
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Catalytic C(sp3)–F bond formation: recent achievements and pertaining challenges
Giulia
Tarantino
 and
Ceri
Hammond
*
and
Ceri
Hammond
*
Department of Chemical Engineering, Imperial College London, London, SW7 2AZ, UK. E-mail: ceri.hammond@imperial.ac.uk
First published on 21st July 2020
Abstract
Increasing demand for fluorine-containing compounds has led to a surge in the development of processes targeting the synthesis of alkyl fluorides. Catalytic methods of fluorination represent an efficient strategy to synthesise high value fluorinated compounds in a selective and efficient manner. Yet, catalytic methods of fluorination have received scant attention compared to those that do not utilise catalysis, despite the significant potential they offer in terms of improved process sustainability. In this review, we summarise the fluorination processes developed so far from a catalytic perspective, in order to highlight the achievements made to date, emphasise the pertaining challenges that continue to be faced, and hence aid the future development of more sustainable methods for the synthesis of C(sp3)–F bonds.
 Giulia Tarantino | Giulia Tarantino obtained her Master Degree in Chemical Sciences from the University of Naples Federico II (Italy) where she specialised in organometallic chemistry and later, organic synthesis. She was awarded a PhD from the Cardiff Catalysis Institute (UK), where she worked under the supervision of Dr Ceri Hammond. Her PhD was focused on the development of catalytic methodologies for C(sp3)–F bond formation with heterogeneous catalysts. She is currently a post-doctoral associate at the Department of Chemical Engineering at Imperial College London (UK). Her current research interests are selective alkane functionalization and investigating reaction mechanisms with in situ and ex situ spectroscopic techniques. |
 Ceri Hammond | Ceri Hammond obtained his PhD in 2012 after working on selective methane oxidation under the guidance of Prof. Graham J. Hutchings at Cardiff University. Following periods at ETH Zürich and Stanford University, he began his independent career at the Cardiff Catalysis Institute following the award of a Royal Society University Research Fellowship in 2015. In 2019, he was offered a tenured post at the Department of Chemical Engineering at Imperial College London, where he and his group are now based. His research group specialises in several aspects of catalysis and sustainable chemistry. |
1. Introduction to organo-fluorine chemistry
In recent decades, fluorinated molecules have rapidly emerged as essential compounds in several fields of chemistry, most notably in medicinal chemistry and materials chemistry. Their utility in these fields arises from the dramatic impact the introduction of the fluorine atom induces in organic compounds.1–8 For example, fluorine is a key element in medicinal chemistry, due to the increased levels of biological activity observed in pharmaceutically active compounds following introduction of fluorine in their backbone. Indeed, the biological, physical and chemical properties of an organic molecule can be significantly altered by the addition of fluorine. Such effects include improved lipophilicity, bioavailability, metabolic stability, and increased protein–ligand interactions.1,2 Inclusion of a radioactive isotope of fluorine, fluorine-18 (18F), also endows organic molecules with a radioactive tracer suitable for Positron Emission Tomography (PET) imaging purposes, which is used to monitor metabolic processes and aid in the identification of diseases.3,4Fluorine is also used to great effect in a variety of other chemical industries, such as the polymer industry. Indeed, the introduction of multiple C–F bonds into monomer building blocks results in the synthesis of final polymers with highly desirable properties, such as hydrophobicity, mechanical stability and low coefficients of friction.5–8 The increasing demand for fluorine in the examples noted above, alongside the use of fluorine in agrochemicals and perfluorinated solvents, has resulted in the increasing importance of fluorinated compounds in modern society.
However, several challenges mean that fluorination processes represent some of the most challenging reactions to perform in a sustainable and green manner. Yet, their importance to society makes their improvement an essential task, and has led to a surge in the development of novel selective fluorination methods.9–12
Amongst the specific challenges in the field of fluorination are those aimed at C(sp3)–F bond formation. This is an especially important synthetic challenge, given that a large fraction of high-value fluorinated compounds contain at least a single C(sp3)–F bond.4,13 Due to the importance of this challenge and the increasing number of reports focused upon it, this review focuses on recent breakthroughs made for C(sp3)–F bond synthesis. However, due to the key role that catalysis can play with respect to increasing the sustainability of a chemical process, this review places particular attention on studies that employ catalysis as a key enabling method during C(sp3)–F bond synthesis, to provide an overview of the current routes and highlight the pertaining challenges present in the field. To better emphasise the role that catalysis can play in the field of fluorination, we place particular attention on radical fluorination processes, which represent the majority of the catalytic fluorination processes developed so far, and where some key catalytic breakthroughs have already been achieved. However, the sustainability principles described herein may also be applied to fluorination reactions of a different nature, such as reductive elimination from Pd(IV) complexes14 and other general methods of fluorination, which have been extensively reviewed elsewhere.15–17 Although novel strategies for difluoromethylation and trifluoromethylation have also been subjected to an increasing growth in the past decade,17–23 the breadth of these additional reports and the prevalence of non-catalytic approaches places them outside the scope of this mini-review.
2. General methods of fluorination
Synthetic methods for introducing fluorine into organic molecules using gaseous fluorine (F2), perchloryl fluoride (FClO3) and xenon difluoride (XeF2) were discovered in the 1960s and 70s.24–27 However, prior to the late 1990s, little progress had been made regarding the improvement of these C–F bond forming methods, primarily due to safety concerns associated to the aggressive nature of these fluorinating reagents. However, an increase in the demand for fluorinated compounds has led to the search for safer fluorinating reagents that are non-explosive, less toxic and preferentially inexpensive. Whilst research is still on-going, breakthroughs have been achieved in this area, and a range of fluorinating agents of improved nature have been designed.28–30 These include fluorinating agents with nucleophilic, electrophilic and/or radical character, such as DAST, Selectfluor® (F-TEDA) and NFSI, which are able to mediate several types of fluorination reactions.31–36 The discovery of such reagents represents a milestone in fluorination chemistry, and has subsequently led to the design of a great deal of new fluorination procedures. As such, a large portion of the modern catalytic methods to synthesise C(sp3)–F bonds typically utilise these “easy-to-handle” fluorinating agents. Irrespective of the formal mechanistic aspects, some of the most common fluorinating agents are reported in Fig. 1.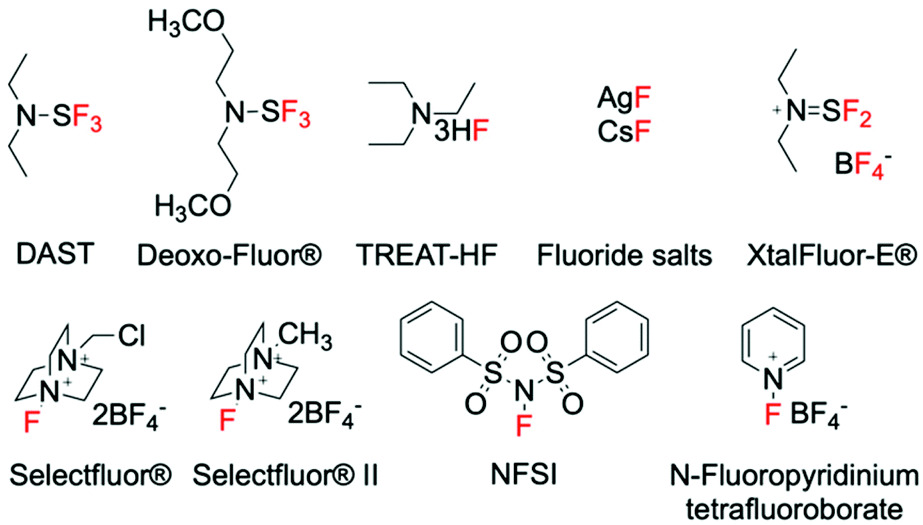 | ||
| Fig. 1 Examples of fluorinating agents. | ||
Depending from the nature of the fluorinating source, these previous studies can be divided into three main categories; nucleophilic, electrophilic, and radical fluorination.31–36 In contrast to nucleophilic reactions, which are generally accepted to involve fluoride ions (F−), electrophilic reactions are not thought to occur via the formation of formal fluoronium cations (F+), which are unlikely to be formed due to their instability and the energy required to their formation.17 For electrophilic fluorination, fluorine transfer is proposed to occur via two possible mechanisms; (i) single electron transfer (SET), or (ii) SN2 nucleophilic substitution, in which a nucleophilic substrate attacks the fluorine atom, yielding the R–F bond.17,35 Considering the ability of some electrophilic fluorinating agents to also undergo SETs in radical reactions, the difference between electrophilic and radical fluorination is a fine line. In fact, competition between the two possible reaction pathways is reportedly dependent upon several parameters, including the choice of reaction conditions, the properties of the nucleophilic substrate and the nature of the electrophilic fluorinating agent.35
2.1. Nucleophilic fluorination
Although fluoride ions (F−) may be identified as the obvious reagent to perform nucleophilic fluorinations, their employment is hampered by the scarce solubility of metal fluorides in organic solvents, and the preference of F− to form hydrogen bonds with H-donors, which undermines the nucleophilicity of F−. Moreover, the double reactivity of F−, which is able to react as both nucleophile and base, makes achieving general applicability challenging.37 Despite these issues, nucleophilic fluorination with F− is still extensively performed in medicinal chemistry for the synthesis of 18F-containing molecules, which are widely used for PET. This is due to the relatively facile synthesis of aqueous 18F−, which can be produced in high yield even from small cyclotrons.38 As such, several studies have focussed on the direct employment of 18F− for the synthesis of radiochemicals.39–42Nucleophilic fluorination involving stoichiometric amounts of safer nucleophilic agents, such as DAST, Deoxofluor, and XtalFluor-E, have also successfully been achieved in an array of non-catalytic reactions, such as the synthesis of high-value allylic fluorides from allylic alcohols (Scheme 1, entries 1–3).43–50 However, as these reagents can also mediate a range of non-fluorinating reactions, including cyclodehydration of hydroxyamides and amidation of carboxylic acids (Scheme 1, entries 4 and 5),51,52 finding the optimal conditions to achieve selective fluorination processes with these reagents is often challenging.
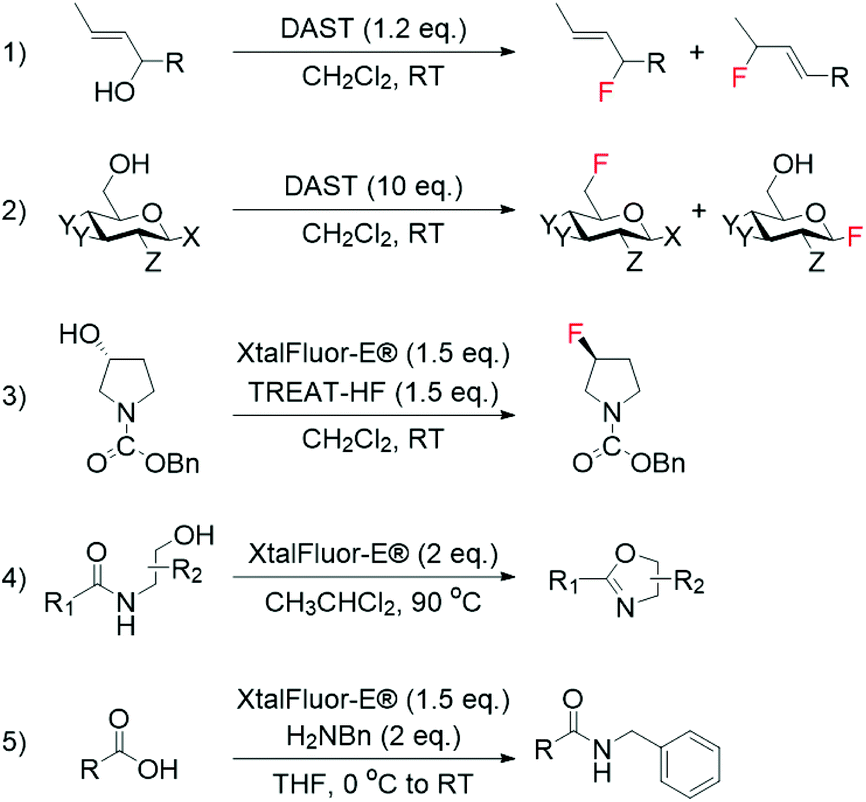 | ||
| Scheme 1 Examples of fluorination reactions employing DAST and XtalFluor-E® as fluorinating reagent: (1) synthesis of allylic fluorides from allylic alcohols,44,46 (2) conversion of sugars to glycosylfluorides,50 and (3) asymmetric deoxyfluorination of alcohols.47 Non-fluorinating reactions mediated by DAST and XtalFluor-E®: (4) cyclodehydration of hydroxyamides,51 and (5) amidation of carboxylic acids.52 | ||
Recently, the Doyle group reported the employment of algorithms to model the reaction outcome i.e. yield of alkyl fluoride, of a series of deoxyfluorination reactions using sulfonyl fluorides with various scaffolds as fluorine sources and different classes of alcohols as substrates (Scheme 2).53 Following tuning of reagent and base, efficient synthesis of a range of alkyl fluorides was successfully predicted, with errors in the range of 7.4–23% calculated through comparison with observed experimental values. This “design of experiment” approach, as opposed to the classical trial and error method, may represent a valid strategy to find the optimal reaction conditions of such processes e.g. amount of base, solvent, temperature, amount and type of fluorinating reagent, in a more efficient manner.
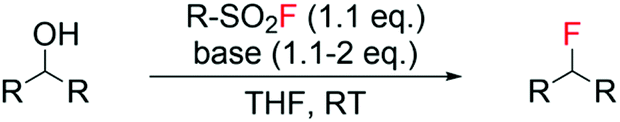 | ||
| Scheme 2 Reaction scheme for deoxyfluorination of alcohols with sulfonyl fluorides.53 | ||
Despite these breakthroughs in stoichiometric nucleophilic fluorination, this strategy is limited to pre-activated substrates, e.g. allylic alcohols, and the functionalisation of less active substrates is generally not possible in the absence of a catalyst.17 Therefore, nucleophilic fluorinations catalysed by transition metals have been investigated in recent years, with interesting results achieved for the synthesis of both C(sp2)–F and C(sp3)–F bonds.37,54,55 Details on the promising catalytic systems developed for the synthesis of simple C–F bonds are provided throughout sections 5–8.
2.2. Electrophilic fluorination
The development of modern electrophilic fluorinating agents, such as Selectfluor® and NFSI,56 has also opened a whole host of research opportunities regarding the selective fluorination of organic compounds.57–61 Amongst these, it has been shown that Selectfluor®, is able to fluorinate a wide range of substrates including aromatics, alkenes and enamines (Scheme 3).58,59 Interesting studies have also shown the successful synthesis of labelled electrophilic fluorinating agents, such as [18F]fluoro-benziodoxole and [18F]NFSI, for radiochemical applications.62,63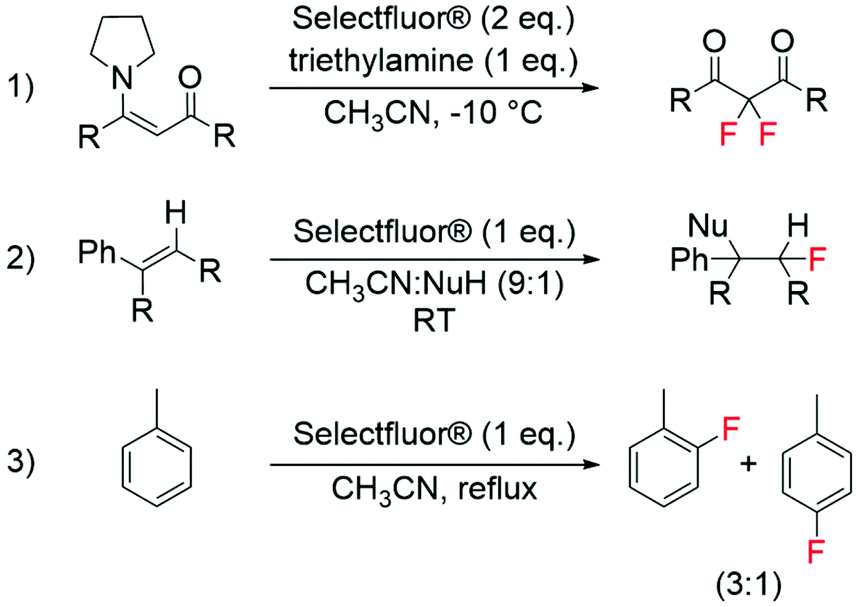 | ||
| Scheme 3 Examples of electrophilic fluorination reactions using Selectfluor®: (1) synthesis of difluorinated carbonyl compounds from enamines,58 (2) fluorination of alkenes,59 and (3) synthesis of aryl fluorides from aromatic compounds.59 | ||
Unfortunately, such stoichiometric, electrophilic fluorinations are only reported for sp2 carbon atoms, and they are not suitable for selective C(sp3)–F bond formation when using the fluorinating reagent alone, i.e. in the absence of a catalyst. For these reasons, the design of catalysts able to perform fluorination reactions in combination with electrophilic fluorinating reagents such as Selectfluor®, is an important challenge (see sections 5–8 for details).
2.3. Radical fluorination
Like several other radical processes, radical fluorination pathways occur rapidly and often proceed with low selectivity and poor reaction control, due to the high reactivity of the radical species in question. Whilst such reactions have long been considered uncontrollable, a small number of examples using F2 and XeF2 have proven to be applicable on a large scale, employing unsaturated hydrocarbons (e.g. olefins and substituted aromatic compounds) or carboxylic acids as substrate.24–26,64 Furthermore, in 2012, the potential of some electrophilic fluorinating agents (NFSI and Selectfluor®) to act as radical fluorine transfer reagents was reported by Rueda-Becerril et al. (Scheme 4, entry 1).65 Their ability to perform in such processes (Scheme 4) arises from the relatively low bond dissociation energies of N–F bonds (63.4 kcal mol−1 and 64 kcal mol−1 for NFSI and Selectfluor®, respectively),66 which are low enough to allow reaction with most alkyl radicals via SET (Scheme 5). These findings have inspired new lines of research, focused on the development of catalysts able to promote the formation of radicals from different types of substrates e.g. carboxylic acids, which are then capable of undergoing SET with the required fluorinating agent (sections 5–8 for details).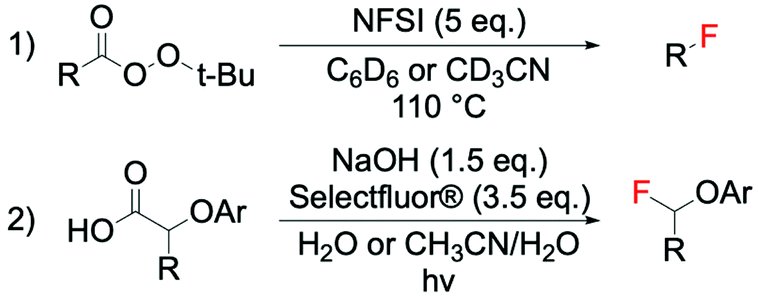 | ||
| Scheme 4 Examples of radical fluorinations using electrophilic fluorinating agents containing N–F bonds: (1) fluorination of tert-butyl peresters to form C(sp3)–F bonds,65 and (2) photo-decarboxylative fluorination of 2-aryloxy and 2-aryl carboxylic acids.67 | ||
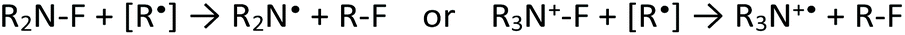 | ||
| Scheme 5 SET process between alkyl radical species and fluorinating agent yielding an R–F species. | ||
Despite all of the progress made in the last decades, a large portion of the fluorination methods reported in the literature are not catalytic,17,47–50,53,57–60,65,68 and could benefit from the use of catalytic technologies. Indeed, catalytic methods provide a number of additional benefits from both a productivity and sustainability standpoint, and may represent a leap forward in the development of more sustainable fluorination routes.
3. Towards more sustainable synthetic processes
During the last decade, the growing knowledge of fluorine science, in addition to the key role played by fluorine-containing compounds in several industrial sectors, have led to continuous search for fluorinating methodologies characterised by improved values of atom economy and E factor (kg waste per kg product), which are key parameters to evaluate the sustainability of a process.69,70 In this context, E factors up to 100 are commonplace in the pharmaceutical industry, where approximately 20–25% of synthetic drugs contain at least one C–F moiety.4,69,71 In contrast, E factors up to 50 are routinely encountered in the agrochemical sector, in which fluorine-containing molecules also represents around 25% of the total number of agrochemicals.72 In addition to producing large amounts of waste, a significant portion of these processes also involve the use of toxic and/or hazardous species, which further undermine their sustainability and scalability.In addition to the substitution of hazardous compounds with safer ones when possible, several other strategies can also be followed to improve the sustainability of current fluorination processes. These include; (i) continuous operation,73–75 (ii) new enabling technologies e.g. mechanochemistry and photochemistry,76–80 (iii) process integration,73,81 and (iv) catalysis.82–84 Amongst these, catalysis is perhaps one of the most influential. In fact, being by definition “a substance that accelerates the rate of a chemical process without itself being consumed”, the employment of small amounts of catalyst can dramatically improve the productivity of a process. Moreover, catalytic routes can also provide increased levels of selectivity toward the desired products, thus reducing the need for additional purification and separation steps, thereby minimising waste and the unnecessary loss of expensive precursor molecules. Therefore, combining fluorination chemistry with catalysis, and potentially with other techniques such as photochemistry or continuous operation, represents an important approach to synthesise high value fluorinated compounds in a more sustainable and scalable way.
A cutting-edge example showing the positive impact of this approach in the pharmaceutical field was reported by Britton and DiRocco.85 They reported a photocatalytic flow system for preparative scale (>45 g) synthesis of an important intermediate used for the synthesis of Odacanatib, which is a drug employed for the treatment of osteoporosis (Scheme 6). Interestingly, the utilisation of a W-based photocatalyst allowed one-pot synthesis of γ-fluoroleucine methyl ester via direct fluorination of leucine methyl ester, substantially increasing the efficiency and minimising the amounts of produced waste, when compared to previous processes.86,87
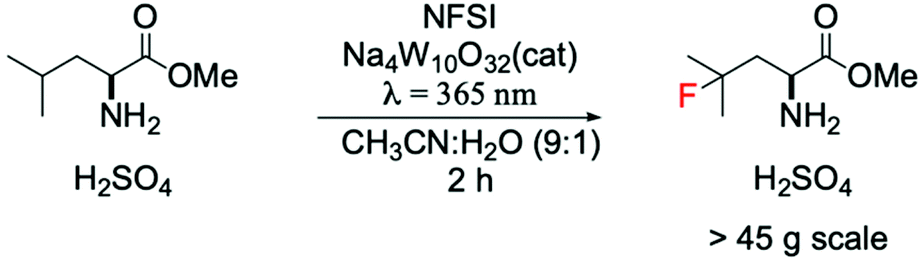 | ||
| Scheme 6 Preparative scale fluorination of leucine methyl ester in a photocatalytic flow system.85 | ||
Due to the key role that catalysis can play with respect to increasing sustainability, this review will focus on catalytic strategies to form C(sp3)–F bonds, providing an overview of the current routes and highlighting the pertaining challenges present in the field. However, to correctly evaluate and compare different catalytic strategies, it is important to first properly assess the catalytic performances of the systems. Yet, this is a challenging task, as several factors require consideration. In this context, although a significant portion of reports present in this field generally describe the performance of a catalyst merely in terms of product yields following an extended period of reaction, other parameters should always be considered and reported. Therefore, the following section will give an introduction of the key parameters required to correctly assess the catalytic performance of a catalyst, particularly in the context of catalytic fluorination.
4. Fundamental principles of catalysis: performance indicators
To design a successful catalyst, its general performance can be evaluated through the employment of the following indicators; (i) activity, (ii) selectivity, (iii) lifetime, and (iv) other parameters such as the cost and industrial applicability of the process.88One of the key performance indicators is the activity of a catalyst, i.e. the amount of product produced per unit time and per unit of catalyst. To describe the catalytic performance of a catalyst, a large percentage of papers report activity in terms of percentage yield of the desired product after extended reaction periods (maximal yield). Although yield – particularly isolated yield – is an essential parameter that must be determined in such experiments, the yield value alone does not provide a complete overview regarding the sustainability of a process. For example, maximal yield values do not provide any information about; (i) the amount of substrate converted at a given unit time, and (ii) the selectivity of the whole process (eqn (1)–(3)). As such, they do not provide important information on the rate or the efficiency of a process, both of which are essential towards determining the green potential of a new process. Hence, yield values should be accompanied by conversion and selectivity values, and should preferentially be recorded at various periods of time, so that a time course of the reaction can be generated.
Nevertheless, both conversion and yield are arbitrary values that depend on the concentration of the reagents and the quantity of catalyst used, and as such, are only related to the reaction conditions employed. For example, a reaction carried out in a solvent at low substrate concentration can give high levels of conversion and yield but may produce far less quantity of product than a solvent-free experiment run at low conversion. Therefore, more rigorous evaluation of catalytic activity can be made by comparing activity in terms of turnover frequency numbers (TOF), which is a parameter indicating the intrinsic activity of a catalyst, expressed in terms of amount of substrate converted per amount of active element of catalyst per unit of time. Notably, measurement of the TOF should be performed during the early stage of the reaction i.e. in the linearity range of a plot of conversion versus time, to minimise the impact of catalyst deactivation on rate evaluation.
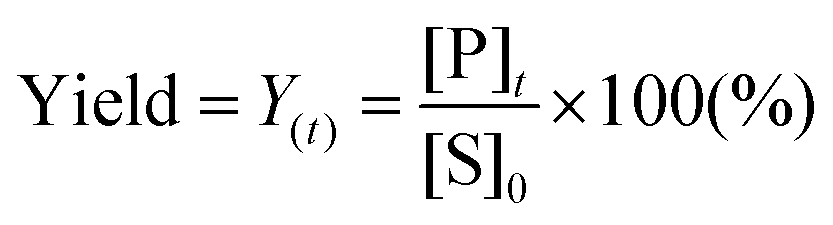 | (1) |
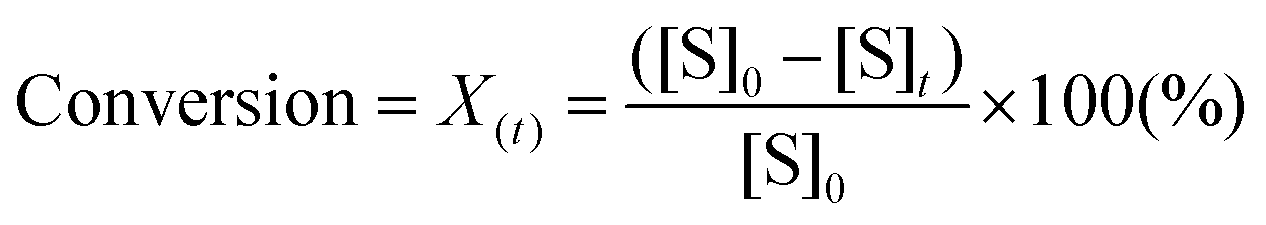 | (2) |
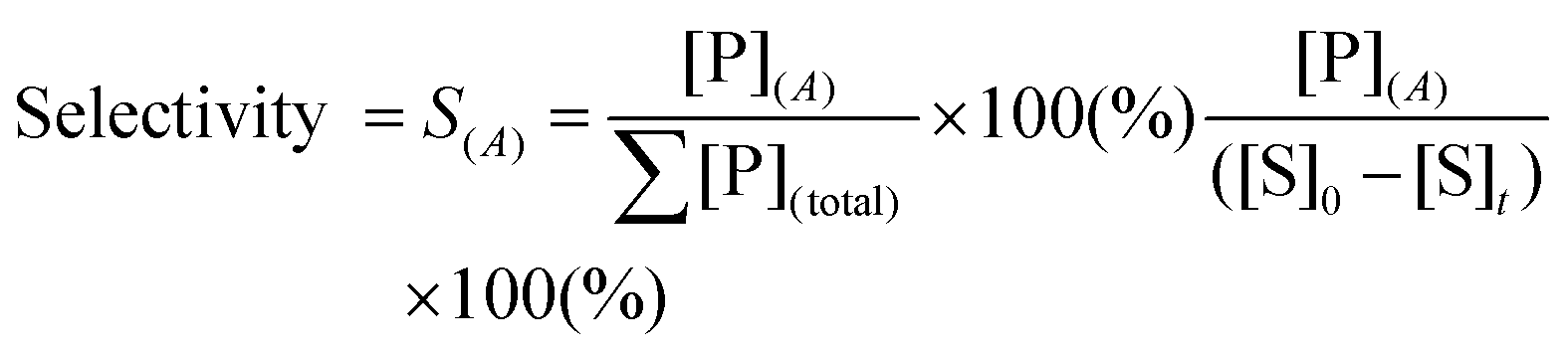 | (3) |
| TOF = moles(substrate converted) moles−1(active element) h−1 | (4) |
Although substrate conversion and product yield are important aspects of catalytic performance of a catalyst, its selectivity is often the performance indicator that best defines its real industrial applicability. Firstly, low selectivity implies large loss of substrate, which is particularly problematic in sectors that use expensive substrate molecules produced in complex, multi-step procedures. Secondly, poor selectivity results in added downstream separation steps, often leading to higher financial and energetic cost, and, therefore, more expensive processes. Hence, several industrial processes employ sub-maximal values of substrate conversion to ensure good selectivity, eventually reusing non-reacted substrate via recycling loops.89
An additional performance indicator is the lifetime of the catalyst. The capability of a catalyst to be efficiently recovered and reused, or continuously used, without losing its overall performance over a certain period of time, is extremely important as it dramatically affects both sustainability and economic viability of a process. A measure of lifetime can be expressed in terms of turnover number (TON), which defines the number of catalytic cycles catalysed by each active element before its deactivation, i.e. its loss of catalytic performance. As such, the TON should be measured at the very end of a reaction, when no more molecules of substrate are converted into the desired product even when unconverted substrate is available in the reaction mixture.
| TON = total moles(substrate converted) moles−1(active element) | (5) |
Despite being important to define the commercial applicability of a catalyst, stability is often ignored or underestimated,90 especially in homogeneous catalysis, where the use of homogeneous metal complexes often results in difficult recovery and reusability procedures.91
In addition to the key performance indicators previously described, other parameters also need to be considered. These include; (i) overall sustainability of the process expressed in terms of toxicity of the chemicals, atom economy and E factor,69,70 (ii) simplicity of catalyst recovery and reuse, when continuous operational mode is not possible, (iii) cost of the catalyst and its preparation, and (iv) scalability. Amongst these, although often overlooked, catalyst preparation also plays an important role, especially in homogeneous catalysis where metal complexes are often subjected to complex synthetic procedures. Indeed, scalability, sustainability and overall industrial feasibility of a process are strongly affected by the availability of the catalyst and its synthetic procedures.92
5. Strategies to catalytically form new C(sp3)–F bonds
Amongst the strategies to catalytically synthesise new C(sp3)–F bonds, two major distinctions can be made: (i) fluorination of pre-activated substrates, and (ii) direct C–H fluorination. Regarding the first route, carboxylic groups have been identified as the most promising class of substrate, mainly due to their low toxicity and large abundance. The second, more elegant route, involves the direct conversion of C–H bonds into C–F bonds. However, whilst many breakthroughs have been achieved for direct allylic and benzylic fluorination,17,37,54,93 reports focused on the direct fluorination from inactivated C(sp3)–H bonds are still scarce, with only a few examples being reported. In light of this, the following sections will focus on the most pursued catalytic routes; (i) decarboxylative fluorination of aliphatic carboxylic acids, (ii) direct benzylic fluorination, and (iii) direct alkane fluorination.6. Decarboxylative fluorination of aliphatic carboxylic acids
Catalytic methods to perform decarboxylative fluorination of aliphatic carboxylic acids may be divided in two main categories; (i) catalytic processes catalysed by transition metals such as Ag and Mn (section 6.1), and (ii) photocatalytic reactions (section 6.2). Notably, the amount of catalyst employed in each process is reported by convention as molar% (mol%) calculated as (moles(active element)/moles(substrate)) × 100, whilst the amount of other reactants is reported as molar equivalents (eq.), where 1 eq. represents the initial moles of substrate. General lack of information on substrate conversion does not allow for accurate calculation of TOF and TON values in many of the reported systems. As such, for comparison of the different catalytic strategies, approximate TON and TOF values have been calculated using reported yields of fluorinated products.6.1. Catalytic radical decarboxylative fluorination
The first example of decarboxylative fluorination with easy to handle fluorinating agents was published in 2012 by Yin et al.,94 who reported a novel and chemo-selective route for aliphatic decarboxylative fluorination using Selectfluor® as fluorinating reagent and the homogeneous silver nitrate salt AgNO3 as catalyst (Scheme 7). To demonstrate the efficiency and the versatility of this system, the authors reported good yields (from 40 to 95%) of fluorinated products from a broad array of carboxylic acids, at relatively mild reaction conditions.94 Whilst the radical nature of this reaction has since been widely accepted by the scientific community, the precise molecular-level mechanism continues to be the subject of much debate.94–96 Indeed, although Yin and colleagues94 hypothesised formation of the C–F bond occurring via SET between an alkyl radical species and a ligand–Ag–F complex, the most accepted mechanism involves SET between the radical species and Selectfluor® itself.95,96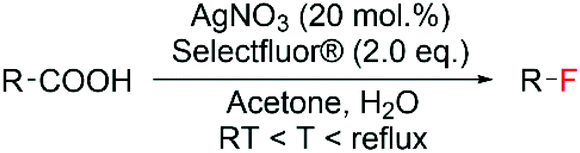 | ||
| Scheme 7 Reaction scheme for Ag-catalysed decarboxylative fluorination.94 | ||
Building on these findings, a few studies have focussed on the development of Ag-containing heterogeneous catalysts to perform this reaction (see section 8 for further details). For example, we found that silver oxides, AgxO, supported on TiO2 are active, stable and reusable catalysts for decarboxylative fluorination of aliphatic carboxylic acids.97 In parallel with our studies, Nguyen et al. reported that delafossite AgFeO2 nanoparticles can also catalyse decarboxylative fluorination, although higher amounts of catalyst (20 mol%) and Selectfluor® (2–2.5 eq.) were required when compared to the AgxO/TiO2 system (see section 8 for further details).98
In addition to the Ag-catalysed route, metalloporphyrins have also been reported to be efficient catalysts for decarboxylative fluorination reactions (Scheme 8). This route, recently discovered by Huang et al.,41 allowed targeted C–18F bond formation to be achieved, although only to moderate levels of yield whilst requiring strongly oxidising conditions.
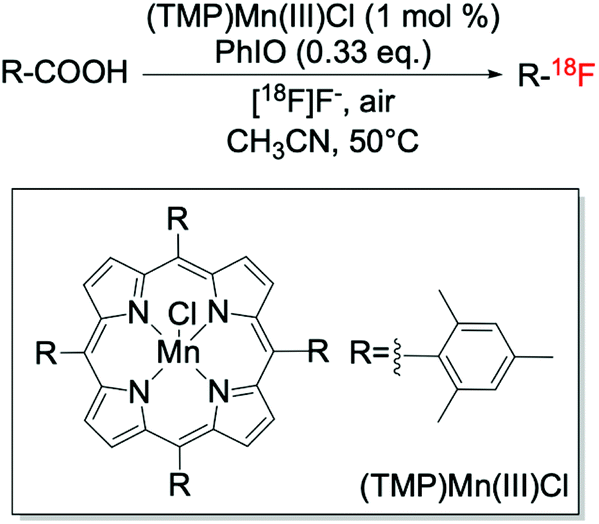 | ||
| Scheme 8 Targeted fluorination via decarboxylative fluorination catalysed by (TMP)Mn(III)Cl.41 | ||
Notably, in all these reports, only moderate numbers of TONs were observed, with the highest value of 34.2 achieved using AgxO supported on TiO2.97
6.2. Radical decarboxylative fluorination activated via photocatalysis
Recent studies have identified photocatalysis as an alternative way to form C(sp3)–F bonds via decarboxylative fluorination using Selectfluor® as fluorine donor. In these processes, a combination of light and photocatalyst is typically employed to remove the carboxylate function of the substrate as CO2, thus, generating the alkyl radical species required for SET-based fluorination.In contrast to classical decarboxylative fluorination, where TONs < 35 have been observed, TONs up to 1960 and TOFs up to 653 h−1 have been achieved for systems photocatalysed by homogeneous Ir and Ru complexes (Scheme 9, entry 1).99,100 Notably, the formation of C(sp3)–F bonds in such short time and with such high TONs is especially important for PET applications, where rapid formation of C(sp3)–18F bonds is essential due to the 109.77 min half-life of 18F, which means that synthesis of radioactive compound and administration to the patient has to occur in less than two hours. Photocatalytic decarboxylative was also achieved by use of organic dyes,101 which in addition to avoiding scarce metals, also required less Selectfluor® (2.0 eq. instead of 3.0 eq.) (Scheme 9, entry 2). However, although providing higher atom economy, lower TONs (up to 20) were reached using organic dyes than homogeneous photocatalysts. Moreover, in contrast to the broad range of substrates converted by the Ag-catalysed routes, several substrate limitations are associated with the employment of organic dyes. For example, poor activity has been reported for decarboxylative fluorination of primary aliphatic carboxylic acids, with only moderate yields achieved in the presence of heteroatoms in the proximity of the carboxylic group. Although some reaction mechanisms have been proposed for this class of reaction, which suggest SET between the Selectfluor® and alkyl radical species formed in situ,100,101 the reaction mechanisms that are predominating during such processes have not yet been clearly identified.
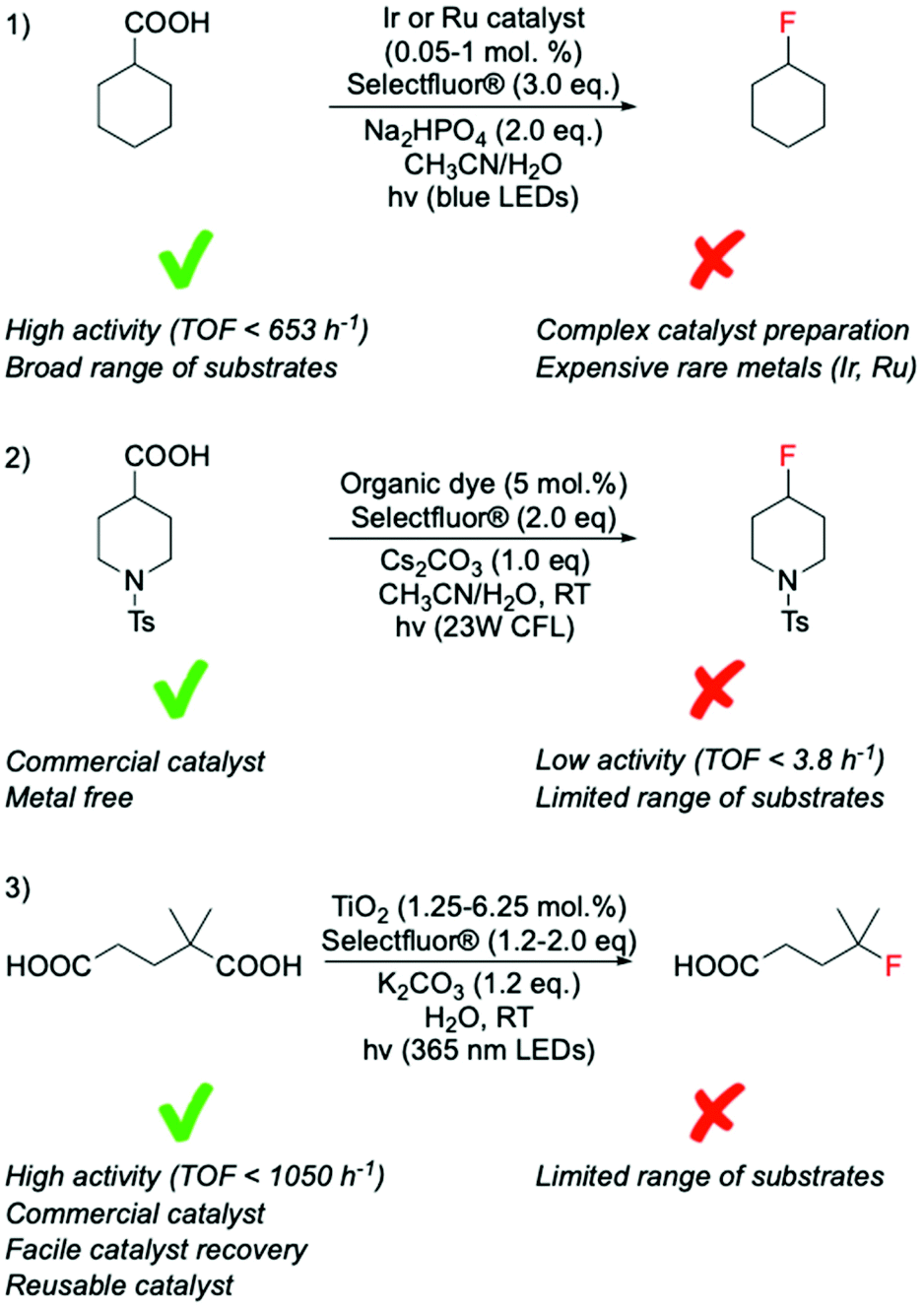 | ||
| Scheme 9 Comparison of photocatalytic fluorination of carboxylic acids mediated by catalysts of different nature; (1) transition metal complexes,99,100 (2) organic dyes,101 and (3) heterogenous catalysts.102 CLF = compact fluorescent lamp, LED = light-emitting diode. | ||
Along these lines, we recently found that TiO2, a commercially available semiconductor, was able to perform decarboxylative fluorination with TOF up to 1050 h−1, under irradiation with a solar light simulator or a LED torch (365 nm wavelength) (Scheme 9, entry 3).102 Notably, the employment of a relatively inexpensive heterogeneous catalyst, with high intrinsic activity, good durability and easily recoverability from the reaction mixture, substantially increases the sustainability of this process (see section 8 for further detail).
7. Direct C(sp3)–H bonds fluorination
Despite the promising results reported for the conversion of carboxylic acids to alkyl fluorides, direct functionalisation of C(sp3)–H to yield the corresponding C(sp3)–F species is the most ideal and desirable target. This is mostly due to (i) the broad range of easily available substrates and (ii) the avoidance of unnecessary pre-activation steps, resulting in processes with shorter numbers of synthetic steps for the preparation of fluorinated compounds, and hence lower overall E-factors. Therefore, the following sections will focus on direct C(sp3)–H fluorinations from (i) benzylic substrates, which possess weaker C–H bonds (Bond-Dissociation Energy (BDE) values ∼85–90 kcal mol−1), and (ii) alkanes, with stronger C–H bonds (BDEs > −95 kcal mol−1).7.1. Benzylic fluorination
Amongst the different strategies to perform direct C(sp3)–H fluorination using activated substrates, benzylic fluorination represents one of the most explored routes. Indeed, it represents a good model system for selective direct C(sp3)–H fluorination, due to the activated benzylic C–H bonds requiring milder reaction conditions for activation. This route is also particularly desirable in medicinal chemistry as it represents a useful tool for selective insertion of F and 18F in bioactive compounds.103The first example of catalytic benzylic fluorination was reported by Hull et al.93 in 2006, employing Pd(OAc)2 as a catalyst and an electrophilic fluorine source, 2,4,6-Me3-NFPy·BF4 (Scheme 10, entry 1). However, only in recent years has benzylic fluorination started to gain increased attention, proving to be a powerful tool for site-specific C(sp3)–F bond formation.93,104–114 Generally, amongst these studies, different fluorination routes can be identified: (i) electrophilic fluorination employing Selectfluor®, N-fluoropiridinium salts and NFSI as fluorinating sources,93,104–107 (ii) nucleophilic fluorination with triethylamine trihydrofluoride, TREAT·HF, and fluoride salts,108,109 and (iii) thermal- or photocatalysed radical fluorination.110–113 Amongst these, benzylic fluorination catalysed by transition metal complexes using electrophilic fluorinating reagents represents the most exploited (see examples in Scheme 10, entries 1–3).93,104–107
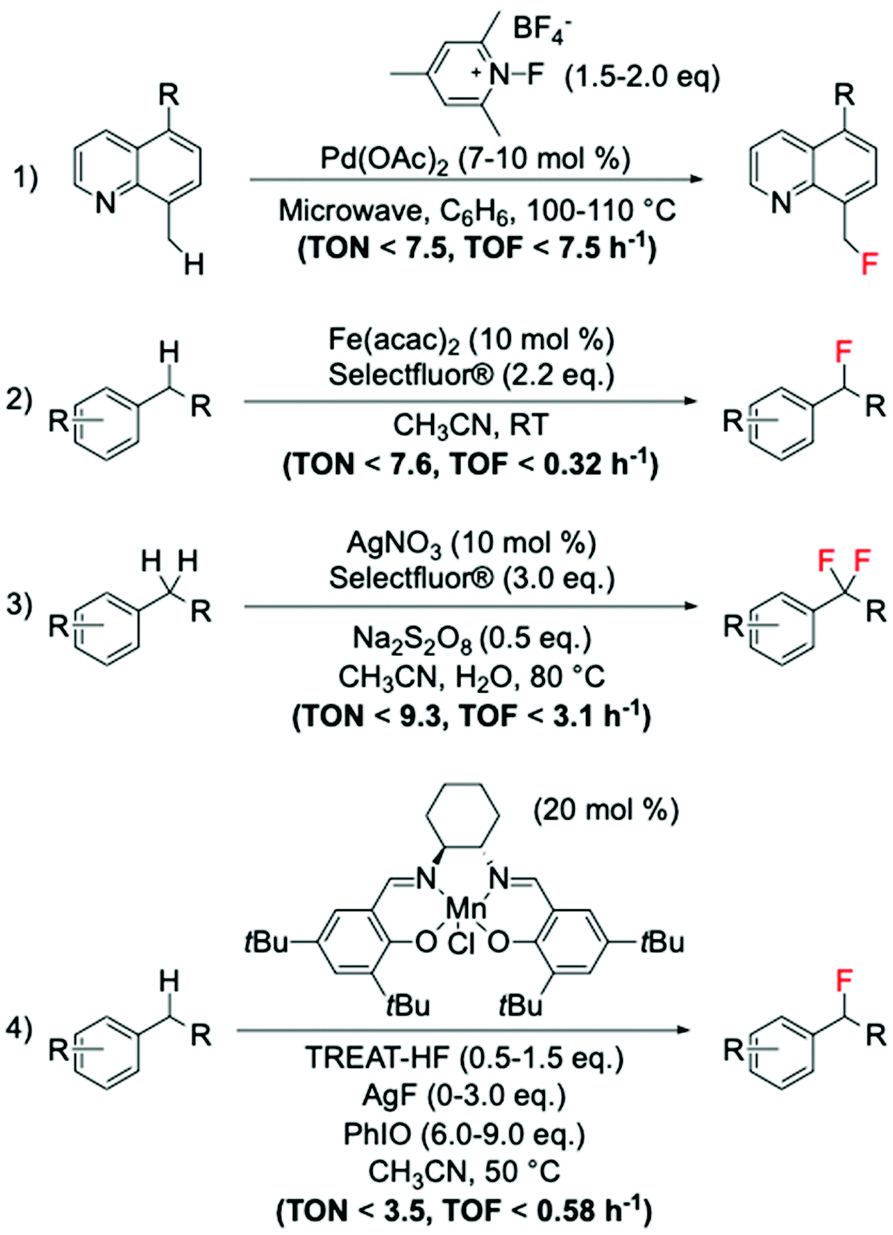 | ||
| Scheme 10 Examples of benzylic fluorinations reported by: (1) Sanford group,93 (2) Lectka group,104,106 (3) Tang group,105 and (4) Groves group.108,109 | ||
Despite the difficulties associated with the activation of C–H bonds, which may require stronger reaction conditions i.e. more powerful oxidants or radical initiators, than those typically used for pre-activated substrates such as carboxylic acids, moderate TONs (up to 10) have been achieved via this route.
Along with this route, the suitability of nucleophilic fluorinating reagents has also been investigated and reported. One of the most promising studies involves the employment of Mn(salen) complexes, which have been reported by the Groves group to be active catalysts for benzylic fluorinations using nucleophilic fluorine sources, such as TREAT·HF and AgF (Scheme 10, entry 4).108,109 Unfortunately, when nucleophilic sources are employed for benzylic fluorination with Mn complexes (including the Mn porphyrin, TMPMn(III)Cl, Scheme 8), the formation of oxidised by-products is observed, suggesting competition between fluorination and oxidation.108
As previously reported in section 2.3, electrophilic fluorinating reagents can also undergo to SET with radicals, through a radical fluorination mechanism. Therefore, several strategies have been developed based on the formation of radicals from various free radical initiator species. In 2013, Amaoka et al.113 reported a metal-free benzylic fluorination route catalysed by N-oxyl radicals thermally formed by N,N-dihydroxypyromellitimide (NDHPI, Scheme 11, entry 1). This system exhibits higher efficiency than those reported in Scheme 10, with TONs up to 34.4 reported.
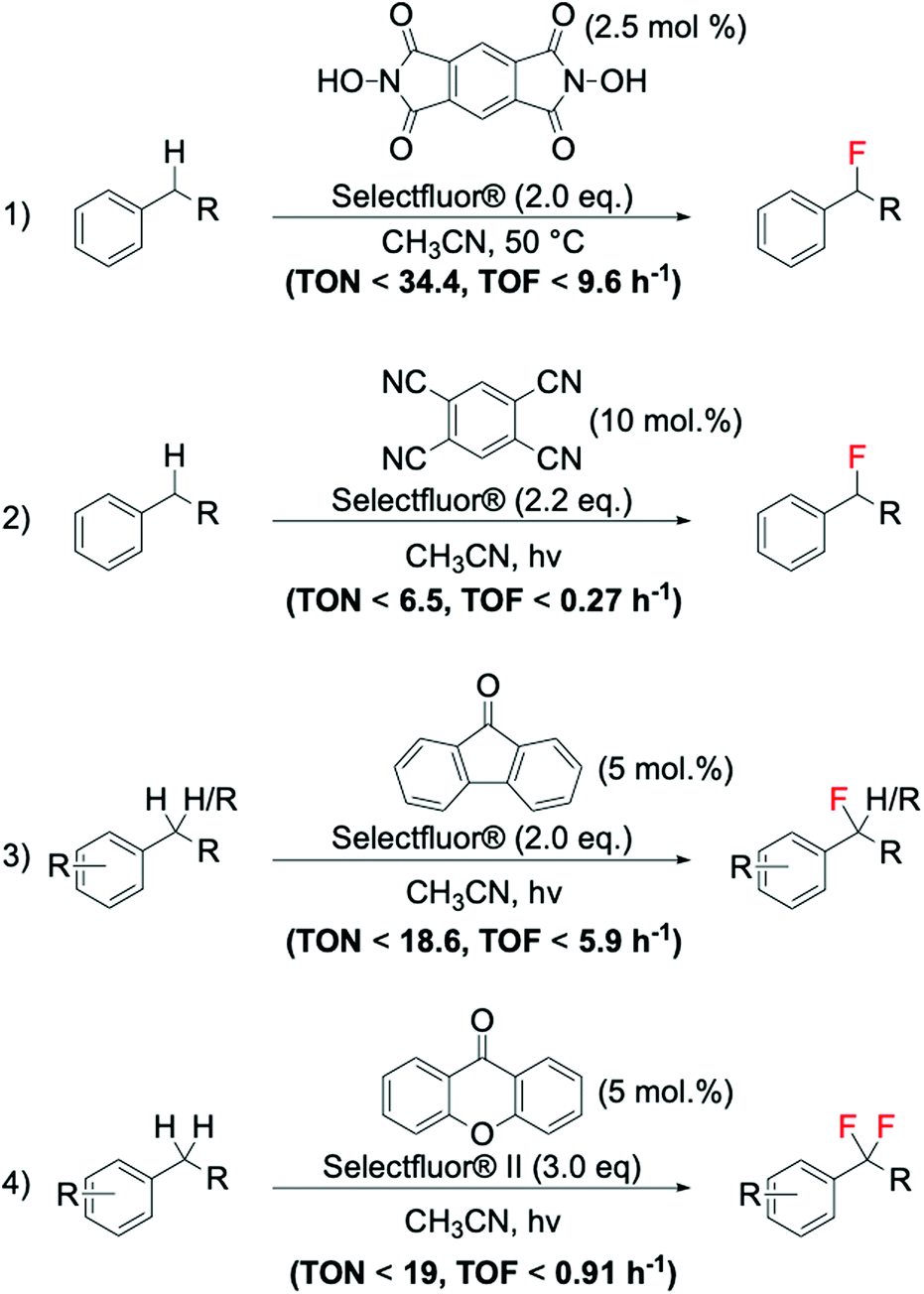 | ||
| Scheme 11 Examples of benzylic fluorinations reported by: (1) Inoue group,113 (2) Lectka group,111 and (3–4) Chen group.110 | ||
In contrast to the thermally activated routes, novel routes involving the employment of light as radical initiator have also been successfully reported for benzylic fluorination. Noteworthy examples of photocatalytic benzylic fluorination are the system developed by Bloom et al. (Scheme 11, entry 2),111 and the system reported by Xia et al. (Scheme 11, entries 3 and 4),110 employing 9-fluorenone as photosensitive catalyst.
Considering the high value of fluorinated compounds, and, thus, the need to maximise their production, outstanding breakthroughs have been achieved in the field of flow chemistry, with some of the previously described systems performed in flow and microflow reactors. Not only does moving from batch to continuous flow typically represent an improvement from a productivity perspective, yielding to higher amounts of benzyl fluoride produced per time and volume, but it also benefits processes from a safety standpoint, by utilisation of smaller reactor volumes.115–118 In this context, Cantillo et al. reported an efficient way to achieve benzylic fluorination in flow photo-reactors, irradiating the reactor with a black-light household compact fluorescent lamp (CFL).112 Using xanthone as organic catalyst and Selectfluor® as fluorinating agent, efficient synthesis of a range of benzylic fluorides was achieved at photocatalytic conditions.112
Although representing a significant breakthrough, it should be added that continuous flow systems can suffer from limited light penetration depth, which typically results in the employment of reaction solutions with low catalyst concentrations, limiting productivity and scalability. Moreover, continuous systems employing homogeneous catalysts do not easily permit long term use or recycle of the soluble catalysts. In fact, the potential of heterogeneous catalysts to operate continuously and be readily recovered post reaction makes them more suitable catalysts for flow applications.
7.2. Direct alkane fluorination
Although challenging to achieve because of the high energy required to cleave C–H bonds, which are characterised by pKa values greater than 50 and bond dissociation energies larger than 95 kcal mol−1,118 a few examples of direct alkane fluorination have been reported in the literature. Amongst these, three different strategies can be identified: (i) a biomimetic approach, involving H-atom abstraction with high valent transition metals, (ii) radical mechanisms involving the use of radical initiators, and (iii) an ion pairing hydrogen bonding mechanism.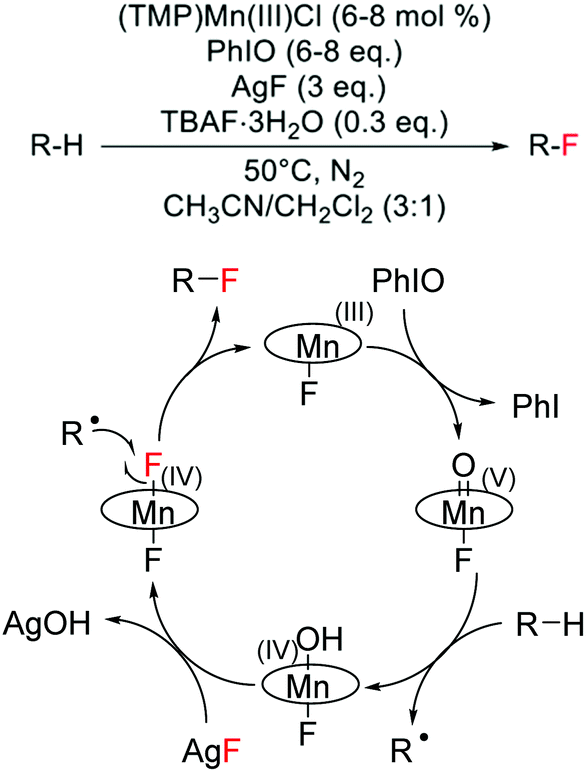 | ||
| Scheme 12 Reaction scheme and proposed reaction mechanism for direct alkane fluorination catalysed by a Mn(III)-porphyrin.119 Reaction scheme adapted from ref. 119. | ||
The unique reactivity of Mn-porphyrins, which are able to achieve selective alkane hydroxylation or selective alkane fluorination under specific reaction conditions, is of great scientific interest. However, additional mechanistic studies are required to fully elucidate the reasons behind the tuneable reactivity of some Mn-porphyrins to yield C(sp3)–F bonds, including C(sp3)–18F bonds, in place of C(sp3)–O bonds.
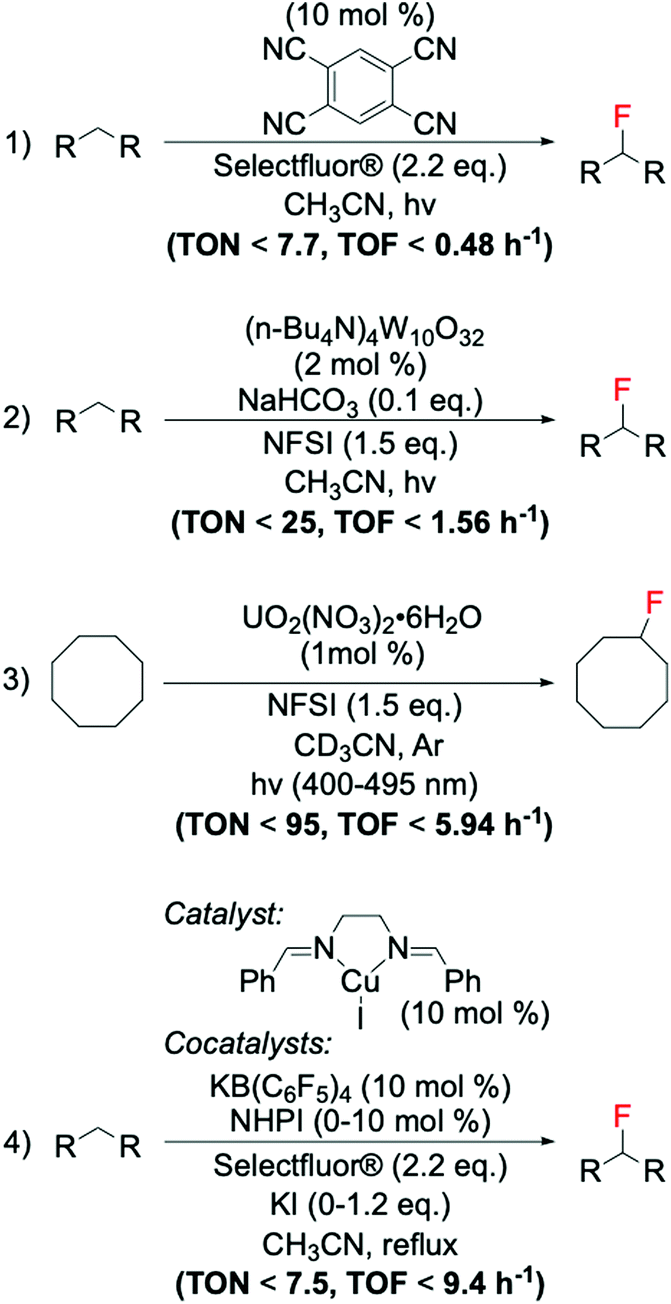
8. Fluorination reactions catalysed by heterogeneous catalysts
Despite the great number of studies focussed on the development of new strategies for the formation of C(sp3)–F bonds, the catalytic capabilities of heterogeneous catalysts have barely been explored. This is despite the potential advantages they offer over homogeneous analogues, such as facile recovery and reuse, both of which are desirable for process intensification and for process economy and environmental impact. Therefore, heterogeneously catalysed fluorination processes represent an attractive challenge for both academic and industrial researchers.The scarce effort to design heterogeneous catalysts for fluorination reactions is mainly attributed to the difficulty of stabilising solid materials at the conditions required for fluorination, which is essential to ensure good catalytic lifetime (see section 4 for further details). In fact, the highly acidic and/or oxidative reaction conditions typically involved in the systems reported so far represent an incredible challenge from a stability perspective. Another major challenge includes the presence of hydrogen fluoride (HF), generated as a by-product in these systems.129 Each of these factors can easily induce restructuring, leaching and thus, deactivation, of solid catalysts.88,133,134 Amongst them, metal leaching is especially undesirable as it leads to the dispersion of metal into the environment, and irreversible deactivation of the catalyst.88,133,134 Traces of metals in the reaction mixture may also behave as homogeneous catalysts and contribute to the reaction kinetics in a non-controlled manner.88
Despite these challenges, recent reports have demonstrated that heterogeneous materials can efficiently be employed for fluorination reactions. Following the findings of Yin et al., who identified the Ag catalysed decarboxylative fluorination of aliphatic carboxylic acids,94 different studies have been conducted on the design of Ag containing heterogeneous catalyst for this reaction. Notably, although unsupported Ag2O has been shown to be a very active material for this reaction, Ag2O does not behave as a stable heterogeneous material. In fact, when employed for decarboxylative fluorinations, we detected a dramatic percentage of Ag dissolving into solution, resulting in a high homogeneous contribution to the reaction rate.97 However, supported Ag catalysts, such as AgFeO2 and mixed Ag oxides supported on TiO2 (AgxO/TiO2), have been found to be efficient, stable and reusable materials for catalytic C(sp3)–F bond formation via decarboxylative fluorination of aliphatic carboxylic acids (Scheme 14, entries 1 and 2). Notably, using mechanochemically prepared AgxO/TiO2 results in higher TONs and TOFs being achieved compared to AgFeO2, even when using less Selectfluor®.97,98
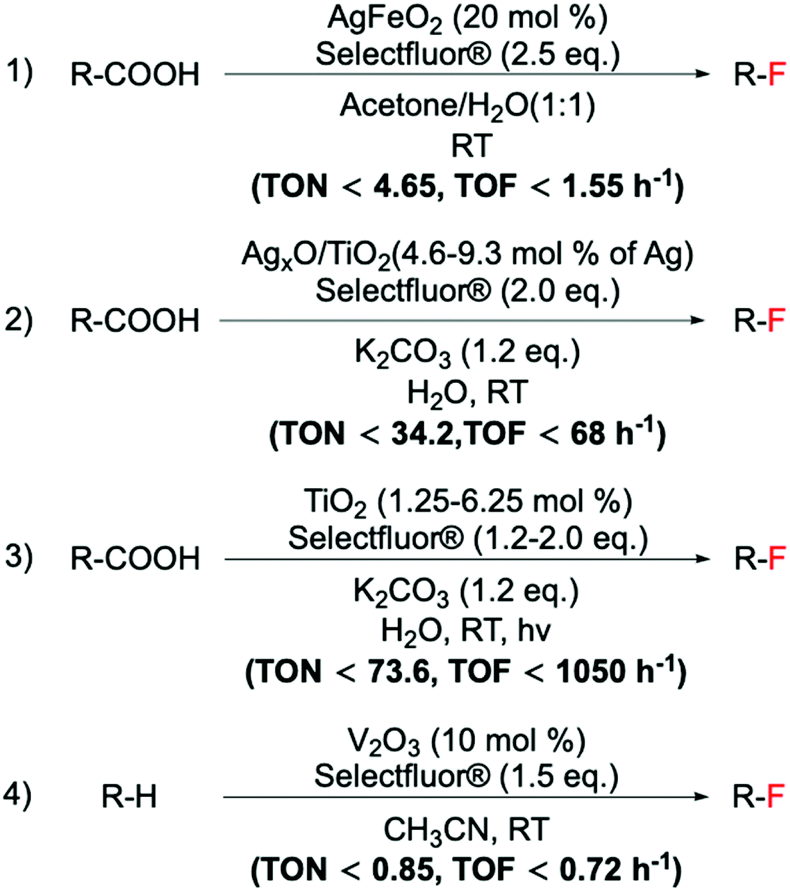 | ||
Scheme 14 Examples of a range of fluorination reactions catalysed by heterogeneous catalysts. C(sp3)–F bond formations catalysed by: (1) AgFeO2,98 (2) AgxO/TiO2,97 (3) TiO2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 102 and (4) V2O3.135 102 and (4) V2O3.135 | ||
Alongside Ag-based materials, other heterogeneous catalysts have proven to be active for fluorination reactions. For example, we recently developed a novel photocatalytic decarboxylative fluorinating route, using commercially available titanium oxide, TiO2 (P25) (Scheme 14, entry 3). This new route shows that even in the absence of expensive metals (such as Ag, Ir, Ru), simple semiconductors like TiO2 can efficiently catalyse fluorination reactions.102 In addition to the high activity exhibited by TiO2, which demonstrated TOFs larger than the ones observed with homogeneous catalysts (<1050 h−1), this method is also characterised by good light efficiency, and proceeds via a mechanism that only requires a catalytic number of photons i.e. a photon efficiency greater than 1 was observed.102 Light efficiency is particularly important in photocatalysis as it reveals whether the incident light acts as a catalyst or merely as a photosensitiser. Good reusability of TiO2 has also been reported, showing potential for further exploration.102 Notably, the proposed reaction mechanism involves contribution from a radical chain process (Scheme 15), in analogy to the mechanism proposed by Lectka for Cu-catalysed aliphatic fluorination.129
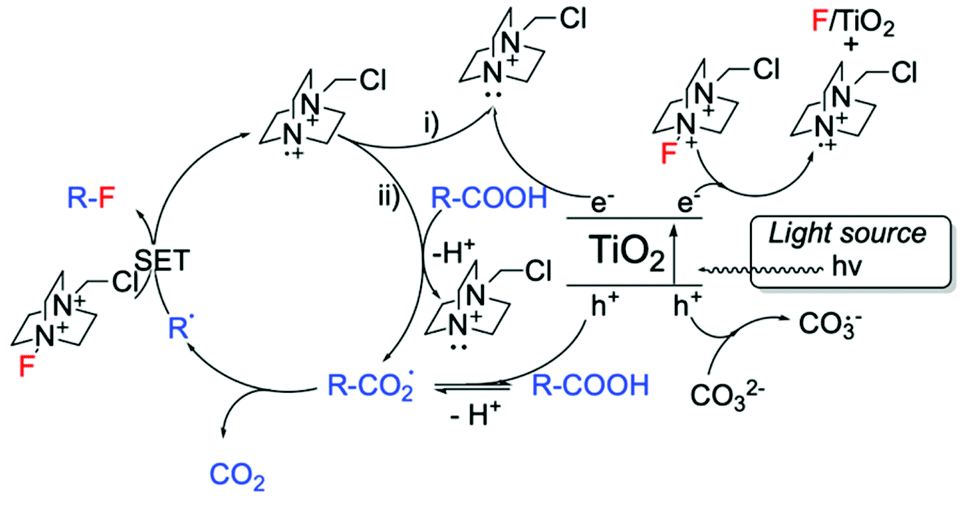 | ||
| Scheme 15 Proposed reaction mechanism for TiO2-photocatalysed decarboxylative fluorinations.102 Scheme adapted with permission from ref. 102 (American Chemical Society, https://pubs.acs.org/doi/10.1021/acscatal.8b02844). | ||
In spite of the greater challenges associated to the direct C(sp3)–H fluorinations, Xia et al. reported that this type of reaction can be achieved using vanadium oxides (Scheme 14, entry 4).135 Interestingly, by employing this cheap, commercial solid, they reported the synthesis of a range of fluorinated compounds, i.e. secondary and tertiary alkyl-fluorides and benzylic fluorides. However, stability studies i.e. leaching and “hot” filtration experiments, and reusability studies, essential to establish the heterogeneous nature of the system, have not yet been reported.88
In contrast to the electrophilic fluorination routes, heterogeneous catalysts for nucleophilic fluorinations have been known since 2006, with several examples of nucleophilic fluorination catalysed by polymer-supported ionic liquids, being present in literature (Scheme 16). Nevertheless, only low TONs can be achieved via this route, with 25–50 mol% of polymer typically employed.136–138
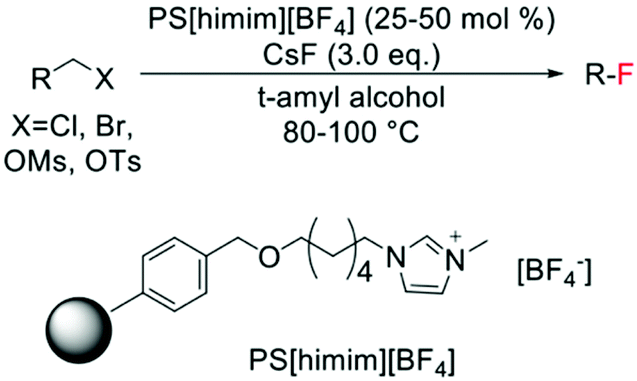 | ||
| Scheme 16 Example of nucleophilic fluorination reported by Kim et al.136 | ||
Additionally, in 2013, Sergeev et al. successfully immobilised the fluorinase enzyme discovered by O'Hagan (vide supra),124 onto a poly(GMA-EDMA) monolith.139 The supported enzyme was shown to be an efficient and reusable catalyst for both fluorination and radiofluorination of the specific substrate, SAM, to 5-FDA and 5-18FDA.139
9. Conclusions and pertaining challenges
Despite the plethora of research published on fluorination strategies within the last fifteen years, several challenges in the field of fluorination remain, and should form the basis of future research studies. Firstly, despite the progresses made, catalytic methods for C(sp3)–F formation are still characterised by limited levels of productivity, and/or poor atom economy, with large excesses of expensive fluorinating reagents and/or fluorine salts typically required. As such, future studies should address the development of catalytic fluorination methods able to achieve C(sp3)–F synthesis in a more sustainable and efficient manner.Several recent works have identified continuous flow systems as a solution to maximise productivity and at the same time, decrease the hazards associated with the employment of toxic chemicals, including HF, potentially generated in situ. In fact, continuous flow processes could provide several advantages from both an efficiency and a productivity perspective, representing a way to produce fluorinated compounds on a larger scale. However, the continuous flow processes developed so far are either non-catalytic, and/or employ soluble catalysts that must be continuously fed into the system. Amongst these, interesting results have been achieved by combining continuous flow methods with photocatalytic fluorination and homogeneous catalysis. However, these systems do not easily permit long-term use or recycle of the soluble catalysts, resulting in heterogenous catalysts being more suitable materials for such flow applications. Irrespective of the nature of the catalyst, challenges associated with light penetration are still prevalent in photocatalytic systems. Accordingly, engineering studies should be addressed towards design of novel photocatalytic reactors able to maximise light efficiency, and, thus, process productivity.
The employment of robust heterogeneous catalysts in a packed bed reactor would also represent an improvement for the productivity and the sustainability of a system, due to immobilising the catalyst from the liquid solution. Despite such benefits, most of the catalytic systems that have been developed up to now for C(sp3)–F formation employ homogeneous catalysts, with few exceptions. Although scarcely employed for fluorination processes, few promising reports indicate that C(sp3)–F bond synthesis can be efficiently achieved with heterogeneous catalysts. Therefore, future studies should be directed toward the design of active and stable heterogeneous materials able to perform a range of fluorination reactions in both batch and continuous flow systems.
Alongside the need for improved productivity and sustainability, more fundamental studies on C(sp3)–F synthesis are still required. Indeed, despite good levels of catalytic performance achieved using pre-activated substrates (i.e. carboxylic acids), direct C(sp3)–H functionalisation is still hampered by several factors, including poor activity, limited substrate scope and the utilisation of toxic catalysts. Moreover, despite the breakthroughs achieved for benzylic fluorinations, the number of catalysts able to perform alkane fluorination is still limited and the mechanistic details of such reactions are still open to debate. Therefore, future fundamental studies in this field should focus on; (i) the development of more sustainable and more efficient routes for direct C(sp3)–H fluorination, and (ii) thorough mechanistic studies of the current catalytic processes, consequently enabling the design of novel catalysts able to unlock this reaction with increased catalytic performance.
We hope that our mini-review will encourage collaboration amongst scientists with various backgrounds (homogeneous and heterogeneous catalysis, catalysis engineering, spectroscopy and material science, amongst others), to develop greener strategies with industrial applicability for the synthesis of fine chemicals containing C(sp3)–F moiety.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
CH gratefully appreciates the support of The Royal Society, for provision of a University Research Fellowship (UF140207, URF\R\201003) and enhanced research grant funding (RGF/EA/180314).References
- B. E. Smart, J. Fluorine Chem., 2001, 109, 3–11 CrossRef CAS.
- M. Shimizu and T. Hiyama, Angew. Chem., Int. Ed., 2005, 44, 214–231 CrossRef CAS PubMed.
- P. Miller, N. Long, R. Vilar and A. Gee, Angew. Chem., Int. Ed., 2008, 47, 8998–9033 CrossRef CAS PubMed.
- S. Purser, P. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320–330 RSC.
- F. Babudri, G. M. Farinola, F. Naso and R. Ragni, Chem. Commun., 2007, 1003–1022 RSC.
- M. Pagliaro and R. Ciriminna, J. Mater. Chem., 2005, 15, 4981–4991 RSC.
- J. T. Robinson, J. S. Burgess, C. E. Junkermeier, S. C. Badescu, T. L. Reinecke, F. K. Perkins, M. K. Zalalutdniov, J. W. Baldwin, J. C. Culbertson, P. E. Sheehan and E. S. Snow, Nano Lett., 2010, 10, 3001–3005 CrossRef CAS PubMed.
- M. Dubecký, J. Phys. Chem. Lett., 2015, 6, 1430–1434 CrossRef PubMed.
- D. O'Hagan and H. S. Rzepa, Chem. Commun., 1997, 645–652 RSC.
- D. O'Hagan, Chem. Soc. Rev., 2008, 37, 308–319 RSC.
- D. W. Zhu, Synthesis, 1993, 953–954 CrossRef CAS.
- P. Jeschke, ChemBioChem, 2004, 5, 571–589 CrossRef PubMed.
- D. E. Yerien, S. Bonesi and A. Postigo, Org. Biomol. Chem., 2016, 14, 8398–8427 RSC.
- J. M. Racowski, J. B. Gary and M. S. Sanford, Angew. Chem., 2012, 124, 3470–3473 CrossRef.
- R. Szpera, D. F. J. Moseley, L. B. Smith, A. J. Sterling and V. Gouverneur, Angew. Chem., Int. Ed., 2019, 58, 14824–14848 CrossRef CAS.
- H. Egami and Y. Hamashima, in Frontiers of Organofluorine Chemistry, ed. I. Ojima, World Scientific, Singapore, 2020, ch. 1, pp. 3–91 Search PubMed.
- T. Liang, C. N. Neumann and T. Ritter, Angew. Chem., Int. Ed., 2013, 52, 8214–8264 CrossRef CAS PubMed.
- J. A. Ma and D. Cahard, J. Fluorine Chem., 2007, 128, 975–996 CrossRef CAS.
- S. Barata-Vallejo, B. Lantaño and A. Postigo, Chem. – Eur. J., 2014, 20, 16806–16829 CrossRef CAS PubMed.
- H. Liu, Z. Gu and X. Jiang, Adv. Synth. Catal., 2013, 355, 617–626 CrossRef CAS.
- Z. Jin, G. B. Hammond and B. Xu, Aldrichimica Acta, 2012, 45, 67–83 CAS.
- J. Charpentier, N. Früh and A. Togni, Chem. Rev., 2015, 115, 650–682 CrossRef CAS PubMed.
- T. Furuya, A. S. Kamlet and T. Ritter, Nature, 2011, 473, 470–477 CrossRef CAS PubMed.
- R. F. Merritt and T. E. Stevens, J. Am. Chem. Soc., 1966, 88, 1822–1823 CrossRef CAS.
- V. Grakauskas, J. Org. Chem., 1969, 34, 2446–2450 CrossRef CAS.
- T. Ido, C.-N. Wan, J. S. Fowler and A. P. Wolf, J. Org. Chem., 1977, 42, 2341–2342 CrossRef CAS.
- D. R. MacKenzie and J. Fajer, J. Am. Chem. Soc., 1970, 92, 4994–4996 CrossRef CAS.
- R. E. Banks, S. N. Mohialdin-Khaffaf, G. S. Lal, l. Sharif and R. G. Syvret, J. Chem. Soc., Chem. Commun., 1992, 595–596 RSC.
- W. J. Middleton, J. Org. Chem., 1975, 40, 574–578 CrossRef CAS.
- D. Meyer, H. Jangra, F. Walther, H. Zipse and P. Renaud, Nat. Commun., 2018, 9, 4888 CrossRef PubMed.
- M. C. Pacheco, S. Purser and V. Gouverneur, Chem. Rev., 2008, 108, 1943–1981 CrossRef CAS PubMed.
- R. P. Singh and J. M. Shreeve, Acc. Chem. Res., 2004, 37, 31–44 CrossRef CAS PubMed.
- G. Valero, X. Companyó and R. Rios, Chem. – Eur. J., 2011, 17, 2018–2037 CrossRef CAS PubMed.
- F. J. Aguilar Troyano, K. Merkens and A. Gómez-Suárez, Asian J. Org. Chem., 2020, 9, 992–1007 CrossRef CAS.
- P. T. Nyffeler, S. G. Durón, M. D. Burkart, S. P. Vincent and C.-H. Wong, Angew. Chem., Int. Ed., 2005, 44, 192–212 CrossRef CAS PubMed.
- M. P. Sibi and Y. Landais, Angew. Chem., Int. Ed., 2013, 52, 3570–3572 CrossRef CAS PubMed.
- C. Hollingworth and V. Gouverneur, Chem. Commun., 2012, 48, 2929–2942 RSC.
- T. L. Ross and H. J. Wester, in Handbook of Nuclear Chemistry, ed. A. Vértes, S. Nagy, Z. Klencsár, R. G. Lovas and F. Rösch, Springer, Boston, MA, 2011, pp. 2021–2071 Search PubMed.
- W. A. Szarek, G. W. Hay and M. M. Perlmutter, J. Chem. Soc., Chem. Commun., 1982, 1253–1254 RSC.
- P. A. Beeley, W. A. Szarek, G. W. Hay and M. M. Perlmutter, Can. J. Chem., 1984, 62, 2709–2711 CrossRef CAS.
- X. Huang, W. Liu, J. M. Hooker and J. T. Groves, Angew. Chem., Int. Ed., 2015, 54, 5241–5245 CrossRef CAS PubMed.
- W. Liu, X. Huang, M. S. Placzek, S. W. Krska, P. McQuade, J. M. Hooker and J. T. Groves, Chem. Sci., 2018, 9, 1168–1172 RSC.
- R. P. Singh and J. M. Shreeve, Synthesis, 2002, 2561–2578 CAS.
- W. Middleton and E. Bingham, J. Org. Chem., 1980, 45, 2883–2887 CrossRef CAS.
- G. Posner and S. Haines, Tetrahedron Lett., 1985, 26, 5–8 CrossRef CAS.
- A. Boukerb, D. Grée, M. Laabassi and R. Grée, J. Fluorine Chem., 1998, 88, 23–27 CrossRef CAS.
- F. Beaulieu, L. Beauregard, G. Courchesne, M. Couturier and A. L'Heureux, Org. Lett., 2009, 11, 5050–5053 CrossRef CAS PubMed.
- A. L'Heureux, F. Beaulieu, C. Bennett, D. R. Bill, S. Clayton, F. LaFlamme, M. Mirmehrabi, S. Tadayon, D. Tovell and M. Couturier, J. Org. Chem., 2010, 75, 3401–3411 CrossRef PubMed.
- P. Adler, C. J. Teskey, D. Kaiser, M. Holy, H. H. Sitte and N. Maulide, Nat. Chem., 2019, 11, 329–334 CrossRef CAS PubMed.
- P.-C. Lin, A. K. Adak, S.-H. Ueng, L.-D. Huang, K.-T. Huang, J.-a. A. Ho and C.-C. Lin, J. Org. Chem., 2009, 74, 4041–4048 CrossRef CAS PubMed.
- M. Pouliot, L. Angers, J. Hamel and J. Paquin, Tetrahedron Lett., 2012, 53, 4121–4123 CrossRef CAS.
- A. Orliac, D. Gomez Pardo, A. Bombrun and J. Cossy, Org. Lett., 2013, 15, 902–905 CrossRef CAS PubMed.
- M. K. Nielsen, D. T. Ahneman, O. Riera and A. G. Doyle, J. Am. Chem. Soc., 2018, 140, 5004–5008 CrossRef CAS PubMed.
- M.-G. Braun and A. G. Doyle, J. Am. Chem. Soc., 2013, 135, 12990–12993 CrossRef CAS PubMed.
- C. Hollingworth, A. Hazari, M. N. Hopkinson, M. Tredwell, E. Benedetto, M. Huiban, A. D. Gee, J. M. Brown and V. Gouverneur, Angew. Chem., Int. Ed., 2011, 50, 2613–2617 CrossRef CAS PubMed.
- E. Differding and H. Ofner, Synlett, 1991, 187–189 CrossRef CAS.
- R. Banks, N. Lawrence and A. Popplewell, J. Chem. Soc., Chem. Commun., 1994, 343–344 RSC.
- W. Peng and J. M. Shreeve, J. Org. Chem., 2005, 70, 5760–5763 CrossRef CAS PubMed.
- G. S. Lal, J. Org. Chem., 1993, 58, 2791–2796 CrossRef CAS.
- M. Zupan, J. Iskra and S. Stavber, J. Fluorine Chem., 1995, 70, 7–8 CrossRef CAS.
- S. Lectard, Y. Hamashima and M. Sodeoka, Adv. Synth. Catal., 2010, 352, 2708–2732 CrossRef CAS.
- M. A. Cortés González, P. Nordeman, A. Bermejo Gómez, D. N. Meyer, G. Antoni, M. Schou and K. J. Szabó, Chem. Commun., 2018, 54, 4286–4289 RSC.
- H. Teare, E. G. Robins, E. Åstad, S. K. Luthra and V. Gouverneur, Chem. Commun., 2007, 2330–2332 RSC.
- J. Hutchinson and G. Sandford, in Elemental Fluorine in Organic Chemistry, ed. R. D. Chambers, Springer, Berlin, Heidelberg, 1997, Top. Curr. Chem., vol. 193, pp. 1–43 Search PubMed.
- M. Rueda-Becerril, C. Chatalova-Sazepin, J. Paquin and G. Sammis, J. Am. Chem. Soc., 2012, 134, 4026–4029 CrossRef CAS PubMed.
- J.-D. Yang, Y. Wang, X.-S. Xue and J.-P. Cheng, J. Org. Chem., 2017, 82, 4129–4135 CrossRef CAS PubMed.
- J. C. T. Leung, C. Chatalova-Sazepin, J. G. West, M. Rueda-Becerril, J.-F. Paquin and G. M. Sammis, Angew. Chem., Int. Ed., 2012, 51, 10804–10807 CrossRef CAS PubMed.
- P. A. Champagne, J. Desroches, J.-D. Hamel, M. Vandamme and J.-F. Paquin, Chem. Rev., 2015, 115, 9073–9174 CrossRef CAS.
- R. H. Sheldon, Green Chem., 2017, 19, 18–43 RSC.
- R. H. Sheldon, Green Chem., 2007, 9, 1273–1283 RSC.
- D. O'Hagan, J. Fluorine Chem., 2010, 131, 1071–1081 CrossRef.
- T. Fujiwara and D. O'Hagan, J. Fluorine Chem., 2014, 167, 16–29 CrossRef CAS.
- R. Porta, M. Benaglia and A. Puglisi, Org. Process Res. Dev., 2016, 20, 2–25 CrossRef CAS.
- C. Wiles and P. Watts, Green Chem., 2012, 14, 38–54 RSC.
- T. H. Rehm, C. Hofmann, D. Reinhard, H.-J. Kost, P. Löb, M. Besold, K. Welzel, J. Barten, A. Didenko, D. V. Sevenard, B. Lix, A. R. Hillson and S. D. Riegel, React. Chem. Eng., 2017, 2, 315–323 RSC.
- A. Kirschning, W. Solodenko and K. Mennecke, Chem. – Eur. J., 2006, 12, 5972–5990 CrossRef CAS PubMed.
- D. L. Browne, M. Baumann, B. H. Harji, I. R. Baxendale and S. V. Ley, Org. Lett., 2011, 13, 3312–3315 CrossRef CAS PubMed.
- S. L. James, C. J. Adams, C. Bolm, D. Braga, P. Collier, T. Friščić, F. Grepioni, K. D. M. Harris, G. Hyett, W. Jones, A. Krebs, J. Mack, L. Maini, A. G. Orpen, I. P. Parkin, W. C. Shearouse, J. W. Steedk and D. C. Waddelli, Chem. Soc. Rev., 2012, 41, 413–447 RSC.
- R. Carrasquillo-Flores, M. Käldström, F. Schüth, J. A. Dumesic and R. Rinaldi, ACS Catal., 2013, 3, 993–997 CrossRef CAS.
- J. Twilton, C. Le, P. Zhang, M. H. Shaw, R. W. Evans and D. W. C. MacMillan, Nat. Rev. Chem., 2017, 1(0052), 1–18 Search PubMed.
- S. Mascia, P. L. Heider, H. Zhang, R. Lakerveld, B. Benyahia, P. I. Barton, R. D. Braatz, C. L. Cooney, J. M. B. Evans, T. F. Jamison, K. F. Jensen, A. S. Myerson and B. L. Trout, Angew. Chem., Int. Ed., 2013, 52, 12359–12363 CrossRef CAS PubMed.
- R. A. Sheldon, I. Arends and U. Hanefeld, Green Chemistry and Catalysis, Wiley-VCH, Weinheim, 2007 Search PubMed.
- R. A. Sheldon, Chem. Soc. Rev., 2012, 41, 1437–1451 RSC.
- P. T. Anastas, M. M. Kirchhoff and T. C. Williamson, Appl. Catal., A, 2001, 221, 3–13 CrossRef CAS.
- S. D. Halperin, D. Kwon, M. Holmes, E. L. Regalado, L.-C. Campeau, D. A. DiRocco and R. Britton, Org. Lett., 2015, 17, 5200–5203 CrossRef CAS PubMed.
- V. L. Truong, J. Y. Gauthier, M. Boyd, B. Roy and J. Scheigetz, Synlett, 2005, 1279–1280 CrossRef CAS.
- C. Nadeau, F. Gosselin, P. D. O'Shea, I. W. Davies and R. P. Volante, Synlett, 2006, 291–295 CrossRef CAS.
- C. Hammond, Green Chem., 2017, 19, 2711–2728 RSC.
- J.-P. Lange, CATTECH, 2001, 5, 82–95 CrossRef CAS.
- T. J. Schwartz, B. J. O'Neill, B. H. Shanks and J. A. Dumesic, ACS Catal., 2014, 4(6), 2060–2069 CrossRef CAS.
- P. W. N. M. van Leeuwen and J. C. Chadwick, Homogeneous Catalysis: Activity, Stability and Deactivation, Wiley, Weinheim, 2011 Search PubMed.
- D. Rodríguez-Padrón, A. R. Puente-Santiago, A. M. Balu, M. J. Muñoz-Batista and R. Luque, ChemCatChem, 2019, 11, 18–38 CrossRef.
- K. L. Hull, W. Q. Anani and M. S. Sanford, J. Am. Chem. Soc., 2006, 128, 7134–7135 CrossRef CAS.
- F. Yin, Z. Wang, Z. Li and C. Li, J. Am. Chem. Soc., 2012, 134, 10401–10404 CrossRef CAS PubMed.
- N. Patel and R. Flowers II, J. Org. Chem., 2015, 80, 5834–5841 CrossRef CAS PubMed.
- X. Zhang, Comput. Theor. Chem., 2016, 1082, 11–20 CrossRef CAS.
- G. Tarantino, L. Botti, N. Dimitratos and C. Hammond, RSC Adv., 2017, 7, 30185–30190 RSC.
- T. V. Nguyen, V. T. Pham, T. V. T. Nguyen, N. T. S. Phan and T. Truong, J. Catal., 2018, 360, 270–276 CrossRef CAS.
- J. A. Porras, I. N. Mills, W. J. Transue and S. Bernhard, J. Am. Chem. Soc., 2016, 138, 9460–9472 CrossRef CAS PubMed.
- S. Ventre, F. R. Petronijevic and D. W. C. MacMillan, J. Am. Chem. Soc., 2015, 137, 5654–5657 CrossRef CAS PubMed.
- X. Wu, C. Meng, X. Yuan, X. Jia, X. Qian and J. Ye, Chem. Commun., 2015, 51, 11864–11867 RSC.
- G. Tarantino and C. Hammond, ACS Catal., 2018, 8, 10321–10330 CrossRef CAS.
- E. P. Gillis, K. J. Eastman, M. D. Hill, D. J. Donnelly and N. A. Meanwell, J. Med. Chem., 2015, 58, 8315–8359 CrossRef CAS PubMed.
- S. Bloom, C. R. Pitts, R. Woltornist, A. Griswold, M. G. Holl and T. Lectka, Org. Lett., 2013, 15, 1722–1724 CrossRef CAS.
- P. Xu, S. Guo, L. Wang and P. Tang, Angew. Chem., Int. Ed., 2014, 53, 5955–5958 CrossRef CAS PubMed.
- S. Bloom, A. Sharber, M. G. Holl, J. L. Knippel and T. Lectka, J. Org. Chem., 2013, 78, 11082–11086 CrossRef CAS PubMed.
- S. Bloom, C. R. Pitts, D. C. Miller, N. Haselton, M. G. Holl, E. Urheim and T. Lectka, Angew. Chem., Int. Ed., 2012, 51, 10580–10583 CrossRef CAS PubMed.
- W. Liu and J. T. Groves, Angew. Chem., Int. Ed., 2013, 52, 6024–6027 CrossRef CAS PubMed.
- X. Huang, W. Liu, H. Ren, R. Neelamegam, J. M. Hooker and J. T. Groves, J. Am. Chem. Soc., 2014, 136, 6842–6845 CrossRef CAS PubMed.
- J.-B. Xia, C. Zhu and C. Chen, J. Am. Chem. Soc., 2013, 135, 17494–17500 CrossRef CAS PubMed.
- S. Bloom, M. McCann and T. Lectka, Org. Lett., 2014, 16, 6338–6341 CrossRef CAS PubMed.
- D. Cantillo, O. de Frutos, J. A. Rincon, C. Mateos and C. O. Kappe, J. Org. Chem., 2014, 79, 8486–8490 CrossRef CAS PubMed.
- Y. Amaoka, M. Nagatomo and M. Inoue, Org. Lett., 2013, 15, 2160–2163 CrossRef CAS.
- A. Koperniku, H. Liu and P. B. Hurley, Eur. J. Org. Chem., 2016, 871–886 CrossRef CAS.
- M. Movsisyan, E. I. P. Delbeke, J. K. E. T. Berton, C. Battilocchio, S. V. Ley and C. V. Stevens, Chem. Soc. Rev., 2016, 45, 4892–4928 RSC.
- D. Cantillo and C. O. Kappe, React. Chem. Eng., 2017, 2, 7–19 RSC.
- B. Gutmann, D. Cantillo and C. O. Kappe, Angew. Chem., Int. Ed., 2015, 54, 6688–6728 CrossRef CAS PubMed.
- M. Zboray, A. T. Bell and E. Iglesia, J. Phys. Chem. C, 2009, 113, 12380–12386 CrossRef CAS.
- W. Liu, X. Y. Huang, M. J. Cheng, R. J. Nielsen, W. A. Goddard and J. T. Groves, Science, 2012, 337, 1322–1325 CrossRef CAS PubMed.
- G. Li, A. K. Dilger, P. T. Cheng, W. R. Ewing and J. T. Groves, Angew. Chem., Int. Ed., 2018, 57, 1251–1255 CrossRef CAS.
- W. Liu and J. T. Groves, Acc. Chem. Res., 2015, 48, 1727–1735 CrossRef CAS.
- P. R. Ortiz de Montellano, Chem. Rev., 2010, 110, 932–948 CrossRef CAS PubMed.
- J. T. Groves, J. Chem. Educ., 1985, 62, 928–931 CrossRef CAS.
- D. O'Hagan, C. Schaffrath, S. L. Cobb, J. T. G. Hamilton and C. D. Murphy, Nature, 2002, 416, 279–279 CrossRef PubMed.
- X. Zhu, D. A. Robinson, A. R. McEwan, D. O'Hagan and J. H. Naismith, J. Am. Chem. Soc., 2007, 129, 14597–14604 CrossRef CAS PubMed.
- S. Bloom, J. L. Knippel and T. Lectka, Chem. Sci., 2014, 5, 1175–1178 RSC.
- S. D. Halperin, H. Fan, S. Chang, R. E. Martin and R. Britton, Angew. Chem., Int. Ed., 2014, 53, 4690–4693 CrossRef CAS PubMed.
- J. G. West, T. A. Bedell and E. G. Sorensen, Angew. Chem., Int. Ed., 2016, 55, 8923–8927 CrossRef CAS PubMed.
- C. R. Pitts, S. Bloom, R. Woltornist, D. J. Auvenshine, L. R. Ryzhkov, M. A. Siegler and T. Leckta, J. Am. Chem. Soc., 2014, 136, 9780–9791 CrossRef CAS PubMed.
- V. Rauniyar, A. D. Lackner, G. L. Hamilton and F. D. Toste, Science, 2011, 334, 1681–1684 CrossRef CAS PubMed.
- G. Pupo, F. Ibba, D. M. H. Ascough, A. C. Vicini, P. Ricci, K. E. Christensen, L. Pfeifer, J. R. Morphy, J. M. Brown, R. S. Paton and V. Gouverneur, Science, 2018, 360, 638–642 CrossRef CAS PubMed.
- G. Pupo, A. C. Vicini, D. M. H. Ascough, F. Ibba, K. E. Christensen, A. L. Thompson, J. M. Brown, R. S. Paton and V. Gouverneur, J. Am. Chem. Soc., 2019, 141, 2878–2883 CrossRef CAS PubMed.
- I. Sadaba, M. L. Granados, A. Riisager and E. Taarning, Green Chem., 2015, 17, 4133–4145 RSC.
- T. Mallat and A. Baiker, Chem. Rev., 2004, 104, 3037–3058 CrossRef CAS.
- J.-B. Xia, Y. Ma and C. Chen, Org. Chem. Front., 2014, 1, 468–472 RSC.
- D. W. Kim, H.-J. Jeong, S. T. Lim, M.-H. Sohn and D. Y. Chi, Tetrahedron, 2008, 64, 4209–4214 CrossRef CAS.
- D. W. Kim, D. J. Hong, K. S. Jang and D. Y. Chi, Adv. Synth. Catal., 2006, 348, 1719–1727 CrossRef CAS.
- V. H. Jadhav, S. H. Jang, H.-J. Jeong, S. T. Lim, M.-H. Sohn, D. Y. Chi and D. W. Kim, Org. Lett., 2010, 12, 3740–3743 CrossRef CAS PubMed.
- M. E. Sergeev, F. Morgia, M. R. Javed, M. Doi and P. Y. Keng, J. Mol. Catal. B: Enzym., 2013, 92, 51–56 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2020 |