
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA near-infrared fluorogenic dimer enables background-free imaging of endogenous GPCRs in living mice†
Lucie Esteoulle‡
a,
François Daubeuf‡a,
Mayeul Collot *b,
Stéphanie Richéa,
Thierry Durrouxc,
David Brassed,
Patrice Marchandd,
Julie Karpenko*a,
Andrey S. Klymchenko*b and
Dominique Bonnet*a
*b,
Stéphanie Richéa,
Thierry Durrouxc,
David Brassed,
Patrice Marchandd,
Julie Karpenko*a,
Andrey S. Klymchenko*b and
Dominique Bonnet*a
aLaboratoire d’Innovation Thérapeutique, LabEx MEDALIS, UMR7200, CNRS, Université de Strasbourg, 74 route du Rhin, 67401 Illkirch-Graffenstaden, France. E-mail: dominique.bonnet@unistra.fr; julie.karpenko@unistra.fr
bLaboratoire de Bioimagerie et Pathologies, UMR 7021, CNRS/Université de Strasbourg, 74 route du Rhin, 67401 Illkirch-Graffenstaden, France. E-mail: andrey.klymchenko@unistra.fr; mayeul.collot@unistra.fr
cCNRS, UMR 5203, Institut de Génomique Fonctionnelle, INSERM, U. 1191, Université de Montpellier, Montpellier, France
dUniversité de Strasbourg, CNRS, IPHC UMR 7178, F-67000 Strasbourg, France
First published on 17th June 2020
Abstract
Fluorescent probes are commonly used in studying G protein-coupled receptors in living cells; however their application to the whole animal receptor imaging is still challenging. To address this problem, we report the design and the synthesis of the first near-infrared emitting fluorogenic dimer with environment-sensitive folding. Due to the formation of non-fluorescent H-aggregates in an aqueous medium, the near-infrared fluorogenic dimer displays a strong turn-on response (up to 140-fold) in an apolar environment and exceptional brightness: 56% quantum yield and ≈444![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 M−1 cm−1 extinction coefficient. Grafted on a ligand of the oxytocin receptor, it allows the unprecedented background-free and target-specific imaging of the naturally expressed receptor in living mice.
000 M−1 cm−1 extinction coefficient. Grafted on a ligand of the oxytocin receptor, it allows the unprecedented background-free and target-specific imaging of the naturally expressed receptor in living mice.
Introduction
G protein-coupled receptors (GPCRs) are the largest family of the transmembrane receptors in humans. GPCRs are involved in virtually all aspects of human physiology in health and disease.1 Not surprisingly, GPCRs are molecular targets of more than 30% of currently available drugs on the market.2 Each member of the GPCR family has a unique tissue-dependent expression and localization pattern which is crucial for playing its physiological and physiopathological roles. Therefore, to correlate the expression level of GPCRs to a disease, it is crucial to access their spatial distribution at the cell and also the organismal levels.Regarding the whole-organism molecular imaging techniques, fluorescence-based contrast agents emit non-ionizing radiation and have longer shelf lifetime compared with radioisotope-based probes. Moreover, the fluorescence properties of organic dyes can be modulated in a wide range by chemical modifications.3–5 Although fluorescent probes are readily used in studying GPCRs in living cells,6–8 the whole animal fluorescence imaging of receptors is still in its infancy. A few reports have been focused on the imaging of transgenic GPCRs in living mice. For instance, Ma et al.9 imaged the α1-AR receptor in a xenograft model using a far-red dye–ligand conjugate. More recently, Alcobia et al. used the β2-adrenergic receptor fused to the bioluminescent reporter NanoLuc enabling the detection of the receptor–ligand binding by BRET.10 However, to the best of our knowledge, no example of fluorescence imaging of endogenous GPCRs has been reported in mice, mostly due to the low expression level of many endogenous GPCRs and the lack of appropriate fluorescent probes.
Ideally, a fluorescent probe for the in vivo imaging of endogenous GPCRs should meet the following requirements: (1) high affinity and selectivity for its target; (2) absorption and emission of the probe in the near-infrared (NIR) region, which is an optical window minimizing the light scattering and absorption in tissues and blood to enhance tissue penetration;11–13 and (3) a fluorogenic character to “turn on” its fluorescence after binding to the target receptor to ensure a high signal-to-noise ratio. Fluorogenic dyes have been successfully used for background-free detection and imaging of various analytes,14–16 and have been developed by us for the detection of ligand–GPCR binding in living cells.17,18 Of particular interest are probes operating by the self-quenching mechanism, where closely connected dyes undergo proximal orbital overlap and form non-emissive H-aggregates, which turn on their emission after target recognition or enzymatic cleavage.19–22 Recently, we have reported the concept of fluorogenic squaraine dimers with environment-sensitive folding which allowed for the visualization of GPCRs in living cells under no-wash conditions with a high signal-to-noise ratio.23 In an aqueous medium, the formation of the dimer of the H-aggregate type resulted in complete fluorescence quenching of the probe. In contrast, once bound to the receptor, the fluorophores were exposed to the hydrophobic environment of the biomembrane, which led to dissociation of the dimer and recovery of fluorescence. Although the squaraine dimer is a powerful tool for receptor labelling in living cells, it displays absorption and emission in the far-red region which is not optimal for the in vivo imaging.
Here, based on the dimer concept, we developed a NIR fluorogenic probe with unprecedented brightness allowing for the first time the background-free detection of an endogenous GPCR, the oxytocin receptor (OTR), in living mice. The OTR is known to be involved in the modulation of complex social behavior such as social recognition, attachment, empathy, and trust,24 and is proposed as a potential therapeutic target for the treatment of the autistic spectrum disorders.25 In mice, the OTR is highly expressed in the uterus during pregnancy and in the mammary glands during late pregnancy and lactation.26 However, direct in vivo optical imaging of this receptor and GPCRs in general has remained a challenge so far.
Results and discussion
The key element in the design of the NIR fluorogenic dimer probe for the OTR is the choice of the fluorophore. In addition to operating in the NIR window (700–950 nm), the fluorophore should be bright, photostable and sufficiently water-soluble. For this purpose, we designed and synthesized an original cyanine derivative 2 (Cy5.5) decorated with a PEG8 chain (Scheme 1 and the ESI†), which is intended to compensate for the lipophilic character of the dye and to avoid non-specific interactions.17,23,27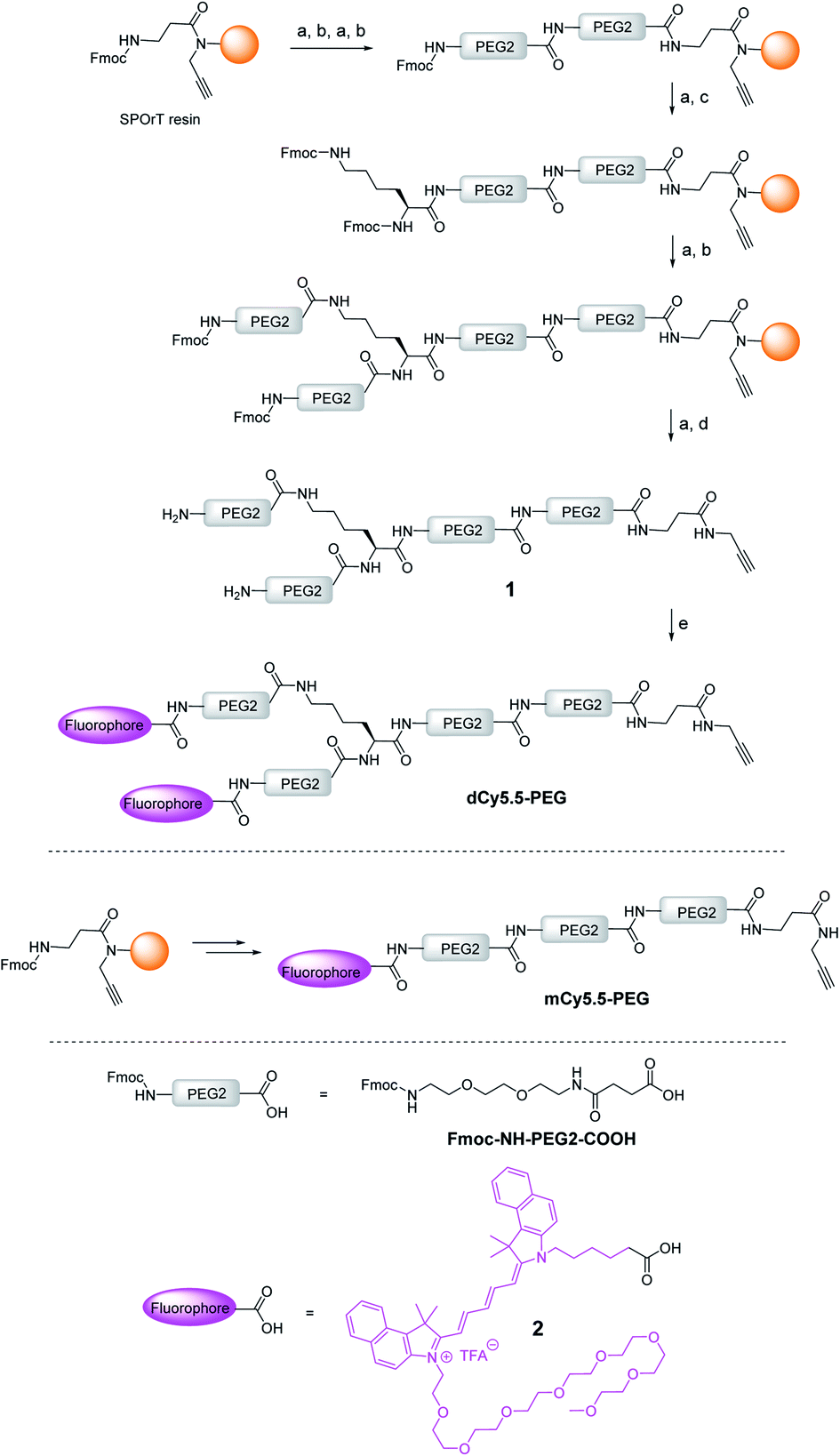 | ||
| Scheme 1 Synthesis of the NIR fluorogenic dimer dCy5.5-PEG and the monomer mCy5.5-PEG. Conditions: (a) piperidine/DMF, (b) Fmoc–NH–PEG2–COOH, HBTU, HOBt, DIPEA, DMF, (c) Fmoc–L–Lys(Fmoc)–OH, HBTU, HOBt, DIPEA, DMF, (d) TFA/TIS/H2O, and (e) 2, PyBOP, DIPEA, DMF. | ||
The construct bearing two fluorophore copies and the OTR ligand was built using PEG linker 1, presenting two amino groups and an alkyne moiety (Scheme 1). PEG units were selected to ensure enough flexibility to the linker in order to facilitate the formation of intramolecular aggregates between both dyes and to improve the water solubility of the probe. The alkyne was introduced by performing the synthesis of 1 on SPOrT resin, developed previously by our group,28 using a Fmoc/tBu approach and HBTU/HOBt activation. First, two PEG2 units were assembled using the Fmoc–NH–PEG2–COOH amino acid, which was obtained as described by Soriano, et al.29 Then, the Fmoc–L-Lys(Fmoc)–OH amino acid was introduced to create the bifurcation. Finally, the chain was elongated with another PEG2 unit, and the terminal Fmoc protecting groups were removed. The resulting dimeric chain 1 was cleaved from the solid phase in a mixture of TFA/H2O/TIS 95/2.5/2.5 (v/v) and isolated by semi-preparative HPLC. Under these acidic conditions, the alkyne moiety was found to be fully stable. The introduction of the fluorophore 2 was then performed in solution using PyBOP in situ activation, yielding the NIR dimer dCy5.5-PEG. The dye was coupled in solution to avoid the use of an excess of the costly fluorophore usually required by solid-phase reactions and to preserve the dye from potential degradations during the final solid-phase acidic cleavage step. In a similar way, by skipping the introduction of Fmoc–L-Lys(Fmoc)–OH, the monomeric PEG chain was synthesized and coupled to the fluorophore 2 to yield mCy5.5-PEG (Scheme 1).
To characterize the fluorogenicity resulting from the dimerization of NIR cyanine, the absorption and fluorescence properties of the dimer dCy5.5-PEG and the monomer mCy5.5-PEG were evaluated in solvents of different polarities. Both the dimer and the monomer were highly fluorescent in organic solvents, with fluorescence quantum yields (QY) ranging from 26 to 59% (Table 1) and the fluorescence maxima situated around 710 nm. However, in contrast to the monomer mCy5.5-PEG which was fluorescent in water and PBS (QY = 22% and 21%, respectively), the fluorescence in the aqueous medium of the dimer dCy5.5-PEG was almost negligible (QY = 0.4% in water and 0.3% in PBS).
| Solvent | mCy5.5-PEG | dCy5.5-PEG | ||||
|---|---|---|---|---|---|---|
| λ abs, nma | λ em, nmb | QY% | λ abs, nma | λ em, nmb | QY% | |
| a Position of the absorption maximum. b Position of the emission maximum. c Fluorescence quantum yield. | ||||||
| 1,4–Dioxane | 689 | 712 | 59 | 689 | 718 | 28 |
| DMF | 686 | 709 | 50 | 685 | 709 | 56 |
| EtOH | 686 | 709 | 34 | 686 | 710 | 26 |
| MeOH | 681 | 705 | 32 | 682 | 705 | 26 |
| Water | 679 | 700 | 22 | 629 | 700 | 0.4 |
| PBS | 679 | 700 | 21 | 629 | 700 | 0.3 |
Although the absorption spectrum of the dimer in MeOH was identical to that of the monomer having the maximum at 682 nm, its absorption spectrum in water presented a blue-shifted maximum at 629 nm and a long-wavelength shoulder. This new band can be assigned to the non-fluorescent intramolecular dimer of the H-aggregate type, which is highly favored in an aqueous medium.23 Indeed, the intramolecular H-aggregate in dCy5.5-PEG quickly disappeared upon the addition of MeOH to water, which resulted in the shift of the absorption maximum to 682 nm (Fig. 1B) and the recovery of the fluorescence (Fig. 1C). As a consequence, dCy5.5-PEG presented excellent fluorogenic properties, with up to 140-fold higher QY in organic solvents than in water (Fig. 1D). For comparison, the monomer mCy5.5-PEG is characterized by only <2.7-fold difference in QY between water and organic solvents. As the absorption spectrum of dCy5.5-PEG in the open form (in MeOH) is the same as that of the cyanine 2 (Fig. S3, ESI†), the extinction coefficient of the dimer should be ca. double that of the monomer. The extinction coefficient for the cyanine 2 was measured to be 222000 M−1 cm−1 in MeOH, which allows estimation of the extinction coefficient for the dimer dCy5.5-PEG: 222000 M−1 cm−1 × 2 = 444000 M−1 cm−1. Then, given its strong QY (56% in DMF), dCy5.5-PEG is, to the best of our knowledge, one of the brightest fluorogenic NIR dyes reported to date.
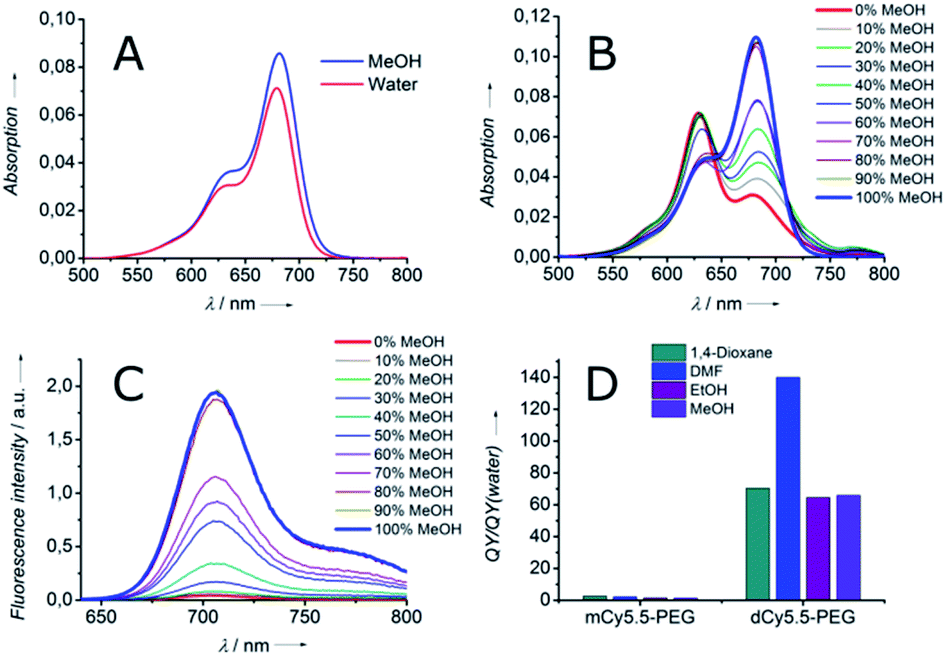 | ||
| Fig. 1 Spectroscopic properties of monomeric and dimeric Cy5.5 derivatives: (A) absorption spectra of mCy5.5-PEG in MeOH and water. Absorption (B) and fluorescence (C) spectra of dCy5.5-PEG in water–MeOH mixtures. (D) Ratio of QY in organic solvents to that in water for mCy5.5-PEG and dCy5.5-PEG. | ||
Being encouraged by the strong fluorogenicity of the Cy5.5 dimer, we envisaged its grafting to a ligand targeting the OTR (Fig. 2). Recently, we demonstrated that carbetocin (CBT), a peptidic ligand for the OTR, could be modified with bulky fluorophores with or without PEG chains at position 8 while preserving the binding properties for its receptor.17 In order to link CBT to the fluorogenic dimer, we performed the synthesis of the azide derivative of CBT, Lys(N3)–CBT, on a solid-phase by introducing Fmoc–L-Lys(N3)–OH at position 8. Finally, the conjugation between dCy5.5-PEG and Lys(N3)–CBT was performed by copper(I)-catalyzed azide alkyne cycloaddition (CuAAC) click chemistry in solution, and the conjugate dCy5.5-PEG–CBT was isolated by semi-preparative HPLC. In a similar manner, by coupling mCy5.5-PEG to Lys(N3)–CBT, the monomeric conjugate mCy5.5-PEG–CBT was obtained.
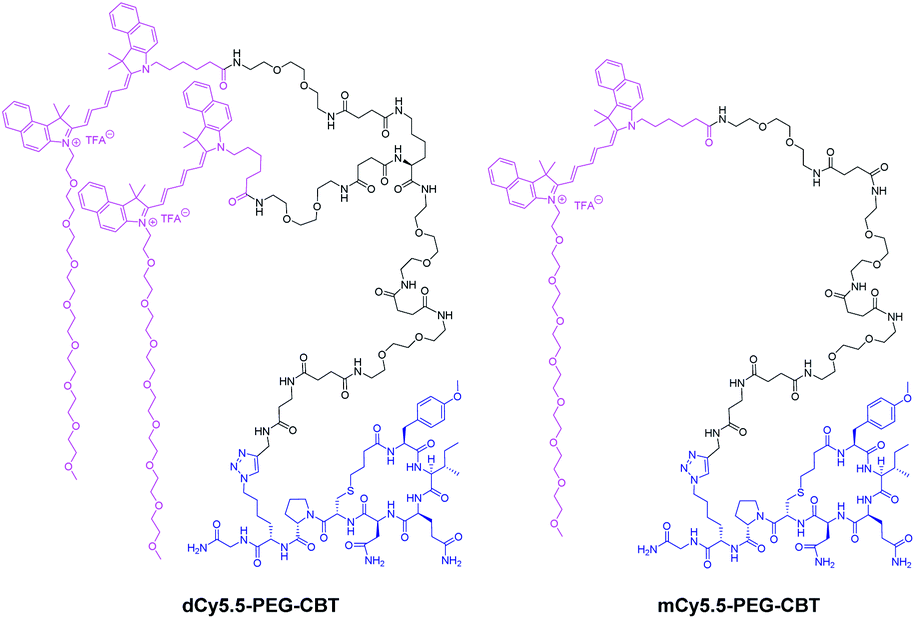 | ||
| Fig. 2 Structures of the conjugates of the NIR fluorogenic dimer and the corresponding monomer with the carbetocin ligand. | ||
To address the capacity of the NIR fluorogenic dimer dCy5.5-PEG–CBT to detect the OTR in living cells, we performed confocal microscopy experiments under no-wash conditions on HEK 293 cells overexpressing the OTR fused to GFP. As shown in Fig. 3, the addition of as low as a 10 nM solution of dCy5.5-PEG–CBT revealed the OTR at the cell membrane. The competition experiment performed in the presence of a large excess of the unlabeled CBT ligand did not reveal any fluorescence membrane staining, demonstrating the absence of non-specific interactions of dCy5.5-PEG–CBT with cell membranes and its specific binding to the OTR. The specificity of the probe for the receptor was also confirmed by confocal microscopy experiments on HEK 293 cells which do not express the OTR. As shown in Fig. S4,† no membrane staining was observed with dCy5.5-PEG–CBT, in the presence or absence of CBT.
 | ||
| Fig. 3 Confocal microscopy studies of dCy5.5-PEG–CBT and mCy5.5-PEG–CBT on living HEK 293 cells expressing OTR-GFP fusion under no-wash conditions. Cells were incubated with the ligands for 5 min at room temperature prior to the imaging. Excitation/emission wavelengths: 488 nm/500–550 nm for GFP and 635 nm/680–750 nm for Cyanine 5.5. | ||
To highlight the advantage of using fluorogenic dyes in biological sensing, the OTR imaging was performed either in the presence of fluorogenic dCy5.5-PEG–CBT or non-fluorogenic mCy5.5-PEG–CBT at a 500 nM concentration under no-wash conditions. The excess of unbound non-fluorogenic mCy5.5-PEG–CBT was highly fluorescent in aqueous solution, creating a strong background (Fig. 3). In sharp contrast, the background of the image with dCy5.5-PEG–CBT remained completely dark, probably because in solution the dimeric probe existed in the form of the non-fluorescent H-aggregate.
The ultimate challenge was to visualize the endogenous OTR on a lactating mouse model using the new probe. dCy5.5-PEG–CBT (7.5 nmol) was injected by a tail intravenous (i.v.) route in lactating Swiss mice 11 days after delivery. After 30 min, the mice were imaged in a small animal living imaging system. As shown in Fig. 4A, strong fluorescence in mammary glands was detected, with a practically negligible off-target signal, except for the liver, the organ expected to accumulate the injected dyes.
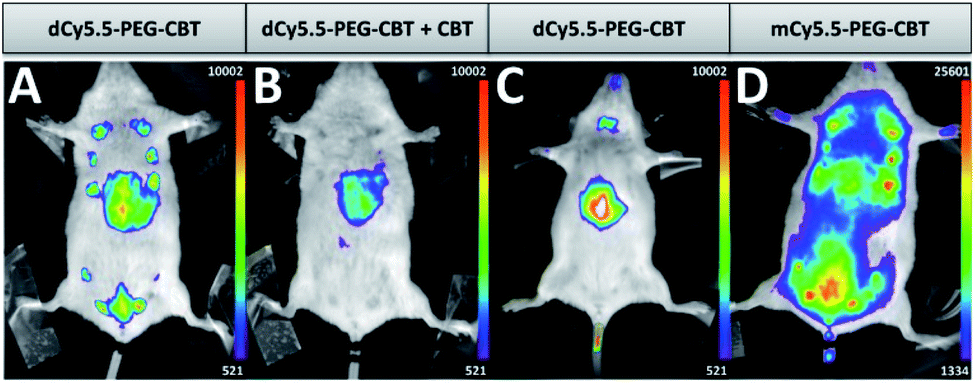 | ||
| Fig. 4 In vivo images of lactating (A, B and D) or naïve (C) mice injected i.v. with 7.5 nmol of dCy5.5-PEG–CBT (A and C), 7.5 nmol of dCy5.5-PEG–CBT and 450 nmol of CBT (B) or 7.5 nmol of mCy5.5-PEG–CBT (D) 30 min prior to the imaging. Representative images of at least 3 biological replicates. | ||
Noteworthily, no toxicity was observed even after seven days post-injection of the probe. To demonstrate the specific labelling of the OTR, dCy5.5-PEG–CBT was injected in the presence of a 60-fold excess of non-fluorescent CBT (Fig. 4B). In this case, only the liver of the mice was fluorescent, leaving the mammary glands non-labelled. The absence of mammary gland labelling was also observed in naïve mice (Fig. 4C), which is not expected to overexpress the oxytocin GPCR in the gland region. Finally, the administration of the monomeric probe mCy5.5-PEG–CBT resulted in a strong off-target fluorescence, which can be seen in the image (Fig. 4D) using the equivalent intensity scale (maximum value of this scale is 20-fold larger than the minimum value). These results highlight the advantage of using a fluorogenic dimer probe to increase the signal-to-noise ratio for the in vivo imaging.
Conclusions
In conclusion, based on the concept of fluorogenic dimers with environment-sensitive folding, we developed a bright fluorogenic NIR probe allowing the specific, background-free and unprecedented imaging of the endogenous OTR in living mice. We anticipate that this concept can be readily implemented for other GPCRs and other NIR dyes, which therefore opens up fascinating perspectives of non-invasive and non-ionizing fluorescence cartography of GPCRs in living animals.Experimental section
Chemical synthesis
Small animal fluorescence imaging
Animal experimentation was conducted with the approval of the French Ministry of Agriculture and the Ethics local committee for animal experimentation of Strasbourg University (CREMEAS) under authorization number #11974-2017103010101372. More detailed information about animal handling and in vivo fluorescence imaging is provided in the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Agence Nationale de la Recherche (FluoroPEP, ANR-16-CE18-0030), the IdEx Unistra supported by the program “investments for the future” of the French Government, the LabEx MEDALIS (ANR-10-LABX-0034), the ERC Consolidator grant BrightSens 648528, the Centre National de la Recherche Scientifique, and the University of Strasbourg. L. E. was supported by a fellowship from the Ministère de l’Education Nationale, de l'Enseignement Supèrieur et de la Recherche. We are grateful to Dr Dominique Bagnard (INSERM 1119 – BMNTS Lab) for giving us access to the NightOwl luminograph, Christel Valencia and Dr Pascal Villa (Plate-forme de Chimie Biologique Intégrative de Strasbourg, UMS3286) for prodiving us with the HEK293 Cells, Dr Delphine Garnier and Dr Estefania Oliva for mass spectrometry (PACSI platform GDS3670) and Dr Dmytro Dziuba for fruitful suggestions and proofreading the manuscript.Notes and references
- D. M. Rosenbaum, S. G. F. Rasmussen and B. K. Kobilka, Nature, 2009, 459, 356–363 CrossRef CAS PubMed.
- R. Santos, O. Ursu, A. Gaulton, A. P. Bento, R. S. Donadi, C. G. Bologa, A. Karlsson, B. Al-Lazikani, A. Hersey, T. I. Oprea and J. P. Overington, Nat. Rev. Drug Discovery, 2017, 16, 19–34 CrossRef CAS PubMed.
- J. B. Grimm, B. P. English, J. Chen, J. P. Slaughter, Z. Zhang, A. Revyakin, R. Patel, J. J. Macklin, D. Normanno, R. H. Singer, T. Lionnet and L. D. Lavis, Nat. Methods, 2015, 12, 244–250 CrossRef CAS PubMed.
- G. Lukinavičius, L. Reymond, K. Umezawa, O. Sallin, E. D'Este, F. Göttfert, H. Ta, S. W. Hell, Y. Urano and K. Johnsson, J. Am. Chem. Soc., 2016, 138, 9365–9368 CrossRef PubMed.
- A. N. Butkevich, G. Yu. Mitronova, S. C. Sidenstein, J. L. Klocke, D. Kamin, D. N. H. Meineke, E. D'Este, P.-T. Kraemer, J. G. Danzl, V. N. Belov and S. W. Hell, Angew. Chem., Int. Ed., 2016, 55, 3290–3294 CrossRef CAS PubMed.
- Z. Ma, L. Du and M. Li, J. Med. Chem., 2014, 57, 8187–8203 CrossRef CAS PubMed.
- F. Ciruela, K. A. Jacobson and V. Fernández-Dueñas, ACS Chem. Biol., 2014, 9, 1918–1928 CrossRef CAS PubMed.
- C. Iliopoulos-Tsoutsouvas, R. N. Kulkarni, A. Makriyannis and S. P. Nikas, Expert Opin. Drug Discovery, 2018, 13, 933–947 CrossRef CAS.
- Z. Ma, Y. Lin, Y. Cheng, W. Wu, R. Cai, S. Chen, B. Shi, B. Han, X. Shi, Y. Zhou, L. Du and M. Li, J. Med. Chem., 2016, 59, 2151–2162 CrossRef CAS PubMed.
- D. C. Alcobia, A. I. Ziegler, A. Kondrashov, E. Comeo, S. Mistry, B. Kellam, A. Chang, J. Woolard, S. J. Hill and E. K. Sloan, iScience, 2018, 6, 280–288 CrossRef CAS PubMed.
- L. Yuan, W. Lin, K. Zheng, L. He and W. Huang, Chem. Soc. Rev., 2013, 42, 622–661 RSC.
- S. A. Hilderbrand and R. Weissleder, Curr. Opin. Chem. Biol., 2010, 14, 71–79 CrossRef CAS PubMed.
- S. Stolik, J. A. Delgado, A. Pérez and L. Anasagasti, J. Photochem. Photobiol., B, 2000, 57, 90–93 CrossRef CAS.
- A. S. Klymchenko, Acc. Chem. Res., 2017, 50, 366–375 CrossRef CAS PubMed.
- A. Nadler and C. Schultz, Angew. Chem., Int. Ed., 2013, 52, 2408–2410 CrossRef CAS PubMed.
- G. S. Loving, M. Sainlos and B. Imperiali, Trends Biotechnol., 2010, 28, 73–83 CrossRef CAS PubMed.
- I. A. Karpenko, R. Kreder, C. Valencia, P. Villa, C. Mendre, B. Mouillac, Y. Mély, M. Hibert, D. Bonnet and A. S. Klymchenko, ChemBioChem, 2014, 15, 359–363 CrossRef CAS PubMed.
- I. A. Karpenko, A. S. Klymchenko, S. Gioria, R. Kreder, I. Shulov, P. Villa, Y. Mély, M. Hibert and D. Bonnet, Chem. Commun., 2015, 51, 2960–2963 RSC.
- A. Okamoto, Chem. Soc. Rev., 2011, 40, 5815 RSC.
- R. Weissleder, C.-H. Tung, U. Mahmood and A. Bogdanov, Nat. Biotechnol., 1999, 17, 375–378 CrossRef CAS PubMed.
- J. M. Ellard, T. Zollitsch, W. J. Cummins, A. L. Hamilton and M. Bradley, Angew. Chem., Int. Ed. Engl., 2002, 41, 3233–3236 CrossRef PubMed.
- F. Bouhedda, K. T. Fam, M. Collot, A. Autour, S. Marzi, A. Klymchenko and M. Ryckelynck, Nat. Chem. Biol., 2020, 16, 69–76 CrossRef CAS PubMed.
- I. A. Karpenko, M. Collot, L. Richert, C. Valencia, P. Villa, Y. Mély, M. Hibert, D. Bonnet and A. S. Klymchenko, J. Am. Chem. Soc., 2015, 137, 405–412 CrossRef CAS PubMed.
- K. M. Kendrick, A. J. Guastella and B. Becker, in Behavioral Pharmacology of Neuropeptides: Oxytocin, ed. R. Hurlemann and V. Grinevich, Springer International Publishing, Cham, 2017, vol. 35, pp. 321–348 Search PubMed.
- H. Yamasue and G. Domes, in Behavioral Pharmacology of Neuropeptides: Oxytocin, ed. R. Hurlemann and V. Grinevich, Springer International Publishing, Cham, 2017, vol. 35, pp. 449–465 Search PubMed.
- G. Gimpl and F. Fahrenholz, Physiol. Rev., 2001, 81, 629–683 CrossRef CAS PubMed.
- F. Ponsot, W. Shen, P. Ashokkumar, E. Audinat, A. S. Klymchenko and M. Collot, ACS Sens., 2017, 2, 1706–1712 CrossRef CAS PubMed.
- D. Bonnet, S. Riché, S. Loison, R. Dagher, M. Frantz, L. Boudier, R. Rahmeh, B. Mouillac, J. Haiech and M. Hibert, Chem.–Eur. J., 2008, 14, 6247–6254 CrossRef CAS PubMed.
- A. Soriano, R. Ventura, A. Molero, R. Hoen, V. Casadó, A. Cortés, F. Fanelli, F. Albericio, C. Lluís, R. Franco and M. Royo, J. Med. Chem., 2009, 52, 5590–5602 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthesis of pegylated Cy5.5, photophysical characterisation, fluorescence microscopy imaging and small animal in vivo fluorescence imaging experiments. See DOI: 10.1039/d0sc01018a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |