
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOne-pot synthesis of indoles and quinolinones from ortho-tosylaminophenyl-substituted para-quinone methides†
Junwei Wang‡
 *,
Xiang Pan‡,
Quanjin Rong,
Lei Zhao,
Lin Zhao,
Weichen Dai,
Kun Zhao* and
Lihong Hu*
*,
Xiang Pan‡,
Quanjin Rong,
Lei Zhao,
Lin Zhao,
Weichen Dai,
Kun Zhao* and
Lihong Hu*
Jiangsu Key Laboratory for Functional Substances of Chinese Medicine, School of Pharmacy, Nanjing University of Chinese Medicine, Nanjing, P. R. China. E-mail: jwwang@njucm.edu.cn; kunzhao13@gmail.com; lhhu@njucm.edu.cn
First published on 10th September 2020
Abstract
A facile one-pot synthesis has been developed through alkylation/acylation of ortho-tosylaminophenyl-substituted para-quinone methides followed by an intramolecular 1,6-conjugate addition and oxidation sequence. This cascade reaction occurs readily in good yield (up to 95%), providing a divergent synthetic approach to structurally diverse 2,3-disubstituted indoles and 3,4-diaryl-substituted quinolinones.
Nitrogen-containing heterocycles as privileged structural motifs are widely found in natural products and small molecule pharmaceuticals.1 In particular, indoles and quinolinones are of great significance in drug discovery because of their diverse biological activities.2,3 For example, indometacin (I), a synthetic indole acetic acid derivative, is an effective non-steroidal anti-inflammatory drug.2a Fluvastatin sodium (II) and compound III, two 3-aryl-substituted indole derivatives, have been designed as a HMG-CoA reductase inhibitor2b and carbonic anhydrase inhibitor,2c respectively. 4-Phenyl-substituted quinolinone derivatives such as IV, V and VI have been demonstrated to possess excellent antitumor activity (Fig. 1).3b–d Thus, it is highly desirable to develop more efficient and facile methods to construct these nitrogen-containing heterocycles.
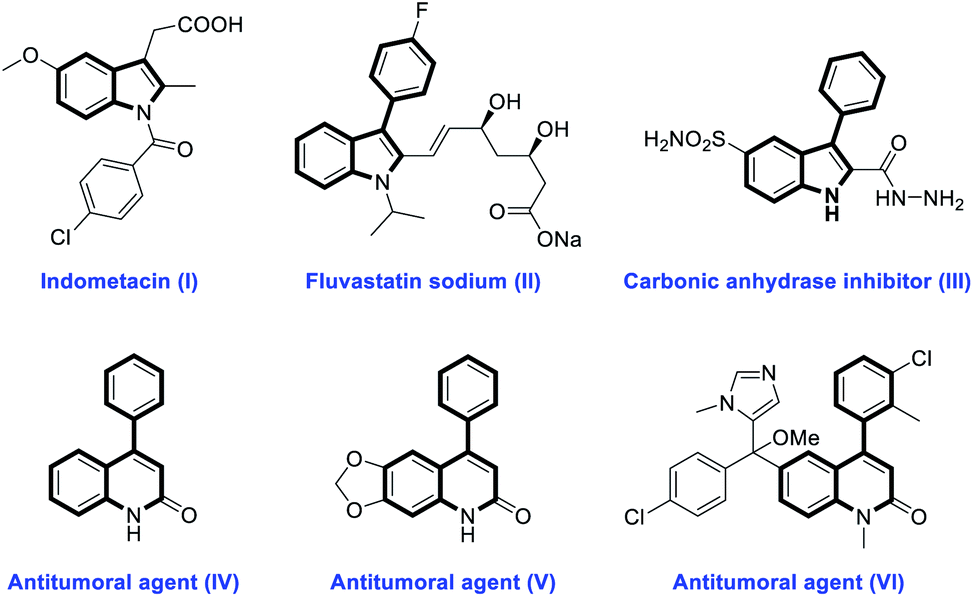 | ||
| Fig. 1 Selected natural products and synthetic compounds containing indole and quinolinone frameworks. | ||
Recently, para-quinone methides (p-QMs) have emerged as versatile building blocks due to their intrinsic reactivities.4 A large number of transformations based on p-QMs have been achieved since the seminal reports by Fan5 and Jørgensen.6 For example, annulation reactions based on simple p-QMs and vinyl p-QMs have been reported by Yao, Fan, Zhao, and Waser groups.7 In 2016, Enders and co-workers pioneered the design and application of ortho-hydroxyphenyl-substituted p-QMs in [4 + 2] cyclizations.8 After that the use of ortho-hydroxyphenyl-substituted p-QMs in [4 + 1], [4 + 2] and [4 + 3] cyclizations reactions was extensively investigated,9–12 allowing the synthesis of diverse oxygenous heterocyclic motifs (Scheme 1a). Quite recently our group designed in situ generated ortho-tosylaminophenyl-substituted p-QMs and successfully applied this class of substrates in [4 + 2] and [4 + 1] annulation reactions to synthesize tetrahydroquinolines and 2,3-dihydroindoles, respectively (Scheme 1b).13
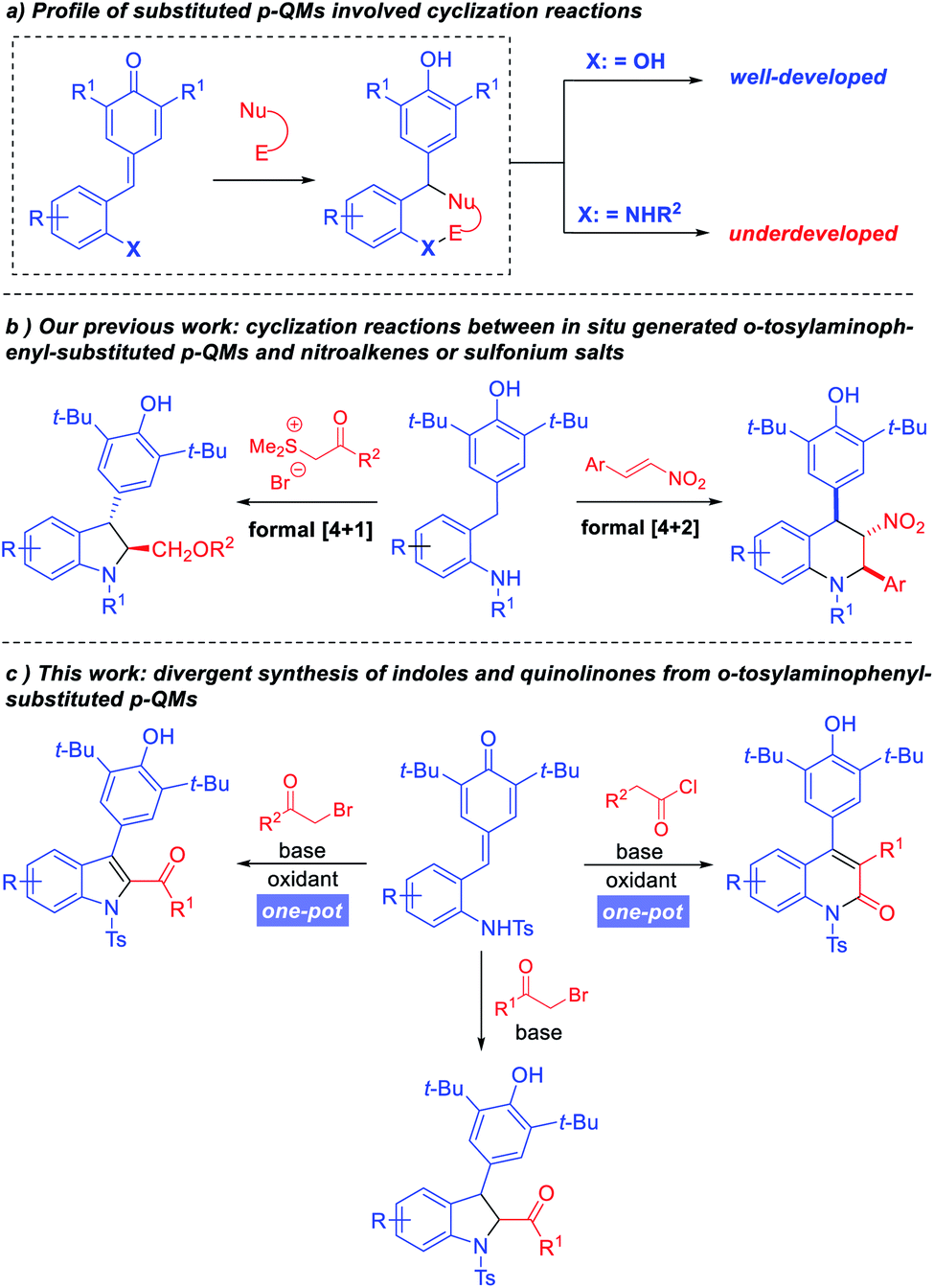 | ||
| Scheme 1 Reported reactions based on p-QMs and our design. | ||
Although great progress has been made in the synthesis of oxygen-containing heterocyclic frameworks, the application of p-QMs in the construction of nitrogen-containing heterocyclic frameworks remains underdeveloped. Cyclizations using ortho-tosylaminophenyl-substituted p-QMs as building blocks are still rather limited. Whereas, the privileged status of indoles and quinolinones in organic synthesis and biological applications demands more efficient strategies for their preparation. In 2019, Anand and co-workers reported a one-pot synthesis of oxygen-based heterocycles from 2-hydroxyphenyl-substituted p-QMs, which provided an efficient method for the construction of 2,3-disubstituted benzo[b]furans, 2,3-dihydrobenzofurans and diaryl-substituted coumarin derivatives.10 Inspired by this work, we wondered whether this strategy could be extended to ortho-tosylaminophenyl-substituted p-QMs, so that we might establish a powerful divergent cascade reaction to synthesize 2,3-dihydroindoles, indoles and quinolinone derivatives.
We hypothesized that assembly of 2,3-dihydroindoles could be realized through the union of ortho-tosylaminophenyl-substituted p-QMs and α-halo ketones via N-alkylation followed by intramolecular cyclization. Then, a suitable oxidant can promote the in situ generation of indoles. Although we have reported the synthesis of 2,3-dihydroindoles through a formal [4 + 1] annulation of ortho-tosylaminophenyl-substituted p-QMs with sulfur ylides,13b the direct one-pot synthesis of 2,3-disubstituted indoles and 3,4-diaryl-substituted quinolinone derivatives from ortho-tosylaminophenyl-substituted p-QMs through N-alkylation/acylation followed by intramolecular 1,6-conjugate addition and oxidation strategy has not been reported yet (Scheme 1c).
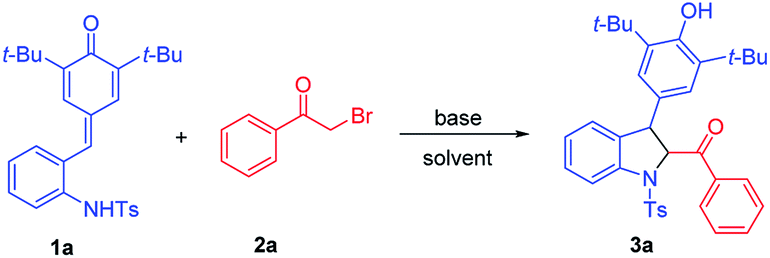
To verify our hypothesis, we initially tried to identify the optimal conditions for the synthesis of 2,3-dihydroindoles before exploring the one-pot synthesis of indoles. ortho-Tosylaminophenyl-substituted p-QMs 1a and 2-bromoacetophenone 2a were chosen as the model substrates to optimize the reaction conditions (Table 1). To our delight, in the presence of Et3N (1.5 equiv.) in CH3CN at 20 °C, the expected reaction occurred and the desired product 3a could be obtained, albeit with moderate yield (Table 1, entry 1). Encouraged by this promising result, different bases were evaluated, and we found that the inorganic base Cs2CO3 performed best, offering 3a in 82% yield (Table 1, entry 6). Striving for higher efficiency of this reaction, the effect of solvent was next investigated and we found that other solvents (e.g. CH2Cl2, CHCl3, acetone, toluene and DCE) failed to improve the yield (entries 7–11). After establishing the optimal base and solvent, the dosage of the base was then evaluated and it was found that increasing or decreasing the amount of base is not beneficial for the reaction (entries 12 and 13). We further examined the reaction time and the reaction temperature (entries 14–16). Notably, when the temperature reached 50 °C, the expected product 3a could be afforded in a good yield of 92%. Finally, the optimal reaction conditions were selected as those shown in entry 16 (1.5 equiv. of Cs2CO3 as base, CH3CN as solvent and reaction at 50 °C for 1.5 h).
|
||||
|---|---|---|---|---|
| Entry | Base | T (°C) | Solvent | Yieldb (%) |
a All reactions were conducted with 1a (0.11 mmol), 2a (0.10 mmol), base (1.5 equiv.), solvent (1.5 mL), 1.5 h.b Determined by 1H NMR using 1,3,5-trimethoxybenzene as an internal standard; dr > 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.c 1.0 equiv. of base was used.d 2.0 equiv. of base was used.e t = 3 h. :1.c 1.0 equiv. of base was used.d 2.0 equiv. of base was used.e t = 3 h. |
||||
| 1 | Et3N | 20 | CH3CN | 54 |
| 2 | DBU | 20 | CH3CN | 38 |
| 3 | iPr2NH | 20 | CH3CN | 7 |
| 4 | Na2CO3 | 20 | CH3CN | 4 |
| 5 | K2CO3 | 20 | CH3CN | 5 |
| 6 | Cs2CO3 | 20 | CH3CN | 82 |
| 7 | Cs2CO3 | 20 | CH2Cl2 | 60 |
| 8 | Cs2CO3 | 20 | CHCl3 | 57 |
| 9 | Cs2CO3 | 20 | Acetone | 80 |
| 10 | Cs2CO3 | 20 | Toluene | 53 |
| 11 | Cs2CO3 | 20 | DCE | 50 |
| 12c | Cs2CO3 | 20 | CH3CN | 80 |
| 13d | Cs2CO3 | 20 | CH3CN | 76 |
| 14e | Cs2CO3 | 20 | CH3CN | 83 |
| 15 | Cs2CO3 | 35 | CH3CN | 85 |
| 16 | Cs2CO3 | 50 | CH3CN | 92 |
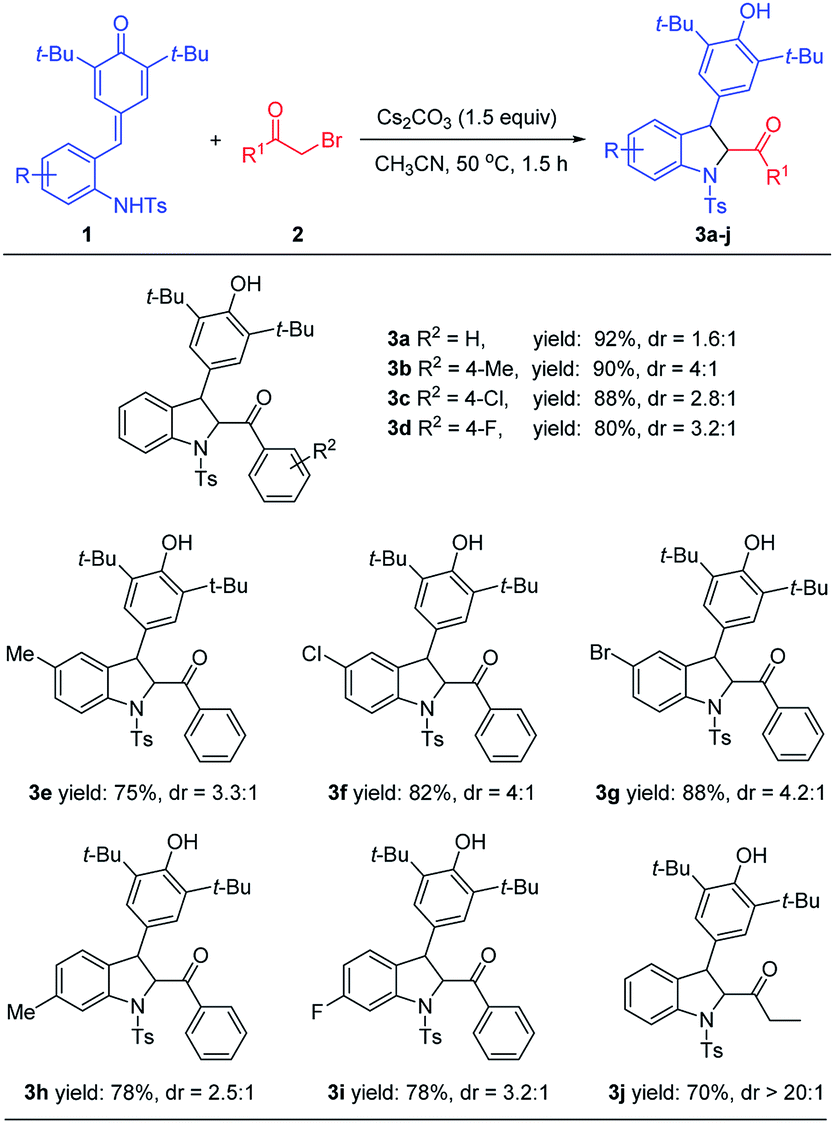
After establishing the optimal reactions conditions for 2,3-dihydroindoles, the substrate scope was explored. As shown in Table 2, a wide range of p-QMs 1 bearing different substituents on the benzene ring and different substituted bromomethyl aryl ketones and bromomethyl alkyl ketones could be smoothly processed to afford the expected products 3a–3j in 70–92% yields. The reaction efficiency was less affected by the variations of the electronic properties or the position of substituents on the benzene ring. Based on this good result, we continued to investigate the one-pot synthesis of indoles, directly from the in situ dehydrogenative oxidation of 2,3-dihydroindoles by using suitable oxidant. After several attempts (e.g. Ag2CO3, Ag2O, DDQ, PhI(OAc)2 and MnO2), we found that DDQ performed best and the corresponding product 4a could be obtained in a total yield of 83% (not shown).
| a All reactions were conducted with 1 (0.11 mmol), 2 (0.10 mmol), Cs2CO3 (1.5 equiv.), CH3CN (1.5 mL). Yields are those of isolated products 3 after column chromatography. |
|---|
 |
With the optimal one-pot reaction conditions in hand, we began to explore the substrate scope of this annulation reaction. The scope of bromomethyl ketones part was examined firstly and we were pleased to find that a wide range of bromomethyl aryl ketones 2 could readily react with ortho-tosylaminophenyl-substituted p-QM 1a to afford 4a–4o in good yields (Table 3). In detail, bromomethyl aryl ketones with electron-neutral (R = H), electron-donating (R = OMe, Me, Ph), or electron-withdrawing (R = Br, Cl, F, NO2) groups at the C3 or C4 position of the benzene ring easily underwent this cascade reaction to provide the desired products 4a–4i in 71–87% yields. Besides, the bromomethyl aryl ketones containing disubstituted groups on the benzene ring were also suitable substrates, and the corresponding products (4j–4m) were obtained in uniform high yields (83–91%). Moreover, the bromomethyl aryl ketones bearing a naphthyl or pyridinyl could also readily take part in this [4 + 1] annulation reaction to give the expected products 4n and 4o in 87% and 89% yields, respectively. In addition, bromomethyl alkyl ketones such as 1-bromo-2-butanone and bromopinacolone could also participate in the process to give the desired products 4p and 4q in 61% and 30% yields, respectively. However, bromomethyl alkyl ketone with an electron-withdrawing group such as 1-bromo-3,3,3-trifluoroacetone was not suitable for this transformation.
| a All reactions were conducted with 1 (0.11 mmol), 2 (0.10 mmol), Cs2CO3 (1.5 equiv.), DDQ (1.5 equiv.), CH3CN (1.5 mL). Yields are those of isolated products 4 after column chromatography. |
|---|
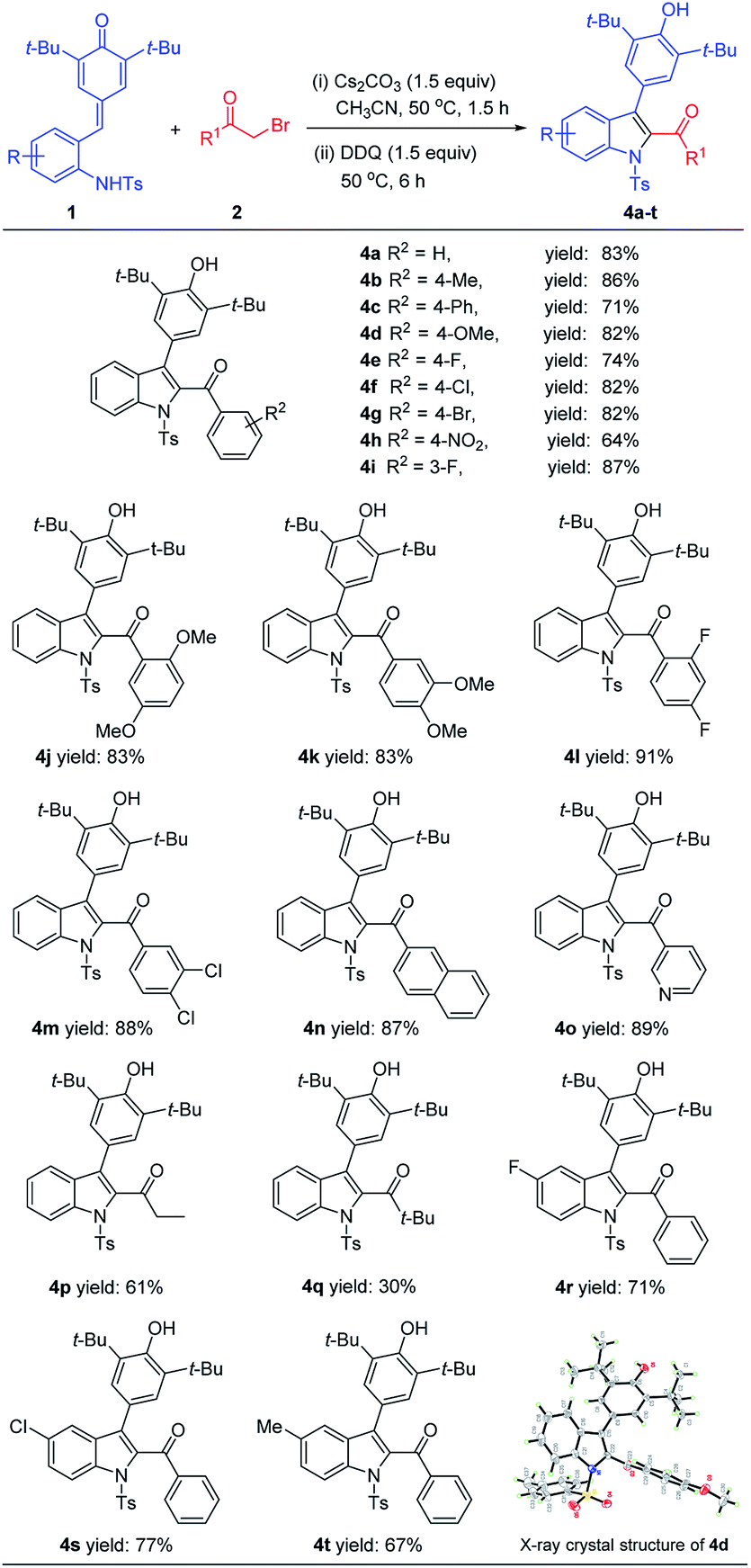 |
Subsequently, the generality of this reaction was further evaluated by varying another reaction partner ortho-tosylaminophenyl-substituted p-QMs 1. It was found that p-QMs bearing different substituents on the benzene ring could be smoothly converted into the expected products 4r–4t in 67–77% yields, and the reaction efficiency was less affected by the electronic properties of substituents. The structure and relative configuration of 4d was determined based on its HRMS, NMR spectroscopy and single-crystal X-ray analyses (Table 3).14
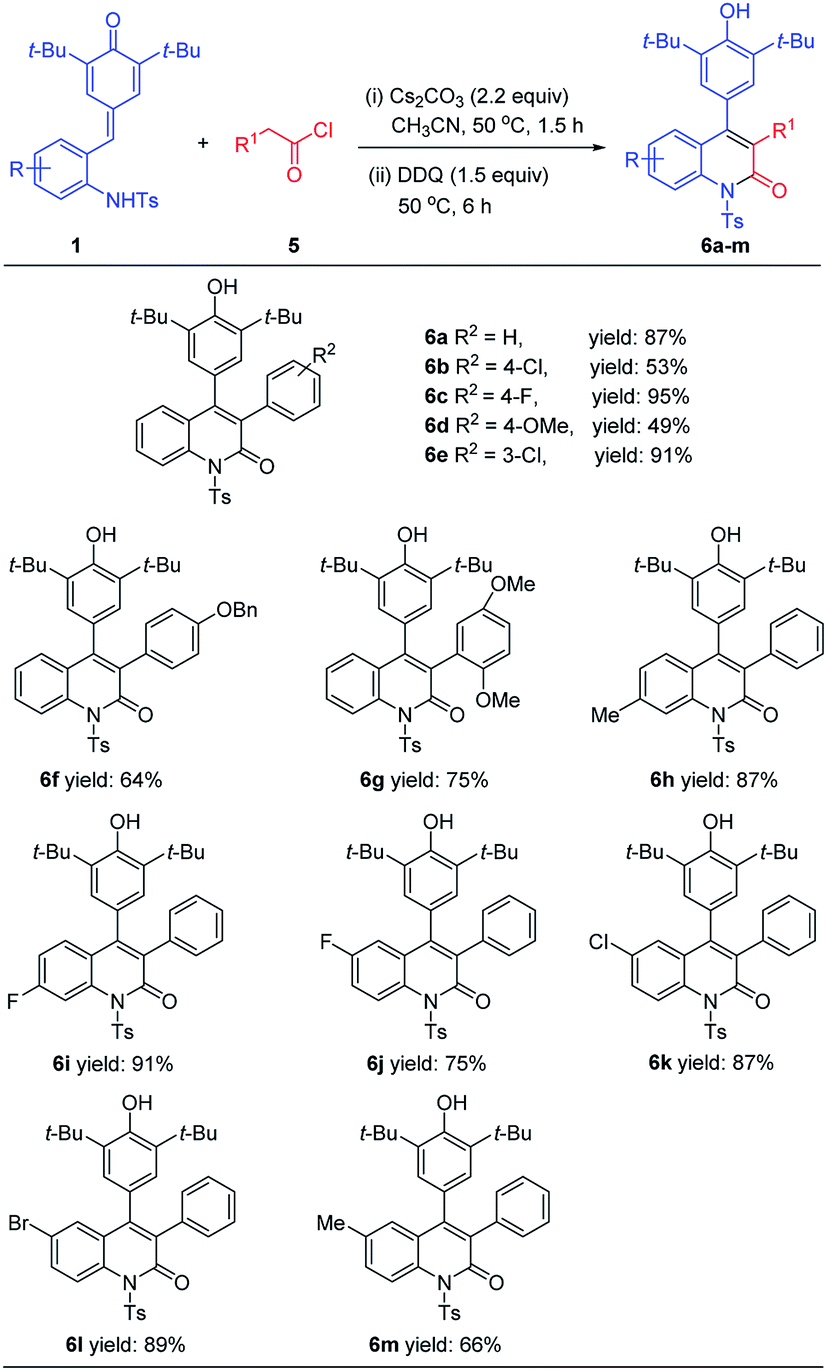
After accomplishing the [4 + 1] annulation reaction of ortho-tosylaminophenyl-substituted p-QMs with bromomethyl ketones for the synthesis of 2,3-disubstituted indole derivatives, we further tried to apply this methodology for the synthesis of other nitrogen-containing heterocycles. We envisioned that assembly of 3,4-diaryl-substituted quinolinone derivatives could be realized by treating ortho-tosylaminophenyl-substituted p-QMs with arylacetyl halides followed by one-pot dehydrogenative oxidation with DDQ. To verify the feasibility of our hypothesis, an initial experiment was carried out by treating 1a with phenylacetyl chloride 5a (1.2 equiv.) in the presence of Cs2CO3 (2.2 equiv.) in CH3CN for 1.5 h followed by the addition of DDQ (1.5 equiv.) and reaction for another 6 h. As expected, the desired product 6a was obtained in 87% isolated yield. Encouraged by this result, the substrate scope of this [4 + 2] annulation reaction was explored. As shown in Table 4, a wide range of arylacetyl chlorides 5 could readily react with 1a to afford products 6a–6g in acceptable yields. For instance, arylacetyl chlorides bearing electron-neutral (R = H), electron-donating (R = OMe, OBn), or electron-withdrawing (R = Cl, F) groups at the C3 or C4 position of the benzene ring were well-tolerated and delivered the desired products 6a–6f in 49–95% yields. Even arylacetyl chlorides bearing disubstituted groups on the benzene ring, the reaction also proceeded smoothly, giving the corresponding product 6g in 75% yield. Furthermore, it was found that a variety of p-QMs 1 bearing different substituents on the benzene ring could be smoothly processed to afford the expected products 6h–6m in 66–91% yields, and the reaction efficiency was less affected by variations of the electronic properties or the position of substituents on the benzene ring.
| a All reactions were conducted with 1 (0.1 mmol), 5 (0.12 mmol), Cs2CO3 (2.2 equiv.), DDQ (1.5 equiv.), CH3CN (1.5 mL). Yields are those of isolated products 6 after column chromatography. |
|---|
 |
To evaluate the general utility and robustness of this novel protocol, we conducted a 1 mmol scale reaction under the standard conditions, and the products 4a and 6a could be isolated in 88% and 90% yields (Scheme 2a). Furthermore, the synthetic transformation of 6a was carried out by treating it with excess of AlCl3 (10 equiv.) in toluene at 60 °C, and the expected de-tert-butylation product 7 was obtained in 78% yield (Scheme 2b).
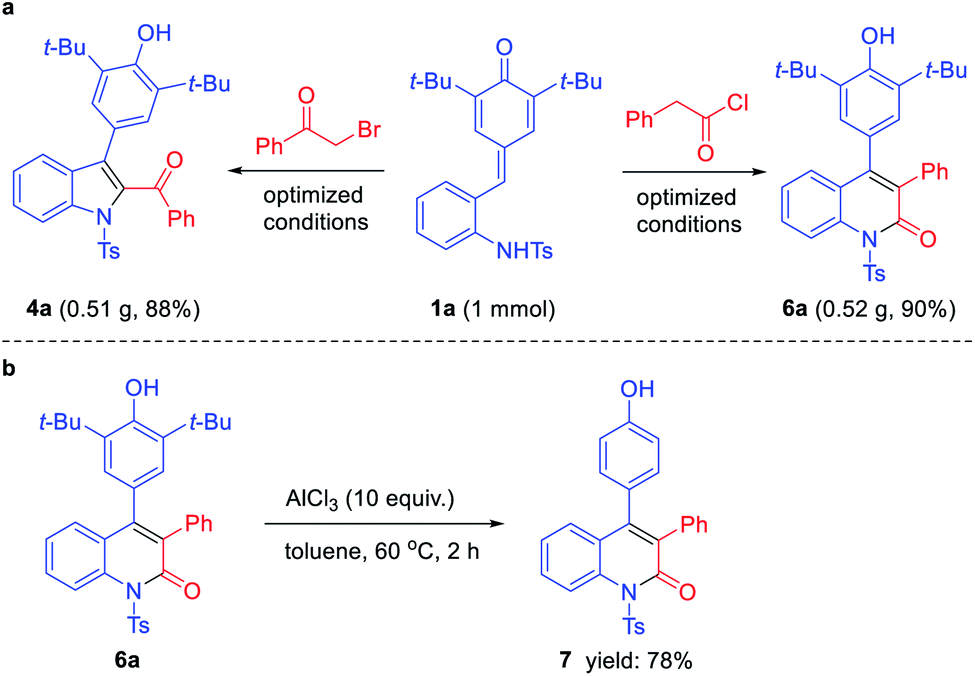 | ||
| Scheme 2 Scale-up synthesis (a) and synthetic transformation (b). | ||
Finally, to understand the mechanism of this reaction, some control experiments were carried out (Scheme 3). Firstly, the reactions of phenyl substituted p-QM 8 with phenacyl bromide and phenylacetyl chloride were carried out individually under standard conditions. However, the corresponding products 9 and 10 were not detected, and the starting material 8 was not transformed in both cases. Further, in exploring the substrate scope of bromomethyl ketones, we obtained some by-products which may help explain the mechanism of this reaction. When treating 1a with 1-bromo-2-butanone under standard conditions, in addition to product 4p, N-alkylated product 11 was isolated in 20% yield. The similar phenomenon was observed when treating 1a with bromopinacolone, N-alkylated product 12 was isolated in 55% yield, while the corresponding 1,6-adduct was only obtained in 30% yield. Based on above results, a plausible mechanism was suggested as shown in Scheme 4. The reaction proceeds through N-alkylation followed by intramolecular 1,6-conjugate addition/cyclization to form 2,3-dihydroindoles, which underwent the in situ dehydrogenative oxidation for the one-pot synthesis of 2,3-disubstituted indoles (Scheme 4a). Also, we can speculate that quinolinone derivatives were formed through N-acylation followed by intramolecular cyclization and one-pot oxidation (Scheme 4b).
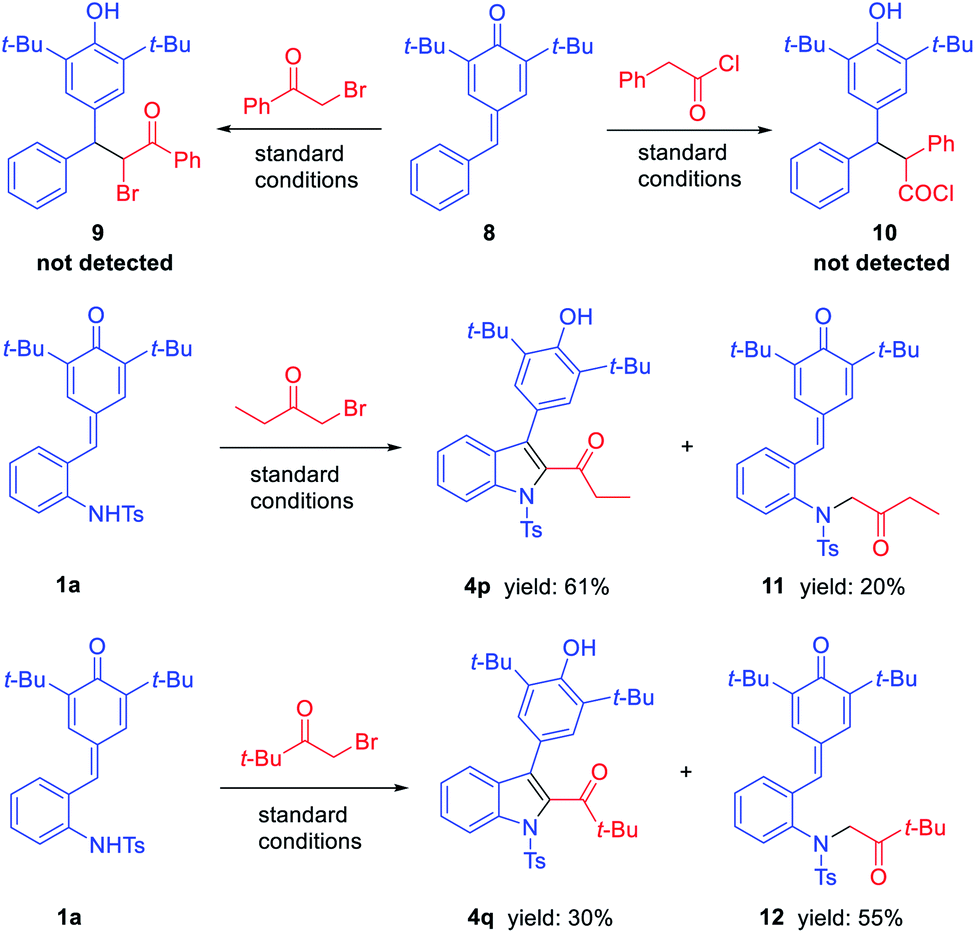 | ||
| Scheme 3 Control experiments. | ||
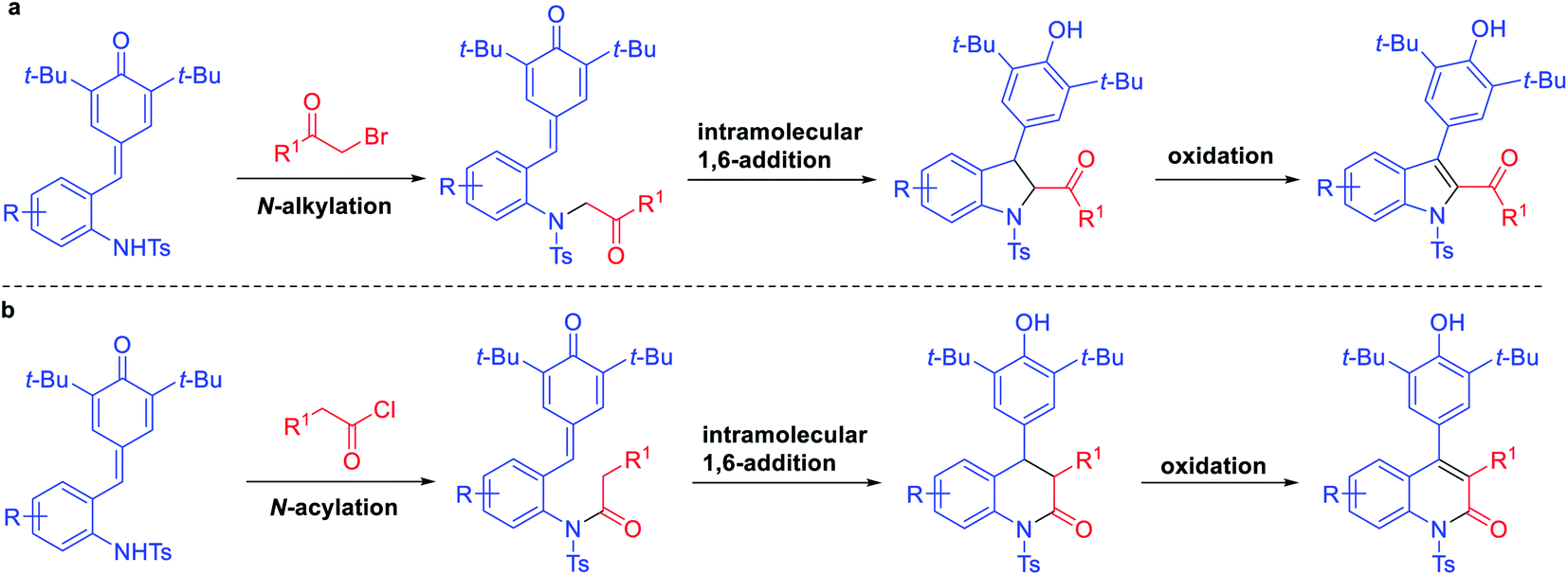 | ||
| Scheme 4 The plausible reaction mechanism for the one-pot synthesis of 2,3-disubstituted indoles (a) and 3,4-diaryl-substituted quinolinones (b). | ||
In summary, we have developed an efficient and facile one-pot method for the synthesis of 2,3-disubstituted indoles and 3,4-diaryl-substituted quinolinones through alkylation/acylation of ortho-tosylaminophenyl-substituted p-QMs followed by intramolecular 1,6-conjugate addition/cyclization and oxidation sequence. This protocol could not only fulfill the task of developing new cyclization reactions of ortho-tosylaminophenyl-substituted p-QMs but also provide an easy access to structurally diverse nitrogen-containing heterocycles.
Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was supported by the National Natural Science Foundation of China (grant: 81803342), the Natural Science Foundation of Jiangsu Province (grant: BK20180826), the Natural Science Research of Jiangsu Higher Education Institutions of China (grants: 18KJB350008, 18KJD360001), the Graduate Research and Innovation Projects of Jiangsu Province (KYCX19_1259).Notes and references
- For recent selected examples, see: (a) W. L. Huang, X. Q. Song, S. X. Peng and Z. Y. Huang, Acta Pharmacol. Sin., 1990, 25, 815 CAS; (b) G. W. Rewcastle, B. D. Palmer, A. J. Bridges, H. D. H. Showalter, L. Sun, J. Nelson, A. Mcmichael, A. J. Kraker, D. W. Fry and W. A. Denny, J. Med. Chem., 1996, 39, 918 CrossRef; (c) S. L. Cao, Y. P. Feng, Y. Y. Jiang, S. Y. Liu, G. Y. Ding and R. T. Li, Bioorg. Med. Chem. Lett., 2005, 36, 1915 CrossRef; (d) M. N. Noolvi, H. M. Patel, V. Bhardwaj and A. Chauhan, Eur. J. Med. Chem., 2011, 46, 2327 CrossRef CAS; (e) K. B. Harikandei, P. Salehi, S. N. Ebrahimi, M. Bararjanian and A. Al-Harrasi, Bioorg. Chem., 2019, 91, 103116 CrossRef.
- (a) H. Sawaoka, S. Tsuji, M. Tsujii, E. S. Gunawan and M. Hori, Lab. Invest., 1999, 79, 1469 CAS; (b) K. Suzumura, M. Yasuhara, K. Tanaka and T. Suzuki, Biochem. Pharmacol., 1999, 57, 697 CrossRef CAS; (c) O. Guzela, A. Innocenti, D. Vullo, A. Scozzafava and C. T. Supuran, Curr. Pharm. Des., 2010, 16, 3317 CrossRef; (d) A. Srivastava and S. N. Pandeya, Asian J. Pharm. Clin. Res., 2011, 4, 5 Search PubMed; (e) C. R. Prakash and S. Raja, Mini-Rev. Med. Chem., 2012, 12, 98 CrossRef CAS.
- (a) E. Ramesh, R. D. R. S. Manian, R. Raghunathan, S. Sainath and M. Raghunathan, Bioorg. Med. Chem., 2009, 17, 660 CrossRef CAS; (b) J. M. Kraus, C. L. M. J. Verlinde, M. Karimi, G. I. Lepesheva, M. H. Gelb and F. S. Buckner, J. Med. Chem., 2009, 52, 1639 CrossRef CAS; (c) Y. F. Chen, Y. C. Lin, P. K. Huang, H. C. Chan, S. C. Kuo, K. H. Lee and L. J. Huang, Bioorg. Med. Chem., 2013, 21, 5064 CrossRef CAS; (d) B. X. Huang, K. Newcomer, K. Kevala, E. Barnaeva, W. Zheng, X. Hu, S. Patnaik, N. Southall, J. Marugan and M. Ferrer, Sci. Rep., 2017, 7, 11673 CrossRef.
- For selected reviews on p-QMs, see: (a) M. M. Toteva and J. P. Richard, Adv. Phys. Org. Chem., 2011, 45, 39 CrossRef CAS; (b) C. Lorenzo, F. Mariafrancesca and B. Luca, Molecules, 2015, 20, 11733 CrossRef; (c) A. Parra and M. Tortosa, ChemCatChem, 2015, 7, 1524 CrossRef CAS; (d) P. Chauhan, U. Kaya and D. Enders, Adv. Synth. Catal., 2017, 359, 888 CrossRef CAS.
- W. D. Chu, L. F. Zhang, X. Bao, X. H. Zhao, C. Zeng, J. Y. Du, G. B. Zhang, F. X. Wang, X. Y. Ma and C. A. Fan, Angew. Chem., Int. Ed., 2013, 52, 9399 CrossRef.
- L. Caruana, F. Kniep, T. K. Johansen, P. H. Poulsen and K. A. Jorgensen, J. Am. Chem. Soc., 2014, 136, 15929 CrossRef CAS.
- For selected examples, see: (a) Y. Lou, P. Cao, T. Jia, Y. Zhang, M. Wang, J. Liao, Y. Lou, P. Cao, T. Jia and Y. Zhang, Angew. Chem., Int. Ed., 2015, 54, 12134 CrossRef CAS; (b) Z. Wang, Y. F. Wong and J. Sun, Angew. Chem., Int. Ed., 2015, 54, 13711 CrossRef CAS; (c) Z. Yuan, X. Fang, X. Li, J. Wu, H. Yao and A. Lin, J. Org. Chem., 2015, 80, 11123 CrossRef CAS; (d) N. Dong, Z. P. Zhang, X. S. Xue, X. Li and J. P. Cheng, Angew. Chem., Int. Ed., 2016, 55, 1460 CrossRef CAS; (e) F. He, J. Jin, Z. Yang, X. Yu, J. S. Fossey and W. Deng, ACS Catal., 2016, 6, 652 CrossRef CAS; (f) S. Gao, X. Xu, Z. Yuan, H. Zhou, H. Yao and A. Lin, Eur. J. Org. Chem., 2016, 3006 CrossRef CAS; (g) X. Li, X. Xu, W. Wei, A. Lin and H. Yao, Org. Lett., 2016, 18, 428 CrossRef CAS; (h) C. Yang, S. Gao, H. Yao and A. Lin, J. Org. Chem., 2016, 81, 11956 CrossRef CAS; (i) Y.-H. Deng, X.-Z. Zhang, K.-Y. Yu, X. Yan, J.-Y. Du, H. Huang and C.-A. Fan, Chem. Commun., 2016, 52, 4183 RSC; (j) X.-Z. Zhang, Y.-H. Deng, X. Yan, K.-Y. Yu, F.-X. Wang, X.-Y. Ma and C.-A. Fan, J. Org. Chem., 2016, 81, 5655 CrossRef CAS; (k) X.-Z. Zhang, J.-Y. Du, Y.-H. Deng, W.-D. Chu, X. Yan, K.-Y. Yu and C.-A. Fan, J. Org. Chem., 2016, 81, 2598 CrossRef CAS; (l) C. Ma, Y. Huang and Y. Zhao, ACS Catal., 2016, 6, 6408 CrossRef CAS; (m) Z. Yuan, L. Liu, R. Pan, H. Yao and A. Lin, J. Org. Chem., 2017, 82, 8743 CrossRef CAS; (n) L. Roiser and M. Waser, Org. Lett., 2017, 19, 2338 CrossRef CAS; (o) Z. Yuan, K. Gai, Y. Wu, J. Wu, A. Lin and H. Yao, Chem. Commun., 2017, 53, 3485 RSC; (p) X.-Z. Zhang, Y.-H. Deng, K.-J. Gan, X. Yan, K.-Y. Yu, F.-X. Wang and C.-A. Fan, Org. Lett., 2017, 19, 1752 CrossRef CAS.
- K. Zhao, Y. Zhi, T. Shu, A. Valkonen, K. Rissanen and D. Enders, Angew. Chem., Int. Ed., 2016, 55, 12104 CrossRef CAS.
- For selected examples of [4 + 1] cyclization, see: (a) K. Zhao, D. Enders, Y. Zhi, C. von Essen and K. Rissanen, Org. Chem. Front., 2018, 5, 1348 RSC; (b) B. Jiang, Y.-J. Xiong, S.-Q. Shi, W.-J. Hao and S.-J. Tu, Org. Chem. Front., 2018, 5, 3483 RSC; (c) L. Liu, Z. Yuan, R. Pan, Y. Zeng, A. Lin, H. Yao and Y. Huang, Org. Chem. Front., 2018, 5, 623 RSC; (d) Z. Jing, G. Liang, X. Hu, L. Zhou and Z. Hui, Tetrahedron, 2018, 74, 1492 CrossRef; (e) X. M. Chen, K. X. Xie, D. F. Yue, X. M. Zhang, X. Y. Xu and W. C. Yuan, Tetrahedron, 2018, 74, 600 CrossRef CAS; (f) H. Lu, H.-X. Zhang, C.-Y. Tan, J.-Y. Liu, H. Wei and P.-F. Xu, J. Org. Chem., 2019, 84, 10292 CrossRef CAS; (g) J.-P. Tan, P. Yu, J.-H. Wu, Y. Chen, J. Pan, C. Jiang, X. Ren, H.-S. Zhang and T. Wang, Org. Lett., 2019, 21, 7298 CrossRef CAS.
- G. Singh, S. Kumar, A. Chowdhury and R. V. Anand, J. Org. Chem., 2019, 84, 15978 CrossRef CAS.
- For selected examples of [4 + 2] cyclization, see: (a) G. J. Mei, S. L. Xu, W. Q. Zheng, C. Y. Bian and F. Shi, J. Org. Chem., 2018, 83, 1414 CrossRef CAS; (b) X.-L. Jiang, S.-F. Wu, J.-R. Wang, G.-J. Mei and F. Shi, Adv. Synth. Catal., 2018, 360, 4225 CrossRef CAS; (c) J.-R. Zhang, H.-S. Jin, R.-B. Wang and L.-M. Zhao, Adv. Synth. Catal., 2019, 361, 4811 CrossRef CAS; (d) Y.-C. Cheng, C.-S. Wang, T.-Z. Li, F. Gao, Y. Jiao and F. Shi, Org. Biomol. Chem., 2019, 17, 6662 RSC; (e) Z. Li, W. Wang, H. Jian, W. Li, B. Dai and L. He, Chin. Chem. Lett., 2019, 30, 386–388 CrossRef CAS; (f) Y. You, T.-T. Li, S.-P. Yuan, K.-X. Xie, Z.-H. Wang, J.-Q. Zhao, M.-Q. Zhou and W.-C. Yuan, Chem. Commun., 2020, 56, 439 RSC; (g) B. Jiang, H.-M. Huang, X.-Y. Wu, B.-R. Leng, X.-C. Meng, Y. Hong, B. Jiang and D.-C. Wang, Org. Chem. Front., 2020, 7, 414 RSC.
- For selected examples of [4 + 3] cyclization, see: (a) F. Jiang, F. Yuan, L. Jin, G. Mei and F. Shi, ACS Catal., 2018, 8, 10234 CrossRef CAS; (b) L. Qiang, L. Sun, C. Xiang-Yu, R. Kari and E. Dieter, Org. Lett., 2018, 20, 3622 CrossRef; (c) W. Li, H. Yuan, Z. Liu, Z. Zhang, Y. Cheng and P. Li, Adv. Synth. Catal., 2018, 360, 2460 CrossRef CAS; (d) B. Jiang, K. Chen, W.-J. Hao and S.-J. Tu, Green Chem., 2019, 21, 675 RSC.
- For selected examples of ortho-tosylaminophenyl-substituted p-QMs, see: (a) J. Wang, X. Pan, J. Liu, L. Zhao, Y. Zhi, K. Zhao and L. Hu, Org. Lett., 2018, 20, 5995 CrossRef CAS; (b) J. Wang, X. Pan, L. Zhao, L. Zhao, J. Liu, Y. Zhi, A. Wang, K. Zhao and L. Hu, Org. Biomol. Chem., 2019, 17, 10158 RSC.
- CCDC 2009636 (4d) contains the supplementary crystallographic data for this paper.†.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2009636. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ra05497f |
| ‡ Equally contributing authors. |
| This journal is © The Royal Society of Chemistry 2020 |