
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHydrosilylation and hydroboration in a sustainable manner: from Earth-abundant catalysts to catalyst-free solutions
Krzysztof
Kuciński
 * and
Grzegorz
Hreczycho
* and
Grzegorz
Hreczycho
Faculty of Chemistry, Adam Mickiewicz University in Poznań, Ul. Uniwersytetu Poznańskiego 8, 61-614 Poznań, Poland. E-mail: kucinski.k@amu.edu.pl
First published on 3rd August 2020
Abstract
Hydroelementation enables a facile reduction or functionalization of several unsaturated systems, and thus activation of such bonds like B–H and Si–H is a powerful synthetic tool. Furthermore, the addition of these moieties is in line with the main aspects of green chemistry, in particular with regard to atom economy. In view of the above, several effective methods have been developed in recent years. This critical review will mainly detail solutions utilizing Earth-abundant catalysts (based on s-block elements), although their potential will be particularly discussed in relation to sustainability problems. Besides, there is increasing interest in catalyst-free and solvent-free approaches, which will also be included. This review highlights recent developments in hydroboration and hydrosilylation of unsaturated carbon–heteroatom (O, N) moieties and covers the literature from the last ten years.
 Krzysztof Kuciński | Krzysztof Kuciński has been an Assistant Professor in the Department of Chemistry and Technology of Silicon Compounds at Adam Mickiewicz University in Poznan (Poland) since March 2019. He studied chemistry at AMU Poznan, and did his PhD with Prof. G. Hreczycho at the same institution (2018). During the studies, he was an intern for eminent scientists such as Professor Claudio Palomo (Universidad Del Pais Vasco San Sebastian, Spain) and Professor Lutz Ackermann (Georg-August-Universität Göttingen, Germany). His research interests include organoboron and organosilicon chemistry with a strong emphasis on the sustainability of the processes. |
 Grzegorz Hreczycho | Grzegorz Hreczycho received his Ph.D. (2007) and Habilitation (2015) in chemistry from Adam Mickiewicz University in Poznan (Poland). His research interests cover novel applications of silicon, boron, and germanium compounds and in addition coupling reactions catalyzed by transition metal complexes and Lewis acid catalysts. More than 60 research publications and patents document his activity in the fields of organometallic chemistry, homogeneous catalysis, and organic synthesis. |
Introduction
In a period of increasing society's awareness in terms of sustainable developments and risks arising from irresponsible resource management, scientists pay more and more attention to the development of greener synthetic solutions. One of the main directions is gradual, although noticeable resignation from the use of highly expensive and quite often toxic coordination compounds of transition metals (e.g., platinum, ruthenium, rhodium, etc.). It is not an easy process due to the highest catalytic activity and selectivity of most of them. Nevertheless, an increasing number of research teams focus on catalysis based on main-group elements or Earth-abundant transition metals (Fe, Mn, Ni, Co). Moreover, catalyst-free and solvent-free approaches are attracting more and more attention too.Hydroboration and hydrosilylation are used to reduce unsaturated moieties, as well as for synthesizing sila- and bora-functionalized derivatives. Both processes have been described in innumerable articles and have been covered in several reviews in recent years.1–27 In this respect, it should be noted that the purpose of the presented summary was to include selected recent literature in the field (since 2010) concerning both hydroboration and hydrosilylation of unsaturated carbon–heteroatom moieties, with an emphasis on aspects related to green and sustainable chemistry. To the best of our knowledge, this is the first example of such coverage and the authors hope that this account will stimulate further development of greener alternatives.
Hydrosilylation and hydroboration – a short overview
Undoubtedly, hydrosilylation is to the most useful and applicable reaction in organosilicon chemistry, being simultaneously one of the most important homogeneous processes in industry.28 In spite of a multitude of efforts to replace noble metals as catalysts, it should be made clear that platinum-based systems are still the most useful for both large29 and small-scale30–37 productions of organosilicon derivatives, which were quite nicely summarized by Marciniec and co-workers.38 However, an extremely high price of these catalysts and often also harsh reaction conditions have caused an extensive exploration of less expensive and milder alternatives.Hydrosilylation may lead to different products depending on the type of unsaturated system. In the case of carbonyls (aldehydes and ketones), imines, and nitriles, it leads (via reduction) to the formation of the corresponding silyl ethers, which might be easily hydrolyzed to give alcohols or amines (Fig. 1).
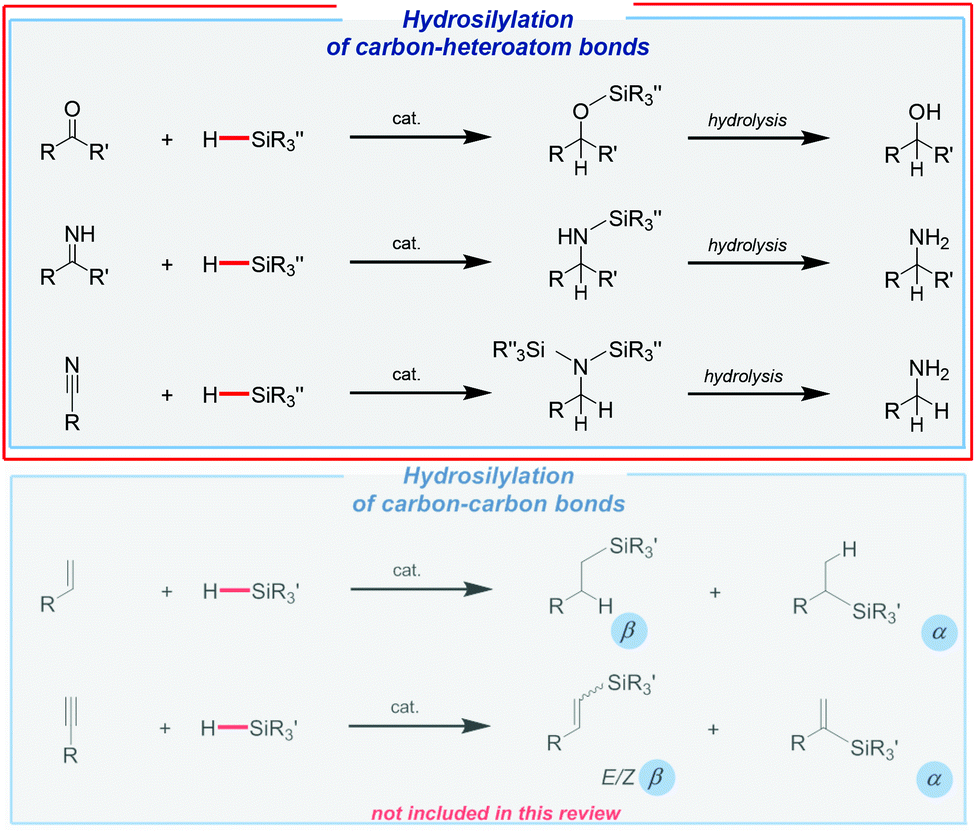 | ||
| Fig. 1 Hydrosilylation reaction of various unsaturated systems. | ||
On the other hand, hydrosilylation of terminal alkenes may give an α or β-isomer (Fig. 1). Whilst the formation of the latter one, known as anti-Markovnikov regioselectivity, is the most common, it should be noted that Markovnikov regioisomers are the subject of increasing interest, and they have been recently reviewed by Zaranek and Pawluc.39 In the case of alkynes, a whole range of possible products can be observed. Monohydrosilylation of terminal alkynes leads to vinyl-substituted silanes (E/Z-β-isomers or α-isomers), which are one of the most important organosilicon species because of their unique physical properties and specific reactivity (Fig. 1). Moreover, their use as synthetic intermediates or building blocks has been confirmed by many scientists. Furthermore, the double hydrosilylation can also be observed in some cases.
The applicability of hydroboration is much more specific and targeted to the preparation of some fine chemicals or useful reagents.4 Its industrial potential is incomparably lower compared to hydrosilylation. Although transition metal complexes are still essential components of this powerful hydroelementation pathway, the research into Earth-abundant catalysts has grown and evolved in extraordinary ways. Perhaps more importantly, the addition of B–H moieties can also be performed under catalyst-free conditions, which is virtually uncommon in the case of Si–H bond addition.40,41 This extremely high reactivity mainly applies to non-hindered boranes. Unfortunately, such non-catalytic synthesis requires special precautions and often also harsh reaction conditions. Furthermore, it may sometimes lead to mixtures of products, including those of dihydroboration. However, in general, the hydroboration of unsaturated bonds gives access to analogous products to those of hydrosilylation, as shown in Fig. 2.
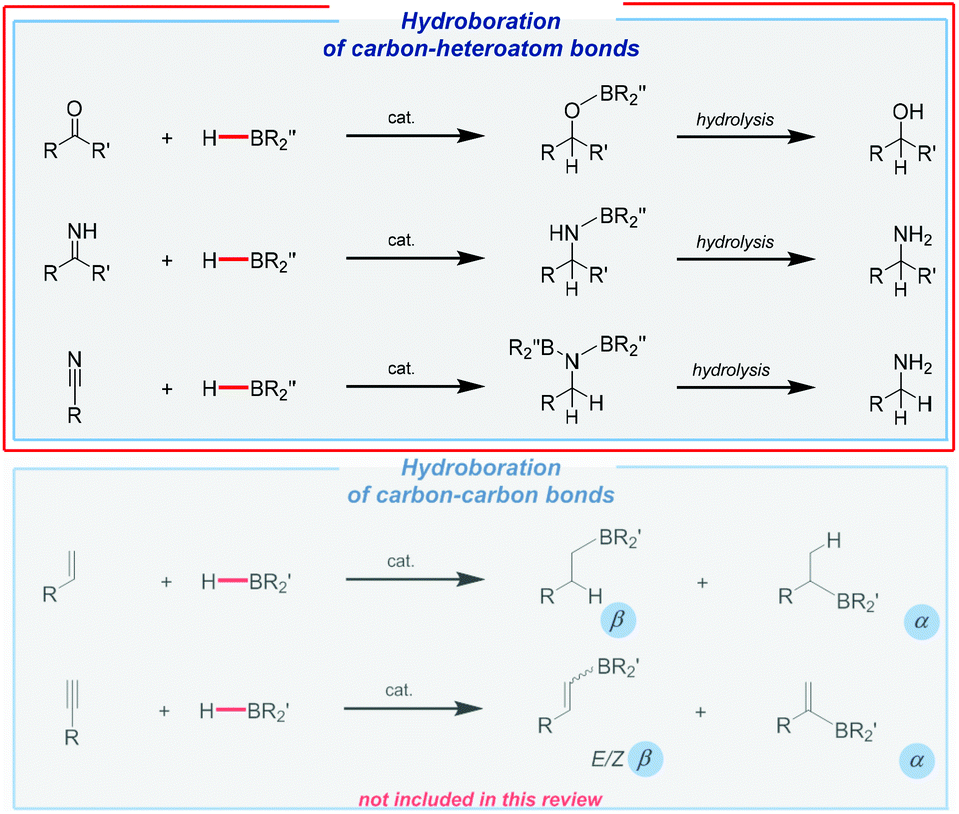 | ||
| Fig. 2 Hydroboration of various unsaturated systems. | ||
To sum up, the last ten years have witnessed considerable advances in hydrosilylation and hydroboration, in terms of sustainability and economy. The key to success was the use of Earth-abundant species, which are less expensive, in contrast to classical noble metal catalysts. In this review, the most interesting implementations from the perspective of green chemistry will be discussed, without a detailed description of mechanical studies.
s-Block element catalysts
Introduction
The main and undeniable advantage of alkali and alkaline earth metals is their high abundance in Earth's crust. These compounds are relatively inexpensive and widely accessible. In this regard, it should be mentioned that the general development of humanity and modern synthetic chemistry would not have been possible without simple salts of these metals. What is more, from the perspective of organic chemists, the key role of organolithium42 and organomagnesium43 compounds is also not to be underestimated. Nowadays, there has been considerable progress in the development of s-block compounds that can replicate catalytic activity associated with transition metal complexes. However, financially speaking, the main key is to find the most simple, environmentally benign and inexpensive solution.Hydrosilylation of unsaturated carbon–heteroatom (O, N) moieties
Historically, the pioneering reports by Ojima (in the early 1970s), on the rhodium-catalyzed hydrosilylation of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and CN bonds, are commonly recognized as a step forward for the use of hydrosilanes as reducing agents.44,45 These early studies were conducted by several efficient procedures utilizing transition-metal catalysts46–55 and main-group species.56–62 In this context, our attention was drawn in particular to the procedures utilizing s-block element species. As a result, selected examples were discussed with an emphasis on sustainability problems.
O and CN bonds, are commonly recognized as a step forward for the use of hydrosilanes as reducing agents.44,45 These early studies were conducted by several efficient procedures utilizing transition-metal catalysts46–55 and main-group species.56–62 In this context, our attention was drawn in particular to the procedures utilizing s-block element species. As a result, selected examples were discussed with an emphasis on sustainability problems.
In 2010, Beller and co-workers disclosed a convenient base-mediated protocol that used simple bases such as sodium hydroxide or potassium tert-butoxide as catalysts (Scheme 1).63
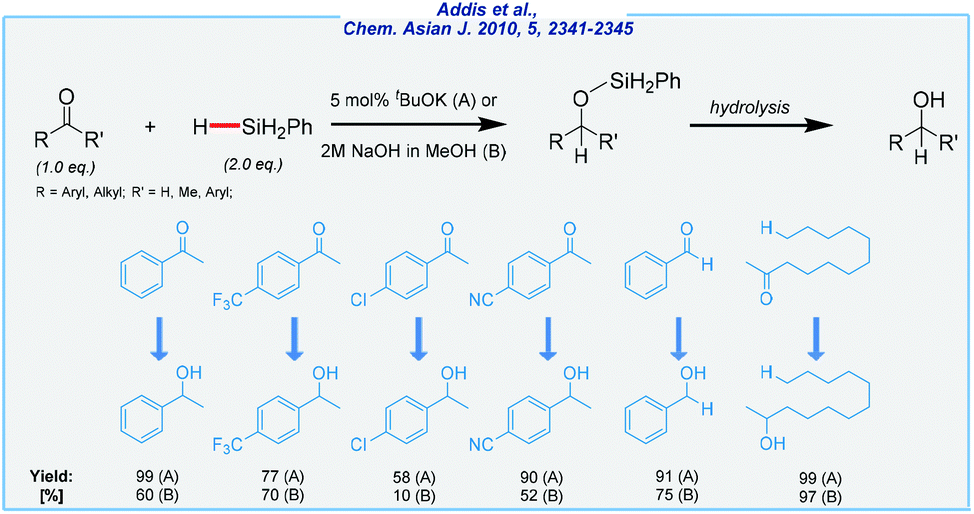 | ||
| Scheme 1 General scheme and substrate scope for tBuOK-mediated hydrosilylation of carbonyls. | ||
The authors tested a few reducing agents, including phenyl- and diphenylsilane, as well as diethoxymethylsilane. All of them are commercially available, but they are not regarded as inexpensive reagents. In general, the tBuOK-mediated pathway gives better yields than that carried out in the presence of NaOH. The halogenated substrates are well tolerated, as well as nitrile and nitro groups, which remain untouched during the process. Summing up, it should be said that the scope of substrates was quite narrow and was mostly limited to aryl methyl ketones. All transformations were carried out in toluene at rt, and under an argon atmosphere. Finally, the crude products were purified by extraction and column chromatography. It should be noted that the concept of base-mediated reduction of carbonyls is not new, and there were already known similar synthetic solutions, headed by Kobayashi work utilizing fluoride ions.64 To conclude, the method developed by Beller has many advantages, including a simple catalyst and quite mild conditions. Of the major drawbacks should be noted are a fairly high price of silane reducing agents, moderate isolation yields, and the use of toluene as the solvent.
Next, the Nolan group has expanded the application of simple potassium bases to the reduction of esters and tertiary amides (Scheme 2).65 Phenylsilane was used as a reducing agent once more and the reaction was carried out at rt under solvent-free conditions.
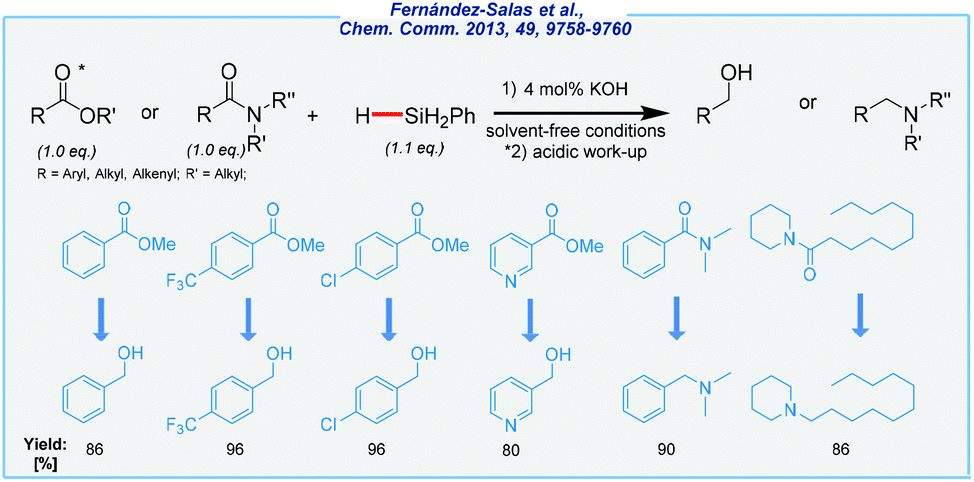 | ||
| Scheme 2 General scheme and substrate scope for KOH-mediated hydrosilylation of esters and amides. | ||
Within these studies, the authors demonstrated that several functional groups such as chloro, methoxy, and trifluoromethyl were shown to be tolerated, regardless of their position in the benzene ring. What is more, a wide range of tertiary amides was reduced, including aryl, heterocyclic, and alkyl groups. During the investigation, primary and secondary amides were non-reactive. From a green chemistry perspective, the authors accomplished the synthesis of alcohols and tertiary amines in very good isolated yields (up to 96%) at rt. In contrast to the analogous tBuOK-catalyzed hydrosilylation of simple carbonyls, this procedure avoided the use of a solvent and an argon atmosphere. Unfortunately, these reactions are not compatible with other, less expensive silanes.
Literally, at the same time as Nolan and his co-workers, the Nikonov group has investigated base-catalyzed hydrosilylation of CO bonds in esters and ketones (Scheme 3).66 The main idea was to implement much more economic reducing agents in the form of polymethylhydrosiloxane (PMHS). Apart from the obvious difference in the price, PMHS is easy to use, very stable and non-toxic. A broad scope of substrates was remarkable, including several aldehydes, ketones, and esters. In the latter case, the reaction takes place mostly at elevated temperatures (70 °C). The conversion rates were excellent, albeit the isolated yields were less satisfactory, as well as there is a need to ensure an inert atmosphere during the process. Furthermore, the authors investigated the reducing source in detail and provided very useful mechanistic information for the formation of very reactive hydrosilanes, which served as the true reducing agents.
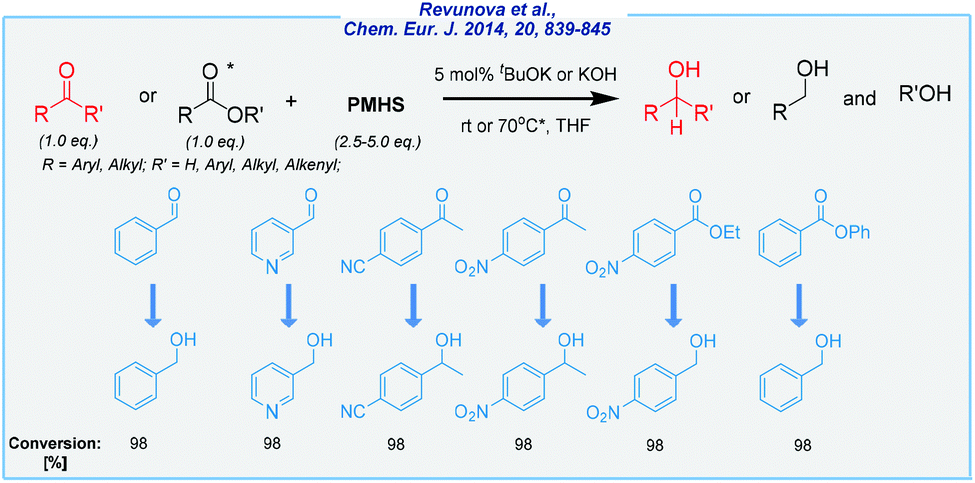 | ||
| Scheme 3 General scheme and substrate scope for tBuOK-mediated hydrosilylation of ketones and esters with PMHS. | ||
Meanwhile, further investigations on the tBuOK-catalyzed reduction of tertiary amides utilizing an excess of trialkoxyhydrosilanes set the stage for the formation of the corresponding enamines in excellent yields (Scheme 4).67 In sharp contrast to Nolan and Nikonov reports, the α-proton is removed, which finally leads to the formation of carbon–carbon double bonds. A high chemoselectivity and efficiency of this approach are remarkable, but it must be emphasized that trialkoxyhydrosilanes are quite toxic and their use requires special precaution (i.e., they may cause eye damage).
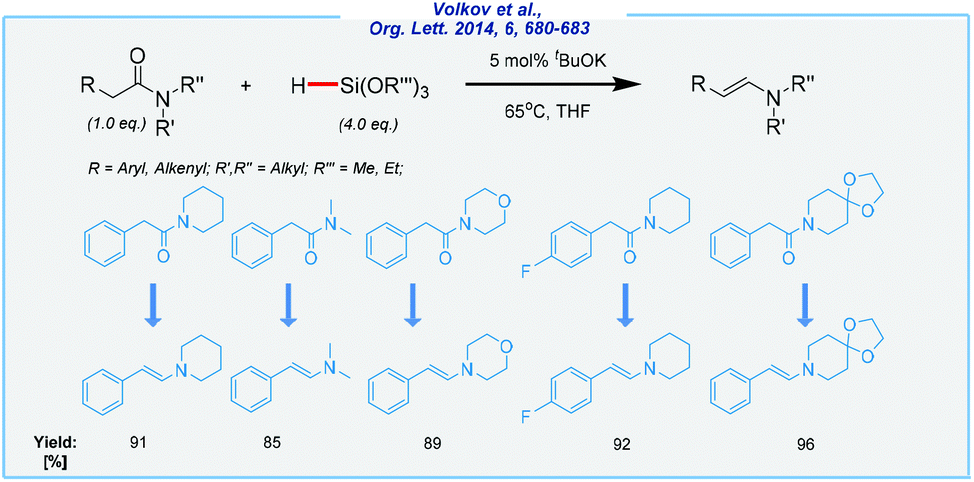 | ||
| Scheme 4 General scheme and substrate scope for tBuOK-mediated reduction of tertiary amides with trialkoxyhydrosilanes. | ||
In independent studies, the Cui group developed an expedient synthetic solution for the preparation of several alcohols from aldehydes and ketones (Scheme 5).68 Here, the combination of catalytic amounts of Cs2CO3 and expensive Ph2SiH2 as a reducing agent enabled this transformation under solvent-free conditions. Cesium is far less common in the Earth's crust, but its compounds are still relatively inexpensive. All operations were carried out at rt. Various functional groups were well-tolerated and finally, a wide range of alcohols was produced in very good yields. Notably, the α,β-unsaturated aldehydes were selectively reduced to the corresponding 1,2-addition products. Furthermore, the Yang group applied carbonate-mediated hydrosilylation of carbonyls to the selective reduction of biobased derivatives like furfural and 5-hydroxymethylfurfural (HMF).69,70 In the case of HMF, the hydrosilylation was achieved by using Ph2SiH2 in the presence of potassium carbonate at rt. As a result, 2,5-bis(hydroxymethyl)furan was isolated in excellent yield (94%). On the other hand, furfural was reduced to furfuryl alcohol over cesium carbonate in yields up to 99%. This time, PMHS was used as an eco-friendly and inexpensive reducing agent. However, DMF was applied as the solvent, to ensure homogeneity and high conversion.
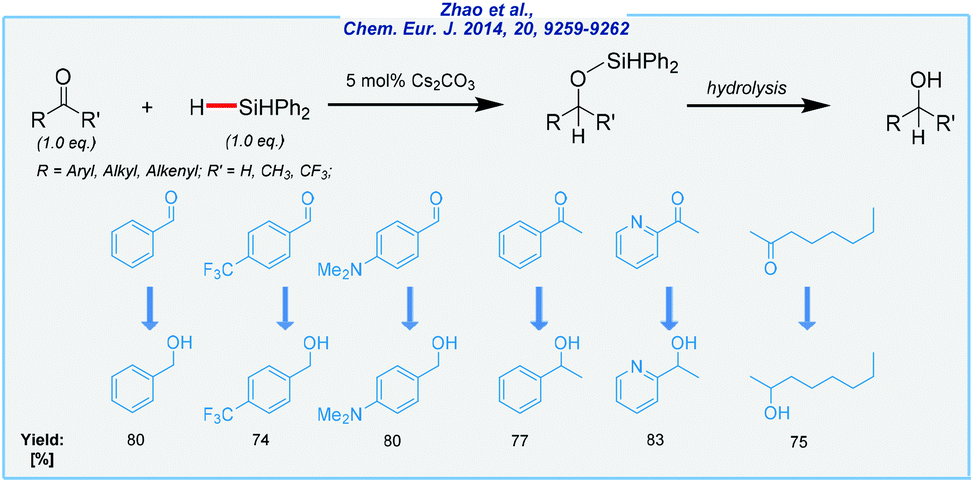 | ||
| Scheme 5 Cesium carbonate-catalyzed hydrosilylation of aldehydes and ketones. | ||
In 2014, the Crabtree group demonstrated that a wide range of unsaturated compounds, including aldehydes, ketones, esters, amides, imines, and N-heterocycles, can be easily reduced in the presence of lithium and sodium borohydrides (Scheme 6).71 This extremely simple hydrosilylation procedure is based on commercially available catalysts and inexpensive silyl reagents (PMHS). All transformations are carried out under mild conditions (rt–60 °C) and give desired products in good yields. Furthermore, the authors also investigated the mechanism in detail and presented very useful information.
 | ||
| Scheme 6 Metal borohydride-catalyzed reduction of various unsaturated systems. | ||
Next, Sekar and co-workers reported K3PO4-mediated hydrosilylation of α-ketoamides to form α-hydroxyamides and 3-phenyloxindoles at rt.72 Various substituents, both electron-donating and electron-withdrawing, are compatible with the reaction conditions and led to very good yields of products. PMHS ensures cost-effectiveness of this transformation. Notably, this methodology offers convenient access to several Passerini adducts by avoiding harmful isocyanides. Later, the same research group improved their method by utilizing cesium carbonate as the catalyst. What is more, 2-methyltetrahydrofuran was used as the solvent in exchange for a carcinogen 1,4-dioxane (Scheme 7).73
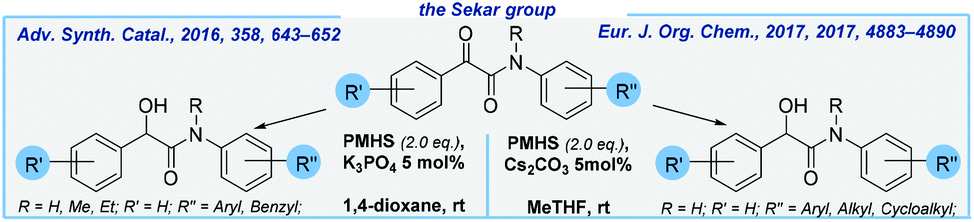 | ||
| Scheme 7 General schemes for base-mediated formation of α-hydroxyamides. | ||
Subsequently, Ding et al. exploited the KOH-catalyzed system for the reduction of cyclic imides (Scheme 8).74 Upon the investigation of the reaction scope, several ω-hydroxylactams and aryl lactams were obtained in quite good yields, utilizing PMHS or Ph2SiH2 as reducing agents. However, the main drawback of this approach is the need to use DMF as the solvent.
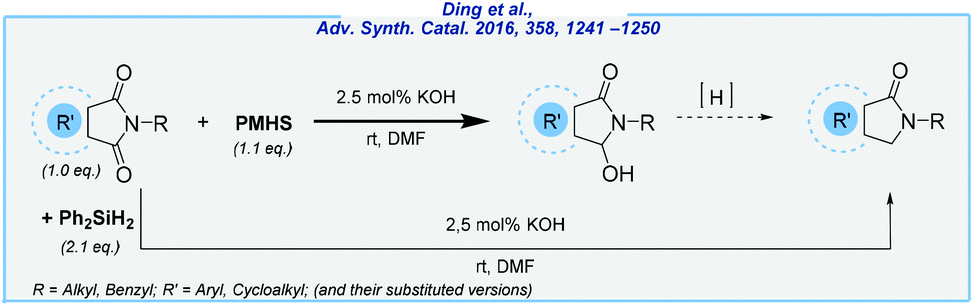 | ||
| Scheme 8 General scheme for KOH-mediated reduction of cyclic imides with PMHS or Ph2SiH2. | ||
Next, the Adler group has reported an unusual approach enabling the reduction of carbonyls with air-stable hydrosilatranes (Fig. 3).75,76 This is a very interesting alternative to simple hydrosilanes. 1-Hydrosilatrane is very expensive in the commercial market, but it might be individually synthesized from cheap and easily accessible reagents, and finally, it can be stored for several months without any special handling. Of the major drawbacks should be noted are an extremely high excess of a base activator and the use of DMF as the solvent.
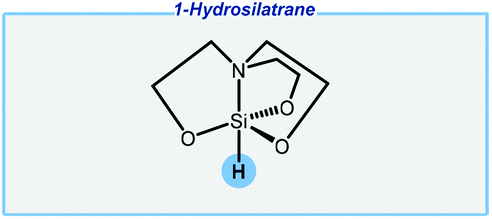 | ||
| Fig. 3 Structure of 1-hydrosilatrane. | ||
Meanwhile, the hydrosilylation of aldehydes and ketones under base catalysis also became possible by the utilization of potassium carbonate at rt (Scheme 9).77 In this process, both carbonyls were effectively reduced with inexpensive PMHS to deliver corresponding alcohols. Under an oxygen atmosphere, the reaction was carried out in DMF and offered satisfactory product yields. The optimization studies showed that efficient reduction of aldehydes is possible with 25 mol% of K2CO3, whilst ketones required 2 eq. of potassium salt as the activator.
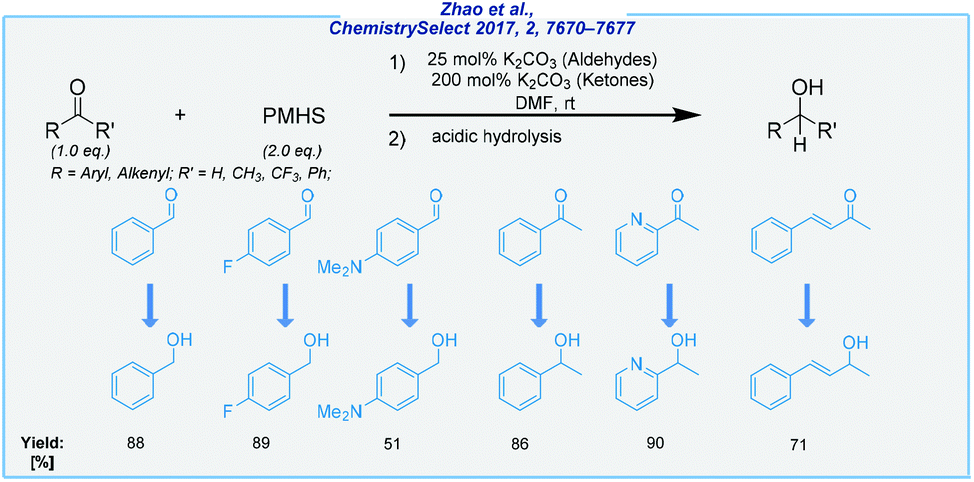 | ||
| Scheme 9 Potassium carbonate-catalyzed hydrosilylation of aldehydes and ketones. | ||
Later, the Xie group developed an efficient strategy for the formation of isoindoles and isoquinolines (Scheme 10).
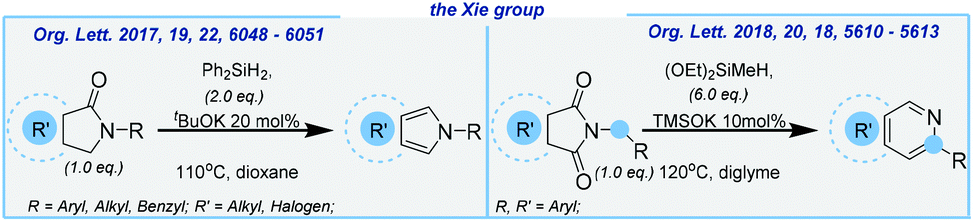 | ||
| Scheme 10 General schemes for base-mediated formation of isoindoles and isoquinolines. | ||
In the latter, a quite expensive diethoxymethylsilane, sensitive potassium silanolate and very toxic diglyme solvent were used. Both reactions delivered the corresponding products in moderate yields.
In continuation of Crabtree's idea, the Yao group has developed the selective cleavage of inert C–N bonds in tertiary amides with triethoxysilane as the reducing agent.78 The reactions were carried out under mild conditions, using a simple NaBEt3H catalyst in the absence of any additives. Most recently, the same research group also reported a controlled reduction of amides by using different hydrosilanes (Scheme 11).79
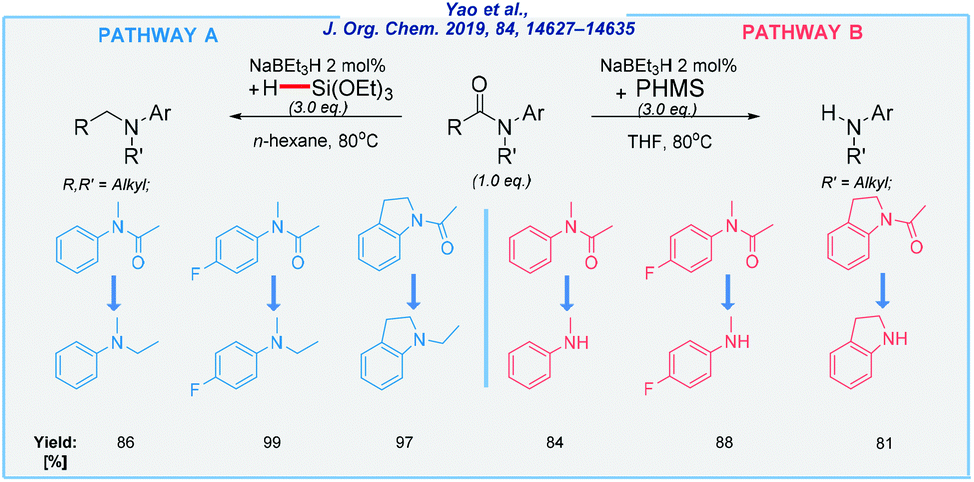 | ||
| Scheme 11 NaBEt3H-catalyzed reduction of various amides. | ||
This method is compatible with a wide range of functional groups, including halides, alkoxy, and trifluoromethyl moieties. All of them can be reduced in a controlled way to deliver corresponding tertiary or secondary amines in extremely high yields, depending on the type of reductant. All reactions were carried out at elevated temperature (80 °C) in the presence of n-hexane or THF as the solvent. According to pathway A, the amide undergoes C–O bond dissociation. In pathway B, the selectivity was switched to the C–N bond scission (see Scheme 11).
Harder and co-workers reported that potassium, calcium, strontium and barium-based catalysts can be used for an efficient reduction of imines.80,81 Notably, the authors showed that the incorporation of well-defined ligands is not necessary, and Si–H bonds might be easily activated by simple amides such as alkali and alkaline earth metal bis(trimethylsilyl)amides. One amongst them is easy to use potassium hexamethyldisilazide, which is commercially available. In each case, an expensive phenylsilane was used as a reducing agent and the reactions were performed in deuterated benzene. However, it is worth pointing out in the end that any of the optimization studies has been performed, which opens a wide field for further investigations.
In contrast, there are also more sophisticated synthetic solutions utilizing more complex s-block species. Certainly, the discoveries done by the pioneers such as Harder, Hill, Okuda, and Sadow might be brought forward here.80–88 They are less sustainable, and the installation of special ligands requires a number of steps, during which undesired wastes are generated. In this respect, it should however be noted that such research investigations are extremely interesting and provide a wide range of theoretical answers that may have an impact on practical approaches, especially in order to ensure high selectivity for the synthesis of fine chemicals. Thus, given the fact that all of these protocols mainly concern mechanistic matters, we decided to not discuss them.
Hydroboration of unsaturated carbon–heteroatom (O, N) moieties
A substantial amount of work related to the hydroboration of all unsaturated systems was carried out by Herbert C. Brown, who is recognized as the godfather of organoboron chemistry. His contribution opened up a new method for preparing several boron derivatives.89–93 The innumerable catalytic solutions have been implemented over many years, starting with those based on transition metals,94–102 through those utilizing rare-earth element complexes,103–108 up to the main group species-catalyzed approaches.109–113 Once more, our attention was drawn in particular to the procedures utilizing Earth-abundant s-block species. As a result, selected examples were discussed with an emphasis on sustainability problems.In 2011, Query et al. reported on the pioneering example of the reduction of ketones in the presence of basic sodium tert-butoxide at rt (Scheme 12).114 The authors suggested the formation of trialkoxyborohydride derived from the catalyst and pinacolborane (HBPin), which can subsequently serve as the active hydride source in the hydroboration of ketones. The scope of substrates was quite narrow, but the reaction proceeded with very good functional tolerance. To conclude, the method developed by the Clark group has many advantages, including a simple catalyst, mild conditions, and very good isolated yields. Of the major drawbacks should be noted is the use of toluene as the solvent.
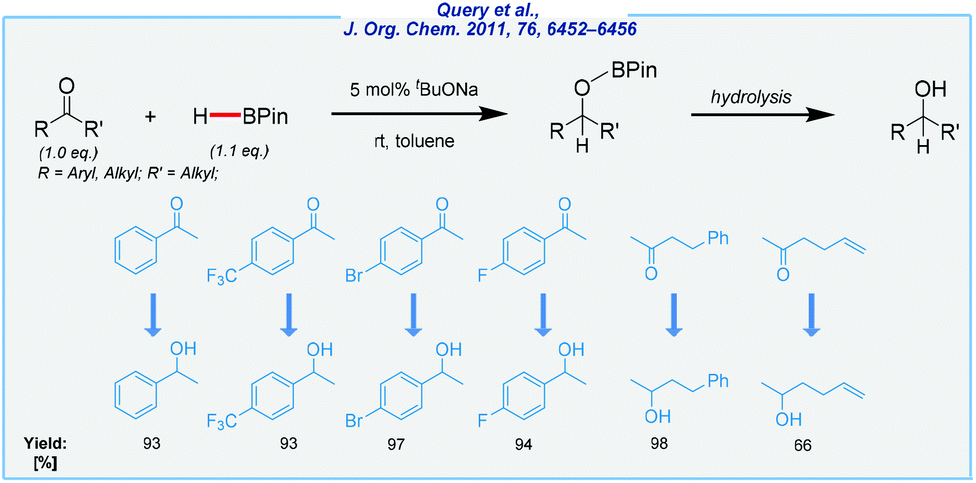 | ||
| Scheme 12 Alkoxide-catalyzed hydroboration of ketones. | ||
After these remarkable findings, we had to wait relatively long for the next versions of base-catalyzed hydroboration protocols. In 2017, Nembenna reported the amide-catalyzed selective reduction of esters with HBPin (Scheme 13).115
 | ||
| Scheme 13 Mg(HMDS)2-catalyzed hydroboration of esters. | ||
Initially, the authors tested both simple and much more complex magnesium amides. As a result, magnesium bis(trimethylsilyl)amide [Mg(HMDS)2] turns out to be a very active catalyst, and a variety of esters was effectively reduced to provide the corresponding alcohols, even in the presence of other functional groups (e.g., nitro, alkynes, alkenes, etc.). Interestingly, in the case of liquid substrates, the reactions were carried out under solvent-free conditions. Notably, the catalyst loading was remarkably low (0.1–0.5 mol%). Of the major drawbacks should be noted are a high price and sensitivity of Mg(HMDS)2, which implies the need for an inert atmosphere.
Literally, at the same time as Nembenna and his co-workers, Wu et al. achieved base-catalyzed hydroboration of various unsaturated systems (aldehydes, ketones, alkenes, alkynes, and imines), in the presence of sodium hydroxide as the catalyst (Scheme 14).116
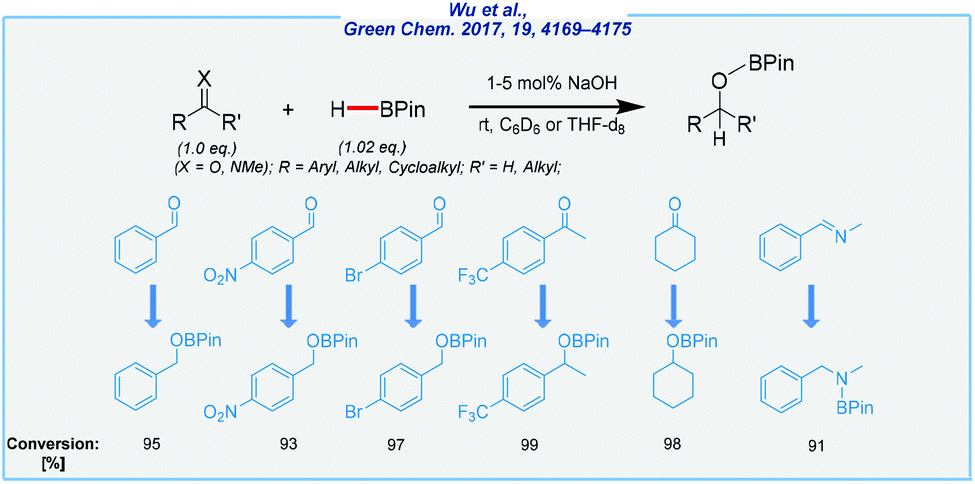 | ||
| Scheme 14 General scheme and substrate scope for NaOH-mediated hydroboration of aldehydes, ketones, and imines with pinacolborane. | ||
Taking into account the combination of desirable features, such as operation simplicity, the high yields, benign reaction conditions, and the utilization of inexpensive and commercially available NaOH as the catalyst, this method is expected to provide a promising alternative and attractive approach to a series of organoboron compounds. Well, on the one hand, the authors were able to extend the substrate scope to a wide range of unsaturated compounds. On the other hand, the hydrolysis of the obtained boronate esters was not performed and all reactions were carried out in deuterated benzene. The authors also tested a less toxic alternative in the form of deuterated tetrahydrofuran. This shows further opportunities to improve the described approach such as the development of a one-pot procedure for the hydroboration/hydrolysis processes.
Subsequently, several efficient protocols have been devised for the hydroboration of related substrates with catalytic amounts of various compounds. Sen and his co-workers studied different lithium compounds such as 2,6-di-tert-butylphenoxylithium (1c), 1,10-dilithioferrocene (2c) and the nacnac lithium complex (3c) (Fig. 4).117
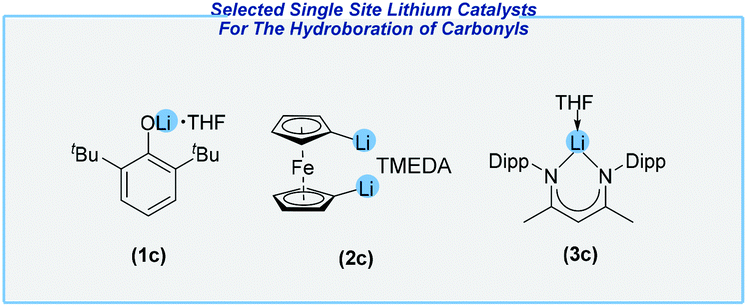 | ||
| Fig. 4 Structures of catalytically active single site lithium compounds. | ||
All of them have been found to be efficient catalysts for the reduction of carbonyls at ambient temperature, with HBPin as the reducing agent. Later, however, the most Lewis acidic Li center (3c) revealed the highest catalytic activity. Both ketones and aldehydes were efficiently converted, and thus delivered several boronate esters with different functional groups in very good yields. Despite their low toxicity, neither of them is commercially available, which is a significant practical problem. Concurrently, the Bao and An groups reported that the readily accessible n-butyllithium reagent is an extremely active catalyst for the hydroboration of a variety of aldehydes and ketones (Scheme 15).118,119
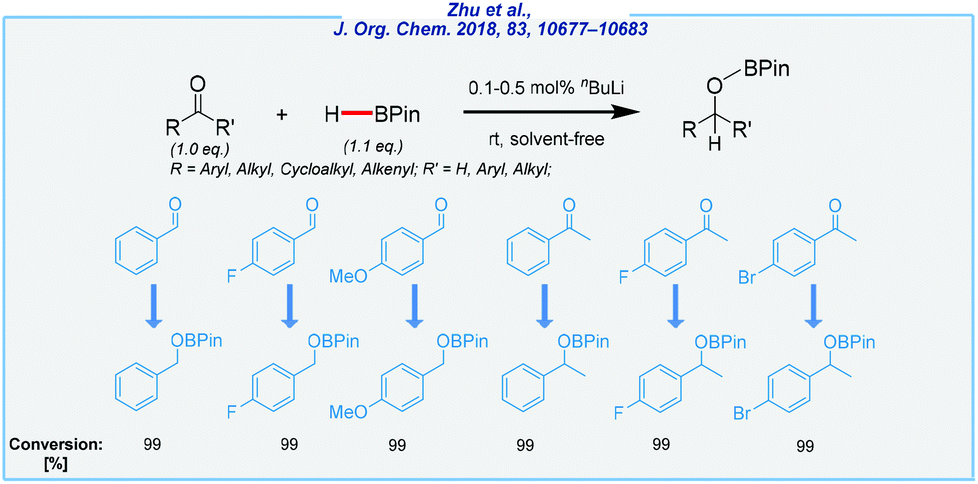 | ||
| Scheme 15 n BuLi-catalyzed hydroboration of aldehydes and ketones. | ||
The low catalyst loading is remarkable (even to 0.1 mol%, under solvent-free conditions). However, it should be noted that commercially available nBuLi is always marketed as a solution in alkanes. Despite their many virtues, and in the light of other methods, the practicability of these protocols is questionable, due to the high sensitivity of n-butyllithium. Nevertheless, Bao and Shi recently expanded its utility in the hydroboration of allylic alcohols,120 alkenes, alkynes, and imines.121,122
Meanwhile, An and co-workers disclosed an example of hydroboration of aldehydes and ketones in the presence of sodium hydride at rt, to give corresponding alcohols (after hydrolysis) in almost quantitative yields (Scheme 16).123
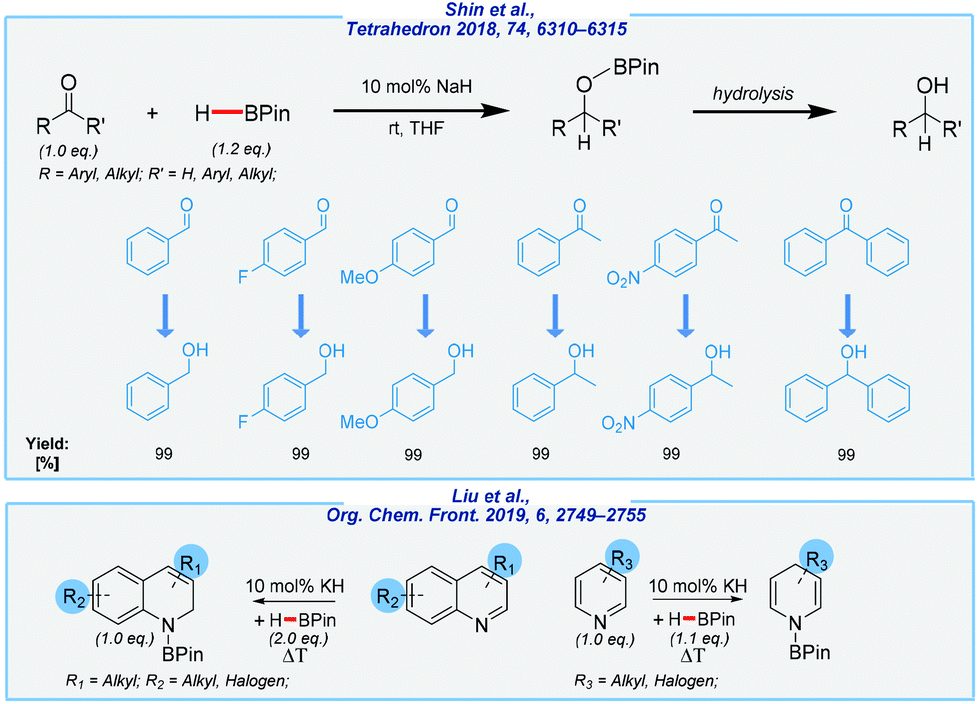 | ||
| Scheme 16 Alkali metal hydride-catalyzed hydroboration of various unsaturated systems. | ||
A considerable drawback of this protocol is again connected to the sensitivity of the catalyst. A reaction involving its utilization requires special care and is extremely hazardous. Admittedly, the authors also tested a commercially available calcium hydride, which is easier to use than similar alkali hydrides, but it showed lower activity (conversion up to 77%). In our opinion, the use of CaH2 seems to be much more practical, and further research is needed to determine its usefulness. The outstanding activity of alkali metal hydrides in B–H additions was recently extended by the Zhang group (Scheme 16).124 Here, the authors devised a regioselective KH-mediated 1,2 or 1,4-hydroboration of N-heteroarenes with pinacolborane, which provided access to N-borylated-1,2-reduction products (up to 99% yield) and N-borylated-1,4-reduction products (up to 95% yield). Despite its roughly similar disadvantages, the simplicity of this approach should be made clear.
At almost the same time, the Chang group reported a similar procedure utilizing less sensitive potassium alkoxide as the catalyst (Scheme 17).125 Here, the reduction occurs selectively at the 1,4-position under an argon atmosphere, and in the presence of expensive 18-crown-6 ether. In addition, the authors disclosed detailed mechanistic studies. Finally, the same research group also explored the related transformation using imidazolium salts as a catalyst precursor, which proceeds with remarkably high regioselectivity at the 1,2-position. This exploration goes far beyond the scope of this review but deserves particular attention.126
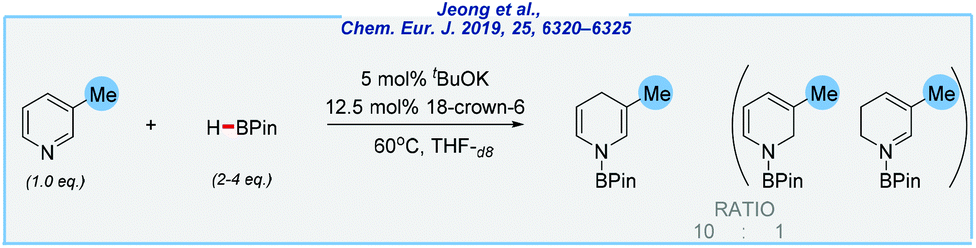 | ||
| Scheme 17 The example of tBuOK-catalyzed hydroboration of heterocycles. | ||
Another illustrative example of alkoxide-mediated hydroboration is the utilization of lithium tert-butoxide.127 In contrast to Clark's work, the An group was able to extend the substrate scope to include aldehydes and esters (Scheme 18). What is more, less toxic tetrahydrofuran was used as the solvent. In the case of simple carbonyls, the catalyst loading was remarkably low (1 mol%). The reduction of esters was more challenging and required 40 mol% of tBuOLi and a significant excess of HBPin (3.0 eq.).
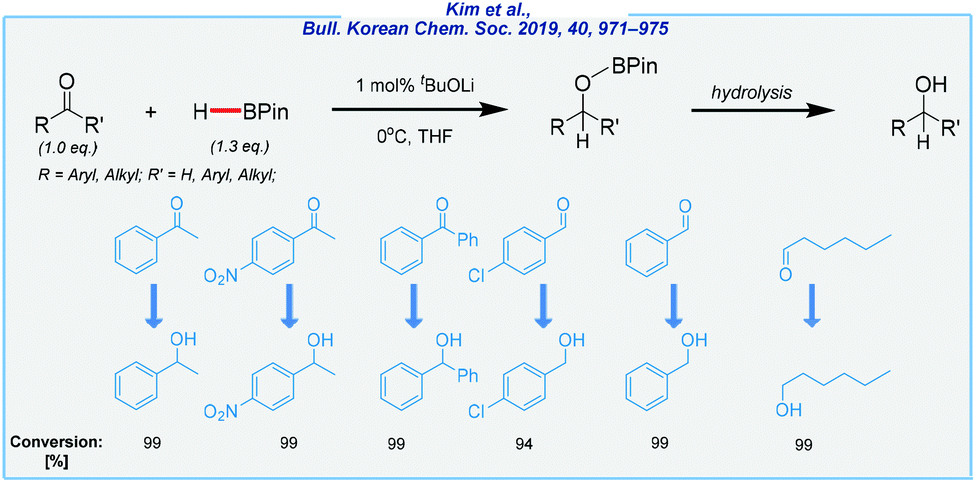 | ||
| Scheme 18 t BuOLi-catalyzed hydroboration of aldehydes and ketones. | ||
Next, the same group developed an efficient protocol for hydroboration of aldehydes, ketones, and alkenes using potassium carbonate as the catalyst (Scheme 19).128 This salt is widely accessible, non-toxic, inexpensive, and not so sensitive as Grignard reagents. In the case of carbonyls, the reaction proceeds under mild conditions (rt) in tetrahydrofuran, and a low amount of the catalyst is required (0.5 mol% for aldehydes, 5 mol% for ketones). The reduction of alkenes was more challenging and required elevated temperature (110 °C).
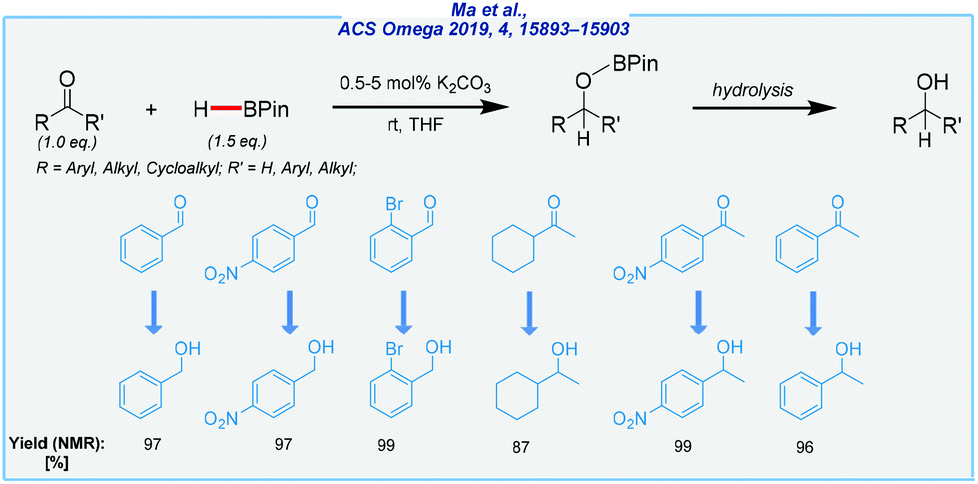 | ||
| Scheme 19 K2CO3-catalyzed hydroboration of carbonyls. | ||
On the other hand, it was conducted under neat conditions. It should be noted that the analogous method utilizing hydrosilanes as reductants in the presence of K2CO3 was less efficient and required higher amounts of the catalyst (25 mol% for aldehydes, 200 mol% for ketones) and hazardous DMF as the solvent. Considering this, the presented approach has strong potential and might be useful for large-scale reductions.
Furthermore, Kuciński and Hreczycho have also developed the B–H activation strategy for the synthesis of several primary and secondary alcohols (Scheme 20).129 A wide range of aldehydes, and more importantly also ketones, was reduced with pinacolborane in the presence of lithium triethylborohydride under mild conditions (air atmosphere, rt). Most of these transformations proceed under solvent-free conditions at very low catalyst loading (0.1–0.3 mol%). The key advantages of this process are its exclusive aldehyde selectivity over ketones, wide reducible functional group tolerance, the possibility of one-pot hydrolysis to desired alcohols, and wide availability of the catalyst, as well as its low price.
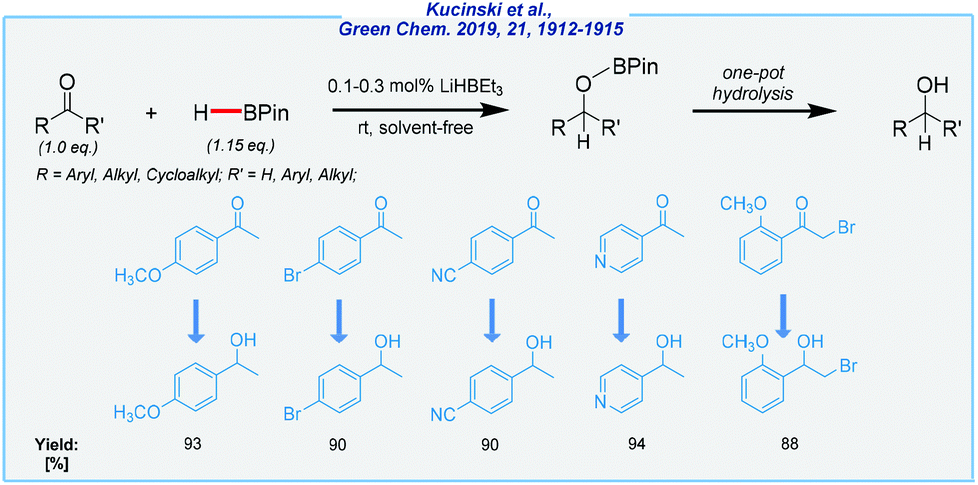 | ||
| Scheme 20 General scheme and substrate scope for LiHBEt3-mediated hydroboration of aldehydes and ketones with pinacolborane. | ||
The group of Rueping reported a series of examples that involve hydroboration of ketones, carbonates, and alkynes (Scheme 21).130–133 In these processes, a commercially available, non-toxic, and quite inexpensive di-n-butylmagnesium solution has been applied as the catalyst. An enantioselective version provides products with excellent yields (up to 99%) and enantioselectivities (er up to 99![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). Here, the chiral magnesium complex with a BINOL backbone was found to be the most efficient one. As has been shown, the alkali salt additive (20 mol%) increases the enantiomeric ratio, as well as decreased temperature (−40 °C), using toluene as the solvent. In our opinion, without a doubt, this method represents a valuable and benign alternative to TM-catalyzed versions. Mostly the same concept also enabled a chemoselective Luche-type reduction of α,β-unsaturated ketones. Besides simple enones and propargylic ketones, some natural derivatives were also feasible, and a broad substrate scope proved the versatility of this method. Finally, an efficient reduction of linear and cyclic organic carbonates was also possible. All of these approaches are characterized by mild conditions and low catalyst loading (especially in the case of α,β-unsaturated ketones; 0.2 mol%).
:1). Here, the chiral magnesium complex with a BINOL backbone was found to be the most efficient one. As has been shown, the alkali salt additive (20 mol%) increases the enantiomeric ratio, as well as decreased temperature (−40 °C), using toluene as the solvent. In our opinion, without a doubt, this method represents a valuable and benign alternative to TM-catalyzed versions. Mostly the same concept also enabled a chemoselective Luche-type reduction of α,β-unsaturated ketones. Besides simple enones and propargylic ketones, some natural derivatives were also feasible, and a broad substrate scope proved the versatility of this method. Finally, an efficient reduction of linear and cyclic organic carbonates was also possible. All of these approaches are characterized by mild conditions and low catalyst loading (especially in the case of α,β-unsaturated ketones; 0.2 mol%).
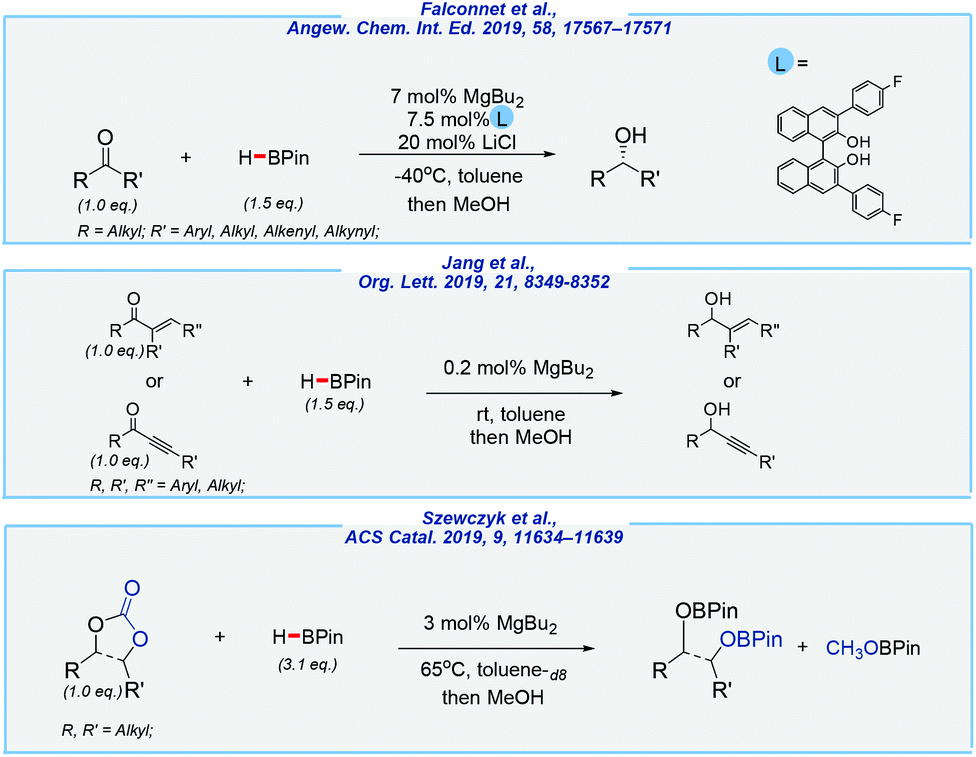 | ||
| Scheme 21 MgBu2-catalyzed hydroboration of various unsaturated derivatives. | ||
Almost at the same time, the Ma group utilized less expensive methylmagnesium iodide as the catalyst for the hydroboration of a wide range of aldehydes and ketones (Scheme 22).134 The reaction proceeds at remarkably low catalyst loadings (aldehydes = 0.05 mol%, ketones = 0.5 mol%), and requires extremely short time (aldehydes = 10 min, ketones = 20 min). What is more, it is conducted under the neat conditions at room temperature.
 | ||
| Scheme 22 MgMeI-catalyzed hydroboration of ketones and aldehydes. | ||
Among the most recent accomplishments, the Gade group reported highly enantioselective hydroboration of ketones in the presence of the magnesium dialkyl precursor [Mg(CH2SiMe3)2THF] and boxmi-H ligand (Fig. 5).135 The reactions were performed with high enantioselectivity, but commercial inaccessibility of some reagents is a major problem.
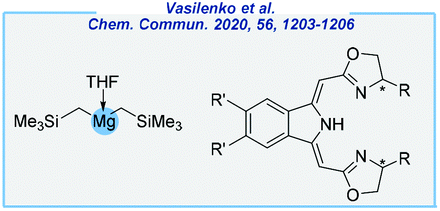 | ||
| Fig. 5 Catalytic system for asymmetric ketone hydroboration reported by the Gade group. | ||
In this context, the Melen group recently reported on the use of commercially available lithium amide in combination with BINOL-derived ligands for the enantioselective hydroboration of acetophenones at rt (Fig. 6).136 In our opinion, the asymmetric catalysis for this reaction is very challenging, and the application of s-block based catalysts is a significant achievement that would be of great interest to other scientists. However, the main drawback of this approach, within the context of green chemistry, is the use of carcinogen 1,4-dioxane as the solvent. Furthermore, there is still considerable room for improvement with regard to the optimal selection of chiral ligands, in particular in view of their prices.
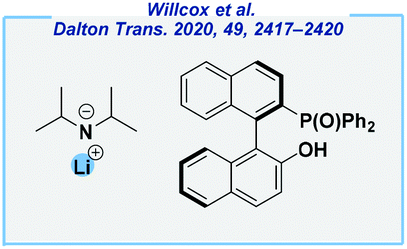 | ||
| Fig. 6 Catalytic system for asymmetric ketone hydroboration reported by the Melen group. | ||
Coming back to the theme of sustainable solutions, just recently, Kuciński et al. developed the KF-mediated B–H addition to several carbonyls (Scheme 23).137
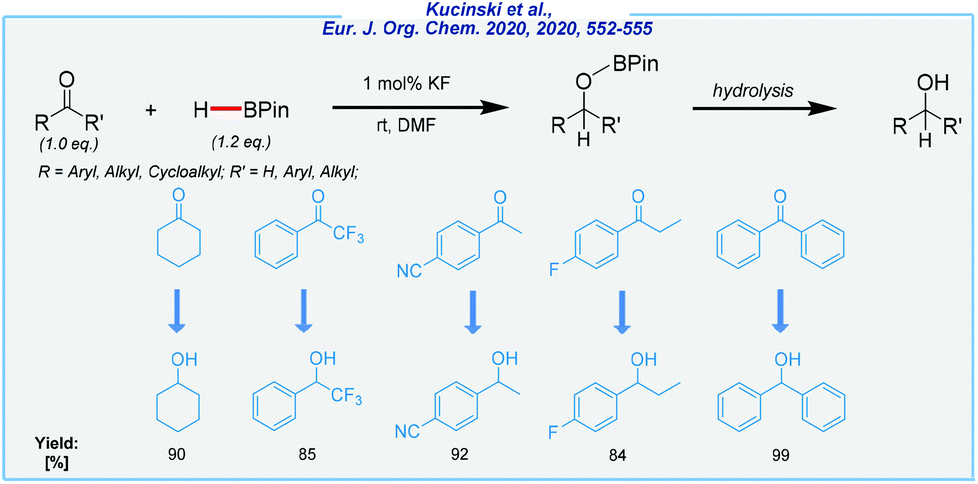 | ||
| Scheme 23 KF-catalyzed hydroboration of carbonyls. | ||
Unlike the hydrosilylation,138–140 this was the first study about the utilization of simple inorganic salts containing fluoride anions (potassium and cesium fluorides) in such transformation. Hydroboration occurs under mild conditions (air atmosphere, room temperature). As a result, the numerous ketones and aldehydes, bearing a wide array of electron-withdrawing and electron-donating groups, were successfully reduced to the corresponding alcohols (after the hydrolysis step). Of the major drawbacks should be noted is the use of DMF as the solvent.
Next, the Findlater group achieved highly efficient hydroboration of organic nitriles with HBPin, in the presence of sodium triethylborohydride (Scheme 24).141
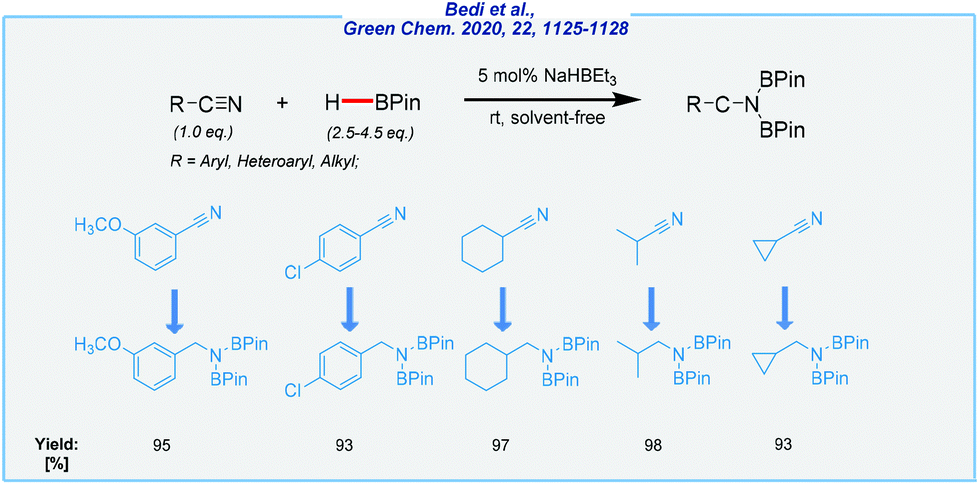 | ||
| Scheme 24 General scheme and substrate scope for NaHBEt3-mediated hydroboration of nitriles with pinacolborane. | ||
The power of this catalytic system was reflected by solvent-free synthesis under extremely mild conditions (rt), which is quite difficult in the case of nitriles. Moreover, it resolves several problems related to waste generation. This combined with wide accessibility of the catalyst and its price could be seen as a step in the right direction.
In a related process, von Wangelin and his co-workers achieved the double hydroboration of nitriles with a remarkably low amount of pinacolborane (2.1 equiv.) as the reductant (Scheme 25).142 The reaction occurred under mild conditions (20–70 °C) in the presence of lithium bis(trimethylsilyl)amide (10 mol%). Despite the fact that the applied catalyst is commercially available, its high sensitivity to moisture should be noted. Moreover, all reactions were investigated in deuterated benzene.
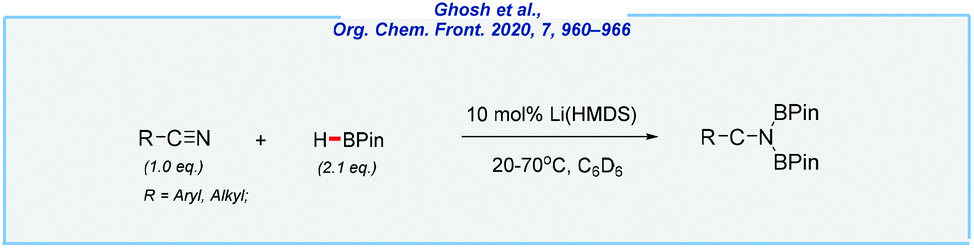 | ||
| Scheme 25 Li(HMDS)-catalyzed hydroboration of nitriles. | ||
Very recently, the Mandal group further expanded the capacity of disilazide salts.143 They employed a potassium complex consisting of potassium bis(trimethylsilyl)amide and abnormal N-heterocyclic carbene (Fig. 7). Such a catalytic system allows a selective reduction of aliphatic, aromatic and heterocyclic primary amides with excellent functional group tolerance. The reaction proceeds at low catalyst loading (2 mol%) in toluene, under mild conditions (40 °C). Despite some critical drawbacks in the context of green chemistry, the presented discovery brings many interesting mechanistic conclusions, including the Lewis acidic activation by potassium cations.
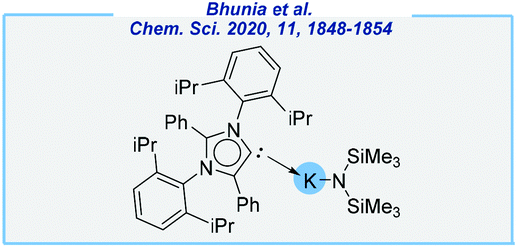 | ||
| Fig. 7 aNHC-based potassium complex used by the Mandal group. | ||
Most recently, the Panda group reported the hydroboration of aldimines with pinacolborane (Scheme 26).144 Here, catalytic amounts of potassium benzyl (5 mol%) enabled the synthesis of N-borylated products, which upon hydrolysis furnishes a wide range of secondary amines. The key advantages of this process are mild conditions (rt), and the solvent-free synthesis. Of the major drawbacks should be noted is an extremely high sensitivity of C6H5CH2K, which implies the need for the inert atmosphere.
 | ||
| Scheme 26 C6H5CH2K-catalyzed hydroboration of aldimines. | ||
Also this time, the contemporary developments in the discovery of sophisticated alkali and alkaline earth metal catalysts for the hydroboration are not presented in detail here.145–168 The majority of these approaches requires further investigations that will allow for a better understanding of these metal complexes, while taking into account the aspects of green chemistry.
At this point, we would like to raise an interesting issue. Most recently, the Thomas group published a brilliant article on the subject of nucleophile-mediated decomposition of pinacolborane.169 The authors showcased strong evidence that most commonly used nucleophiles (intended to be the catalysts) only promote the formation of BH3 which serves as a true catalyst in the hydroboration of alkynes and alkenes. However, in the case of ketones (and most likely for other carbonyl derivatives), the formation of triorganoborohydride species is proposed, which is in line with previous Clark's discoveries.114 The mechanistic matters are not dealt with in this review; however, Thomas and his co-workers touched on a vital issue. In consequence, future studies need to provide more insights regarding the nature of the process.
Catalyst-free approaches
Conversely, in contrast to hydrosilylation, the hydroboration of various unsaturated moieties can proceed in the absence of any catalyst. In a historical context, when the boron source is highly reactive diborane (B2H6) or borane adducts (e.g., BH3·THF), the reaction occurs rapidly at room temperature, in the absence of any catalyst.170–172 There are also some pioneering studies reported by Knochel and Piers, which used different boranes in the catalyst-free hydroboration of alkenes and alkynes.173–175 But before we get to recent examples, it should be noted that absolute exclusion of even traces of any catalytic species is extremely difficult. These words might be confirmed by a recent article written by Ananikov – a world-class specialist in the field of catalysis.176 This amazing publication has shown the importance of every little detail even like defects of magnetic stir bars. However, in the case of boranes (and despite the potential contaminations of substrates and equipment), the nature of these compounds (e.g., Lewis acidity, extremely high oxophilicity,177etc.) allows assuming the possibility of the catalyst-free pathways.In 2018, the Hreczycho group reported on the pioneering example of the hydroboration of aldehydes with pinacolborane, under catalyst-free and solvent-free conditions at rt–60 °C (Scheme 27).178 They were encouraged by their previous results and the work disclosed by the Bertrand group, which concerned uncatalyzed borylation of various O-nucleophiles.179–181 It should be noted that the authors of several previous publications have unanimously claimed that catalyst-free hydroboration of benzaldehyde displays very low conversion into the boronic ester. However, all of them performed the reactions in the presence of the solvent. Thus, the absence of any solvent seems to be crucial for the entire process and allows the target compounds to be obtained in excellent yields and in a short period of time. Following aldehydes, the hydroboration of ketones was also investigated, although this was found to be less efficient. The described results became an inspiration for other researchers, which led to significant progress in this field.
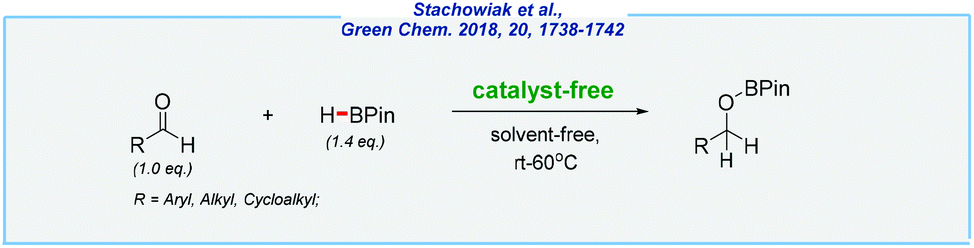 | ||
| Scheme 27 Catalyst-free and solvent-free hydroboration of aldehydes. | ||
Likewise, Wang et al. extended this idea to reduce a wide range of ketones (Scheme 28).182 This was made possible by slight changes in the reaction conditions. Therefore, by raising the temperature to 80 °C (or even 140 °C), the authors were able to overcome an energy barrier, which resulted in the desired boronic esters. The presented approach has also strong potential and might be useful for large-scale reductions. As an example, 5.50 g of 1-phenylethanol has been obtained in high yield (90%).
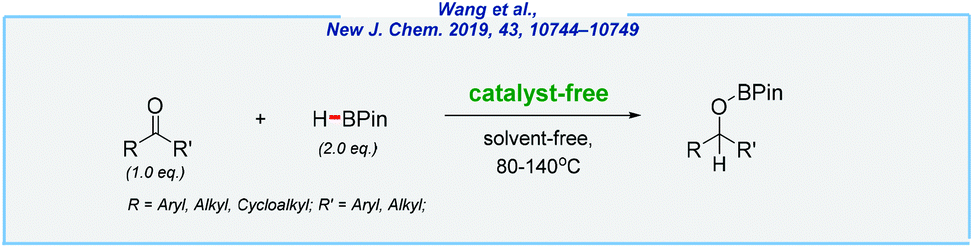 | ||
| Scheme 28 Catalyst-free and solvent-free hydroboration of ketones. | ||
Almost at the same time, three independent procedures have been reported for the hydroboration of carboxylic acids in a catalyst-free manner by the groups of Panda,183 Ma,184 and Xue185 (Scheme 29).
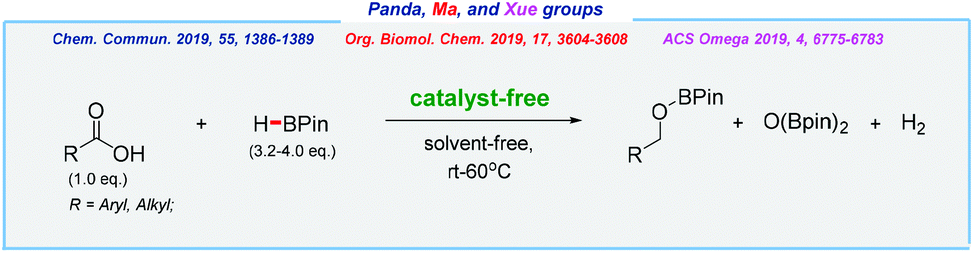 | ||
| Scheme 29 Catalyst-free and solvent-free hydroboration of carboxylic acids. | ||
Both aromatic and aliphatic carboxylic acids were reduced to boronate esters, which upon hydrolysis furnishes a wide range of primary alcohols. Of the major drawbacks should be noted is the formation of two by-products – diboraxane derivative, as well as hazardous dihydrogen molecules.
Finally, the Rit group developed an expedient approach for the synthesis of secondary amines via the hydroboration of imines (Scheme 30).186
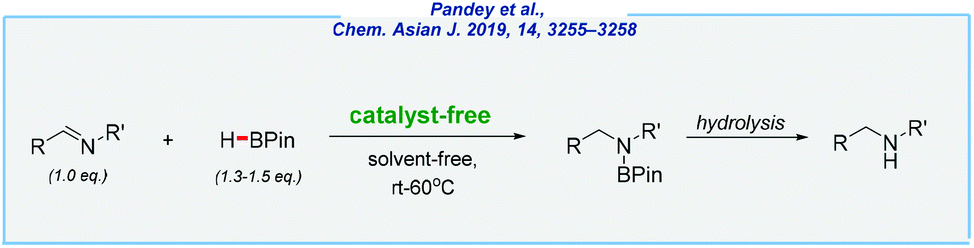 | ||
| Scheme 30 Catalyst-free and solvent-free hydroboration of imines. | ||
Here, all reactions were carried out in a catalyst-free and solvent-free manner, at room temperature. This simple protocol enables the reduction of diverse imines including several aldimines as well as ketimines and shows very good functional tolerance (chemoselective conversion of imines in the presence of alkene, alkyne, ketone, nitro, and nitrile moieties). The presented approach has also strong potential and might be useful for gram-scale reductions. As an example, 0.86 g of N-benzylaniline has been obtained in high yield (85%). Very recently, the same research group extended this idea to perform reductive amination under catalyst-free and solvent-free conditions.187
Conclusions
Over the past ten years, s-block metal-catalyzed hydroelementations have attracted considerable attention. Several developed methods have proved their usefulness, also taking into account the aspects of green chemistry. In some cases, they were even able to outperform their precious metal counterparts. Given the sustainable nature of these catalysts, further exciting advances are expected in this fascinating research arena. It is hoped that further achievements will meet the criteria of broadly understood green synthesis, which will ensure the sustainable development of our societies.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We owe a very special thanks to Renata Żbel for painting the graphical abstract. This work was supported by a National Science Centre Grant UMO-2018/30/E/ST5/00045 (GH).Notes and references
- H. Maciejewski, C. Pietraszuk, P. Pawluć and B. Marciniec, Hydrosilylation - A Comprehensive Review on Recent Advances, Springer, Berlin, 2009, vol. 1 Search PubMed.
- A. G. M. Barrett, M. R. Crimmin, M. S. Hill and P. A. Procopiou, Proc. R. Soc. A, 2010, 466, 927–963 CAS.
- R. J. Hofmann, M. Vlatkovic and F. Wiesbrock, Polymers, 2017, 9, 534–570 CrossRef PubMed.
- J. V. Obligacion and P. J. Chirik, Nat. Rev. Chem., 2018, 2, 15–34 CrossRef CAS PubMed.
- D. Wei and C. Darcel, Chem. Rev., 2019, 119, 2550–2610 CrossRef CAS PubMed.
- T. Hackel and N. A. McGrath, Molecules, 2019, 24, 432 CrossRef PubMed.
- M. L. Shegavi and S. K. Bose, Catal. Sci. Technol., 2019, 9, 3307–3336 RSC.
- A. Y. Li and A. Moores, ACS Sustainable Chem. Eng., 2019, 7, 10182–10197 CrossRef CAS.
- S. R. Tamang and M. Findlater, Molecules, 2019, 24, 3194 CrossRef CAS PubMed.
- M. Iglesias, F. J. Fernández-Alvarez and L. A. Oro, Coord. Chem. Rev., 2019, 386, 240–266 CrossRef CAS.
- H. Wen, G. Liu and Z. Huang, Coord. Chem. Rev., 2019, 386, 138–153 CrossRef CAS.
- S. Harder, in Early Main Group Metal Catalysis Concept, ed. S. Harder, Wiley-VCH Verlag GmbH & Co. KGaA., 1st edn., 2020, pp. 151–173 Search PubMed.
- K. Riener, M. P. Högerl, P. Gigler and F. E. Kühn, ACS Catal., 2012, 2, 613–621 CrossRef CAS.
- A. D. Sadow, in Early Main Group Metal Catalysis, ed. S. Harder, Wiley-VCH Verlag GmbH & Co. KGaA., 1st edn., 2020, pp. 201–224 Search PubMed.
- M. Bhunia, P. Sreejyothi and S. K. Mandal, Coord. Chem. Rev., 2020, 405, 213110 CrossRef CAS.
- S. J. Geier, C. M. Vogels and S. A. Westcott, in Boron Reagents in Synthesis, ed. A. Coca, American Chemical Society, New Haven, 2016, pp. 209–225 Search PubMed.
- B. Chatterjee and C. Gunanathan, J. Chem. Sci., 2019, 131, 1–22 CrossRef.
- S. Park, ChemCatChem, 2020, 12, 3170–3185 CrossRef CAS.
- H. Liu and M. S. Eisen, Synthesis, 2020, 52, 629–644 CrossRef CAS.
- Á. Raya-Barón, P. Oña-Burgos and I. Fernández, ACS Catal., 2019, 9, 5400–5417 CrossRef.
- Y. Nakajima and S. Shimada, RSC Adv., 2015, 5, 20603–20616 RSC.
- K. Revunova and G. I. Nikonov, Dalton Trans., 2015, 44, 840–866 RSC.
- C. C. Chong and R. Kinjo, ACS Catal., 2015, 5, 3238–3259 CrossRef CAS.
- B. Li, J.-B. Sortais and C. Darcel, RSC Adv., 2016, 6, 57603–57625 RSC.
- A. Volkov, F. Tinnis, T. Slagbrand, P. Trillo and H. Adolfsson, Chem. Soc. Rev., 2016, 45, 6685–6697 RSC.
- M. S. Hill, D. J. Liptrot and C. Weetman, Chem. Soc. Rev., 2016, 45, 972–988 RSC.
- M. Zaranek, B. Marciniec and P. Pawluć, Org. Chem. Front., 2016, 3, 1337–1344 RSC.
- T. K. Meister, K. Riener, P. Gigler, J. Stohrer, W. A. Herrmann and F. E. Kühn, ACS Catal., 2016, 6, 1274–1284 CrossRef CAS.
- S. C. Shit and P. Shah, Natl. Acad. Sci. Lett., 2013, 36, 355–365 CrossRef CAS.
- K. Kuciński and G. Hreczycho, Inorg. Chim. Acta, 2017, 461, 233–238 CrossRef.
- C. Xu, B. Huang, T. Yan and M. Cai, Green Chem., 2018, 20, 391–397 RSC.
- M. Jankowska-Wajda, O. Bartlewicz, A. Walczak, A. R. Stefankiewicz and H. Maciejewski, J. Catal., 2019, 374, 266–275 CrossRef CAS.
- A. Walczak, H. Stachowiak, G. Kurpik, J. Kaźmierczak, G. Hreczycho and A. R. Stefankiewicz, J. Catal., 2019, 373, 139–146 CrossRef CAS.
- P. Żak, M. Bołt and C. Pietraszuk, Eur. J. Inorg. Chem., 2019, 2019, 2455–2461 CrossRef.
- V. Pandarus, R. Ciriminna, G. Gingras, F. Béland, S. Kaliaguine and M. Pagliaro, Green Chem., 2019, 21, 129–140 RSC.
- M. Grzelak, R. Januszewski and B. Marciniec, Inorg. Chem., 2020, 59, 7830–7840 CrossRef CAS PubMed.
- J. Chang, F. Fang, J. Zhang and X. Chen, Adv. Synth. Catal., 2020 DOI:10.1002/adsc.202000166.
- B. Marciniec, H. Maciejewski, C. Pietraszuk and P. Pawluc, in Applied Homogeneous Catalysis with Organometallic Compounds, ed. B. Cornils, W. A. Herrmann, M. Beller and R. Paciello, Wiley-VCH Verlag GmbH & Co. KGaA., 3rd edn, 2017, pp. 569–620 Search PubMed.
- M. Zaranek and P. Pawluc, ACS Catal., 2018, 8, 9865–9876 CrossRef CAS.
- R. Rodriguez, D. Gau, Y. Contie, T. Kato, N. Saffon-Merceron and A. Baceiredo, Angew. Chem., Int. Ed., 2011, 50, 11492–11495 CrossRef CAS PubMed.
- R. Jambor and A. Lyčka, Eur. J. Inorg. Chem., 2017, 2017, 4887–4898 CrossRef CAS.
- U. Wietelmann and J. Klett, Z. Anorg. Allg. Chem., 2018, 644, 194–204 CrossRef CAS PubMed.
- D. Seyferth, Organometallics, 2009, 2, 1598–1605 CrossRef.
- I. Ojima, M. Nihonyanagi and Y. Nagai, J. Chem. Soc., Chem. Commun., 1972, 938 RSC.
- I. Ojima, T. Kogure and Y. Nagai, Tetrahedron Lett., 1973, 14, 2475–2478 CrossRef.
- A. K. Roy, Adv. Organomet. Chem., 2007, 55, 1–59 CrossRef.
- S. Díez-González and S. P. Nolan, Acc. Chem. Res., 2008, 41, 349 CrossRef PubMed.
- J. Yang and T. D. Tilley, Angew. Chem., Int. Ed., 2010, 49, 10186–10188 CrossRef CAS PubMed.
- N. Castellanos-Blanco, M. Flores-Alamo and J. J. García, Inorg. Chim. Acta, 2017, 466, 324–332 CrossRef CAS.
- B. Yiğit, M. Yiğit and İ. Özdemir, Inorg. Chim. Acta, 2017, 467, 75–79 CrossRef.
- F. Ritter, D. Mukherjee, T. P. Spaniol, A. Hoffmann and J. Okuda, Angew. Chem., Int. Ed., 2019, 58, 1818–1822 CrossRef CAS PubMed.
- N. U. D. Reshi, L. Kathuria and A. G. Samuelson, Inorg. Chim. Acta, 2019, 486, 119–128 CrossRef CAS.
- O. Martínez-Ferraté, B. Chatterjee, C. Werlé and W. Leitner, Catal. Sci. Technol., 2019, 9, 6370–6378 RSC.
- B. Ghaffari, J. Mendes-Burak, K. W. Chan and C. Copéret, Chem. – Eur. J., 2019, 25, 13869–13873 CrossRef CAS PubMed.
- Q. Zhao, J. Zhang and M. Szostak, Chem. Commun., 2019, 55, 9003–9006 RSC.
- D. J. Parks and W. E. Piers, J. Am. Chem. Soc., 1996, 118, 9440–9441 CrossRef CAS.
- M. X. Tan and Y. Zhang, Tetrahedron Lett., 2009, 50, 4912–4915 CrossRef CAS.
- B. Assoah, J. R. Vale, E. Kalenius, L. F. Veiros and N. R. Candeias, Eur. J. Org. Chem., 2018, 2910–2917 CrossRef CAS.
- A. Chardon, J. Rouden and J. Blanchet, Eur. J. Org. Chem., 2019, 995–998 CrossRef CAS.
- R. J. Andrews, S. S. Chitnis and D. W. Stephan, Chem. Commun., 2019, 55, 5599–5602 RSC.
- S. Rawat, M. Bhandari, V. K. Porwal and S. Singh, Inorg. Chem., 2020, 59, 7195–7203 CrossRef CAS PubMed.
- R. Kannan, S. Balasubramaniam, S. Kumar, R. Chambenahalli, E. D. Jemmis and A. Venugopal, Chem. – Eur. J., 2020 DOI:10.1002/chem.202002006.
- D. Addis, S. Zhou, S. Das, K. Junge, H. Kosslick, J. Harloff, H. Lund, A. Schulz and M. Beller, Chem. – Asian J., 2010, 5, 2341–2345 CrossRef CAS PubMed.
- Y. Kobayashi, E. Takahisa, M. Nakano and K. Watatani, Tetrahedron, 1997, 53, 1627–1634 CrossRef CAS.
- J. A. Fernández-Salas, S. Manzini and S. P. Nolan, Chem. Commun., 2013, 49, 9758–9760 RSC.
- K. Revunova and G. I. Nikonov, Chem. – Eur. J., 2014, 20, 839–845 CrossRef CAS PubMed.
- A. Volkov, F. Tinnis and H. Adolfsson, Org. Lett., 2014, 16, 680–683 CrossRef CAS PubMed.
- M. Zhao, W. Xie and C. Cui, Chem. – Eur. J., 2014, 20, 9259–9262 CrossRef CAS PubMed.
- J. Long, W. Zhao, Y. Xu, H. Li and S. Yang, Catalysts, 2018, 8, 633–644 CrossRef.
- J. Long, W. Zhao, Y. Xu, W. Wu, C. Fang, H. Li and S. Yang, RSC Adv., 2019, 9, 3063–3071 RSC.
- M. G. Manas, L. S. Sharninghausen, D. Balcells and R. H. Crabtree, New J. Chem., 2014, 38, 1694–1700 RSC.
- A. Muthukumar, N. C. Mamillapalli and G. Sekar, Adv. Synth. Catal., 2016, 358, 643–652 CrossRef CAS.
- G. Kumar, A. Muthukumar and G. Sekar, Eur. J. Org. Chem., 2017, 4883–4890 CrossRef CAS.
- G. Ding, C. Li, Y. Shen, B. Lu, Z. Zhang and X. Xie, Adv. Synth. Catal., 2016, 358, 1241–1250 CrossRef CAS.
- V. Skrypai, J. J. M. Hurley and M. J. Adler, Eur. J. Org. Chem., 2016, 2207–2211 CrossRef CAS.
- S. E. Varjosaari, V. Skrypai, P. Suating, J. J. M. Hurley, T. M. Gilbert and M. J. Adler, Eur. J. Org. Chem., 2017, 229–232 CrossRef CAS.
- X. X. Zhao, P. Zhang and Z. X. Guo, ChemistrySelect, 2017, 2, 7670–7677 CrossRef CAS.
- W. Yao, R. Li, J. Yang and F. Hao, Catal. Sci. Technol., 2019, 9, 3874–3878 RSC.
- W. Yao, L. He, D. Han and A. Zhong, J. Org. Chem., 2019, 84, 14627–14635 CrossRef CAS PubMed.
- J. Intemann, H. Bauer, J. Pahl, L. Maron and S. Harder, Chem. – Eur. J., 2015, 21, 11452–11461 CrossRef CAS PubMed.
- H. Elsen, C. Fischer, C. Knüpfer, A. Escalona and S. Harder, Chem. – Eur. J., 2019, 25, 16141–16147 CrossRef CAS PubMed.
- N. L. Lampland, A. Pindwal, S. R. Neal, S. Schlauderaff, A. Ellern and A. D. Sadow, Chem. Sci., 2015, 6, 6901–6907 RSC.
- M. D. Anker, M. S. Hill, J. P. Lowe and M. F. Mahon, Angew. Chem., Int. Ed., 2015, 54, 10009–10011 CrossRef CAS PubMed.
- M. Ma, X. Shen, W. Wang, J. Li, W. Yao and L. Zhu, Eur. J. Inorg. Chem., 2016, 2016, 5057–5062 CrossRef CAS.
- M. D. Anker, C. E. Kefalidis, Y. Yang, J. Fang, M. S. Hill, M. F. Mahon and L. Maron, J. Am. Chem. Soc., 2017, 139, 10036–10054 CrossRef CAS PubMed.
- Á. Raya-Barón, P. Oña-Burgos, A. Rodríguez-Diéguez and I. Fernández, Organometallics, 2018, 37, 2682–2689 CrossRef.
- B. Freitag, P. Stegner, K. Thum, C. A. Fischer and S. Harder, Eur. J. Inorg. Chem., 2018, 2018, 1938–1944 CrossRef CAS.
- D. Mukherjee, D. Schuhknecht and J. Okuda, Angew. Chem., Int. Ed., 2018, 57, 9590–9602 CrossRef CAS PubMed.
- H. C. Brown and B. C. Subba Rao, J. Org. Chem., 1957, 22, 1137–1138 CrossRef CAS.
- H. C. Brown and G. Zweifel, J. Am. Chem. Soc., 1960, 82, 4708–4712 CrossRef CAS.
- H. C. Brown and S. K. Gupta, J. Am. Chem. Soc., 1972, 94, 4370–4371 CrossRef CAS.
- H. C. Brown, S. C. Kim and S. Krishnamurthy, J. Org. Chem., 1980, 45, 1–12 CrossRef CAS.
- H. C. Brown and S. C. Kim, J. Org. Chem., 1984, 49, 1064–1071 CrossRef CAS.
- S. R. Tamang and M. Findlater, J. Org. Chem., 2017, 82, 12857–12862 CrossRef CAS PubMed.
- S. R. Tamang, D. Bedi, S. Shafiei-Haghighi, C. R. Smith, C. Crawford and M. Findlater, Org. Lett., 2018, 20, 6695–6700 CrossRef CAS PubMed.
- S. Das, J. Bhattacharjee and T. K. Panda, New J. Chem., 2019, 43, 16812–16818 RSC.
- G. Zhang, J. Cheng, K. Davis, M. G. Bonifacio and C. Zajaczkowski, Green Chem., 2019, 21, 1114–1121 RSC.
- M. Khononov, N. Fridman, M. Tamm and M. S. Eisen, Eur. J. Org. Chem., 2020, 3153–3160 CrossRef CAS.
- G. S. Kumar, A. Harinath, R. Narvariya and T. K. Panda, Eur. J. Inorg. Chem., 2020, 2020, 467–474 CrossRef CAS.
- N. Sarkar, M. Mahato and S. Nembenna, Eur. J. Inorg. Chem., 2020, 2020, 2295–2301 CrossRef CAS.
- I. Hossain and J. A. R. Schmidt, Eur. J. Inorg. Chem., 2020, 2020, 1877–1884 CrossRef CAS.
- S. A. Orr, J. A. Kelly, A. J. Boutland and V. L. Blair, Chem. – Eur. J., 2020, 26, 4947–4951 CrossRef CAS PubMed.
- V. L. Weidner, C. J. Barger, M. Delferro, T. L. Lohr and T. J. Marks, ACS Catal., 2017, 7, 1244–1247 CrossRef CAS.
- S. Chen, D. Yan, M. Xue, Y. Hong, Y. Yao and Q. Shen, Org. Lett., 2017, 19, 3382–3385 CrossRef CAS PubMed.
- W. Wang, X. Shen, F. Zhao, H. Jiang, W. Yao, S. A. Pullarkat, L. Xu and M. Ma, J. Org. Chem., 2018, 83, 69–74 CrossRef CAS PubMed.
- C. J. Barger, A. Motta, V. L. Weidner, T. L. Lohr, T. J. Marks, S. Chimiche, R. La, U. Roma and P. A. Moro, ACS Catal., 2019, 9, 9015–9024 CrossRef CAS.
- S. R. Tamang, A. Singh, D. Bedi, A. R. Bazkiaei, A. A. Warner, K. Glogau, C. McDonald, D. K. Unruh and M. Findlater, Nat. Catal., 2020, 3, 154–162 CrossRef CAS.
- C. J. Barger, R. D. Dicken, V. L. Weidner, A. Motta, T. L. Lohr and T. J. Marks, J. Am. Chem. Soc., 2020, 142, 8019–8028 CrossRef CAS PubMed.
- J. R. Lawson, L. C. Wilkins and R. L. Melen, Chem. – Eur. J., 2017, 23, 10997–11000 CrossRef CAS PubMed.
- Q. Yin, Y. Soltani, R. L. Melen and M. Oestreich, Organometallics, 2017, 36, 2381–2384 CrossRef CAS.
- D. M. C. Ould and R. L. Melen, Chem. – Eur. J., 2018, 24, 15201–15204 CrossRef CAS PubMed.
- J. L. Carden, L. J. Gierlichs, D. F. Wass, D. L. Browne and R. L. Melen, Chem. Commun., 2019, 55, 318–321 RSC.
- M. W. Stanford, A. Bismuto and M. J. Cowley, Chem. – Eur. J., 2020 DOI:10.1002/chem.202000897.
- I. P. Query, P. A. Squier, E. M. Larson, N. A. Isley and T. B. Clark, J. Org. Chem., 2011, 76, 6452–6456 CrossRef CAS PubMed.
- M. K. Barman, A. Baishya and S. Nembenna, Dalton Trans., 2017, 46, 4152–4156 RSC.
- Y. Wu, C. Shan, J. Ying, J. Su, J. Zhu, L. L. Liu and Y. Zhao, Green Chem., 2017, 19, 4169–4175 RSC.
- M. K. Bisai, T. Das, K. Vanka and S. S. Sen, Chem. Commun., 2018, 54, 6843–6846 RSC.
- Z. Zhu, X. Wu, X. Xu, Z. Wu, M. Xue, Y. Yao, Q. Shen and X. Bao, J. Org. Chem., 2018, 83, 10677–10683 CrossRef CAS PubMed.
- S. J. Yang, A. K. Jaladi, J. H. Kim, S. Gundeti and D. K. An, Bull. Korean Chem. Soc., 2019, 40, 34–38 CAS.
- Z. C. Wang, D. Shen, J. Gao, X. Jia, Y. Xu and S. L. Shi, Chem. Commun., 2019, 55, 8848–8851 RSC.
- D. Yan, X. Wu, J. Xiao, Z. Zhu, X. Xu, X. Bao, Y. Yao, Q. Shen and M. Xue, Org. Chem. Front., 2019, 6, 648–653 RSC.
- Z. C. Wang, M. Wang, J. Gao, S. L. Shi and Y. Xu, Org. Chem. Front., 2019, 6, 2949–2953 RSC.
- W. K. Shin, H. Kim, A. K. Jaladi and D. K. An, Tetrahedron, 2018, 74, 6310–6315 CrossRef CAS.
- T. Liu, J. He and Y. Zhang, Org. Chem. Front., 2019, 6, 2749–2755 RSC.
- E. Jeong, J. Heo, S. Park and S. Chang, Chem. – Eur. J., 2019, 25, 6320–6325 CrossRef CAS PubMed.
- J. Jeong, J. Heo, D. Kim and S. Chang, ACS Catal., 2020, 10, 5023–5029 CrossRef CAS.
- J. H. Kim, A. K. Jaladi, H. T. Kim and D. K. An, Bull. Korean Chem. Soc., 2019, 40, 971–975 CrossRef CAS.
- D. H. Ma, A. K. Jaladi, J. H. Lee, T. S. Kim, W. K. Shin, H. Hwang and D. K. An, ACS Omega, 2019, 4, 15893–15903 CrossRef CAS PubMed.
- K. Kuciński and G. Hreczycho, Green Chem., 2019, 21, 1912–1915 RSC.
- A. Falconnet, M. Magre, B. Maity, L. Cavallo and M. Rueping, Angew. Chem., Int. Ed., 2019, 58, 17567–17571 CrossRef CAS PubMed.
- Y. K. Jang, M. Magre and M. Rueping, Org. Lett., 2019, 21, 8349–8352 CrossRef CAS PubMed.
- M. Szewczyk, M. Magre, V. Zubar and M. Rueping, ACS Catal., 2019, 9, 11634–11639 CrossRef CAS.
- M. Magre, B. Maity, A. Falconnet, L. Cavallo and M. Rueping, Angew. Chem., Int. Ed., 2019, 58, 7025–7029 CrossRef CAS PubMed.
- W. Wang, K. Lu, Y. Qin, W. Yao, D. Yuan, S. A. Pullarkat, L. Xu and M. Ma, Tetrahedron, 2020, 76, 131145 CrossRef CAS.
- V. Vasilenko, C. K. Blasius, H. Wadepohl and L. H. Gade, Chem. Commun., 2020, 56, 1203–1206 RSC.
- D. Willcox, J. L. Carden, A. J. Ruddy, P. D. Newman and R. L. Melen, Dalton Trans., 2020, 49, 2417–2420 RSC.
- K. Kuciński and G. Hreczycho, Eur. J. Org. Chem., 2020, 552–555 CrossRef.
- R. J. P. Corriu, R. Perz and C. Reye, Tetrahedron, 1983, 39, 999–1009 CrossRef CAS.
- M. Fujita and T. Hiyama, Tetrahedron Lett., 1987, 28, 2263–2264 CrossRef CAS.
- Y. Goldberg, E. Abele, M. Shymanska and E. Lukevics, J. Organomet. Chem., 1991, 410, 127–133 CrossRef CAS.
- D. Bedi, A. Brar and M. Findlater, Green Chem., 2020, 22, 1125–1128 RSC.
- P. Ghosh and A. Jacobi von Wangelin, Org. Chem. Front., 2020, 7, 960–966 RSC.
- M. Bhunia, S. R. Sahoo, A. Das, J. Ahmed, P. Sreejyothi and S. K. Mandal, Chem. Sci., 2020, 11, 1848–1854 RSC.
- T. K. Panda, I. Banerjee and S. Sagar, Appl. Organomet. Chem., 2020 DOI:10.1002/aoc.5765.
- M. Arrowsmith, M. S. Hill, T. Hadlington, G. Kociok-Köhn and C. Weetman, Organometallics, 2011, 30, 5556–5559 CrossRef CAS.
- M. Arrowsmith, T. J. Hadlington, M. S. Hill and G. Kociok-Köhn, Chem. Commun., 2012, 48, 4567–4569 RSC.
- M. Arrowsmith, M. S. Hill and G. Kociok-Köhn, Chem. – Eur. J., 2013, 19, 2776–2783 CrossRef CAS PubMed.
- D. Mukherjee, A. Ellern and A. D. Sadow, Chem. Sci., 2014, 5, 959–964 RSC.
- J. Intemann, M. Lutz and S. Harder, Organometallics, 2014, 33, 5722–5729 CrossRef CAS.
- N. L. Lampland, M. Hovey, D. Mukherjee and A. D. Sadow, ACS Catal., 2015, 5, 4219–4226 CrossRef CAS.
- C. Weetman, M. S. Hill and M. F. Mahon, Chem. – Eur. J., 2016, 22, 7158–7162 CrossRef CAS PubMed.
- K. Manna, P. Ji, F. X. Greene and W. Lin, J. Am. Chem. Soc., 2016, 138, 7488–7491 CrossRef CAS PubMed.
- C. Weetman, M. D. Anker, M. Arrowsmith, M. S. Hill, G. Kociok-Köhn, D. J. Liptrot and M. F. Mahon, Chem. Sci., 2016, 7, 628–641 RSC.
- D. Mukherjee, S. Shirase, T. P. Spaniol, K. Mashima and J. Okuda, Chem. Commun., 2016, 52, 13155–13158 RSC.
- H. Osseili, D. Mukherjee, K. Beckerle, T. P. Spaniol and J. Okuda, Organometallics, 2017, 36, 3029–3034 CrossRef CAS.
- R. McLellan, A. R. Kennedy, R. E. Mulvey, S. A. Orr and S. D. Robertson, Chem. – Eur. J., 2017, 23, 16853–16861 CrossRef CAS PubMed.
- M. Ma, J. Li, X. Shen, Z. Yu, W. Yao and S. A. Pullarkat, RSC Adv., 2017, 7, 45401–45407 RSC.
- S. Yadav, S. Pahar and S. S. Sen, Chem. Commun., 2017, 53, 4562–4564 RSC.
- L. E. Lemmerz, T. P. Spaniol and J. Okuda, Dalton Trans., 2018, 47, 12553–12561 RSC.
- J. Li, M. Luo, X. Sheng, H. Hua, W. Yao, S. A. Pullarkat, L. Xu and M. Ma, Org. Chem. Front., 2018, 5, 3538–3547 RSC.
- A. Harinath, J. Bhattacharjee, H. P. Nayek and T. K. Panda, Dalton Trans., 2018, 47, 12613–12622 RSC.
- M. Luo, J. Li, Q. Xiao, S. Yang, F. Su and M. Ma, J. Organomet. Chem., 2018, 868, 31–35 CrossRef CAS.
- V. A. Pollard, S. A. Orr, R. McLellan, A. R. Kennedy, E. Hevia and R. E. Mulvey, Chem. Commun., 2018, 54, 1233–1236 RSC.
- S. Yadav, R. Dixit, M. K. Bisai, K. Vanka and S. S. Sen, Organometallics, 2018, 37, 4576–4584 CrossRef CAS.
- X. Cao, W. Wang, K. Lu, W. Yao, F. Xue and M. Ma, Dalton Trans., 2020, 49, 2776–2780 RSC.
- T. T. Nguyen, J.-H. Kim, S. Kim, C. Oh, M. Flores, T. L. Groy, M.-H. Baik and R. J. Trovitch, Chem. Commun., 2020, 56, 3959–3962 RSC.
- S. Brand, A. Causero, H. Elsen, J. Pahl, J. Langer and S. Harder, Eur. J. Inorg. Chem., 2020, 2020, 1728–1735 CrossRef CAS.
- X. Liu, B. Li, X. Hua and D. Cui, Org. Lett., 2020, 22(13), 4960–4965 CrossRef CAS PubMed.
- A. D. Bage, T. A. Hunt and S. P. Thomas, Org. Lett., 2020, 22, 4107–4112 CrossRef CAS PubMed.
- A. M. Carroll, T. P. O'Sullivan and P. J. Guiry, Adv. Synth. Catal., 2005, 347, 609–631 CrossRef CAS.
- X. Yang, T. Fox and H. Berke, Tetrahedron, 2011, 67, 7121–7127 CrossRef CAS.
- K. Smith, A. A. Balakit, R. T. Pardasani and G. A. El-Hiti, J. Sulfur Chem., 2011, 32, 287–295 CrossRef CAS.
- C. E. Tucker, J. Davidson and P. Knochel, J. Org. Chem., 1992, 57, 3482–3485 CrossRef CAS.
- D. J. Parks and W. E. Piers, Tetrahedron, 1998, 54, 15469–15488 CrossRef CAS.
- D. J. Parks, W. E. Piers and G. P. A. Yap, Organometallics, 1998, 17, 5492–5503 CrossRef CAS.
- E. O. Pentsak, D. B. Eremin, E. G. Gordeev and V. P. Ananikov, ACS Catal., 2019, 9, 3070–3081 CrossRef CAS.
- K. P. Kepp, Inorg. Chem., 2016, 55, 9461–9470 CrossRef CAS PubMed.
- H. Stachowiak, J. Kaźmierczak, K. Kuciński and G. Hreczycho, Green Chem., 2018, 20, 1738–1742 RSC.
- E. A. Romero, J. L. Peltier, R. Jazzar and G. Bertrand, Chem. Commun., 2016, 52, 10563–10565 RSC.
- K. Kuciński and G. Hreczycho, ChemSusChem, 2017, 10, 4695–4698 CrossRef PubMed.
- J. Kaźmierczak, K. Kuciński, H. Stachowiak and G. Hreczycho, Chem. – Eur. J., 2018, 24, 2509–2514 CrossRef PubMed.
- W. Wang, M. Luo, W. Yao, M. Ma, S. A. Pullarkat, L. Xu and P. H. Leung, New J. Chem., 2019, 43, 10744–10749 RSC.
- T. K. Panda, A. Harinath and J. Bhattacharjee, Chem. Commun., 2019, 55, 1386–1389 RSC.
- W. Wang, M. Luo, D. Zhu, W. Yao, L. Xu and M. Ma, Org. Biomol. Chem., 2019, 17, 3604–3608 RSC.
- X. Xu, D. Yan, Z. Zhu, Z. Kang, Y. Yao, Q. Shen and M. Xue, ACS Omega, 2019, 4, 6775–6783 CrossRef CAS PubMed.
- A. Rit, V. K. Pandey and S. N. R. Donthireddy, Chem. – Asian J., 2019, 14, 3255–3258 CrossRef PubMed.
- V. K. Pandey, S. Bauri and A. Rit, Org. Biomol. Chem., 2020, 18, 3853–3857 RSC.
| This journal is © The Royal Society of Chemistry 2020 |