
Total synthesis and biological evaluation of simplified aplyronine analogues as synthetically tractable anticancer agents†
Talia R.
Pettigrew‡
a,
Rachel J.
Porter‡
a,
Stephen J.
Walsh
 ab,
Michael P.
Housden
a,
Nelson Y. S.
Lam
a,
Jason S.
Carroll
b,
Jeremy S.
Parker
c,
David R.
Spring
a and
Ian
Paterson
*a
ab,
Michael P.
Housden
a,
Nelson Y. S.
Lam
a,
Jason S.
Carroll
b,
Jeremy S.
Parker
c,
David R.
Spring
a and
Ian
Paterson
*a
aUniversity Chemical Laboratory, Lensfield Road, Cambridge, CB2 1EW, UK
bCancer Research UK Cambridge Institute, University of Cambridge, Cambridge, CB2 0RE, UK
cEarly Chemical Development, Pharmaceutical Sciences, R&D, AstraZeneca, Macclesfield, UK
First published on 3rd January 2020
Abstract
The aplyronines are a family of highly cytotoxic marine natural products with potential application in targeted cancer chemotherapy. To address the severe supply issue, function-oriented molecular editing of their macrolactone scaffold led to the design of a series of simplified aplyronine analogues. Enabled by a highly convergent aldol-based route, the total synthesis of four analogues was achieved, with a significant improvement in step economy versus previous compounds, and their cancer cell growth inhibition in the HeLa cell line was determined. The modular strategy presented offers a means for significantly shortening their chemical synthesis to facilitate the continued development of this promising class of anticancer agent.
Isolated in 1993 by Yamada and co-workers from the sea hare Aplysia kurodai,1 the aplyronines are a family of complex polyketides comprised of a 24-membered macrolactone and an elaborate side chain terminating in an N-vinyl-N-formamide moiety.1–3 Aplyronines A (1, Fig. 1) and C (2) bear a C29 N,N-dimethylalanine (DMAla) residue, while aplyronine D (3) contains a N,N-dimethylglycine (DMGly). Aplyronines A and D further feature a N,N,O-trimethylserine (TMSer) residue at C7. Aplyronine A was found to display sub-nanomolar growth inhibitory activity (mean GI50 = 0.2 nM) in the NCI 60 cell line panel, prompting investigations into its mode of action in disrupting cytoskeletal dynamics.4 The 11-carbon side chain of the aplyronines is conserved across several related anticancer macrolides (reidispongiolide A (4), scytophycin C (5) and rhizopodin (6)), which all bind to actin as their protein target.5 Additionally, aplyronine A was found to bind to tubulin through the formation of an unprecedented actin–aplyronine–tubulin heterotrimeric complex.6 Interestingly, actin binding was shown to be a necessary but insufficient marker for their cytotoxicity, where aplyronine A is roughly 50 times more cytotoxic than aplyronine C, despite registering similar actin depolymerising ability. This structure–activity relationship (SAR) highlighted the synergistic role of the macrolactone, specifically the C1–C13 region, with the C7 TMSer important in forming the crucial interaction with tubulin in aplyronines A and D.7
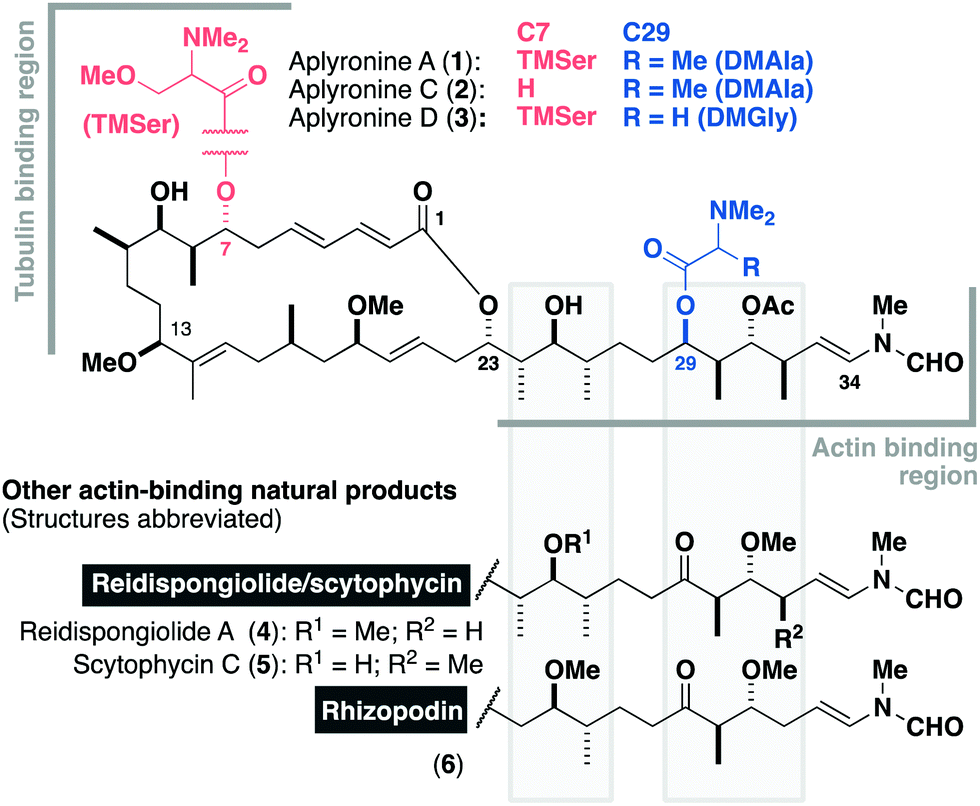 | ||
| Fig. 1 The aplyronine family of cytotoxic marine macrolides, highlighting the structural differences between the congeners from C7 and C29 acyl substitution. Key regions implicated in protein binding are denoted, as well as their structural homology to related actin-binding macrolides. | ||
Their extraordinary potency and novel mode of action mark the aplyronines out as promising candidates as novel payloads for antibody–drug conjugates.8,9 While recent total syntheses of the aplyronines have been achieved in 29–38 steps LLS (and up to 80 total steps),9,10 their structural complexity and natural scarcity (2.6 mg of aplyronine D was isolated from 300 kg of Aplysia kurodai)1b adversely impact their further development. To address these challenges, we turned towards a function-oriented molecular editing of the aplyronine scaffold to help improve the step economy while retaining the biological activity.11 Herein, we report the concise total synthesis of a series of simplified aplyronine analogues in 24–26 steps LLS (37–39 total steps) and their preliminary biological evaluation.
Inspection of the aplyronine A–actin complex crystal structure,5 and taking previous SAR studies3 into account, indicated that the C16–C22 region of the macrolactone did not appear to form any significant interaction with the protein binding site (see the ESI† for discussion). As outlined in Scheme 1, we proposed that this region was primarily acting as a conformational constraint for the macrocycle and that its replacement by a simple (CH2)7 unit should preserve the 3D structure and critical C1–C13 binding region, and hence maintain its biological activity. Additionally, our proposed aplyrologues involved replacing the C25 hydroxyl and C31 acetoxy groups with methyl ethers, which simplifies the protecting group strategy, as well as removing the propensity for the acetate to eliminate (Scheme 1).9 Moreover, these latter modifications mirror the functionalities present in the equivalent side chains (Fig. 1) present in reidispongiolide A (4), scytophycin C (5) and rhizopodin (6). We envisaged that these designed aplyrologues12 would preserve the core pharmacophore, while significantly improving the synthetic tractability for this promising class of anticancer agent.
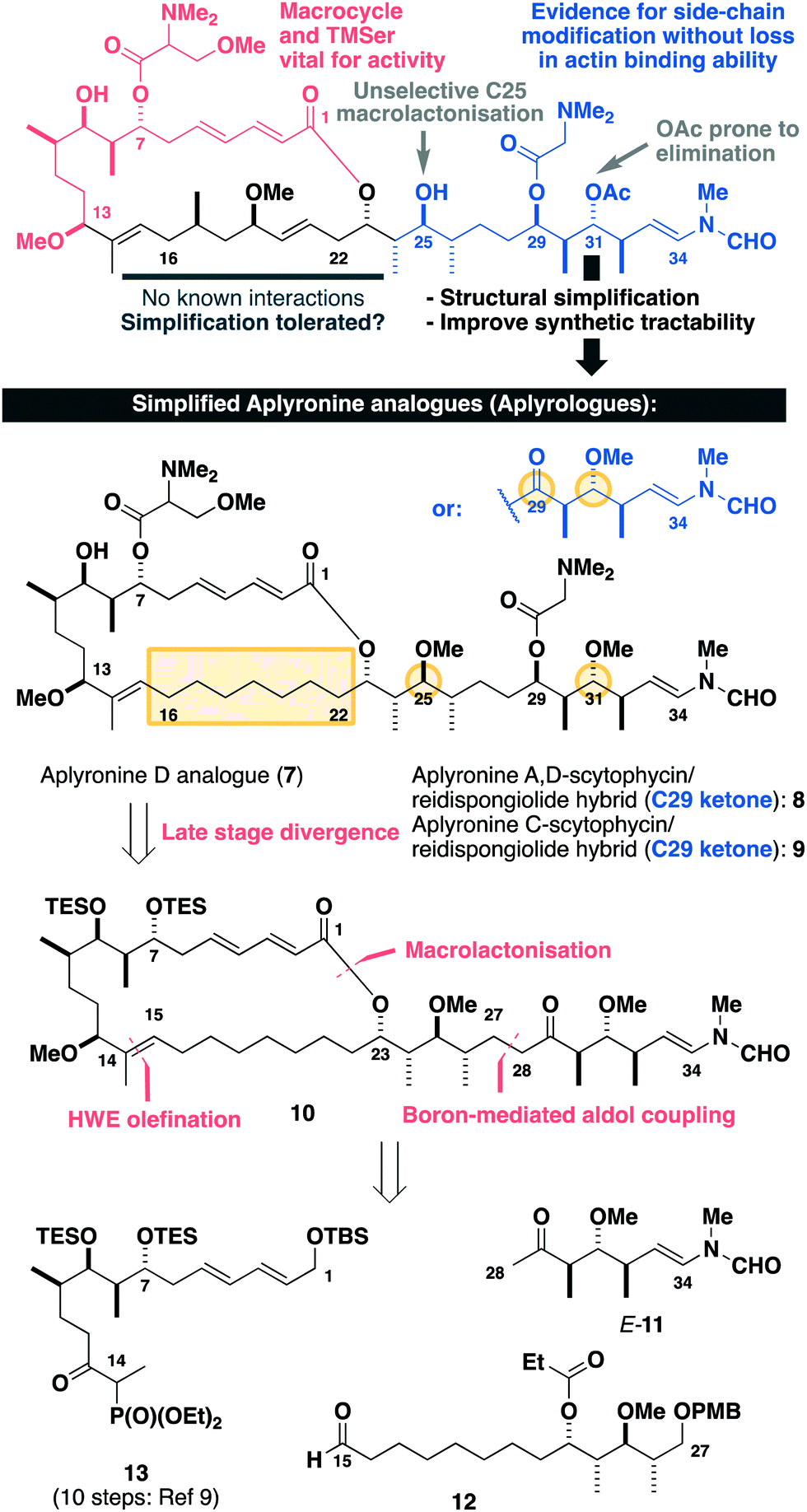 | ||
| Scheme 1 Summary of SAR studies (see the ESI†) for the aplyronines and opportunities for molecular editing, leading to the design of simplified aplyronine analogues 7–9 containing macrocyclic and side chain modifications. Our planned synthesis invokes a modular strategy using side chain E-11, aldehyde 12 and phosphonate 13 as the key fragments. | ||
Building on our recent total synthesis of aplyronines A and D,9 we planned a late-stage diversification to construct aplyrologues bearing the aplyronine D side chain, as in 7 (Scheme 1), as well as the scytophycin–aplyronine A/D (bearing the C7 TMSer: 8) and C (9) analogues. This analysis leads back to the common scaffold 10, which would be forged through an aldol coupling sequence with the C28–C34 ketone E-11 and a macrocyclic aldehyde. The macrocycle itself can then be constructed by a Horner–Wadsworth–Emmons (HWE) olefination with the simplified C15–C27 aldehyde 12 and the previously reported C1–C14 phosphonate 13.9a
Construction of the C15–C27 aldehyde 12 (Scheme 2) commenced with a Ti(OiPr)Cl3-mediated aldol reaction13 between the Roche ester-derived ketone S-1314 and the aldehyde 14 to give the 1,4-syn, 3,4-syn adduct 15. An Evans–Tishchenko reduction15 then delivered the 1,3-anti configured alcohol 16, which was methylated, desilylated and oxidised to afford the aldehyde 12 in five steps from 13 and 14. Notably, this expedient sequence represented more than a three-fold reduction in the step count relative to the analogous fragment required for the aplyronines.9
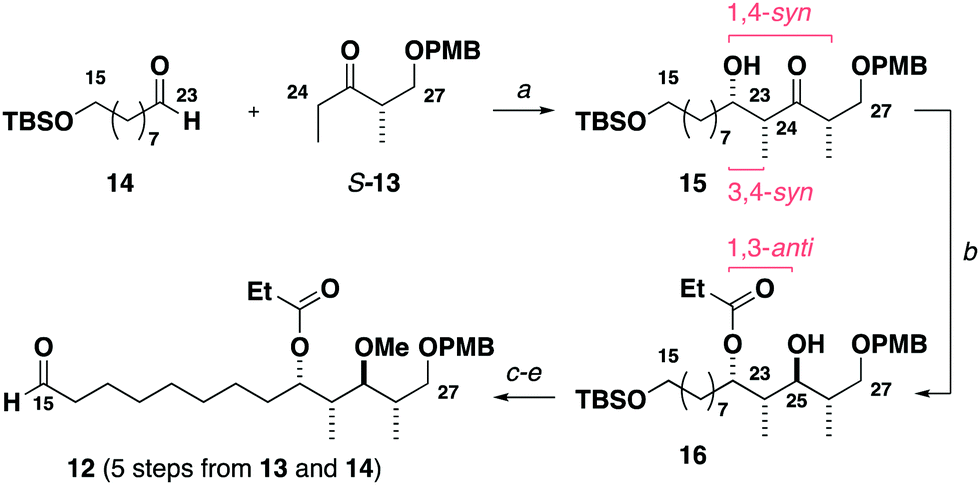 | ||
Scheme 2 Synthesis of the C15–C27 aldehyde 12. Reagents and conditions: (a) S-13, Ti(OiPr)Cl3, DIPEA, CH2Cl2, −78 °C; 14, CH2Cl2, −78 °C, 85%, 18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr, (b) SmI2, EtCHO, THF, −20 °C, 95%, >20:1 dr, (c) Me3O·BF4, Proton Sponge®, CH2Cl2, r.t., 89%, 99% brsm, (d) HCl (3 M)/THF/H2O (5:2:1), r.t., 99%, (e) (COCl)2, DMSO, CH2Cl2, −78 °C; Et3N, −78 °C to r.t., 94%. :1 dr, (b) SmI2, EtCHO, THF, −20 °C, 95%, >20:1 dr, (c) Me3O·BF4, Proton Sponge®, CH2Cl2, r.t., 89%, 99% brsm, (d) HCl (3 M)/THF/H2O (5:2:1), r.t., 99%, (e) (COCl)2, DMSO, CH2Cl2, −78 °C; Et3N, −78 °C to r.t., 94%. | ||
Synthesis of the modified C28–C34 side chain (Scheme 3) commenced with an analogous titanium aldol reaction between the ketone R-13 and acetaldehyde to give 17. Adduct 17 was reduced to give alcohol 18 followed by methylation, PMB ether cleavage and ester reduction to afford the diol 19. At this point, a double oxidation under Swern conditions gave ketoaldehyde 20, which was directly subjected to a Wittig olefination16 with phosphonium salt 21 to generate the enamides E/Z-11. Final completion of the configurationally pure C28–C34 ketone E-11 was accomplished by I2-mediated isomerisation.
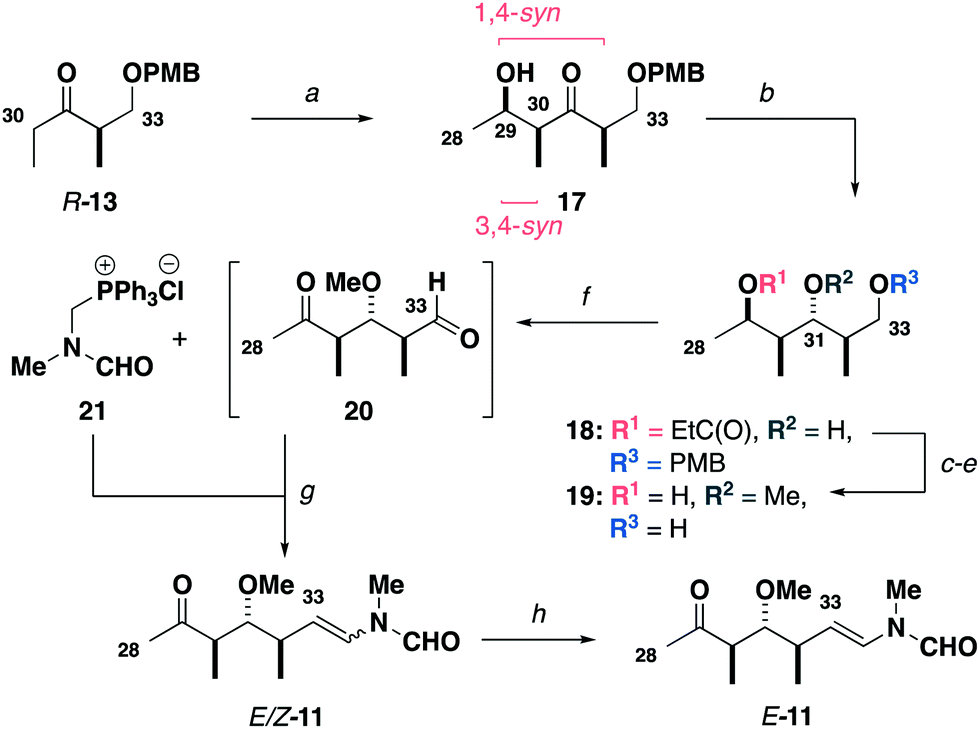 | ||
| Scheme 3 Synthesis of the C28–C34 ketone E-11. Reagents and conditions: (a) R-13, Ti(OiPr)Cl3, DIPEA, CH2Cl2, −78 °C; acetaldehyde, CH2Cl2, −78 °C, 77%, 17:1 dr, (b) SmI2, EtCHO, THF, −20 °C, 97%, >20:1 dr, (c) Me3O·BF4, Proton Sponge®, CH2Cl2, r.t., 90%, (d) DDQ, CH2Cl2/pH 7 buffer (2:1), r.t., 93%, (e) DIBAL, CH2Cl2, −78 °C, 87%, (f) (COCl)2, DMSO, CH2Cl2, −78 °C; Et3N, −78 °C to r.t., (g) 21, LiHMDS, THF, −78 °C; 20, THF −78 to −20 °C, 60% over two steps, 3:1 Z/E, (h) I2 (5 mol%), CH2Cl2, r.t., 86%, >20:1 E/Z. | ||
With all three fragments in hand, attention now turned towards the assembly of the full carbon skeleton of the targeted analogues. Aldehyde 12 was first coupled with the phosphonate 13 (10 steps)9avia an HWE olefination17 to generate the C1–C27 fragment 22 (Scheme 4). A CBS reduction18 of the C13 ketone was then followed by methylation to give 23. Next, the C23 ester was cleaved and a DDQ-mediated deprotection/allylic oxidation19 afforded the seco acid 24. The macrolactonisation reaction of 24 proceeded smoothly under Yamaguchi conditions20 to give 25. Moving forward, oxidative cleavage of the PMB ether in 25 followed by Dess–Martin oxidation of the resulting alcohol afforded the aldehyde 26. This was then subjected to a three-step sequence involving a Cy2BCl-mediated aldol coupling9b with the C28–C34 ketone E-11, a Burgess elimination followed by a copper-catalysed conjugate reduction of the resulting enone gave 10, corresponding to a protected aplyrologue bearing the scytophycin/reidispongiolide-type side chain. Additionally, the C29 ketone in 10 could be reduced and acylated with DMGly under Keck conditions21 to give the 1,3-diol 27 after deprotection, which corresponded to the aplyrologue scaffold bearing the aplyronine-type side chain. The advanced intermediate 10 was also deprotected to give the scytophycin–aplyronine C hybrid 9. In turn, this could be site-selectively acylated at C7 with the required enantiomer of TMSer to generate the scytophycin–aplyronine A hybrids S-8 and R-8. Noting that aplyronine D bearing the C7 TMSer and C29 DMGly residues was reported to be the most active congener,1b the corresponding aplyrologue D was targeted. Thus, the diol 27 was site-selectively acylated with S-TMSer to generate 7.
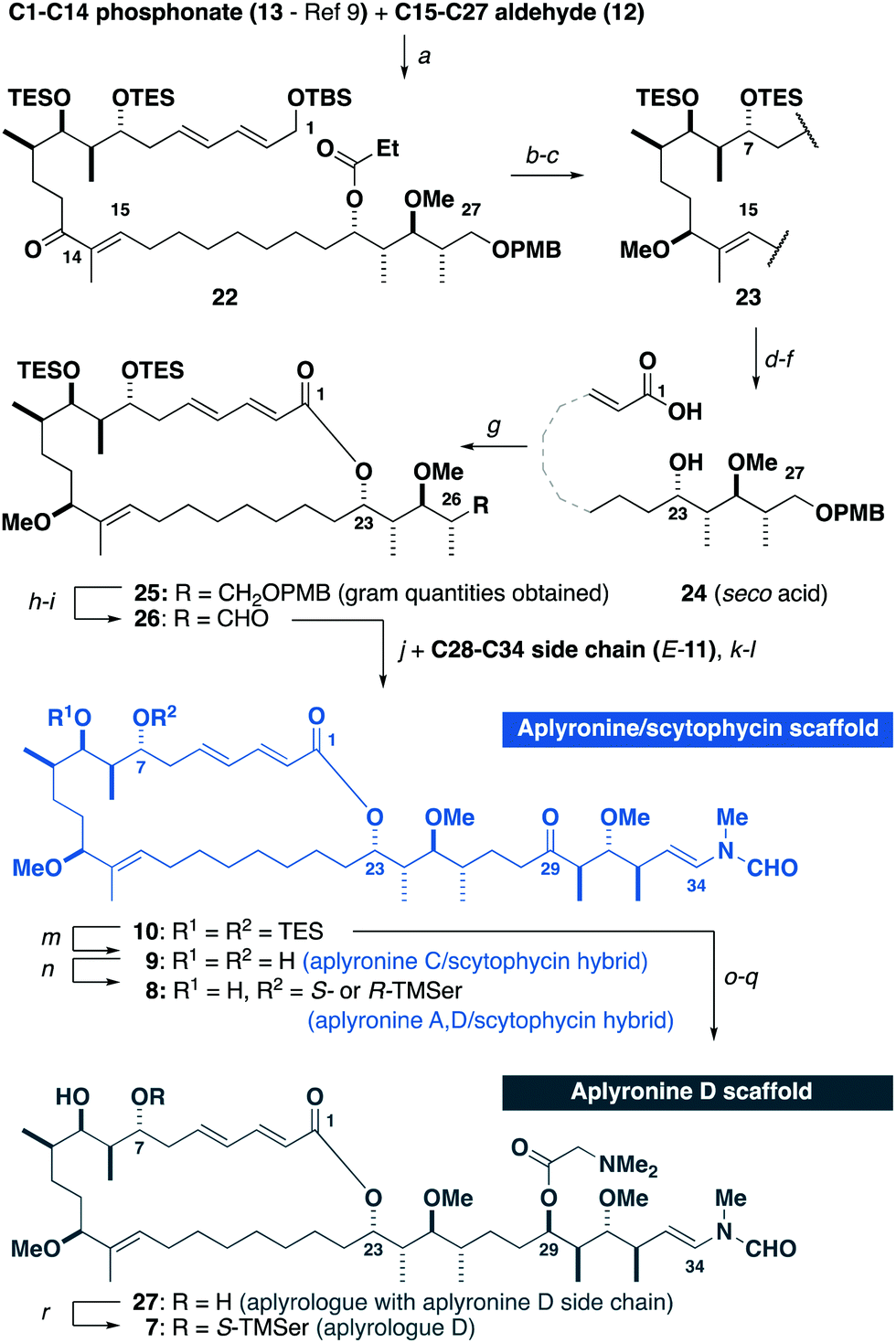 | ||
| Scheme 4 Synthesis of aplyrologues. Reagents and conditions: (a) Ba(OH)2, THF/H2O (40:1), r.t., 85%, >20:1 E/Z, (b) R-MeCBS, BH3·SMe2, −10 °C, 93%, >20:1 dr, (c) Me3O·BF4, Proton Sponge®, CH2Cl2, r.t., 87%, (d) DIBAL, CH2Cl2, −78 °C, 99%, (e) DDQ, CH2Cl2/pH 9.2 buffer (4:1), 0 °C to r.t., 75%, 91% brsm, (f) NaClO2, NaH2PO4·H2O, 2-methyl-but-2-ene, H2O/tBuOH, 0 °C to r.t., (g) TCBC, Et3N; DMAP, PhMe, r.t., 70% over two steps, (h) DDQ, CH2Cl2/pH 7.0 buffer (2:1), r.t., 82%, (i) DMP, NaHCO3, CH2Cl2, r.t., (j) E-11, Cy2BCl, Et3N, Et2O, 0 °C; 26, Et2O, −78 to −10 °C, (k) Burgess reagent, THF, r.t., 65% over three steps, (l) Cu(OAc)2, PPh3, TMDS, PhMe, r.t., 89%. (m) HF·py/py (1:2), THF, 0 °C to r.t., 85%, (n) TCBC, R- or S-TMSer, Et3N, DMAP, PhH, 62% for S-8, 73% for R-8, (o) Zn(BH)4, Et2O, 0 °C, 58%, 2:1 dr, (p) DMGly, DCC, DMAP, DMAP·HCl, r.t., (q) HF·py/py (1:2), THF, 0 °C to r.t., 69% over two steps, (r) TCBC, S-TMSer, Et3N, DMAP, PhH, 65%. CBS: Corey–Bakshi–Shibata catalyst. | ||
With this first series of aplyrologues in hand, attention now turned towards determining their in vitro cytotoxicity. It was considered prudent to initially examine the activity of our previously synthesised samples of aplyronines A (1), C (2) and D (3), with discodermolide included as a control (Table 1). While the expected sub-nanomolar potencies were found for aplyronines A and D, we did not find any significant difference between these congeners in the same HeLa cell line (Table 1, entries 1–3). However, our synthetic aplyrologues did not register the same exquisite potency compared with the native aplyronines. Nevertheless, the compounds R- and S-8 (entries 5 and 6), bearing the scytophycin side chain, as well as 7 (entry 4) still gave inhibitory activities in the nM range. Interestingly, both the configuration of the TMSer residue and the nature of the C29 functionality as the ketone or DMGly ester did not appear to greatly influence the biological activity, suggesting that the deleterious effect may be attributable to an oversimplification of the southern hemisphere region of the macrolactone.
| Entry | Compound | IC50a (nM) |
|---|---|---|
| a n = 3. See the ESI for cytotoxicity curves and further experimental details. | ||
| 1 | Aplyronine A (1) | 0.31 |
| 2 | Aplyronine C (2) | 3.67 |
| 3 | Aplyronine D (3) | 0.26 |
| 4 | 7 (aplyrologue D) | 124 |
| 5 | S-8 (aplyronine A, D/scytophycin C hybrid) | 164 |
| 6 | R-8 (aplyronine A, D/scytophycin C hybrid) | 192 |
| 7 | 9 (aplyronine C/scytophycin C hybrid) | >1000 |
| 8 | Discodermolide | 3.40 |
In conclusion, the preliminary biological results obtained for this first series of aplyrologues suggests that the southern hemisphere of the macrocycle may play a subtle but vital role in reinforcing the proposed actin–tubulin interaction that the aplyronines uniquely mediate. Additionally, the biological data obtained for our synthetic aplyronines does not replicate the greater cytotoxicity previously reported for a natural sample of aplyronine D relative to aplyronine A,1b signifying the scope for structural diversification of the side chain. In future work, further judicious molecular editing of the southern hemisphere region can be flexibly adapted towards the incorporation of more elaborate macrocyclic tethers. We envisage that our modular synthetic platform offers a means for enhancing the synthetic tractability of the aplyronines to facilitate the continued development of this promising class of anticancer agent.22
We thank Trinity College (TRP), AstraZeneca (RJP and SJW), EPSRC (RJP and SJW), Downing College (MPH) and the Woolf Fisher Trust (NYSL) for financial support, Dr Nika Anžicek and Dr Simon Williams for helpful discussions, and the National Mass Spectrometry Centre (Swansea) for mass spectra.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) K. Yamada, M. Ojika, T. Ishigaki, Y. Yoshida, H. Ekimoto and M. Arakawa, J. Am. Chem. Soc., 1993, 115, 11020 CrossRef CAS; (b) M. Ojika, H. Kigoshi, K. Suenaga, Y. Imamura, K. Yoshikawa, T. Ishigaki, A. Sakakura, T. Mutou and K. Yamada, Tetrahedron, 2012, 68, 982 CrossRef CAS.
- (a) M. Ojika, H. Kigoshi, T. Ishigaki, I. Tsukada, T. Tsuboi, T. Ogawa and K. Yamada, J. Am. Chem. Soc., 1994, 116, 7441 CrossRef CAS; (b) H. Kigoshi, M. Ojika, T. Ishigaki, K. Suenaga, T. Mutou, A. Sakakura, T. Ogawa and K. Yamada, J. Am. Chem. Soc., 1994, 116, 7443 CrossRef CAS.
- (a) H. Kigoshi, K. Suenaga, M. Takagi, A. Akao, K. Kanematsu, N. Kamei, Y. Okugawa and K. Yamada, Tetrahedron, 2002, 58, 1075 CrossRef CAS; (b) M. Ojika, H. Kigoshi, Y. Yoshida, T. Ishigaki, M. Nisiwaki, I. Tsukada, M. Arakawa, H. Ekimoto and K. Yamada, Tetrahedron, 2007, 63, 3138 CrossRef CAS.
- K. Hirata, S. Muraoka, K. Suenaga, T. Kuroda, K. Kato, H. Tanaka, M. Yamamoto, M. Takata, K. Yamada and H. Kigoshi, J. Mol. Biol., 2006, 356, 945 CrossRef CAS PubMed.
- (a) K.-S. Yeung and I. Paterson, Angew. Chem., Int. Ed., 2002, 41, 4632 CrossRef CAS PubMed; (b) J. S. Allingham, V. A. Klenchin and I. Rayment, Cell. Mol. Life Sci., 2006, 63, 2119 CrossRef CAS PubMed.
- M. Kita, Y. Hirayama, K. Yoneda, K. Yamagishi, T. Chinen, T. Usui, E. Sumiya, M. Uesugi and H. Kigoshi, J. Am. Chem. Soc., 2013, 135, 18089 CrossRef CAS PubMed.
- M. Kita and H. Kigoshi, Nat. Prod. Rep., 2015, 32, 534 RSC.
- (a) R. V. J. Chari, M. L. Miller and W. C. Widdison, Angew. Chem., Int. Ed., 2014, 53, 3796 CrossRef CAS PubMed; (b) K. C. Nicolaou and S. Rigol, Angew. Chem., Int. Ed., 2019, 58, 11206 CrossRef CAS PubMed; (c) D. J. Newman, Mar. Drugs, 2019, 17, 324 CrossRef CAS PubMed.
- (a) N. Anžiček, S. Williams, M. P. Housden and I. Paterson, Org. Biomol. Chem., 2018, 16, 1343 RSC; (b) I. Paterson, S. J. Fink, L. Y. W. Lee, S. J. Atkinson and S. B. Blakey, Org. Lett., 2013, 15, 3118 CrossRef CAS PubMed.
- I. Hayakawa, K. Saito, S. Matsumoto, S. Kobayashi, A. Taniguchi, K. Kobayashi, Y. Fujii, T. Kaneko and H. Kigoshi, Org. Biomol. Chem., 2017, 15, 124 RSC.
- P. A. Wender, R. V. Quiroz and M. C. Stevens, Acc. Chem. Res., 2015, 48, 752 CrossRef CAS PubMed.
- (a) K. Futaki, M. Takahashi, K. Tanabe, A. Fujiedo, H. Kigoshi and M. Kita, ACS Omega, 2019, 4, 8598 CrossRef CAS PubMed; (b) T. Ohyoshi, A. Takano, M. Namiki, T. Ogura, Y. Miyazaki, Y. Ebihara, K. Takeno, I. Hawakawa and H. Kigoshi, Chem. Commun., 2018, 54, 9537 RSC.
- J. G. Solsona, J. Nebot, P. Romea and F. Urpí, J. Org. Chem., 2005, 70, 6533 CrossRef CAS PubMed.
- I. Paterson, G. J. Florence, K. Gerlach, J. P. Scott and N. Sereinig, J. Am. Chem. Soc., 2001, 123, 9535 CrossRef CAS PubMed.
- D. A. Evans and A. H. Hoveyda, J. Am. Chem. Soc., 1990, 112, 6447 CrossRef CAS.
- I. Paterson, C. J. Cowden, V. S. Rahn and M. D. Woodrow, Synlett, 1998, 915 CrossRef CAS.
- I. Paterson, K.-S. Yeung and J. B. Smaill, Synlett, 1993, 774 CrossRef CAS.
- E. J. Corey, R. K. Bakshi, S. Shibata, C. P. Chen and V. K. Singh, J. Am. Chem. Soc., 1987, 109, 7925 CrossRef CAS.
- I. Paterson, C. J. Cowden, V. S. Rahn and M. D. Woodrow, Synlett, 1998, 915 CrossRef CAS.
- J. Inanaga, K. Hirata, H. Saeki, T. Katsuki and M. Yamaguchi, Bull. Chem. Soc. Jpn., 1979, 52, 1989 CrossRef CAS.
- E. P. Boden and G. E. Keck, J. Org. Chem., 1985, 50, 2394 CrossRef CAS.
- For other recent work from our group, see: (a) I. Paterson and N. Y. S. Lam, J. Antibiot., 2017, 71, 215 CrossRef PubMed; (b) N. Y. S. Lam and I. Paterson, Eur. J. Org. Chem. DOI:10.1002/ejoc.201901243; (c) N. Y. S. Lam, G. Muir, V. R. Challa, R. Britton and I. Paterson, Chem. Commun., 2019, 55, 9717 RSC; (d) M. Anketell, T. Sharrock and I. Paterson, Angew. Chem., Int. Ed. DOI:10.1002/acie.201914042.
Footnotes |
| † Electronic supplementary information (ESI) available: Full experimental procedures and discussion. See DOI: 10.1039/c9cc09050a |
| ‡ Authors contributed to this work equally. |
| This journal is © The Royal Society of Chemistry 2020 |