
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNickel-catalyzed decarbonylation of N-acylated N-heteroarenes†
Toshifumi
Morioka
,
Syun
Nakatani
,
Yuki
Sakamoto
,
Takuya
Kodama
 ,
Sensuke
Ogoshi
,
Naoto
Chatani
* and
Mamoru
Tobisu
*
,
Sensuke
Ogoshi
,
Naoto
Chatani
* and
Mamoru
Tobisu
*
Department of Applied Chemistry, Faculty of Engineering, Osaka University, Suita, Osaka 565-0871, Japan. E-mail: tobisu@chem.eng.osaka-u.ac.jp; chatani@chem.eng.osaka-u.ac.jp
First published on 29th May 2019
Abstract
Nickel-catalyzed decarbonylation of N-acylated N-heteroarenes is developed. This method can be used to produce a variety of N-aryl heteroarenes, including pyrroles, indoles, carbazoles and phenoxazines, using benzoic acid derivatives as arylating reagents. Arylnickelamide intermediates that are relevant to the catalytic reaction were characterized by X-ray crystallography. When N-acylated benzimidazoles are used as substrates, decarbonylation accompanied 1,2-migration to form 2-arylated benzimidazoles.
Introduction
Because of their widespread utility in medicinal and materials chemistry,1 methodology for preparing N-arylated heteroarenes has continued to be a subject of intense studies. Currently, the most powerful approach to these compounds involves the catalytic cross-coupling of aryl halides with NH-heteroarenes (Scheme 1a).2,3 If aromatic acids, in place of aryl halides, could be used as aryl sources for the synthesis of N-aryl N-heteroarenes, several benefits would be expected. For example, aromatic acids are widely available feedstock materials, often with substitution patterns that are difficult to access from the corresponding aryl halides, and therefore the flexibility of synthetic planning would be enhanced. In addition, a new synthetic strategy, such as orthogonal and late-stage transformation, would be possible if a carboxyl group can serve as a handle with a reactivity profile different from halides. However, despite significant progress made in the decarboxylative cross-coupling,4 decarboxylative N-arylation of NH-heteroarenes has not been reported, to the best of our knowledge.5 We hypothesized that the use of an aromatic acid in the N-arylation of NH-heteroarenes would be possible formally through a two-step sequence, namely, amide formation (N-acylation) and decarbonylation of the resulting amide (Scheme 1b). Since a number of reliable methods are available for amide formation,6,7 the key issue is the development of the latter decarbonylation reaction. Except for 2-azinecarboxamides,8 catalytic decarbonylation of amides is an unattained transformation, indicating that C(aryl)–N bond formation via decarbonylation is clearly not a trivial task. We report herein on the realization of this objective.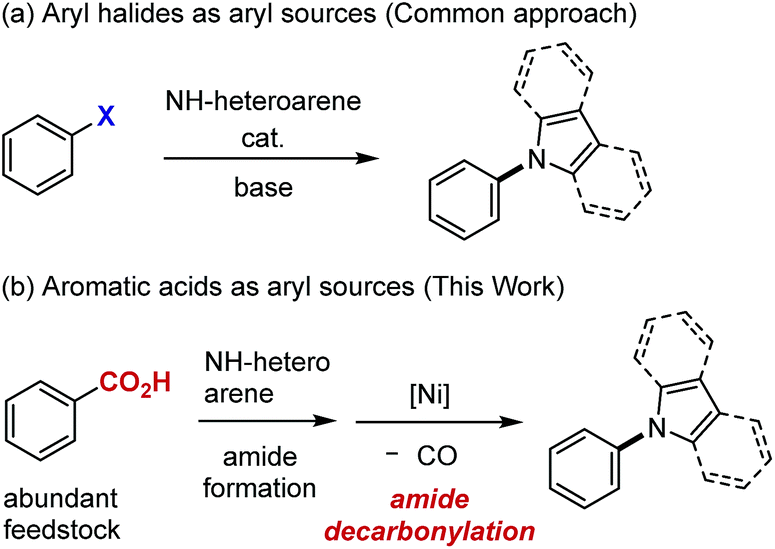 | ||
| Scheme 1 Approaches to the N-arylation of heteroarenes. | ||
Results and discussion
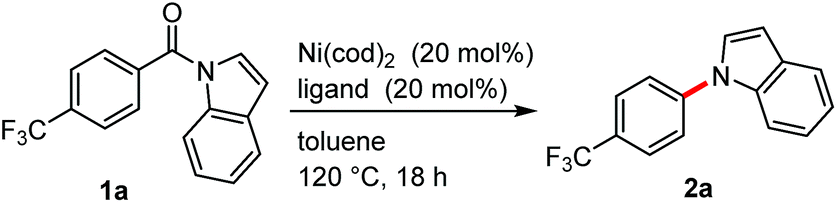
We initiated our study by examining the decarbonylation of N-acylindole 1a. To promote this reaction, we focused on the use of a nickel catalyst, since it has been successfully used in the activation of amides9 and in the ketone decarbonylation reaction.10 A brief screening of ligands revealed that strong σ-donor ligands, such as alkylphosphines and N-heterocyclic carbenes (NHCs), actually afforded the desired decarbonylation product 2a in 12–31% yield (Table 1, entries 2–7). Although the difference in the yield of 2a was not significant between the ligands examined, a larger amount of 1a was recovered from the reactions when phosphine ligands were used than when NHCs were used. Therefore, further optimization was pursued using PCy3 and 1,2-bis(dicyclohexylphosphino)ethane (dcype). When 1 equiv. of Ni(cod)2 and PCy3 were used, the decarbonylation proceeded quantitatively, even at 80 °C (entry 8). The catalytic decarbonylation was accomplished by running the reaction at 180 °C using dcype as the ligand (entry 11). The reaction was not effective when conducted at 180 °C using 20 mol% of Ni(cod)2/PCy3 (entry 9) nor at 80 °C using 1 equiv. of Ni(cod)2/dcype (entry 10). We therefore decided to use two sets of conditions for investigating the scope and limitations of the reaction: 20 mol% of Ni(cod)2/dcype at 180 °C (condition A) and 1 equiv. of Ni(cod)2/PCy3 at 80 °C (condition B).|
|
|||
|---|---|---|---|
| Entry | Ligandb | GC yields (%) | |
| 2a | 1a | ||
| a Reaction conditions: 1a (0.25 mmol), Ni(cod)2 (0.050 mmol), ligand (0.050 mmol), in toluene (1.0 mL) for 18 h at 120 °C. b N-Heterocyclic carbene ligands were generated in situ from the corresponding imidazolium salts by treatment with NaOtBu (25 mol%). c Ni(cod)2 (0.25 mmol) and the ligand (0.25 mmol) were used at 80 °C. d Run at 180 °C. e Isolated yield. | |||
| 1 | PPh3 | 0 | 71 |
| 2 | PCy3 | 31 | 60 |
| 3 | dcype | 16 | 52 |
| 4 | ICy | 0 | 46 |
| 5 | IMes | 28 | 29 |
| 6 | IMesMe | 27 | 27 |
| 7 | IPr | 12 | 25 |
| 8c | PCy3 | >99 (88)e | 0 |
| 9d | PCy3 | 19 | 65 |
| 10c | dcype | 4 | 86 |
| 11d | dcype | 93 (84)e | 0 |
|
|||
The scope of the N-acyl group was initially investigated to identify the electronic requirement for this nickel-catalyzed decarbonylation reaction (Table 2). Under the catalytic conditions (condition A), N-acylindoles bearing an electron-withdrawing group (i.e., 1a and 1b) readily underwent decarbonylation, while electron-neutral (1c and 1d) and electron-rich (1e and 1f) amides were much less reactive.11 The yields of these less reactive amides could be improved by using condition B, in which Ni(cod)2/PCy3 (1 equiv. each) was used.
|
|
|||
|---|---|---|---|
| R | σ p | Isolated yield [%] | |
| A | B | ||
| a Condition A: amide (0.25 mmol), Ni(cod)2 (0.050 mmol), dcype (0.050 mmol) in toluene (1.0 mL) for 18 h at 180 °C; condition B: amide (0.25 mmol), Ni(cod)2 (0.25 mmol), PCy3 (0.25 mmol) in toluene (1.0 mL) for 18 h at 80 °C. b Substituent constants from ref. 11. c GC yield. d NMR yield. e 3,4-Bis(dicyclohexylphosphino)thiophene (100 mol%) was used instead of PCy3 and run at 180 °C. | |||
| CF3 (1a) | 0.54 | 84 | — |
| CO2Me (1b) | 0.45 | 85 | — |
| Ph (1c) | −0.01 | 30c | 79 |
| H (1d) | 0 | 23c | 71 |
| Me (1e) | −0.17 | 18c | 63 |
| OMe (1f) | −0.27 | 8c | 14d (33)c,e |
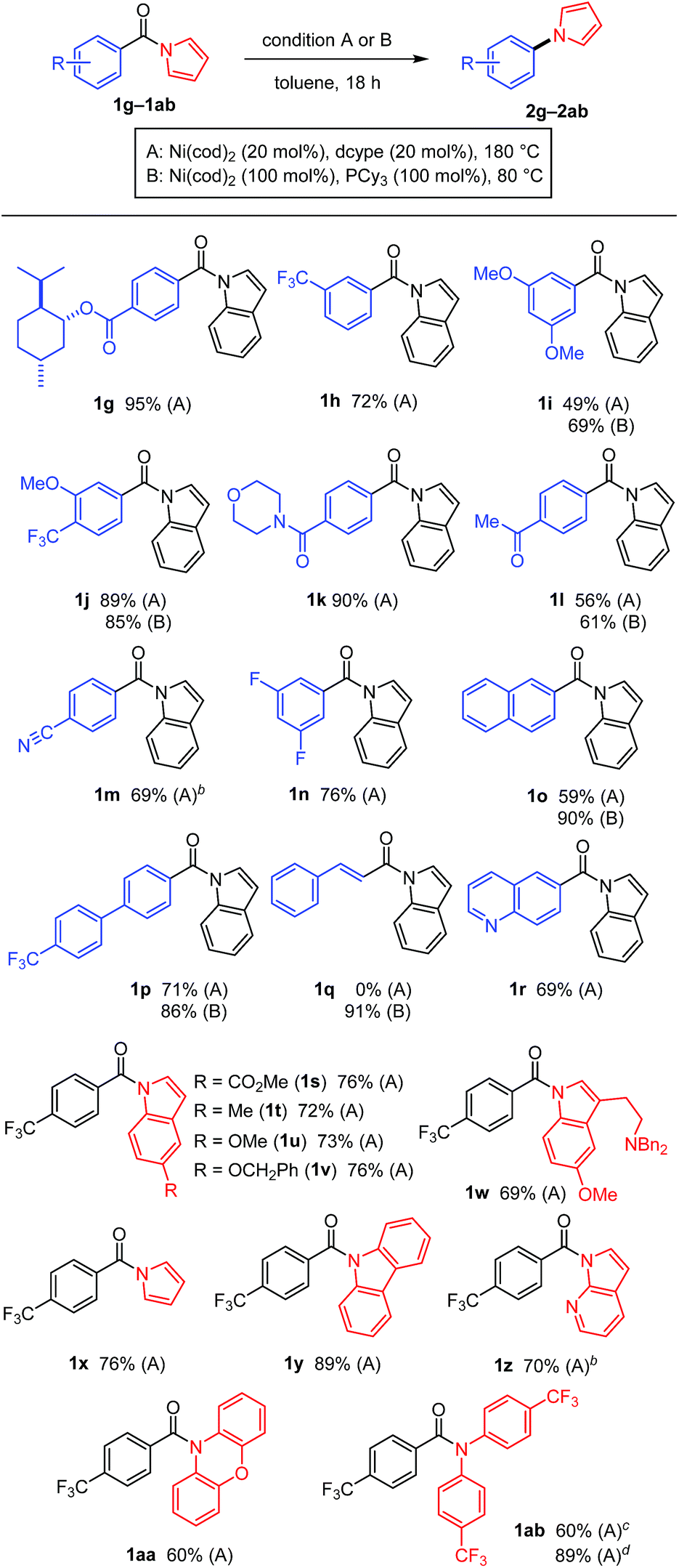
A diverse array of N-acylated N-heteroarenes could be decarbonylated using our nickel-mediated protocol (Table 3). Although a methoxy group can be activated by Ni(0) catalysis,12 it was compatible with this decarbonylation reaction, as exemplified by efficient reactions when 1i and 1j were used. Several other common functional groups, including esters 1g, amides 1k, ketones 1l, nitriles 1m, and fluorides 1n, were found to be tolerated and decarbonylation occurred chemoselectively at the indole amide moiety. In addition to benzoic acid derivatives, π-extended analogues, such as 1o and 1p, also served as good substrates for forming N-arylated products. The alkenyl amide 1q could be used to produce N-alkenylindole 2q in excellent yield under condition B. Heteroarene-based carboxylic acids can also be used in this decarbonylation reaction, as demonstrated in the reaction of 1r. The scope of the N-heteroarene moiety was also investigated. Substituents on the indole ring had no significant effect on the efficiency of the reaction, thus permitting a variety of indole derivatives to be produced via decarbonylation (1s–1ab). This decarbonylation is not limited to indole derivatives and pyrroles 1x, carbazoles 1y and azaindoles 1z also participated in this decarbonylation reaction, allowing rapid access to a diverse array of arylated heteroarenes from the corresponding carboxylic acids. Moreover, the non-aromatic six-membered phenoxazine derivative 1aa was decarbonylated successfully, whereas the N,N-diphenyl amide was much less reactive (10% yield, see ESI† for details). These results indicate that there is a threshold for the pKa value of the parent amine in determining whether or not the reaction will proceed (pKa in DMSO: pyrrole 23, indole 21, carbazole 20, phenoxazine 22, and diphenylamine 25).13 This seems to be reasonable since the C(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)–N bond becomes stronger as the parent amine becomes more basic, leading to a lower reactivity in this decarbonylation reaction. In agreement with this conclusion, a substrate derived from an electron-deficient diphenylamine (i.e., 1ab) exhibited a significantly improved reactivity for this decarbonylation reaction.
O)–N bond becomes stronger as the parent amine becomes more basic, leading to a lower reactivity in this decarbonylation reaction. In agreement with this conclusion, a substrate derived from an electron-deficient diphenylamine (i.e., 1ab) exhibited a significantly improved reactivity for this decarbonylation reaction.
| a Condition A: amide (0.25 mmol), Ni(cod)2 (0.050 mmol), dcype (0.050 mmol) in toluene (1.0 mL) for 18 h at 180 °C; condition B: amide (0.25 mmol), Ni(cod)2 (0.25 mmol), PCy3 (0.25 mmol) in toluene (1.0 mL) for 18 h at 80 °C. b 1,3-Bis(dicyclohexylphosphino)propane (0.050 mmol) was used instead of dcype. c Run for 48 h. d Ni(cod)2 (0.25 mmol) and dcype (0.25 mmol) were used. |
|---|
|
This decarbonylative N-arylation allows for the orthogonal introduction of azoles into halogen and carboxylic acid functionalities. For example, the N-arylation of carbazole using ethyl 3-iodobenzoate (3) occurs at the iodide site when the common copper-catalyzed method is used,2 and the resulting carboxylic acid moiety can be converted to another azole ring through an amidation/decarbonylation protocol (Scheme 2).
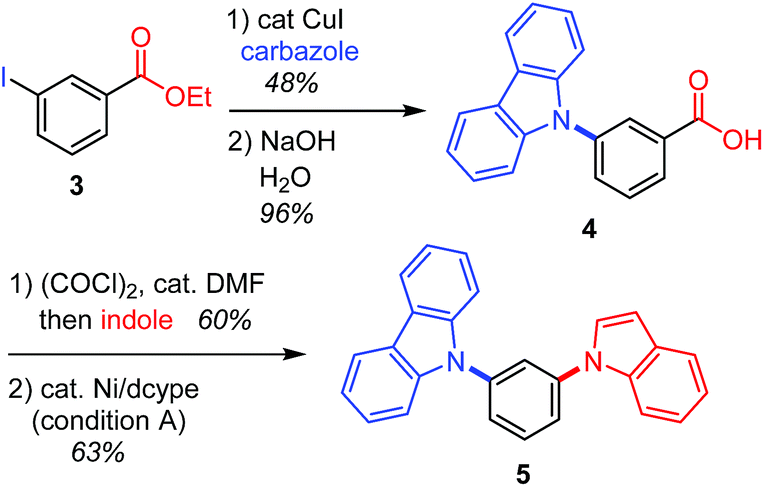 | ||
| Scheme 2 Sequential C–N bond formation using halogen and carboxyl groups as orthogonal handles. | ||
Several mechanistic experiments were conducted. A nickel-catalyzed reaction using a mixture of 1a and 1ac provided the corresponding decarbonylation products 2a and 2ac without the formation of any crossover products, indicating that this reaction proceeds via an intramolecular pathway (Scheme 3a). The reaction of amide 1a with 1 equivalent of Ni(cod)2 and dcype was monitored by 31P NMR, which revealed that a signal derived from the Ni(CO)2(dcype) (64.0 ppm, singlet) and two sets of doublets (57.9 ppm and 60.7 ppm, J = 13.1 Hz) appeared upon heating the mixture at 80 °C.14 These results indicate that a relatively stable organometallic intermediate is formed. Additional stoichiometric experiments resulted in the isolation of the arylnickelamide complex 6a, the structure of which was unambiguously determined by X-ray crystallography (Scheme 3b).15 Interestingly, the corresponding arylnickelamide complex 6f can also be formed from a methoxy-substituted amide 1f under similar conditions, although the amide 6f was much less reactive under the catalytic conditions used. These results indicate that, the reductive elimination from the arylnickelamide species to form a C–N bond has higher activation barrier than the prior C–N and C–C(O) bond cleavage processes. Indeed, a higher temperature (>120 °C) was required for this reductive elimination to occur to form an N-arylated product.16 Since Ni(CO)2(dcype) is also formed as the decarbonylation reaction proceeds, its catalytic activity was investigated (Scheme 3c). The Ni(CO)2(dcype) was found to catalyze the decarbonylation of 1a at 180 °C, whereas only a trace amount of the product was formed at 120 °C. These results suggest that heating at 180 °C is necessary for the dissociation of a CO ligand to occur to generate a catalytically active nickel species.
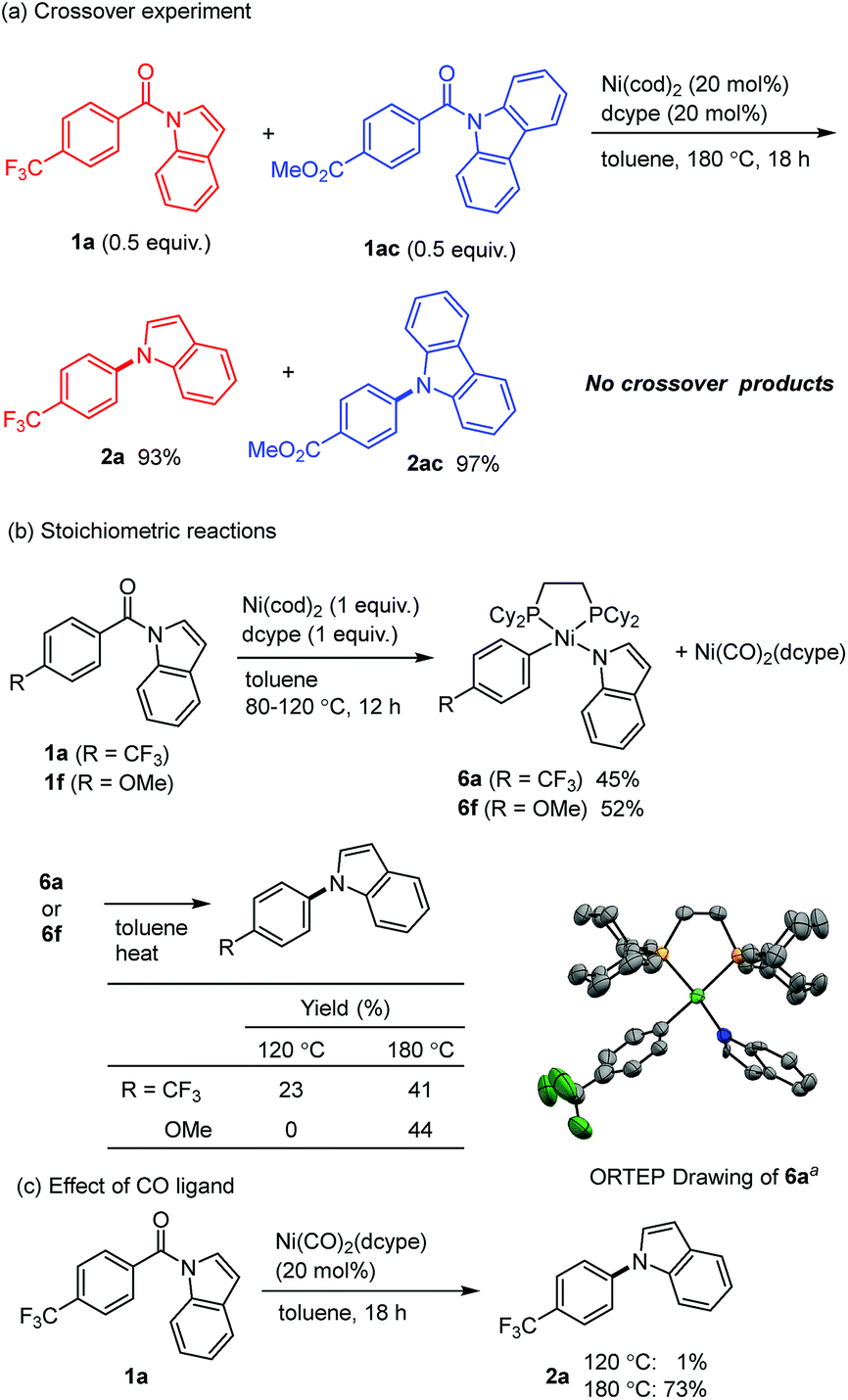 | ||
| Scheme 3 Mechanistic Studies. aThermal ellipsoids set at the 50% probability level. All hydrogen atoms are omitted for clarity.15 | ||
Based on the mechanistic studies described in Scheme 3, we propose a catalytic cycle shown in Scheme 4. In situ generated Ni(dcype) A17 serves as the catalytically active species, which initially activates a C(acyl)–N bond of the amide 1dvia oxidative addition18 to form an acylnickelamide intermediate B. The subsequent extrusion of CO from the acyl ligand in B leads to the formation of the carbonylnickel species C, in which one of the phosphorus atoms of dcype is dissociated. The dissociation of the CO ligand in C is facilitated by the recovery of the bidentate coordination of dcype to form the arylnickelamide complex D, which is one of the resting states of the catalyst. C–N bond forming reductive elimination from D is one of the difficult steps of this catalytic cycle. The higher reactivity of electron deficient amides is in agreement with the general trend in which C(aryl)–N bond reductive elimination proceeds more efficiently with an aryl ligand bearing an electron-withdrawing group.19 The dissociated CO is captured by A to form Ni(CO)2(dcype), which is catalytically inactive but can regenerate A upon heating to temperatures over 180 °C. The use of a PCy3 ligand instead of dcype generates a more active complex that can more easily mediate all the elementary steps required for the conversion of the amide 1d to the amine 2d (i.e., oxidative addition, extrusion of CO, and reductive elimination). However, unlike the case of dcype, there is no driving force available for the CO dissociation step, which hampers catalyst turnover when PCy3 is used. A similar contrast between PCy3 and dcype was also reported in the catalytic decarbonylative cross-coupling of phenyl esters.20
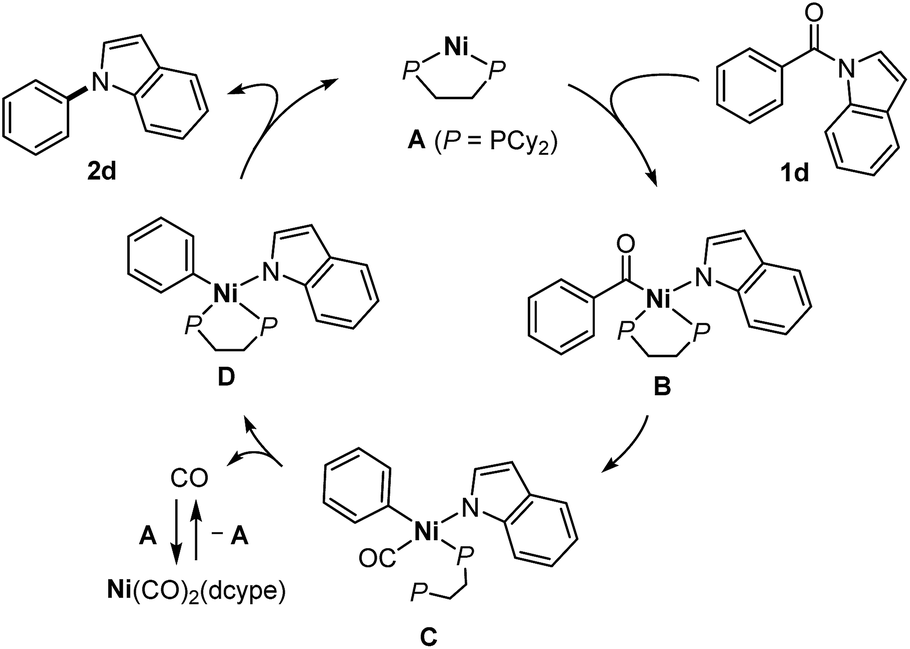 | ||
| Scheme 4 Proposed mechanism. | ||
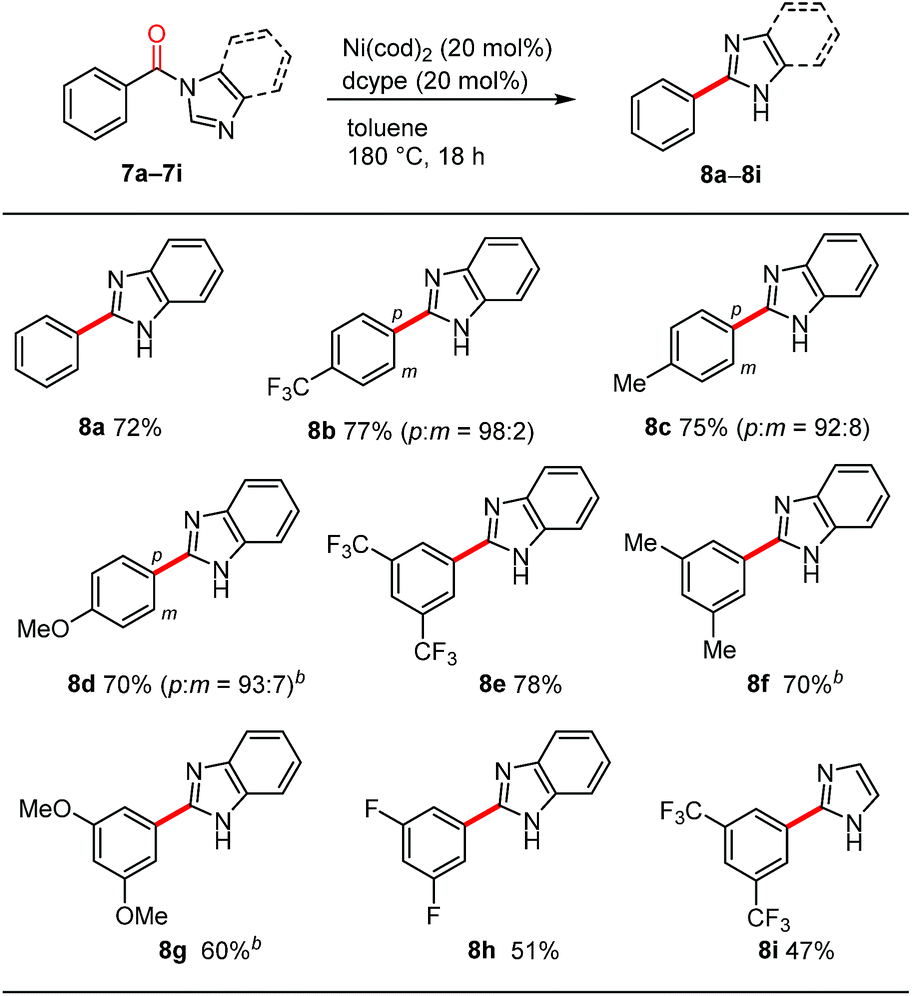
During the course of our investigations into the decarbonylation of N-acylated N-heteroarenes, we examined the benzimidazole derivative 7a. To our surprise, an N-arylation product was not formed in the Ni/dcype-catalyzed reaction of 7a but rather, C2-arylation occurred to afford 8a (Table 4). The overall reaction involves decarbonylation and 1,2-migration of the N-substituent.21,22 Although the detailed mechanism for this intriguing transformation remains elusive at present, the results of several preliminary mechanistic studies are noteworthy. First, a crossover experiment using two different N-acylated benzimidazole derivatives resulted in the formation of crossover products, suggesting that, unlike the decarbonylation of N-acylated indoles shown in Scheme 3a, an intermolecular pathway is likely to be operative. Second, when benzimidazoles bearing a para-substituted benzoyl group on the nitrogen atom, such as 8b, 8c and 8d, were used as substrates, small amounts of meta isomers were also formed in addition to the expected para-substituted 2-arylbenzimidazoles.23 This type of rearrangement was not observed in the case of the other heteroarene-based substrates shown in Table 3. Third, neither N-phenylbenzimidazole (S17) nor 2-benzoylbenzimidazole (S18) were converted into the 2-phenylbenzimidazole (8a) under these nickel-catalyzed conditions, excluding the intermediacy of these compounds in this migrative decarbonylation reaction. Finally, labelling experiments using a substrate having deuterium at the 2-position of a benzimidazole ring revealed that the cleavage of the C(2)–H bond is not involved in a turnover-limiting step.24meta-Disubstituted substrates 8e–8h underwent this decarbonylation to form the corresponding 2-arylated products without forming regioisomers. The reaction can also be applied to N-acylimidazole 8i, albeit in a lower yield.
Conclusions
In conclusion, we have developed the first decarbonylation of N-acylated N-heteroarenes, which allows benzoic acid derivatives to be used as arylating agents for N-heteroarenes. Arylnickelamide complexes were successfully isolated and characterized as viable catalytic intermediates. 1,2-Migratory decarbonylation occurred when N-acylated benzimidazoles were used as substrates. We expect that the capacity of employing feedstock chemicals in synthesizing N-arylated heteroarenes will find utility across a number of research fields. Further studies directed at the development of new catalytic decarbonylation reactions using simple carbonyl compounds are currently underway in our laboratory.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by JSPS KAKENHI (15H03811) and Scientific Research on Innovative Area “Precisely Designed Catalysts with Customized Scaffolding” (18H04259) from MEXT, Japan. MT thanks Toray Science Foundation and Hoansha Foundation for financial support. TM thanks the Program for Leading Graduate Schools: “Interactive Materials Science Cadet Program.” for their support. We also thank the Instrumental Analysis Center, Faculty of Engineering, Osaka University, for assistance with HRMS.Notes and references
- (a) P. Ruiz-Castillo and S. L. Buchwald, Chem. Rev., 2016, 116, 12564 CrossRef CAS PubMed; (b) G. Evano, N. Blanchard and M. Toumi, Chem. Rev., 2008, 108, 3054 CrossRef CAS PubMed.
- (a) D. S. Surry and S. L. Buchwald, Chem. Sci., 2010, 1, 13 RSC; (b) I. P. Beletskaya and A. V. Cheprakov, Organometallics, 2012, 31, 7753 CrossRef CAS.
- Oxidative cross-coupling between azoles and arylboronic acids (Chan–Lam–Evans reaction) represents another common method. K. Sanjeeva Rao and T.-S. Wu, Tetrahedron, 2012, 68, 7735 CrossRef CAS.
- Selected reviews: (a) J. Schwarz and B. König, Green Chem., 2018, 20, 323 RSC; (b) Y. Wei, P. Hu, M. Zhang and W. Su, Chem. Rev., 2017, 117, 8864 CrossRef CAS PubMed; (c) W. I. Dzik, P. P. Lange and L. J. Gooßen, Chem. Sci., 2012, 3, 2671 RSC; (d) N. Rodríguez and L. J. Goossen, Chem. Soc. Rev., 2011, 40, 5030 RSC; (e) W. I. Dzik, P. P. Lange and L. J. Gooßen, Chem. Sci., 2012, 3, 2671 RSC.
- Although several decarboxylative amination reactions have been reported to date, NH-heteroarenes cannot be used as a nitrogen-based nucleophile. Selected examples: (a) M. Pichette Drapeau, J. Bahri, D. Lichte and L. J. Gooßen, Angew. Chem., Int. Ed., 2019, 58, 892 CrossRef CAS PubMed; (b) W. Zhao, R. P. Wurz, J. C. Peters and G. C. Fu, J. Am. Chem. Soc., 2017, 139, 12153 CrossRef CAS PubMed; (c) Z.-J. Liu, X. Lu, G. Wang, L. Li, W.-T. Jiang, Y.-D. Wang, B. Xiao and Y. Fu, J. Am. Chem. Soc., 2016, 138, 9714 CrossRef CAS PubMed; (d) Q. Dai, P. Li, N. Ma and C. Hu, Org. Lett., 2016, 18, 5560 CrossRef CAS PubMed; (e) S. B. Lang, K. C. Cartwright, R. S. Welter, T. M. Locascio and J. A. Tunge, Eur. J. Org. Chem., 2016, 2016, 3331 CrossRef CAS PubMed; (f) Y. Zhang, S. Patel and N. Mainolfi, Chem. Sci., 2012, 3, 3196 RSC.
- The N-acylated N-heteroarenes used in this study was readily synthesized by condensation via acid chlorides. See the ESI† for details.
- Synthetic utility of N-acylated azoles: A. M. Goldys and C. S. P. McErlean, Eur. J. Org. Chem., 2012, 2012, 1877 CrossRef CAS.
- X. Liu, H. Yue, J. Jia, L. Guo and M. Rueping, Chem.–Eur. J., 2017, 23, 11771 CrossRef CAS PubMed.
- (a) R. Takise, K. Muto and J. Yamaguchi, Chem. Soc. Rev., 2017, 46, 5864 RSC; (b) J. E. Dander and N. K. Garg, ACS Catal., 2017, 7, 1413 CrossRef CAS PubMed; (c) G. Meng and M. Szostak, Org. Biomol. Chem., 2016, 14, 5690 RSC; (d) C. Liu and M. Szostak, Org. Biomol. Chem., 2018, 16, 7998 RSC.
- (a) T. Morioka, A. Nishizawa, T. Furukawa, M. Tobisu and N. Chatani, J. Am. Chem. Soc., 2017, 139, 1416 CrossRef CAS PubMed; (b) T.-T. Zhao, W.-H. Xu, Z.-J. Zheng, P.-F. Xu and H. Wei, J. Am. Chem. Soc., 2018, 140, 586 CrossRef CAS PubMed.
- C. Hansch, A. Leo and R. W. Taft, Chem. Rev., 1991, 91, 165 CrossRef CAS.
- (a) M. Tobisu and N. Chatani, Acc. Chem. Res., 2015, 48, 1717 CrossRef CAS PubMed; (b) J. Cornella, C. Zarate and R. Martin, Chem. Soc. Rev., 2014, 43, 8081 RSC.
- F. G. Bordwell, Acc. Chem. Res., 1988, 21, 456 CrossRef CAS.
- We also conducted a similar stoichiometric reaction using PCy3, instead of dcype, and observed several new resonances that might be assignable to relevant nickel species. See the ESI† for details.
- Crystal data for 6a, monoclinic, space group P21/c (no. 14), a = 16.8932(6) Å, b = 36.2820(11) Å, c = 13.0079(5) Å, β = 103.787(4)°, V = 7743.1(5) Å3, T = 123 K, Z = 4, R1 (wR2) = 0.0825 (0.2308) for 899 parameters and 19
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 566 unique reflections. GOF = 1.050. CCDC 1901697.†.
566 unique reflections. GOF = 1.050. CCDC 1901697.†. - (a) Reductive elimination from the MeO-substituted complex 6f required a higher temperature (180 °C) than that needed for reaction of the CF3-substituted complex 6a; (b) Under catalytic conditions, the rate of product formation is expected to be much less since 6f is generated only in a catalytic amount; (c) The yield of 2a by the reductive elimination of 6a increased to 51% when the reaction was conducted in the presence of Ni(CO)2(dcype) (1 equiv.), indicating that CO could accelerate the reductive elimination under catalytic conditions.
- The formation of Ni(dcype)(cod) under the reaction conditions was confirmed by 31P NMR (δ = 60.3 ppm, s).
- Catalytic reactions involving the cleavage of a C(O)–N bond of acylpyrroles:
(a) G. Meng, R. Szostak and M. Szostak, Org. Lett., 2017, 19, 3596 CrossRef CAS PubMed;
(b) P.-Q. Huang and H. Chen, Chem. Commun., 2017, 53, 12584 RSC.
- M. S. Driver and J. F. Hartwig, J. Am. Chem. Soc., 1997, 119, 8232 CrossRef CAS.
- X. Hong, Y. Liang and K. N. Houk, J. Am. Chem. Soc., 2014, 136, 2017 CrossRef CAS PubMed.
- The thermal isomerization of 1-carbamylimidazole to a 2-substituted isomer was reported to occur (>200 °C), possibly via the formation of isocyanate and imidazole. In contrast, we confirmed that 2-benzoylbenzimidazole (S18) remained unchanged after heating at 180 °C for 18 h in the absence of the catalyst. K. Mitsuhashi, E.-I. Itho, T. Kawahara and K. Tanaka, J. Heterocycl. Chem., 1983, 20, 1103 CrossRef CAS.
- 1-Acylimidazoles was reported to isomerize to form a mixture of 2- and 4-acylated imidazoles photochemically. 4-Substituted isomers were not observed in our decarbonylation reaction. S. Iwasaki, Helv. Chim. Acta, 1976, 59, 2738 CrossRef CAS PubMed.
- This type of formation of regioisomers were reported in palladium-catalyzed fluorination of aryl triflates: A. C. Sather and S. L. Buchwald, Acc. Chem. Res., 2016, 49, 2146 CrossRef CAS PubMed.
- A KIE value of 1.16 was obtained. See the ESI† for details.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1901697 and 1901702. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc02035g |
| This journal is © The Royal Society of Chemistry 2019 |