
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Structure–activity prediction networks (SAPNets): a step beyond Nano-QSAR for effective implementation of the safe-by-design concept†
Anna
Rybińska-Fryca
 a,
Alicja
Mikolajczyk
ab and
Tomasz
Puzyn
*ab
a,
Alicja
Mikolajczyk
ab and
Tomasz
Puzyn
*ab
aQSAR Lab Ltd., Aleja Grunwaldzka 190/102, 80-266 Gdansk, Poland. E-mail: t.puzyn@qsarlab.com
bUniversity of Gdansk, Faculty of Chemistry, Wita Stwosza 63, 80-308 Gdansk, Poland
First published on 13th October 2020
Abstract
A significant number of experimental studies are supported by computational methods such as quantitative structure–activity relationship modeling of nanoparticles (Nano-QSAR). This is especially so in research focused on design and synthesis of new, safer nanomaterials using safe-by-design concepts. However, Nano-QSAR has a number of important limitations. For example, it is not clear which descriptors that describe the nanoparticle physicochemical and structural properties are essential and can be adjusted to alter the target properties. This limitation can be overcome with the use of the Structure–Activity Prediction Network (SAPNet) presented in this paper. There are three main phases of building the SAPNet. First, information about the structural characterization of a nanomaterial, its physical and chemical properties and toxicity is compiled. Then, the most relevant properties (intrinsic/extrinsic) likely to influence the ENM toxicity are identified by developing “meta-models”. Finally, these “meta-models” describing the dependencies between the most relevant properties of the ENMs and their adverse biological properties are developed. In this way, the network is built layer by layer from the endpoint (e.g. toxicity or other properties of interest) to descriptors that describe the particle structure. Therefore, SAPNets go beyond the current standards and provide sufficient information on what structural features should be altered to obtain a material with desired properties.
Introduction
Precise manipulation and control of the structure of matter on the nanoscale brings an opportunity to design nanomaterials (ENMs) that are safe for humans and the environment in addition to obtaining their maximal efficiency in the context of the required application. This concept is called ‘safe-by-design’.1 The crucial aspect of ‘safe-by-design’ is gathering knowledge on the relationships between the nanomaterials’ structure, physicochemical properties and toxicity. Thus, the designer knows precisely how to modify the structure to get the expected change in the activity.The search for the structure–property and/or structure–activity relationships can be significantly supported by computational techniques (e.g. quantitative structure–activity/property relationship (QSAR/QSPR) modeling). The application of the QSAR helps in reducing the expenses and the number of necessary experiments. Moreover, it provides quantitative description of the relationships, in the form of mathematical equations. Thus, the developer is able to calculate to what extent the property/toxicity would increase/decrease in response to the considered structural change.
The idea of QSAR modeling for nanomaterials (Nano-QSAR) was introduced by Puzyn et al. in 2009.2 Since then, it has been used to improve the existing models, to develop descriptors for nanomaterials, and to support experimental studies. The most important directions of further developments of Nano-QSAR have been discussed in a joint “EU-US Nanoinformatics 2030 Roadmap” and such EU Horizon 2020 projects as NanoSolveIT (http://www.nanosolveit.eu) and NanoInformaTIX (http://www.nanoinformatix.eu) that work on the introduction of an innovative Integrated Test and Evaluation Approach (IATA) for environmental health and nanomaterial safety and promoting the use of Nano-QSAR methods as a part of the safe-by-design approach.3
To the best of our knowledge, there is a limited number of Nano-QSAR/QSPR models that directly express toxicity or a selected property as a function of the structural features (descriptors expressing structural attributes). Roy et al. presented a nano-QSTR approach based on periodic table based descriptors.4 The authors developed a series of linear regression models for predicting the toxicity of heterogeneous TiO2-based NPs towards the Chinese hamster ovary cell line. However, only one model was based on a descriptor that directly describes the structure (amount of silver metal). The other variables (electrochemical equivalent, 2nd ionization potential, covalent radius, and thermal conductivity) can be considered as properties that are consequences of the structure. Another example is the model for the prediction of zeta potential based on grouping of the NMs according to their nearest neighbors developed by Varsou et al.5 The model utilizes three variables: the type of the core (metal oxide or pure metal), the main elongation expressing the lengthening of the particle, and the pH where the zeta potential was measured.5 There are also models based on quasi-SMILES, which are character-based representations derived from traditional SMILES.6 They can encode the structural features, the physicochemical properties, and the exposure conditions such as cell lines.7–11 Another way to take into account factors such as changes in chemical compositions, assay organisms, or exposure time is to apply the QSAR-perturbation approach.12,13 All of the mentioned models are very useful tools. However, they do not provide specific information on the dependency between the structural features and properties of ENMs that subsequently influences the biological activity.
Majority of the contributions present the predictions of ENMs’ biological activity based on the intrinsic physico-chemical properties (i.e. features independent of the environments that characterize the nanoparticle as an effect of having a given structure).14–16 The intrinsic properties applied as toxicity predictors allow us to explain the toxicity mode of action, but do not provide knowledge about the influence of the structural features on the toxic effect. As a consequence, such models are insufficient from a “designer” point of view (i.e. a designer does not know how to modify the structure to obtain the expected effect). On the other hand, in many discussions nanotoxicologists raise an issue that the influence of the environment (e.g. solvent and pH) on the toxicity is of high importance. Therefore, it might be impossible to predict the toxicity directly from the structure. Instead, the toxicity can be predicted from the system-dependent and/or intrinsic properties and such properties can be linked to the purely structural features.
Here, we propose to replace the traditional Nano-QSAR modeling practice with the use of Structure–Activity Prediction Networks (SAPNets) – an approach that effectively links the description of ENMs’ structure with their toxicity through a series of layers built from nodes that correspond to predictive “meta-models” developed with machine learning techniques as well as Artificial Intelligence (AI) (Fig. 1).
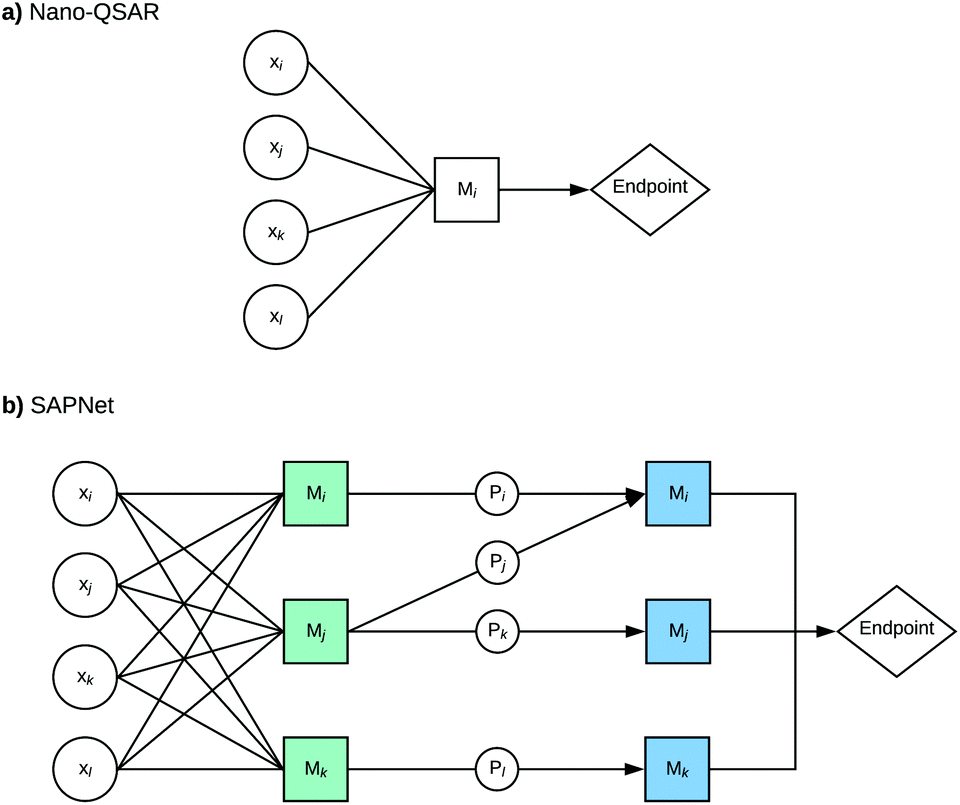 | ||
| Fig. 1 The schematic representation of the structure–activity prediction networks for modeling of ENMs’ toxic activity; x – descriptor that describes either structural attributes (D) or properties (P) of ENMs, M – meta-model (meta-models of the 1st layer – indicated in green, and meta-models of the 2nd layer – indicated in blue). | ||
Idea of structure activity prediction networks (SAPNets)
The first step of building a network is to gather information about structural characterization of a nanomaterial, its physical and chemical properties and toxicity. In the case of chemically diverse ENMs, their properties can be derived from experimental as well as computational studies. The advantage of the theoretical/computational approach is the possibility of retrieving characterization for a larger number of ENMs than in the case of experimental studies.The second step is the identification of relevant properties directly influencing the ENMs’ toxicity by developing “meta-models”. However, these models should not be limited to the Nano-QSAR or more precisely quantitative property–activity relationship (Nano-QPAR) approach. One should consider other methods like read-across, knowledge-based decision rules, models based on omics data, and other types of models based on AI and machine learning techniques.
The subsequent steps are to build the next layers of “meta-models” that describe the relationships between the selected properties of the studied ENMs and their structural features (descriptors that express structural attributes). In this way, the network is built layer by layer from the endpoint (e.g. toxicity or other properties of interest) to the structure. It is worth noting that SAPNets can be used not only for predicting adverse effects (toxicity) but also for optimizing the desired properties of the newly designed ENMs, such as photocatalytic activity, higher electrical conductivity, etc.
Although the development of structure–activity prediction networks requires extensive knowledge and experience in computational nanotoxicology, SAPNets may be further easily applied for predicting the toxicity and properties of ENMs by non-specialists. This is because they are based on descriptors that are understandable for non-specialists (e.g. size, shape, aspect-ratio, and type of coating) and do not require additional computational calculations – the user provides only the values of such descriptors and then the predictions are made. Moreover, the user can see precisely how the modification of the descriptor values (e.g. size) will influence the predicted endpoint.
We do believe that the proposed new approach responds to the needs of the scientific community focused on research on nanomaterials. Here, we present three case studies in which the structure–activity prediction networks methodology is used to estimate the photocatalytic activity as well as the biological activity of ENMs.
Materials and methods
Case study details: predicting the properties of ENMs with SAPNets with the example of the phenol degradation efficiency
The presented case study was based on the publicly available data.17 To assure the reliability of the model, we split the studied data into the training (T, 15 samples) and external validation (V, 6 samples) sets. First, the samples were sorted based on the increasing endpoint value (percentage of phenol degradation, τOH). Then, the ones with the highest and the lowest value were arbitrarily assigned to the training set. The rest of the samples were randomly assigned to the validation set. Therefore, the points from the validation set were evenly distributed within the range of the endpoint of the training set. Details can be found in Table 1S in the ESI.† The training set was used to find an equation that links the properties of samples with phenol degradation (τOH) by titanium dioxide. The considered properties were the efficiency of charge carrier trapping, migration, and transfer expressed as the intensity of photoluminescence, and the band gap reduction described through UV-Vis absorption spectra (Tables 2S and 3S in the ESI†). The relationship between the categorical (τOH) and independent variables was described by the logistic regression model. The stepwise selection approach was used to find an optimal combination of descriptors. The quality of the model was determined on the basis of parameters such as accuracy, sensitivity, specificity, precision, and misclassification error, calculated for both the training and validation set. Therefore, we were able to verify the goodness-of-fit of the model and ability to predict the endpoint value for external samples. Additionally, the internal validation of the model (leave-one-out cross-validation) was performed to measure its robustness.| accuracy = TP + TN/TP + TN + FP + FN |
| sensitivity = TP/TP + FN |
| specificity = TN/TN + FP |
| misclassification = FP + FN/TP + TN + FP + FN |
TP, TN, FP, and FN stand for true positive, true negative, false positive, false negative respectively. The borders of the applicability domain (AD) of the model are defined by the minimal and maximal values of descriptors characterizing samples from the training set. Moreover, the developed model should be applied only to TiO2-based nanophotocatalysts synthesized in the presence of ionic liquids according to the protocol described in the source publication.17
All of the analyses in the case study were carried out using packages available in the R statistical program (R version 3.6.2).
Examples of implementation of the SAPNet methodology
Predicting the properties of ENMs with SAPNets with the example of the phenol degradation efficiency
According to the proposed workflow, the developed SAPNet should finally correlate the structure of a nanomaterial to the selected endpoint. Here, we present an example of how the photodegradation of phenol (τOH) by titanium dioxide can be determined in line with the SAPNet methodology (Fig. 2). In this case study, for developing SAPNets, we used experimental data from our previous publication.17 The analyzed dataset contains experimental characterization of 23 samples of titanium dioxide synthesized in the presence of ionic liquids (IL) and information about the photocatalytic degradation of phenol for each sample (Table 1S in the ESI†). Since the values of photoluminescence (PL) had been unavailable for two samples, the dataset used in this case was reduced to 21 observations. | ||
Fig. 2 Determination of photodegradation of phenol by titanium dioxide (τOH) in line with the SAPNet methodology (BET – Brunauer–Emmett–Teller surface area analysis; N, C – the amount of nitrogen and carbon atoms; xratio – molar ratio (IL![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :TiO2 precursor); ΔIL – decomposition of the ionic liquid during the solvothermal reaction; cation – the type of cation in the IL; anion – type of anion in IL; PL398 – the intensity of the photoluminescence signal at 398 nm). :TiO2 precursor); ΔIL – decomposition of the ionic liquid during the solvothermal reaction; cation – the type of cation in the IL; anion – type of anion in IL; PL398 – the intensity of the photoluminescence signal at 398 nm). | ||
The last node of the network defines the relationship between the photocatalytic activity of the TiO2-IL semiconductors and experimentally measured properties. Photocatalytic degradation of phenol (τOH) was determined in a model reaction of the compound decomposition in an aqueous solution under visible irradiation and expressed on a continuous scale in the range from 0 to 100%. Since phenol degradation in the case of typical TiO2-based semiconductors is around 30%, the samples were assigned to one of the categories: with high photoactivity or with low photoactivity (percent of degradation less than 35%).18,19 Then, the dataset was divided into training and validation sets. Finally, several machine learning techniques were used to find a property based on which the level of photoactivity can be estimated. The best model was obtained by using logistic regression with one property (intensity of photoluminescence at 398 nm) as an independent variable (predictor). The quality of the model was determined on the basis of parameters such as accuracy, sensitivity, specificity, precision, and misclassification error, calculated for the training and validation sets.20 Details can be found in Table 1 and the Materials and Methods section.
| Intercept and coefficient × predictor | |||
|---|---|---|---|
| Model type: logistic regression | |||
| 4.32(±2.10) – 0.051(±0.03)(PL 398 ) | |||
| Calibration | Internal validation | External validation | |
| Accuracy | 0.80 | 0.80 | 0.83 |
| Sensitivity | 0.91 | — | 1 |
| Specificity | 0.5 | — | 0.5 |
| Precision | 0.83 | — | 0.83 |
| Misclassification error | 0.20 | 0.20 | 0.17 |
| AUC | 0.91 | — | 0.75 |
| PL398 − emission intensity at 398 nm (photoluminescence signal) | |||
In the second step, we have been trying to find the dependency between the property used in the first model and the structure of nanomaterials (meta-model). Each sample is described by surface area, the amount of nitrogen and carbon atoms, ionic liquid decomposition rate (ΔIL), molar ratio and the type of cations and anions. We decided to use Principal Component Analysis (PCA) to visualize the TiO2-IL samples in the multidimensional space of predictors describing the structure of nanomaterials (Fig. 3). The first two principal components explain 59.66% of the variance. Based on the loading's values (coefficients of the linear combination of the original variables which construct the PCs) we were able to estimate how much each predictor contributes to a particular principal component. These are surface area and the amount of nitrogen and carbon atoms for PC1 and molar ratio and the type of ions for PC2 (Table 4S in the ESI†). To identify the possible hidden patterns and relationships the information about photoluminescence intensity at 398 nm was added to the PCA plot. Unfortunately, after visual analysis, we couldn't distinguish groups of samples that are similar in terms of experimental characterization, as well as property. We can assume that available experimental characterization is not sufficient and should be extended to find relevant variables.
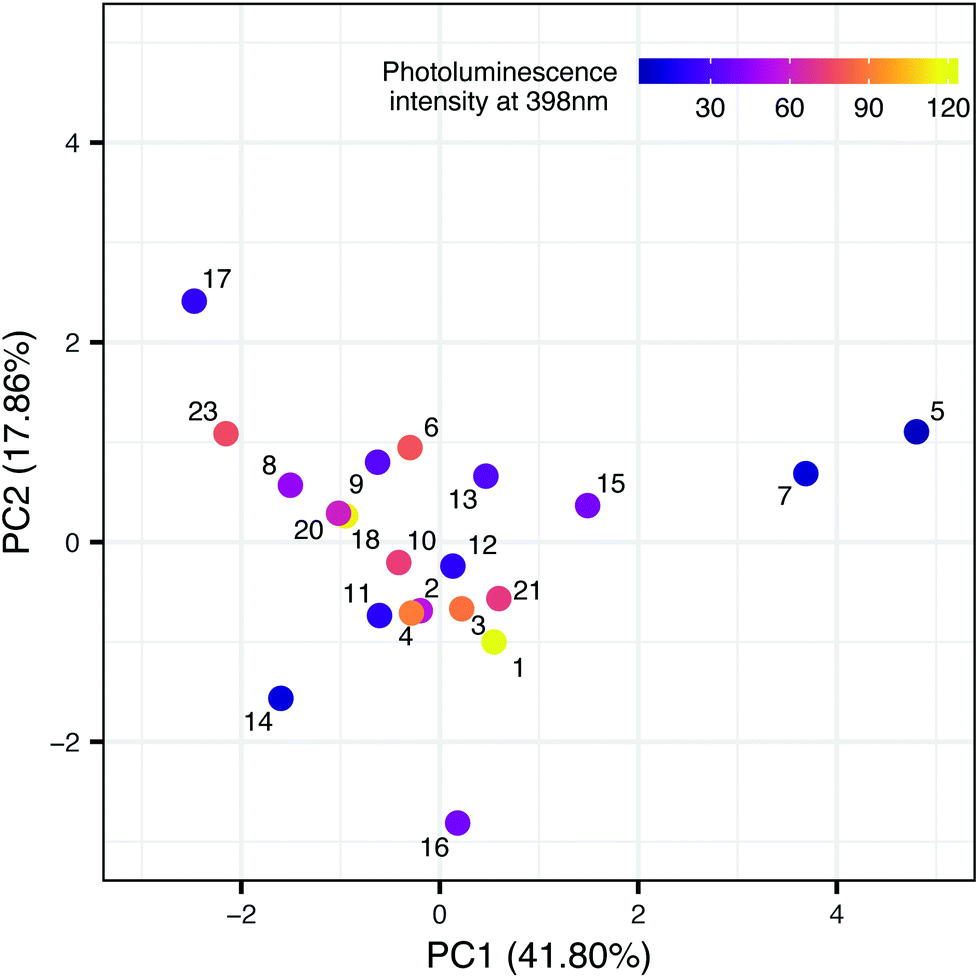 | ||
| Fig. 3 TiO2-IL samples in the space of the experimental predictors. | ||
Nevertheless, the presented case study highlights one of the most crucial issues related to the Nano-QSAR method – the comprehensive characterization of physical and chemical properties of nanoparticles. Building a model based on structure information that can be easily interpreted requires access to a suitably prepared data set. Each experimental study that provides publicly available data, subsequently used for model development, should meet the standards of findability, accessibility, interoperability, and reusability (FAIR idea).21,22 Thus, collecting additional data from various sources and filling the gaps in developed datasets will be possible. Moreover, the presented SAPNet includes a meta-model that links phenol degradation with experimentally measured photoluminescence. Hence, the predictive potential of the network is limited. This issue can be overcome by adding a node dedicated to the theoretical simulation of signal intensity at a certain wavelength.
Predicting the toxicity of ENMs in line with the SAPNet methodology
The modern approach to the introduction of new ENMs should involve synthesis targeted to the specific structure, morphology, and physical and chemical properties, as well as procedure of safety assessment. The evaluation of possible adverse outcomes toward humans and the environment can be supported by computational methods such as Nano-QSAR. Unfortunately, the majority of available models are based on the predictors that require knowledge and experience in computational chemistry. For example, one of the first Nano-QSAR models links the cytotoxicity effect towards E. coli with theoretical predictors for 17 metal oxide nanoparticles derived from molecular models that were optimized at the semiempirical level of theory (PM6). However, the theoretical predictors can be derived from calculations conducted at different levels of theory, i.e. (i) electronic level; (ii) atomistic level; (iii) mesoscopic level; or (iv) continuum level.14,23 The theoretical predictors have been widely applied in the development of different types of Nano-QSAR models.24–28 Unfortunately, their application in toxicity prediction requires time-consuming computational resources and specialized knowledge.In 2019, Mikolajczyk et al.29 used a set of 29 TiO2-based nanomaterials modified with Au, Ag, Pt and Pd nanoclusters (Memix-TiO2) to develop a model capable of predicting toxicity towards the CHO-K1 cell line (epithelial cells obtained from the Chinese hamster ovary, ATCC® CCL-61™).29 The published Nano-QSAR model utilized only one descriptor, which represents additive electronegativity (χmix):
| pEC50 = 6.37(±0.07) + 0.56(±0.02) × χmix |
It may seem that the calculation of this particular predictor requires experience in the Nano-QSAR methodology and computational chemistry. Nevertheless, it is strictly related to the designed and synthesized nanostructure. The type and concentration of a certain metal in the mixture (%molMe) as well as electronegativity of a particular metal (χMe) are necessary to estimate the additive electronegativity:
| χmix = %molMe1 × χMe1+ ⋯ + %molMen) × χMen |
Therefore, information about the structure can be used to calculate a property that is relevant for a selected biological activity. However, to emphasize these relations, the developed model should be presented according to the SAPNet scheme (Fig. 4).
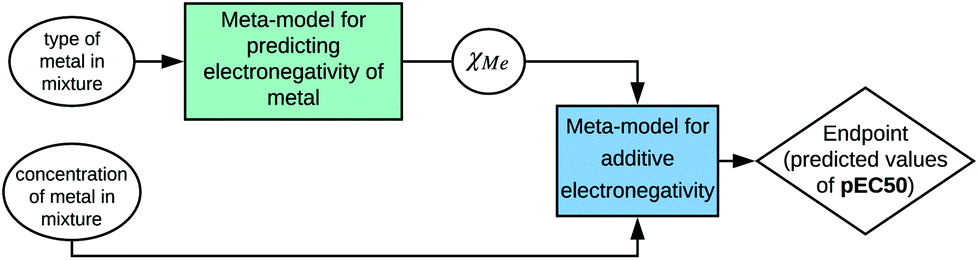 | ||
| Fig. 4 Example of presenting a model according to the SAPNet scheme; χMe is the electronegativity of a particular metal. | ||
Incorporation of the existing models into SAPNets
The presented approach is not limited to building new predictive models for nanomaterials. It can be used to combine already published models into a network that will be useful in the context of designing new advanced materials to be “tailored” for specific needs. One of the most important characteristics of a nanoparticle is its stability in different media. The ability to form agglomerates is one of the physical factors that affect the environmental fate and behavior of ENMs.30,31 For example, agglomeration can change the sedimentation process; therefore, it indirectly influences the effective doses responsible for potential toxicity towards living organisms.30,31 The agglomerate formation is linked with surface charge; however, it cannot be measured directly. Hence, the zeta potential (ζ) in a given medium is a commonly used parameter to express the surface charge.In 2015 Mikolajczyk et al.32 published a Nano-QSPR model for predicting the zeta potential (ζ) of metal oxide nanoparticles (NPs) in a medium. The authors collected the experimental values of zeta potentials of selected 15 MeOx NPs. Every nanoparticle was described by 11 image-based descriptors and 17 properties calculated with quantum-mechanical methods (at the level of semi-empirical theory). The best combination of the most relevant input variables was selected with use of the Genetic Algorithm (GA). Finally, the authors used multiple linear regression and obtained the following model:
| ζ = −11.26 − 4.46ψ − 2.39 εHOMO/nMe |
Another model was presented by Toropov et al. in 2018.33 In this case, the dataset contained 87 data points of zeta potential measurements in aqueous solutions (ζH20) for nanomaterials made of silica and metal oxides having various sizes. Each nanoparticle was described by a modified version of the simplified molecular input line entry system (quasi-SMILES) that represents all available information on the structure.34 Moreover, the nominal sizes of NPs, as well as sizes in water, were used as input variables. Nano-QSPR models were constructed by using the Monte Carlo approach.
The values of zeta potential in water (ζH20) for different nanoparticles obtained by the application of the mentioned model can be used as the input to the model proposed by Wyrzykowska et al.:
| ζKCl = 3.98 + 21.68 ζH2O + 7.88 PN |
 | ||
| Fig. 5 Example of incorporation of two existing models for predicting zeta potential in different environmental conditions developed by Toropov et al.33 and Wyrzykowska et al.35 into the SAPNet scheme. | ||
Discussion and conclusions
The three presented case studies illustrate the high potential of SAPNets to serve as a valuable tool for developing new nanoparticles in line with the idea of safe-by-design. This is because the user obtains precise information on which structural features of the studied nanoparticles should be modified to acquire the material with desired properties and/or low toxicity. In addition, the designing process can be done fully virtually, without the necessity to synthesize the particle first and measure the experimental predictors, formerly used as input variables to Nano-QSAR models. Moreover, in the case of predicting toxicity, SAPNets may include a series of meta-models that allow considering the influence of the environment (such conditions as different pHs, solvents, etc.) on the nanomaterials’ property and behavior. This was an important limitation of simple Nano-QSARs in cases where it may be related to system-dependent properties.36–38The concept of the SAPNets can be described as a “series of mutually dependent predictive models”. A similar approach can be noticed in the case of toxicity-toxicity relationship studies (QTTR). For example, Roy et al. developed several QSTR models for both rat and mouse oral toxicity of carbamate derivatives. Then, the QTTR models were developed by taking each of the predicted responses as independent variables.39 Therefore, the specific information about the structure (expressed by descriptors) can be used to estimate the toxicity towards a particular organism. Then, the predicted value can be used as an input in the QTTR model to fill the data gaps for another species. The approach was used in the cases of various types of chemicals including ionic liquids and metal oxide nanoparticles.40,41
An important aspect of using networks of predictive models is possible propagation of uncertainty, along the network. This is a well-known phenomenon in statistics42 and may affect SAPNets, in which the output variables from one model are used as input variables to the next one. The importance of the uncertainty propagation in the endpoint prediction should be further investigated.
Among the challenges of building classic Nano-QSAR models, one is especially often raised by authors: the limited number of observations in the data set. The issue can be overcome by the development of new modeling algorithms dedicated to small training sets. In 2017, Gajewicz et al. presented the read-across algorithm (Nano-QRA) that can be used to fill data gaps in a quantitative manner.43 The predictions are based on interpolation and extrapolation approaches: one-point-slope, two-point formula, or the equation of a plane passing through three points to predict a particular activity for an unknown chemical(s). Such prediction models as Nano-QRA can be easily implemented as nodes in SAPNets.
As mentioned, the development of nanoinformatics tools should be in line with published guidelines and standards established by the scientific community; especially, the outcomes from projects focused on the regulations are significant for this matter. A recent contribution by Giusti et al.36 summaries existing approaches for nanomaterial grouping and provides a new approach with further recommendations. The authors suggest that risk assessment should be based on three crucial statements: “what they are”, “where they go” and “what they do”. Thus, one should gather information about (i) physico-chemical characterization of pristine materials (as synthesized); (ii) changes of properties in various conditions (system-dependent properties), toxicokinetics, and fate; (iii) physical hazards, human toxicity, and ecotoxicity. The presented “NanoReg2 approach” requires a more detailed description of nanoforms. However, the difference between descriptors (that describe the structure/morphology) of ENMs and the properties (that result from the structure) was not clearly distinguished. Thus, a precise analysis of the relationship between the structure of ENMs and their properties may be challenging. Building the structure–activity prediction networks that include various methods, such as the presented NanoReg2 approach, could be a way to extract all crucial information and point out dependencies between the structure of a NM and its properties and biological activity.
Finally, it is worth mentioning that the additional benefits of using structure–activity prediction networks are more effective incorporation of the computational safety assessment at the stage of design of new materials and better collaboration between experts focused on different aspects of research on nanomaterials. This is especially important in the context of extending the SAPNet with layers related to the synthesis stage.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The concept of the SAPNets was developed under funding that has been received from the European Union's Horizon 2020 research and innovation programme via NanoSolveIT Project under grant agreement no. 814572.Prediction of phenol degradation efficiency with the SAPNet concept and generation of the data required to perform the first case study were funded by the National Science Centre within program SONATA 8 (grant entitled: “Influence of the ionic liquid structure on interactions with TiO2 particles in ionic liquid assisted hydrothermal synthesis”), contract No. 2014/15/D/ST5/0274.
References
- I. van de Poel and Z. Robaey, Nanoethics, 2017, 11, 297–306 CrossRef.
- T. Puzyn, D. Leszczynska and J. Leszczynski, Small, 2009, 5, 2494–2509 CrossRef CAS.
- A. Afantitis, G. Melagraki, P. Isigonis, A. Tsoumanis, D. D. Varsou, E. Valsami-Jones, A. Papadiamantis, L.-J. A. Ellis, H. Sarimveis, P. Doganis, P. Karatzas, P. Tsiros, I. Liampa, V. Lobaskin, D. Greco, A. Serra, P. A. S. Kinaret, L. A. Saarimäki, R. Grafström, P. Kohonen, P. Nymark, E. Willighagen, T. Puzyn, A. Rybinska-Fryca, A. Lyubartsev, K. Alstrup Jensen, J. G. Brandenburg, S. Lofts, C. Svendsen, S. Harrison, D. Maier, K. Tamm, J. Jänes, L. Sikk, M. Dusinska, E. Longhin, E. Rundén-Pran, E. Mariussen, N. El Yamani, W. Unger, J. Radnik, A. Tropsha, Y. Cohen, J. Leszczynski, C. Ogilvie Hendren, M. Wiesner, D. Winkler, N. Suzuki, T. H. Yoon, J.-S. Choi, N. Sanabria, M. Gulumian and I. Lynch, Comput. Struct. Biotechnol. J., 2020, 18, 583–602 CrossRef CAS.
- J. Roy, P. K. Ojha and K. Roy, Nanotoxicology, 2019, 13, 701–716 CrossRef CAS.
- D. Varsou, A. Afantitis, A. Tsoumanis, A. Papadiamantis, E. Valsami-Jones, I. Lynch and G. Melagraki, Small, 2020, 16, 1906588 CrossRef CAS.
- A. P. Toropova, A. A. Toropov, A. M. Veselinović, J. B. Veselinović, D. Leszczynska and J. Leszczynski, Multi-Scale Approaches Drug Discov., 2017, pp. 191–221 Search PubMed.
- S. Ahmadi, Chemosphere, 2020, 242, 125192 CrossRef CAS.
- T. X. Trinh, J.-S. Choi, H. Jeon, H.-G. Byun, T.-H. Yoon and J. Kim, Chem. Res. Toxicol., 2018, 31, 183–190 Search PubMed.
- J.-S. Choi, T. X. Trinh, T.-H. Yoon, J. Kim and H.-G. Byun, Chemosphere, 2019, 217, 243–249 CrossRef CAS.
- K. Jafari and M. H. Fatemi, J. Therm. Anal. Calorim., 2020 DOI:10.1007/s10973-019-09215-3.
- K. Jafari and M. H. Fatemi, Adv. Powder Technol., 2020, 31, 3018–3027 CrossRef CAS.
- V. V. Kleandrova, F. Luan, H. González-Díaz, J. M. Ruso, A. Melo, A. Speck-Planche and M. N. D. S. Cordeiro, Environ. Int., 2014, 73, 288–294 CrossRef CAS.
- F. Luan, V. V. Kleandrova, H. González-Díaz, J. M. Ruso, A. Melo, A. Speck-Planche and M. N. D. S. Cordeiro, Nanoscale, 2014, 6, 10623 RSC.
- T. Puzyn, B. Rasulev, A. Gajewicz, X. Hu, T. P. Dasari, A. Michalkova, H.-M. Hwang, A. Toropov, D. Leszczynska and J. Leszczynski, Nat. Nanotechnol., 2011, 6, 175–178 CrossRef CAS.
- S. Kar, A. Gajewicz, T. Puzyn and K. Roy, Toxicol. In Vitro, 2014, 28, 600–606 CrossRef CAS.
- S. Kar, A. Gajewicz, T. Puzyn, K. Roy and J. Leszczynski, Ecotoxicol. Environ. Saf., 2014, 107, 162–169 CrossRef CAS.
- A. Rybińska-Fryca, A. Mikołajczyk, J. Łuczak, M. Paszkiewicz-Gawron, M. Paszkiewicz, A. Zaleska-Medynska and T. Puzyn, J. Colloid Interface Sci., 2020, 572, 396–407 CrossRef.
- A. Mikolajczyk, A. Malankowska, G. Nowaczyk, A. Gajewicz, S. Hirano, S. Jurga, A. Zaleska-Medynska and T. Puzyn, Environ. Sci.: Nano, 2016, 3, 1425–1435 RSC.
- G. Veréb, L. Manczinger, G. Bozsó, A. Sienkiewicz, L. Forró, K. Mogyorósi, K. Hernádi and A. Dombi, Appl. Catal., B, 2013, 129, 566–574 CrossRef.
- K. Roy, S. Kar and R. N. Das, Understanding the basics of QSAR for applications in pharmaceutical sciences and risk assessment, Elsevier Academic Press, Amsterdam, Boston, 2015 Search PubMed.
- M. D. Wilkinson, M. Dumontier, Ij. J. Aalbersberg, G. Appleton, M. Axton, A. Baak, N. Blomberg, J.-W. Boiten, L. B. da Silva Santos, P. E. Bourne, J. Bouwman, A. J. Brookes, T. Clark, M. Crosas, I. Dillo, O. Dumon, S. Edmunds, C. T. Evelo, R. Finkers, A. Gonzalez-Beltran, A. J. G. Gray, P. Groth, C. Goble, J. S. Grethe, J. Heringa, P. A. ‘t Hoen, R. Hooft, T. Kuhn, R. Kok, J. Kok, S. J. Lusher, M. E. Martone, A. Mons, A. L. Packer, B. Persson, P. Rocca-Serra, M. Roos, R. van Schaik, S.-A. Sansone, E. Schultes, T. Sengstag, T. Slater, G. Strawn, M. A. Swertz, M. Thompson, J. van der Lei, E. van Mulligen, J. Velterop, A. Waagmeester, P. Wittenburg, K. Wolstencroft, J. Zhao and B. Mons, Sci. Data, 2016, 3, 160018 CrossRef.
- S. Bonaretti and E. Willighagen, bioRxiv, 2019, 739334 Search PubMed.
- S. Kmiecik, D. Gront, M. Kolinski, L. Wieteska, A. E. Dawid and A. Kolinski, Chem. Rev., 2016, 116, 7898–7936 CrossRef CAS.
- D. Fourches, D. Pu, C. Tassa, R. Weissleder, S. Y. Shaw, R. J. Mumper and A. Tropsha, ACS Nano, 2010, 4, 5703–5712 CrossRef CAS.
- C. Kaweeteerawat, A. Ivask, R. Liu, H. Zhang, C. H. Chang, C. Low-Kam, H. Fischer, Z. Ji, S. Pokhrel, Y. Cohen, D. Telesca, J. Zink, L. Mädler, P. A. Holden, A. Nel and H. Godwin, Environ. Sci. Technol., 2015, 49, 1105–1112 CrossRef CAS.
- E. Burello, Environ. Sci.: Nano, 2015, 2, 454–462 RSC.
- H. Zhang, Z. Ji, T. Xia, H. Meng, C. Low-Kam, R. Liu, S. Pokhrel, S. Lin, X. Wang, Y.-P. Liao, M. Wang, L. Li, R. Rallo, R. Damoiseaux, D. Telesca, L. Mädler, Y. Cohen, J. I. Zink and A. E. Nel, ACS Nano, 2012, 6, 4349–4368 CrossRef CAS.
- A. Gajewicz, N. Schaeublin, B. Rasulev, S. Hussain, D. Leszczynska, T. Puzyn and J. Leszczynski, Nanotoxicology, 2015, 9, 313–325 CrossRef CAS.
- A. Mikolajczyk, N. Sizochenko, E. Mulkiewicz, A. Malankowska, B. Rasulev and T. Puzyn, Nanoscale, 2019, 11, 11808–11818 RSC.
- W. J. G. M. Peijnenburg, M. Baalousha, J. Chen, Q. Chaudry, F. Von der kammer, T. A. J. Kuhlbusch, J. Lead, C. Nickel, J. T. K. Quik, M. Renker, Z. Wang and A. A. Koelmans, Crit. Rev. Environ. Sci. Technol., 2015, 45, 2084–2134 CrossRef CAS.
- M. K. Ha, Y. J. Shim and T. H. Yoon, Environ. Sci.: Nano, 2018, 5, 446–455 RSC.
- A. Mikolajczyk, A. Gajewicz, B. Rasulev, N. Schaeublin, E. Maurer-Gardner, S. Hussain, J. Leszczynski and T. Puzyn, Chem. Mater., 2015, 27, 2400–2407 CrossRef CAS.
- A. Toropov, N. Sizochenko, A. Toropova and J. Leszczynski, Nanomaterials, 2018, 8, 243 CrossRef.
- A. P. Toropova and A. A. Toropov, Mol. Divers., 2019, 23, 403–412 CrossRef CAS.
- E. Wyrzykowska, A. Mikolajczyk, C. Sikorska and T. Puzyn, Nanotechnology, 2016, 27, 445702 CrossRef.
- A. Giusti, R. Atluri, R. Tsekovska, A. Gajewicz, M. D. Apostolova, C. L. Battistelli, E. A. J. Bleeker, C. Bossa, J. Bouillard, M. Dusinska, P. Gómez-Fernández, R. Grafström, M. Gromelski, Y. Handzhiyski, N. R. Jacobsen, P. Jantunen, K. A. Jensen, A. Mech, J. M. Navas, P. Nymark, A. G. Oomen, T. Puzyn, K. Rasmussen, C. Riebeling, I. Rodriguez-Llopis, S. Sabella, J. R. Sintes, B. Suarez-Merino, S. Tanasescu, H. Wallin and A. Haase, NanoImpact, 2019, 16, 100–182 CrossRef.
- European Commission, Commission Regulation (EU) 2018/1881 of 3 December 2018 amending Regulation (EC) No 1907/2006 of the European Parliament and of the Council on the Registration, Evaluation, Authorisation and Restriction of Chemicals (REACH) as regards Annexes I, III,VI, V, 2018 Search PubMed.
- European Chemical Agency, Guidance on information requirements and chemical safety assessment. Appendix R.6-1 for nanoforms applicable to the Guidance on QSARs and Grouping of Chemicals, 2019 Search PubMed.
- P. P. Roy, P. Banjare, S. Verma and J. Singh, Mol. Inf., 2019, 38, 1800151 CrossRef CAS.
- R. N. Das, K. Roy and P. L. Popelier, Ecotoxicol. Environ. Saf., 2015, 122, 497–520 CrossRef CAS.
- S. Kar, A. Gajewicz, K. Roy, J. Leszczynski and T. Puzyn, Ecotoxicol. Environ. Saf., 2016, 126, 238–244 CrossRef CAS.
- L. Zhao, W. Wang, A. Sedykh and H. Zhu, ACS Omega, 2017, 2, 2805–2812 CrossRef CAS.
- A. Gajewicz, K. Jagiello, M. T. D. Cronin, J. Leszczynski and T. Puzyn, Environ. Sci.: Nano, 2017, 4, 346–358 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0nr05220e |
| This journal is © The Royal Society of Chemistry 2020 |