocenes. | ||
The plausible catalytic cycle involving a Pd(II)/Pd(0) catalysis is depicted in Scheme 24b. The selective C–H bond cleavage of ferrocene with Boc-L-Val-OH and the Pd catalyst provides the cyclic Pd(II) intermediate A. Subsequent transmetalation between intermediate A and aryl boronic acid forms the intermediate B. The reductive elimination of the intermediate B affords the desired chiral product.
Additionally, they developed another efficient method for the synthesis of planar chiral arylated-ferrocenes through the Pd(II)-catalyzed asymmetric oxidative annulation of N,N-disubstituted dialkylaminomethyl ferrocenes with diphenylacetylene by using a chiral ligand, L24 (Scheme 25a).41 The reaction mechanism was also proposed as shown in Scheme 25b. First, the cyclic Pd(II) intermediate A is generated through coordination of the N-atom of the ferrocene derivative with the chiral Pd(II) catalyst followed by site-selective C–H palladation. Next, syn-insertion of diphenylacetylene into intermediate A gives intermediate B, which can be further transformed into its trans-isomer B′. Subsequently, the second diphenylacetylene molecule is inserted, affording intermediate C. Finally, the desired product is formed through a sequential intramolecular 5-exo-dig insertion, migration and reductive elimination process. Moreover, You and coworkers utilized a Pd (OAc)2/Boc-L-Ile-OH (L25) catalytic system to achieve the highly efficient asymmetric oxidative cross-coupling reaction between ferrocenes and electron-rich heteroarenes in 2016 (Scheme 26).42
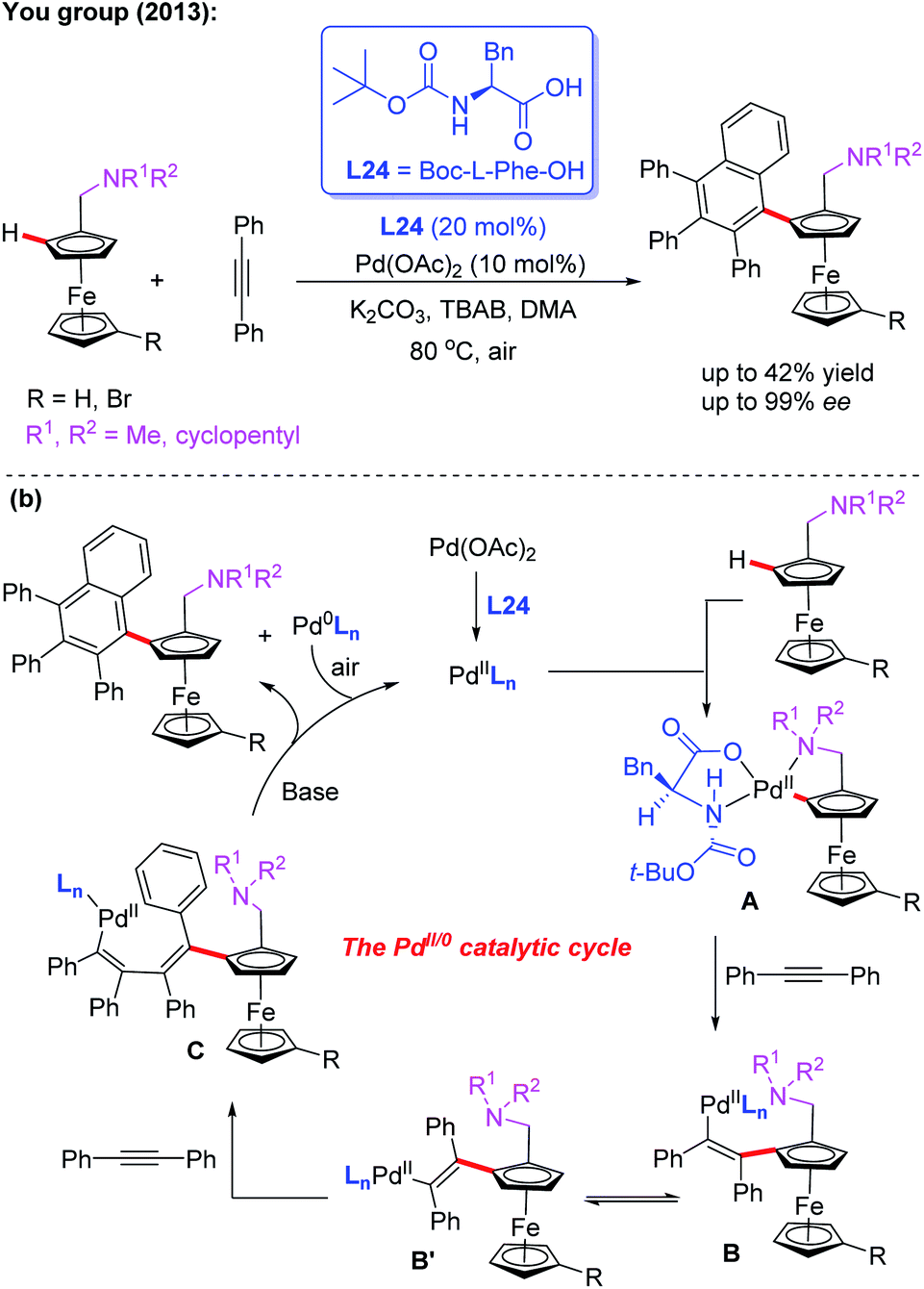 | ||
| Scheme 25 Pd(II)-catalyzed asymmetric oxidative annulation of N,N-disubstituted dialkylaminomethyl ferrocenes with diphenylacetylene. | ||
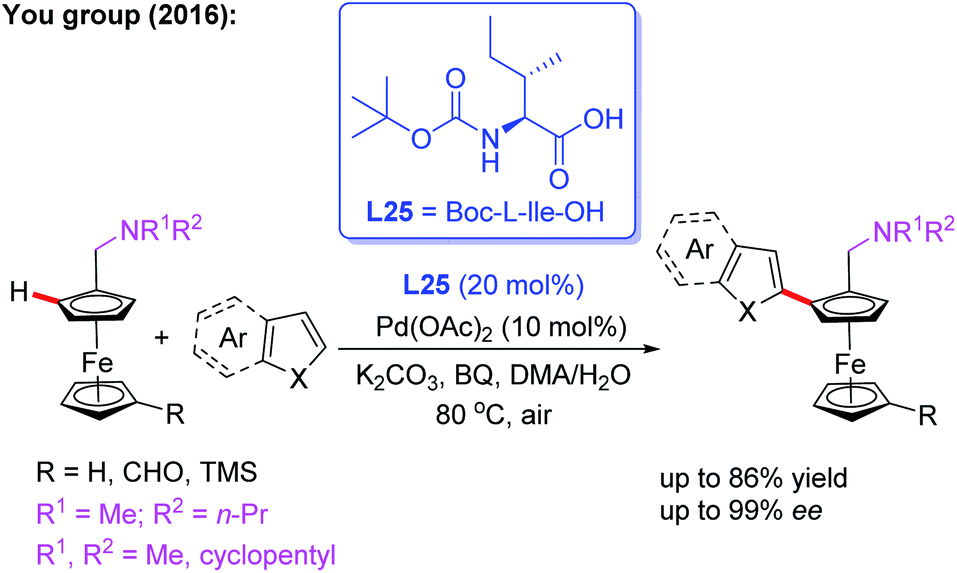 | ||
| Scheme 26 Pd(II)-catalyzed asymmetric oxidative cross-coupling reaction between ferrocenes and heteroarenes. | ||
In 2013, the Pd(II)-catalyzed asymmetric oxidative Heck reaction for the efficient synthesis of planar chiral ferrocenes in the presence of a chiral ligand, L24, was also developed by the Cui and Wu group (Scheme 27a).43 In this process, a variety of olefins, such as acrylates, substituted styrenes, vinyl cyclohexanes, and acrylamides, were used to prepare the alkenylation products in excellent yields and enantioselectivities.
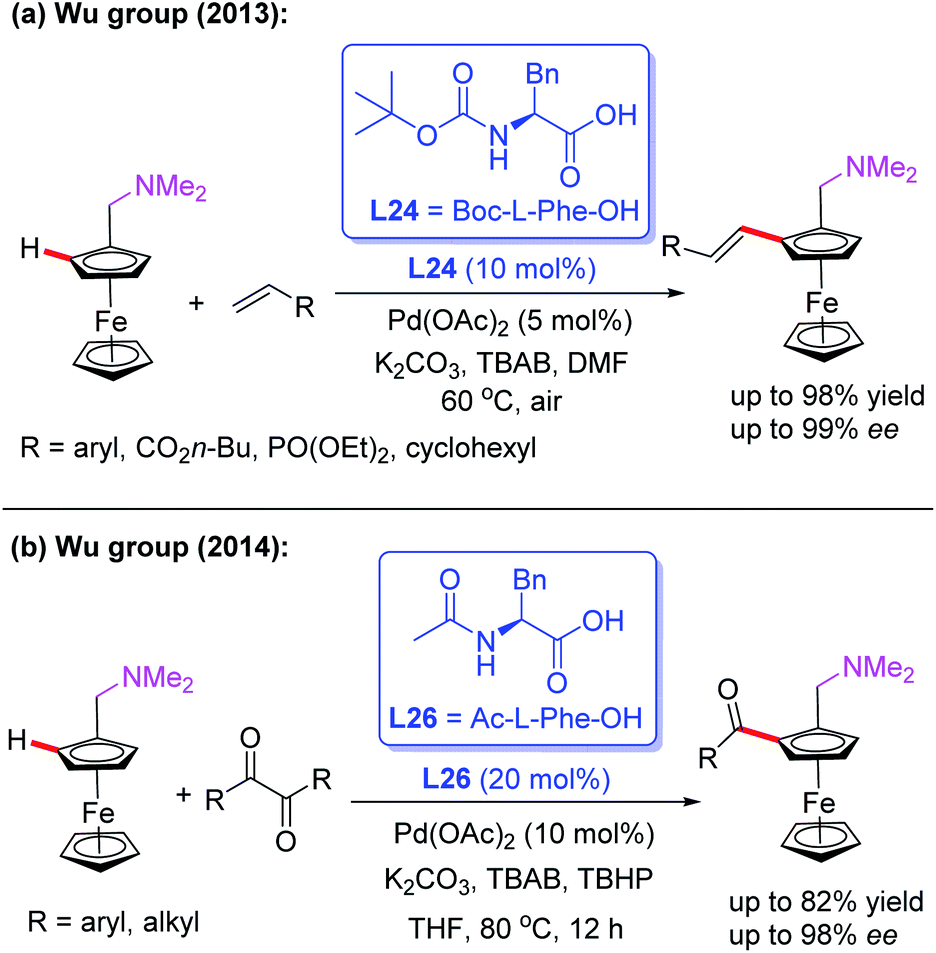 | ||
| Scheme 27 Pd(II)-catalyzed asymmetric oxidative Heck and acylation reaction. | ||
Later, they demonstrated a novel catalytic asymmetric C–H acylation reaction in the presence of a Pd(OAc)2 catalyst and Ac-L-Phe-OH (L26) (Scheme 27b).44 Diaryldiketones bearing either an electron-withdrawing or electron-donating group were well suited for this strategy and produced various planar chiral ferrocenes in excellent yields and enantioselectivities. During the investigation of the reaction mechanism, they found that the radical scavenger TEMPO inhibited the process. Thus, a radical mechanistic pathway is proposed in Scheme 28. The initiated selective C–H bond cleavage of N,N-dimethyl aminomethyl ferrocene in the presence of the Pd(II) catalyst and Ac-L-Phe-OH provides the cyclopalladated intermediate A. Meanwhile, the reaction of diaryldiketone with TBHP also provides radical B. The subsequent radical addition reaction of species B to intermediate A generates the Pd (III or VI) intermediate C. The desired chiral acylated product was then produced via a reductive elimination process.
 | ||
| Scheme 28 The proposed mechanistic pathway. | ||
As discussed above, significant progress has been made on monodentate directing group assisted asymmetric C–H functionalization using chiral amino acids or aminomethyl heterocycle ligands. However, only a few applicable direct enantioselective functionalization reactions of linear methylene C(sp3)–H bonds have been reported. In 2016, the direct Pd-catalyzed enantioselective arylation of a linear methylene C(sp3)–H bond was achieved by the Yu group (Scheme 29).45 The combination of a weakly coordinating 4-trifluoromethyltetrafluorophenyl amide directing group with a chiral acetyl-protected aminoethyl quinolone (APAQ) ligand, L27, proved to be crucial and produced the desired chiral products in good yields (up to 89% yield) and enantioselectivity (up to 92% ee). Moreover, density functional theory (DFT) studies on the observed enantioselectivity in the CMD step were carried out and the results indicated that TS-R is more favorable than TS-S.
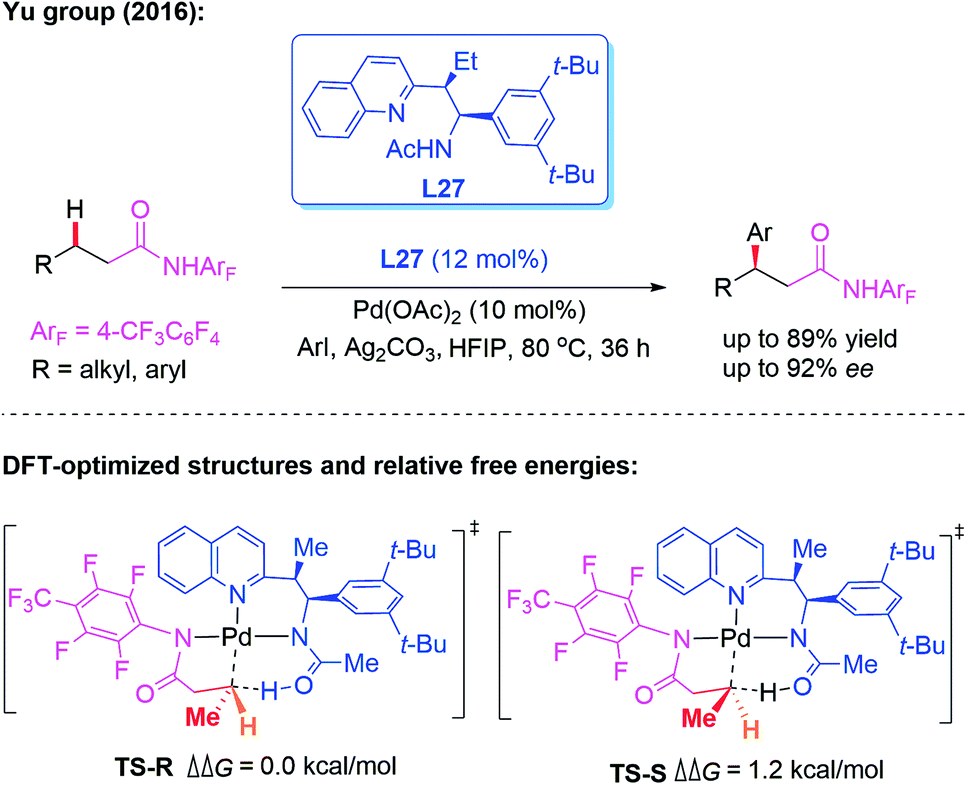 | ||
| Scheme 29 Pd(II)-catalyzed enantioselective arylation of the linear methylene C(sp3)–H bond activation. | ||
3. Bidentate directing group-enabled asymmetric C–H functionalization
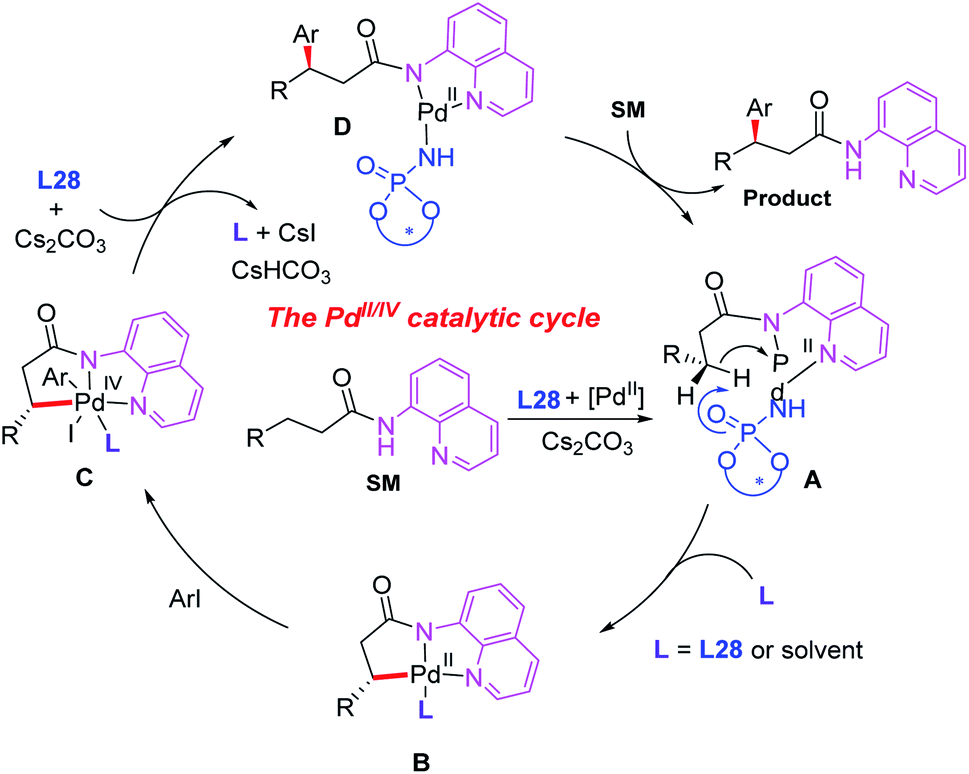
Since N,N-bidentate aminoquinoline (8-aminoquinoline) was first introduced by the Daugulis group, a wide range of transition metal-catalyzed C(sp3)–H functionalization reactions have been developed in recent years.46 A bidentate directing group often exhibits higher reactivity than a monodentate directing group. However, it is much more difficult to exert chiral induction because there is only a single available coordination site on the metal center.In 2015, the first example of Pd(II)-catalyzed 8-aminoquinoline (AQ)-assisted enantioselective methylene C(sp3)–H arylation of 3-substituted propanamides was reported by Duan and co-workers employing a chiral BINOL phosphoramide ligand, L28. An array of chiral β,β-diaryl carboxylic acid derivatives were generated in moderate to good enantioselectivities. The mechanistic studies indicated that the chiral phosphoric amide L28 could accelerate the reaction rate and control the reaction enantioselectivity (Scheme 30).47
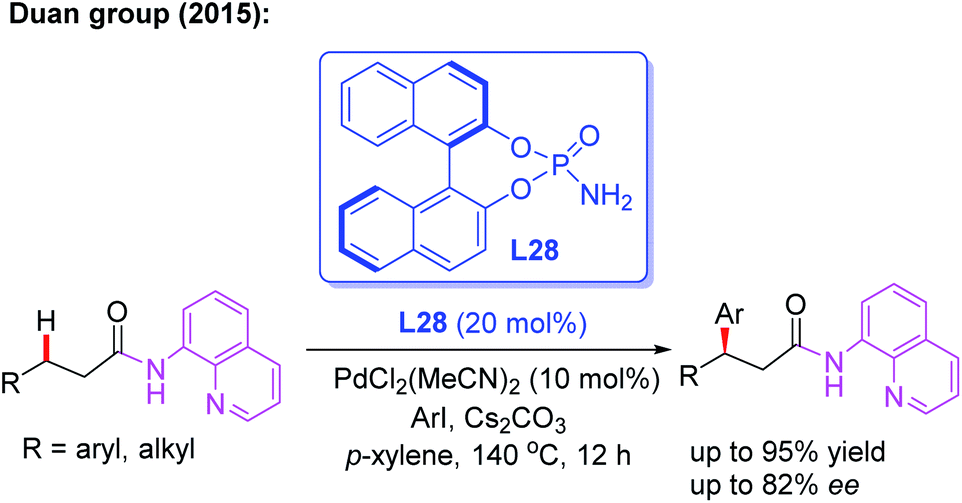 | ||
| Scheme 30 Pd(II)-catalyzed and 8-aminoquinoline-assisted enantioselective methylene C(sp3)–H arylation of 3-substituted propanamides. | ||
A plausible catalytic cycle is proposed in Scheme 31. The ligand exchange of the Pd(II) catalyst with 3-substituted propanamide in the presence of a chiral phosphoric amide, L28, and the base Cs2CO3 provides the cyclic Pd(II) intermediate A. Diastereoselective cyclometalation of this intermediate gives rise to a [5,5]-bicyclic Pd(II) intermediate Bvia a site-selective C–H bond activation process. Oxidative addition of intermediate B with an aryl iodide generates the Pd(IV) species C. Subsequent reductive elimination of the palladium complex C produces intermediate D, which produces the desired product via ligand exchange with 3-substituted propanamide.
 | ||
| Scheme 31 A plausible catalytic cycle. | ||
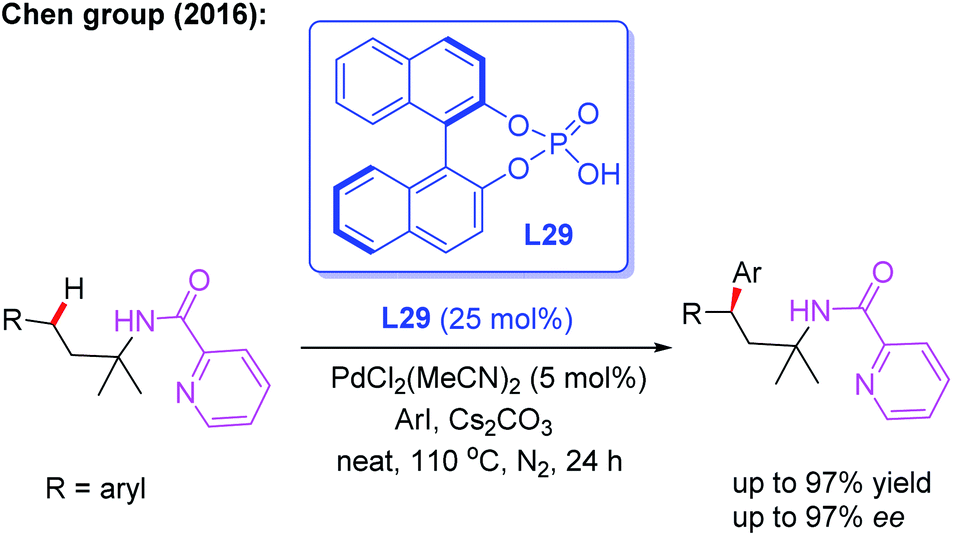
In 2016, Chen and co-workers illustrated picolinamide (PA)-directed Pd(II)-catalyzed enantioselective γ-C(sp3)–H arylation of 3-arylpropylamines with aryl iodides using the BINOL phosphoric acid ligand L29 (Scheme 32).48 For the first time, the chiral β,β-diaryl-3-propylamines were achieved in excellent yields (up to 97%) and enantioselectivities (up to 97% ee). Mechanistic studies indicated that combination of the BINOL phosphoric acid ligand L29 and Cs2CO3 in the absence of solvents gave the highly optically enriched products.
 | ||
| Scheme 32 PA-directed and Pd(II)-catalyzed enantioselective γ-C(sp3)–H arylation of 3-arylpropylamines. | ||
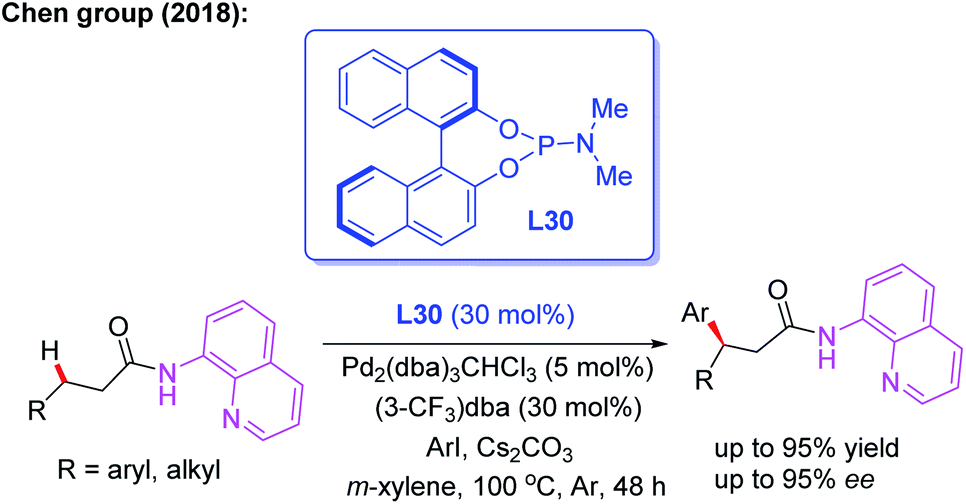
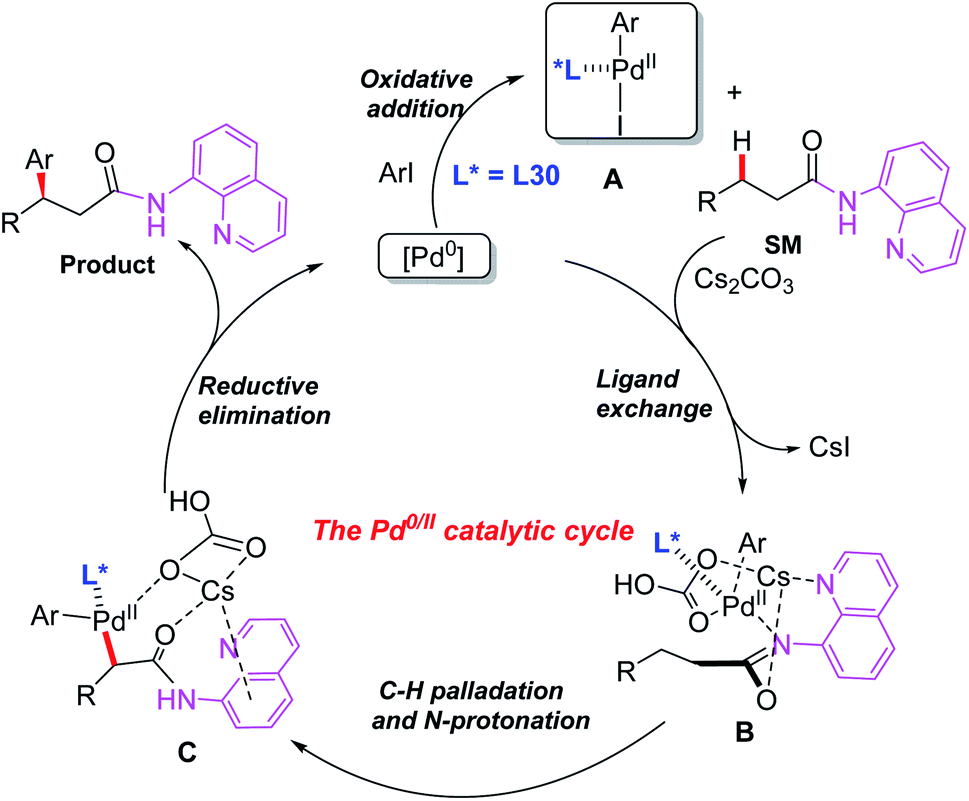
Later, the same group introduced a novel protocol for AQ-mediated Pd(0)-catalyzed enantioselective β-C(sp3)–H arylation of 3-substituted propanamides in the presence of the BINOL phosphoramidite ligand L30 (Scheme 33).49 Compared with Duan's work, this is the first example of AQ-directed processes involving a Pd(0/II) catalytic cycle, providing the chiral desired products in good yields and enantioselectivities (up to 95% ee). Furthermore, the control reactions and DFT calculations showed that both the BINOL phosphoramidite ligand L30 and Cs2CO3 were involved in the enantio-determining C(sp3)–H palladation step. Additionally, a plausible Pd(0/II) catalytic cycle was proposed (Scheme 34). Oxidation of the Pd(0) catalyst with ArI in the presence of the chiral phosphoric amide L30 produces the Pd(II) intermediate A. Subsequently, intermediate B is formed through a ligand exchange process of intermediate A with the AQ-coupled substrate in the presence of Cs2CO3. The diastereoselective C–H palladation and protonation affords the palladium intermediate C which provides the desired product upon reductive elimination.
 | ||
| Scheme 33 AQ-mediated and Pd(0)-catalyzed enantioselective β-C(sp3)–H arylation of 3-arylpropanamides. | ||
 | ||
| Scheme 34 A plausible Pd(0/II) catalytic cycle. | ||
Very recently, the Chen group reported a Pd(II)-catalyzed enantioselective intramolecular β-C(sp3)–H amidation reaction of 3-substituted propanamides for the synthesis of chiral β-aryl-β-lactams. With this novel method, the desired products could be obtained in up to 94% ee in the presence of the chiral 3,3′-F2-BINOL ligand L31. The control experiments suggested that 2-methoxy-5-chlorophenyl iodide, as the critical oxidant, controls the competing C–C versus C–N reductive elimination pathway of the Pd(IV) intermediate due to its steric and electronic effect (Scheme 35).50
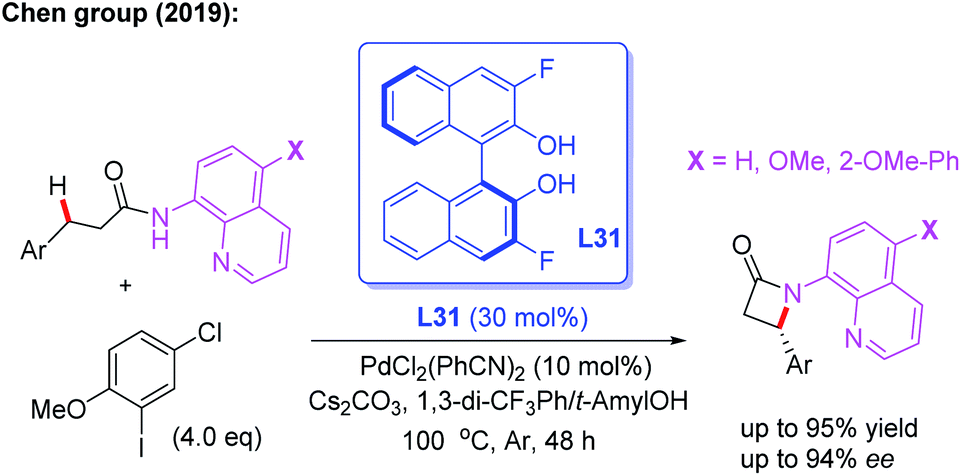 | ||
| Scheme 35 Pd(II)-catalyzed enantioselective intramolecular β-C(sp3)–H amidation of 3-substituted propanamides. | ||
The bidentate directing group, 2-(pyridine-yl)isopropyl amine (PIP), was originally designed by the Shi group for the activation of the methylene C(sp3)–H bond.51 Very recently, the group developed the first example of Pd(II)-catalyzed enantioselective functionalization reactions of linear methylene C(sp3)–H bonds by using the cooperative effects between the PIP group and chiral phosphoric acid ligand L32 (Scheme 36a).52 With aryl bromides as the less reactive arylating reagents, a variety of 3-alkylpropanamides were readily arylated and provided the desired product in excellent yields (up to 96%) and good enantioselectivities (up to 90% ee). Later, the same group realized the first example of PIP-assisted Pd(II)-catalyzed enantioselective methylene C(sp3)–H alkynylation of 3-alkyl and 3-aryl propanamides employing the chiral 3,3′-F2-BINOL ligand L31. The control experiments suggested that the superior effect of PIP than of AQ existed in this alkynylation reaction (Scheme 36b).53
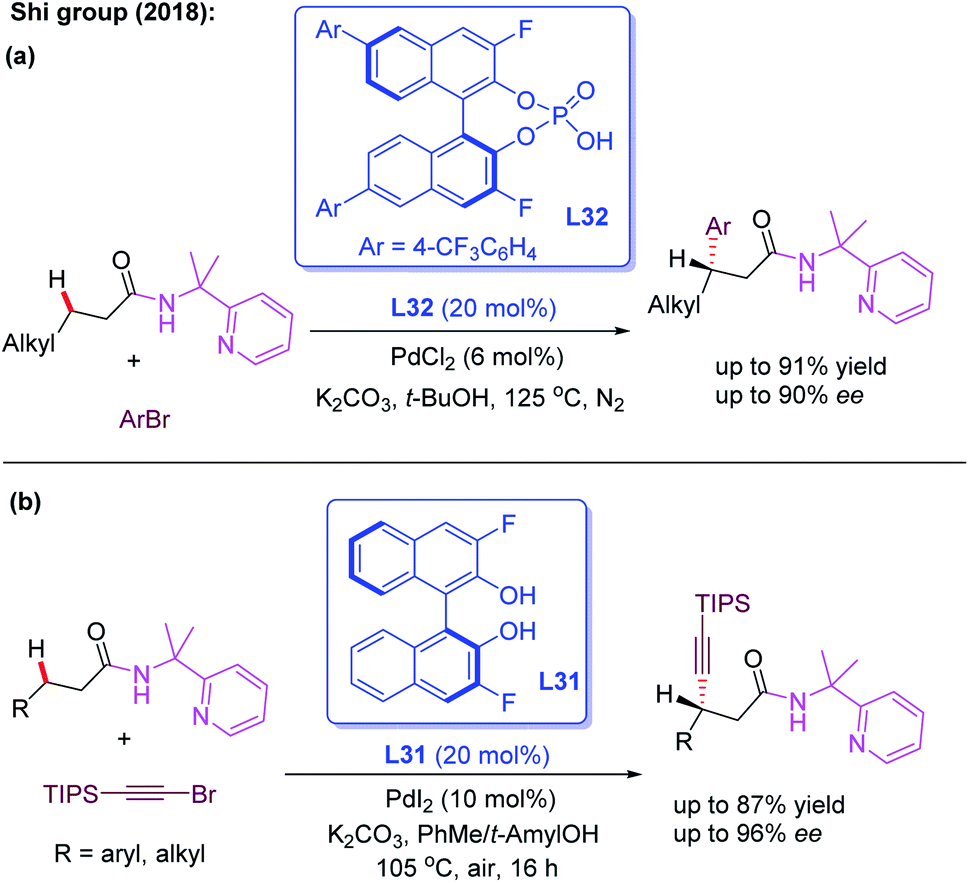 | ||
| Scheme 36 Pd(II)-catalyzed enantioselective functionalization of linear methylene C(sp3)–H bonds enabled by a PIP group. | ||
4. Transient directing group-enabled asymmetric C–H functionalization
Pd-catalyzed asymmetric C(sp2)–H and C(sp3)–H bond functionalization reactions have been well documented by employing a directing group and external chiral ligand. However, a major issue exists in this strategy: pre-installation and removal of the directing group.8–11 Thus, a promising approach of developing the Pd-catalyzed asymmetric C–H functionalization would be the utilization of a chiral transient directing group. In order to achieve this asymmetric process, two issues need to be taken into consideration: (1) the chiral center is usually far away from the target C–H bond, which may result in low enantioselectivity; (2) the free chiral transient ligand could coordinate with the metal center, which may lead to the opposite asymmetric induction.8bThe first example of Pd(II)-catalyzed chiral transient ligand-enabled asymmetric C(sp3)–H bond functionalization of ortho-alkylbenzaldehydes was developed by Yu and co-workers in 2016 (Scheme 37a).7 Using L-tert-leucine (L33) as the chiral transient directing group, Pd(II)-catalyzed enantioselective benzylic C(sp3)–H arylation was demonstrated. Under optimal conditions, 2-ethyl-5-(trifluoromethyl)-benzaldehyde was arylated with methyl 4-iodobenzoate to provide the desired chiral product with 96% ee. Subsequently, the same group revealed a Pd(II)-catalyzed enantioselective C(sp3)–H fluorination of ortho-alkylbenzaldehydes in the presence of the chiral directing group L34. The detailed mechanistic studies showed that L34 bearing a bulky group played an important role in promoting C–F reductive elimination and achieving a high enantioselectivity (Scheme 37b).54
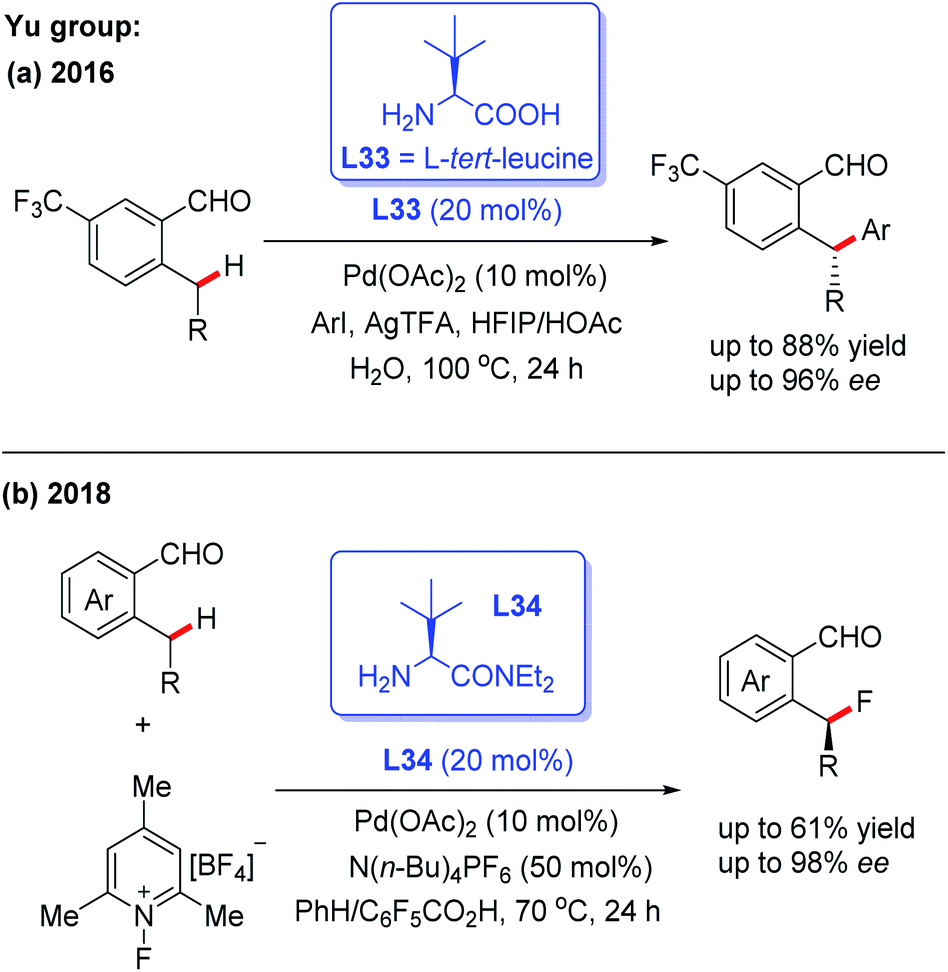 | ||
| Scheme 37 Pd(II)-catalyzed asymmetric C(sp3)–H bond functionalization of ortho-alkylbenzaldehydes. | ||
Very recently, the first example of Pd(II)-catalyzed asymmetric C(sp3) –H arylation of ketones employing D-valine (L35) as a chiral transient directing group was reported by the Yu group. The control experiments suggested that both the 3-nitro-5-trifluoromethyl-2-pyridone ligand and Ag3PO4 additive play crucial roles in this asymmetric desymmetrization of cyclobutyl ketones. However, when using cyclopentyl ketones and linear ketones as the starting materials, the results were not satisfactory (Scheme 38a).55 A plausible mechanism is proposed in Scheme 38b. It was envisioned that the process is initiated with reversible imine formation from 1-cyclobutylethanone with the chiral directing group D-valine, providing the imine intermediate A. Coordination of intermediate A with the Pd(II) species followed by a ligand exchange process with 3-nitro-5-trifluoromethyl-2-pyridone generates the palladium complex B. Cyclopalladation of intermediate B generates the [5,5]-bicyclic intermediate C through a concerted metallation-deprotonation (CMD) process. Oxidative addition of intermediate C with an aryl iodide affords the Pd(IV) complex D. In the presence of Ag3PO4, reductive elimination of this Pd complex provides intermediate F, which releases the final chiral product and D-valine.
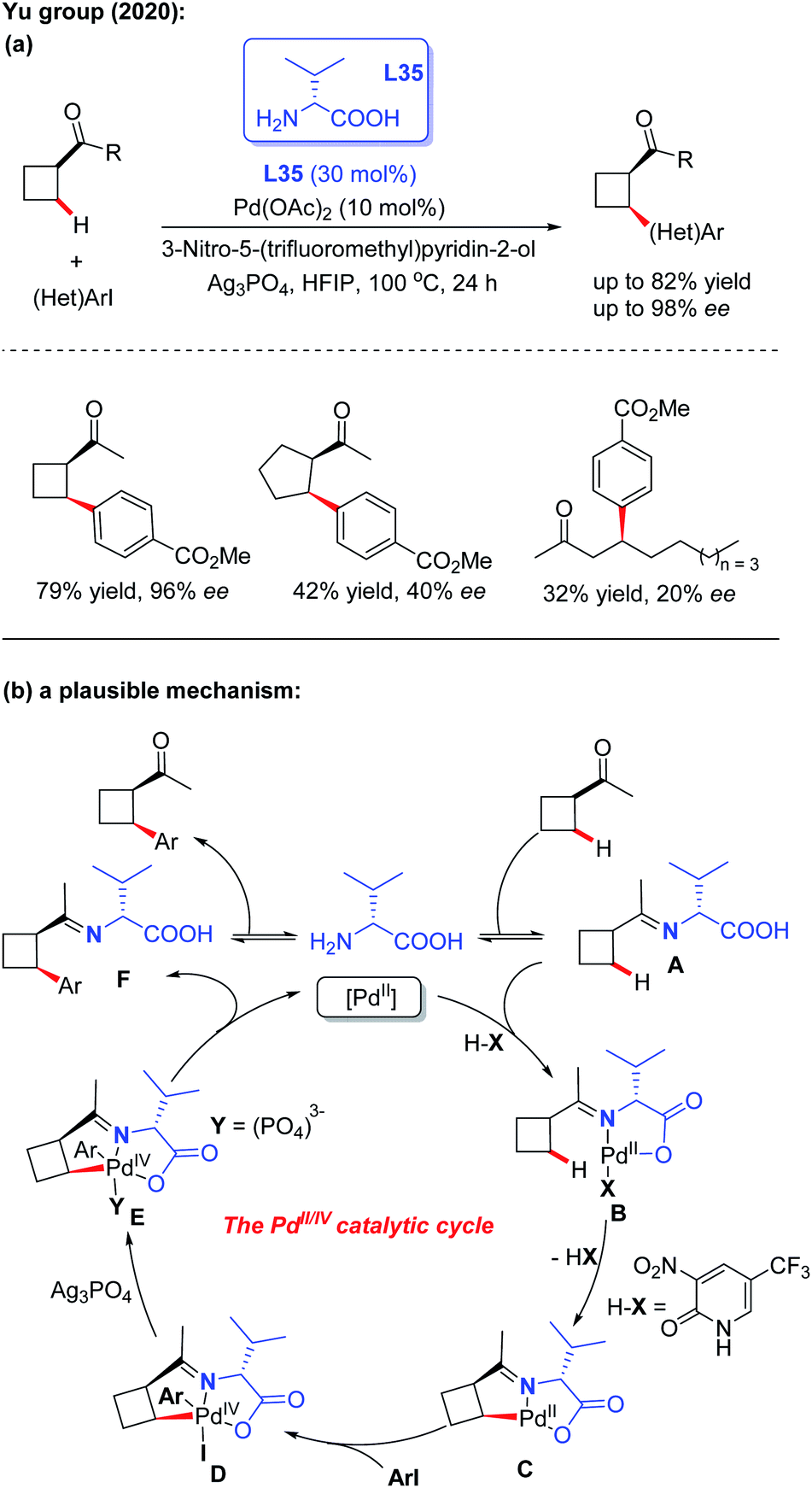 | ||
| Scheme 38 Pd(II)-catalyzed asymmetric C(sp3)–H arylation of ketones employing D-valine as a chiral transient directing group. | ||
Inspired by Yu's work, the Xu and Jin group demonstrated the Pd-catalyzed asymmetric C–H arylation of ferrocenyl ketones through a dynamic kinetic resolution process by using commercially available L-tert-leucine (L33) as the chiral transient directing group (Scheme 39).56 The absolute configuration of chiral arylated ferrocenyl ketones was assigned to Rp through single crystal X-ray diffraction analysis. This strategy provides a complementary approach to the synthesis of various novel and important planar chiral ferrocenyl-phosphine ligands. In the presence of stoichiometric amounts of PPh3, a di-cyclopalladated intermediate was isolated from the reaction of acetylferrocene, Pd(OAc)2 and 2-(aminooxy)acetic acid hydrochloride salt, and the structure of this intermediate was further confirmed by X-ray analysis.
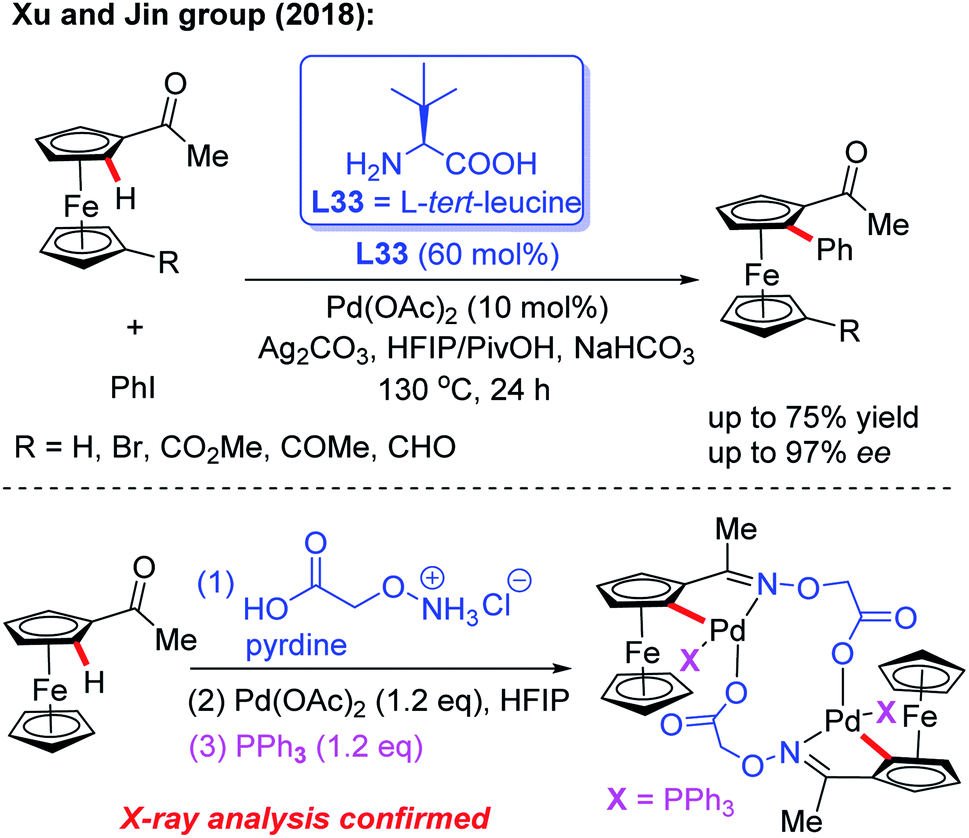 | ||
| Scheme 39 Pd(II)-catalyzed asymmetric C–H arylation of ferrocenyl ketones enabled by a transient directing group L-tert-leucine. | ||
In the meantime, using the same chiral transient directing group L33, Shi and co-workers realized an efficient method for the construction of axially chiral biaryl aldehydes via a Pd(II)-catalyzed asymmetric C–H bond olefination process (Scheme 40a).57 Later, they utilized L-tert-leucine (L33) as the efficient chiral transient ligand for the Pd(II)-catalyzed asymmetric C–H alkynylation and allylation of biaryl aldehydes (Scheme 40b and c).58,59 Additionally, they developed the Pd(II)-catalyzed atroposelective C–H alkynylation, allylation and alkenylation for the synthesis of axially chiral heteroaryls by using L33 as a chiral transient ligand. A wide range of five-membered heteroarenes, including pyrroles, thiophenes, benzothiophenes, and benzofurans, were all compatible with this strategy, providing the axially chiral heteroaryls in good enantioselectivities (Scheme 41 and 42).60,61
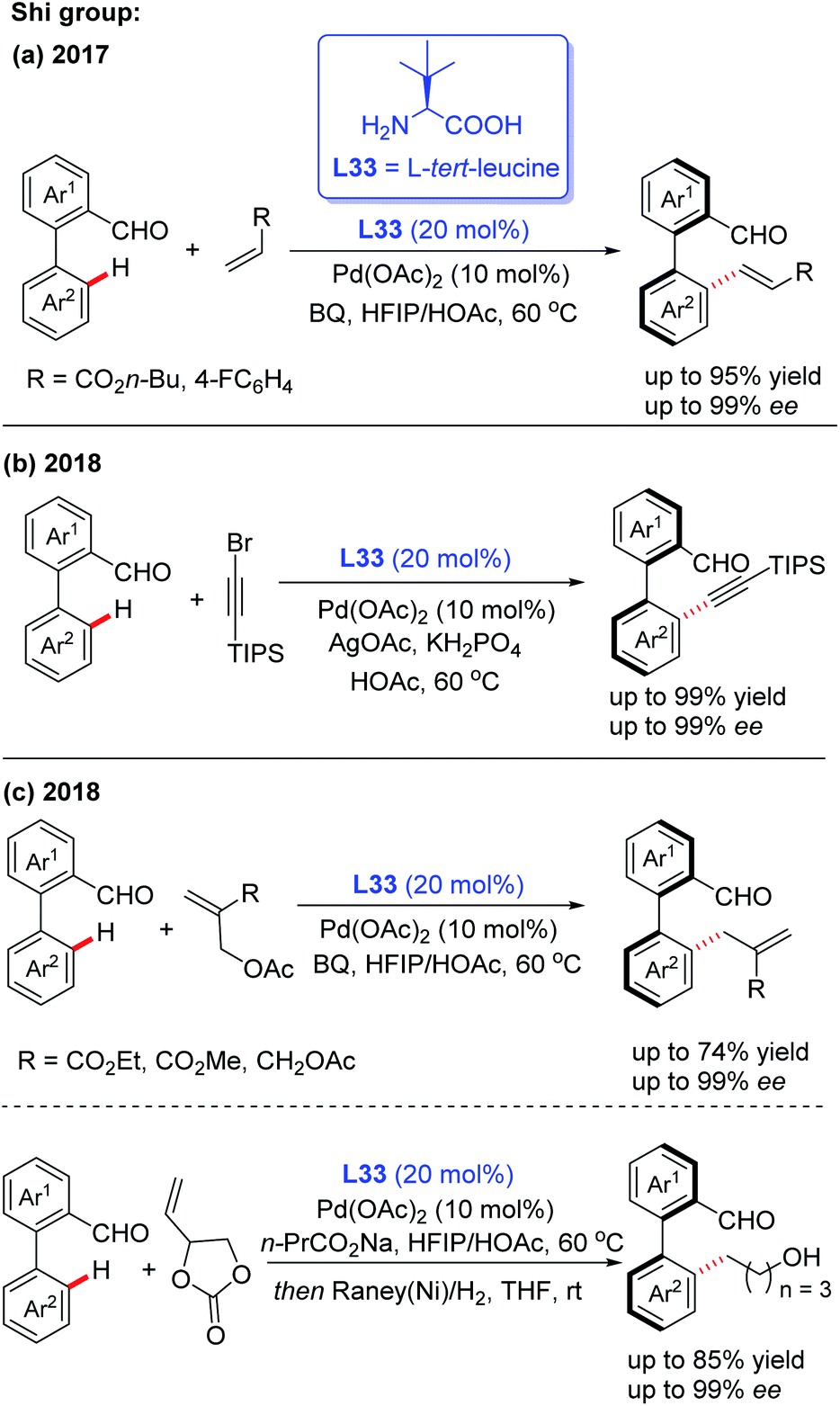 | ||
| Scheme 40 Pd(II)-catalyzed asymmetric C–H bond functionalization for the construction of axially chiral biaryl aldehydes. | ||
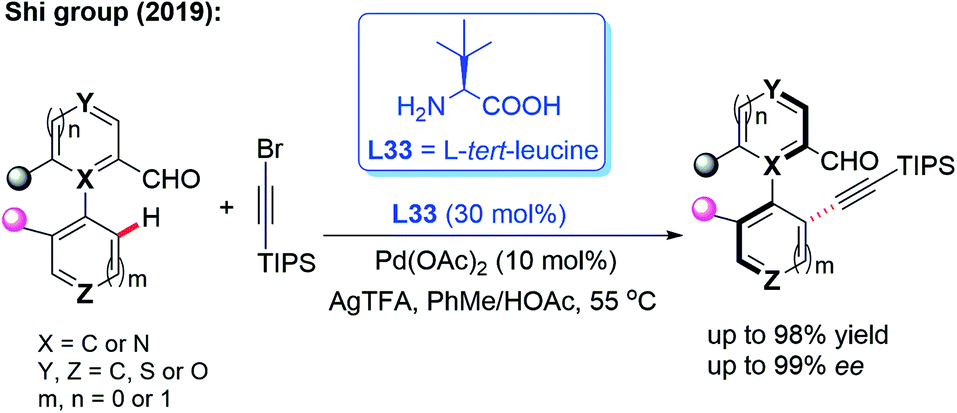 | ||
| Scheme 41 Pd(II)-catalyzed atroposelective C–H alkynylation for the synthesis of axially chiral heteroaryls. | ||
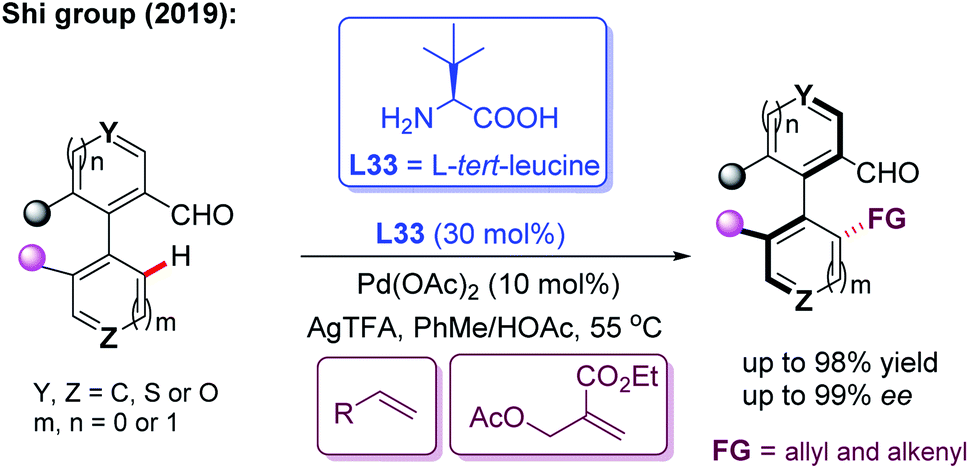 | ||
| Scheme 42 Pd(II)-catalyzed atroposelective C–H allylation and alkenylation for the synthesis of axially chiral heteroaryls. | ||
In 2019, the Shi group used this novel asymmetric C–H functionalization strategy for the synthesis of various axially chiral biaryl aldehydes. In the presence of Pd(OAc)2 and L-tert-leucine (L33), the atroposelective C–H naphthylation with 7-oxabenzonorbornadienes was achieved in good yields and excellent enantioselectivities (up to 99% ee). Using these synthetic axially chiral aldehydes as the chiral catalysts, better catalytic activity was exhibited in the asymmetric reaction of glycine derived amides and dipeptides (Scheme 43).62 A plausible mechanism is proposed in Scheme 44. First, the reversible imine formation between the biaryl aldehyde and the chiral directing group L-tert-leucine affords the imine intermediate A. The selective C–H cyclopalladation of intermediate A gives rise to the enantioenriched palladacycle intermediate B. Migratory insertion of palladacycle B with 1,4-dihydro-1,4-epoxynaphthalene and subsequent β-oxygen elimination generates intermediate D which releases intermediate E, L-tert-leucine, and the Pd(II) catalyst through a protonolysis process in the presence of HOAc. Dehydration of intermediate E finally provides the desired product.
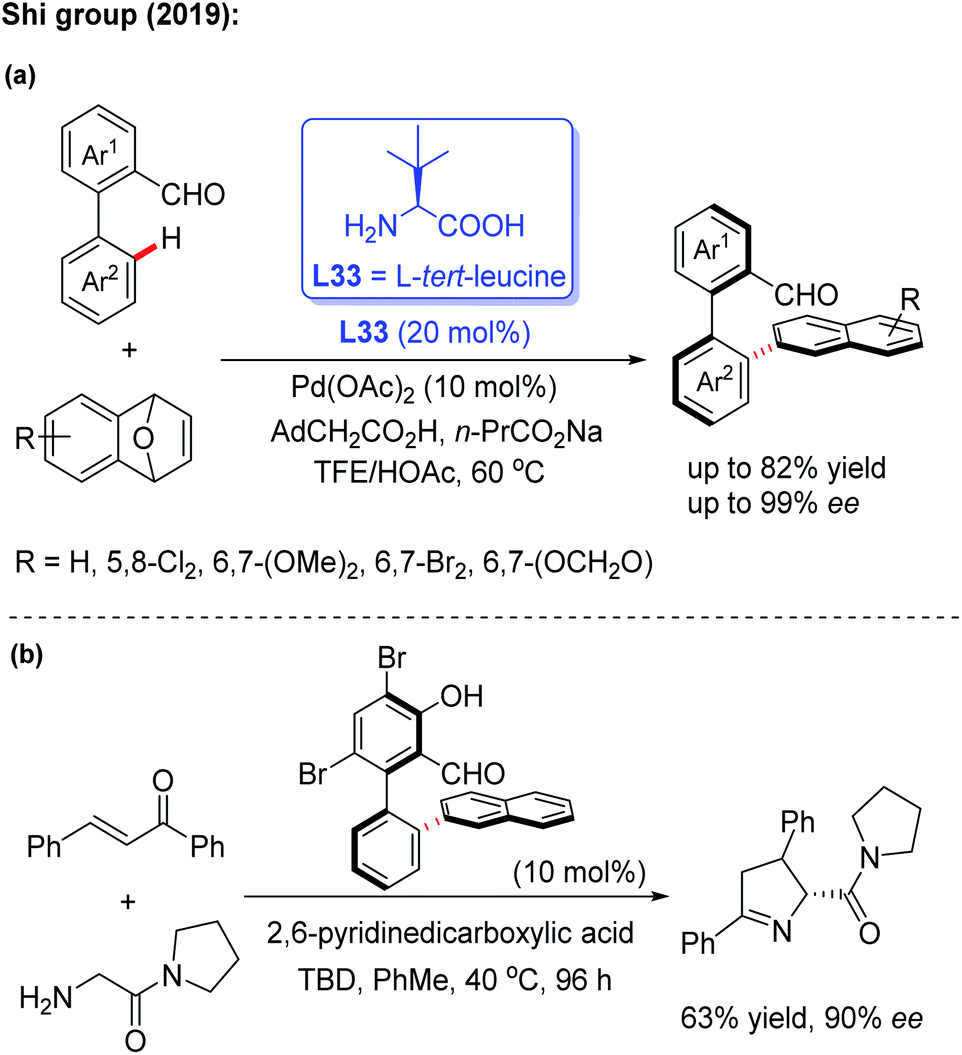 | ||
| Scheme 43 Pd(II)-catalyzed asymmetric C–H naphthylation with 7-oxabenzonorbornadienes and their application. | ||
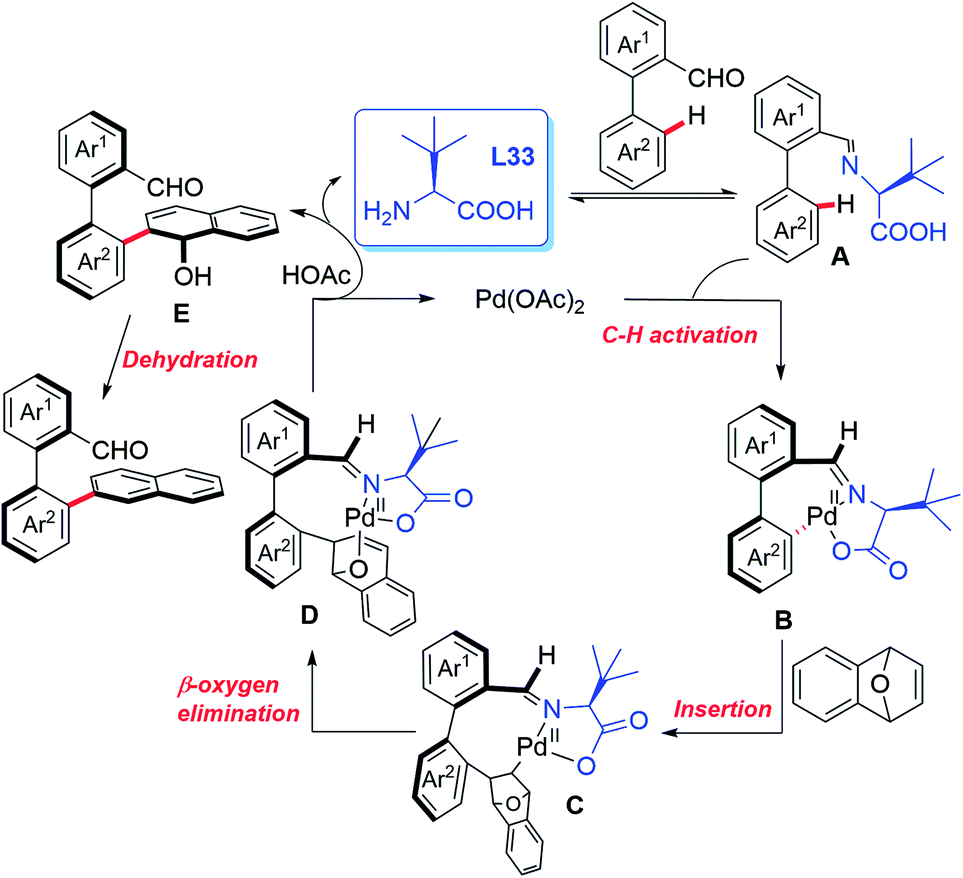 | ||
| Scheme 44 The proposed mechanism. | ||
Very recently, the same group reported an efficient and practical method to construct a novel class of axially chiral styrenes through a Pd-catalyzed asymmetric C–H olefination reaction using the bulky amino amide L36 as a modified chiral transient directing group (Scheme 45).63 This novel method provides a simple, efficient and fast way to synthesize a variety of axially chiral styrene ligands. Moreover, this kind of ligand could also be used to prepare the corresponding chiral acid ligand for the Co(III)-catalyzed asymmetric C(sp3)–H amidation of thioamide.
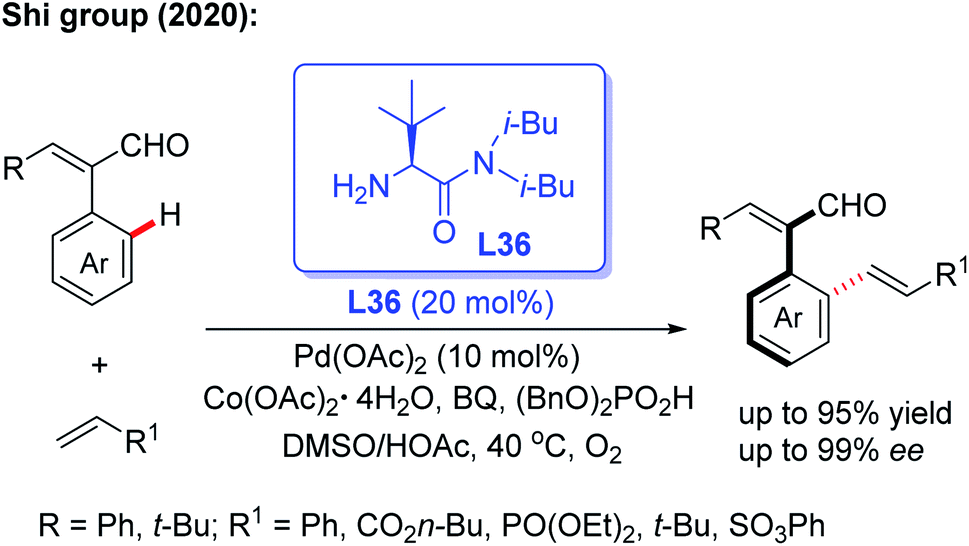 | ||
| Scheme 45 Pd(II)-catalyzed asymmetric C–H olefination using a bulky chiral amino amide, L36, for the synthesis of axially chiral styrenes. | ||
5. Conclusions and outlook
In the past decade, transition metal-catalyzed asymmetric C–H functionalization has become a direct and effective method to access various chiral skeletons. In this mini-review, we provided a robust discussion of recent advances in Pd-catalyzed asymmetric C–H functionalization by using different directing groups, including a monodentate, bidentate, and transient directing group. A range of optically active building blocks, such as chiral heterocyclic compounds, chiral amides, chiral carboxylic acids, chiral amines, axially chiral biaryls, and planar chiral ferrocenes, have been obtained through Pd-catalyzed asymmetric C(sp2)–H and C(sp3)–H functionalization with a chiral ligand as a monodentate directing group. Bidentate directing groups (AQ, PA and PIP) exhibit a higher reactivity and could be used in asymmetric linear methylene C(sp3)–H functionalization in combination with a chiral BINOL ligand. In general, these directing groups often need to be pre-installed on the substrates to promote the asymmetric C–H functionalization reactions, which severely limits the efficiency of the process. More recently, the transient directing strategy has been developed in Pd-catalyzed asymmetric C–H functionalization. While some significant work has been realized by using chiral transient directing groups, there are still many opportunities for improvement and utilization, such as the use of other transition metals and non-covalent bonding modes. We hope this mini-review will provide some insights for readers and inspire them to discover more innovative strategies in the transition metal-catalyzed asymmetric C–H functionalization reactions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the NSF (CHE-2029932), Robert A. Welch Foundation (D-2034-20200401), and Texas Tech University for financial support. Ke Yang is grateful for the financial support from the NSFC (No. 21702019) and Advanced Catalysis and Green Manufacturing Collaborative Innovation Center, Changzhou University.Notes and references
- (a) Y. Yang, J. Lan and J. You, Chem. Rev., 2017, 117, 8787 CrossRef CAS PubMed; (b) J. F. Hartwig, Acc. Chem. Res., 2017, 50, 549 CrossRef CAS PubMed; (c) H. M. L. Davies and D. Morton, ACS Cent. Sci., 2017, 3, 936 CrossRef CAS PubMed; (d) Q.-Z. Zheng and N. Jiao, Chem. Soc. Rev., 2016, 45, 4590 RSC; (e) H. Kim and S. Chang, ACS Catal., 2016, 6, 2341 CrossRef CAS; (f) J. Miao and H. Ge, Eur. J. Org. Chem., 2015, 2015, 7859 CrossRef CAS; (g) Z. Huang, H. N. Lim, F. Mo, M. C. Young and G. Dong, Chem. Soc. Rev., 2015, 44, 7764 RSC; (h) C. S. Yeung and V. M. Dong, Chem. Rev., 2011, 111, 1215 CrossRef CAS PubMed; (i) U. Dutta, S. Maiti, S. Pimparkar, S. Maiti, L. R. Gahan, E. H. Krenske, D. W. Lupton and D. Maiti, Chem. Sci., 2019, 10, 7426 RSC; (j) S. Maity, P. Dolui, R. Kancherla and D. Maiti, Chem. Sci., 2017, 8, 5181 RSC.
- (a) J. He, M. Wasa, K. S. L. Chan, Q. Shao and J.-Q. Yu, Chem. Rev., 2017, 117, 8754 CrossRef CAS PubMed; (b) T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147 CrossRef CAS PubMed; (c) X. Chen, K. M. Engle, D.-H. Wang and J.-Q. Yu, Angew. Chem., Int. Ed., 2009, 48, 5094 CrossRef CAS.
- (a) S. Guin, P. Dolui, X. Zhang, S. Paul, V. K. Singh, S. Pradhan, H. B. Chandrashekar, S. S. Anjana, R. S. Paton and D. Maiti, Angew. Chem., Int. Ed., 2019, 58, 5633 CrossRef CAS PubMed; (b) K. Ramakrishna, J. P. Biswas, S. Jana, T. K. Achar, S. Porey and D. Maiti, Angew. Chem., Int. Ed., 2019, 58, 13808 CrossRef CAS PubMed; (c) C. Sambiagio, D. Schönbauer, R. Blieck, T. Dao-Huy, G. Pototschnig, P. Schaaf, T. Wiesinger, M. F. Zia, J. Wencel-Delord, T. Besset, B. U. W. Maes and M. Schnürch, Chem. Soc. Rev., 2018, 47, 6603 RSC; (d) W.-B. Ma, P. Gandeepan, J. Li and L. Ackermann, Org. Chem. Front., 2017, 4, 1435 RSC; (e) A. Maji, B. Bhaskararao, S. Singha, R. B. Sunoj and D. Maiti, Chem. Sci., 2016, 7, 3147 RSC; (f) Z.-K. Chen, B.-J. Wang, J.-T. Zhang, W.-L. Yu, Z.-X. Liu and Y.-H. Zhang, Org. Chem. Front., 2015, 2, 1107 RSC.
- (a) B. Niu, K. Yang, B. Lawrence and H. Ge, ChemSusChem, 2019, 12, 2955 CrossRef CAS PubMed; (b) Q. Zhao, T. Polsson, X. Pannecouke and T. Besset, Synthesis, 2017, 49, 4808 CrossRef CAS; (c) P. Gandeepan and L. Ackermann, Chem, 2018, 4, 199 CrossRef CAS; (d) T. Bhattacharya, S. Pimparkar and D. Maiti, RSC Adv., 2018, 8, 19456 RSC; (e) S. St John-Campbell, J. Campbell and J. A. Bull, Org. Biomol. Chem., 2018, 16, 4582 RSC.
- (a) J. Loup, U. Dhawa, F. Pesciaioli, J. Wencel-Delord and L. Ackermann, Angew. Chem., Int. Ed., 2019, 58, 2 CrossRef; (b) Q. Lu and F. Glorius, Angew. Chem., Int. Ed., 2017, 56, 49 CrossRef CAS PubMed; (c) Y. Qin, L. Zhu and S. Luo, Chem. Rev., 2017, 117, 9433 CrossRef CAS PubMed; (d) Y.-N. Ma, S.-X. Li and S.-D. Yang, Acc. Chem. Res., 2017, 50, 1480 CrossRef CAS PubMed; (e) C. Zheng and S.-L. You, RSC Adv., 2014, 4, 6173 RSC; (f) S. K. Murphy and V. M. Dong, Chem. Commun., 2014, 50, 13645 RSC.
- B.-F. Shi, N. Maugel, Y.-H. Zhang and J.-Q. Yu, Angew. Chem., Int. Ed., 2008, 47, 4882 CrossRef CAS PubMed.
- F.-L. Zhang, K. Hong, T.-J. Li, H. Park and J.-Q. Yu, Science, 2016, 351, 252 CrossRef CAS PubMed.
- (a) Q. Shao, K. Wu, Z. Zhuang, S. Qian and J.-Q. Yu, Acc. Chem. Res., 2020, 53, 833 CrossRef CAS PubMed; (b) T. G. Saint-Denis, R.-Y. Zhu, G. Chen, Q.-F. Wu and J.-Q. Yu, Science, 2018, 359, 759 CrossRef CAS PubMed.
- (a) J. Huang, Q. Gu and S. You, Chin. J. Org. Chem., 2018, 38, 51 CrossRef CAS; (b) D.-W. Gao, Q. Gu, C. Zheng and S.-L. You, Acc. Chem. Res., 2017, 50, 351 CrossRef CAS PubMed.
- G. Liao, T. Zhou, Q.-J. Yao and B.-F. Shi, Chem. Commun., 2019, 55, 8514 RSC.
- C. G. Newton, S.-G. Wang, C. C. Oliveira and N. Cramer, Chem. Rev., 2017, 117, 8908 CrossRef CAS PubMed.
- X.-P. Zeng, Z.-Y. Cao, Y.-H. Wang, F. Zhou and J. Zho, Chem. Rev., 2016, 116, 7330 CrossRef CAS PubMed.
- (a) R. Giri, X. Chen and J.-Q. Yu, Angew. Chem., Int. Ed., 2005, 44, 2112 CrossRef CAS PubMed; (b) R. Giri, J. Liang, J.-G. Lei, J.-J. Li, D.-H. Wang, X. Chen, I. C. Naggar, C. Guo, B. M. Foxman and J.-Q. Yu, Angew. Chem., Int. Ed., 2005, 44, 7420 CrossRef CAS PubMed; (c) B. Bhaskararao, S. Singh, M. Anand, P. Verma, P. Prakash, A. C, S. Malakar, H. F. Schaefer and R. B. Sunoj, Chem. Sci., 2020, 11, 208 RSC; (d) N. Rodriguez and L. J. Goossen, Chem. Soc. Rev., 2011, 40, 5030 RSC.
- B.-F. Shi, Y.-H. Zhang, J. K. Lam, D.-H. Wang and J.-Q. Yu, J. Am. Chem. Soc., 2010, 132, 460 CrossRef CAS PubMed.
- M. Wasa, K. M. Engle, D. W. Lin, E. J. Yoo and J.-Q. Yu, J. Am. Chem. Soc., 2011, 133, 19598 CrossRef CAS PubMed.
- K.-J. Xiao, D. W. Lin, M. Miura, R.-Y. Zhu, W. Gong, M. Wasa and J.-Q. Yu, J. Am. Chem. Soc., 2014, 136, 8138 CrossRef CAS PubMed.
- J. He, Q. Shao, Q. Wu and J.-Q. Yu, J. Am. Chem. Soc., 2017, 139, 3344 CrossRef CAS PubMed.
- Q.-F. Wu, X.-B. Wang, P.-X. Shen and J.-Q. Yu, ACS Catal., 2018, 8, 2577 CrossRef CAS PubMed.
- Q.-F. Wu, P.-X. Shen, J. He, X.-B. Wang, F. Zhang, Q. Shao, R.-Y. Zhu, C. Mapelli, J. X. Qiao, M. A. Poss and J.-Q. Yu, Science, 2017, 355, 499 CrossRef CAS PubMed.
- G. Xu, C. H. Senanayake and W. Tang, Acc. Chem. Res., 2019, 52, 1101 CrossRef CAS PubMed.
- Z.-J. Du, J. Guan, G.-J. Wu, P. Xu, L.-X. Gao and F.-S. Han, J. Am. Chem. Soc., 2015, 137, 632 CrossRef CAS PubMed.
- P. X. Shen, L. Hu, Q. Shao, K. Hong and J.-Q. Yu, J. Am. Chem. Soc., 2018, 140, 6545 CrossRef CAS PubMed.
- L. Hu, P.-X. Shen, Q. Shao, K. Hong, J. X. Qiao and J.-Q. Yu, Angew. Chem., Int. Ed., 2019, 58, 2134 CrossRef CAS PubMed.
- R. Hili and A. K. Yudin, Nat. Chem. Biol., 2006, 2, 284 CrossRef CAS PubMed.
- L. Chu, X.-C. Wang, C. E. Moore, A. L. Rheingold and J.-Q. Yu, J. Am. Chem. Soc., 2013, 135, 16344 CrossRef CAS PubMed.
- K. S. L. Chan, H.-Y. Fu and J.-Q. Yu, J. Am. Chem. Soc., 2015, 137, 2042 CrossRef CAS PubMed.
- P. Jain, P. Verma, G. Xia and J.-Q. Yu, Nature Chem., 2016, 9, 140 CrossRef PubMed.
- Q. Shao, Q.-F. Wu, J. He and J.-Q. Yu, J. Am. Chem. Soc., 2018, 140, 5322 CrossRef CAS PubMed.
- K.-J. Xiao, L. Chu and J.-Q. Yu, Angew. Chem., Int. Ed., 2016, 55, 2856 CrossRef CAS PubMed.
- K.-J. Xiao, L. Chu, G. Chen and J.-Q. Yu, J. Am. Chem. Soc., 2016, 138, 7796 CrossRef CAS PubMed.
- (a) J. E. Smyth, N. M. Butler and P. A. Keller, Nat. Prod. Rep., 2015, 32, 1562 RSC; (b) G. Bringmann, T. Gulder, T. A. M. Gulder and M. Breuning, Chem. Rev., 2011, 111, 563 CrossRef CAS PubMed; (c) S. R. LaPlante, L. D. Fader, K. R. Fandrick, D. R. Fandrick, O. Hucke, R. Kemper, S. P. F. Miller and P. J. Edwards, J. Med. Chem., 2011, 54, 700 CrossRef PubMed.
- T. P. Yoon and E. N. Jacobsen, Science, 2003, 299, 1691 CrossRef CAS PubMed.
- F. Kakiuchi, P. L. Gendre, A. Yamada, H. Ohtaki and S. Murai, Tetrahedron: Asymmetry, 2000, 11, 2647 CrossRef CAS.
- D.-W. Gao, Q. Gu and S.-L. You, ACS Catal., 2014, 4, 2741 CrossRef CAS.
- S.-X. Li, Y. N. Ma and S.-D. Yang, Org. Lett., 2017, 19, 1842 CrossRef CAS PubMed.
- J. Luo, T. Zhang, L. Wang, G. Liao, Q.-J. Yao, Y.-J. Wu, B.-B. Zhan, Y. Lan, X.-F. Lin and B.-F. Shi, Angew. Chem., Int. Ed., 2019, 58, 6708 CrossRef CAS PubMed.
- B.-B. Zhan, L. Wang, J. Luo, X.-F. Lin and B.-F. Shi, Angew. Chem., Int. Ed., 2020, 59, 3568 CrossRef CAS PubMed.
- L. Jin, Q.-J. Yao, P.-P. Xie, Y. Li, B.-B. Zhan, Y.-Q. Han, X. Hong and B.-F. Shi, Chem, 2020, 6, 497 CAS.
- T. Noël and J. Van der Eycken, Green Process. Synth., 2013, 2, 297 Search PubMed.
- D.-W. Gao, Y.-C. Shi, Q. Gu, Z.-L. Zhao and S.-L. You, J. Am. Chem. Soc., 2013, 135, 86 CrossRef CAS PubMed.
- Y.-C. Shi, R.-F. Yang, D.-W. Gao and S.-L. You, Beilstein J. Org. Chem., 2013, 9, 1891 CrossRef PubMed.
- D.-W. Gao, Q. Gu and S.-L. You, J. Am. Chem. Soc., 2016, 138, 2544 CrossRef CAS PubMed.
- C. Pi, Y. Li, X.-L. Cui, H. Zhang, Y.-B. Han and Y.-J. Wu, Chem. Sci., 2013, 4, 2675 RSC.
- C. Pi, X.-L. Cui, X.-Y. Liu, M. X. Guo, H.-Y. Zhang and Y.-J. Wu, Org. Lett., 2014, 16, 5164 CrossRef CAS PubMed.
- G. Chen, W. Gong, Z. Zhuang, M. S. Andrä, Y.-Q. Chen, X. Hong, Y.-F. Yang, T. Liu, K. N. Houk and J.-Q. Yu, Science, 2016, 353, 1023 CrossRef CAS PubMed.
- (a) S. Rej, Y. Ano and N. Chatani, Chem. Rev., 2020, 120, 1788 CrossRef CAS PubMed; (b) G. Rouquet and N. Chatani, Angew. Chem., Int. Ed., 2013, 52, 11726 CrossRef CAS PubMed; (c) D. Shabashov and O. Daugulis, J. Am. Chem. Soc., 2010, 132, 3965 CrossRef CAS PubMed.
- S.-B. Yan, S. Zhang and W.-L. Duan, Org. Lett., 2015, 17, 2458 CrossRef CAS PubMed.
- (a) H. Wang, H.-R. Tong, G. He and G. Chen, Angew. Chem., Int. Ed., 2016, 55, 15387 CrossRef CAS PubMed; (b) O. Daugulis, J. Roane and L. D. Tran, Acc. Chem. Res., 2015, 48, 1053 CrossRef CAS PubMed.
- H.-R. Tong, S. Zheng, X. Li, Z. Deng, H. Wang, G. He, Q. Peng and G. Chen, ACS Catal., 2018, 8, 11502 CrossRef CAS.
- H.-R. Tong, W. Zheng, X. Lv, G. He, P. Liu and G. Chen, ACS Catal., 2020, 10, 114 CrossRef CAS.
- Q. Zhang and B.-F. Shi, Chin. J. Chem., 2019, 37, 647 CrossRef CAS.
- S.-Y. Yan, Y.-Q. Han, Q.-J. Yao, X.-L. Nie, L. Liu and B.-F. Shi, Angew. Chem., Int. Ed., 2018, 57, 9093 CrossRef CAS PubMed.
- Y.-Q. Han, Y. Ding, T. Zhou, S.-Y. Yan, H. Song and B.-F. Shi, J. Am. Chem. Soc., 2019, 141, 4558 CrossRef CAS PubMed.
- H. Park, P. Verma, K. Hong and J.-Q. Yu, Nature Chem., 2018, 10, 755 CrossRef CAS PubMed.
- L.-J. Xiao, K. Hong, F. Luo, L. Hu, W. R. Ewing, K.-S. Yeung and J.-Q. Yu, Angew. Chem., Int. Ed., 2020, 59, 9594 CrossRef CAS PubMed.
- J.-C. Xu, Y. Liu, J.-L. Zhang, X.-H. Xu and Z. Jin, Chem. Commun., 2018, 54, 689 RSC.
- J. Yao, S. Zhang, B.-B. Zhan and B.-F. Shi, Angew. Chem., Int. Ed., 2017, 56, 6617 CrossRef PubMed.
- G. Liao, B. Li, H.-M. Chen, Q.-J. Yao, Y.-N. Xia, J. Luo and B.-F. Shi, Angew. Chem., Int. Ed., 2018, 57, 17151 CrossRef CAS PubMed.
- G. Liao, Q.-J. Yao, Z.-Z. Zhang, Y.-J. Wu, D.-Y. Huang and B.-F. Shi, Angew. Chem., Int. Ed., 2018, 57, 3661 CrossRef CAS PubMed.
- H.-M. Chen, S. Zhang, G. Liao, Q.-J. Yao, X.-T. Xu, K. Zhang and B.-F. Shi, Organometallics, 2019, 38, 4022 CrossRef CAS.
- S. Zhang, Q.-J. Yao, G. Liao, X. Li, H. Li, H.-M. Chen, X. Hong and B.-F. Shi, ACS Catal., 2019, 9, 1956 CrossRef CAS.
- G. Liao, H.-M. Chen, Y.-N. Xia, B. Li, Q.-J. Yao and B.-F. Shi, Angew. Chem., Int. Ed., 2019, 58, 11464 CrossRef CAS PubMed.
- H. Song, Y. Li, Q.-J. Yao, L. Jin, L. Liu, Y.-H. Liu and B.-F. Shi, Angew. Chem., Int. Ed., 2020, 59, 6576 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2020 |