
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDiversity-oriented synthesis of peptide-boronic acids by a versatile building-block approach†
Stefan P. A.
Hinkes
 ,
Severin
Kämmerer
and
Christian D. P.
Klein
*
,
Severin
Kämmerer
and
Christian D. P.
Klein
*
Department of Medicinal Chemistry, Institute of Pharmacy and Molecular Biotechnology (IPMB), Heidelberg University, Im Neuenheimer Feld 364, 69120 Heidelberg, Germany. E-mail: c.klein@uni-heidelberg.de
First published on 21st August 2020
Abstract
A new strategy for the synthesis of peptide-boronic acids (PBAs) is presented. 20 Fmoc-protected natural amino acids with orthogonal side-chain protection were straightforwardly converted into their corresponding boron analogues in three simple steps. Subsequent immobilisation on commercially available 1-glycerol polystyrene resin and on-resin transformations yielded a diversity of sequences in high purity. The strategy eliminates various synthetic obstacles such as multi-step routes, low yields, and inseparable impurities. The described method comprises great potential to be implemented in automated combinatorial approaches by markedly facilitating the access to a variety of PBAs. The coupling of amino acids or other building blocks with α-aminoboronates allows the creation of hybrid molecules with significant potential in various scientific disciplines, such as medicinal chemistry, structural biology, and materials science.
Introduction
In the past years, boronic acids have gained importance within numerous research fields, with an unmet versatility as synthetic intermediates and various other applications.1,2 Peptide-boronic acids (PBAs) in particular are used as protease inhibitors3–5 and as covalent ligands in structural biology.4,6 Bortezomib is the first FDA-approved PBA and concurrently the first-in-class proteasome inhibitor.7–9 Numerous other investigational and approved PBA drugs followed, among these are ixazomib,10 delanzomib,11 vaborbactam12 and taniborbactam.13 However, synthesis and structure–activity relationship studies of PBAs remain tedious work, in which the construction of the α-aminoboronate partial structure and deprotection of boronate esters constitute the crucial steps.The established synthetic approaches towards PBAs and related compounds can be subdivided into late-stage borylations and building block approaches (Fig. 1). The most commonly used building block strategy remains the Matteson homologation, with chiral α-chloroboronic esters as key intermediates.14,15 While this transformation is highly stereocontrolled, the harsh reaction conditions restrict the substrate scope and necessitate various transformations after introduction of the boronic ester. An elegant substrate-controlled asymmetric borylation of imines was developed by Ellman and coworkers in 2008,16 and optimised in 2014.17 However, this approach remains underexploited towards the synthesis of boron equivalents of proteinogenic amino acids, likely due to the scarcity of suitable aldehyde precursors.
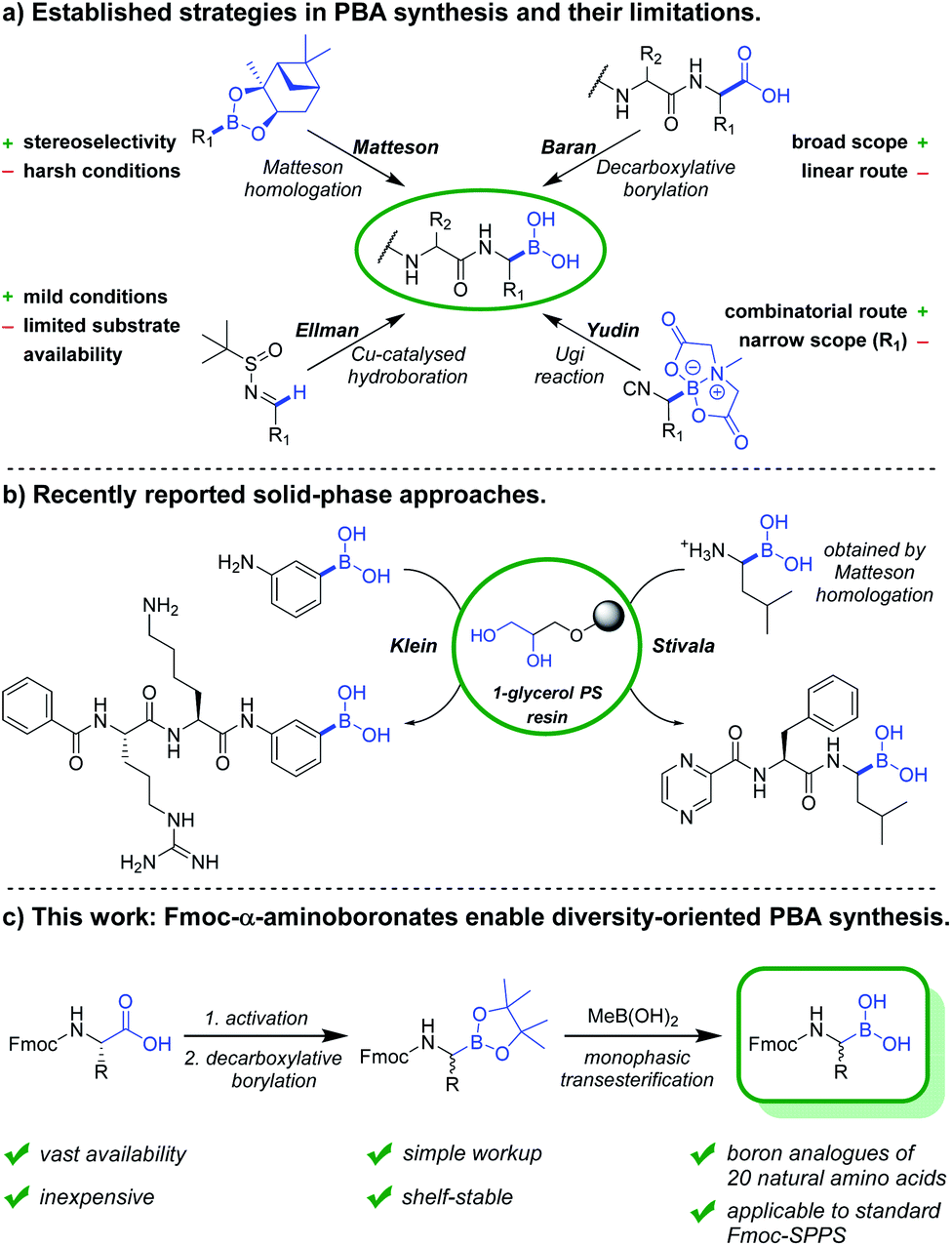 | ||
| Fig. 1 Strategies for the synthesis of peptide-boronic acids. (a) Commonly used solution-phase approaches and their limitations. (b) Recently reported solid-phase approaches. (c) This work: facilitated access to a diversity of PBAs by introducing N-Fmoc-α-aminoboronic acids as versatile building blocks. | ||
Recently, Baran and coworkers reported the Ni-catalysed decarboxylative borylation of redox-active esters.18 Along with its Cu-catalysed variant,19 it constituted a milestone in boronic acid synthesis due to its broad scope. Although primary, secondary, tertiary and peptidic carboxylic acids were readily converted into their boron counterparts, N-Fmoc-α-aminoboronates have not been reported, probably due to their poor stability in presence of silica gel. The Yudin group recently reported the design of boron-based peptidomimetics in an elegant multicomponent approach using α-boryl isocyanides (Fig. 1).5,20 However, only derivatives of boro-Phe and boro-Leu have been reported. In 2020, Reyes et al. described the asymmetric Rh-catalysed synthesis of α-aminoboronates using a chiral monophosphite ligand.21 Although the concept is very promising, the reaction scope is restricted by the necessity of a hydrophobic interaction between aromatic moieties of substrate and chiral ligand. While late-stage borylations can be superior in linear synthesis routes, versatile building blocks are obviously indispensable for convergent and diversity-oriented reaction sequences.
To date, several on-resin transformations of aromatic and aliphatic boronates have been described.22–26 Our group recently reported commercially available 1-glycerol polystyrene resin to be compatible with Fmoc-based chemistry,27 which has been applied to the solid-phase synthesis of bortezomib and ixazomib by Daniels and Stivala.28 Obviously, a combinatorial approach consisting of routine and automatable steps would grant access to complex libraries, thereby allowing the exploration of novel chemical space and biomolecular interactions. However, no route has yet been described for the synthesis of Fmoc-α-aminoboronates. We hereby report a strategy to obtain these versatile building blocks, which can be readily employed in solid-phase synthesis to obtain a diversity of PBA sequences.
Results and discussion
Considering the recent achievements in decarboxylative borylation reactions,18,19,29,30 Fmoc-α-aminocarboxylic acids derived from natural amino acids appeared to be prime starting materials for an innovative building block strategy due to their diversity, wide availability and applicability to solid-phase synthesis.We initially investigated the formation and stability of redox-active N-hydroxyphthalimide (NHPI) esters as substrates in decarboxylative borylations. Although only few examples of Fmoc-protected NHPI esters are described in the literature,31,32 we found that transformations went smoothly in most cases and provided stable products in high yields (Table 1). Only some compounds showed instabilities and side reactions were observed, for example δ-lactam formation33 with all tested monoprotected (–Pbf, –Mtr, –Tos, –NO2) arginine derivatives. Fortunately, the NHPI ester of bis-Boc protected arginine (2q) could be easily obtained. Fmoc-His(Trt)-ONHPI (2u) was found to be of limited stability towards silica gel, but decomposition could be minimised by using deactivated silica gel (SiO2/H2O 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :35, m/m) in the purification step.
:35, m/m) in the purification step.
|
|
|---|
| a Isolated yields. b Reaction conditions: 1 (0.5–1.0 mmol scale, 1.0 equiv.), NHPI (1.0 equiv.), DIC (1.1 equiv.), DMAP (0.1 equiv.), DCM, 0 °C, 1 h; rt, 2–16 h. c Reaction conditions: NiCl2·6H2O (10 mol%), 4,4′-dimethoxy-2,2′-bipyridyl (13 mol%), MgBr2·OEt2 (1.5 equiv.), [B2pin2Me]Li (3.0 equiv.), THF, 0 °C, 1 h; rt, 1 h; sIBX (6.0 equiv.), EtOAc, 40 °C, 2 h (see the ESI for details). d Without sIBX workup, therefore obtained as a crude material containing B2pin2 impurities. |

|

The obtained redox-active esters were examined for their viability as substrates in the recently published Ni-catalysed decarboxylative borylation. To our delight, the vast majority of building blocks could be transformed into their corresponding pinacolyl boronates, however accompanied by the expected loss of stereoinformation at the α-carbon due to the radical reaction mechanism.18 For all tested cysteine derivatives (3v, 3w and 3x), only trace amounts could be identified, indicating that the presence of a sulfur atom in β-position is detrimental. No appreciable side reactions were observed during the borylation of Fmoc-Met-ONHPI (2g), where sulfur is located in γ-position. It should be noted that we were unable to obtain acceptable yields of Fmoc-α-aminoboronates by applying the operationally simpler Cu-catalysed decarboxylative borylation method.19
Workup of the crude borylation products proved demanding at first attempts. α-Amino pinacolyl boronates are known to lack stability on silica gel or alumina columns, and it should be noted that the tested compounds even decomposed partially on deactivated silica gel and product fractions were often found to be contaminated with phthalimide and B2pin2. Therefore, chromatography did not seem feasible and the workup procedure was optimised to circumvent chromatographic purification: after filtration of the quenched reaction mixture, the crude residue was treated with stabilised 2-iodoxybenzoic acid (sIBX).34 Delightfully, the addition of sIBX oxidatively destroyed the excess B2pin2 while leaving the α-amino pinacolyl boronates unaffected, thereby providing evidence of their relative stability towards mild oxidants. A short extraction procedure was appended to simultaneously remove phthalimide, B2pin2 degradation products and IBX stabilisers (see the ESI† for details).
Applying the optimised workup procedure allowed us to obtain the desired N-Fmoc-α-pinacolyl boronates in good yields (Table 1). Most transformations went smoothly and without impairment of the respective side-chain functionalities, confirming the findings of Li et al.18 and highlighting the broad scope of the Ni-catalysed decarboxylative borylation. Fmoc-Met-Bpin (3g) was found to be partially oxidised to its sulfoxide by IBX under the tested conditions as it is described for other thioethers.35 Therefore, the IBX workup was bypassed in the case of 3g to obtain the compound with B2pin2 impurities, which could be removed in the subsequent step (vide infra).
Initial attempts to deprotect the pinacolyl boronates with established methods failed.36–38 However, our recently published monophasic transesterification method39 enabled the deprotection of pinacolyl building blocks in a straightforward fashion. The procedure was further optimised to allow a direct lyophilisation after complete conversion to avoid elevated temperatures. Reactants were transesterified with volatile methylboronic acid in mixtures of acetonitrile and dilute aqueous hydrochloric acid to give solutions that could be freeze-dried to obtain the desired compounds in very high yields (see the ESI† for details). While conditions A were expedient for acid-insensitive compounds and superior in terms of methylboronic acid equivalents, reducing the amount of dilute hydrochloric acid was essential to prevent premature side-chain deprotection of some acid-labile compounds (conditions B). Unexpectedly, partial side-chain deprotection was observed for compounds 4n, 4q and 4t even when binary water/acetonitrile mixtures were used as a solvent, presumably due to the acidity of methylboronic acid. Therefore, a buffer system using phosphate buffer pH 7.0 and a short extraction step were employed in the transesterification step (conditions C). With these three conditions in hand, all 20 investigated compounds could be obtained in high yields (Table 2). We emphasise that this is the first method to straightforwardly obtain a library of α-aminoboronic acid building blocks that are readily employable in diversity-oriented synthesis approaches.
|
|
|---|
|
a Isolated yields.
b Reaction conditions (0.1–0.6 mmol scale): compound 3 (1.0 equiv.), MeB(OH)2 (5.0 equiv.), MeCN/0.1 N HCl (1:1, v/v), rt, 16 h.
c Reaction conditions (0.1–0.6 mmol scale): compound 3 (1.0 equiv.), MeB(OH)2 (10.0 equiv.), MeCN/0.1 N HCl (9:1, v/v), rt, 2 h.
d Reaction conditions (0.1–0.6 mmol scale): compound 3 (1.0 equiv.), MeB(OH)2 (10.0 equiv.), MeCN/phosphate buffer pH 7.0 (1:1, v/v), rt, 2 h.
e Applying the same transesterification conditions to crude compound 3 according to route 2, Scheme 1.
|

|

Our recent findings indicated that B2pin2 could be converted into volatile or water-soluble degradation products by double transesterification with MeB(OH)2 under acidic aqueous conditions.39 These results prompted us to circumvent IBX workup in the case of oxidation-sensitive derivative 3g. Delightfully, the crude compound could be transesterified smoothly to give a product with boric acid contaminants that were easily removed by addition of methanol and evaporation of the formed trimethyl borate under reduced pressure (see the ESI† for details).
Inspired by these results, an optimised protocol with superior overall atom economy was established (Scheme 1, route 2) and tested for selected compounds (Table 2). While the oxidative destruction of excess B2pin2 with IBX (route 1) allows yield determination and full characterisation of Fmoc-α-aminopinacolyl boronates, the simultaneous transesterification of pinacolyl boronates and B2pin2 is considerably faster and less expensive. Additionally, IBX is a hazardous and potentially explosive reagent.40 Route 2 is therefore preferable for large-scale syntheses and industrial applications.
 | ||
| Scheme 1 Process optimisation for the preparation of Fmoc-α-aminoboronates. Route 1: oxidative removal of B2pin2 contaminants with IBX. Route 2: simultaneous transesterification of pinacolyl boronates and B2pin2 contaminants (see the ESI† for details). | ||
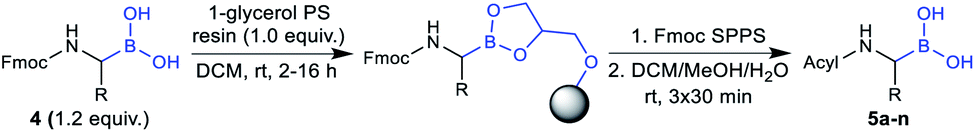
The utility of the synthesised Fmoc-α-aminoboronates for solid-phase synthesis approaches was subsequently investigated. The compounds were readily soluble in methylene chloride, thereby enabling their straightforward immobilisation onto 1-glycerol polystyrene resin. No additives, e.g. bases, were necessary to efficiently immobilise the building blocks under these conditions. It should be noted that the use of tetrahydrofuran as in literature-known protocols27,28 led to diminished yields, most likely due to its hygroscopy or its propensity to form peroxides that would be detrimental to boronic acids. Mild and efficient cleavage from solid support was achieved by treating the resin with DCM/MeOH/H2O (5:4:1, v/v/v). All 20 building blocks were tested regarding their loading efficiency and were shown to be readily immobilised and cleaved from solid support (Table 3).
|
|
|||
|---|---|---|---|
| Entry | 4 | Compound | Loading efficiencyb |
|
a Reaction conditions: 1-glycerol polystyrene resin (100.0 mg, B = 0.60 mmol g−1, 0.06 mmol, 1.0 equiv.), compound 4 (0.072 mmol, 1.2 equiv.), DCM (1.5 mL), rt, 2 h. Cleavage was performed with DCM/MeOH/H2O (5:4:1, v/v/v) for 3 × 30 min (see the ESI for details).
b Calculated by the molar ratio of cleaved compound 4 and the stated loading capacity of 1-glycerol polystyrene resin (B = 0.60 mmol g−1).
|
|||
| 1 | (±)-4a | boro-Gly | 66% |
| 2 | (±)-4b | boro-Ala | 81% |
| 3 | (±)-4c | boro-Val | 46% |
| 4 | (±)-4d | boro-Leu | 60% |
| 5 | 4e | boro-Ile | 64% |
| 6 | (±)-4f | boro-Pro | 60% |
| 7 | (±)-4g | boro-Met | 43% |
| 8 | (±)-4h | boro-Phe | 46% |
| 9 | (±)-4i | boro-Asn(Trt) | 44% |
| 10 | (±)-4j | boro-Gln(Trt) | 62% |
| 11 | (±)-4k | boro-Asp(tBu) | 71% |
| 12 | (±)-4l | boro-Glu(tBu) | 84% |
| 13 | (±)-4m | boro-Ser(tBu) | 67% |
| 14 | 4n | boro-Thr(tBu) | 66% |
| 15 | (±)-4o | boro-Orn(Boc) | 84% |
| 16 | (±)-4p | boro-Lys(Boc) | 66% |
| 17 | (±)-4q | boro-Arg(bis-Boc) | 54% |
| 18 | (±)-4r | boro-Tyr(tBu) | 26% |
| 19 | (±)-4s | boro-Trp(Boc) | 49% |
| 20 | (±)-4t | boro-His(Boc) | 70% |

As further proof of concept, a selection of building blocks was applied in standard Fmoc solid-phase synthesis protocols. Sequences could be obtained in high purity with full conservation of side-chain protecting groups. Due to the non-acidic cleavage conditions, the described method provides access to synthetic intermediates readily applicable to late-stage diversifications, e.g. petasis reactions41–43 or cross-coupling reactions.44,45
Final compounds were obtained by side-chain deprotection using TFA-based cleavage solutions and subsequent HPLC purification. Syntheses were successful for building blocks with nonpolar (5a–e), polar (5f–l) and aromatic (5m–n) side-chains, underlining the general versatility of the described approach (Table 4).
|
|
|---|
|
a Isolated yields.
b Loading step: compound 4 (1.2 equiv.), DCM, rt, 2–16 h. Peptide elongation was performed using standard Fmoc-SPPS protocols (see the ESI for details). Cleavage step: DCM/MeOH/H2O 5:4:1 (v/v/v), 3 × 30 min.
c Reaction conditions: 40% TFA in methylene chloride, rt, 2 h.
d Reaction conditions: MeCN/TFA/H2O (7:2:1, v/v/v), rt, 2 h.
|

|
Conclusions
We hereby present a straightforward method for the conversion of Fmoc-α-aminocarboxylic acids into their boron equivalents in three simple steps. Additionally, the efficient deprotection of boronic esters under neutral, buffered conditions was established in the course of our investigations. Considering the utility of the intermediates for automated synthesis, the process was further optimised by establishing a fast protocol with enhanced atom economy. All building blocks were shown to be employable in standard Fmoc-based solid-phase synthesis protocols and yielded a diversity of peptide-boronic acids. The synthetic toolbox provided here will likely facilitate the diversity-oriented synthesis of PBAs with a multitude of potential applications in drug discovery, life sciences and materials research.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Tobias Timmermann for NMR support, Heiko Rudy and Daniel Wolf for HRMS measurements, Sebastian Fuss and Bettina Metz for their participation in the precursor synthesis. S. K. acknowledges funding by a fellowship of the German National Academic Foundation (Studienstiftung des deutschen Volkes).Notes and references
- For a general overview of the synthesis of aminoboronates, see: A. Šterman, I. Sosič, S. Gobec and Z. Časar, Org. Chem. Front., 2019, 6, 2991–2998 RSC.
- For a recent review about α-aminoboronates, see: P. Andrés, G. Ballano, M. I. Calaza and C. Cativiela, Chem. Soc. Rev., 2016, 45, 2291–2307 RSC.
- For an introduction to boron-containing compounds as protease inhibitors, please see: R. Smoum, A. Rubinstein, V. M. Dembitsky and M. Srebnik, Chem. Rev., 2012, 112, 4156–4220 CrossRef CAS.
- C. Nitsche, L. Zhang, L. F. Weigel, J. Schilz, D. Graf, R. Bartenschlager, R. Hilgenfeld and C. D. Klein, J. Med. Chem., 2017, 60, 511–516 CrossRef CAS.
- J. Tan, J. J. Grouleff, Y. Jitkova, D. B. Diaz, E. C. Griffith, W. Shao, A. F. Bogdanchikova, G. Poda, A. D. Schimmer, R. E. Lee and A. K. Yudin, J. Med. Chem., 2019, 62, 6377–6390 CrossRef CAS.
- J. Lei, G. Hansen, C. Nitsche, C. D. Klein, L. Zhang and R. Hilgenfeld, Science, 2016, 353, 503–505 CrossRef CAS.
- J. Adams, M. Behnke, S. Chen, A. A. Cruickshank, L. R. Dick, L. Grenier, J. M. Klunder, Y.-T. Ma, L. Plamondon and R. L. Stein, Bioorg. Med. Chem. Lett., 1998, 8, 333–338 CrossRef CAS.
- J. Adams, V. J. Palombella, E. A. Sausville, J. Johnson, A. Destree, D. D. Lazarus, J. Maas, C. S. Pien, S. Prakash and P. J. Elliott, Cancer Res., 1999, 59, 2615 CAS.
- A. Paramore and S. Frantz, Nat. Rev. Drug Discovery, 2003, 2, 611–612 CrossRef CAS.
- E. Kupperman, E. C. Lee, Y. Cao, B. Bannerman, M. Fitzgerald, A. Berger, J. Yu, Y. Yang, P. Hales, F. Bruzzese, J. Liu, J. Blank, K. Garcia, C. Tsu, L. Dick, P. Fleming, L. Yu, M. Manfredi, M. Rolfe and J. Bolen, Cancer Res., 2010, 70, 1970 CrossRef CAS.
- R. C. Roemmele and M. A. Christie, Org. Process Res. Dev., 2013, 17, 422–426 CrossRef CAS.
- S. J. Hecker, K. R. Reddy, M. Totrov, G. C. Hirst, O. Lomovskaya, D. C. Griffith, P. King, R. Tsivkovski, D. Sun, M. Sabet, Z. Tarazi, M. C. Clifton, K. Atkins, A. Raymond, K. T. Potts, J. Abendroth, S. H. Boyer, J. S. Loutit, E. E. Morgan, S. Durso and M. N. Dudley, J. Med. Chem., 2015, 58, 3682–3692 CrossRef CAS.
- A. Krajnc, J. Brem, P. Hinchliffe, K. Calvopiña, T. D. Panduwawala, P. A. Lang, J. J. A. G. Kamps, J. M. Tyrrell, E. Widlake, B. G. Saward, T. R. Walsh, J. Spencer and C. J. Schofield, J. Med. Chem., 2019, 62, 8544–8556 CrossRef CAS.
- D. S. Matteson and D. Majumdar, J. Am. Chem. Soc., 1980, 102, 7588–7590 CrossRef CAS.
- D. S. Matteson, R. Ray, R. R. Rocks and D. J. S. Tsai, Organometallics, 1983, 2, 1536–1543 CrossRef CAS.
- M. A. Beenen, C. An and J. A. Ellman, J. Am. Chem. Soc., 2008, 130, 6910–6911 CrossRef CAS.
- A. W. Buesking, V. Bacauanu, I. Cai and J. A. Ellman, J. Org. Chem., 2014, 79, 3671–3677 CrossRef CAS.
- C. Li, J. Wang, L. M. Barton, S. Yu, M. Tian, D. S. Peters, M. Kumar, A. W. Yu, K. A. Johnson, A. K. Chatterjee, M. Yan and P. S. Baran, Science, 2017, 356, eaam7355 CrossRef.
- J. Wang, M. Shang, H. Lundberg, K. S. Feu, S. J. Hecker, T. Qin, D. G. Blackmond and P. S. Baran, ACS Catal., 2018, 8, 9537–9542 CrossRef CAS.
- A. Zajdlik, Z. Wang, J. L. Hickey, A. Aman, A. D. Schimmer and A. K. Yudin, Angew. Chem., Int. Ed., 2013, 52, 8411–8415 CrossRef CAS.
- R. L. Reyes, M. Sato, T. Iwai and M. Sawamura, J. Am. Chem. Soc., 2020, 142, 589–597 CrossRef CAS.
- B. Carboni, C. Pourbaix, F. Carreaux, H. Deleuze and B. Maillard, Tetrahedron Lett., 1999, 40, 7979–7983 CrossRef CAS.
- D. G. Hall, J. Tailor and M. Gravel, Angew. Chem., Int. Ed., 1999, 38, 3064–3067 CrossRef CAS.
- C. Pourbaix, F. Carreaux, B. Carboni and H. Deleuze, Chem. Commun., 2000, 1275–1276 RSC.
- M. Gravel, K. A. Thompson, M. Zak, C. Bérubé and D. G. Hall, J. Org. Chem., 2002, 67, 3–15 CrossRef CAS.
- R. M. Dunsdon, J. R. Greening, P. S. Jones, S. Jordan and F. X. Wilson, Bioorg. Med. Chem. Lett., 2000, 10, 1577–1579 CrossRef CAS.
- M. A. M. Behnam, T. R. Sundermann and C. D. Klein, Org. Lett., 2016, 18, 2016–2019 CrossRef CAS.
- B. E. Daniels and C. E. Stivala, RSC Adv., 2018, 8, 3343–3347 RSC.
- A. Fawcett, J. Pradeilles, Y. Wang, T. Mutsuga, E. L. Myers and V. K. Aggarwal, Science, 2017, 357, 283–286 CrossRef CAS.
- L. Xu, Eur. J. Org. Chem., 2018, 3884–3890 CrossRef CAS.
- G. H. L. Nefkens, G. I. Tesser and R. J. F. Nivard, Recl. Trav. Chim. Pays-Bas, 1962, 81, 683–690 CrossRef CAS.
- J. Cornella, J. T. Edwards, T. Qin, S. Kawamura, J. Wang, C.-M. Pan, R. Gianatassio, M. Schmidt, M. D. Eastgate and P. S. Baran, J. Am. Chem. Soc., 2016, 138, 2174–2177 CrossRef CAS.
- M. H. Cezari and L. Juliano, Pept. Res., 1996, 9, 88–91 CAS.
- A. Ozanne, L. Pouységu, D. Depernet, B. François and S. Quideau, Org. Lett., 2003, 5, 2903–2906 CrossRef CAS.
- V. G. Shukla, P. D. Salgaonkar and K. G. Akamanchi, J. Org. Chem., 2003, 68, 5422–5425 CrossRef CAS.
- S. J. Coutts, J. Adams, D. Krolikowski and R. J. Snow, Tetrahedron Lett., 1994, 35, 5109–5112 CrossRef CAS.
- J. Sun, M. T. Perfetti and W. L. Santos, J. Org. Chem., 2011, 76, 3571–3575 CrossRef CAS.
- D. S. Matteson, T. J. Michnick, R. D. Willett and C. D. Patterson, Organometallics, 1989, 8, 726–729 CrossRef CAS.
- S. P. A. Hinkes and C. D. P. Klein, Org. Lett., 2019, 21, 3048–3052 CrossRef CAS.
- J. B. Plumb and D. J. Harper, Chem. Eng. News, 1990, 68, 3 CAS.
- N. A. Petasis and I. Akritopoulou, Tetrahedron Lett., 1993, 34, 583–586 CrossRef CAS.
- M. G. Ricardo, D. Llanes, L. A. Wessjohann and D. G. Rivera, Angew. Chem., Int. Ed., 2019, 58, 2700–2704 CrossRef CAS.
- A. Yamaguchi, S. J. Kaldas, S. D. Appavoo, D. B. Diaz and A. K. Yudin, Chem. Commun., 2019, 55, 10567–10570 RSC.
- A. Bonet, M. Odachowski, D. Leonori, S. Essafi and V. K. Aggarwal, Nat. Chem., 2014, 6, 584–589 CrossRef CAS.
- S. Laulhé, J. M. Blackburn and J. L. Roizen, Org. Lett., 2016, 18, 4440–4443 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, procedures and characterisation of all compounds, including 1H and 13C NMR spectra. See DOI: 10.1039/d0sc03999c |
| This journal is © The Royal Society of Chemistry 2020 |