DOI:
10.1039/D0RA08068C
(Review Article)
RSC Adv., 2020,
10, 40983-41003
Synthesis of polysubstituted arenes through organocatalytic benzannulation
Received
21st September 2020
, Accepted 3rd November 2020
First published on 10th November 2020
Abstract
Polysubstituted arenes serve as ubiquitous structural cores of aromatic compounds with significant applications in chemistry, biological science, and materials science. Among all the synthetic approaches toward these highly functionalized arenes, organocatalytic benzannulation represents one of the most efficient and versatile transformations in the assembly of structurally diverse arene architectures under mild conditions with exceptional chemo-, regio- or stereoselectivities. Thus, the development of new benzannulation reactions through organocatalysis has attracted much attention in the past ten years. This review systemically presents recent advances in the organocatalytic benzannulation strategies, categorized as follows: (1) Brønsted acid-catalysis, (2) secondary amine catalysis, (3) primary amine catalysis, (4) tertiary amine catalysis, (5) tertiary phosphine catalysis, and (6) N-heterocyclic carbene catalysis. Each part is further classified into several types according to the number of carbon atoms contributed by different synthons participating in the cyclization reaction. The reaction mechanisms involved in different benzannulation strategies were highlighted.
1 Introduction
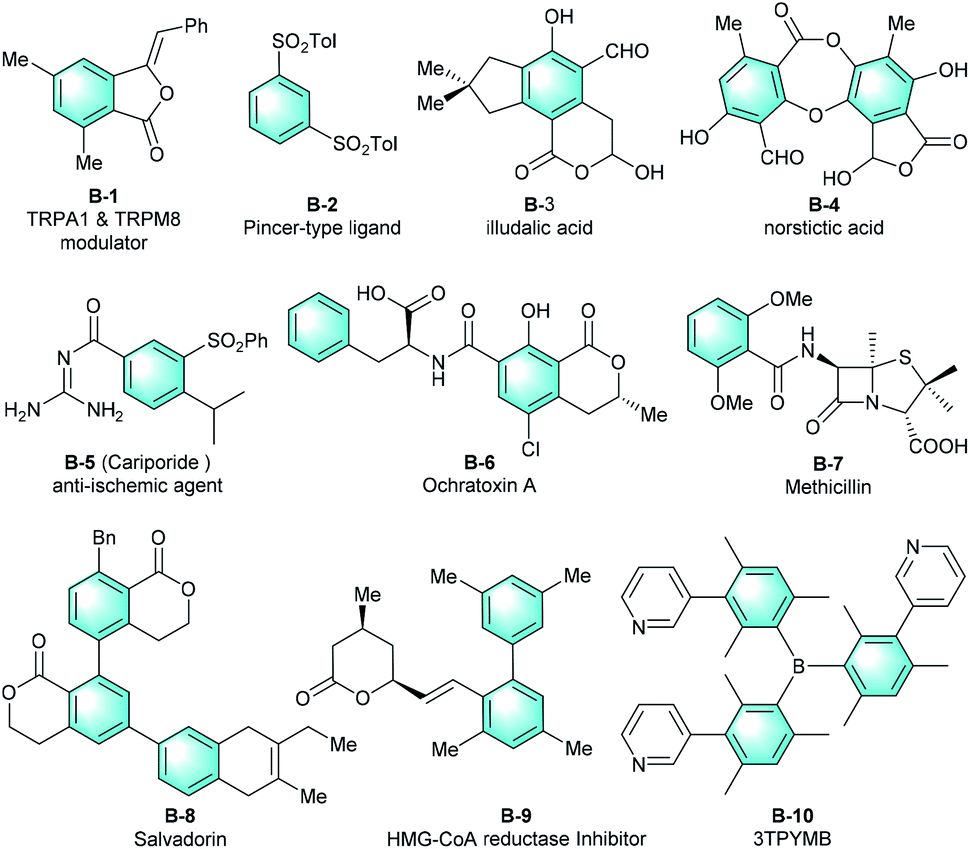
Polysubstituted arenes represent a core structural framework that ubiquitously occurs in various natural products, pharmaceuticals, and agrochemicals. They are also universal synthetic building blocks in preparing most of today's industrial chemicals and functional materials.1 The functions of these molecules are directly associated with the substituents on the benzene core.2 Therefore, flexible substitution patterns are highly desirable for the functionalized benzenes, and the synthesis of various multi-substituted benzenes is a longstanding focus and has drawn much attention in the organic chemistry community.3 Fig. 1 shows several selected molecules containing a highly functionalized benzene.
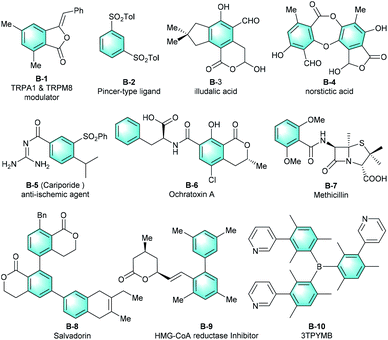 |
| | Fig. 1 Examples of multi-substituted benzenes. | |
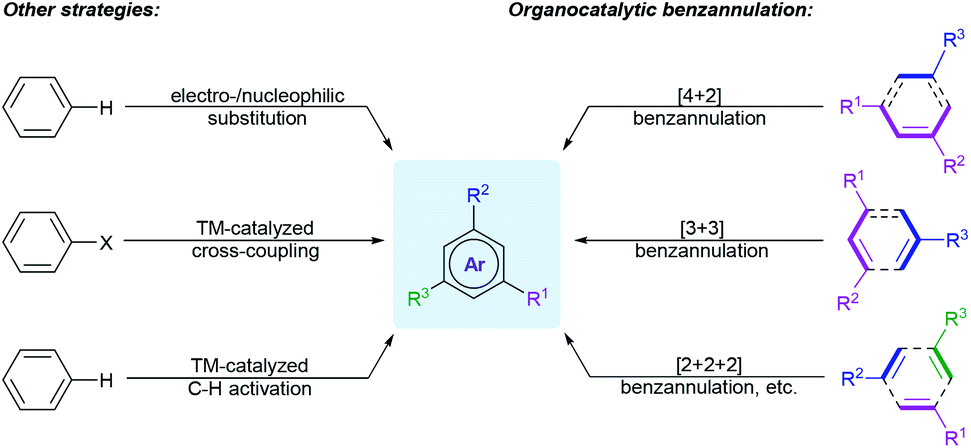
Synthetic methodologies for the construction of polysubstituted benzenes mainly rely on the modification of existing six-membered carbocyclic compounds or arene substrates, such as catalytic dehydrogenation of carbocyclic rings,4 electrophilic and nucleophilic aromatic substitution reactions,5 as well as transition-metal-catalyzed cross-coupling reactions6 or direct C–H functionalization of arenes7 (Scheme 1). Despite providing impressive progresses for benzene substitutions, some of these approaches may suffer from expensive metal catalysts, harsh reaction conditions, multistep synthetic sequences to introduce directing or functional groups into the parent benzene rings, and, in particular, the limited substitution patterns. Besides, different functional groups on the aromatic cores may hugely affect both the selectivity and reactivity of these reactions.
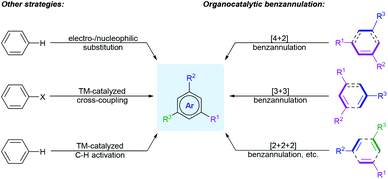 |
| | Scheme 1 Strategies for the synthesis of polysubstituted arenes. | |
Benzannulation is a method to assemble the arene rings through C–C bonds formation between two or more appropriate building blocks. It is a valid alternative strategy for the synthesis of diverse polyfunctionalized arenes, because these approaches could reduce the number of steps and usually exhibit better regioselectivities and flexible substitution patterns. These reactions have been summarized in several reviews.8 In 2008, Wessig and Müller reviewed the dehydro-Diels–Alder reactions, which mainly provided benzannulation products.8a In the same year, Kotha and co-workers comprehensively discussed different types of benzannulation reactions for the creation of benzene rings, including Diels–Alder reaction, ring-closing metathesis (RCM), cycloaddition reactions and other transition-metal-promoted processes.8b In 2017, Dichtel and co-workers summarized the transition-metal-catalyzed benzannulation reactions of alkynes.8i Later, Lee et al. reviewed the various base-mediated benzannulations under transition-metal-free conditions, such as Cs2CO3, t-BuOK, and DBU-catalyzed and -mediated reactions.8m
In recent ten years, from the standpoint of green and sustainable chemistry, the ongoing research on the construction of the functionalized benzene compounds through organocatalytic benzannulation strategies has emerged as a hot topic in organic synthesis. Taking advantage of various organocatalysts, different activation modes, and versatile annulation strategies, the scope of benzannulation approaches were remarkably broadened. Comparing with the conventional benzannulation methods, these approaches feature numerous beneficial attributes, including low economic cost, mild reaction conditions, good functional group compatibility, flexible substitution patterns, high regio- and stereoselectivity (Scheme 1). Organocatalytic benzannulation process can also avoid the problems of removing metal residues or directing groups. On the other hand, organocatalytic benzannulation is a useful tool for the construction of various biaryl scaffolds, which are essential structural motifs found in natural products, biologically active compounds, and functional materials.9 Comparing with the transition-metal-catalyzed cross-coupling or C–H arylation strategies,10 which form the aryl–aryl bonds, these tactics enables the facile access to biaryls from a wide range of acyclic starting materials with more flexible substitution patterns and excellent tolerance of functional groups.
Despite the great advance of organocatalytic benzannulation strategies over the last decade, no comprehensive review has been devoted to this topic.11 In this review, we systemically present recent advances in the organocatalytic benzannulation strategies categorized as follows: (1) Brønsted acid-catalysis, (2) secondary amine catalysis, (3) primary amine catalysis, (4) tertiary amine catalysis, (5) tertiary phosphine catalysis, and (6) N-heterocyclic carbene catalysis. To discuss the reactions in a more logical way, each part is further classified into several types according to the number of carbon atoms contributed by different synthons participating in the cyclization reaction, such as [4 + 2], [3 + 3], [3 + 2 + 1], [2 + 2 + 2] annulation, etc. (Scheme 1). The reaction mechanisms involved in different benzannulation strategies were highlighted and enantioselective versions were emphasized. Organocatalytic strategies for the synthesis of heteroaromatics and the aromatization using quinones as the cyclic precursors would not be mentioned in this review. The literature has been surveyed mainly from 2010 to April of 2020. We believe that this review will be instructive for researchers in different fields and will inspire further reaction design and developments.
2 Brønsted acid-catalyzed benzannulation
Brønsted acids have been widely used as efficient and eco-friendly organocatalysts. Over recent decades, significant progress has been achieved in the Brønsted acid-catalyzed annulation reactions due to their versatile potential in the activation of carbonyl groups, hydroxyl groups, imines, and amines. In particular, the Brønsted acid-catalyzed benzannulation has provided a powerful approach to the synthetically useful polysubstituted arenes. In this section, we will systemically discuss the application of the Brønsted acid-catalysis strategy in benzannulation reactions.
2.1 [4 + 2] benzannulation
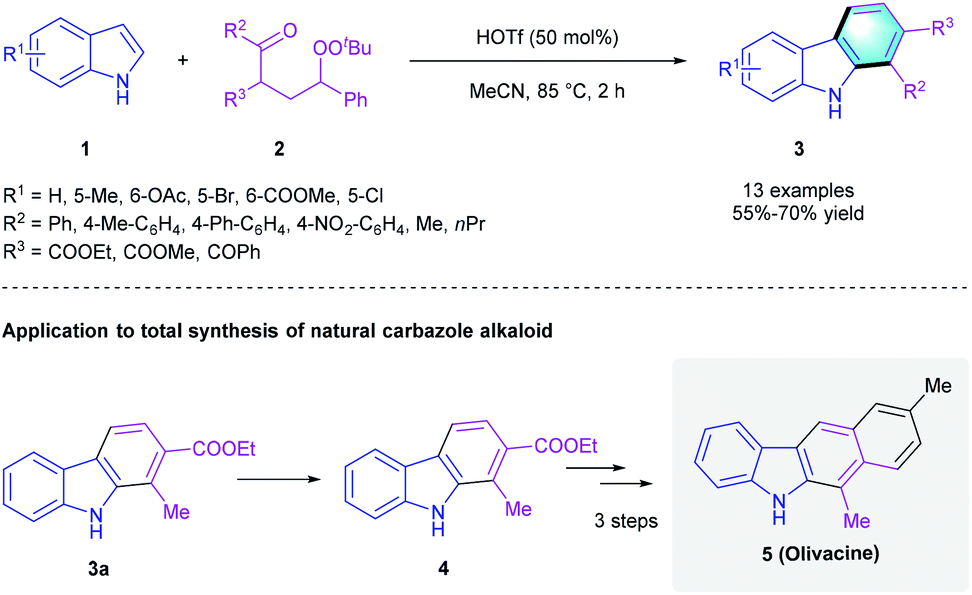
Because of the extensive application of [4 + 2] annulation in the de novo assembly of new arenes, these methods have drawn much attention. Considering the privileged biological activities of highly functionalized aromatic compounds like polysubstituted carbazoles and indolocarbazoles, tremendous efforts have been made to construct these useful building blocks from simple starting materials.12 In 2014, the Li group found that trifluoromethanesulfonic acid (HOTf) could catalyze the cyclization between indole 1 and γ-carbonyl tert-butylperoxide 2 (Scheme 2).13 The corresponding polysubstituted carbazole derivatives 3 were obtained in 55–70% yields. Moreover, this transformation of indole-to-carbazole was successfully applied to the total synthesis of natural carbazole alkaloid olivacine (5).
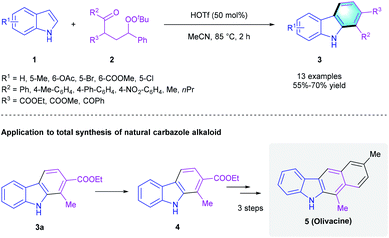 |
| | Scheme 2 HOTf-catalyzed [4 + 2] benzannulation to synthesize carbazoles. | |
In 2015, Gu et al. reported another convenient method to form polysubstituted carbazoles from 2-alkoxy-2,3-dihydrofuran 6 and indoles 7 (Scheme 3).14 In the presence of a catalytic amount of TsOH (5 mol%), carbazole derivatives 8 were delivered with moderate to excellent yields (58–99%) in MeCN. The authors further applied the strategy to the synthesis of other benzoarenes. In the presence of a catalytic amount of TfOH, the reaction of 2-alkoxy-2,3-dihydrofuran 6 with 2-methylfuran 9, thiophene 10 and 1,2-dimethoxybenzene 11 could provide corresponding products 12–14 in 46–66% yields.
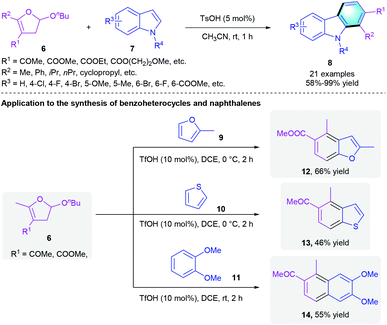 |
| | Scheme 3 Acid-catalyzed [4 + 2] benzannulation to synthesize substituted carbazoles. | |
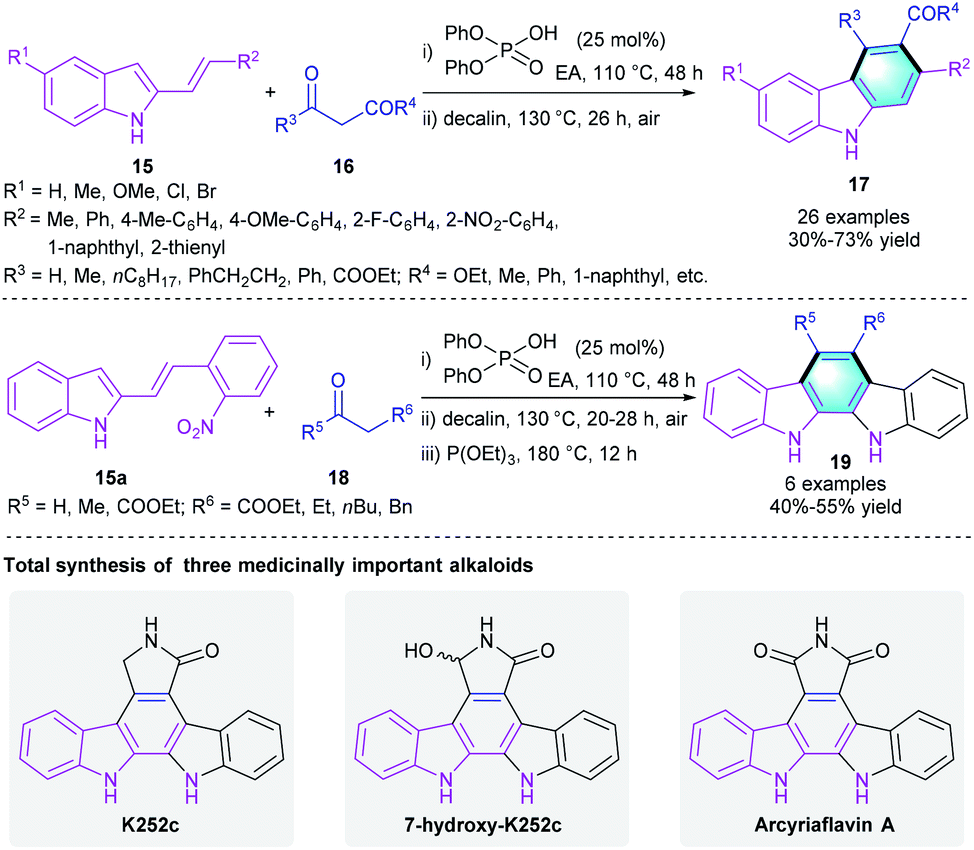
Later in 2018, the Maji group employed Brønsted acid catalysis in constructing polyfunctionalized carbazoles and its derivatives through a protecting-group-free benzannulation of 2-alkenylindoles 15 with 1,3-dicarbonyls 16/18 (Scheme 4).15 Notably, this method using cheap diphenylphosphoric acid as the catalyst and eco-friendly air as an ideal oxidant. Substrates with various substituents were well tolerated in the protocol, and all the reactions smoothly provided the target aromatic molecules 17/19 regardless of their electronic properties. The total synthesis of three carbazole alkaloids further demonstrated the practical applicability of the novel strategy. Besides, the same group successfully expanded the [4 + 2] benzannulation reaction of 15 with aldehydes for the synthesis of other multifunctionalized aromatic compounds.16
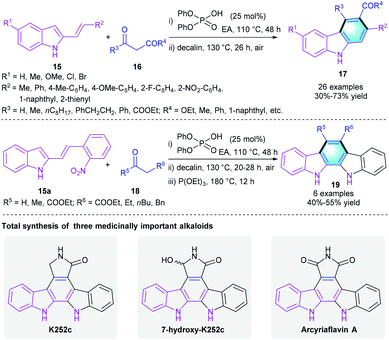 |
| | Scheme 4 Phosphoric acid-catalyzed [4 + 2] benzannulation to synthesize carbazoles and indolocarbazoles. | |
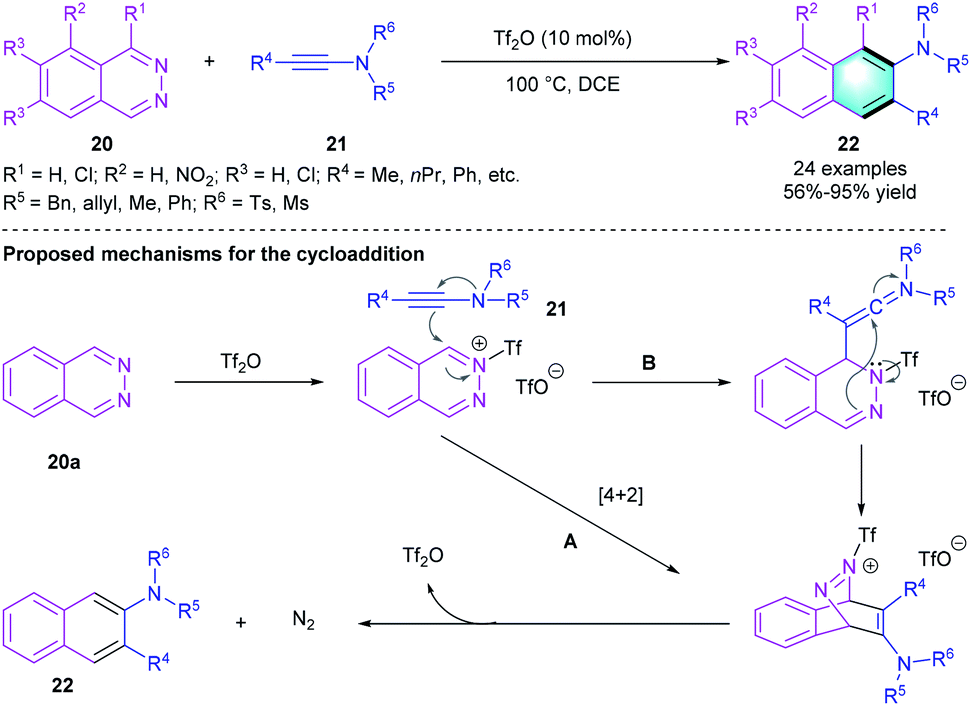
In 2018, Chang's group reported an inverse-electron-demand Diels–Alder reaction between 1,2-diazines 20 and ynamides 21.17 Optimizing the reaction conditions showed that the reaction proceeded well under the catalysis of Tf2O in DCE at 100 °C. The catalytic protocol could tolerate various functional groups, both electron-rich and electron-deficient substituents on 1,2-diazines are compatible in the [4 + 2] benzannulation, giving a series of 2-aminonaphthalenes or 2-aminoanthracenes 22 in moderate to excellent yields (56–95%). As shown in Scheme 5, a possible mechanism of the reaction was proposed.
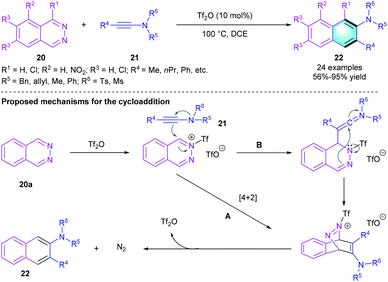 |
| | Scheme 5 Tf2O-catalyzed [4 + 2] benzannulation to synthesize 2-aminonaphthalenes and 2-aminoanthracenes. | |
2.2 [3 + 3] benzannulation
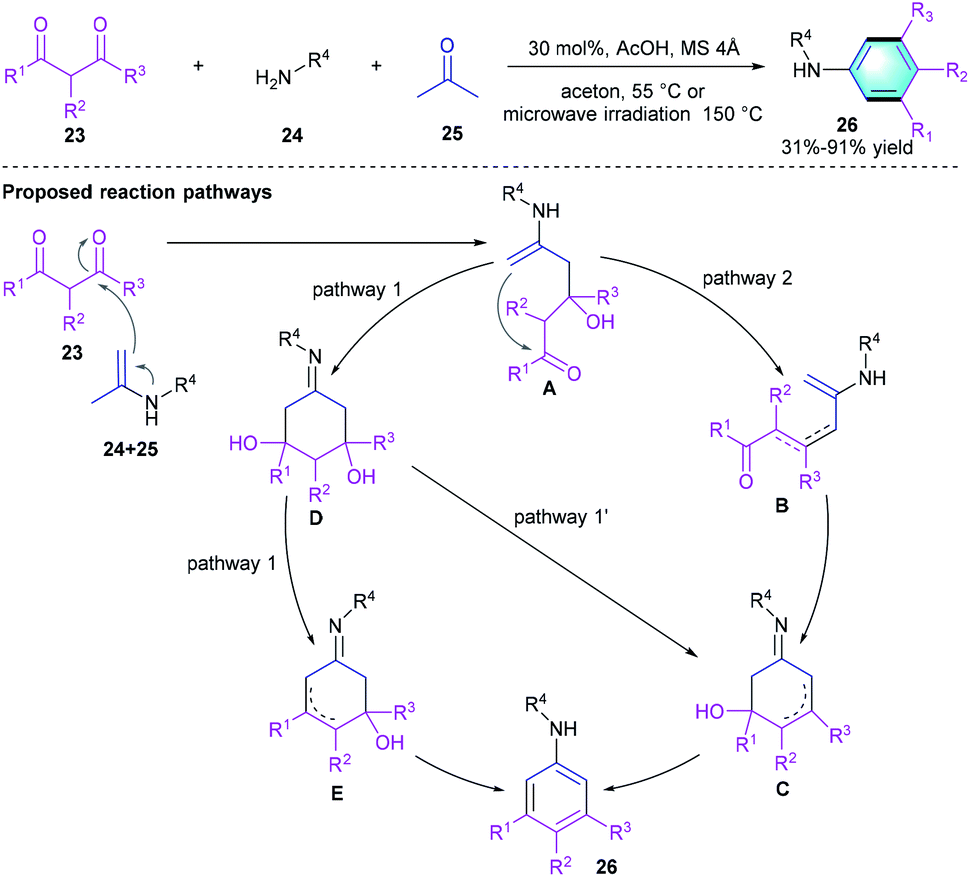
Comparing with the acid-catalyzed [4 + 2] benzannulations, [3 + 3] benzannulation strategy has been much less reported in the literatures. In 2019, Rubin et al. developed a facile [3 + 3] cyclization to access meta-substituted anilines from a three-component acyclic precursor, 1,3-diketones 23, amines 24, and acetone 25 (Scheme 6).18 The optimum reaction condition is using 30 mol% of acetic acid in acetone under conventional heating (55 °C) or microwave irradiation (150 °C). The compatibility of this procedure was well-investigated, and various substrates participate in the reaction successfully to provide the meta-substituted anilines 26 with acceptable-to-excellent yields (31–91%). A subsequent gram-scale reaction afforded the aniline product in favorable yield after crystallization. The proposed mechanism was shown in Scheme 6, the key step of this [4 + 2] benzannulation is the cyclo-condensation/aromatization of the in situ formed imines of acetone 1,3-diketones.
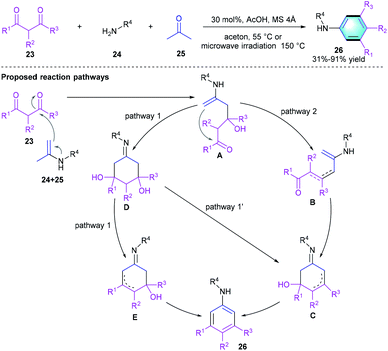 |
| | Scheme 6 AcOH-catalyzed [3 + 3] benzannulation to synthesize meta-substituted anilines. | |
2.3 [2 + 2 + 2] benzannulation
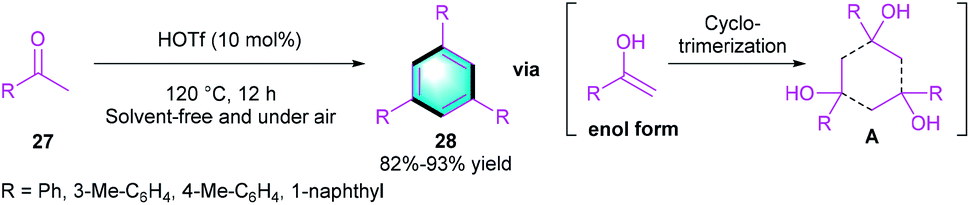
Taking advantage of the unique catalytic activity of HOTf, Yi and co-workers realized a condensation reaction for assembling privileged benzene using ketones 27 as simple substrates (Scheme 7).19 The cyclo-trimerization of ketones in the presence of HOTf (10 mol%) at 120 °C, furnishes a series of tri-substituted arenes 28 in good to high yields (82–93%).
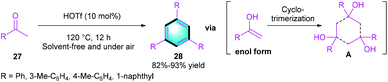 |
| | Scheme 7 HOTf-catalyzed [2 + 2 + 2] benzannulation to synthesize tri-substituted arenes. | |
2.4 Intramolecular benzannulation
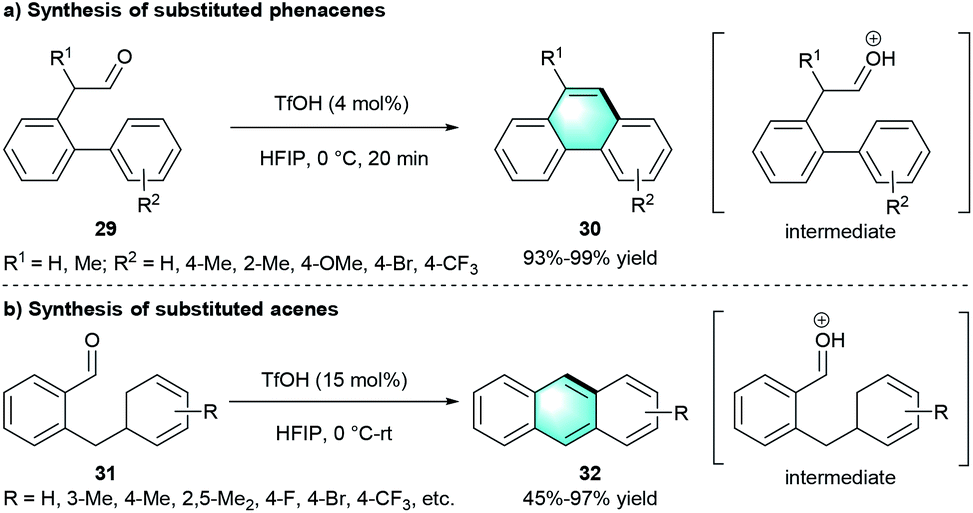
Following the concept of atom economy, the group of Ichikawa disclosed an intramolecular benzannulation of aromatic aldehydes to construct polycyclic aromatic hydrocarbons (Scheme 8).20 Using a catalytic amount of TfOH as catalyst and 1,1,1,3,3,3-hexafluoropropan-2-ol (HFIP) as the solvent enabled access to functionalized phenacenes 30 with high yields (Scheme 8a, 93–99%). Furthermore, the protocol was suitable for the cyclization precursor 2-benzylbenzaldehydes 31, resulting in moderate-to-excellent yields (45–97%) of the corresponding acenes 32 through dehydrative benzannulation (Scheme 8b). The author further proved that HFIP was important for the cyclizations because of its high ionizing power.
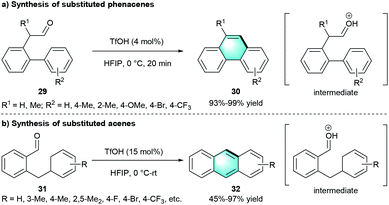 |
| | Scheme 8 TfOH-catalyzed intramolecular benzannulation to synthesize phenacenes and acenes. | |
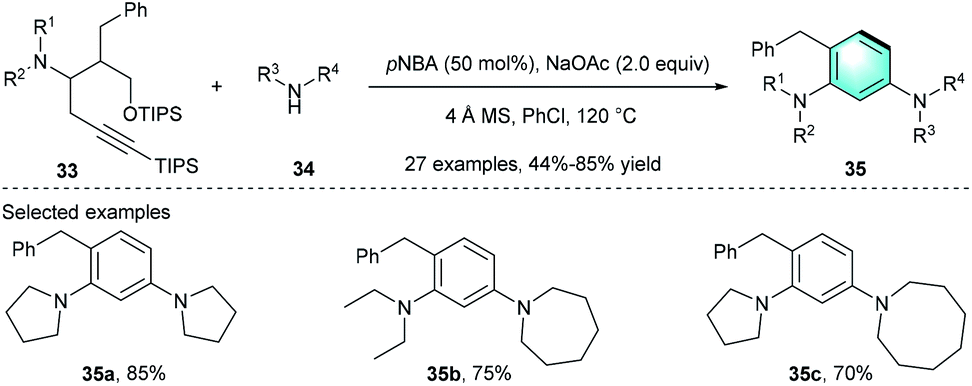
Meanwhile, an intramolecular 6π-electrocyclization–aromatization process of ynedienamine 33 to multi-substituted benzene was reported by Huang's group in 2017 (Scheme 9).21 The investigation of the reaction conditions showed that p-nitrobenzoic acid (pNBA) with NaOAC in chlorobenzene provided product 35a with good yield (85%). Various acyclic or cyclic ynedienamines 33 and five-/six-/seven-/eight-membered cyclic amines 34 endured the cyclization under the optimized conditions, achieving 44–85% yields for the functionalized arenes 35 bearing two distinct meta-amino groups.
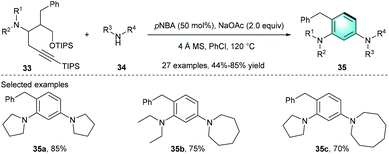 |
| | Scheme 9 pNBA-catalyzed intramolecular benzannulation to synthesize substituted benzenes. | |
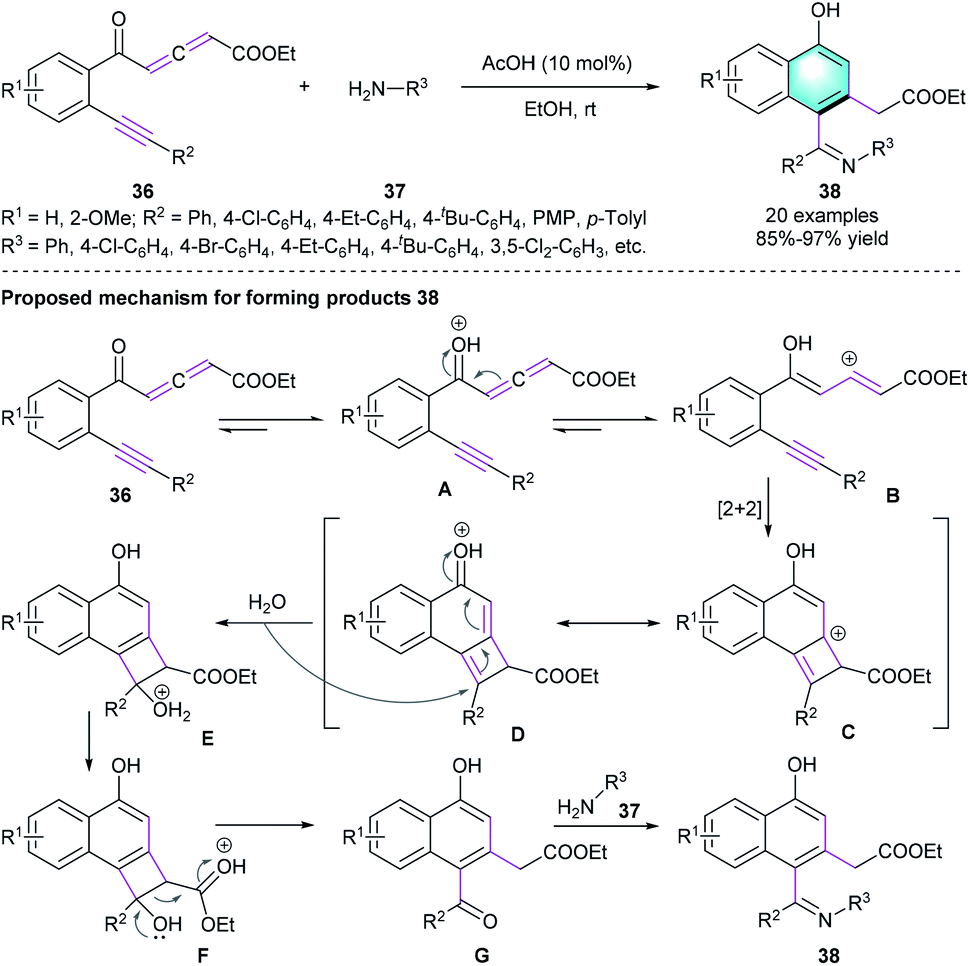
In addition, Jiang and co-workers disclosed that yne-allenone had the potential for constructing 1-naphthols via cleavage/rearrangement of carbon–carbon triple bonds in the presence of equivalent NBS and NCS.22 Encouraged by this work, the same group developed an AcOH-catalyzed intramolecular benzannulation of readily available yne-allenone esters 36, allowing the construction of α-naphthol derivatives 38 in high to excellent yields (85–97%).23 The reaction featured broad substrate scope, functional group tolerance, as well as an atom- and step-economical manner (Scheme 10). The authors also proposed a possible mechanism for the reaction. Initially, vinyl cation intermediates B was generated from 36 in the presence of AcOH. Then, [2 + 2] cycloaddition of B occurred to form intermediate D. Subsequent ring opening and deprotonation/isomerization generated G, which reacted with aromatic amine 37 and delivered the final products 38. In the same year, the group of Jiang presented another novel alternative methodology for the synthesis of substituted α-naphthols via NHC-catalyzed C–C triple bond cleavage of yne-allenone under mild conditions.24
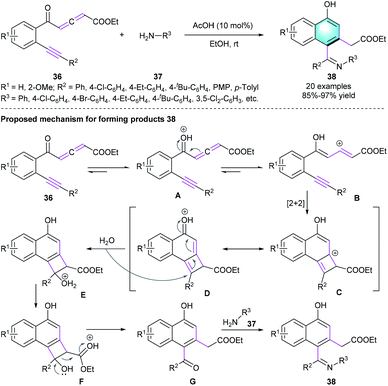 |
| | Scheme 10 AcOH-catalyzed intramolecular benzannulation to synthesize α-naphthol derivatives. | |
3 Secondary amine-catalyzed benzannulation
Secondary amines are considered as the dominant organic amine catalysts. At present, secondary amines, such as Jørgensen–Hayashi catalyst (diphenylprolinol silyl ether), MacMillan's imidazolinone catalyst, and proline, have been commonly applied in the assembling of complex cyclic and polycyclic skeletons via enamine or iminium activation and showed extraordinary ability in stereocontrol.25 In addition, some simple achiral secondary amines (e.g., pyrrolidine and piperidine) have been used in the highly efficient cyclization reactions.26 Taking advantage of these powerful organocatalysts, a series of benzannulation reactions have been developed.
3.1 [4 + 2] benzannulation
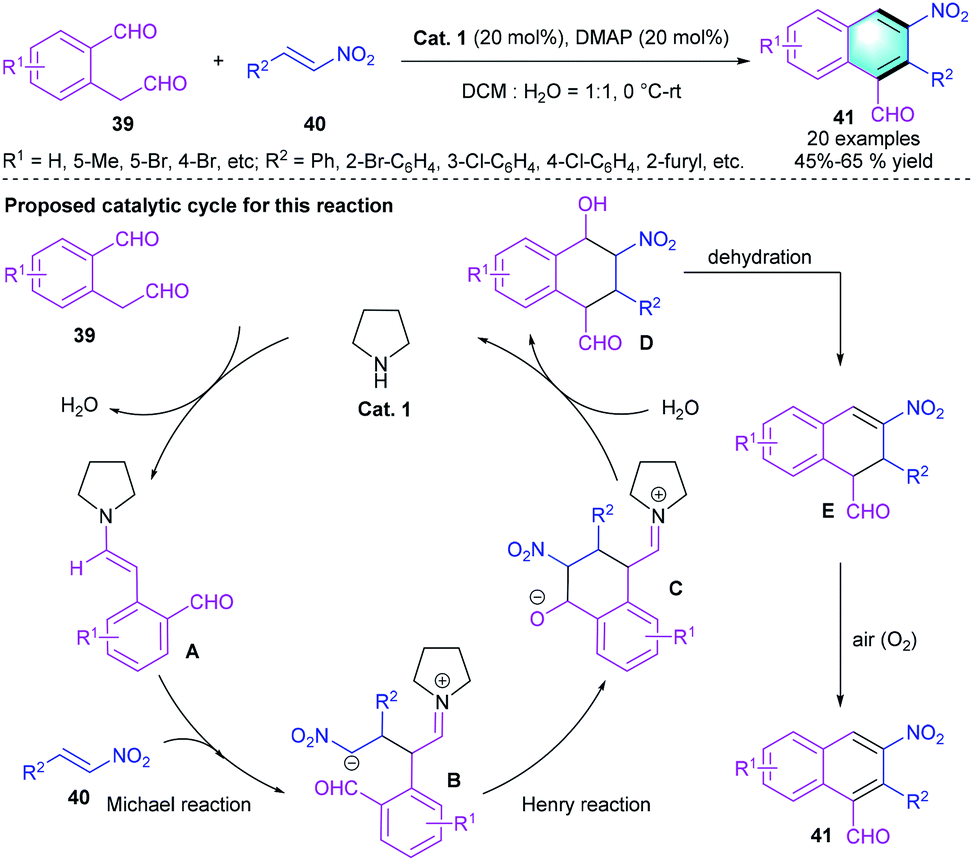
In 2010, Xu and co-workers reported a [4 + 2] cascade reaction between dialdehydes 39 and nitroalkenes 40 via a Michael–Henry–dehydration–aromatization sequence.27 Using pyrrolidine (Cat. 1) as the catalyst and DMAP as the additive in DCM and water (VDCM/VH2O = 1/1) afforded benzene derivatives 41 in 45–65% yields, which bears three adjacent substituents. Notably, nitroalkene with an alkyl group, such as n-propyl, exhibits poor reactivity, and no final product is observed probably because of its instability. Scheme 11 shows the plausible pathway for the reaction. The catalytic cycle begins with enamine activation of dialdehyde 39, the Michael reaction between enamine intermediate A and nitroolefin 40 formed the zwitterion B, which further underwent the intramolecular Henry reaction. Subsequent hydrolysis afforded the unstable intermediate D, followed by dehydration and aromatization to furnish the corresponding product.
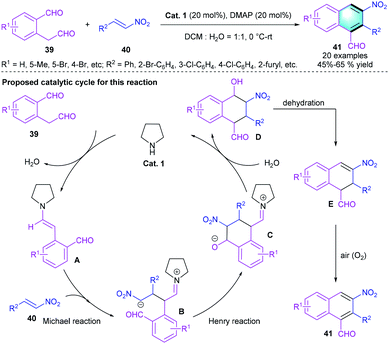 |
| | Scheme 11 Pyrrolidine catalyzed [4 + 2] benzannulation to synthesize polysubstituted naphthalene derivatives. | |
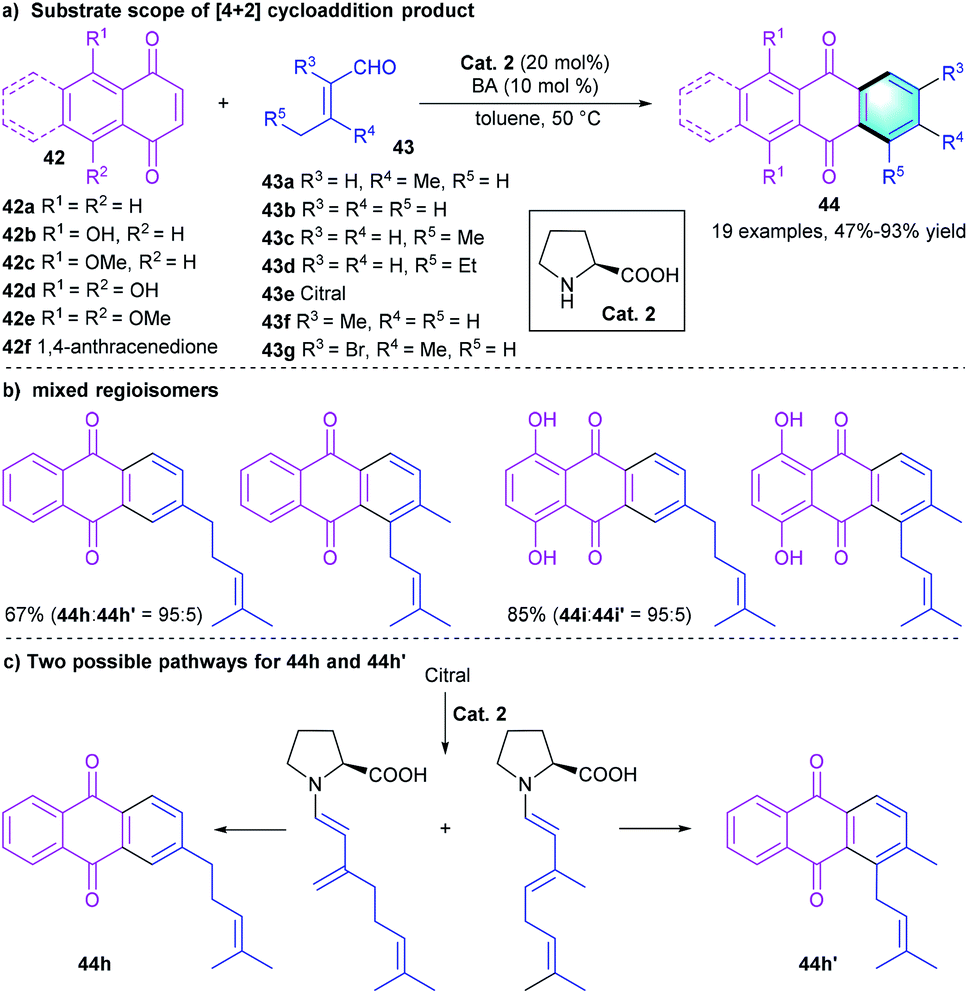
Secondary amine-catalyzed [4 + 2] benzannulation is also considered as an important method to prepare polycyclic aromatic hydrocarbons. For example, in 2015, Lee's group developed a one-pot reaction for the synthesis of anthraquinones through a [4 + 2] annulation between 1,4-naphthoquinones 42 and α,β-unsaturated aldehydes 43 (Scheme 12).28 In the condition screening, using L-proline as the catalyst, benzoic acid as the additive provided the best catalytic efficiency. The tandem benzannulation tolerated a series of substrates with substituents of different electronic properties, affording product 44 in moderate to excellent yields (47–93%). Notably, the reaction of 42a or 42d with citral (43e) under the standard conditions gave a mixture of regioisomers, 44h and 44h′. A plausible mechanism was proposed to explain the formation of these two regioisomers (Scheme 12c).
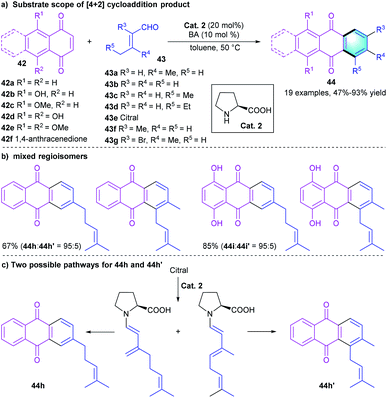 |
| | Scheme 12 L-Proline-catalyzed [4 + 2] benzannulation to synthesize polycyclic arenes. | |
Axially chiral molecules can be found in many natural products and are widely applied in chiral ligands, material chemistry, and drug discovery. Thus, the enantioselective synthesis of axially chiral compounds is of great significance.
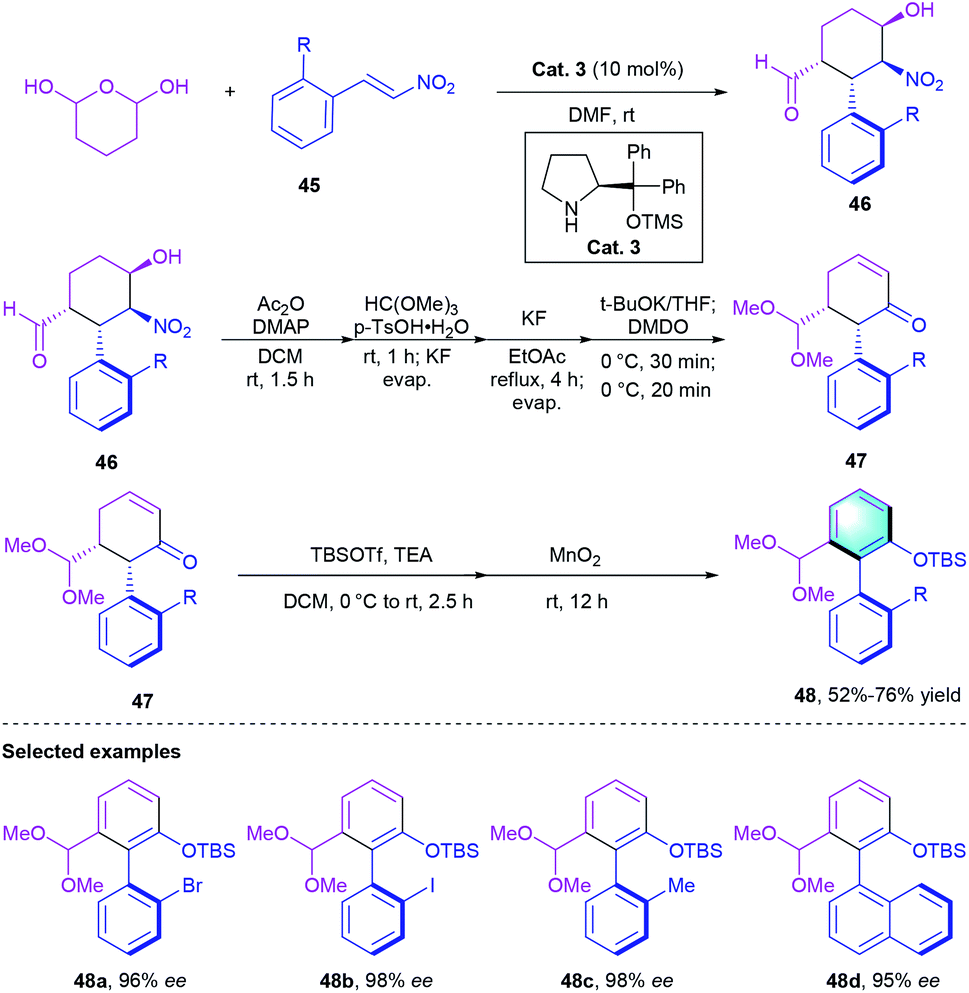
Recently, the de novo construction of new arenes via benzannulation becomes efficient access to synthesize axially chiral scaffolds.29 In this context, Hayashi's group designed a chiral secondary amine-catalyzed [4 + 2] cyclization of tetrahydro-2H-pyran-2,6-diol with nitroalkenes 45 through a domino Michael–Henry reaction. Chiral nitrocyclohexanecarbaldehyde 46 bearing four contiguous stereogenic centers were obtained. Removing the central chirality of cyclohexane moiety, the corresponding enantioenriched biaryl atropisomers 48 could be acquired with up to 98% ee (Scheme 13).
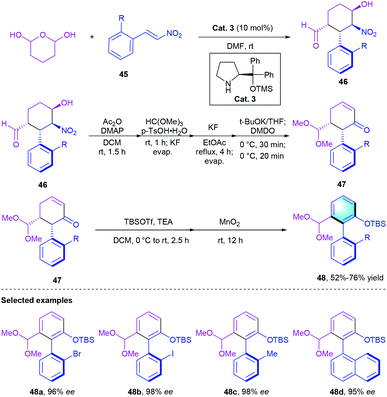 |
| | Scheme 13 Secondary amine-catalyzed [4 + 2] benzannulation to synthesize axially chiral nitrocyclohexanecarbaldehyde. | |
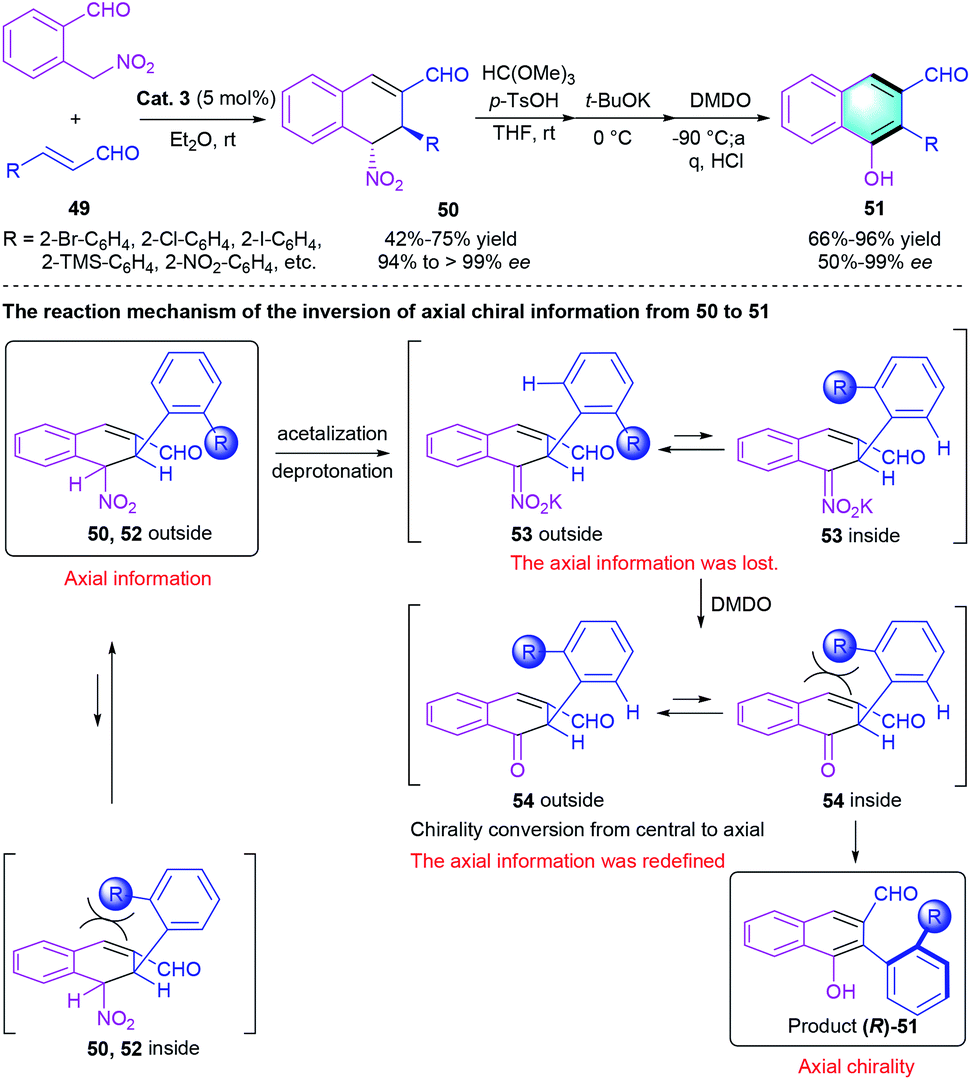
In 2020, the same group described a similar reaction catalyzed by chiral diphenylprolinol silyl ether (Scheme 14).30 In this work, Michael addition between 2-(nitromethyl)–benzaldehyde and α,β-unsaturated aldehyde 49, followed by intramolecular aldol condensation and aromatization, would generate the final atropisomeric biaryl product 51 with axial chirality in moderate-to-excellent yields (66–96%) with up to 99% ee. Notably, a sufficiently bulky nitro group led to a moderate ee value (50%), whereas the less bulky substituent only afforded racemic biaryls probably because of the decrease of rotational barrier around the axis. The author also proposed a possible reaction mechanism to explain the inversion of axial chirality information from 50 to 51.
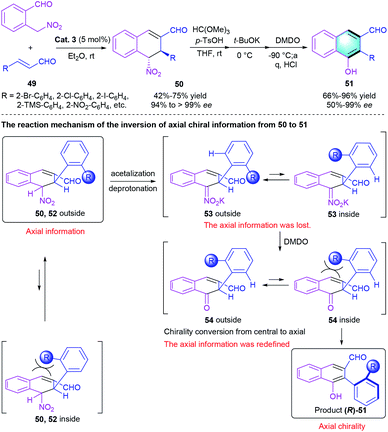 |
| | Scheme 14 Diphenylprolinol silyl ether-catalyzed [4 + 2] benzannulation to synthesize axially chiral biaryls. | |
3.2 [3 + 3] benzannulation
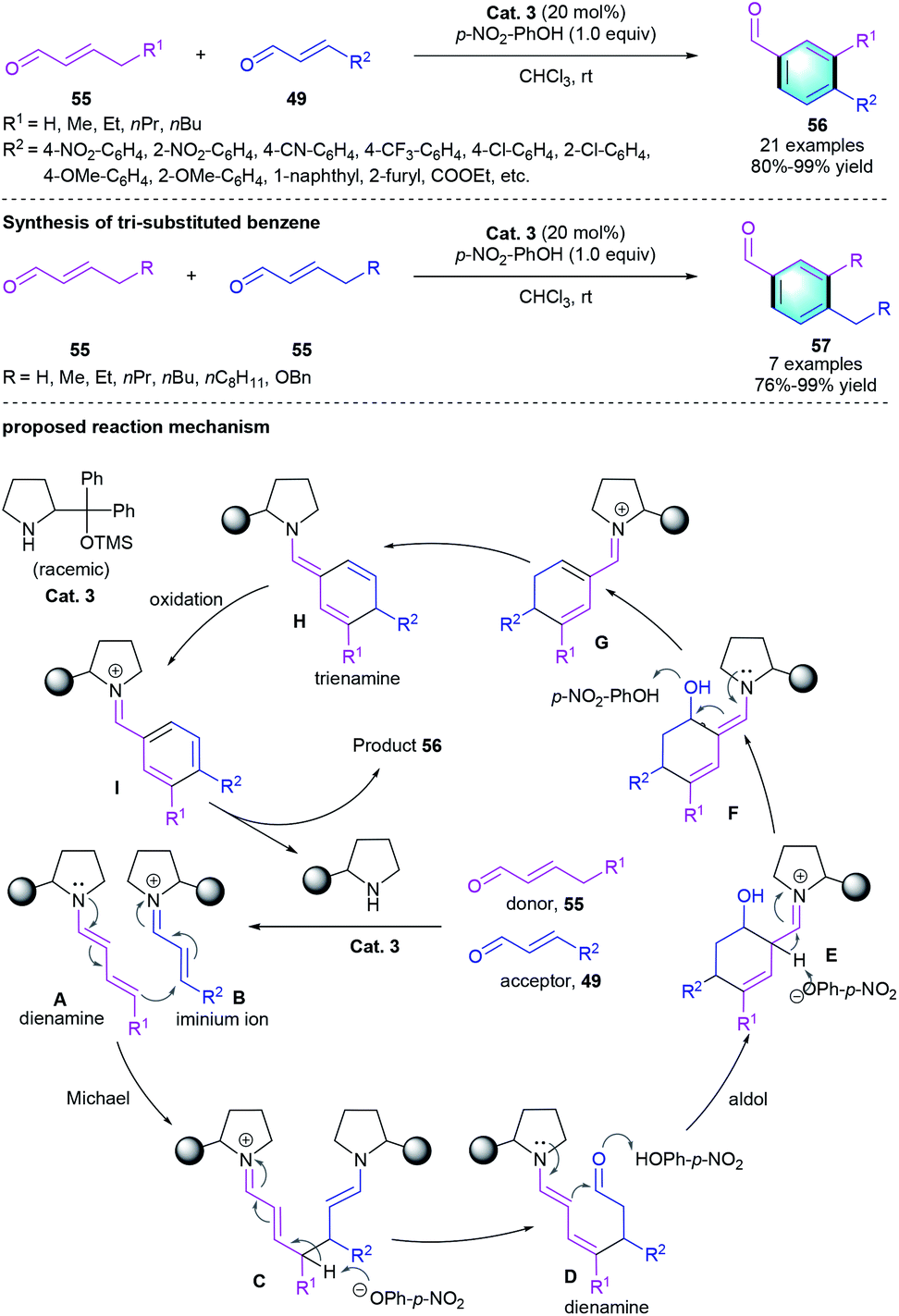
Besides [4 + 2] benzannulation, secondary amine-catalyzed [3 + 3] benzannulation strategy was also successfully applied in the construction of polysubstituted benzenes. For example, in 2012, Wang et al. developed a [3 + 3] benzannulation from easily accessible enals using diphenylprolinol silyl ether Cat. 3 and additive (Scheme 15).31 Two kinds of α,β-unsaturated aldehydes 55/49 were used as substrates in the reaction, which exclusively produced the corresponding polysubstituted aromatic aldehydes 56 in good yields (80–99%) with high chemo- and regioselectivities. Moreover, the self-dimerized homo-coupling processes smoothly with aliphatic 55 under the standard conditions to give the desire products 57. The author also proposed a possible mechanism for this Michael-aldol aromatization process. First, iminium ion B was generated from a certain enal and the catalyst. Then intermediate B underwent the Michael addition with dienamine A, which was derived from another enal and the secondary amine. Subsequently, the intermediate Michael adduct was converted into the new dienamine D in the presence of p-nitrophenoxide. An intramolecular aldol reaction formed enamine E, followed by the formation of electron-rich trienamine H. Finally, oxidation of the trienamine intermediate would afford arene I. Hydrolysis released the catalyst and furnished the tri-substituted benzene product 56.
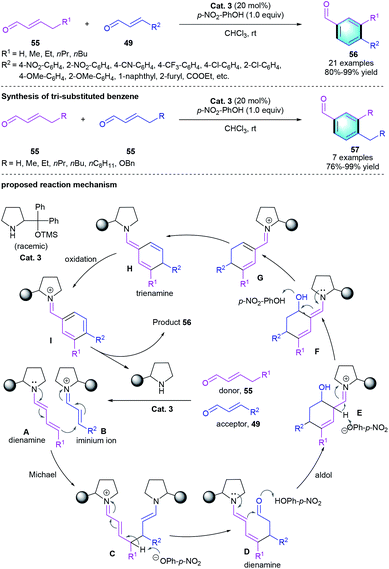 |
| | Scheme 15 Diphenylprolinol silyl ether-catalyzed [3 + 3] benzannulation to synthesize tri-substituted benzenes. | |
Based on the previous work, Jiang and co-workers reported a secondary amine-catalyzed [3 + 3] cascade reaction.32 Different α,β-unsaturated aldehydes 49 and allylic sulfone-containing 1,3-bisnucleophiles 58 were smoothly converted into multi-substituted benzenes 59 in the presence of pyrrolidine (20 mol%) at room temperature (Scheme 16). This reaction involves iminium–enamine–iminium activation, and the variation of substituents on substrate 49 or 58 was well tolerated, providing the desired product in high-to-excellent yields (74–96%) with good regioselectivies. However, using trans-crotonaldehyde as the substrate, only a complex mixture was detected, probably because of the coexistence of dienamine activation compared with aromatic enals.
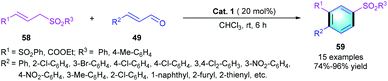 |
| | Scheme 16 Pyrrolidine catalyzed [3 + 3] benzannulation to synthesize tri-substituted benzenes. | |
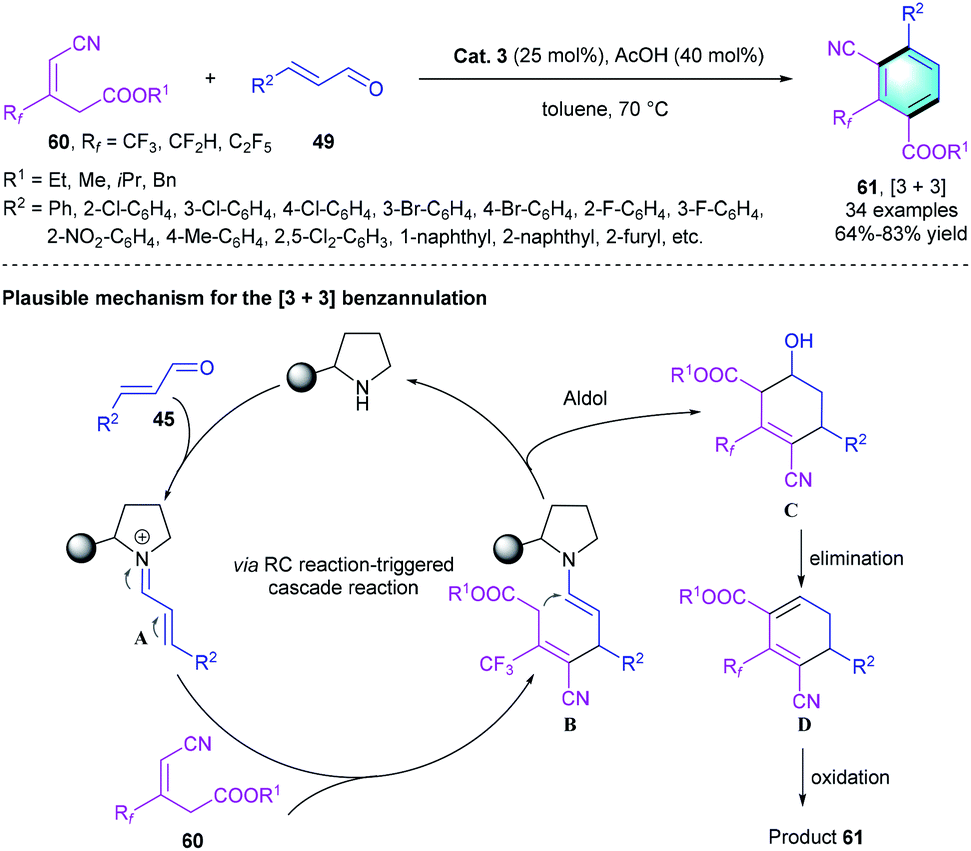
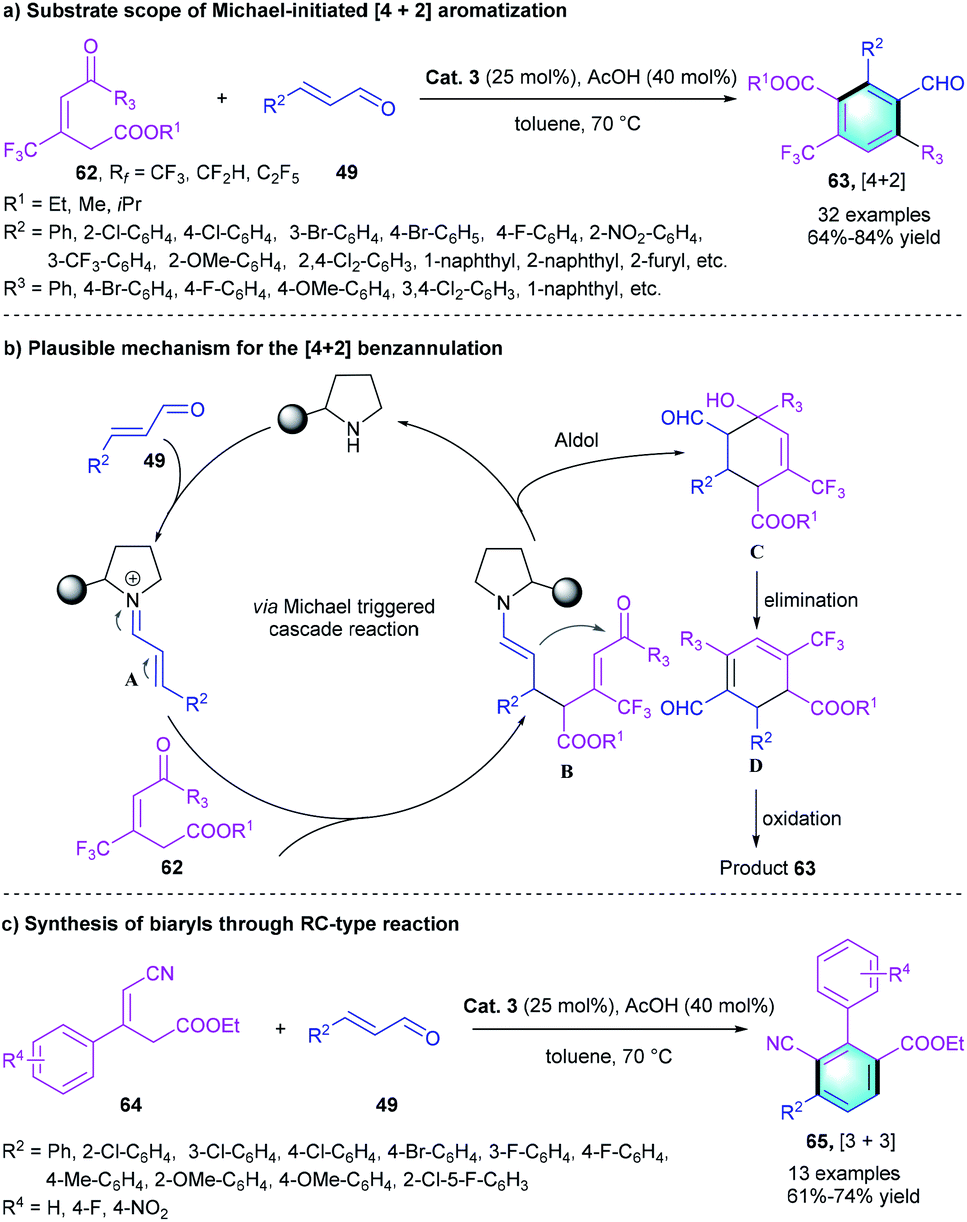
Given our experience to assemble pharmacologically important biologically interesting frameworks and the positive pharmacokinetic effects brought by CF3 group-containing compounds,33 our group recently reported a novel protocol for the construction of trifluoromethylated multi-functionalized benzenes through a secondary amine-catalyzed [3 + 3] benzannulation.34 The tri-substituted alkene with cyano group 60 was used as a nucleophile.35 α,α-Diphenylprolinol OTMS ether Cat. 3 was selected to facilitate the generation of iminium species A with the assistance of AcOH. The subsequent 1,4-addition reaction between 60 and A afforded enamine intermediate B, followed by an intramolecular aldol condensation to produce cyclic intermediate C. Simultaneous elimination and oxidation of C resulted in the [3 + 3] benzannulation product 61, which bears three adjacent substituents (Scheme 17). The investigation of substrate scope showed that the electron-deficient and electron-rich substituents could be well tolerated, producing the corresponding CF3-substituted benzenes in moderate-to-good yields (64–83%). A gram-scale reaction proceeded smoothly under the optimal condition, which verified the practical utility of this novel strategy.
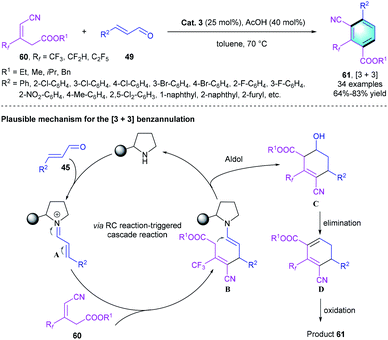 |
| | Scheme 17 Diphenylprolinol silyl ether catalyzed [3 + 3] benzannulation to synthesize tetra-substituted benzenes. | |
Moreover, the regioselectivity of the reaction was controlled by the functional groups of the tri-substituted alkenes. When a benzoyl group was introduced into the alkene substrate instead of the cyano group, a Michael-initialized [4 + 2] aromatization occurred, leading to the formation of a penta-substituted trifluoromethyl benzenes 63 (Scheme 18a). Diverse α,β-unsaturated aldehydes 49 and alkenes 62 with different substituents could participate in the reaction and furnished the target products in good yields (64–84%), which proved the generality of this aromatization. Notably, our strategy also provided a straightforward method for the synthesis of biaryls (Scheme 18c). The reaction between substrate 64 and unsaturated aldehydes 49 produced the desired aryl-substituted benzenes 65 in 61–74% yields.
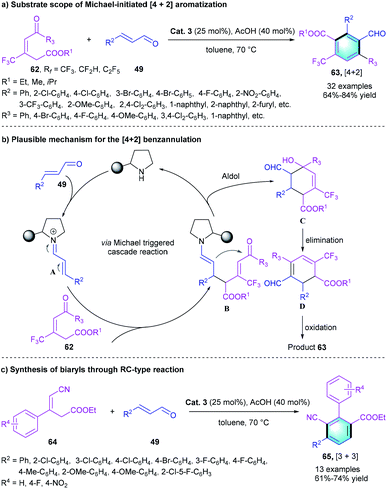 |
| | Scheme 18 Diphenylprolinol silyl ether catalyzed benzannulation to synthesize penta-substituted benzenes and biaryls. | |
3.3 [3 + 2 + 1] benzannulation
In 2006, Ramachary et al. presented a secondary amine-catalyzed multi-component benzannulation process for the construction of highly functionalized arenes.36 The secondary amine, piperidine or pyrrolidine, could smoothly catalyze the Knoevenagel–Michael–aldol condensation–decarboxylation cascade reaction of alkyl acetoacetates 66, aldehydes 67, and nitrosoarenes 69, giving the final substituted benzenes in good yields with excellent chemo- and regioselectivities (Scheme 19). In the reaction, Hagemann's ester 68 was firstly obtained via a [3 + 2 + 1] annulation of 66 and 67 in the presence of piperidine. The postulated reaction pathway of the following steps is illustrated in Scheme 20. The iminium activation of Hagemann's ester 68 formed intermediate A, which was converted into dienamine B. The reaction of B with nitrosoarenes 69 furnished intermediate D and released the hydroxide ion to give the imine product E. Spontaneous hydrolysis and isoaromatization of E generated the penta-substituted products 70.
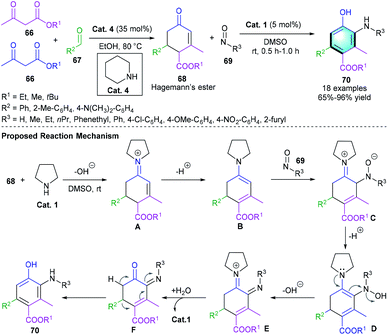 |
| | Scheme 19 Pyrrolidine catalyzed [3 + 2 + 1] benzannulation to synthesize penta-substituted benzenes. | |
 |
| | Scheme 20 Proline derivatives catalyzed intramolecular benzannulation to synthesize axially chiral biaryls. | |
3.4 Intramolecular benzannulation
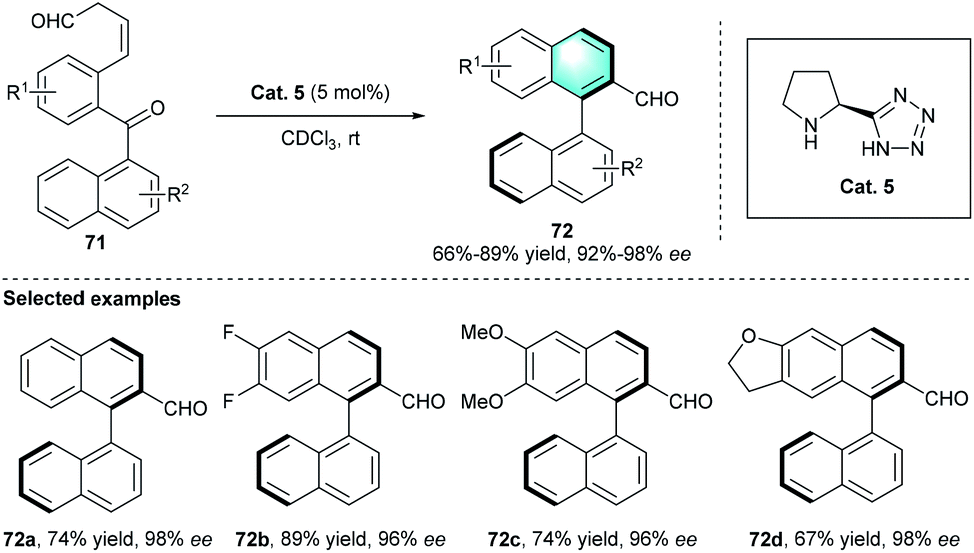
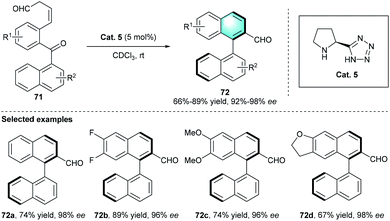
Apart from the above-mentioned strategies, secondary amine-catalyzed intramolecular cyclization is also a powerful route to achieve diverse arenes or biarys. In 2014, Sparr and co-workers presented an efficient method for the preparation of optically active biaryl atropisomer through an intramolecular benzannulation.37 Considering the excellent reactivity of β,γ-unsaturated aldehydes via dienamine catalysis, the exploration was started with ketoaldehyde 71, which could be obtained from a precursor through double oxidation (Scheme 20). The enantiocontrol of the process was determined by the asymmetric catalytic aldol condensation through enamine activation and the following central-to-axial chirality transfer. The evaluation of several privileged secondary amines indicated that proline derivatives Cat. 5 was the optimal catalyst. Using CDCl3 as the reaction medium in the absence of additive at room temperature proved to be the optimal reaction conditions. In the further generality and limitation investigation, various substituents with different electronic properties participated in the reaction successfully, providing the corresponding axially chiral products 72 with excellent enantioselectivities (up to 98%). Based on the literatures, this study was the first report to assemble axially chiral biaryls by aromatic annulation using secondary amine catalyst.
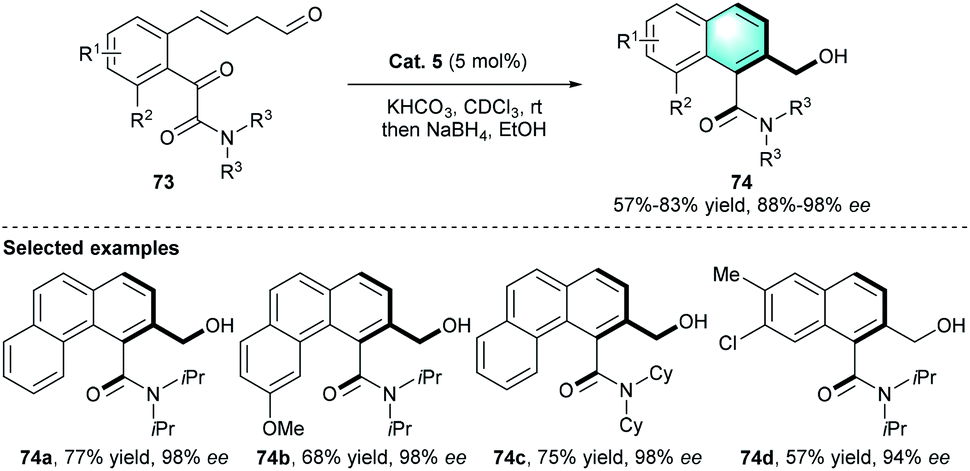
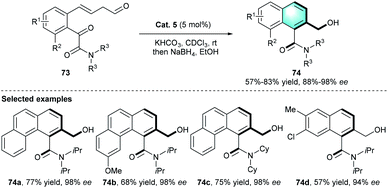
In a follow-up study, the Sparr group developed another stereoselective arene-forming aldol condensation using the chiral secondary amine catalysts mentioned above (Scheme 21).38 Under certain reaction conditions, axially chiral aromatic amides 74 were produced with high stereoselectivities.
 |
| | Scheme 21 Proline derivatives-catalyzed intramolecular benzannulation to synthesize axially chiral aromatic amides. | |
Compared with their previous work, this reaction proceeded faster and could complete within minutes because of the enhanced reactivity of glyoxylic amide substrates.
4 Primary amine-catalyzed benzannulation
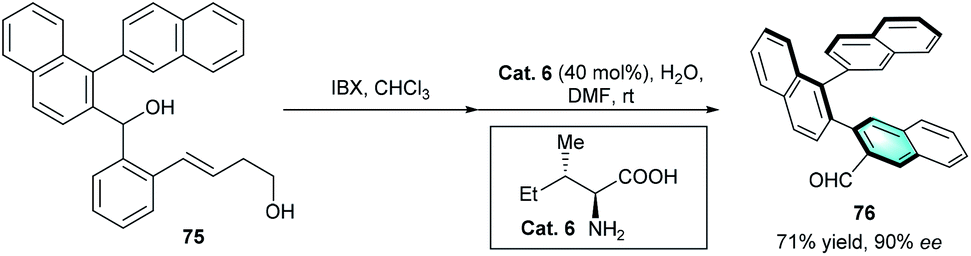
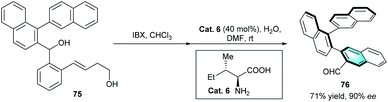
The development of primary amine catalysis has provided a possible solution for the challenging scenarios of catalytic benzannulation.39 Many elegant publications on primary amine catalysis to access various complex pharmaceutically and structurally important cyclic scaffolds have been reported.40 However, primary amine-catalyzed benzannulation is less reported. Up to now, the only example was reported by Sparr and co-workers in 2016.41 In the protocol, the oxidation of binaphthalene derivative 75 led to the corresponding ketoaldehyde, which then underwent a stereoselective intramolecular arene-forming aldol condensation catalyzed by natural primary amine catalyst (Scheme 22). The strategy provided an efficient access to 1,2-naphthalene oligomers 76 with two chiral axes in good yield with excellent enantioselectivity (90% ee).
 |
| | Scheme 22 Primary amine-catalyzed intramolecular benzannulation to synthesize axially chiral 1,2-naphthalene oligomers. | |
5 Tertiary amine-catalyzed benzannulation
Tertiary amine catalysts, which could serve as Lewis or Brønsted base, have been widely applied in facilitating the annulation reactions, thereby synthesizing numerous important carbocyclic and heterocyclic compounds. In this part, tertiary amine-catalyzed benzannulation for the construction of highly functionalized arenes was discussed.
5.1 [4 + 2] benzannulation
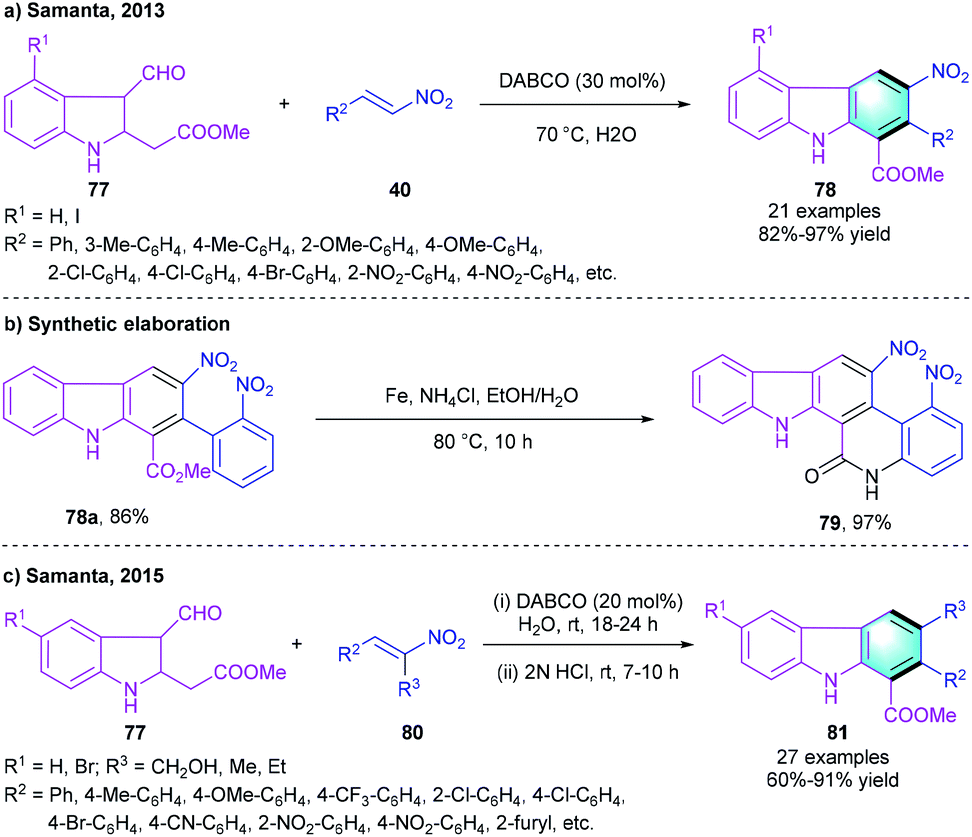
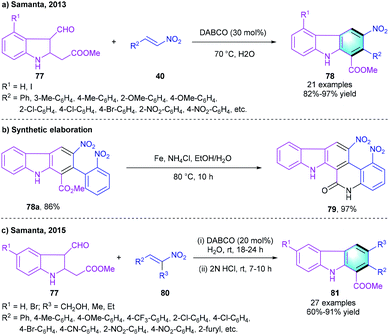
In 2013, the group of Samanta developed a one-pot [4 + 2] cyclization for the preparation of carbazole derivatives using methyl 2-(3-formyl-indolin-2-yl)acetates 77 and nitroolefins 40 as substrates (Scheme 23a).42 This Michael–Henry–aromatization cascade reaction proceeded smoothly in the presence of DABCO, and the optimal reaction efficiency was observed at 70 °C in water. The variation of substitution R1 did not affect the reaction much. 77 with both electron-donating and electron-withdrawing substituents gave the corresponding products 78 in high-to-ideal yields (82–97%). A gram-scale reaction was carried out under standard conditions without loss of yield and efficiency. Moreover, the practicability was proved by using the strategy to synthesize new fused carbazole 79 (Scheme 23b). Later, the same group further reported a one-pot two-step approach to construct highly functionalized carbazole scaffolds. The process was initiated with the DABCO-catalyzed Michael–Henry reaction between 77 and substituted nitrodiene 80. Subsequent elimination occurred under the treatment of 2 N HCl, forming target compound 81 (Scheme 23c).43 This work achieved a flexible and direct synthetic route to multi-substituted carbazole derivatives.
 |
| | Scheme 23 DABCO-catalyzed [4 + 2] benzannulation to synthesize carbazole derivatives. | |
As a versatile synthon to access cyclic and polycyclic compounds, α-substituted allenoates and relevant [4 + 2] reactions have attracted considerable attention. For example, in 2016, Huang's group disclosed an efficient method to attain polysubstituted benzenes 84 via the annulation of dienic sulfones 82 and α-substituted allenoates 83 (Scheme 24).44 The reaction proceeded successfully with DMAP as the catalyst and CH3COONa as the additive in CHCl3 at 60 °C. Various substituents on 82 or 83 were well tolerated, providing product 84 in acceptable-to-good yields (29–72%) with high regioselectivity. However, low yields were observed when the benzene core beard an electron-deficient (NO2) or an electron-rich group (OMe). Moreover, some control experiments were conducted to reveal the mechanism. Isolated intermediates D and F could be converted into 84 efficiently in the presence of CH3COONa, proving the reaction pathway in Scheme 24.
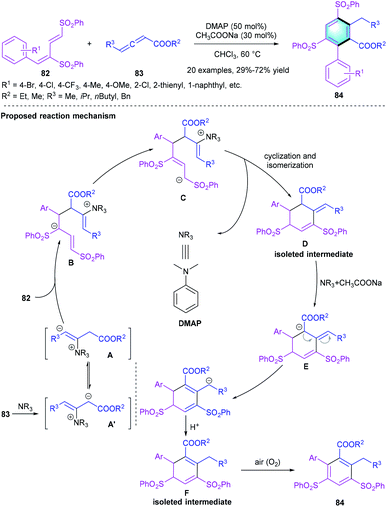 |
| | Scheme 24 DMAP-catalyzed [4 + 2] benzannulation to synthesize penta-substituted benzenes. | |
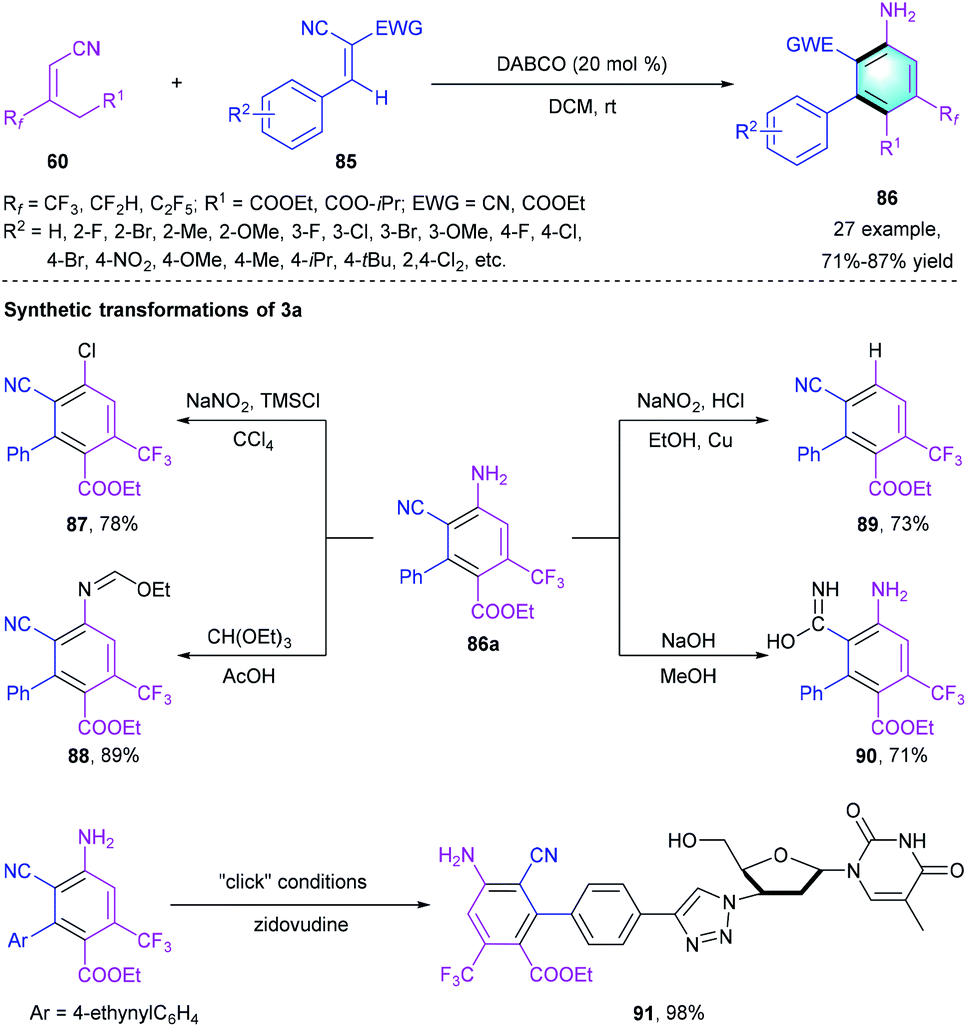
More recently, our group reported a highly chemoselective benzannulation cascade reaction to assemble CF3-functionalized biaryls (Scheme 25).45 The Michael addition-initiated [4 + 2] annulation between electron-deficient alkene 85 and fluoro-substituted 60 occurred uneventfully using DABCO as the catalyst under mild conditions. Diverse R2 substituted on the phenyl ring at different positions were compatible in the reaction, providing polysubstituted biaryls 86 in good-to-high yields (71–87%). A gram-scale reaction proceeded smoothly under the optimal conditions with comparable reaction efficiency. Polysubstituted biaryl 86a could be smoothly converted to diverse derivatives through functional group transformations, which illustrated the synthetic utility of this reaction.
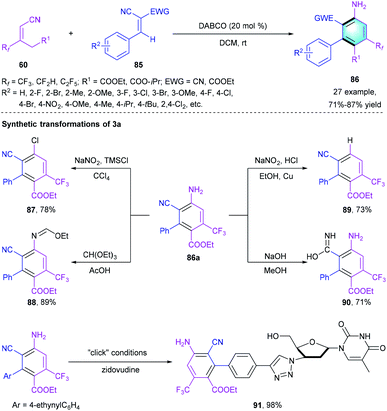 |
| | Scheme 25 DABCO-catalyzed [4 + 2] benzannulation to synthesize biaryls. | |
5.2 [3 + 3] benzannulation
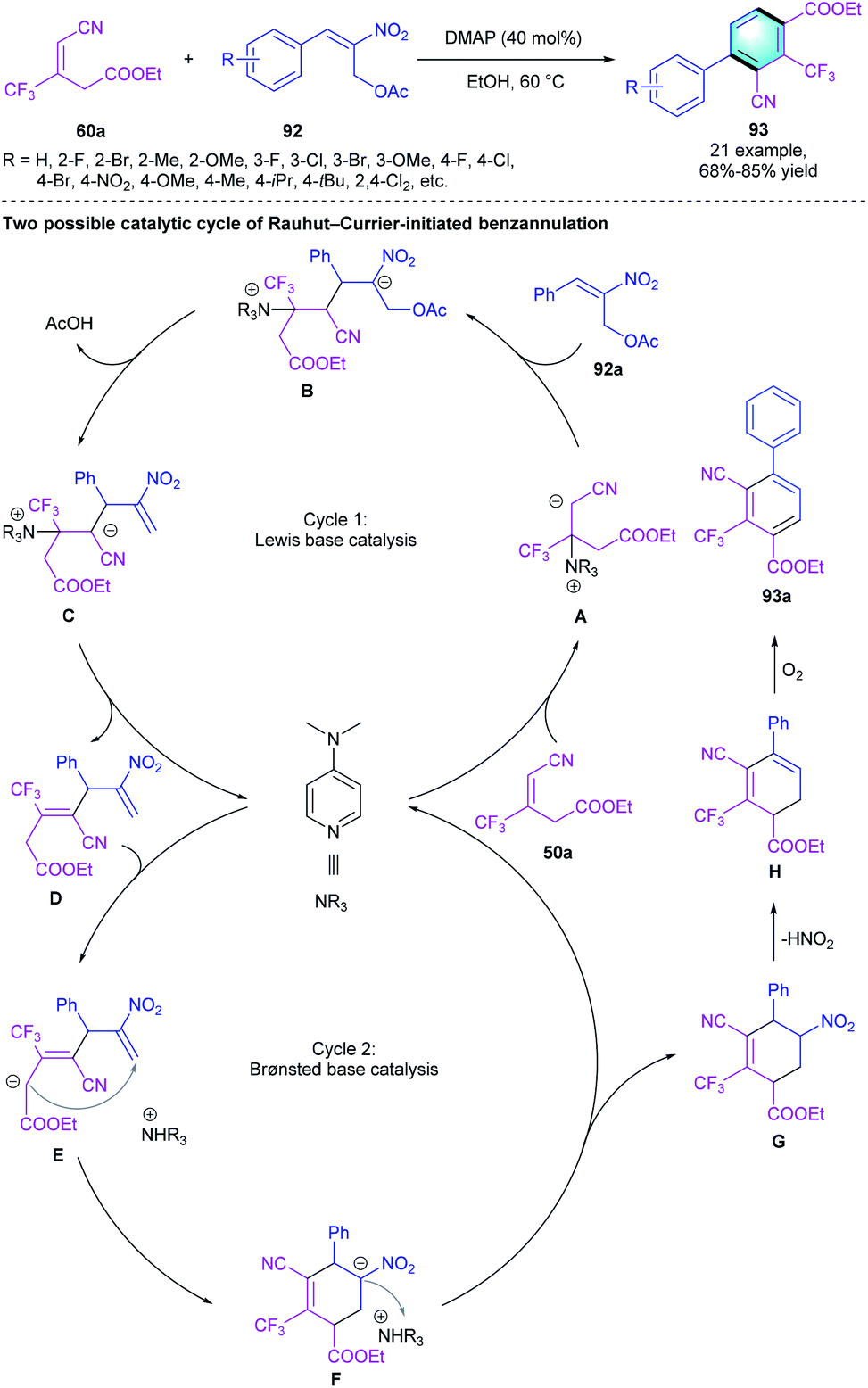
Our group had demonstrated that the trifluoromethylated alkenes 60 could act as useful C4 synthons in the [4 + 2] benzannulation with enals and electron-deficient alkenes. In the following work, an appropriate dielectrophilic synthon was applied to probe whether CF3-alkenes 60 could serve as the dinucleophilic C3 synthons. Consequently, a novel biaryl was obtained through the Rauhut–Currier reaction-initiated [3 + 3] aromatization using 2-nitroallylic acetate 92a as the C3 counterpart. The screening of tertiary amine catalysts indicated that the nucleophilic DMAP has the best efficiency. A broad spectrum of tetrasubstituted trifluoromethylbenzene 93 was obtained regardless of the electronic properties of R. As shown in the proposed mechanism, DMAP served as a Lewis base in the first cycle and as a Brønsted base in cycle 2 (Scheme 26).
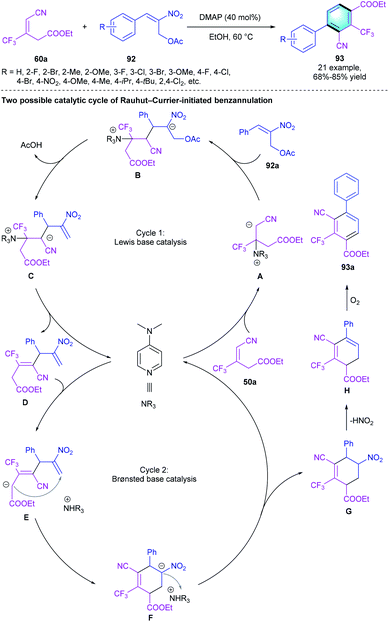 |
| | Scheme 26 DMAP-catalyzed [3 + 3] benzannulation to synthesize biaryls. | |
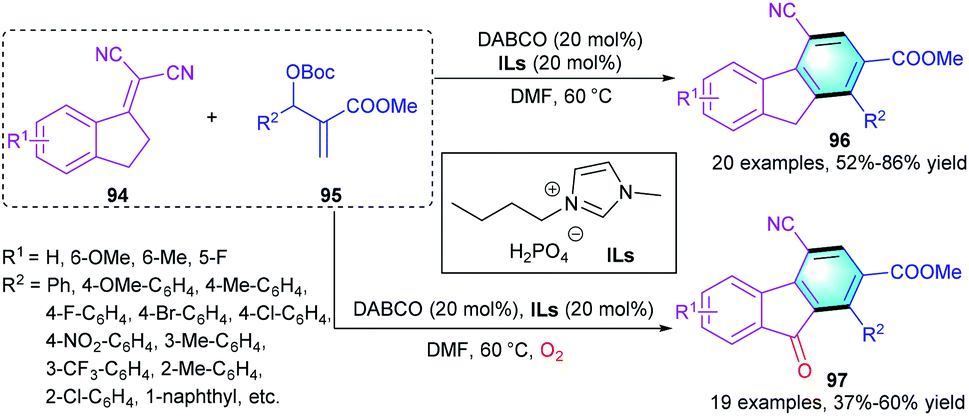
Recently, Yuan's group reported a [3 + 3] benzannulation of 1-indanylidenemalononitrile with MBH carbonate to deliver polyfunctionalized fluorene 96 or fluorenone 97 (Scheme 27).46 Using DABCO in the presence of ionic liquid, a series of functional fluorenes 96 was produced in moderate-to-high yields (52–86%). However, the electronic properties of substituents R1 and R2 showed a remarkable influence on the yields, wherein substrates with the electron-deficient groups providing higher yields than those with the electron-rich groups. Substituents at the ortho-position of 95 led to low yields because of their bulky steric hindrance. Interestingly, when oxygen was introduced to this reaction, a variety of fluorenone derivatives 97 could be obtained in 55–60% yields.
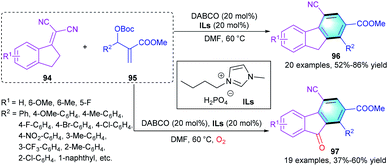 |
| | Scheme 27 DABCO-catalyzed [3 + 3] benzannulation to synthesize fluorine/fluorenone derivatives. | |
5.3 [2 + 2 + 2] benzannulation
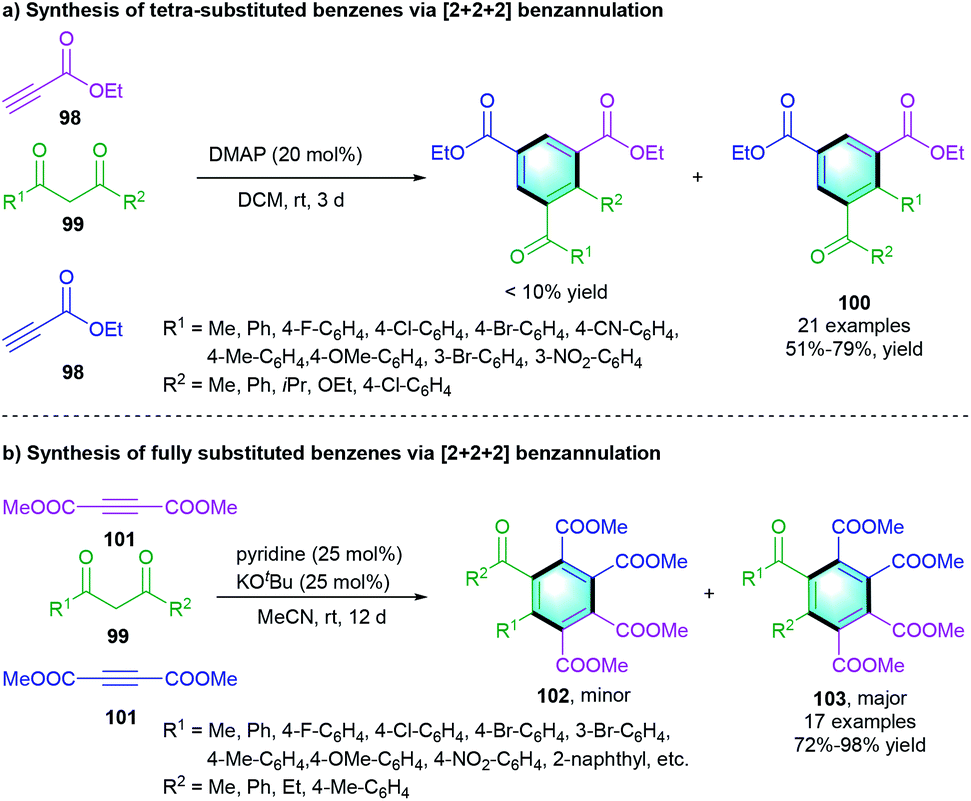
A three-component [2 + 2 + 2] annulation of ethyl propiolate 98 and 1,3-dicarbonyl compounds 99 was reported by Xue and co-workers, enabling the access to highly substituted benzenes 100 (Scheme 28a).47 The favorable yields were obtained in the presence of DMAP as the catalyst. Afterward, the same group developed a similar reaction of 99 with dimethyl acetylenedicarboxylate 101 using pyridine as the catalyst with the assistance of potassium tert-butoxide (Scheme 28b). The fully substituted benzenes 103 were smoothly formed with good-to-excellent yields (72–98%).48
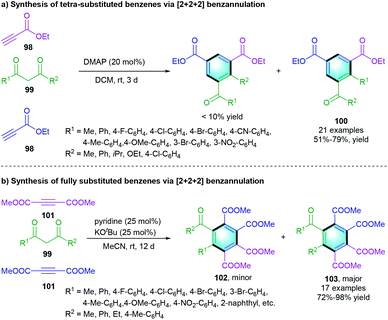 |
| | Scheme 28 DMAP-catalyzed [2 + 2 + 2] benzannulation to synthesize highly substituted benzenes. | |
5.4 Intramolecular benzannulation
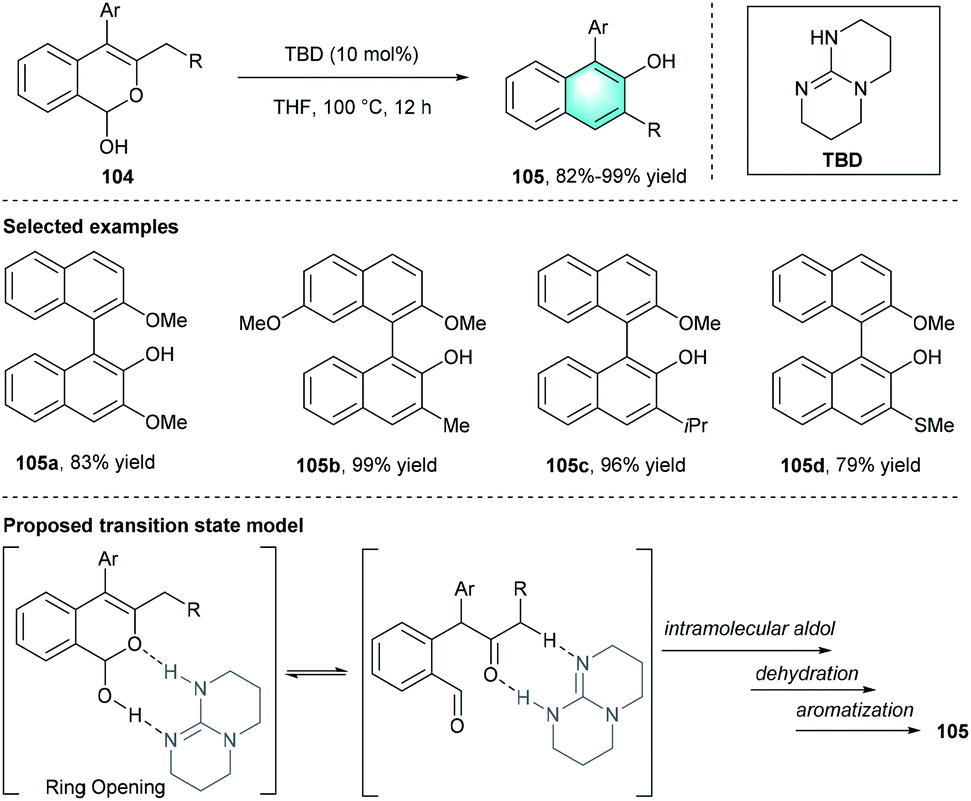
In 2012, Terada's group developed a direct intramolecular method to synthesize polycyclic substituted arenes through the Ferrier-type rearrangement of lactols 104.49 The strong organic base, 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD), was identified to be the optimal catalyst. Further evaluation of the solvents indicated that THF was important for the reaction. Under the standard conditions, the intramolecular multi-step transformation provided unsymmetrically substituted biaryls 105 with high yields (up to 98%). The plausible transition state was depicted in Scheme 29.
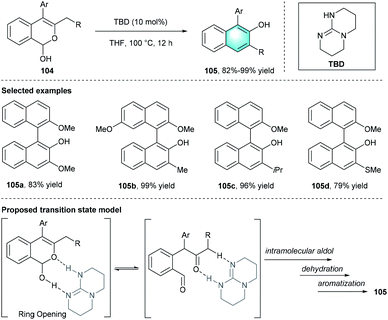 |
| | Scheme 29 TBD-catalyzed intramolecular benzannulation to synthesize substituted biaryls. | |
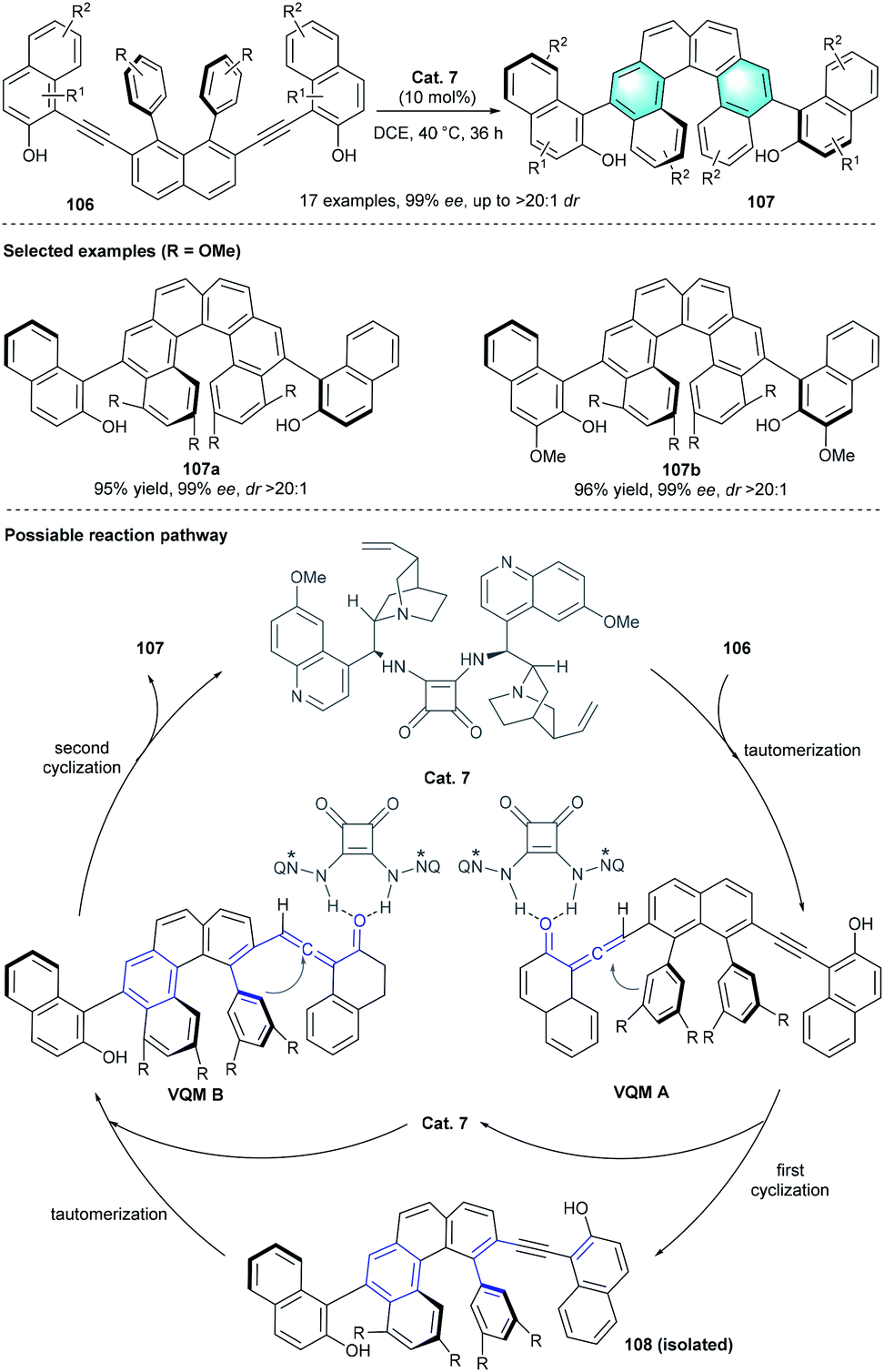
In 2019, Yan et al. reported the first diastereo- and enantioselective synthesis of axially and helically chiral arenes through tertiary amine-catalyzed intramolecular benzannulation.50 After careful investigation of the reaction parameters, the optimal conditions were proven to be using 10 mol% bisquinine squaramide as the catalyst in DCE at 40 °C. The axially and helically chiral polycyclic arenes 107 could be obtained in acceptable-to-high yields (43–97%) with excellent diastereoselectivities and enantioselectivities (99% ee, up to >20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr). Substrates with dimethylamine phenyl or electron-donating heteroaromatics, such as furyl or thienyl, failed to provide the desired product. The authors proposed a plausible pathway for the reaction. As shown in Scheme 30, the vinylidene ortho-quinone methide intermediate A was formed in the presence of Cat. 7, following intramolecular cyclization generated the tetrahelicene-like molecule 108 with a stereogenic axis. The subsequent benzannulation of 108 would provide the corresponding enantioenriched products 107 with helical and axial chirality elements.
:1 dr). Substrates with dimethylamine phenyl or electron-donating heteroaromatics, such as furyl or thienyl, failed to provide the desired product. The authors proposed a plausible pathway for the reaction. As shown in Scheme 30, the vinylidene ortho-quinone methide intermediate A was formed in the presence of Cat. 7, following intramolecular cyclization generated the tetrahelicene-like molecule 108 with a stereogenic axis. The subsequent benzannulation of 108 would provide the corresponding enantioenriched products 107 with helical and axial chirality elements.
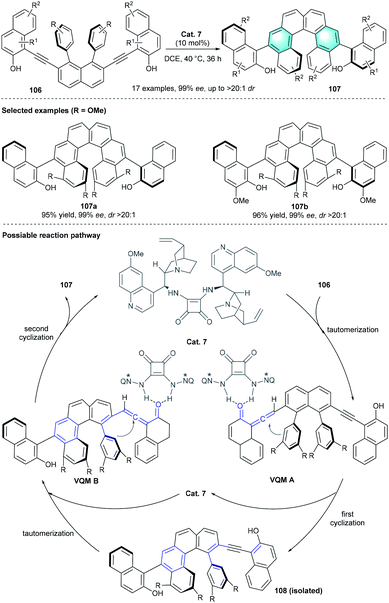 |
| | Scheme 30 Bisquinine squaramide-catalyzed intramolecular benzannulation to synthesize chiral helical arenes. | |
6 Tertiary phosphine-catalyzed benzannulation
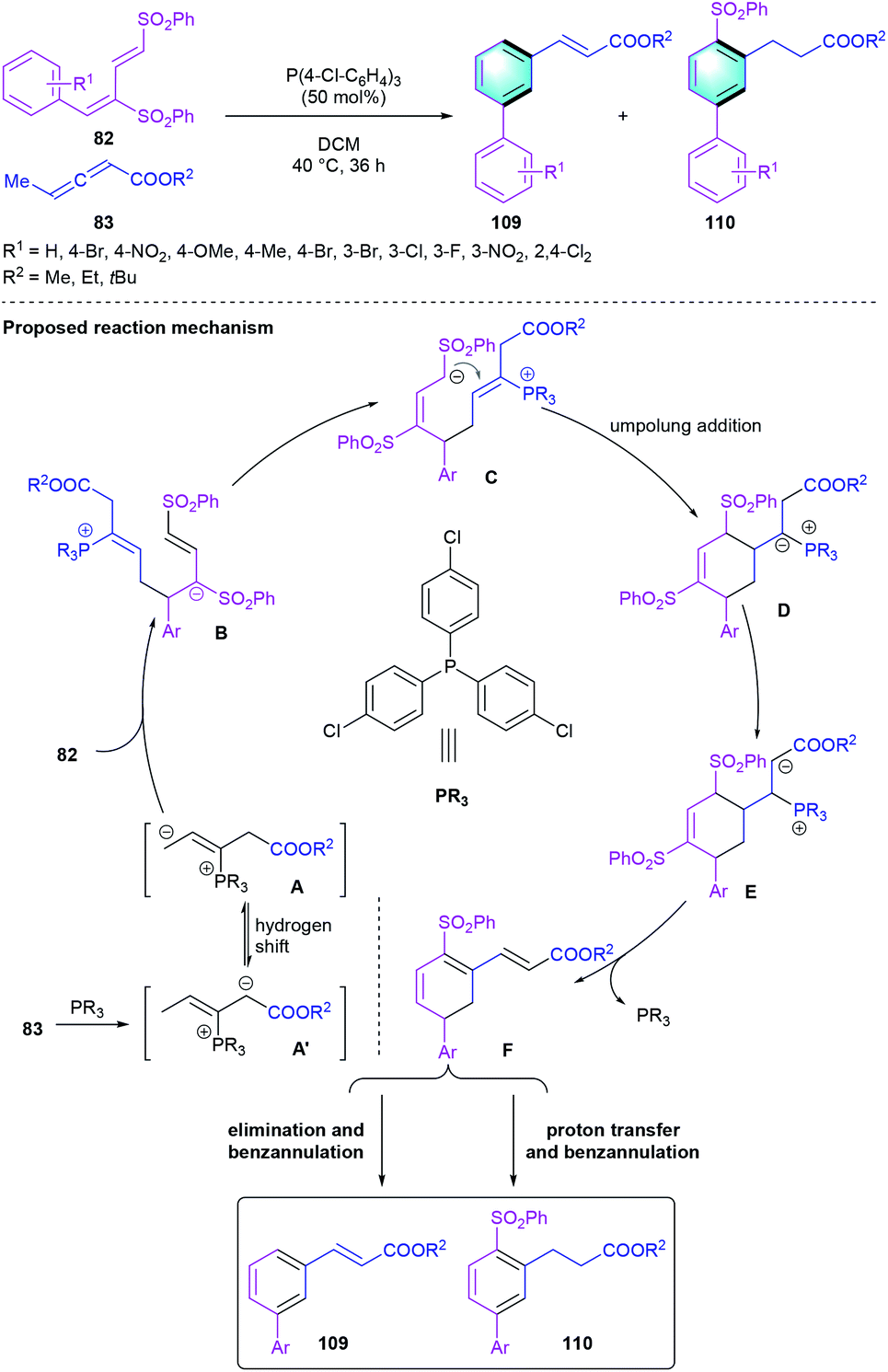
Besides being used as powerful ligands, tertiary phosphine-catalyzed reactions were rapidly developed over the past two decades, which has emerged as a powerful tool for constructing carbon–carbon bonds due to its good nucleophilicity and unique ability as leaving group. In 2013, the first tertiary phosphine-catalyzed [4 + 2] benzannulation between dienic sulfones 82 and allenoates 83 was reported by Huang and co-workers, leading to the formation of biaryls 109 and 110.51 Various phosphine catalysts were evaluated and tris(4-chlorophenyl)phosphine turned out to be the optimal catalyst. Substituent variations on the aryl unit or allenoate were well tolerated to produce the functionalized biaryls 109 and 110 in 42–89% total yields. As illustrated in Scheme 31, the elimination and aromatization of the crucial intermediate F would afford the disubstituted benzene, whereas proton transfer and aromatization generated the tri-substituted aryl compound.
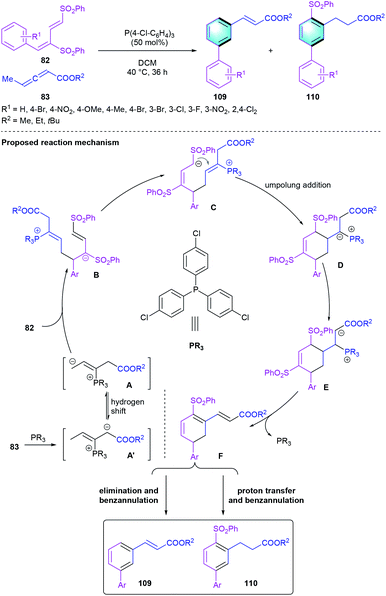 |
| | Scheme 31 Tertiary phosphine-catalyzed [4 + 2] benzannulation to synthesize biaryls. | |
Inspired by the pioneering work of the phosphine-catalyzed asymmetric annulation of allenoates, the Tong group documented a phosphine-catalyzed atroposelective [4 + 2] benzannulation for the first time.52 The cyclization reaction between δ-acetoxy allenoate 111 and 2-hydroxyquinone derivatives 112 was efficiently promoted by the bifunctional ferrocenylphosphine Cat. 8, providing the axially chiral biaryls 113 in satisfying yields (up to 87%) with high optical purity (up to 99%). The excellent stereocontrol might be attributed to the dual activation of both tertiary phosphine and tertiary amine moiety of Cat. 1, which cooperatively improved the reaction activity and enantioselectivity. This strategy has a broad substrate scope, and selected aryl-naphthoquinone products were shown in Scheme 32.
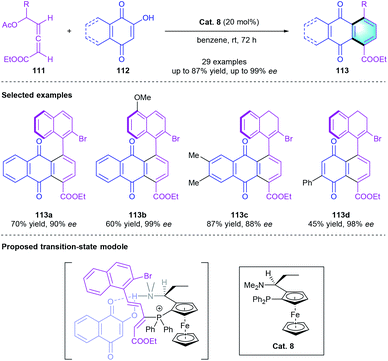 |
| | Scheme 32 Bifunctional ferrocenylphosphine-catalyzed atroposelective [4 + 2] benzannulation to synthesize aryl-naphthaquinones. | |
Given the extensive application of phosphine catalysis in the reaction of allenoates, in 2020, Swamy and co-workers uncovered a [4 + 2] Michael–vinylogous Mannich–aromatization cascade reaction to synthesize the polysubstituted arenes.53 In this protocol, using Ph3P as the catalyst, a series of functionalized phenyl sulfamates 115 was attained from 111 and cyclic sulfamidate imines 114 in 68–85% yields. Notably, if the reaction temperature was raised to 80 °C, the S–O bond would cleaved to afford product 116 (Scheme 33a). Moreover, the [4 + 2] benzannulation could be extended to cyclic sulfonyl imine 117, generating various substituted arenes 118 with a sulfonamide group in 69–82% yield (Scheme 33b).
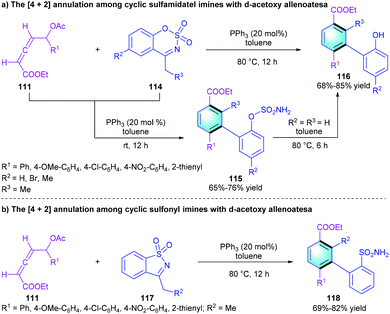 |
| | Scheme 33 Ph3P-catalyzed [4 + 2] benzannulation to synthesize highly functionalized arenes. | |
7 N-Heterocyclic carbene catalyzed-benzannulation
Since the discovery of stable nucleophilic carbenes by the pioneering work of the Bertrand and Arduengo groups,54 NHCs have emerged as exceptional organocatalysts and ligands because of their excellent σ-donor characteristics and moderate π-acidity, leading to the formation of useful metal complexes and active reaction intermediates.55 The ability of NHCs to serve as active organocatalyst enabled access to unique reaction modes, which were extensively explored and applied in catalytic C–C bond formation and cyclization reactions for the synthesis of challenging molecules.56 The following part discussed the achiral or chiral NHC-catalyzed benzannulations as powerful tools in the construction of multi-substituted arenes.
7.1 [4 + 2] benzannulation
Among the NHC-catalyzed cyclization reactions, the [4 + 2] benzannulation has been extensively studied and has emerged as a useful tool to access new aromatic cores. Early in 2011, Lupton's group disclosed a groundbreaking work of the NHC-catalyzed all-carbon [4 + 2] cycloaddition to synthesize 1,3-cyclohexadienes with complete diastereocontrol.57 In the next nine years, various useful NHC-catalyzed [4 + 2] strategies have been developed for constructing highly functionalized arene molecules.
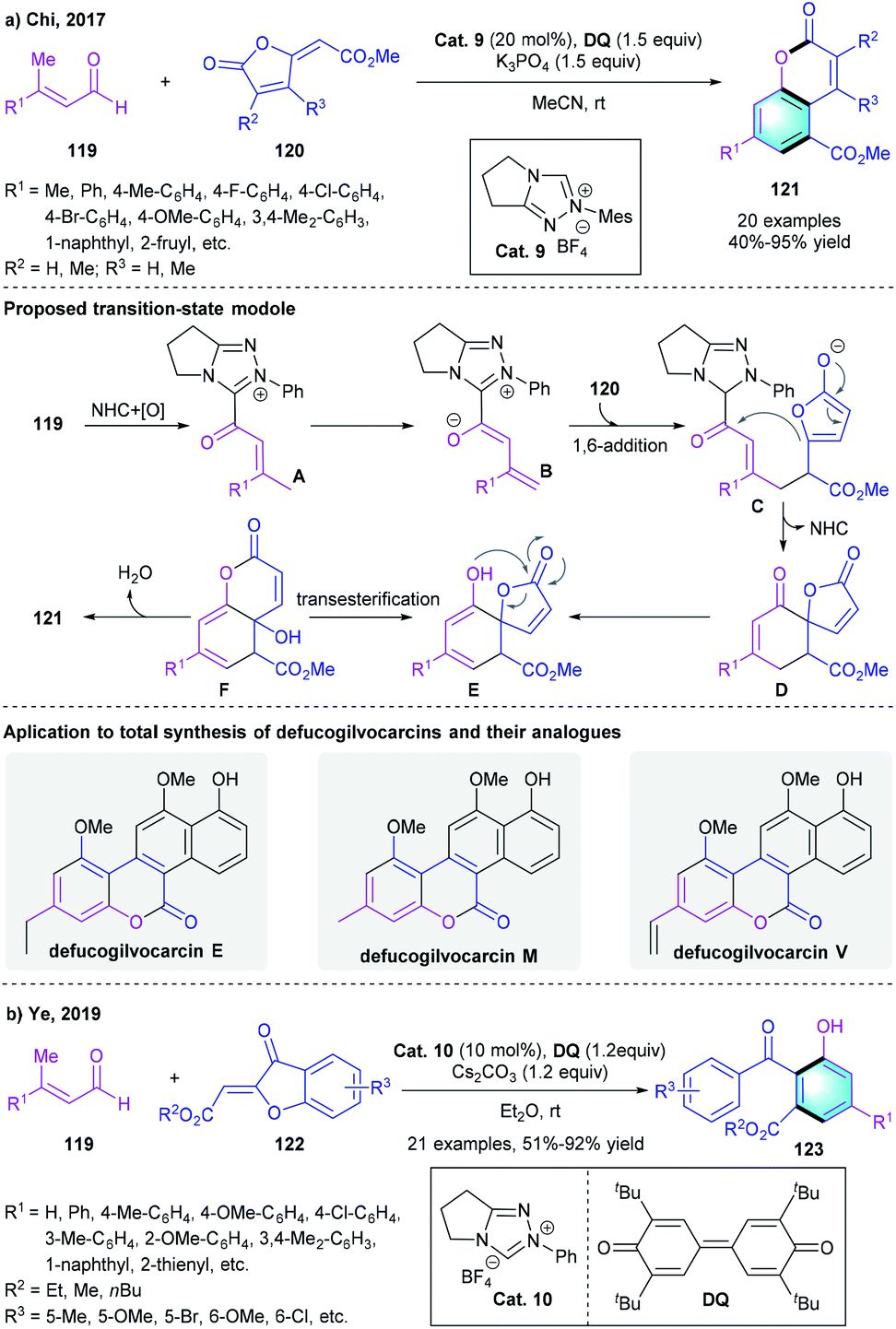
α,β-Unsaturated aldehydes could be used to afford azolium dienolate species under the catalysis of NHCs after oxidation. Based on this useful activation model, in 2017, Chi and co-workers developed a formal [4 + 2] process for the rapid construction of coumarins analogs (Scheme 34a).58 The [4 + 2] reaction of enals 119 with furanones 120 proceeded smoothly in the presence of Cat. 9 and DQ. In the proposed mechanism, the oxidation of Breslow intermediate would generate intermediate A. Deprotonation of A formed intermediate B, which underwent 1,6-addition to afford Intermediate C further underwent an intramolecular vinylogous Claisen-type condensation to generate intermediate D. Spontaneous enolization, transesterification and dehydration would afford the final multi-substituted coumarins 121. This [4 + 2] benzannulation process was found compatible with different substituted enals or α-/β-substituted furanones, producing the corresponding arenes in 40–95% yield. A gram-scale reaction also proceeded efficiently under the standard conditions. Moreover, the author successfully applied the method in the preparation of bioactive compound defucogilvocarcins E, M, and V. In 2019, the group of Ye extended the [4 + 2] benzannulation strategy to the reaction of β-methylenals 119 with aurones 122 for the synthesis of dihydroxybenzophenones (Scheme 34b).59
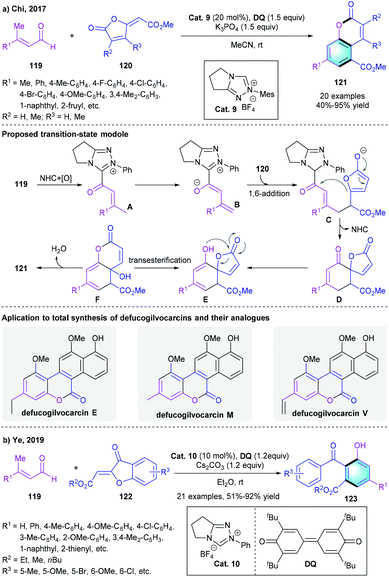 |
| | Scheme 34 NHC-catalyzed formal [4 + 2] benzannulation to synthesize coumarins analogs and dihydroxybenzophenones. | |
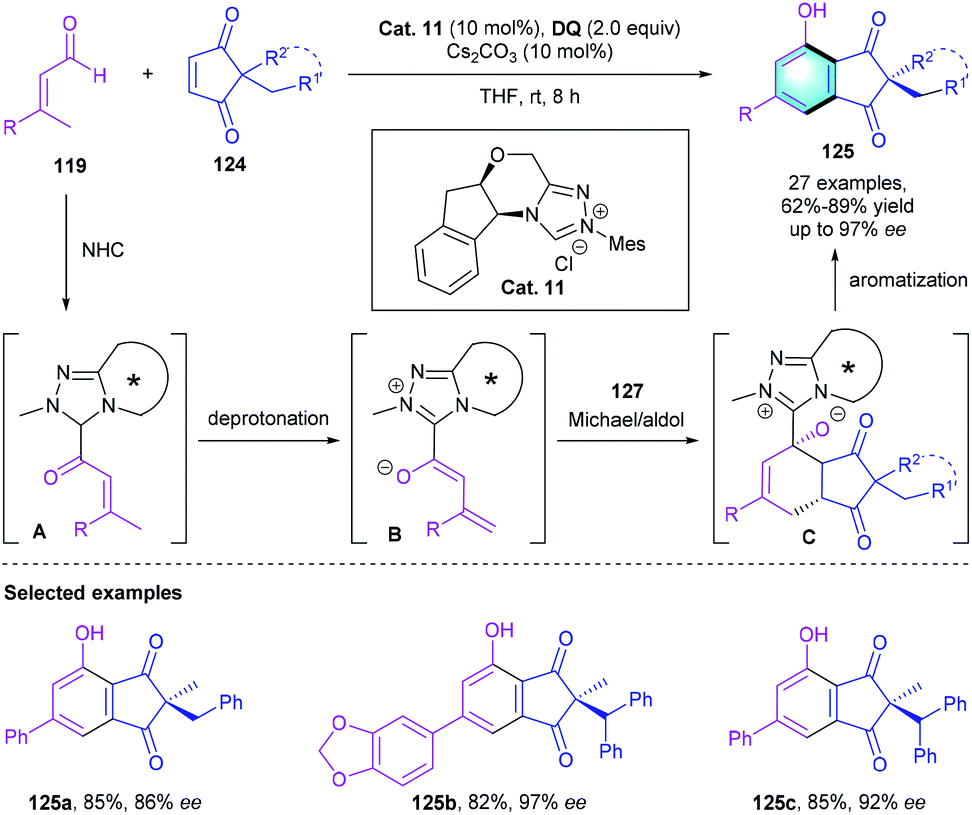
Further exploring the application of NHC catalysis in [4 + 2] benzannulation, the group of Chi described a chiral NHC-catalyzed desymmetrization of easily accessible diketones 124 with enals 119 (Scheme 35).60 In the presence of the NHC catalyst Cat. 11, the reaction proceeded efficiently, affording the corresponding multi-substituted phenols in 62–89% yields, with up to 97% ee. The reaction was supposed to start with the NHC-initiated formation of intermediate A, followed by γ-CH deprotonation, Michael reaction, aldol annulation, and oxidative aromatization to produce the final arenes 125 with an all-carbon quaternary chiral center. Notably, using E- or Z-enals did not affect the reaction outcomes, leading to the same yields and enantioselectivities. Afterward, a similar asymmetric desymmetrization reaction for constructing functionalized indandione derivatives was reported by Wang's group.61
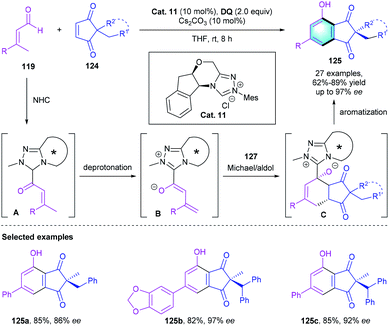 |
| | Scheme 35 NHC-catalyzed [4 + 2] benzannulation to synthesize multisubstituted phenols. | |
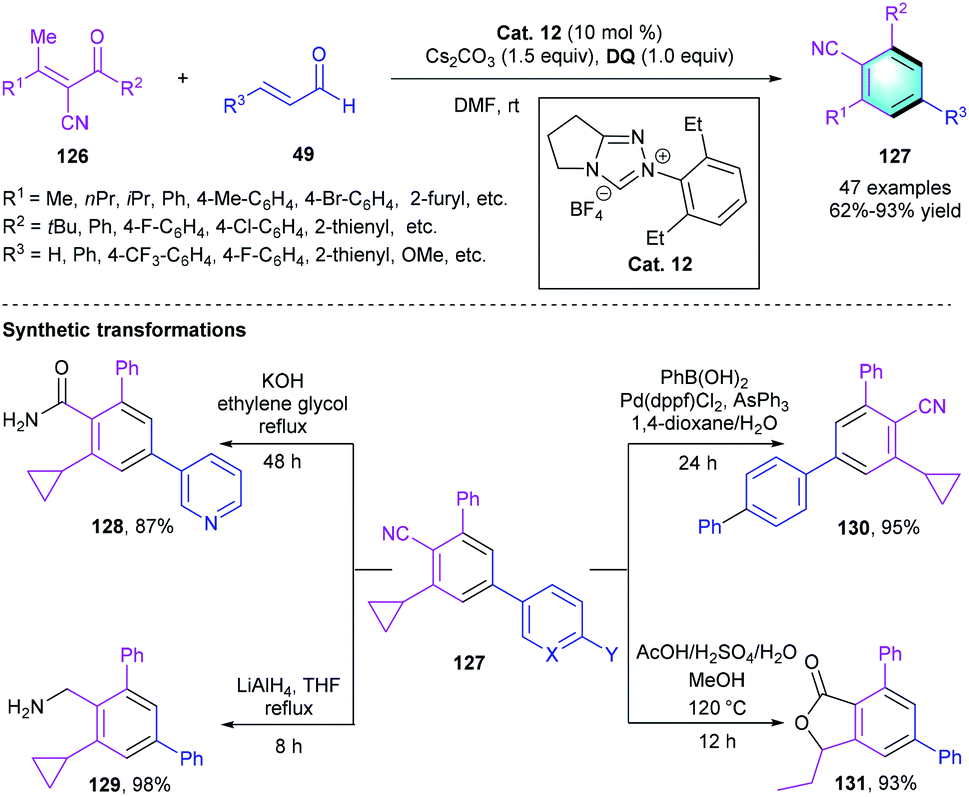
Besides the nucleophilic activation of the γ-position, the electrophilic activation of β-position of α,β-unsaturated aldehyde was also applied in NHC-catalyzed [4 + 2] benzannulations. In 2016, Wang's group presented an NHC-catalyzed [4 + 2] annulation between α-cyano-β-methylenones 126 and α,β-unsaturated aldehydes 49 for assembling benzonitrile unit (Scheme 36).62 The optimization of reaction conditions indicated that NHC precursors and solvents had a significant effect on the reactivity. Enhanced efficiency and yield were observed when using achiral Cat. 12 as the catalyst and tetra-tertbutyldiphenoquinone as the oxidant in the presence of Cs2CO3 in DMF. The substrate scope of enones 126 and enals 49 were carefully evaluated, giving a series of cyano-substituted benzene 127 with up to 93% yield regardless of the electronic properties of the substituents. Transformations of products 127 were carried out to demonstrate the applicability of the method. Later on, Ye and co-workers developed a similar reaction catalyzed by NHC precursors for the concise assembly of polysubstituted arenes.63
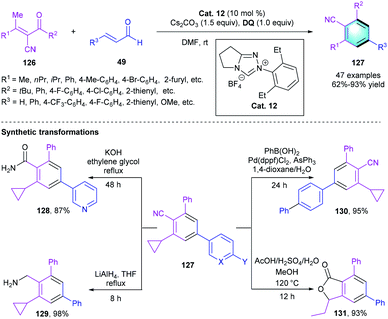 |
| | Scheme 36 NHC-catalyzed [4 + 2] benzannulation to synthesize cyano-bearing functional benzenes. | |
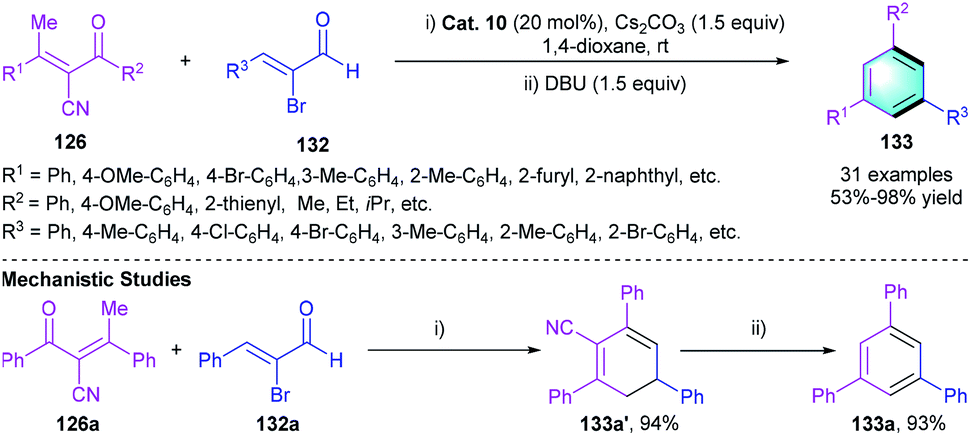
Later in the same year, Ye and co-workers reported another NHC-catalyzed [4 + 2] benzannulation of the above-mentioned enones 126 with α-bromoenals 132 instead of α,β-unsaturated aldehydes (Scheme 37).64 Employing a bicyclic N-phenyltriazolium NHC precursor Cat. 10 with the assistance of base in 1,4-dioxane enables the formation of 1,3,5-trisubstituted arene 133. In the scope investigation, good to excellent yields (77–98%) were attained in almost all cases. Several control experiments were conducted to gain insight into the mechanism. The reaction between 126a and 132a generated intermediate 133a′ followed by DBU-mediated elimination of cyanide, which eventually furnished the desired products.
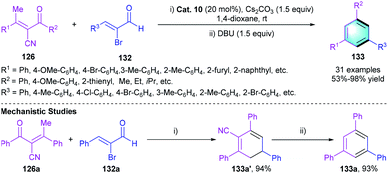 |
| | Scheme 37 NHC-catalyzed [4 + 2] benzannulation to synthesize tri-substituted benzenes. | |
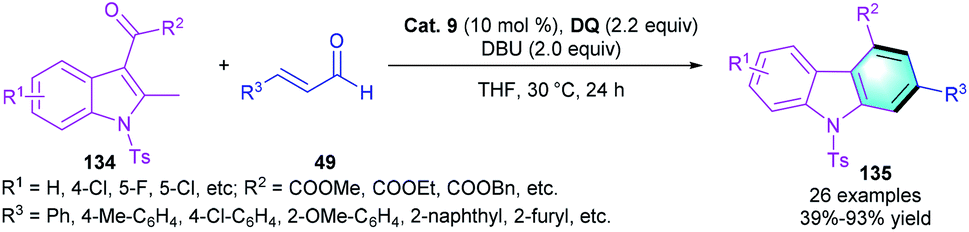
In 2018, Fu and co-workers extended the β-position activation strategy to the reaction of 2-methyl-3-oxoacetate indoles 134 with enals 49 to access multi-substituted carbazoles 135 (Scheme 38).65 The best reaction efficiency was achieved by using Cat. 9 as the catalyst in THF. The reaction has a broad substrate scope and corresponding products could be obtained in good yields (83–93%). Notably, the reaction of β-alkyl enal could also produce the final product with a decreased yield (39%).
 |
| | Scheme 38 NHC-catalyzed [4 + 2] benzannulation to synthesize carbazoles derivatives. | |
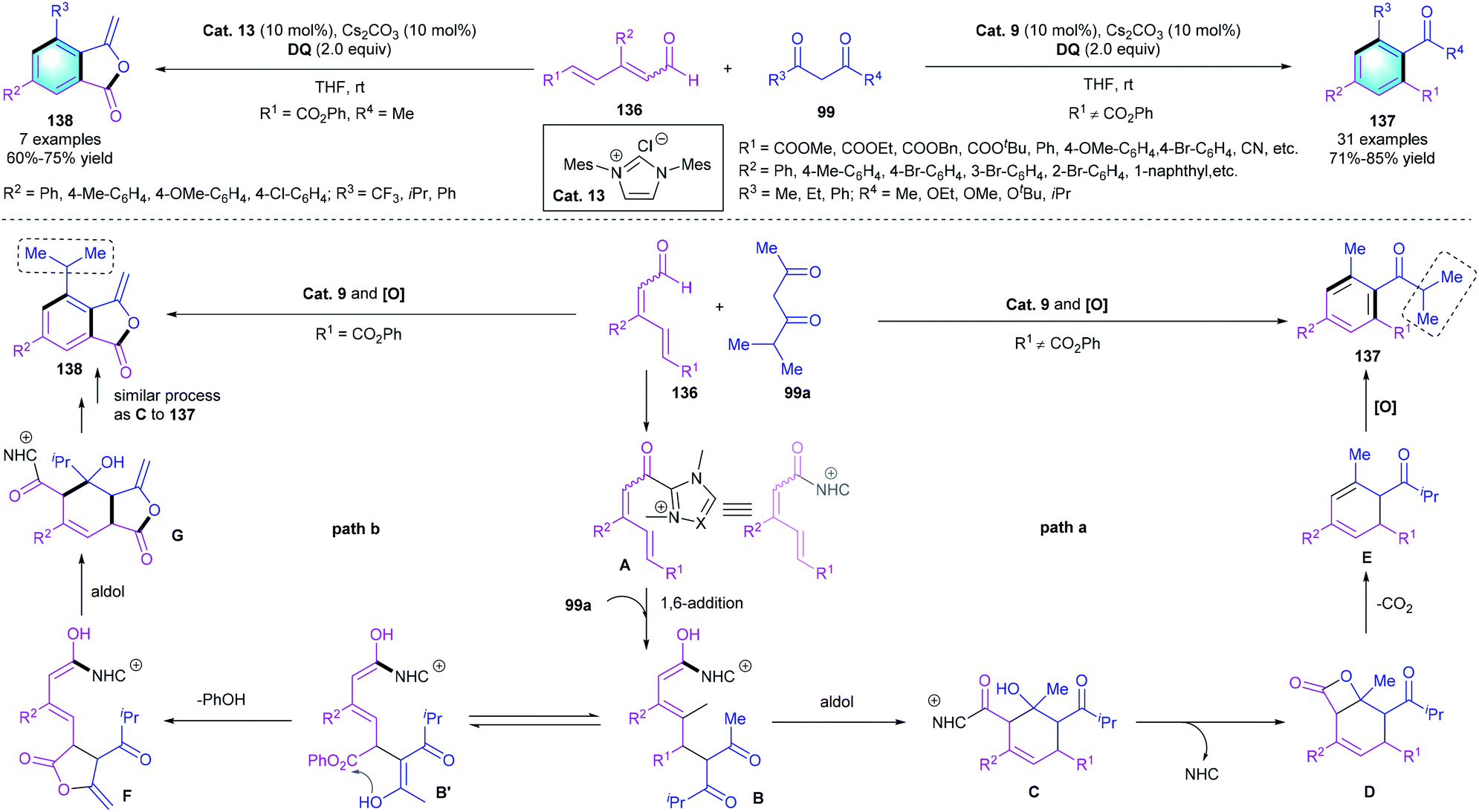
Based on the same activation strategy of enal and the principle of vinylogy, in 2015, Chi and co-workers first reported an NHC-catalyzed approach for the δ-carbon activation of α,β-γ,δ-diunsaturated aldehydes (Scheme 39). The highly efficient [4 + 2] benzannulation involving unsaturated aldehydes 136 and 1,3-diketones 99 proceeded smoothly, resulting in multi-substituted benzenes 137 in 71–85% yield.66 Notably, when 136 bearing a phenol ester group was used as the substrate, 3-ylidenephthalide 138 rather than 137 was delivered under identical conditions. A possible mechanism was proposed to explain the different chemoselectivity. As shown in Scheme 41, the two catalytic cycles initiated with the NHC-catalyzed activation of the remote δ-carbon of 136, and then 1,6-addition formed intermediate B. In pathway a, an intramolecular aldol condensation followed by β-lactone formation would give intermediate D. Subsequent decarboxylation and oxidative aromatization released the desired tetra-substituted arenes 137. On the contrary, in pathway b, R1 is an active aryl ester unit. Transesterification and isomerization would form the 5-membered lactone F, which underwent similar decarboxylation and oxidative aromatization processes to give the corresponding product 138.
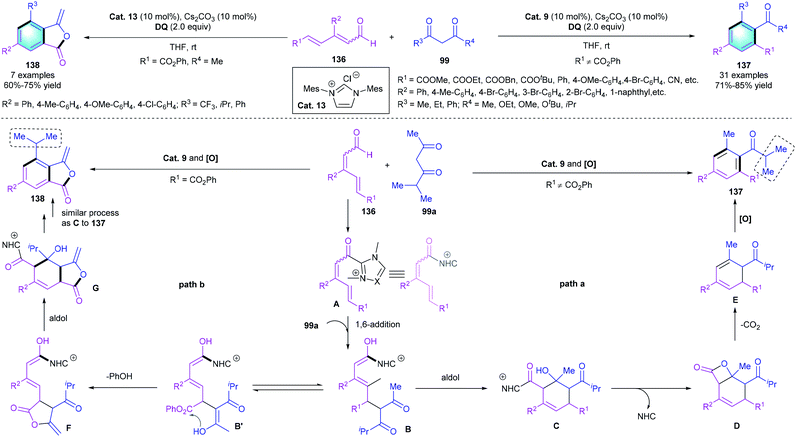 |
| | Scheme 39 NHC-catalyzed and substrate-controlled [4 + 2] benzannulation to synthesize tetra-substituted arenes or 3-ylidenephthalides. | |
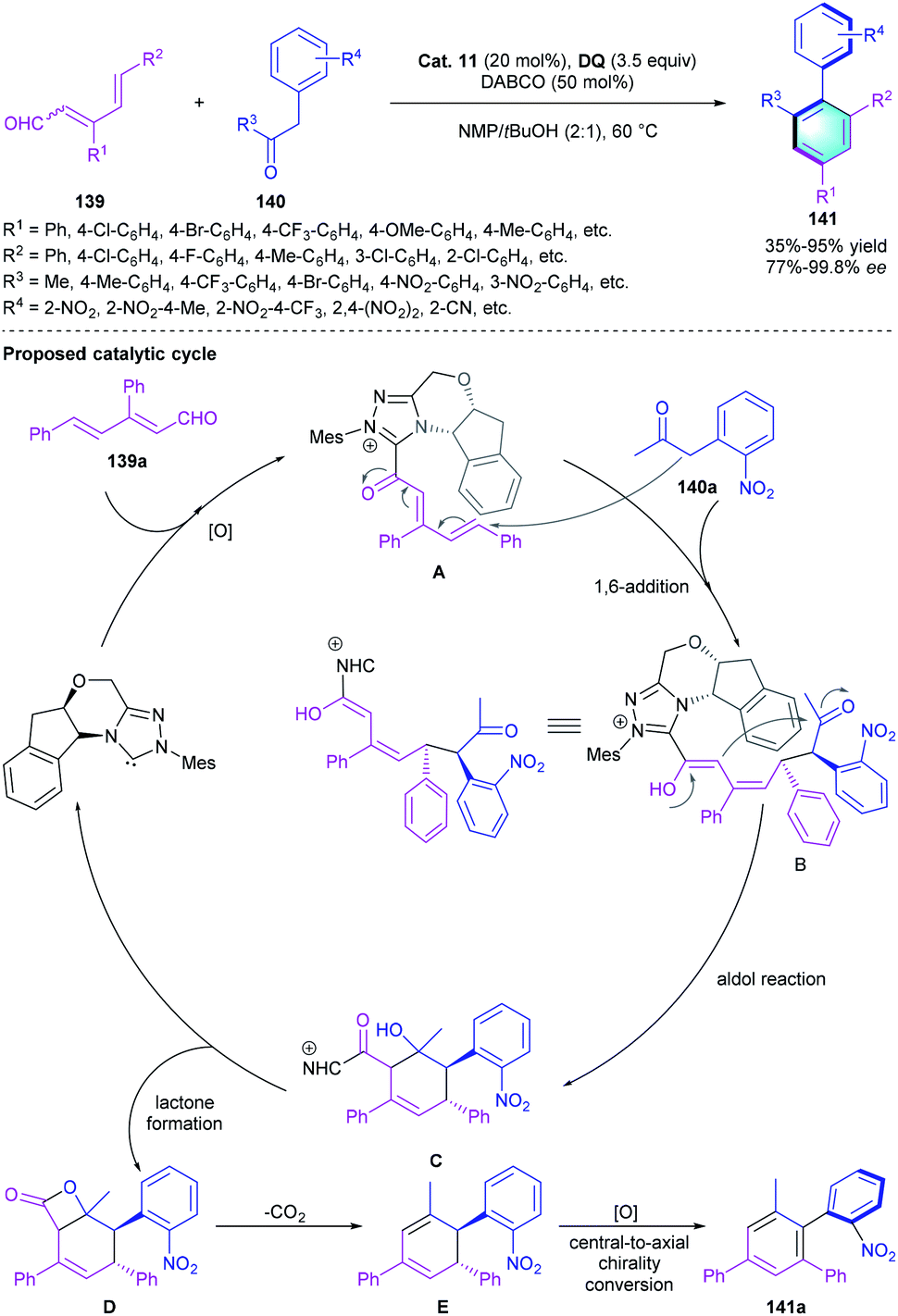
Using the remote activation strategy, Zhu and co-workers demonstrated that chiral NHC catalysts could be used for the asymmetric construction of atropisomeric arenes.67 The [4 + 2] cycloaddition of conjugated dienals 139 with α-aryl ketones 140 was developed to generate the chiral biaryls by using Cat. 11 as the pre-catalyst, DABCO as the base, and DQ as the oxidant (Scheme 40). As shown in the catalytic cycle, the reaction was initiated with the formation of acyl azolium intermediate A through NHC activation and oxidation, followed by the addition of 140a to generate adduct B. Then intermediate B underwent intramolecular aldol condensation to form the cyclic adduct C. Lactonization ended the catalytic cycle and regenerated the NHC catalyst. Thermodynamic controlled decarboxylation and aromatization led to the central-to-axial chirality transfer, providing the axially chiral arene 141. Notably, this study significantly expanded the application of NHC-catalyzed benzannulation in the assembly of enantioenriched atropisomeric biaryls.
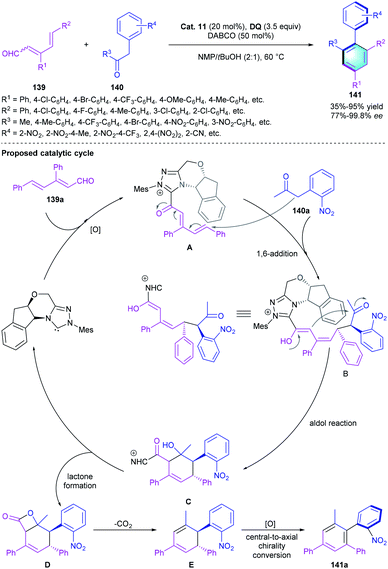 |
| | Scheme 40 NHC-catalyzed [4 + 2] benzannulation to synthesize axially chiral biaryl. | |
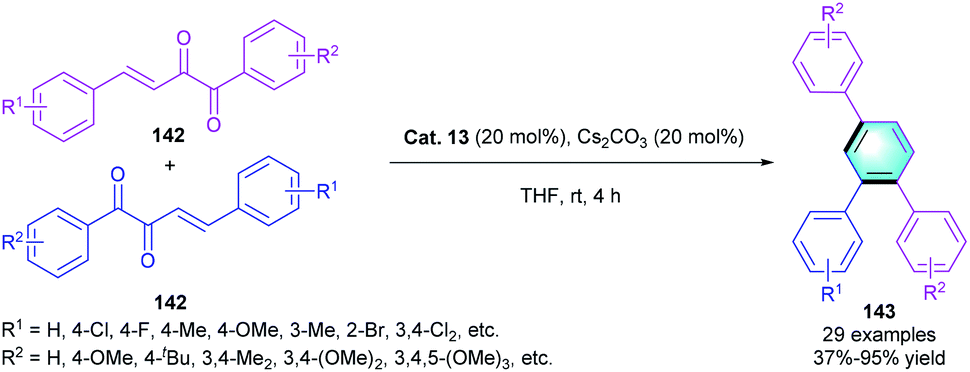
Comparing with the aldehyde, NHC catalysts were much less used to activate ketone derivatives. In 2017, Fang and co-workers developed a direct and efficient method for the synthesis of 1,2,4-triarylbenzenes through NHC-catalyzed umpolung [4 + 2] benzannulation of β,γ-unsaturated diketones 142 (Scheme 41).68 The reaction proceeded smoothly to deliver the corresponding benzene derivatives 143 by using Cat. 13 as the catalyst. Electron-donating or electron-withdrawing substituents at ortho-/meta-/para-positions on the aryl rings were all compatible in the reaction, resulting in the final products with favorable yields (up to 98%).
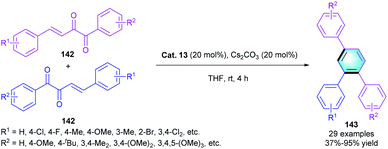 |
| | Scheme 41 NHC-catalyzed [4 + 2] benzannulation to synthesize triaryl substituted benzenes. | |
7.2 [3 + 3] benzannulation
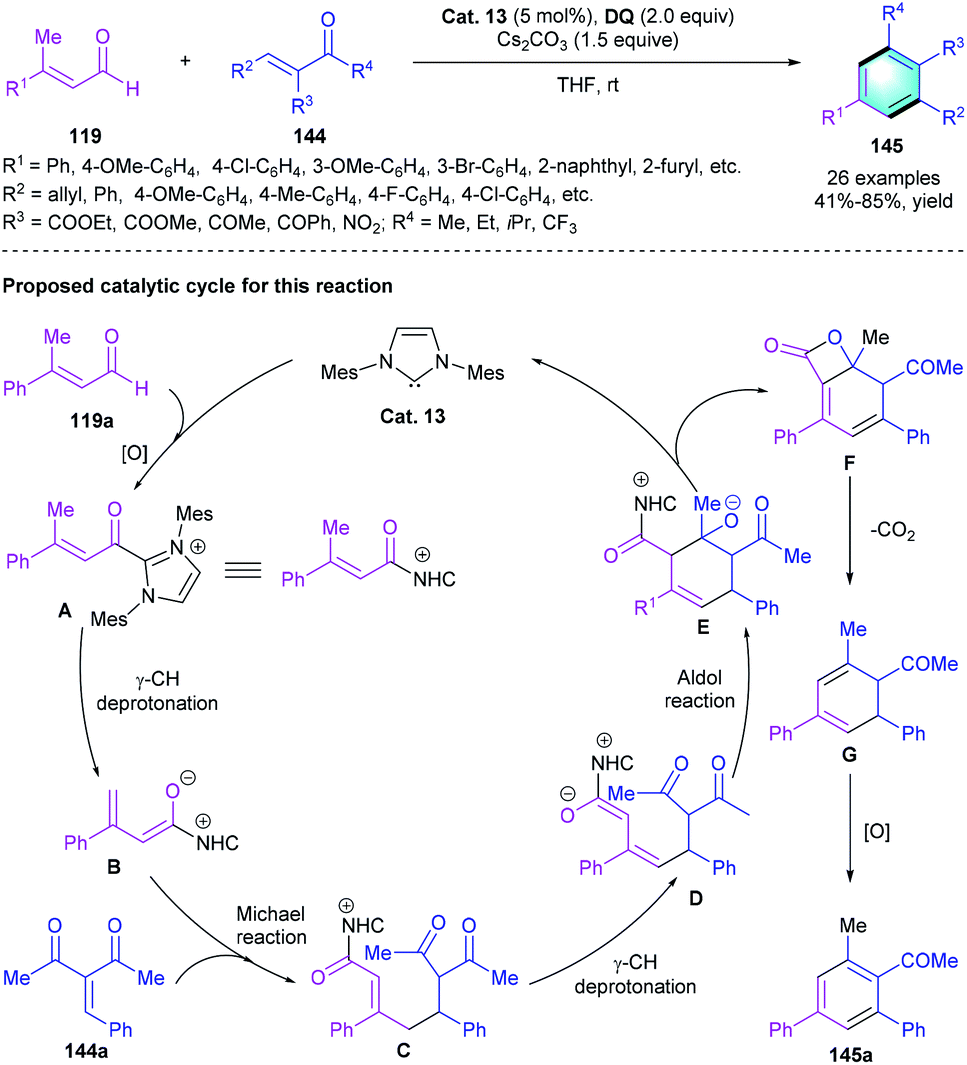
As mentioned above, α,β-unsaturated aldehydes could be used to afford azolium dienolate species in the presence of NHC catalyst. Based on this useful activation model, Chi and co-workers developed an NHC-catalyzed [3 + 3] cyclization of enals with enones (Scheme 42).69 In the reaction, β-methyl enal 119 was used as the precursor to form azolium dienolates, then reacted with enones 144, producing corresponding polyfunctionalized benzenes 145 in medium-to-good yields (41–85%). Different substituents at the β-position (R1) of enals 119 and the α/β-position (R2/R3) of enones 144 were well tolerated. Notably, using Z- or E-isomer of the substrates barely affected the reaction outcomes, which simplified the preparation of the substrates. A possible mechanism for this reaction was proposed. The catalytic cycle started with the generation of A through oxidation. Following γ-deprotonation formed azolium dienolate intermediate B. Subsequent nucleophilic addition of B to 144a resulted in intermediate C, which further underwent an intramolecular aldol reaction to generate intermediate E. Esterification completed the catalytic cycle and released bicyclic adduct F. Spontaneous decarboxylation and oxidation of F generated the tetra-substituted benzene products.
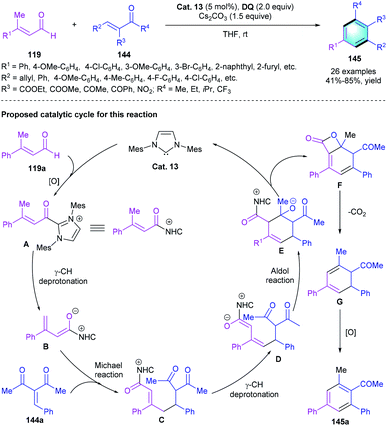 |
| | Scheme 42 NHC-catalyzed [3 + 3] benzannulation to synthesize polyfunctionalized benzenes. | |
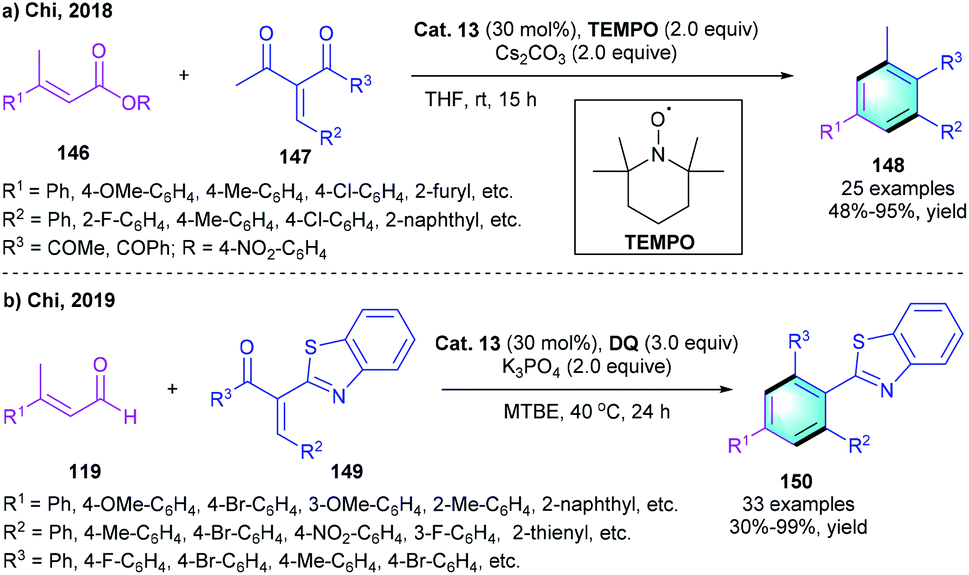
In recent years, esters were found to be applicable substrates in NHC catalysis.70 In 2018, the chi group reported a similar [3 + 3] benzannulation via the direct activation of the ester substrates (Scheme 43a).71 The reaction of α,β-unsaturated carboxylic ester 146 with enones 147 successfully afforded the tetra-substituted benzenes 148 with up to 95% yield under the catalysis of Cat. 13 using TEMPO as the oxidant. However, β-dialkyl ester substrates were not compatible with this process, which might be caused by the insufficient acidity of the C–H bond on the β-alkyl group. Compared with previous work using aldehyde as the substrates, this process lowers the economic cost because of the use of readily available ester substrates and inexpensive TEMPO oxidant. Later in 2019, Chi and co-workers presented another [3 + 3] benzannulation to provide substituted 2-phenylbenzothiazoles 150 in acceptable-to-perfect yields (Scheme 43b).72
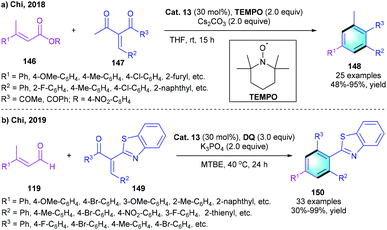 |
| | Scheme 43 NHC-catalyzed [3 + 3] benzannulation to synthesize polyfunctionalized benzenes and 2-phenylbenzothiazoles. | |
7.3 Intramolecular benzannulation
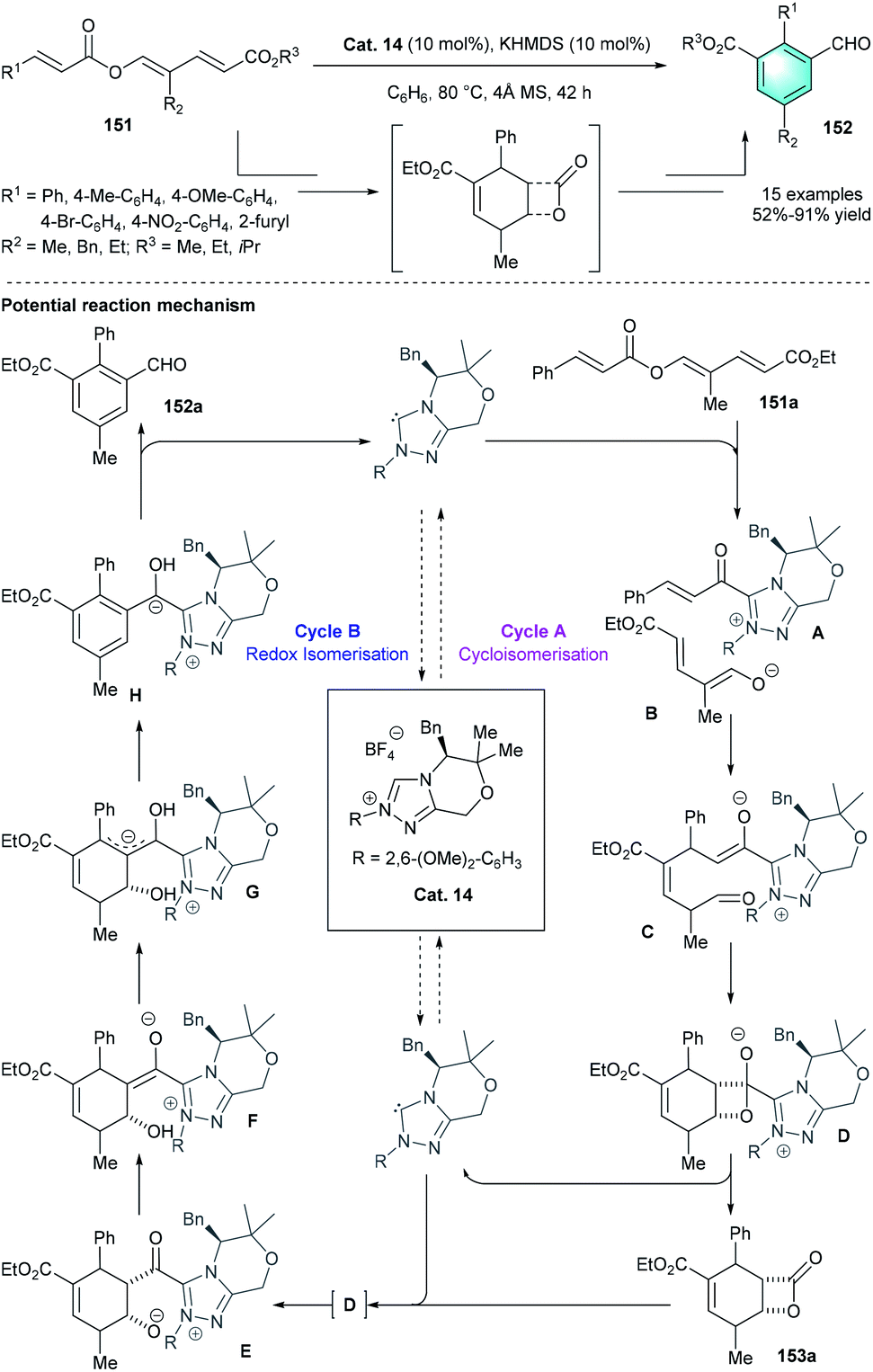
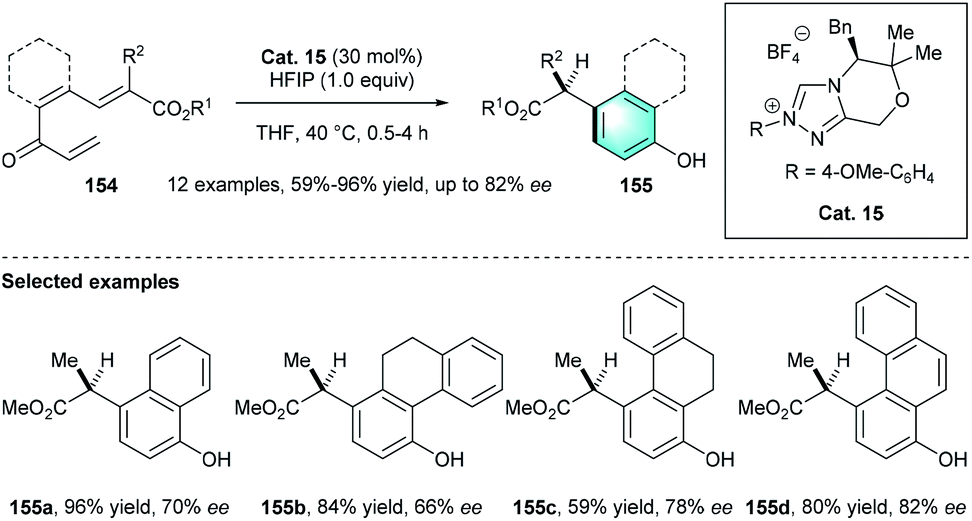
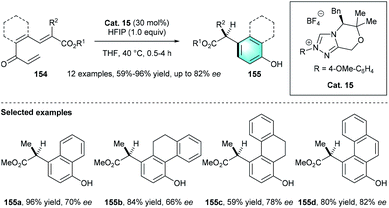
Unlike the above-mentioned conversions, in 2015, the group of Lupton uncovered an elegant NHC-catalyzed redox isomerization process for the synthesis of functionalized benzaldehydes from trienyl esters (Scheme 44).73 By using Cat.14 and KHMDS in benzene at 80 °C, the desired redox-isomerised aldehyde 152 was synthesized in moderate-to-excellent yields (52–91%). The author proposed a possible mechanism, including two linked catalytic cycles. First, acyl azolium A and dienolate B were originated from the fragmentation of 151a. Then a vinylogous Michael addition between A and B generated intermediate C. Following β-lactone formation and release of NHC catalyst would produce bicyclic adduct 153a. The addition of Cat. 14 to 153a resulted in acyl azolium intermediate E, which ultimately led to the generation of tri-substituted benzaldehyde 152. In 2016, by using a similar NHC pre-catalyst Cat. 15, the same group disclosed another intramolecular benzannulation process, realizing the first enantioselective synthesis of highly functionalized phenol product 155 via the umpolung of α, β-unsaturated ketones 154 (Scheme 45).74
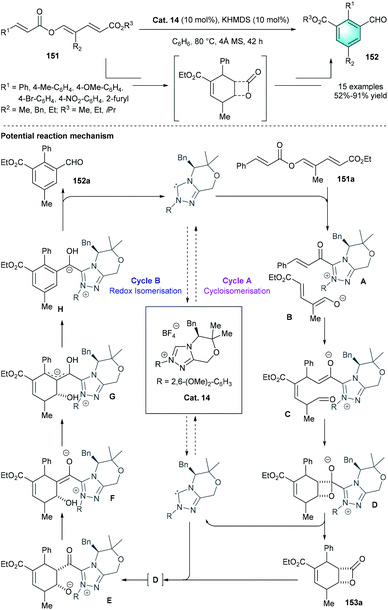 |
| | Scheme 44 NHC-catalyzed intramolecular benzannulation to synthesize tri-substituted benzaldehydes. | |
 |
| | Scheme 45 NHC-catalyzed intramolecular benzannulation to synthesize multi-functionalized phenols. | |
8 Summary and outlook
This review systemically summarized recent advances in the organocatalytic benzannulation strategies, including Brønsted acid-catalyzed benzannulation, secondary amine-catalyzed benzannulation, primary amine-catalyzed benzannulation, tertiary amine-catalyzed benzannulation, tertiary phosphine-catalyzed benzannulation, and NHC-catalyzed benzannulation. As presented in the review, recent ten years have witnessed the impressive progress on the organocatalytic benzannulation, which has become an important tool for the construction of structurally complex and densely functionalized arene systems. This review revealed that the organocatalytic benzannulation is a quite different strategy from the conventional benzannulation methods. The main advantage is that versatile catalysts and diversified activation modes make abundant cyclization reactions possible, thereby allowing the construction of polysubstituted arenes with flexible substitution patterns that other methods cannot provide. In different catalytic systems, various types of substrates can be used in the cyclization reaction. As classified in the review, according to the number of carbon atoms contributed by different synthons participating in the cyclization reaction, benzannulation mainly includes [4 + 2], [3 + 3], [3 + 2 + 1], [2 + 2 + 2], and intramolecular annulations. In addition, low economic cost, mild reaction conditions, good functional group compatibility, and excellent chemo-/regioselectivity enable this strategy to be applied in more scenarios, such as in the multistep synthesis of natural products, biologically active compounds, and functional molecules. Moreover, the intrinsic structural features of organocatalyst have enabled the efficient de novo construction of chiral arenes.
Based on these impressive advances, new challenges are likely to be addressed in future research of this field. For example, although several examples of asymmetric catalysis have been developed, the methodological development to construct a diverse range of aromatic ring systems containing different chiral elements including central chirality, axial chirality, and helical chirality is still highly desired. So far, aminocatalysis is the main strategy applied in asymmetric organocatalytic benzannulation to achieve the construction of chiral arenes. In future research, the application of chiral tertiary amine catalysis, chiral tertiary phosphine catalysis, chiral Brønsted acid catalysis, and chiral NHC catalysis in asymmetric benzannulation are worth exploring. In addition, the application of dual catalysis of organocatalysis with metal catalysis or photocatalysis in benzannulation may break through the limitations of conventional activation modes, and provide a useful solution for the construction of new functionalized aromatic ring systems. We hope that this review will promote future studies in this field and prompt the application of organocatalytic benzannulation strategy in the construction of densely functionalized arenes.
Conflicts of interest
The authors confirm that this article content has no conflicts of interest.
Acknowledgements
We are grateful for financial support from Ministry of Science and Technology of China (2019ZX09721001-008), National Natural Science Foundation of China (81773889, 82073998, 22001024 and 81630101), the “Thousand Talents Program” of Sichuan Province and Xinglin Scholar Research Premotion Project of CDUTCM.
Notes and references
-
(a) D. A. Horton, G. T. Bourne and M. L. Smythe, Chem. Rev., 2003, 103, 893 CrossRef CAS;
(b) A. Bouisseau, A. Roland, F. Reillon, R. Schneider and F. Cavelier, Org. Lett., 2013, 15, 3888 CrossRef CAS;
(c) M. Li, Y. W. Liu, R. H. Duan, X. F. Wei, Y. P. Yi, Y. Wang and C.-F. Chen, Angew. Chem., Int. Ed., 2017, 56, 8818 CrossRef CAS;
(d) C. L. Zhuang, W. Zhang, C. Q. Sheng, W. N. Zhang, C. G. Xing and Z. Y. Miao, Chem. Rev., 2017, 117, 7762–7810 CrossRef CAS;
(e) T. K. Allred, F. Manoni and P. G. Harran, Chem. Rev., 2017, 117, 11994 CrossRef CAS;
(f) E. K. Davison and J. Sperry, J. Nat. Prod., 2017, 80, 3060 CrossRef CAS;
(g) K. Kitamura, Y. Ando, T. Matsumoto and K. Suzuki, Chem. Rev., 2018, 118, 1495 CrossRef CAS;
(h) M. M. Unterlass, Hot Water Generates, Angew. Chem., Int. Ed., 2018, 57, 2292 CrossRef CAS.
-
(a) J. A. Bull, M. G. Hutchings and P. Quayle, Angew. Chem., Int. Ed., 2007, 46, 1869 CrossRef CAS;
(b) B. Crone, S. F. Kirsch and K. D. Umland, Angew. Chem., Int. Ed., 2010, 49, 4661 CrossRef CAS;
(c) V. Snieckus, Beilstein J. Org. Chem., 2011, 7, 1215 CrossRef CAS;
(d) Y. Izawa, D. Pun and S. S. Stahl, Science, 2011, 333, 209 CrossRef CAS.
- For selected reviews, see:
(a) T. B. Poulsen and K. A. Jørgensen, Chem. Rev., 2008, 108, 2903 CrossRef CAS;
(b) W. A. L. van Otterlo and C. B. de Koning, Chem. Rev., 2009, 109, 3743 CrossRef CAS;
(c) S. L. You, Q. Cai and M. Zeng, Chem. Soc. Rev., 2009, 38, 2190 RSC;
(d) T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147 CrossRef CAS;
(e) J. Wencel-Delord, T. Dröge, F. Liu and F. Glorius, Chem. Soc. Rev., 2011, 40, 4740 RSC;
(f) K. M. Engle, T.-S. Mei, M. Wasa and J.-Q. Yu, Acc. Chem. Res., 2012, 45, 788 CrossRef CAS.
- For selected examples of catalytic dehydrogenation of carbocyclic rings, see:
(a) T. Diao and S. S. Stahl, J. Am. Chem. Soc., 2011, 133, 14566 CrossRef CAS;
(b) A. V. Iosub and S. S. Stahl, J. Am. Chem. Soc., 2015, 137, 3454 CrossRef CAS;
(c) A. V. Iosub and S. S. Stahl, ACS Catal., 2016, 6, 8201 CrossRef CAS;
(d) Y. F. Liang, X. Li, X. Wang, M. Zou, C. Tang, Y. Liang, S. Song and N. Jiao, J. Am. Chem. Soc., 2016, 138, 12271 CrossRef CAS.
- For selected examples of electrophilic and nucleophilic aromatic substitution reactions, see:
(a) G. Illuminati and H. Gilman, J. Am. Chem. Soc., 1952, 74, 2896 CrossRef CAS;
(b) T. Nakagome, A. Kobayash and A. Misaki, Chem. Pharm. Bull., 1966, 14, 1074 CrossRef CAS;
(c) W. J. Coates and A. McKillop, J. Org. Chem., 1990, 55, 5418 CrossRef CAS;
(d) J. D. Kirkham, R. J. Butlin and J. P. Harrity, Angew. Chem., Int. Ed., 2012, 51, 6402 CrossRef CAS;
(e) C. Liu, M. Zhang, Z. Zhang, S. B. Zhang, S. Yang, A. Zhang, L. Yin, S. Swarts, S. Vidyasagar, L. Zhang and P. Okunieff, Bioorg. Med. Chem., 2016, 24, 4263 CrossRef CAS;
(f) S. Sivanathan, F. Körber and J. Scherkenbeck, Bioorg. Med. Chem., 2016, 24, 873 CrossRef CAS.
- For selected examples of transition-metal-catalyzed cross-coupling reactions, see:
(a) S. M. Bonesi, M. Fagnoni and A. Albini, Angew. Chem., Int. Ed., 2008, 47, 10022 CrossRef CAS;
(b) D.-G. Yu, M. Yu, B.-T. Guan, B.-J. Li, Y. Zheng, Z.-H. Wu and Z.-J. Shi, Org. Lett., 2009, 11, 3374 CrossRef CAS;
(c) J. L. Gustafson, D. Lim, K. T. Barrett and S. J. Miller, Angew. Chem., Int. Ed., 2011, 50, 5125 CrossRef CAS;
(d) A. Rahimi, J. C. Namyslo, M. H. H. Drafz, J. Halm, E. Huebner, M. Nieger, N. Rautzenberg and A. Schmidt, J. Org. Chem., 2011, 76, 7316 CrossRef CAS;
(e) C. L. Ciana, R. J. Phipps, J. R. Brandt, F. M. Meyer and M. J. Gaunt, Angew. Chem., Int. Ed., 2011, 50, 458 CrossRef CAS;
(f) F. Zhao, Y. F. Zhang, J. Wen, D. G. Yu, J. B. Wei, Z. Xi and Z. J. Shi, Org. Lett., 2013, 15, 3230 CrossRef CAS;
(g) M. Xie, S. Wang, J. Wang, K. Fang, C. Liu, C. Zha and J. Jia, J. Org. Chem., 2016, 81, 3329 CrossRef CAS.
- For selected examples of direct C–H functionalization of arenes, see:
(a) Y. W. Zheng, B. Chen, P. Ye, K. Feng, W. Wang, Q. Y. Meng, L. Z. Wu and C. H. Tung, J. Am. Chem. Soc., 2016, 138, 10080 CrossRef CAS;
(b) S. Bag, R. Jayarajan, U. Dutta, R. Chowdhury, R. Mondal and D. Maiti, Angew. Chem., Int. Ed., 2017, 56, 12538 CrossRef CAS;
(c) B. Ma, Z. Chu, B. Huang, Z. Liu, L. Liu and J. Zhang, Angew. Chem., Int. Ed., 2017, 56, 2749 CrossRef CAS;
(d) H. W. Wang, Y. Lu, B. Zhang, J. He, H. J. Xu, Y. S. Kang, W. Y. Sun and J. Q. Yu, Angew. Chem., Int. Ed., 2017, 56, 7449 CrossRef CAS;
(e) P. Wang, P. Verma, G. Xia, J. Shi, J. X. Qiao, S. Tao, P. T. W. Cheng, M. A. Poss, M. E. Farmer, K. S. Yeung and J. Q. Yu, Nature, 2017, 551, 489 CrossRef CAS;
(f) M. Z. Lu, X. R. Chen, H. Xu, H. X. Dai and J. Q. Yu, Chem. Sci., 2018, 9, 1311 RSC.
- For selected reviews of benzannulation reactions in the recent years,
(a) P. Wessig and G. Müller, Chem. Rev., 2008, 108, 2051 CrossRef CAS;
(b) S. Kotha, S. Misra and S. Halder, Tetrahedron, 2008, 64, 10775 CrossRef CAS;
(c) G. Dominguez and J. Perez-Castells, Chem. Soc. Rev., 2011, 40, 3430 RSC;
(d) K. Tanaka and Y. Shibata, Synthesis, 2012, 44, 323 CrossRef;
(e) R. Fernandes and S. Mulay, Synthesis, 2014, 46, 1836 CrossRef;
(f) C. Holden and M. F. Greaney, Angew. Chem., Int. Ed., 2014, 53, 5746 CrossRef CAS;
(g) A. L. Siva Kumari, A. Siva Reddy and K. C. K. Swamy, Org. Biomol. Chem., 2016, 14, 6651 RSC;
(h) P. Loxq, E. Manoury, R. Poli, E. Deydier and A. Labande, Coord. Chem. Rev., 2016, 308, 131 CrossRef CAS;
(i) S. J. Hein, D. Lehnherr, H. Arslan, F. J. Uribe-Romo and W. R. Dichtel, Acc. Chem. Res., 2017, 50, 2776 CrossRef CAS;
(j) W. Song, S. A. Blaszczyk, J. Liu, S. Wang and W. Tang, Org. Biomol. Chem., 2017, 15, 7490 RSC;
(k) L. Chen, Z. Liu and S. Zhu, Org. Biomol. Chem., 2018, 16, 8884 RSC;
(l) A. Link and C. Sparr, Chem. Soc. Rev., 2018, 47, 3804 RSC;
(m) T. N. Poudel, R. J. I. Tamargo, H. Cai and Y. R. Lee, Asian J. Org. Chem., 2018, 7, 985 CrossRef CAS;
(n) A. D. Senese and W. A. Chalifoux, Molecules, 2018, 24, 118 CrossRef;
(o) X. Zhang and S. Ma, Isr. J. Chem., 2018, 58, 608 CrossRef CAS;
(p) T. Bera, K. Pandey and R. Ali, ChemistrySelect, 2020, 5, 5239 CrossRef CAS;
(q) Q. Jia, Y. Li and Y. Lin, Chin. J. Org. Chem., 2020, 40, 1502 CrossRef;
(r) K. M. Magiera, V. Aryal and W. A. Chalifoux, Org. Biomol. Chem., 2020, 18, 2372 RSC;
(s) T. Mies, T.-K. Ma and A. G. M. Barrett, Russ. Chem. Rev., 2020, 89, 917 CrossRef.
-
(a) T. P. Yoon and E. N. Jacobsen, Privileged Chiral Catalysts, Science, 2003, 299, 1691 CrossRef CAS;
(b) M. C. Kozlowski, B. J. Morgan and E. C. Linton, Chem. Soc. Rev., 2009, 38, 3193 RSC;
(c) G. Bringmann, T. Gulder, T. A. M. Gulder and M. Breuning, Chem. Rev., 2011, 111, 563 CrossRef CAS.
- For selected reviews see:
(a) G. P. McGlacken and L. M. Bateman, Chem. Soc. Rev., 2009, 38, 2447 RSC;
(b) C. M. So and F. Y. Kwong, Chem. Soc. Rev., 2011, 40, 4963 RSC;
(c) N. Kuhl, M. N. Hopkinson, J. Wencel-Delord and F. Glorius, Angew. Chem., Int. Ed., 2012, 51, 10236 CrossRef CAS;
(d) L. T. Ball, G. C. Lloyd-Jones and C. A. Russell, Science, 2012, 337, 1644 CrossRef CAS;
(e) P. Loxq, E. Manoury, R. Poli, E. Deydier and A. Labande, Coord. Chem. Rev., 2016, 308, 131 CrossRef CAS;
(f) P. Y. Choy, S. M. Wong, A. Kapdi and F. Y. Kwong, Org. Chem. Front., 2018, 5, 288 RSC;
(g) F.-X. Felpin and S. Sengupta, Chem. Soc. Rev., 2019, 48, 1150 RSC;
(h) Y. F. Zhang and Z. J. Shi, Acc. Chem. Res., 2019, 52, 161 CrossRef CAS.
- Very recently, after we finished the preparation of the manuscript, we found that a similar review on this topic occurred, which did not include Brønsted acid-catalyzed benzannulation. See: L. Wu, B. Yu and E.-Q. Li, Adv. Synth. Catal., 2020, 362, 4010 CrossRef CAS.
-
(a) W. R. Chao, D. Yean, K. Amin, C. Green and L. Jong, J. Med. Chem., 2007, 50, 3412 CrossRef CAS;
(b) T. Janosik, N. Wahlstrom and J. Bergman, Tetrahedron, 2008, 64, 9159 CrossRef CAS;
(c) J. Roy, A. K. Jana and D. Mal, Tetrahedron, 2012, 68, 6099 CrossRef CAS;
(d) A. W. Schmidt, K. R. Reddy and H. J. Knölker, Chem. Rev., 2012, 112, 3193 CrossRef CAS;
(e) T. Janosik, A. Rannug, U. Rannug, N. Wahlstrom, J. Slätt and J. Bergman, Chem. Rev., 2018, 118, 9058 CrossRef CAS;
(f) J. N. Wang, H. J. Zhang, J. Q. Li, W. J. Ding and Z. J. Ma, J. Nat. Prod., 2018, 81, 949 CrossRef CAS.
- X. Zheng, L. Lv, S. Lu, W. Wang and Z. Li, Org. Lett., 2014, 16, 5156 CrossRef CAS.
- C. Liu, W. Huang, M. Wang, B. Pan and Y. Gu, Adv. Synth. Catal., 2016, 358, 2260 CrossRef CAS.
- S. Saha, A. Banerjee and M. S. Maji, Org. Lett., 2018, 20, 6920 CrossRef CAS.
- A. Banerjee, A. Guin, S. Saha, A. Mondal and M. S. Maji, Org. Biomol. Chem., 2019, 17, 1822 RSC.
- J. Xue, E. Gao, X. N. Wang and J. Chang, Org. Lett., 2018, 20, 6055 CrossRef CAS.
- A. R. Galeev, M. V. Dmitriev, I. G. Mokrushin, I. V. Mashevskaya, A. N. Maslivets and M. Rubin, Org. Biomol. Chem., 2019, 17, 10030 RSC.
- X. Zhang, Z. Wang, K. Xu, Y. Feng, W. Zhao, X. Xu, Y. Yan and W. Yi, Green Chem., 2016, 18, 2313 RSC.
- T. Fujita, I. Takahashi, M. Hayashi, J. Wang, K. Fuchibe and J. Ichikawa, Eur. J. Org. Chem., 2017, 2017, 262 CrossRef CAS.
- X. Li, H. Yu and Y. Huang, Adv. Synth. Catal., 2017, 359, 1379 CrossRef CAS.
- J. Y. Wang, P. Zhou, G. Li, W. J. Hao, S. J. Tu and B. Jiang, Org. Lett., 2017, 19, 6682 CrossRef CAS.
- H.-K. Sha, F. Liu, J. Lu, Z.-Q. Liu, W.-J. Hao, J.-L. Tang, S.-J. Tu and B. Jiang, Green Chem., 2018, 20, 3476 RSC.
- P. Zhou, J. Y. Wang, T. S. Zhang, G. Li, W. J. Hao, S. J. Tu and B. Jiang, Chem. Commun., 2018, 54, 164 RSC.
-
(a) S. P. Brown, N. C. Goodwin, W. C. David and D. W. C. MacMillan, J. Am. Chem. Soc., 2003, 125, 1192 CrossRef CAS;
(b) J. W. Yang, M. T. H. Fonseca and B. List, Angew. Chem., Int. Ed., 2004, 43, 6660 CrossRef;
(c) M. T. H. Fonseca and B. List, Angew. Chem., Int. Ed., 2004, 43, 3958 CrossRef;
(d) A. J. Cobb, D. M. Shaw, D. A. Longbottom, J. B. Gold and S. V. Ley, Org. Biomol. Chem., 2005, 3, 84 RSC;
(e) S. Brandau, A. Landa, J. Franzén, M. Marigo and K. A. Jørgensen, Angew. Chem., Int. Ed., 2006, 45, 4305 CrossRef CAS;
(f) K. R. Knudsen, C. E. T. Mitchell and S. V. Ley, Chem. Commun., 2006, 1, 66 RSC.
-
(a) V. K. Aggarwal, C. Lopin and F. Sandrinelli, J. Am. Chem. Soc., 2003, 125, 7596 CrossRef CAS;
(b) W. Zhuang and A. Jørgensen, Angew. Chem., Int. Ed., 2004, 43, 4476 CrossRef;
(c) A. Armstrong, Angew. Chem., Int. Ed., 2004, 43, 1460 CrossRef CAS;
(d) D. B. Ramachary and C. F. Barbas, J. Org. Chem., 2004, 69, 5838 CrossRef CAS;
(e) N. S. Chowdari and C. F. Barbas, Org. Lett., 2005, 7, 807 CrossRef;
(f) A. McNally, B. Evans and M. J. Gaunt, Angew. Chem., Int. Ed., 2006, 45, 2116 CrossRef CAS.
- S. G. Li, X. Q. Hu, Z. X. Jia and P. F. Xu, Tetrahedron, 2010, 66, 8557 CrossRef CAS.
- K. B. Somai Magar, L. Xia and Y. R. Lee, Chem. Commun., 2015, 51, 8592 RSC.
- Y. Hayashi, A. Takikawa, S. Koshino and K. Ishida, Chem.–Eur. J., 2019, 25, 10319 CrossRef CAS.
- S. Koshino, A. Takikawa, K. Ishida, T. Taniguchi, K. Monde, E. Kwon, S. Umemiya and Y. Hayashi, Chem.–Eur. J., 2020, 26, 4524 CrossRef CAS.
- X. Song, X. Zhang, S. Zhang, H. Li and W. Wang, Chem.–Eur. J., 2012, 18, 9770 CrossRef CAS.
- L. Jiang, H. Li, J.-F. Zhou, M.-W. Yuan, H.-L. Li, Y.-M. Chuan and M.-L. Yuan, Synth. Commun., 2018, 48, 336 CrossRef CAS.
-
(a) F. M. D. Ismail, J. Fluorine Chem., 2002, 118, 27 CrossRef CAS;
(b) D. O'Hagan, Chem. Soc. Rev., 2008, 37, 308 RSC;
(c) S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320 RSC;
(d) T. Furuya, A. S. Kamlet and T. Ritter, Nature, 2011, 473, 470 CrossRef CAS;
(e) T. Liang, C. N. Neumann and T. Ritter, Angew. Chem., Int. Ed., 2013, 52, 8214 CrossRef CAS;
(f) J. Wang, M. Sánchez-Roselló, J. Luis Aceña, C. del Pozo, A. E. Sorochinsky, S. Fustero, V. A. Soloshonok and H. Liu, Chem. Rev., 2014, 114, 2432 CrossRef CAS;
(g) C. Ni, M. Hu and J. Hu, Chem. Rev., 2015, 115, 765 CrossRef CAS;
(h) X. Yang, T. Wu, R. J. Phipps and F. D. Toste, Chem. Rev., 2015, 115, 826 CrossRef CAS;
(i) T. Ahrens, J. Kohlmann, M. Ahrens and T. Braun, Chem. Rev., 2015, 115, 931 CrossRef CAS;
(j) C. Ni and J. Hu, Chem. Soc. Rev., 2016, 45, 5441 RSC;
(k) Q. Zhao, C. Peng, H. Huang, S.-J. Liu, Y.-J. Zhong, W. Huang, G. He and B. Han, Chem. Commun., 2018, 54, 8359 RSC;
(l) X. H. He, Y. L. Ji, C. Peng and B. Han, Adv. Synth. Catal., 2019, 361, 1923 CrossRef CAS;
(m) X. Zhang, X. Li, J.-L. Li, Q.-W. Wang, W.-L. Zou, Y.-Q. Liu, Z.-Q. Jia, F. Peng and B. Han, Chem. Sci., 2020, 11, 2888 RSC;
(n) J.-L. Li, Y. Q. Liu, W. L. Zou, R. Zeng, X. Zhang, Y. Liu, B. Han, Y. He, H. J. Leng and Q. Z. Li, Angew. Chem., Int. Ed., 2020, 59, 1863 CrossRef CAS.
- F. Peng, Q. Zhao, W. Huang, S.-J. Liu, Y.-J. Zhong, Q. Mao, N. Zhang, G. He and B. Han, Green Chem., 2019, 21, 6179 RSC.
- X. H. He, L. Yang, Y. L. Ji, Q. Zhao, M. C. Yang, W. Huang, C. Peng and B. Han, Chem.–Eur. J., 2018, 24, 1947 CrossRef CAS.
- D. B. Ramachary, K. Ramakumar and V. V. Narayana, J. Org. Chem., 2007, 72, 1458 CrossRef CAS.
- A. Link and C. Sparr, Angew. Chem., Int. Ed., 2014, 53, 5458 CrossRef CAS.
- V. C. Faseke and C. Sparr, Angew. Chem., Int. Ed., 2016, 55, 7261 CrossRef.
- L. Zhang, N. Fu and S. Luo, Acc. Chem. Res., 2015, 48, 986 CrossRef CAS.
-
(a) Y.-C. Chen, Synlett, 2008, 1919 CrossRef CAS;
(b) F. Peng and Z. Shao, J. Mol. Catal. A: Chem., 2008, 285, 1 CrossRef CAS;
(c) L.-W. Xu, J. Luo and Y. Lu, Chem. Commun., 2009, 1807 RSC;
(d) P. Melchiorre, Angew. Chem., Int. Ed., 2012, 51, 9748 CrossRef CAS.
- D. Lotter, M. Neuburger, M. Rickhaus, D. Haussinger and C. Sparr, Angew. Chem., Int. Ed., 2016, 55, 2920–2923 CrossRef CAS.
- P. K. Jaiswal, S. Biswas, S. Singh and S. Samanta, Org. Biomol. Chem., 2013, 11, 8410 RSC.
- S. Biswas, A. Dagar, A. Srivastava and S. Samanta, Eur. J. Org. Chem., 2015, 2015, 4493 CrossRef CAS.
- M. Chang, C. Wu, J. Zheng and Y. Huang, Chem.–Asian J., 2016, 11, 1512 CrossRef CAS.
- Y.-L. Ji, X.-H. He, G. Li, Y.-Y. Ai, H.-P. Li, C. Peng and B. Han, Org. Chem. Front., 2020, 7, 563 RSC.
- Y. S. Xie, R. F. Huang, R. Li, C. B. Zhang, J. Y. Fu, L. L. Zhao and J. F. Yuan, Chem. Commun., 2020, 56, 1948 RSC.
- S. Xue, Q.-F. Zhou, F. Yang and Q.-X. Guo, Synlett, 2007, 2073 CrossRef.
- S. Xue, B. Hu, L.-G. Meng, Y.-L. Liu and M. Liang, Synlett, 2009, 24, 4137 Search PubMed.
- M. Terada and K. Dan, Chem. Commun., 2012, 48, 5781 RSC.
- S. Jia, S. Li, Y. Liu, W. Qin and H. Yan, Angew. Chem., Int. Ed., 2019, 58, 18496 CrossRef CAS.
- J. Zheng, Y. Huang and Z. M. Li, Org. Lett., 2013, 15, 5064 CrossRef CAS.
- X. Chen, D. Gao, D. Wang, T. Xu, W. Liu, P. Tian and X. Tong, Angew. Chem., Int. Ed., 2019, 58, 15334 CrossRef CAS.
- S. K. Arupula, A. A. Qureshi and K. C. K. Swamy, J. Org. Chem., 2020, 85, 4130 CrossRef CAS.
-
(a) A. Igau, H. Grutzmacher, A. Baceiredo and G. Bertrand, J. Am. Chem. Soc., 1988, 110, 6463 CrossRef CAS;
(b) A. Igau, A. Baceiredo, G. Trinquier and G. Bertrand, Angew. Chem., Int. Ed., 1989, 28, 621 CrossRef;
(c) A. J. Arduengo, R. L. Harlow and M. Kline, A Stable Crystalline Carbene, J. Am. Chem. Soc., 1991, 113, 361 CrossRef CAS.
- T. Dröge and F. Glorius, Angew. Chem., Int. Ed., 2010, 49, 6940 CrossRef.
-
(a) D. Enders, O. Niemeier and A. Henseler, Chem. Rev., 2007, 107, 5606 CrossRef CAS;
(b) A. Grossmann and D. Enders, Angew. Chem., Int. Ed., 2012, 51, 314 CrossRef CAS;
(c) X. Bugaut and F. Glorius, Chem. Soc. Rev., 2012, 41, 3511 RSC;
(d) S. J. Ryan, L. Candish and D. W. Lupton, Chem. Soc. Rev., 2013, 42, 4906 RSC;
(e) K. P. Jang, G. E. Hutson, R. C. Johnston, E. O. McCusker, P. H. Y. Cheong and K. A. Scheidt, J. Am. Chem. Soc., 2014, 136, 76 CrossRef CAS;
(f) M. N. Hopkinson, C. Richter, M. Schedler and F. Glorius, Nature, 2014, 510, 485 CrossRef CAS;
(g) Q. Zhao, B. Han, B. Wang, H.-J. Leng, C. Peng and W. Huang, RSC Adv., 2015, 5, 26972 RSC;
(h) X. Y. Chen, Q. Liu, P. Chauhan and D. Enders, Angew. Chem., Int. Ed., 2018, 57, 3862 CrossRef CAS.
- S. J. Ryan, L. Candish and D. W. Lupton, J. Am. Chem. Soc., 2011, 133, 4694 CrossRef CAS.
- X. Huang, T. Zhu, Z. Huang, Y. Zhang, Z. Jin, G. Zanoni and Y. R. Chi, Org. Lett., 2017, 19, 6188 CrossRef CAS.
- K. Q. Chen, Z. Luo, Z. H. Gao and S. Ye, Chem.–Eur. J., 2019, 25, 3253 CAS.
- T. Zhu, Y. Liu, M. Smetankova, S. Zhuo, C. Mou, H. Chai, Z. Jin and Y. R. Chi, Angew. Chem., Int. Ed., 2019, 58, 15778 CrossRef CAS.
- J. M. Hu, J. Q. Zhang, B. B. Sun, J. B. Chen, J. Q. Yu, X. P. Yang, H. P. Lv, Z. Wang and X. W. Wang, Org. Lett., 2019, 21, 8582 CrossRef CAS.
- Q. Jia and J. Wang, Org. Lett., 2016, 18, 2212 CrossRef CAS.
- C.-L. Zhang, Z.-H. Gao, Z.-Q. Liang and S. Ye, Adv. Synth. Catal., 2016, 358, 2862 CrossRef CAS.
- C. L. Zhang and S. Ye, Org. Lett., 2016, 18, 6408 CrossRef CAS.
- D. Liu, Y. Gao, J. Huang, Z. Fu and W. Huang, J. Org. Chem., 2018, 83, 14210 CrossRef CAS.
- T. Zhu, C. Mou, B. Li, M. Smetankova, B. A. Song and Y. R. Chi, J. Am. Chem. Soc., 2015, 137, 5658 CrossRef CAS.
- K. Xu, W. Li, S. Zhu and T. Zhu, Angew. Chem., Int. Ed., 2019, 58, 17625 CrossRef CAS.
- J. Liu, D. K. Das, G. Zhang, S. Yang, H. Zhang and X. Fang, Org. Lett., 2018, 20, 64 CrossRef CAS.
- T. Zhu, P. Zheng, C. Mou, S. Yang, B. A. Song and Y. R. Chi, Nat. Commun., 2014, 5, 5027 CrossRef CAS.
- P. Chauhan and D. Enders, Angew. Chem., Int. Ed., 2014, 53, 1485 CrossRef CAS.
- J. Wu, C. Mou and Y. R. Chi, Chin. J. Chem., 2018, 36, 333 CrossRef CAS.
- Z. Ni, C. Mou, X. Zhu, P. Qi, S. Yang, Y. R. Chi and Z. Jin, Eur. J. Org. Chem., 2020, 2020, 492 CrossRef CAS.
- L. Candish, A. Levens and D. W. Lupton, Chem. Sci., 2015, 6, 2366 RSC.
- Y. Nakano and D. W. Lupton, Angew. Chem., Int. Ed., 2016, 55, 3135 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ab,
Gu Zhan*b and
Bo Han
ab,
Gu Zhan*b and
Bo Han