
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSolvent-assisted coordination driven assembly of a supramolecular architecture featuring two types of connectivity from discrete nanocages†
Zheng
Niu
ab,
Lei
Wang
a,
Sheng
Fang
b,
Pui Ching
Lan
a,
Briana
Aguila
 a,
Jason
Perman
a,
Jian-Gong
Ma
*b,
Peng
Cheng
b,
Xiaopeng
Li
a and
Shengqian
Ma
*a
a,
Jason
Perman
a,
Jian-Gong
Ma
*b,
Peng
Cheng
b,
Xiaopeng
Li
a and
Shengqian
Ma
*a
aDepartment of Chemistry, University of South Florida, 4202 E. Fowler Avenue, Tampa, Florida 33620, USA. E-mail: sqma@usf.edu
bDepartment of Chemistry, Institution Key Laboratory of Advanced Energy Materials Chemistry (MOE), Collaborative Innovation Center of Chemical Science and Engineering, Nankai University, Tianjin 300071, P. R. China
First published on 28th May 2019
Abstract
The rapid development of supramolecular chemistry provides a powerful bottom-up approach to construct various well-defined nano-architectures with increasing complexity and functionality. Compared to that of small and simple nanometric objects, the self-assembly of larger and more complex nanometric objects, such as nanocages, remains a significant challenge. Herein, we used a discrete nanocage as the monomer to successfully construct a novel three-dimensional (3D) supramolecular architecture, which comprises two types of nanocage building units with different connectivity, using the solvent-assisted coordination-driven assembly approach. The mechanism of this supramolecular assembly process was investigated by electrospray ionization mass spectrometry (ESI-MS) studies, which identified for the first time the formation of a nanocage dimer intermediate during the assembly process. The assembly of discrete nanocages into a 3D supramolecular architecture led to remarkable enhancement of stability and gas adsorption properties.
Introduction
The construction of structurally well-defined nanoarchitectures starting from discrete nanometric scale objects has quickly become an important route for developing functional materials for magnetics, separation, energy storage, and biological applications.1 From cubes to spheres, various nanometric objects have been used as building blocks to assemble nanoarchitectures for specific applications.2 As one type of nano-object, metal–organic cages (MOCs) that feature pre-designed windows and cavities have captured widespread attention3 in the areas of chemical sensing,4 catalysis,5 molecular encapsulation,6 and separation.7 The abundant metal sites of the nanocage complex8 make MOCs promising candidates for CO2 adsorption. However, most MOC crystals are unstable, which is presumably due to the weak interaction between the discrete cage molecules. This leads to a collapse of the degassing process, thereby largely restricting their application. Alternatively, assembling the discrete MOCs into 1D, 2D, and 3D nanoarchitectures through direct bond-formation would significantly boost the stability as compared to discrete MOCs.1b,9 While extensive studies have been performed on the assembly of MOCs, understanding the intermediate from discrete MOCs to nanocage architectures and assembling these discrete nanocages into more complex hierarchical structures still remain formidable challenges.To date, only three different nanocage architecture nets (4-coordinated (abbreviated 4-c), 6-c and 12-c nets) have been obtained via coordination interactions between additional linkers and nanocages (Scheme 1).10 In these structures, all of the potential junction points on the nanocages can connect with linkers because of the small steric hindrance between the linkers and the nanocages; thus these structures showed simple single node nets. Compared to that in the linker-connected nanocage architectures, the number of potential linkage junctions in “linkerless” nanocage architectures is depended on the steric hindrance of neighboring nanocages, thus affecting the complexity of the nanocage architecture. Until now, all of the reported studies have exhibited only the single node topology structures (4-c or 6-c nets) and the intermediate structure of these assembly processes remains a mystery.9 For most of the MOCs, their high symmetry11,12 restricts the generation of multiple connecting nodes, while multiple connectivity is essential for constructing complex hierarchical structures. To expand the possible complex architectures using one kind of nanocage reagent, multiple types of connectivity are needed for the nanocage building units, which has thus far remained challenging.
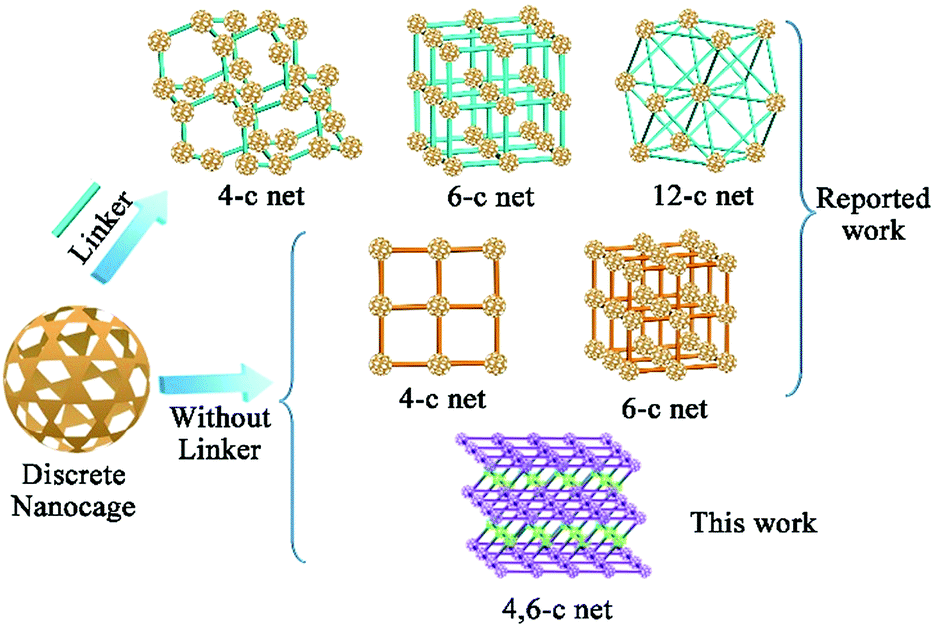 | ||
| Scheme 1 The summary of current nanocage architectures assembled from discrete nanocages. | ||
In this contribution, we present a paradigm to assemble a 3D nanocage architecture with (4,6)-coordinated (abbreviated (4,6)-c) nets from discrete MOCs for the first time. A nanocage dimer intermediate during the supramolecular assembly process was also observed for the first time through electrospray ionization mass spectrometry (ESI-MS) studies. Compared to discrete nanocage materials, the assembly of these nanocage architectures leads to a stable framework with significantly enhanced gas adsorption performance. In particular, the (4,6)-c nanocage architecture demonstrates significantly enhanced gas adsorption performance compared with 4-c and 6-c single node nanocage architectures with similar nanocage units.
Results and discussion
Structure description of nanocage 1 and nanocage architecture 2
Nanocage 1, [Cu24(L)24(DMF)12(H2O)12]·36MeOH·59DMF, was used to construct the novel nanocage architecture and to explore the formation of the intermediate during the assembly process. The structure of 1 as determined from SCXRD measurement shows that the nanocage of 1 is constructed with twelve Cu2(COO)4 paddlewheels and twenty-four 2,4-isophthalate bridging ligands. The 120-degree angle between the two carboxylic acids of the ligand connect the paddlewheels to form a rhombihexahedron cage with eight triangular and six square windows (Fig. 1). The nanocage of 1 has an approximately 3 nm outer diameter and a 1.5 nm inner void diameter. As depicted in Fig. 1a, each outer vertex of the Cu2(COO)4 paddlewheel is occupied by DMF solvent molecules.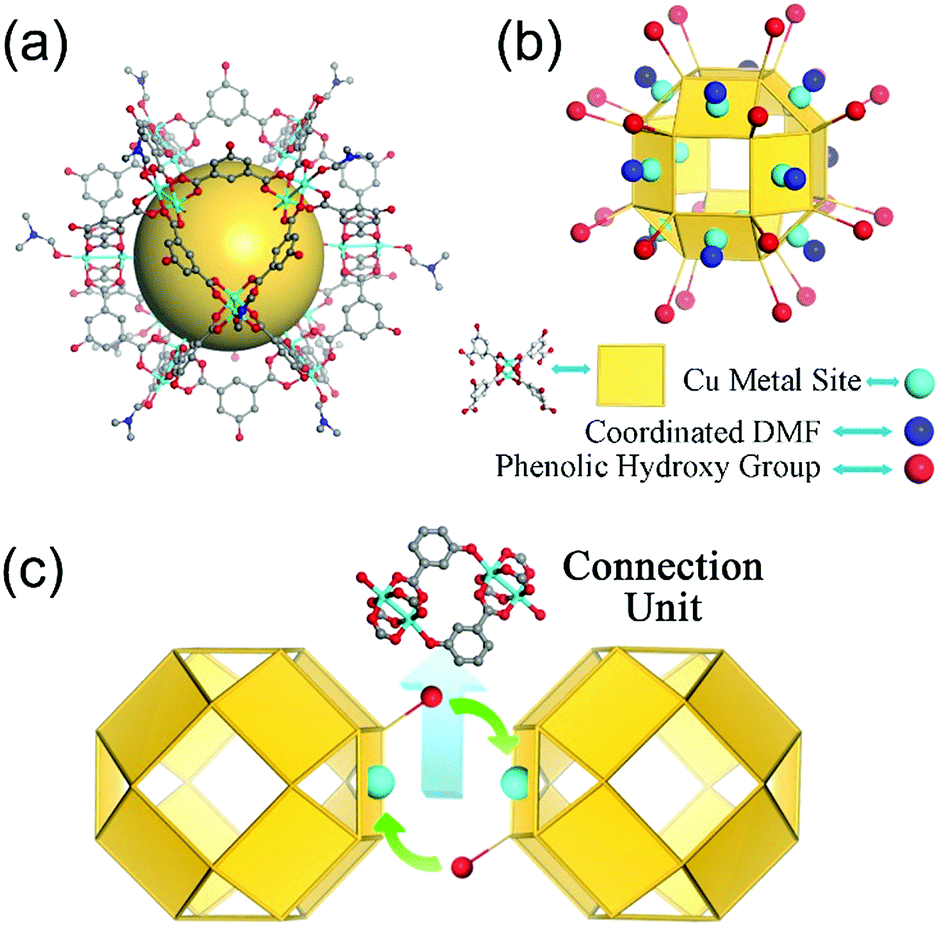 | ||
| Fig. 1 The structure of discrete nanocage 1. (a) The ball–stick representation of nanocage 1; (b) the simplified structure of nanocage 1; (c) the orbicular connection unit between two neighboring nanocages. | ||
The basic nanocage of 1 exhibits good dissolvability in MeOH and the existence of similar discrete cages in the solvent as individual molecules has been proven in previously reported studies.13 The potential metal sites (vertices of Cu2(COO)4 paddlewheels) and phenolic hydroxyl groups give 1 the ability to connect with neighboring cages through coordination interactions. However, the attempt for direct assembly of nanocage 1 was unsuccessful due to the steric hindrance effect of coordinated DMF molecules. To supply a favorable environment for the assembly of the nanocage, we adjusted the temperature and introduced DMSO into the reaction to compete with coordinated DMF molecules.
Following the addition of a small amount of DMSO into the nanocage 1 MeOH solution (DMSO![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :MeOH = 1:4) and after heating the mixture at 120 °C for 1 day, green block crystals were obtained. SCXRD analysis indicates that the nanocages of 1 form a 3D architecture with a formula of {[Cu24(L)24DMSO (MeOH)4(H2O)14]·66MeOH·18DMSO}n (2, the guest molecules were determined through elemental analysis and thermogravimetric analysis). The nanocage architecture 2 was constructed with the small molecule-coordinated [Cu24(L)24] basic nanocage building unit, which comes from the nanocage of 1. In the structure of 2, the nanocage building units connect with each other through a type of orbicular connection unit, which is constructed between two phenolic hydroxyl groups from one nanocage and two Cu2(COO)4 paddlewheel vertices from the neighboring nanocages (Fig. 1c). During the polymerization process, DMSO could substitute the coordinated DMF molecules and reduce the steric hindrance around the metal sites, thus facilitating the assembly of the nanocages.
:MeOH = 1:4) and after heating the mixture at 120 °C for 1 day, green block crystals were obtained. SCXRD analysis indicates that the nanocages of 1 form a 3D architecture with a formula of {[Cu24(L)24DMSO (MeOH)4(H2O)14]·66MeOH·18DMSO}n (2, the guest molecules were determined through elemental analysis and thermogravimetric analysis). The nanocage architecture 2 was constructed with the small molecule-coordinated [Cu24(L)24] basic nanocage building unit, which comes from the nanocage of 1. In the structure of 2, the nanocage building units connect with each other through a type of orbicular connection unit, which is constructed between two phenolic hydroxyl groups from one nanocage and two Cu2(COO)4 paddlewheel vertices from the neighboring nanocages (Fig. 1c). During the polymerization process, DMSO could substitute the coordinated DMF molecules and reduce the steric hindrance around the metal sites, thus facilitating the assembly of the nanocages.
In general, the high symmetry of the MOC leads to a single type of nanocage building unit in most reported nanocage architectures.11,12 Interestingly, the nanocage of 1 exhibits high Oh symmetry; moreover, two types of nanocage building units, nanocage building units A and B (Fig. 2), were observed in the nanocage architecture 2. To the best of our knowledge, this is the first time a nanocage architecture with two types of nanocage building units has been obtained. The basic structure of the nanocage building units A and B are similar, yet their connection numbers are different. As shown in Fig. 2, each nanocage building unit B has six Cu metal sites (turquoise balls) and six phenolic hydroxyl groups (red balls), which can coordinate with neighboring cages, including four coplanar nanocages and two out-of-plane nanocages, thus generating a 6-connected node. In comparison, nanocage building unit A has four Cu metal sites and four phenolic hydroxyl groups coordinating with neighboring nanocages, thus forming a 4-connected node with four neighboring nanocages in the same plane.
 | ||
| Fig. 2 Structure of nanocage architecture 2. The ball–stick representation (left) and simplified model (right) of (a) nanocage building unit A and (b) nanocage building unit B; (c) the interconnection between nanocage building units A and B. (d) The topology of nanocage architecture 2. (The turquoise and red balls stand for coordinated Cu metal sites and phenolic hydroxyl groups between neighboring cages, respectively; the green and purple polyhedra/balls stand for 4-connected nanocage building unit A and 6-connected nanocage building unit B, respectively). | ||
In nanocage architecture 2, nanocage building units A and B alternately connect with each other to construct a 3D polymer of nanocages. As shown in Fig. 2c, each nanocage building unit B (purple polyhedron) connects with four B and two A (green polyhedron); meanwhile each nanocage building unit A connects with two A and two B, thus constructing a 4,6-connected structure. Topologically, 2 features a rare fsg net14 (Fig. 2d). To our knowledge, this is the first time that a fsg net has been observed in a coordination polymer built with cage or cluster blocks.
Studies of the assembly process from nanocage monomer 1 to nanocage architecture 2
To understand the process for the assembly of nanocage architecture 2 from discrete nanocage 1, electrospray ionization mass spectrometry (ESI-MS) experiments were conducted. The existence of the individual nanocage monomers in the reaction solution (DMSO:MeOH = 1:4) was confirmed by ESI-MS studies. As shown in Fig. 3a, the ESI-MS spectrum of the individual nanocage monomer showed a series of doubly charged signals corresponding to the loss of encapsulated solvent molecules including DMF and H2O (i.e., m/z = 3050.81 for [Cu24(C8O5H4)24(DMF)2(H2O)6 + 2H+]2+). Interestingly, after heating the solution at 120 °C for 1 h, the peaks belonging to individual nanocage monomers shifted slightly towards a higher m/z region and a new series of peaks appeared in the ESI-MS spectrum (Fig. S6–S8†). As depicted in Fig. 3b, the doubly charged peaks correspond to the loss of solvent molecules including DMSO and H2O (i.e. m/z = 3055.77 for [Cu24(C8O5H4)24(DMSO)2(H2O)6 + 2H+]2+). These results indicated that the encapsulated DMF molecules were substituted by DMSO molecules during the assembly process. In particular, a new sets of peaks (Fig. 3c) between 4000 and 4300 (m/z) with triple charge was attributed to the nanocage dimer based on calculation (i.e., m/z = 4116.71 for [(Cu24(C8O5H4)24(DMSO)2(H2O)9.5)2 + 3H+]3+). The isotope pattern of all the assigned peaks agreed well with the calculated values (Fig. S9†), indicating the composition of the proposed structure.
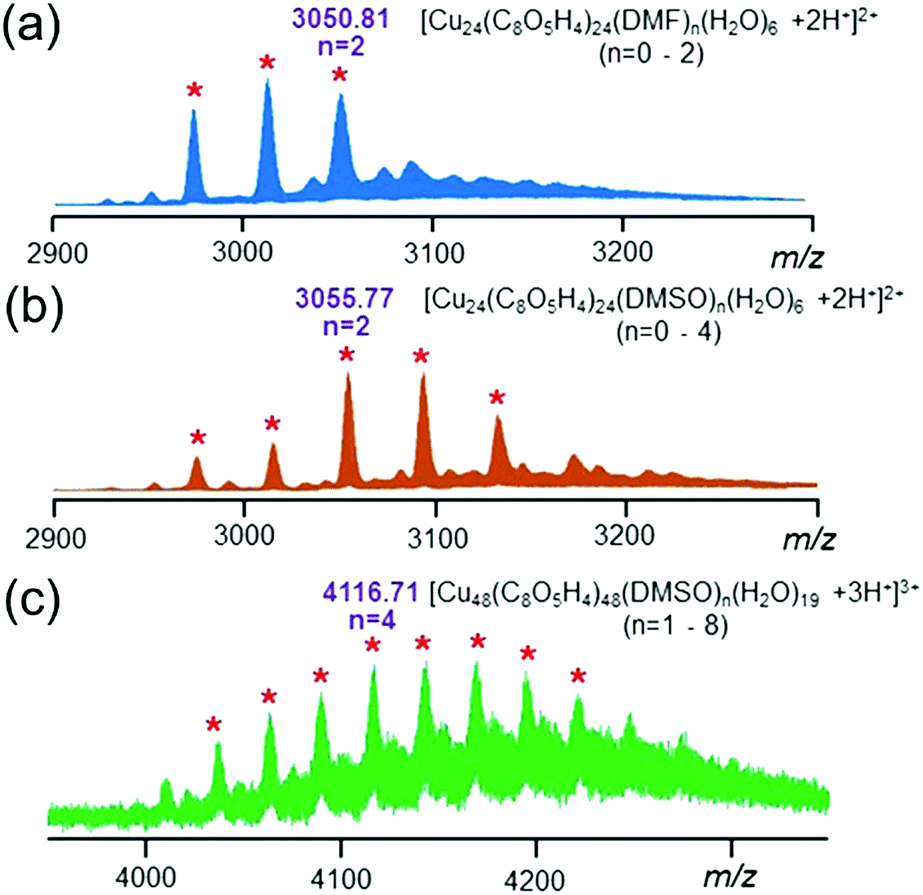 | ||
| Fig. 3 ESI-MS spectra of (a) nanocage monomer (charge state 2+, loss of DMF molecules); (b) DMSO substituted nanocage monomer (charge state 2+, loss of DMSO molecules); (c) dimer nanocage intermediate (charge state 3+, loss of DMSO molecules). | ||
On the basis of SCXRD and ESI-MS results, the detailed assembly process of the nanocage architecture is illustrated in Scheme 2. When the nanocage 1 was dispersed in the DMSO/MeOH solvent mixture, the Cu metal sites were still occupied by DMF molecules. After heating the solution at 120 °C for 1 h, the DMF molecules were substituted by DMSO molecules, which implied that the DMSO coordinated nanocage was favored when increasing the temperature. Meanwhile, two phenolic hydroxyl groups from one nanocage coordinated with two Cu2(COO)4 paddlewheel vertices from the neighboring nanocages to construct an orbicular connection unit, thus forming the novel nanocage dimer intermediate. This intermediate indicated that the substitution of DMF by DMSO on nanocage 1 reduces the steric hindrance effect around the metal sites, thus facilitating the polymerization of the nanocages to form the nanocage architecture 2.
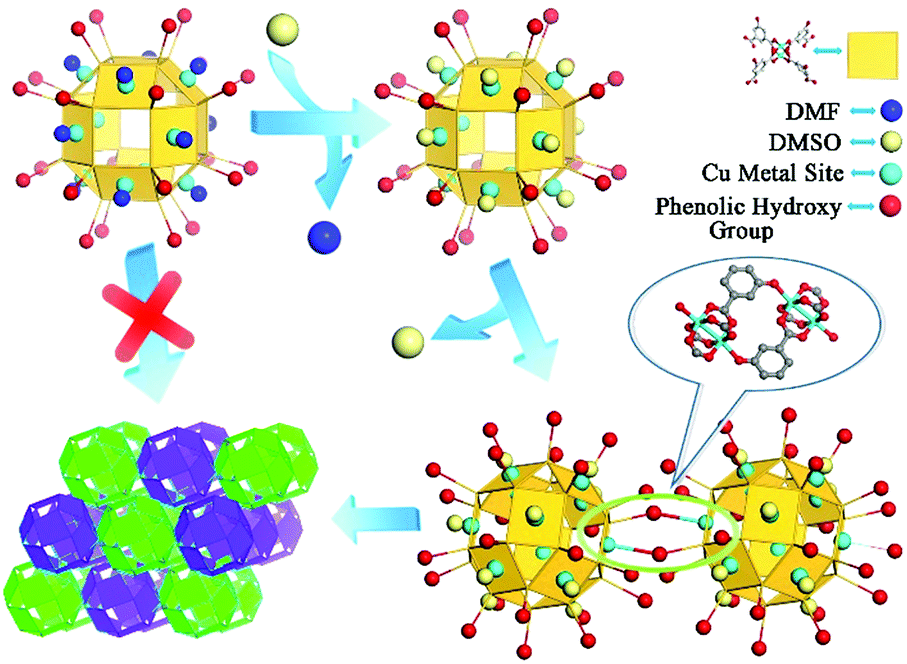 | ||
| Scheme 2 The detailed assembly process of nanocage architecture 2 from discrete nanocage 1 and the nanocage dimer. | ||
Gas adsorption studies of nanocage monomer 1 and nanocage architecture 2
Similar to the 4-c and 6-c nanocage architectures, nanocage architecture 2 is stable in common organic solvents, such as MeOH, EtOH, DMA, DMF, and DMSO. The good stability of nanocage polymer 2 prompted us to examine the gas adsorption properties of the discrete nanocage 1 and the nanocage architecture 2. The Brunauer–Emmett–Teller (BET) surface areas of nanocage 1 and nanocage architecture 2 were derived from the CO2 adsorption isotherms at 195 K. As presented in Fig. 4a, the CO2 isotherms at 195 K of both nanocage 1 and nanocage architecture 2 show type-I adsorption behavior, indicating the microporosity of both. The BET surface area of nanocage architecture 2 reaches 604 m2 g−1, which is 8 times higher than that of nanocage 1 (73 m2 g−1). In particular, the surface area of 2 (4,6-c nanocage architecture) is also much higher than that of 4-c and 6-c nanocage architectures (244 m2 g−1 and 471 m2 g−1, respectively). The higher BET surface area of 2 can be attributed to the 4,6-c net, which provides a more stable 3D framework than the 4-c 2D net as well as more porosity compared to the 6-c net. The CO2 adsorption measurement of the nanocage and nanocage architectures further underscores the advantage of the 4,6-c net in gas adsorption. As shown in Fig. 4b, the CO2 adsorption capacity at 298 K for the nanocage architecture 2 is 35 cm3 g−1, which is not only much higher than that of nanocage 1, but also higher than that of 4-c and 6-c nanocage architectures (17 cm3 g−1 and 27 cm3 g−1, respectively). The gas adsorption results indicate that the assembly of the discrete nanocages into a 3D nanoarchitecture with the 4,6-c net not only drastically increases the surface area, but also shows better CO2 gas adsorption performance compared with the nanoarchitectures with a single node net.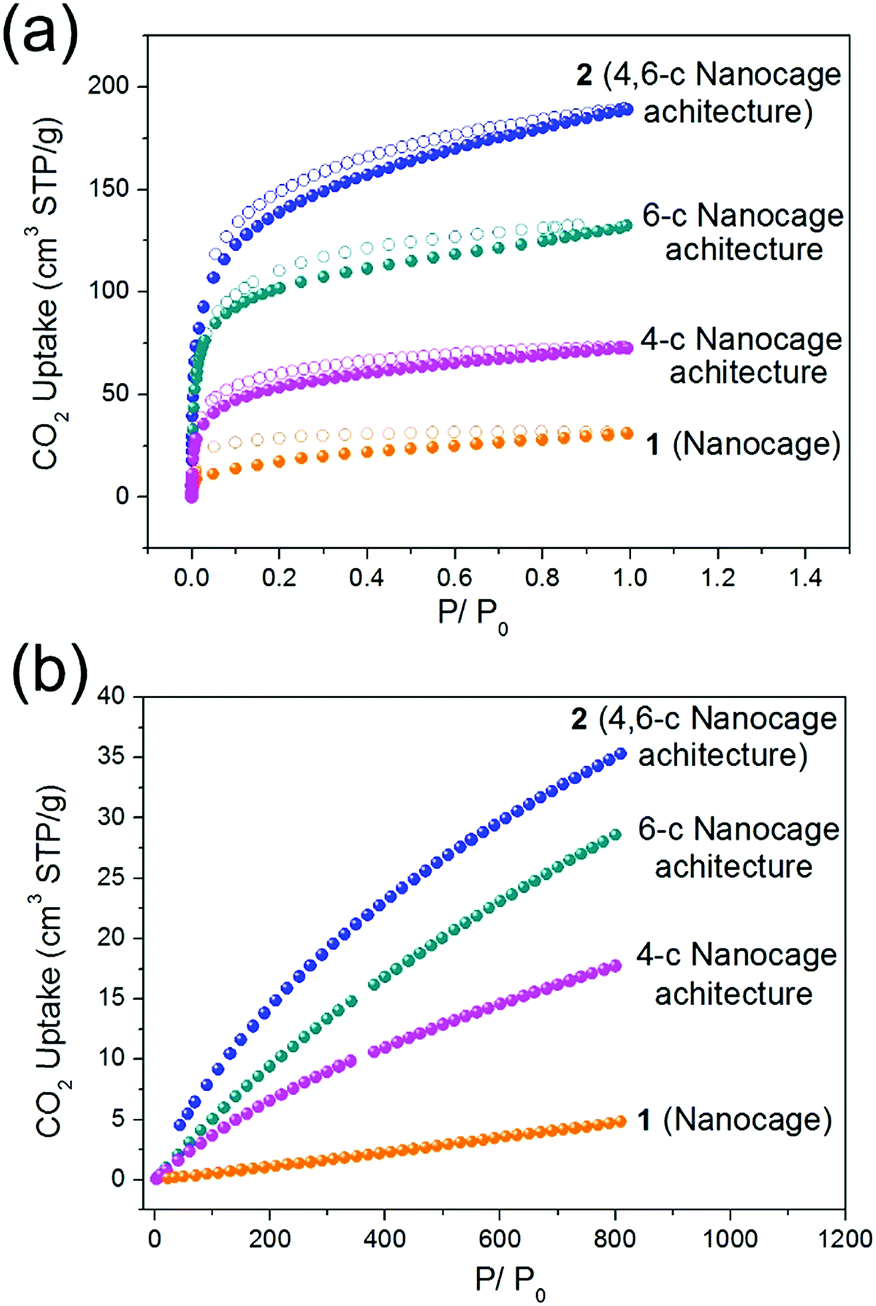 | ||
| Fig. 4 The CO2 adsorption isotherms of the nanocage (1), 4-c nanocage architecture, 6-c nanocage architecture and 4,6-c nanocage architecture (2) at (a) 195 K and (b) 298 K. | ||
Conclusion
In summary, we have successfully constructed a 3D supramolecular architecture featuring dual nanocage building units through solvent-assisted coordination-driven assembly of one type of nanocage for the first time. The nanocage dimer intermediate formed during the assembly process was also identified for the first time via ESI-MS studies. The two types of nanocage building units feature different connectivity, thus enabling the construction of a complex 4,6-c nanocage architecture with the rare fsg topology. The 4,6-c nanocage architecture showed an enhanced surface area and CO2 adsorption performance compared with nanocage 1 and the 4-c and 6-c nanocage architectures. Our work thereby paves the way for controlled construction of complex nano-object architectures from discrete nano-objects for a broader range of applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the NSF (DMR-1352065) and the University of South Florida for financial support of this work. Partial financial support from the National Key R&D Program of China (2016YFB0600902) is also acknowledged.Notes and references
- (a) T. F. Liu, Y. P. Chen, A. A. Yakovenko and H. C. Zhou, J. Am. Chem. Soc., 2012, 134, 17358–17361 CrossRef CAS PubMed; (b) J. Tian, Z.-Y. Xu, D.-W. Zhang, H. Wang, S.-H. Xie, D.-W. Xu, Y.-H. Ren, H. Wang, Y. Liu and Z.-T. Li, Nat. Commun., 2016, 7, 11580–11588 CrossRef CAS PubMed; (c) H. Kumari, C. L. Dennis, A. V. Mossine, C. A. Deakyne and J. L. Atwood, J. Am. Chem. Soc., 2013, 135, 7110–7113 CrossRef CAS PubMed; (d) D. Wang, R. Kou, D. Choi, Z. Yang, Z. Nie, J. Li, L. V. Saraf, D. Hu, J. Zhang, G. L. Graff, J. Liu, M. A. Pope and I. A. Aksay, ACS Nano, 2010, 4, 1587–1595 CrossRef CAS PubMed; (e) G. Zheng, F. Patolsky, Y. Cui, W. U. Wang and C. M. Lieber, Nat. Biotechnol., 2005, 23, 1294–1301 CrossRef CAS PubMed.
- (a) P. Wei, T. R. Cook, X. Yan, F. Huang and P. J. Stang, J. Am. Chem. Soc., 2014, 136, 15497–15500 CrossRef CAS PubMed; (b) J. E. Macdonald, M. Bar Sadan, L. Houben, I. Popov and U. Banin, Nat. Mater., 2010, 9, 810–815 CrossRef CAS PubMed; (c) Z. Y. Li, Y. Zhang, C. W. Zhang, L. J. Chen, C. Wang, H. Tan, Y. Yu, X. Li and H. B. Yang, J. Am. Chem. Soc., 2014, 136, 8577–8589 CrossRef CAS PubMed; (d) K. M. Hutchins, T. P. Rupasinghe, L. R. Ditzler, D. C. Swenson, J. R. Sander, J. Baltrusaitis, A. V. Tivanski and L. R. MacGillivray, J. Am. Chem. Soc., 2014, 136, 6778–6781 CrossRef CAS PubMed.
- (a) T. R. Cook and P. J. Stang, Chem. Rev., 2015, 115, 7001–7045 CrossRef CAS PubMed; (b) H. Kumari, C. A. Deakyne and J. L. Atwood, Acc. Chem. Res., 2014, 47, 3080–3088 CrossRef CAS PubMed; (c) K. Jie, Y. Zhou, Y. Yao, B. Shi and F. Huang, J. Am. Chem. Soc., 2015, 137, 10472–10475 CrossRef CAS PubMed; (d) M. Kieffer, B. S. Pilgrim, T. K. Ronson, D. A. Roberts, M. Aleksanyan and J. R. Nitschke, J. Am. Chem. Soc., 2016, 138, 6813–6821 CrossRef CAS PubMed; (e) B. M. Schmidt, T. Osuga, T. Sawada, M. Hoshino and M. Fujita, Angew. Chem., Int. Ed., 2016, 55, 1561–1564 CrossRef CAS PubMed; (f) F. R. Dai and Z. Wang, J. Am. Chem. Soc., 2012, 134, 8002–8005 CrossRef CAS PubMed; (g) T. D. Hamilton, G. S. Papaefstathiou, T. Friscic, D. K. Bucar and L. R. MacGillivray, J. Am. Chem. Soc., 2008, 130, 14366–14367 CrossRef CAS PubMed; (h) D. J. Tranchemontagne, Z. Ni, M. O'Keeffe and O. M. Yaghi, Angew. Chem., Int. Ed., 2008, 47, 5136–5147 CrossRef CAS PubMed; (i) D. Luo, X. P. Zhou and D. Li, Angew. Chem., Int. Ed., 2015, 54, 6190–6195 CrossRef CAS PubMed; (j) M. H. Alkordi, J. L. Belof, E. Rivera, L. Wojtas and M. Eddaoudi, Chem. Sci., 2011, 2, 1695–1705 RSC; (k) M. Liu, W. Liao, C. Hu, S. Du and H. Zhang, Angew. Chem., Int. Ed., 2012, 51, 1585–1588 CrossRef CAS PubMed; (l) D. Liu, Z. Jiang, M. Wang, X. Yang, H. Liu, M. Chen, C. N. Moorefield, G. R. Newkome, X. Li and P. Wang, Chem. Commun., 2016, 52, 9773–9776 RSC.
- (a) M. L. Saha, X. Yan and P. J. Stang, Acc. Chem. Res., 2016, 49, 2527–2539 CrossRef CAS PubMed; (b) X. Yan, M. Wang, T. R. Cook, M. Zhang, M. L. Saha, Z. Zhou, X. Li, F. Huang and P. J. Stang, J. Am. Chem. Soc., 2016, 138, 4580–4588 CrossRef CAS PubMed; (c) D. Zhao, S. Tan, D. Yuan, W. Lu, Y. H. Rezenom, H. Jiang, L. Q. Wang and H. C. Zhou, Adv. Mater., 2011, 23, 90–93 CrossRef CAS PubMed; (d) M. Zhang, M. L. Saha, M. Wang, Z. Zhou, B. Song, C. Lu, X. Yan, X. Li, F. Huang, S. Yin and P. J. Stang, J. Am. Chem. Soc., 2017, 139, 5067–5074 CrossRef CAS PubMed.
- (a) Y. H. Kang, X. D. Liu, N. Yan, Y. Jiang, X. Q. Liu, L. B. Sun and J. R. Li, J. Am. Chem. Soc., 2016, 138, 6099–6102 CrossRef CAS PubMed; (b) S. Chen, K. Li, F. Zhao, L. Zhang, M. Pan, Y. Z. Fan, J. Guo, J. Shi and C. Y. Su, Nat. Commun., 2016, 7, 13169 CrossRef CAS PubMed; (c) X. Jing, C. He, Y. Yang and C. Duan, J. Am. Chem. Soc., 2015, 137, 3967–3974 CrossRef CAS PubMed; (d) J. Guo, Y. W. Xu, K. Li, L. M. Xiao, S. Chen, K. Wu, X. D. Chen, Y. Z. Fan, J. M. Liu and C. Y. Su, Angew. Chem., Int. Ed., 2017, 56, 3852–3856 CrossRef CAS PubMed.
- (a) Z. J. Wang, C. J. Brown, R. G. Bergman, K. N. Raymond and F. D. Toste, J. Am. Chem. Soc., 2011, 133, 7358–7360 CrossRef CAS PubMed; (b) S. Wang, X. Gao, X. Hang, X. Zhu, H. Han, W. Liao and W. Chen, J. Am. Chem. Soc., 2016, 138, 16236–16239 CrossRef CAS PubMed; (c) D. P. August, G. S. Nichol and P. J. Lusby, Angew. Chem., Int. Ed., 2016, 55, 15022–15026 CrossRef CAS PubMed; (d) J. Mosquera, B. Szyszko, S. K. Ho and J. R. Nitschke, Nat. Commun., 2017, 8, 14882 CrossRef CAS PubMed.
- (a) X. Hang, B. Liu, X. Zhu, S. Wang, H. Han, W. Liao, Y. Liu and C. Hu, J. Am. Chem. Soc., 2016, 138, 2969–2972 CrossRef CAS PubMed; (b) W. Brenner, T. K. Ronson and J. R. Nitschke, J. Am. Chem. Soc., 2017, 139, 75–78 CrossRef CAS PubMed; (c) W. P. Chen, P. Q. Liao, Y. Yu, Z. Zheng, X. M. Chen and Y. Z. Zheng, Angew. Chem., Int. Ed., 2016, 55, 9375–9379 CrossRef CAS PubMed; (d) W. Y. Pei, G. Xu, J. Yang, H. Wu, B. Chen, W. Zhou and J. F. Ma, J. Am. Chem. Soc., 2017, 139, 7648–7656 CrossRef CAS PubMed.
- (a) M. Eddaoudi, J. Kim, J. B. Wachter, H. K. Chae, M. O'Keeffe and O. M. Yaghi, J. Am. Chem. Soc., 2001, 123, 4368–4369 CrossRef CAS PubMed; (b) H. Abourahma, A. W. Coleman, B. Moulton, B. Rather, P. Shahgaldian and M. J. Zaworotko, Chem. Commun., 2001, 2380–2381 RSC.
- (a) C. Zhang, R. S. Patil, C. Liu, C. L. Barnes and J. L. Atwood, J. Am. Chem. Soc., 2017, 139, 2920–2923 CrossRef CAS PubMed; (b) Z. Niu, S. Fang, X. Liu, J. G. Ma, S. Ma and P. Cheng, J. Am. Chem. Soc., 2015, 137, 14873–14876 CrossRef CAS PubMed.
- (a) L. Cao, P. Wang, X. Miao, Y. Dong, H. Wang, H. Duan, Y. Yu, X. Li and P. J. Stang, J. Am. Chem. Soc., 2018, 140, 7005–7011 CrossRef CAS PubMed; (b) J.-R. Li, D. J. Timmons and H.-C. Zhou, J. Am. Chem. Soc., 2009, 131, 6368–6369 CrossRef CAS PubMed; (c) H.-N. Wang, X. Meng, G.-S. Yang, X.-L. Wang, L.-Z. Shao, Z.-M. Su and C.-G. Wang, Chem. Commun., 2011, 47, 7128–7130 RSC.
- (a) Y. Sun, Y. Yao, H. Wang, W. X. Fu, C. Y. Chen, M. L. Saha, M. M. Zhang, S. Datta, Z. X. Zhou, H. X. Yu, X. P. Li and P. J. Stang, J. Am. Chem. Soc., 2018, 140, 12819–12828 CrossRef CAS PubMed; (b) Y. Sun, F. M. Zhang, S. W. Jiang, Z. F. Wang, R. D. Ni, H. Wang, W. D. Zhou, X. P. Li and P. J. Stang, J. Am. Chem. Soc., 2018, 140, 17297–17307 CrossRef CAS PubMed.
- (a) S. R. Seidel and P. J. Stang, Acc. Chem. Res., 2002, 35, 972–983 CrossRef CAS PubMed; (b) J. R. Li and H. C. Zhou, Nat. Chem., 2010, 2, 893–898 CrossRef CAS PubMed; (c) L. Chen, Q. Chen, M. Wu, F. Jiang and M. Hong, Acc. Chem. Res., 2015, 48, 201–210 CrossRef CAS PubMed; (d) S. Zarra, D. M. Wood, D. A. Roberts and J. R. Nitschke, Chem. Soc. Rev., 2015, 44, 419–432 RSC.
- (a) L. B. Sun, J. R. Li, W. Lu, Z. Y. Gu, Z. Luo and H. C. Zhou, J. Am. Chem. Soc., 2012, 134, 15923–15928 CrossRef CAS PubMed; (b) R. W. Larsen, G. J. McManus, J. J. Perry IV, E. Rivera-otero and M. J. Zaworotko, Inorg. Chem., 2007, 46, 5904–5910 CrossRef CAS PubMed.
- J. Wang, Z. J. Lin, Y. C. Ou, Y. Shen, R. Herchel and M. L. Tong, Chem.–Eur. J., 2008, 14, 7218–7235 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1569639 and 1569640. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc01892a |
| This journal is © The Royal Society of Chemistry 2019 |