
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Multi-targeting of functional cysteines in multiple conserved SARS-CoV-2 domains by clinically safe Zn-ejectors†
Karen
Sargsyan‡
 a,
Chien-Chu
Lin‡
b,
Ting
Chen‡
a,
Cédric
Grauffel‡
a,
Yi-Ping
Chen
b,
Wei-Zen
Yang
b,
Hanna S.
Yuan
*b and
Carmay
Lim
*ac
a,
Chien-Chu
Lin‡
b,
Ting
Chen‡
a,
Cédric
Grauffel‡
a,
Yi-Ping
Chen
b,
Wei-Zen
Yang
b,
Hanna S.
Yuan
*b and
Carmay
Lim
*ac
aInstitute of Biomedical Sciences, Academia Sinica, Taipei 115, Taiwan. E-mail: carmay@gate.sinica.edu.tw
bInstitute of Molecular Biology, Academia Sinica, Taipei 115, Taiwan. E-mail: hanna@sinica.edu.tw
cDepartment of Chemistry, National Tsing Hua University, Hsinchu 300, Taiwan
First published on 1st September 2020
Abstract
We present a near-term treatment strategy to tackle pandemic outbreaks of coronaviruses with no specific drugs/vaccines by combining evolutionary and physical principles to identify conserved viral domains containing druggable Zn-sites that can be targeted by clinically safe Zn-ejecting compounds. By applying this strategy to SARS-CoV-2 polyprotein-1ab, we predicted multiple labile Zn-sites in papain-like cysteine protease (PLpro), nsp10 transcription factor, and nsp13 helicase. These are attractive drug targets because they are highly conserved among coronaviruses and play vital structural/catalytic roles in viral proteins indispensable for virus replication. We show that five Zn-ejectors can release Zn2+ from PLpro and nsp10, and clinically-safe disulfiram and ebselen can not only covalently bind to the Zn-bound cysteines in both proteins, but also inhibit PLpro protease. We propose combining disulfiram/ebselen with broad-spectrum antivirals/drugs to target different conserved domains acting at various stages of the virus life cycle to synergistically inhibit SARS-CoV-2 replication and reduce the emergence of drug resistance.
New infectious viruses causing epidemics/pandemics such as the SARS-CoV-2 require near-term effective and practical strategies to treat virus-infected patients. This is because developing specific antiviral drugs/vaccines takes time and in the interim, lives are lost/disrupted. One short-term strategy is to leverage the non-specificity of some FDA-approved drugs to target critical viral proteins.1 Here, we present a multi-targeting strategy combining evolutionary (conserved protein domains) and physical (factors controlling Zn2+-bound Cys reactivity) principles to identify new drug targets in conserved viral domains and applied it to the SARS-CoV-2. We show that clinically safe Zn-ejector drugs, disulfiram and ebselen, can target highly conserved Zn2+-binding and/or catalytic cysteines (Fig. 1) in multiple conserved viral domains essential for SARS-CoV-2 replication.
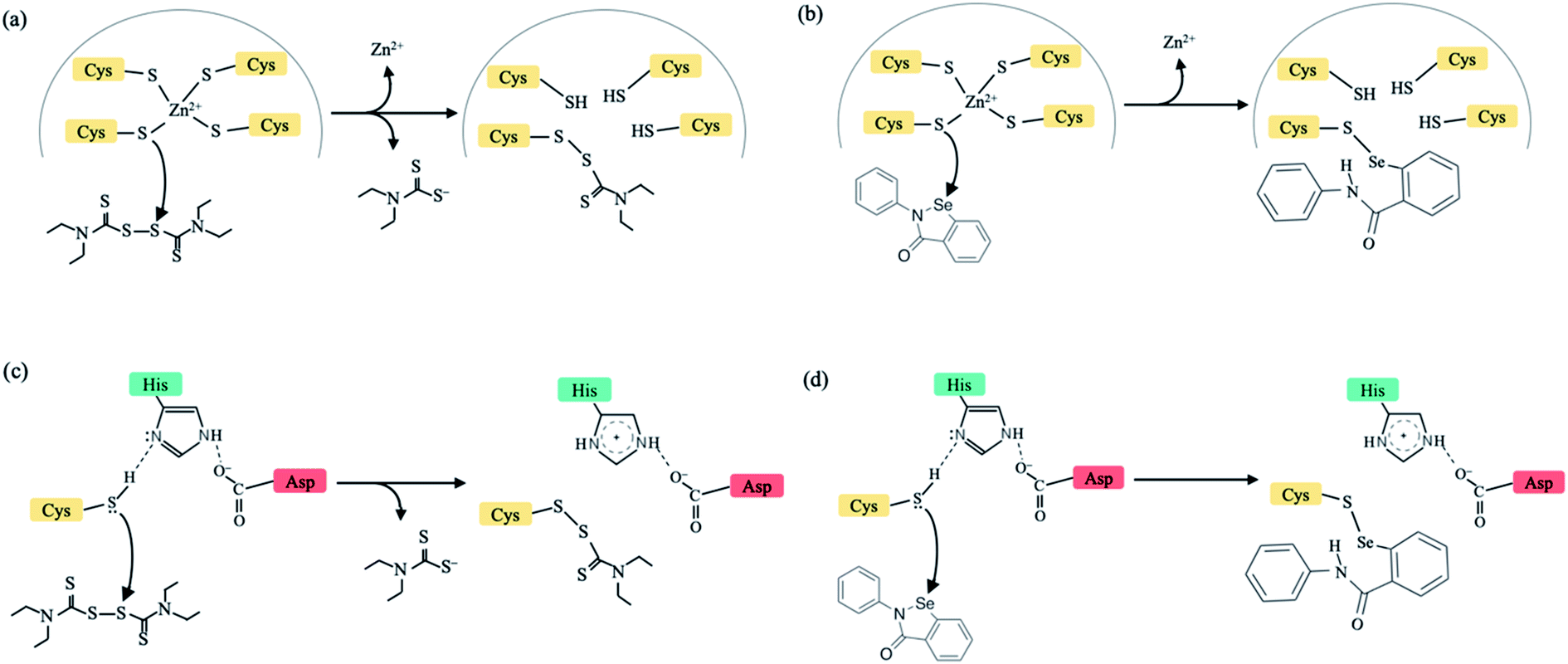 | ||
| Fig. 1 Schematic diagram to illustrate the mechanism of action by disulfiram (left) and ebselen (right) to release Zn2+ (a and b) or form a covalent adduct with a catalytic cysteine (c and d). In (a) and (c), half of disulfiram, diethyl-dithiol-carbamate, is covalently bonded to a Zn2+-bound/catalytic Cys. | ||
Conserved cysteines and Zn2+ play crucial roles in viral infections.2 Cysteines can serve catalytic and/or structural roles (by binding metal ions and forming disulfide bridges) in viral enzymes/proteins. Zn2+ is an essential cofactor of many viral proteins, as it induces protein folding and stabilizes the local structure.3,4 Viral Zn-sites containing reactive cysteines bound to structural Zn2+ cations (termed labile Zn-sites) may serve as drug targets, as the Zn2+-bound thiolates can react with Zn-ejectors, leading to loss of Zn2+ and viral protein structure/function.5–8 Our previous studies7,9 had revealed the key factors determining which Zn2+-bound cysteines in proteins are reactive: labile Zn-sites are likely to be Zn-Cys4 or Zn-Cys3His sites lacking hydrogen bonds to any Zn-bound thiolate, which would reduce the Zn-bound thiolate's negative charge and reactivity (ESI Fig. S1†). We then used these guidelines to identify drug-target proteins containing labile structural Zn-sites.8,10
To circumvent toxicity due to undesirable targeting of essential human proteins, we had proposed using clinically safe (FDA-approved or in phase II/III clinical trials) Zn-ejecting agents that do not affect crucial host proteins (ESI Table S1†) to target putative labile Zn-sites in viral proteins.8 We showed that disulfiram, an anti-alcoholism drug, can eject Zn2+ from the predicted labile Zn-Cys4 site in the hepatitis C virus NS5A protein, inhibiting viral replication, and that inhibition was enhanced when disulfiram was combined with interferon-α.8 Following our work, disulfiram was found to eject Zn2+ and inhibit replication in other viruses, notably SARS- and MERS-CoV papain-like proteases (PLpro),11 which along with the main protease (Mpro) cleave the pp1a and pp1ab replicase polyproteins into 16 nonstructural proteins (nsps).12 Since SARS-CoV PLpro remained inactivated after removing unbound disulfiram, but was reactivated by reductant, disulfiram may also form a covalent adduct with the catalytic cysteine.11
The above findings suggest that a possible strategy to combat infectious coronaviruses with no approved drugs/vaccines is to employ clinically safe Zn-ejector drugs to target multiple essential Zn2+-bound and/or catalytic cysteines in conserved viral domains. Coronaviruses belonging to the order Nidovirales are amenable to our proposed strategy since they employ cysteine proteases (Mpro, PLpro) and share conserved core replicase domains.12 Thus, we analyzed the SARS-CoV-2 genome (GenBank: MN908947.3) comprising 5′-methylated cap, genes encoding nonstructural and structural proteins, and 3′-polyadenylated tail. We focused on the large replicase polyprotein pp1ab because its cleavage products (nsp7–nsp16) are involved in viral replication.12 By searching the pp1ab sequence for conserved domains using the Conserved Domain Database,13 we found 18 such domains (ESI Table S2†). For each conserved domain found, we searched the Protein Data Bank (PDB)14 for <3 Å structures from other coronaviruses sharing similar function and high sequence identity using BLASTp15 (ESI Scheme S1†). We then checked each structure for Zn-(Cys4/Cys3His) sites, lacking hydrogen bonds to the Zn-bound thiolates. Although nsp12 (6nur, 6nus) and nsp14 (5c8s, 5c8t, 5c8u) structures have Zn-(Cys4/Cys3His) sites, their poor (≥3.1 Å) resolution prohibited reliable hydrogen-bond analysis of these Zn-sites.
Putative labile Zn-sites were found in the SARS-CoV structures of PLpro subdomain of nsp3, nsp10 Zn-finger protein, and nsp13 helicase (Table 1). To obtain the corresponding SARS-CoV-2 sequences, we aligned the SARS-CoV PLpro/nsp10/nsp13 and the SARS-CoV-2 pp1ab sequences using BLASTp15 and obtained excellent alignment (ESI Table S3†). The SARS-CoV-2 PLpro structure was homology-modeled from the SARS-CoV 4m0w_A structure using MODELLER,16 whereas the SARS-CoV-2 nsp10 and nsp13 structures were derived from the respective SARS-CoV structures (2xyq_B and 6jyt_B) by point mutations using SCRWL4 (ref. 17) since their sequences differ by only 1–2 residues. These modeled structures confirm the absence of hydrogen bonds to the Zn-bound thiolates (ESI Fig. S2†). Furthermore, model structures of the SARS-CoV-2 PLpro/Mpro with the catalytic cysteine covalently modified by disulfiram/ebselen obtained using QM/MM energy minimization show that the active site can accommodate a covalent adduct, thus inhibiting enzyme activity (ESI Fig. S3†).
| SARS-CoV-2 domaina | SARS-CoV protein | PDB structuresb | Zn-ligandsc |
|---|---|---|---|
| a Conserved domain found by Conserved Domain Database.13 b PDB entry_chain ID of the SARS-CoV protein; that in bold was used to model the respective SARS-CoV-2 protein structure. c Residue numbers correspond to those of the respective SARS-CoV-2 protein. | |||
| PLpro subdomain of nsp3 | PLpro | 4m0w _A, 3e9s_A, 5tl7_B | C189, C192, C224, C226 |
| nsp10 | nsp10 | 2xyq_B, 2fyg_A, 5c8u_A, 5nfy_O, 5nfy_P, 2ga6_F, 2ga6_R, 5c8t_C | C117, C120, C128, C130 |
| nsp13 | Helicase | 6jyt_B | C50, C55, C72, H75 |
Among the three predicted SARS-CoV-2 targets, we chose nsp10 and the PLpro subdomain of nsp3 to validate our predictions. We subcloned the cDNAs of SARS-CoV-2 PLpro and nsp10 (residues 1541–1855 and 4231–4362 of pp1a/pp1ab, respectively) and expressed them in E. coli. Each purified recombinant protein (5 μM) was incubated with ten Zn2+-ejecting compounds (5 μM), including disulfiram and ebselen, and Zn2+ release was monitored by the increase in fluorescence emission signal from the zinc-specific fluorophore, FluoZin-3 (1 μM). Among the ten Zn2+-ejectors tested, disulfiram, ebselen, 5,5′-dithiobis(2-nitrobenzoic acid) (DTNB), 2,2′-dithiodipyridine, and 2,2′-dithiobis(benzothiazole) could eject Zn2+ from PLpro (Fig. 2a) and nsp10 (Fig. 2b).
 | ||
| Fig. 2 Zn2+ release from SARS-CoV-2 PLpro and nsp10 by Zn2+-ejecting compounds. Upon adding each Zn2+-ejecting agent (5 μM) to PLpro (5 μM) (a), or GST-fused nsp10 (5 μM) (b), Zn2+ release was detected by the increase of the fluorescence signal from FluoZin-3 (1 μM) with emission and excitation wavelengths of 494 nm and 516 nm, respectively. The MWs of PLpro (c) and nsp10 (d) before (top) and after adding disulfiram (middle) or ebselen (bottom) were measured by MALTI-TOF mass spectrometry. Three independent experiments gave similar results hence only spectra from one of the experiments are shown. | ||
To confirm that disulfiram and ebselen are covalently bound to the cysteines in PLpro and nsp10, the molecular weights (MWs) of PLpro and nsp10 before and after adding these two zinc-ejectors were measured by mass spectrometry. The PLpro MALTI-TOF mass spectrum (Fig. 2c, top panel) revealed a major peak with a measured MW of 37![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 120 Da close to PLpro's calculated MW (37125 Da). Disulfiram-treated PLpro (Fig. 2c, middle panel) had an additional peak at 37269 Da, suggesting half of disulfiram was bound in PLpro. Ebselen-treated PLpro (Fig. 2c, bottom panel) had additional peaks at 37394 and 37669 Da, indicating one and two ebselen molecules were bound, respectively. For nsp10 with a calculated MW of 14066 Da, the MALTI-TOF mass spectra (Fig. 2d) exhibited additional peaks at 14202 Da (corresponding to 0.5 disulfiram-bound nsp10) and 14339 Da (corresponding to one ebselen-bound nsp10), suggesting that both drugs were covalently bound to cysteines in nsp10. To further verify that the drug was attached to cysteines involved in Zn2+-binding in PLpro, disulfiram- and ebselen-treated PLpro were digested by trypsin, and analyzed by liquid chromatography-tandem mass spectrometry (LC-MS/MS). The MWs of the PLpro peptides, 183RVLNVVCK190 (1076.6 Da) and 191TCGQQQTTLK200 (1253.6 Da) with an additional MW of 148 Da, confirm that the Zn2+-bound C189 and C192 were covalently linked to half of disulfiram. Likewise, the MWs of the PLpro peptides, 184VLNVVCK190 (1048.4 Da) and 191TCGQQQTTLK200 (1381.5 Da) with an additional MW of 274 Da, show that the Zn2+-bound C189 and C192 were bonded to ebselen. These results confirm that disulfiram and ebselen can covalently bind Zn2+-bound cysteines in SARS-CoV-2 PLpro, resulting in the loss of Zn2+.
120 Da close to PLpro's calculated MW (37125 Da). Disulfiram-treated PLpro (Fig. 2c, middle panel) had an additional peak at 37269 Da, suggesting half of disulfiram was bound in PLpro. Ebselen-treated PLpro (Fig. 2c, bottom panel) had additional peaks at 37394 and 37669 Da, indicating one and two ebselen molecules were bound, respectively. For nsp10 with a calculated MW of 14066 Da, the MALTI-TOF mass spectra (Fig. 2d) exhibited additional peaks at 14202 Da (corresponding to 0.5 disulfiram-bound nsp10) and 14339 Da (corresponding to one ebselen-bound nsp10), suggesting that both drugs were covalently bound to cysteines in nsp10. To further verify that the drug was attached to cysteines involved in Zn2+-binding in PLpro, disulfiram- and ebselen-treated PLpro were digested by trypsin, and analyzed by liquid chromatography-tandem mass spectrometry (LC-MS/MS). The MWs of the PLpro peptides, 183RVLNVVCK190 (1076.6 Da) and 191TCGQQQTTLK200 (1253.6 Da) with an additional MW of 148 Da, confirm that the Zn2+-bound C189 and C192 were covalently linked to half of disulfiram. Likewise, the MWs of the PLpro peptides, 184VLNVVCK190 (1048.4 Da) and 191TCGQQQTTLK200 (1381.5 Da) with an additional MW of 274 Da, show that the Zn2+-bound C189 and C192 were bonded to ebselen. These results confirm that disulfiram and ebselen can covalently bind Zn2+-bound cysteines in SARS-CoV-2 PLpro, resulting in the loss of Zn2+.
Since disulfiram and ebselen are in clinical use, we further determined if they could inhibit PLpro by measuring protease activity in cleaving a fluorogenic substrate (Dabcy-FTLKGG↓APTKVTE-Edans) in the absence and presence of varying concentrations of each Zn2+-ejector drug. We found that disulfiram and ebselen inhibited PLpro activity with IC50 of 7.52 ± 2.13 μM and 2.36 ± 0.16 μM, respectively (Fig. 3a and b). Interestingly, compared to disulfiram, ebselen displayed slightly stronger inhibition of PLpro activity despite its less potent Zn-ejecting ability (Fig. 2a). In SARS-CoV-2 Mpro that lacks a Zn2+-site, ebselen (IC50 = 0.67 ± 0.09 μM) also showed stronger inhibition than disulfiram (IC50 = 9.35 ± 0.18 μM).18 This suggests that ebselen may be more effective in targeting the catalytic cysteine than disulfiram.
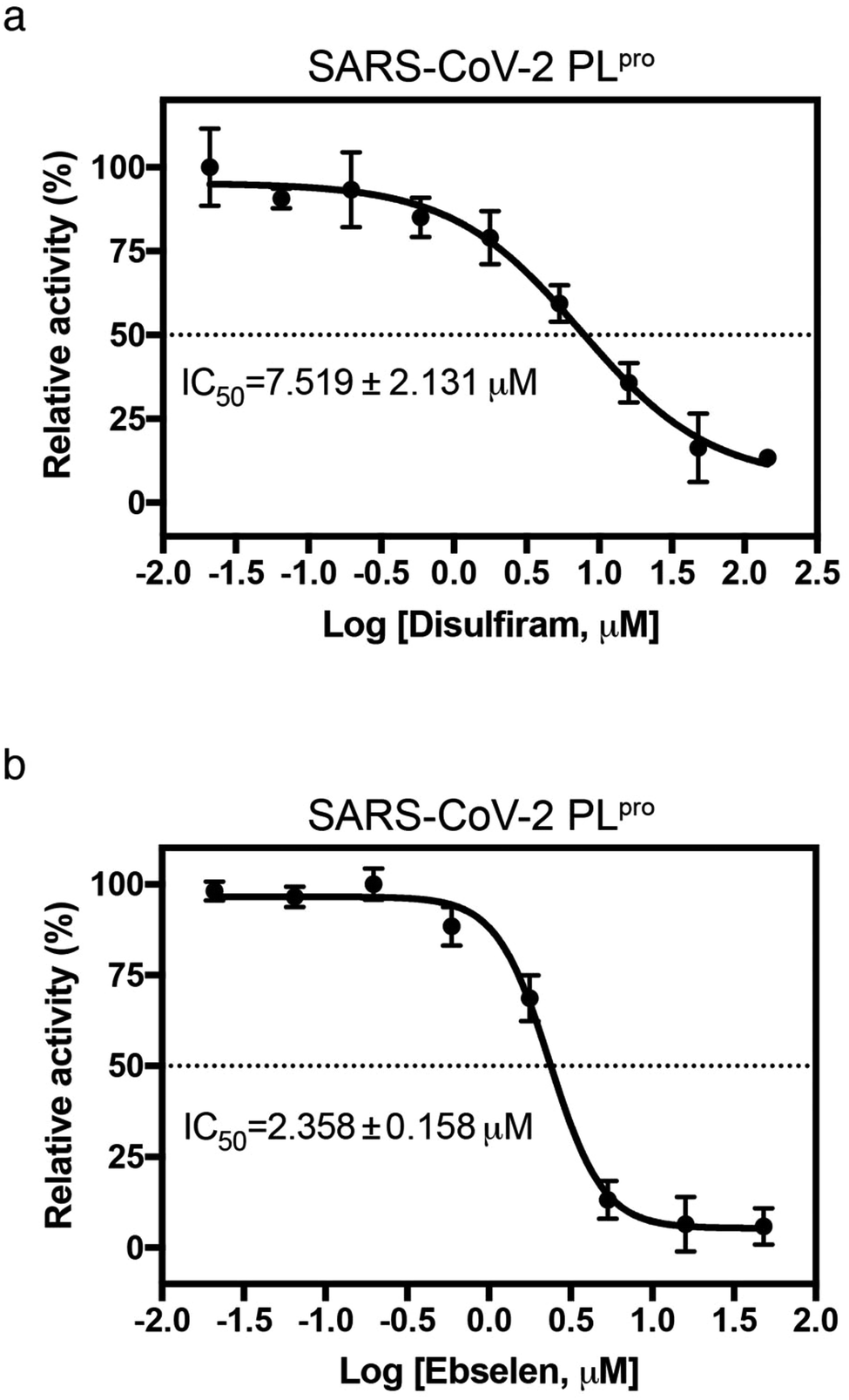 | ||
| Fig. 3 Inhibition of SARS-CoV-2 PLpro by disulfiram and ebselen. The protease activity of PLpro (0.5 μM) in the presence of 0–144 μM disulfiram (a) or 0–48 μM ebselen (b) was assayed using a fluorogenic substrate (50 μM, Dabcy-FTLKGGAPTKVTE-Edans-NH2). The IC50 value was determined by sigmoidal nonlinear regression logistic four parameter analyses using GraphPad Prim software (n = 3, error bars denote standard deviation). | ||
To our knowledge, we are the first to combine knowledge of conserved coronavirus domains and the key factors controlling Zn-bound cysteine reactivity9 to identify previously unknown druggable Zn-sites in multiple SARS-CoV-2 domains. The labile Zn-sites discovered in SARS-CoV-2 are attractive drug targets, as they are highly conserved among coronaviruses and play vital structural/catalytic roles in key proteins in the viral life cycle: the Zn-binding ability of PLpro is crucial for structural integrity and protease activity.19 PLpro not only cleaves the viral polyproteins, but also helps SARS/MERS-CoV to evade the host innate immune response through deubiquitinating and deISGylating enzymatic activities.19,20 The labile Zn-site in nsp10, a crucial cofactor for multiple replicative enzymes such as nsp14 and nsp16,21 plays an important structural role.22 The Zn-binding domain of nsp13 helicase, which catalyzes dsRNA/dsDNA unwinding, is vital for helicase activity.23
Whereas most studies focus on designing inhibitors or repurposing drugs to target a specific viral enzyme/protein such as Mpro,1 our study shows that the same Zn-ejecting drug can attack highly conserved cysteines in multiple viral targets. Disulfiram, used since 1951 with a recommended daily dose of ≤500 mg, and ebselen are considered to be clinically safe.24,25 Both may serve as multi-targeting drugs acting at various stages of the virus life cycle: they can target Zn-bound cysteines in PLpro, nsp10, and possibly nsp13, and/or catalytic cysteines in PLpro and Mpro enzyme. Crippling both PLpro and Mpro enzymes needed to cleave the replicase polyprotein 1a and pp1ab would likely inhibit SARS-CoV-2 replication. Indeed, both disulfiram and ebselen were found to decrease the number of viral RNA copies at 10 μM concentration in SARS-CoV-2-infected Vero E6 cells and ebselen was further shown to inhibit SARS-CoV-2 with a EC50 of 4.67 ± 0.80 μM in plaque-reduction assay.18 Disulfiram/ebselen may serve as a broad-spectrum anti-viral since the domains containing the labile Zn-sites are highly conserved across several types of coronaviruses: disulfiram can inhibit SARS-CoV, MERS-CoV, and SARS-CoV-2 PLproin vitro, whereas most SARS-CoV PLpro inhibitors are inactive against MERS-CoV PLpro.26
A possible advantage of targeting multiple conserved domains is that the virus has to undergo simultaneous appropriate mutations of the different targeted domains to develop drug resistance.8 We propose combining disulfiram/ebselen with other FDA-approved drugs, which have immune-modulatory/anti-inflammatory properties and/or anti-viral effect, to potentially inhibit SARS-CoV-2 replication synergistically. To test this possibility, we chose the zinc ionophore, hydroxychloroquine, because it can downregulate pro-inflammatory cytokines27 and can increase the Zn2+ level inside a cell.28 Increasing intracellular Zn2+ concentration has been shown to inhibit SARS-CoV nsp12 RNA-dependent RNA polymerase, the core enzyme of a multiprotein replication and transcription complex.29 We evaluated antiviral synergy between disulfiram/ebselen and hydroxychloroquine by pretreating Vero E6 cells with the two drugs at various concentrations for 1 h at 37 °C, followed by incubation with SARS-CoV-2 for 1 day at 37 °C (see Methods). We then determined the anti-SARS-CoV-2 activity of each drug or drug combination using immunofluorescence assay to detect SARS-CoV-2 N protein expression (green in Fig. 4a). Consistent with previous results,18 disulfiram and ebselen exhibit an estimated IC50 of 17.5 μM and 23.3 μM, respectively, based on the quantification of SARS-CoV-2 N protein expression (Fig. 4b). The SARS-CoV-2 infection rate in Vero E6 cells treated with a given concentration of hydroxychloroquine and/or disulfiram/ebselen is shown as the mean and corresponding standard deviation of three replicates in Fig. 4c. Disulfiram/ebselen combined with hydroxychloroquine exhibited enhanced antiviral effect compared to each drug alone with p values < 0.05. For example, 12.5 μM disulfiram combined with 5 μM hydroxychloroquine did not affect cell viability, but reduced viral infection compared to disulfiram alone (p value = 0.007) or hydroxychloroquine alone (p value = 0.014). This provides proof-of-concept for combining disulfiram/ebselen with other safe drugs to synergistically inhibit SARS-CoV-2 by targeting multiple conserved viral regions/pathways.
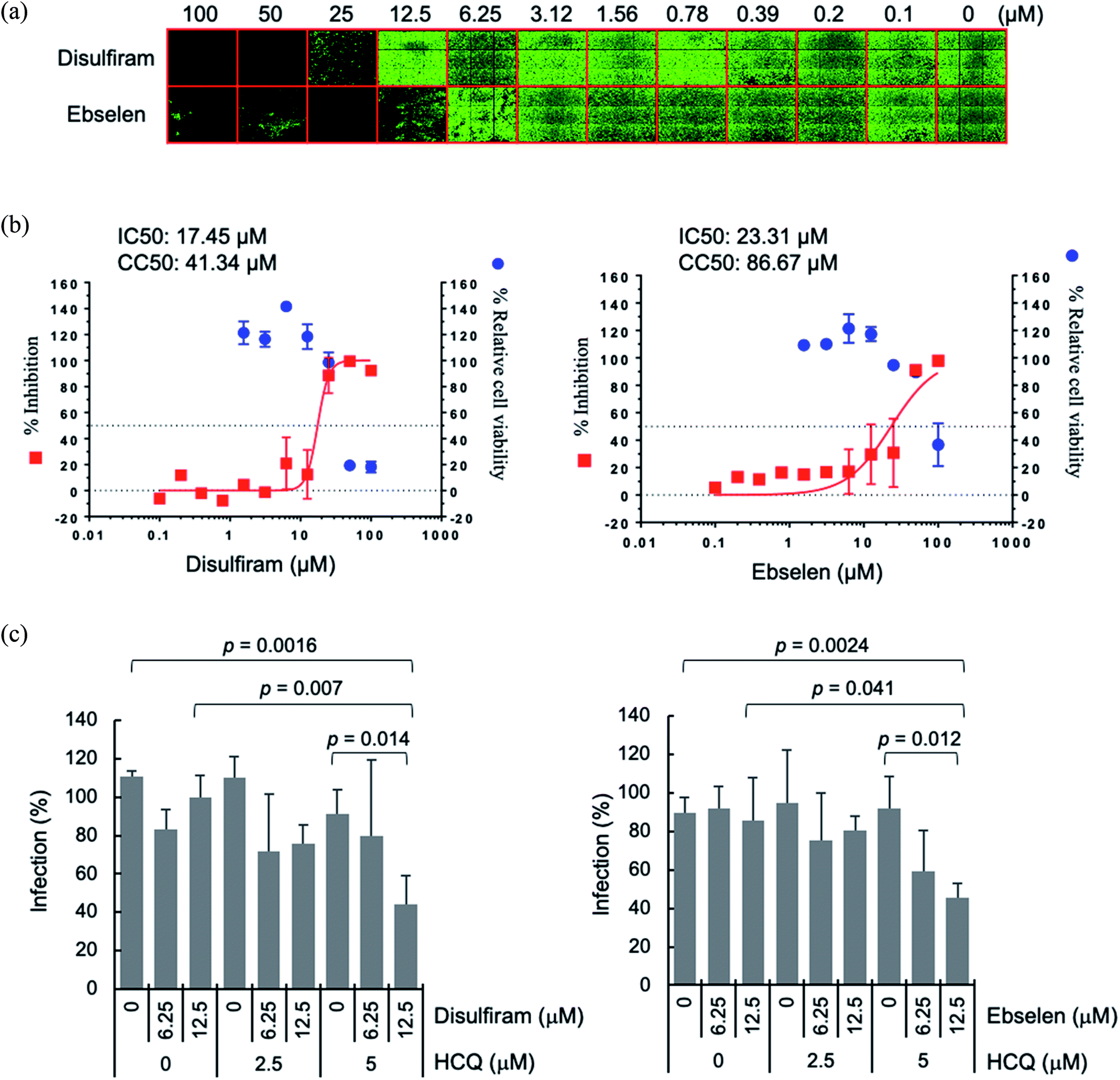 | ||
| Fig. 4 Synergistic antiviral potential of disulfiram/ebselen and hydroxychloroquine (HCQ). (a) The antiviral activities of disulfiram and ebselen against SARS-CoV-2 were determined on Vero E6 cells using immunofluorescence assay to detect SARS-CoV-2 N protein expression (green). (b) Viral infection was quantified by a high-content image analysis system and the average infection rate of no drug treatment was set as 100% for calculation of the 50% inhibitory concentration (IC50). For the 50% cytotoxic concentration (CC50), Vero E6 cells treated with the indicated compound were assayed by Cell Counting Kit-8. IC50 and CC50 were calculated by Prism software. (c) The SARS-CoV-2 infection rates in Vero E6 cells treated with hydroxychloroquine (HCQ) plus disulfiram or ebselen were determined as described above, and shown as means and standard deviations (n = 3). The average infection rate of six sets of experiments with no drug treatment was set as 100%. The p values were calculated by student's t test. | ||
In summary, this study offers a possible strategy to tackle outbreaks of coronaviruses by leveraging the non-specificity of clinically safe Zn-ejector drugs combined with broad-spectrum antivirals to target multiple conserved domains essential for viral replication. Our general strategy based on evolutionary and physical principles can be used to identify druggable Zn-sites in other non-coronaviruses employing essential cysteines and Zn2+ in conserved viral domains.
Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
The cell-based experiments were performed by Dr Jian-Jong Liang (E-mail: jjliang1234@yahoo.com.tw; ) and Dr Chun-Che Liao (jfliao@ibms.sinica.edu.tw) in Dr Yi-Ling Lin's laboratory at the Institute of Biomedical Sciences (IBMS), Academia Sinica, Taipei, Taiwan. We thank Taiwan CDC for providing SARS-CoV-2 (TCDC#4) and funding support for the P3 facility in IBMS from Academia Sinica (AS-CFII-108-102) and the Ministry of Science and Technology, Taiwan (MOST 109-3114-Y-001-001). The MS/MS data analyzed by Orbitrap Fusion mass spectrometer were acquired at the Academia Sinica Common Mass Spectrometry Facilities for Proteomics and Protein Modification Analysis at the Institute of Biological Chemistry, Academia Sinica, supported by Academia Sinica Core Facility and Innovative Instrument Project (AS-CFII-108-107). We thank Dr Tse Wen Chang and Dr Zhu Tong for helpful discussion. We also thank the Biophysics Core and Genomics Core Facilities at the Institute of Molecular Biology, Academia Sinica for assistance in fluorescence and MALDI-TOF MASS assays, respectively. This work was supported by the Ministry of Science & Technology (MOST-107-2113-M-001-018 to C. L.) and Academia Sinica (AS-IA-107-L03 to C. L. and AS-IA-105-L04 to H. S. Y.) Taiwan.References
- G. Li and E. D. Clercq, Nat. Rev. Drug Discovery, 2020, 19, 149–150 CrossRef CAS.
- M. Kar, N. A. Khan, A. Panwar, S. S. Bais, S. Basak, R. Goel, S. Sopory and G. R. Medigeshi, Front. Immunol., 2019, 10, 2347 CrossRef CAS.
- G. Parraga, S. Horvath, A. Eisen and W. E. Taylor, Science, 1988, 241, 1489–1492 CrossRef CAS.
- T. Dudev and C. Lim, J. Am. Chem. Soc., 2007, 129, 12497–12504 CrossRef CAS.
- M. Huang, A. Maynard, J. A. Turpin, L. Graham, G. M. Janini, D. G. Covell and W. G. Rice, J. Med. Chem., 1998, 41, 1371–1381 CrossRef CAS.
- K. Briknarova, C. J. Thomas, J. York and J. H. Nunberg, J. Biol. Chem., 2011, 286, 1528 CrossRef CAS.
- Y.-M. Lee, Y.-F. Lin and C. Lim, J. Chin. Chem. Soc., 2014, 61, 142–150 CrossRef CAS.
- Y.-M. Lee, Y. Duh, S.-T. Wang, M. M. C. Lai, H. S. Yuan and C. Lim, J. Am. Chem. Soc., 2016, 138, 3856–3862 CrossRef CAS.
- Y.-M. Lee and C. Lim, J. Am. Chem. Soc., 2011, 133, 8691–8703 CrossRef CAS.
- Y.-M. Lee, Y.-T. Wang, Y. Duh, H. S. Yuan and C. Lim, J. Am. Chem. Soc., 2013, 135, 14028–14031 CrossRef CAS.
- M.-H. Lin, D. C. Moses, C.-H. Hsieh, S.-C. Cheng, Y.-H. Chen, C.-Y. Sun and C.-Y. Chou, Antiviral Res., 2018, 150, 155–163 CrossRef CAS.
- E. J. Snijder, E. Decroly and J. Ziebuhr, Adv. Virus Res., 2016, 96, 59–126 CAS.
- A. Marchler-Bauer, Y. Bo, L. Han, J. He, C. J. Lanczycki, S. Lu, F. Chitsaz, M. K. Derbyshire, R. C. Geer, N. R. Gonzales, M. Gwadz, D. I. Hurwitz, F. Lu, G. H. Marchler, J. S. Song, N. Thanki, Z. Wang, R. A. Yamashita, D. Zhang, C. Zheng, L. Y. Geer and S. H. Bryant, Nucleic Acids Res., 2017, 45, D200–D203 CrossRef CAS.
- H. M. Berman, T. Battistuz, T. N. Bhat, W. F. Bluhm, P. E. Bourne, K. Burkhardt, Z. Feng, G. L. Gilliland, L. Iype, S. Jain, P. Fagan, J. Marvin, D. Padilla, V. Ravichandran, B. Schneider, N. Thanki, H. Weissig, J. D. Westbrook and C. Zardecki, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2002, 58, 899–907 CrossRef.
- C. Camacho, G. Coulouris, V. Avagyan, N. Ma, J. Papadopoulos, K. Bealer and T. L. Madden, BMC Bioinf., 2009, 10, 421 CrossRef.
- B. Webb and A. Sali, Curr. Protoc. Bioinf., 2016, 54, 5.6.1–5.6.37 CrossRef.
- A. A. Canutescu, A. A. Shelenkov and R. L. Dunbrack Jr, Protein Sci., 2003, 12, 2001–2014 CrossRef CAS.
- Z. Jin, X. Du, Y. Xu, Y. Deng, M. Liu, Y. Zhao, B. Zhang, X. Li, L. Zhang, C. Peng, Y. Duan, J. Yu, L. Wang, K. Yang, F. Liu, R. Jiang, X. Yang, T. You, X. Liu, X. Yang, F. Bai, H. Liu, X. Liu, L. W. Guddat, W. Xu, G. Xiao, C. Qin, Z. Shi, H. Jiang, Z. Rao and H. Yang, Nature, 2020, 582, 289–293 CrossRef CAS.
- N. Barretto, D. Jukneliene, K. Ratia, Z. Chen, A. D. Mesecar and S. C. Baker, J. Virol., 2005, 79, 15189–15198 CrossRef CAS.
- K. Ratia, K. S. Saikatendu, B. D. Santarsiero, N. Barretto, S. C. Baker, R. C. Stevens and A. D. Mesecar, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 5717–5722 CrossRef CAS.
- E. Decroly, C. Debarnot, F. Ferron, M. Bouvet, B. Coutard, I. Imbert, L. Gluais, N. Papageorgiou, A. Sharff, G. Bricogne, M. Ortiz-Lombardia, J. Lescar and B. Canard, PLoS Pathog., 2011, 7, e1002059 CrossRef CAS.
- J. S. Joseph, K. S. Saikatendu, V. Subramanian, B. W. Neuman, A. Brooun, M. Griffith, K. Moy, M. K. Yadav, J. Velasquez, M. J. Buchmeier, R. C. Stevens and P. Kuhn, J. Virol., 2006, 7894–7901 CrossRef CAS.
- Z. Jia, L. Yan, Z. Ren, L. Wu, J. Wang, J. Guo, L. Zheng, Z. Ming, L. Zhang, Z. Lou and Z. Rao, Nucleic Acids Res., 2019, 47, 6538–6550 CrossRef CAS.
- T. N. Srinivasan, T. R. Suresh and J. Vasantha, Indian J. Psychiatry, 1996, 38, 47–50 CAS.
- N. Singh, A. C. Halliday, J. M. Thomas, O. V. Kuznetsova, R. L. Baldwin, E. C. Y. Woon, P. K. Aley, I. Antoniadou, T. Sharp, S. R. Vasudevan and G. C. Churchill, Nat. Commun., 2013, 4, 1332 CrossRef.
- A. Zumla, J. F. W. Chan, E. I. Azhar, D. S. C. Hui and K.-Y. Yuen, Nat. Rev. Drug Discovery, 2016, 15, 327–347 CrossRef CAS.
- R. Willis, A. M. Seif, G. McGwin Jr, L. A. Martinez-Martinez, E. B. González, N. Dang, E. Papalardo, J. Liu, L. M. Vilá, J. D. Reveille, G. S. Alarcón and S. S. Pierangeli, Lupus, 2012, 21, 830–835 CrossRef CAS.
- J. Xue, A. Moyer, B. Peng, J. Wu, B. N. Hannafon and W.-Q. Ding, PLoS One, 2014, 9, e109180 CrossRef.
- A. J. W. t. Velthuis, S. H. E. v. d. Worm, A. C. Sims, R. S. Baric, E. J. Snijder and M. J. v. Hemert, PLoS Pathog., 2010, 6, e1001176 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Fig. S1, S2, and S3 showing, respectively, factors suppressing Zn-bound Cys reactivity, 3d-structures of drug target proteins/sites, and protease active sites after disulfiram/ebselen attack. Tables S1 and S2 and Scheme S1, showing, respectively, clinically used Zn-ejectors, SARS-CoV-2 conserved domains, and the flowchart for predicting labile Zn-sites. Description of experimental methods. See DOI: 10.1039/d0sc02646h |
| ‡ The first four authors made equal contributions. |
| This journal is © The Royal Society of Chemistry 2020 |