
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnantioselective intramolecular C–H amination of aliphatic azides by dual ruthenium and phosphine catalysis†
Jie
Qin
,
Zijun
Zhou
,
Tianjiao
Cui
,
Marcel
Hemming
and
Eric
Meggers
 *
*
Fachbereich Chemie, Philipps-Universität Marburg, Hans-Meerwein-Strasse 4, 35043 Marburg, Germany. E-mail: meggers@chemie.uni-marburg.de
First published on 29th January 2019
Abstract
The catalytic enantioselective intramolecular C(sp3)-H amination of aliphatic azides represents an efficient method for constructing chiral saturated cyclic amines which constitute a prominent structural motif in bioactive compounds. We report a dual catalytic system involving a chiral-at-metal bis(pyridyl-NHC) ruthenium complex and tris(4-fluorophenyl)phosphine (both 1 mol%), which facilitates the cyclization of aliphatic azides to chiral α-aryl pyrrolidines with enantioselectivities of up to 99% ee, including a pyrrolidine which can be converted to the anti-tumor alkaloid (R)-(+)-crispine. Mechanistically, the phosphine activates the organic azide to form an intermediate iminophosphorane and transfers the nitrene unit to the ruthenium providing an imido ruthenium intermediate which engages in the highly stereocontrolled C–H amination. This dual catalysis combines ruthenium catalysis with the Staudinger reaction and provides a novel strategy for catalyzing enantioselective C–H aminations of unactivated aliphatic azides.
Introduction
Pyrrolidines constitute a prominent structural motif in bioactive compounds such as natural products and pharmaceuticals (Fig. 1a).1,2 Their synthesis through direct intramolecular C(sp3)-H amination is particularly appealing due to the lack of necessity for preinstalled functional groups which provides the prospect of an efficient synthesis with high atom economy.3 In this respect, organic azides are attractive functionalities for metal-mediated nitrene C(sp3)-H insertion reactions4,5 because no additional oxidant is required and molecular nitrogen is the only by-product. Even though the amination of saturated C(sp3)-H bonds with aryl, sulfonyl, acyl and phosphoryl azides has been well established,6 the use of non-activated, aliphatic azides is only a recent accomplishment.7,8 In addition to their lower reactivity, a major pitfall for C(sp3)-H aminations of primary aliphatic azides is the competing unproductive 1,2-hydride shift of the intermediate alkyl nitrenoid leading to the irreversible formation of undesirable imines.9 Betley introduced an elegant dipyrrinato-iron(II)-catalyzed ring-closing C(sp3)-H amination of aliphatic azides but the reported turnover numbers were modest with TONs < 10.8b,c Subsequently reported MOF-functionalized Fe(II)-β-diketiminate,8d iron(III)-coordinated redox-active pyridine–aminophenol,8e and cobalt(II) porphyrin8g catalysts by Lin, van der Vlugt, and de Bruin, respectively, provided improved catalytic performances for this challenging transformation. Finally, Che very recently reported an N-heterocyclic carbene iron(III) porphyrin complex exhibiting high activity for this transformation under microwave conditions.8h However, pyrrolidines as part of bioactive compounds are typically chiral,2 but only a single example of a catalytic enantioselective reaction has been reported using a chiral cobalt(II) porphyrin achieving low yields and very low enantiomeric excess (Fig. 1b).8g,10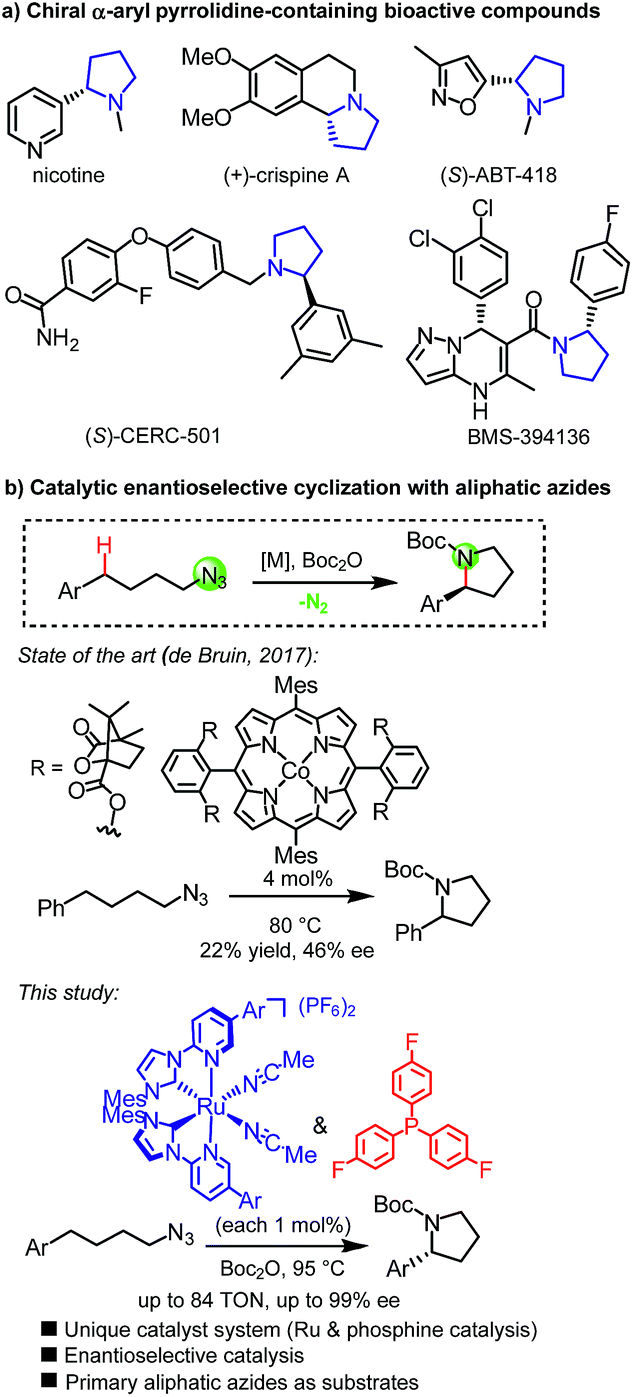 | ||
| Fig. 1 (a) Examples of bioactive compounds containing α-aryl pyrrolidines. (b) Asymmetric ring-closing C(sp3)–H aminations of aliphatic azides to provide non-racemic pyrrolidines. | ||
Ruthenium complexes are well-established for catalyzing C(sp3)-H activation11 and C(sp3)-H aminations of organic azides such as aryl, acyl, and sulfonyl azides,6a,d,f,l–n,12 but applying simple primary aliphatic azides has remained elusive and this has been attributed at least in part to a very efficient 1,2-hydrogen shift of intermediate Ru–imido complexes.9c–e Our group recently reported a new class of “chiral-at-metal” ruthenium catalysts in which two bidentate N-(2-pyridyl)-substituted N-heterocyclic carbenes and two acetonitrile ligands are coordinated to the central ruthenium in a C2-symmetric fashion.13 Despite all ligands being achiral, the overall helical chirality originates from a stereogenic ruthenium center.14,15 We recently demonstrated that such complexes can indeed serve as catalysts for activating aliphatic azides towards enantioselective C–H amination, however, unfortunately, only in a very restricted structural context of converting 2-azidoacetamides into chiral imidazolidin-4-ones.16 By discovering a novel ruthenium and phosphine dual catalysis scheme to activate aliphatic azides towards C–H amination, we here report a highly enantioselective ring-closing C(sp3)-H amination of simple 4-azidobutylarenes to provide chiral α-aryl pyrrolidines with enantioselectivities of up to 99% ee (Fig. 1b).
Results and discussion
Initial optimization
We initiated our study by investigating the intramolecular C–H amination of 4-azidobutylbenzene (1a) to Boc-protected 2-phenylpyrrolidine (2a). Unfortunately, despite extensive screening of the reaction conditions, the results were unsatisfactory (see details in Tables S1–S4†). For example, heating 1a under optimized conditions in 1,2-dichlorobenzene at 95 °C for 40 hours using Λ-Ru1 (1 mol%) in the presence of Boc2O (1 equiv.) provided the desired pyrrolidine 2a only in 26% NMR yield at a conversion of 57% but at least with an encouraging enantioselectivity of 81% ee (Table 1, entry 1). At the same time, Boc-protected amine 3 was detected as a side-product in 14% yield. To our surprise, we finally discovered that the reaction was rapidly improved when performed in the presence of catalytic amounts of PPh3, providing 2a in 44% yield with 79% conversion at an enantioselectivity of 82% ee (entry 2). An even slightly better yield was obtained with tris(4-fluorophenyl)phosphine (entry 3), while other phosphines provided inferior results (Table S5†). The amount of phosphine was also investigated, and it was found that 1 mol% provided the optimal results (Table S6†).|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Cat. | Additive | Conv.b (%) | NMR yieldb (%) | eec (%) | |
| 2a | 3 | |||||
| a Standard conditions: 1a (0.2 mmol), Boc2O (0.2 mmol, 1 equiv.), catalyst (0.002 mmol, 1 mol%), and additive (0.002 mmol, 1 mol%) in 1,2-dichlorobenzene (0.5 mL) at 95 °C for 60 h under N2 unless otherwise noted. b Determined by 1H NMR of crude products using Cl2CHCHCl2 as the internal standard. c Determined by HPLC of the crude main product on a chiral stationary phase. d Reaction time was 40 h. e Isolated yield in parentheses. f 0.5 mol% Λ-Ru7 was used. g Without Boc2O. h Refers to cpds without Boc-protection. i n.a. = not applicable. | ||||||
| 1d | Λ-Ru1 | No | 57 | 26 | 14 | 81 |
| 2d | Λ-Ru1 | P(Ph)3 | 79 | 44 | 17 | 82 |
| 3d | Λ-Ru1 | P(4-F-Ph)3 | 81 | 46 | 18 | 82 |
| 4 | Λ-Ru2 | P(4-F-Ph)3 | 77 | 46 | 18 | 89 |
| 5 | Λ-Ru3 | P(4-F-Ph)3 | 79 | 44 | 16 | 90 |
| 6 | Λ-Ru4 | P(4-F-Ph)3 | 75 | 44 | 20 | 87 |
| 7 | Λ-Ru5 | P(4-F-Ph)3 | 70 | 43 | 17 | 89 |
| 8 | Λ-Ru6 | P(4-F-Ph)3 | 65 | 23 | 18 | 80 |
| 9 | Λ-Ru7 | P(4-F-Ph)3 | 77 | 54 (51)e | 18 | 95 |
| 10 | Λ-Ru7f | P(4-F-Ph)3 | 65 | 42 | 16 | 94 |
| 11 | Λ-Ru7 | No | 45 | 12 | 14 | 95 |
| 12g | Λ-Ru7 | P(4-F-Ph)3 | <3 | 0h | 0h | n.a.i |
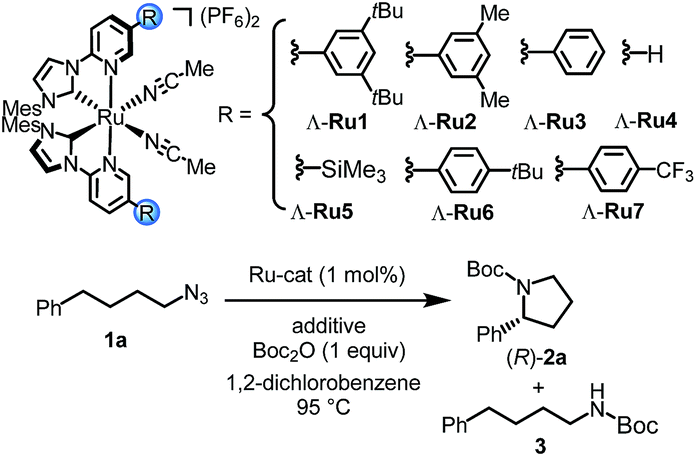
Next, we optimized the ruthenium catalyst for this transformation to improve the enantioselectivity. Our initial experiments were performed with the chiral-at-ruthenium catalyst Λ-Ru1 which bears two very bulky 3,5-di(tert-butyl)phenyl substituents at the coordinating pyridine ligands. Interestingly, Λ-Ru2 with less bulky 3,5-(dimethyl)phenyl substituents at the pyridyl moieties provided an even higher enantioselectivity of 89% ee (entry 4). The phenyl-modified catalyst Λ-Ru3 afforded a further slightly increased enantioselectivity of 90% ee (entry 5), whereas a plain catalyst devoid of additional substituents (Λ-Ru4) yielded the Boc-protected pyrrolidine with reduced 87% ee (entry 6). Furthermore, a trimethylsilyl (TMS)-functionalized ruthenium complex, Λ-Ru5, which is the optimal catalyst in our previous work on enantioselective C–H aminations of 2-azidoacetamides,16 did not provide better results here (entry 7). Adding a tBu-moiety at the 4-position of the phenyl groups (Λ-Ru6) decreased the enantioselectivity to 80% ee (entry 8). However, the best result was obtained with a 4-(CF3)Ph modification (Λ-Ru7) which afforded (R)-2a in 54% NMR yield at 77% conversion and with 95% ee (entry 9). Interestingly, even at a catalyst loading of just 0.5 mol%, the Boc-protected pyrrolidine (R)-2a was still formed with 42% yield (65% conversion) and 94% ee, reflecting a turnover number of 84 (entry 10). Thus, a careful optimization of steric and electronic effects provided a ruthenium catalyst (Λ-Ru7) which, in the presence of tris(4-fluorophenyl)phosphine, effectively discriminates between the two benzylic C–H bonds of 4-azidobutylbenzene to provide the corresponding chiral pyrrolidine with a modest yield but outstanding enantioselectivity.17,18
Control experiments revealed that tris(4-fluorophenyl)phosphine is crucial for obtaining a satisfactory yield. In its absence, the yield diminished to merely 12% even after an extended reaction time, while the enantioselectivity was not affected, thus implying that the phosphine is not involved in the stereocontrolling step (entry 11). Finally, Boc2O is also required for this reaction to proceed (entry 12).16
Substrate scope investigation
With the optimized catalyst Λ-Ru7 and optimized reaction conditions in hand, we investigated the substrate scope of this transformation. As shown in Fig. 2, methyl groups at para or meta positions of the benzene moiety are well tolerated (pyrrolidines 2b, c), but a sterically demanding ortho-methyl group leads to a vastly diminished yield of 15%. Both electron-donating (leading to pyrrolidines 2e–k) and electron-accepting substituents (leading to pyrrolidines 2l, m) are tolerated, although electron-accepting substituents led to decreased yields. The phenyl moiety can also be replaced by a naphthyl moiety (pyrrolidine 2n) and by heteroaromatic moieties (pyrrolidines 2o–s). For example, a carbazole moiety provides the Boc-protected pyrrolidine 2s in 52% yield and 93% ee. However, replacing the aryl group with alkyl, alkenyl, or alkynyl groups suppresses the C–H amination reaction (Table S8,† entries 1–3). Further substrates including bridging aryl groups could also be transformed into their corresponding pyrrolidine products (2t, u). Finally, a racemic substrate with a tertiary C–H group only provided a very modest kinetic resolution (Table S8,† entry 4).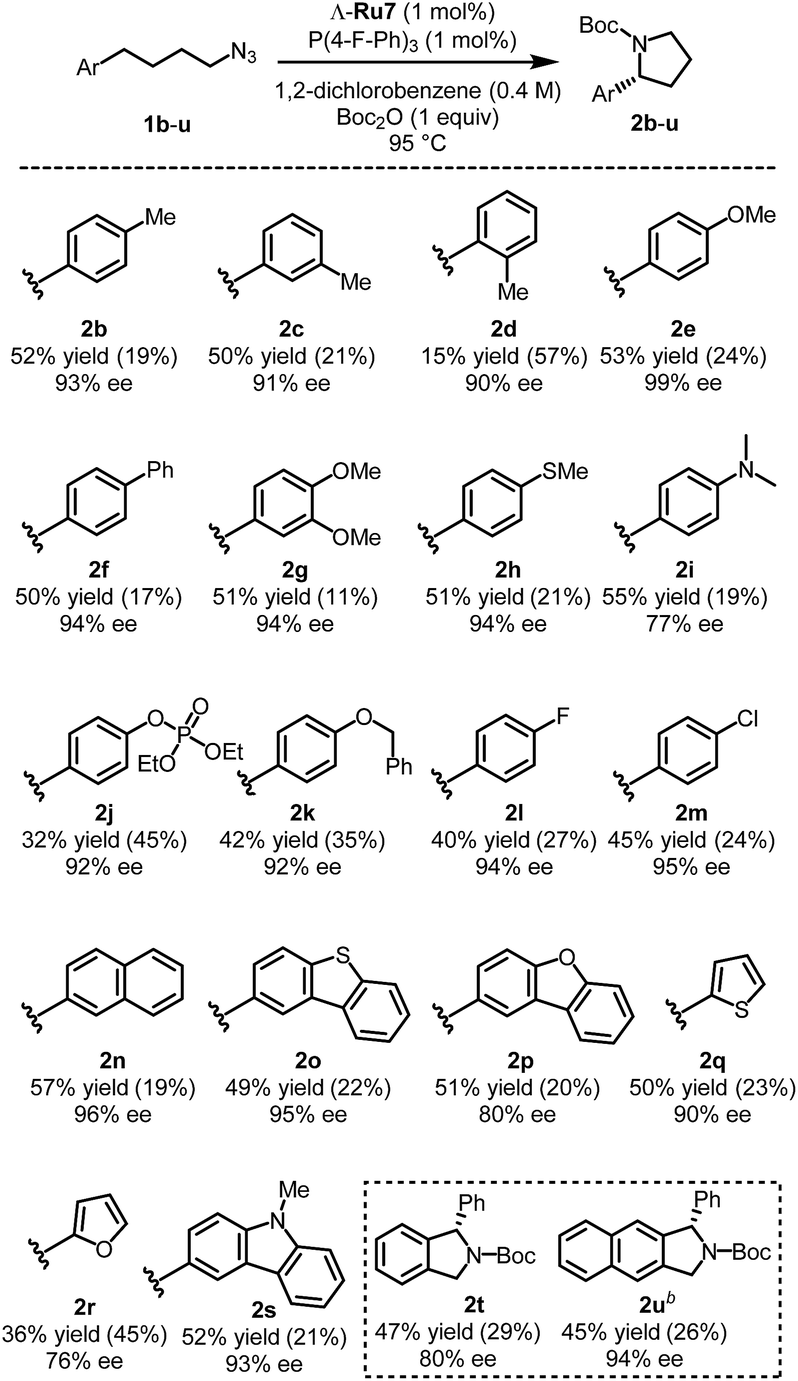 | ||
| Fig. 2 Substrate scope with isolated yieldsa. aRecovered starting materials are shown in parentheses. bLower substrate concentration (0.1 M) and 85 °C was used instead. | ||
Overall, enantioselectivities of 76–99% ee and isolated yields of 15–57% were observed. However it has to be noted that none of the reactions proceed to complete conversion and thus allow reisolation of unreacted starting materials (Fig. 2). Despite the modest yields, it is remarkable that chiral α-aryl pyrrolidines with outstanding enantioselectivities of up to 99% ee can be obtained through this challenging ring-closing C(sp3)-H amination of aliphatic azides.19 Importantly, chiral α-aryl pyrrolidines are prominent structural motifs in bioactive compounds.2 For example, pyrrolidine 2g can be converted to the anti-tumor alkaloid (R)-(+)-crispine in 4 steps using the Pummerer cyclization (Fig. S1†).2c
Mechanism study
We performed experiments to gain insight into the reaction mechanism and started with the unusual function of the phosphine. The role of the phosphine as a co-catalyst to activate the organic azide is advocated by the well-established reactivity of azides towards phosphines.20 Indeed, P(4-F-Ph)3 starts to react with (4-azidobutyl)benzene already at room temperature and complete conversion is obtained at 95 °C for 2 hours to form the corresponding iminophosphorane 4 (Fig. 3a). We also confirmed that iminophosphorane 4 is catalytically competent by itself (Fig. 3b). Interestingly, although such a role of phosphines in the activation of organic azides towards C–H amination has not been reported to our knowledge, the opposite reaction namely the phosphine-induced extraction of a nitrene from a metal imido complex was disclosed independently by McElwee-White21a and Sundermeyer.21b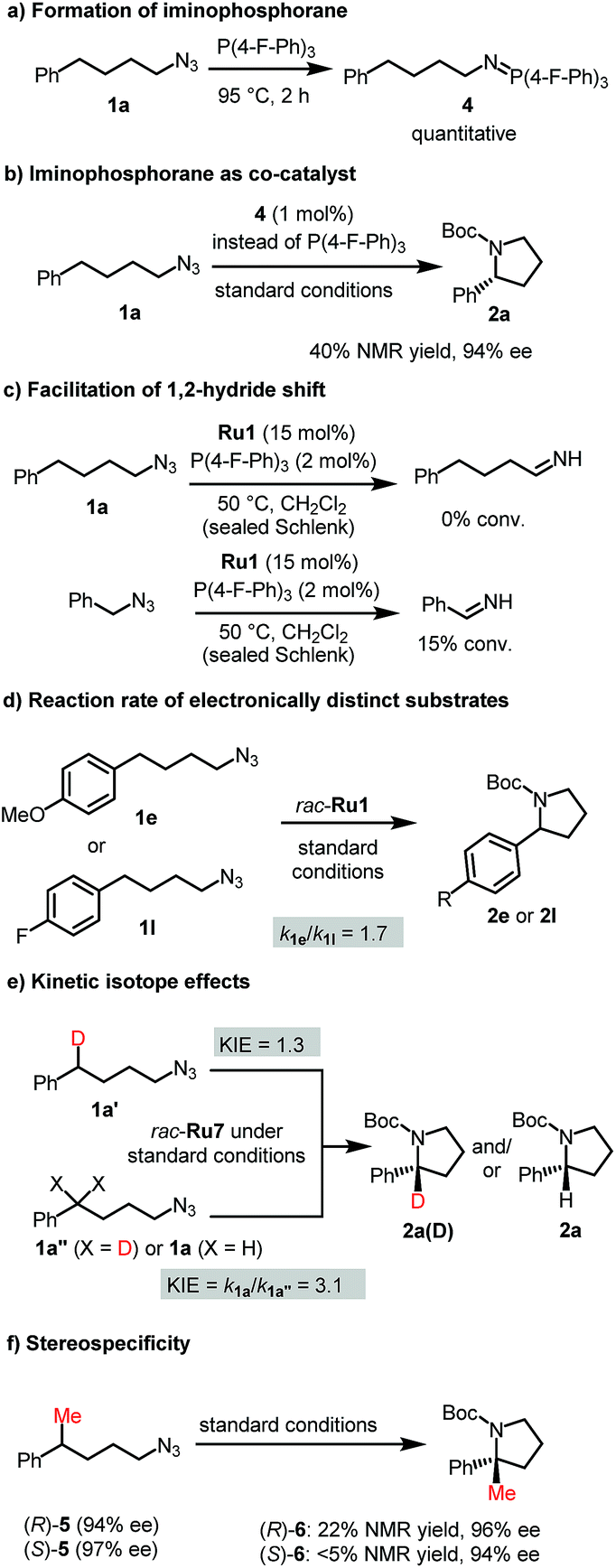 | ||
| Fig. 3 Mechanistic experiments. (a) Iminophosphorane formation, (b) verification of the iminophosporane as co-catalyst, (c) investigation of 1,2-hydride shift, (d) elucidation of electronic effects on reaction rate, (e) determination of kinetic isotope effects, and (f) evaluation of the stereospecificity of the C–H amination. | ||
Next, we attempted to gain insight into the competing 1,2-hydride shift by comparing (4-azidobutyl)benzene and benzyl azide as substrates at a temperature where C–H amination does not yet occur. As a result, only benzyl azide provided significant amounts of imine product which can be traced back to the higher activity of the benzylic C–H group in the ruthenium imido intermediate towards 1,2-H shift (Fig. 3c). This is consistent with a recent report by Park who showed that the degree of 1,2-H shift correlates with the nature of the α-C–H bond.9c
To understand the electronic nature of the proposed ruthenium nitrenoid intermediate, initial C–H amination rates of electronically distinct substrates 1e and 1l were determined. As a result, the cyclization rate of electron-rich 1e was 1.7 times faster than that of electron-deficient 1l suggesting the electrophilic nature of the ruthenium nitrene (Fig. 3d). This is consistent with the substrate scope in which electron-rich substrates provided better yields compared with electron-deficient substrates. The C–H amination with mono-deuterated substrate 1a′ using a racemic catalyst provided an intramolecular kinetic isotope effect (KIE) of 1.3 (Fig. 3e), which is much lower than the value reported for iron8b,e and cobalt8g catalytic systems. This might suggest that the C–H amination appears to occur by a concerted-insertion mechanism or a hydrogen abstraction mechanism with fast radical recombination.8h However, the interpretation of the intramolecular KIE is complicated by the fact that the ruthenium catalyst is intrinsically chiral, although used as a racemic mixture for this experiment, and the monodeuterated substrate 1a′ as well. Indeed the cyclization of the chiral substrate (R)-5 to (R)-6 but not (S)-5 to (S)-6 demonstrates the high stereospecificity of the C–H amination (Fig. 3f). Finally, we determined a pronounced (noncompetitive) intermolecular KIE value of 3.1 by measuring initial C–H amination rates of non-deuterated (1a) and bis-deuterated (1a′′) substrates (Fig. 3e). Using alternatively a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of non-deuterated (1a) and bis-deuterated (1a′′) substrates provides a (competitive) intermolecular KIE of 3.9 (see Fig. S6†). This observation of a significant intermolecular KIE reveals that the C–H amination is the rate limiting step in the overall process.
:1 mixture of non-deuterated (1a) and bis-deuterated (1a′′) substrates provides a (competitive) intermolecular KIE of 3.9 (see Fig. S6†). This observation of a significant intermolecular KIE reveals that the C–H amination is the rate limiting step in the overall process.
Based on previous studies8b–h on the ring-closing C–H amination of (4-azidobutyl)arenes and our mechanism study, the following mechanism is proposed (Fig. 4). P(4-F-Ph)3 activates the organic azide to form an intermediate iminophosphorane (I) through the well-known Staudinger reaction which then transfers a nitrene to the ruthenium center to afford a ruthenium imido complex (intermediate II), followed by a stereo-controlled insertion of the nitrene moiety into the δ-C–H bond (transition state III) to provide a ruthenium-coordinated pyrrolidine (intermediate IV). Alternatively, a stepwise process through H-atom transfer cannot be excluded at this point. Finally, the product is released after Boc-protection.
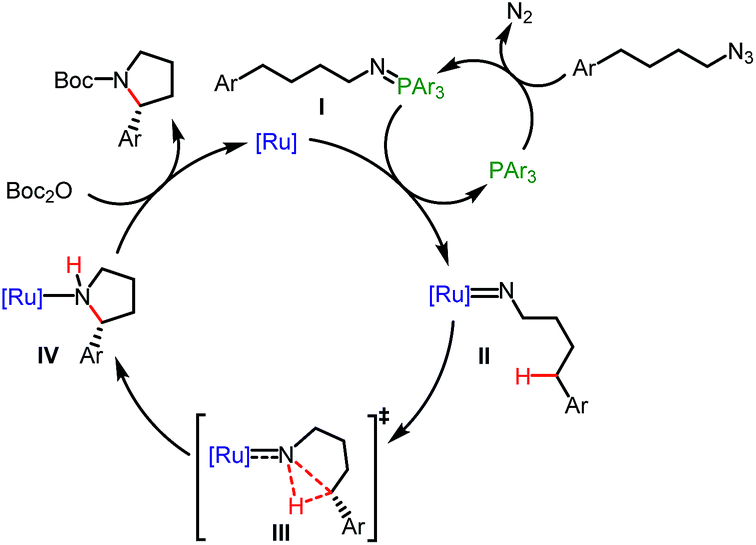 | ||
| Fig. 4 Proposed mechanism. | ||
Conclusions
We here present a highly enantioselective catalytic ring-closing benzylic C(sp3)-H amination of primary aliphatic azides to provide chiral 2-aryl pyrrolidines by combining chiral-at-metal transition metal catalysis with nucleophilic phosphine catalysis. In this unique dual catalysis system, the phosphine activates the organic azide and transfers a nitrene to the ruthenium complex, which then executes the enantioselective C–H amination. This combination of ruthenium catalysis and the Staudinger reaction introduces a novel direction for C–H amination of unactivated aliphatic azides which are very desirable but challenging substrates for this transformation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Support of this work by the Deutsche Forschungsgemeinschaft is gratefully acknowledged (ME1805/15-1).Notes and references
- (a) D. O'Hagan, Nat. Prod. Rep., 2000, 17, 435 RSC; (b) R. D. Taylor, M. MacCoss and A. D. G. Lawson, J. Med. Chem., 2014, 57, 5845 CrossRef CAS PubMed.
- For chiral α-aryl pyrrolidine-containing bioactive molecules, see: (a) J. Lloyd, H. J. Finlay, W. Vacarro, T. Hyunh, A. Kover, R. Bhandaru, L. Yan, K. Atwal, M. L. Conder, T. Jenkins-West, H. Shi, C. Huang, D. Li, H. Sun and P. Levesque, Bioorg. Med. Chem., 2010, 20, 1436 CrossRef CAS PubMed; (b) C. H. Mitch, S. J. Quimby, N. Diaz, C. Pedregal, M. G. de la Torre, A. Jimenez, Q. Shi, E. J. Canada, S. D. Kahl, M. A. Statnick, D. L. McKinzie, D. R. Benesh, K. S. Rash and V. N. Barth, J. Med. Chem., 2011, 54, 8000 CrossRef CAS PubMed; (c) S. C. K. Rotte, A. G. Chittiboyina and I. A. Khan, Eur. J. Org. Chem., 2013, 6355 CrossRef CAS.
- (a) G. He, Y. Zhao, S. Zhang, C. Lu and G. Chen, J. Am. Chem. Soc., 2012, 134, 3 CrossRef CAS PubMed; (b) M. Yang, B. Su, Y. Wang, K. Chen, X. Jiang, Y.-F. Zhang, X.-S. Zhang, G. Chen, Y. Cheng, Z. Cao, Q.-Y. Guo, L. Wang and Z.-J. Shi, Nat. Commun., 2014, 5, 4707 CrossRef CAS PubMed; (c) C. Q. O'Broin, P. Fernández, C. Martínez and K. Muñiz, Org. Lett., 2016, 18, 436 CrossRef PubMed; (d) D. Meng, Y. Tang, J. Wei, X. Shi and M. Yang, Chem. Commun., 2017, 53, 5744 RSC; (e) P. Becker, T. Duhamel, J. C. Stein, M. Reiher and K. Muñiz, Angew. Chem., Int. Ed., 2017, 56, 8004 CrossRef CAS PubMed; (f) P. Becker, T. Duhamel, C. Martínez and K. Muñiz, Angew. Chem., Int. Ed., 2018, 57, 5166 CrossRef CAS PubMed; (g) S. Herold, D. Bafaluy and K. Muñiz, Green Chem., 2018, 20, 3191 RSC.
- For reviews on catalytic C(sp3)-H aminations, see: (a) F. Collet, R. H. Dodd and P. Dauban, Chem. Commun., 2009, 5061 RSC; (b) C.-M. Che, V. K.-Y. Lo, C.-Y. Zhou and J.-S. Huang, Chem. Soc. Rev., 2011, 40, 1950 RSC; (c) F. Collet, C. Lescot and P. Dauban, Chem. Soc. Rev., 2011, 40, 1926 RSC; (d) J. L. Roizen, M. E. Harvey and J. Du Bois, Acc. Chem. Res., 2012, 45, 911 CrossRef CAS PubMed; (e) G. Dequirez, V. Pons and P. Dauban, Angew. Chem., Int. Ed., 2012, 51, 7384 CrossRef CAS PubMed; (f) J. L. Jeffrey and R. Sarpong, Chem. Sci., 2013, 4, 4092 RSC; (g) Y. Park, Y. Kim and S. Chang, Chem. Rev., 2017, 117, 9247 CrossRef CAS PubMed; (h) B. Darses, R. Rodrigues, L. Neuville, M. Mazurais and P. Dauban, Chem. Commun., 2017, 53, 493 RSC; (i) J. C. K. Chu and T. Rovis, Angew. Chem., Int. Ed., 2018, 57, 62 CrossRef CAS PubMed.
- For reviews on C(sp3)-H aminations with organic azides, see: (a) T. G. Driver, Org. Biomol. Chem., 2010, 8, 3831 RSC; (b) T. Uchida and T. Katsuki, Chem. Rec., 2014, 14, 117 CrossRef CAS PubMed; (c) D. Intrieri, P. Zardi, A. Caselli and E. Gallo, Chem. Commun., 2014, 50, 11440 RSC; (d) P. F. Kuijpers, J. I. van der Vlugt, S. Schneider and B. de Bruin, Chem.–Eur. J., 2017, 23, 13819 CrossRef CAS PubMed.
- (a) S.-M. Au, J.-S. Huang, W.-Y. Yu, W.-H. Fung and C.-M. Che, J. Am. Chem. Soc., 1999, 121, 9120 CrossRef CAS; (b) S. Cenini, E. Gallo, A. Penoni, F. Ragaini and S. Tollari, Chem. Commun., 2000, 2265 RSC; (c) F. Ragaini, A. Penoni, E. Gallo, S. Tollari, C. L. Gotti, M. Lapadula, E. Mangioni and S. Cenini, Chem.–Eur. J., 2003, 9, 249 CrossRef CAS PubMed; (d) S. K.-Y. Leung, W.-M. Tsui, J.-S. Huang, C.-M. Che, J.-L. Liang and N. Zhu, J. Am. Chem. Soc., 2005, 127, 16629 CrossRef CAS PubMed; (e) J. V. Ruppel, R. M. Kamble and X. P. Zhang, Org. Lett., 2007, 9, 4889 CrossRef CAS PubMed; (f) S. Fantauzzi, E. Gallo, A. Caselli, F. Ragaini, N. Casati, P. Macchi and S. Cenini, Chem. Commun., 2009, 3952 RSC; (g) K. Sun, R. Sachwani, K. J. Richert and T. G. Driver, Org. Lett., 2009, 11, 3598 CrossRef CAS PubMed; (h) H. Lu, V. Subbarayan, J. Tao and X. P. Zhang, Organometallics, 2010, 29, 389 CrossRef CAS; (i) H. Lu, J. Tao, J. E. Jones, L. Wojtas and X. P. Zhang, Org. Lett., 2010, 12, 1248 CrossRef CAS PubMed; (j) M. Ichinose, H. Suematsu, Y. Yasutomi, Y. Nishioka, T. Uchida and T. Katsuki, Angew. Chem., Int. Ed., 2011, 50, 9884 CrossRef CAS PubMed; (k) Q. Nguyen, K. Sun and T. G. Driver, J. Am. Chem. Soc., 2012, 134, 7262 CrossRef CAS PubMed; (l) W. Xiao, J. Wei, C.-Y. Zhou and C.-M. Che, Chem. Commun., 2013, 49, 4619 RSC; (m) Y. Nishioka, T. Uchida and T. Katsuki, Angew. Chem., Int. Ed., 2013, 52, 1739 CrossRef CAS PubMed; (n) K.-H. Chan, X. Guan, V. K.-Y. Lo and C.-M. Che, Angew. Chem., Int. Ed., 2014, 53, 2982 CrossRef CAS PubMed; (o) T. Kang, Y. Kim, D. Lee, Z. Wang and S. Chang, J. Am. Chem. Soc., 2014, 136, 4141 CrossRef CAS PubMed; (p) I. T. Alt, C. Guttroff and B. Plietker, Angew. Chem., Int. Ed., 2017, 56, 10582 CrossRef CAS PubMed; (q) C. Li, K. Lang, H. Lu, Y. Hu, X. Cui, L. Wojtas and X. P. Zhang, Angew. Chem., Int. Ed., 2018, 57, 16837 CrossRef CAS PubMed.
- For an intermolecular C–H amination of 1-adamantylazide, see: Y. M. Badiei, A. Dinescu, X. Dai, R. M. Palomino, F. W. Heinemann, T. R. Cundari and T. H. Warren, Angew. Chem., Int. Ed., 2008, 47, 9961 CrossRef CAS PubMed.
- For intramolecular C–H aminations of alkyl azides, see: (a) E. R. King and T. A. Betley, Inorg. Chem., 2009, 48, 2361 CrossRef CAS PubMed; (b) E. T. Hennessy and T. A. Betley, Science, 2013, 340, 591 CrossRef CAS PubMed; (c) D. A. Iovan, M. J. T. Wilding, Y. Baek, E. T. Hennessy and T. A. Betley, Angew. Chem., Int. Ed., 2017, 56, 15599 CrossRef CAS PubMed; (d) N. C. Thacker, Z. Lin, T. Zhang, J. C. Gilhula, C. W. Abney and W. Lin, J. Am. Chem. Soc., 2016, 138, 3501 CrossRef CAS PubMed; (e) B. Bagh, D. L. J. Broere, V. Sinha, P. F. Kuijpers, N. P. van Leest, B. de Bruin, S. Demeshko, M. A. Siegler and J. I. van der Vlugt, J. Am. Chem. Soc., 2017, 139, 5117 CrossRef CAS PubMed; (f) D. L. J. Broere, B. de Bruin, J. N. H. Reek, M. Lutz, S. Dechert and J. I. van der Vlugt, J. Am. Chem. Soc., 2014, 136, 11574 CrossRef CAS PubMed; (g) P. F. Kuijpers, M. J. Tiekink, W. B. Breukelaar, D. L. J. Broere, N. P. van Leest, J. I. van der Vlugt, J. N. H. Reek and B. de Bruin, Chem.–Eur. J., 2017, 23, 7945 CrossRef CAS PubMed; (h) K.-P. Shing, Y. Liu, B. Cao, X.-Y. Chang, T. You and C.-M. Che, Angew. Chem., Int. Ed., 2018, 57, 11947 CrossRef CAS PubMed.
- (a) G. Albertin, S. Antoniutti, D. Baldan, J. Castro and S. García-Fontán, Inorg. Chem., 2008, 47, 742 CrossRef CAS PubMed; (b) G. Albertin, S. Antoniutti and J. Castro, J. Organomet. Chem., 2010, 695, 574 CrossRef CAS; (c) J. H. Lee, S. Gupta, W. Jeong, Y. H. Rhee and J. Park, Angew. Chem., Int. Ed., 2012, 51, 10851 CrossRef CAS PubMed; (d) H. K. Pak, J. Han, M. Jeon, Y. Kim, Y. Kwon, J. Y. Park, Y. H. Rhee and J. Park, ChemCatChem, 2015, 7, 4030 CrossRef CAS; (e) J. Y. Park, Y. Kim, D. Y. Bae, Y. H. Rhee and J. Park, Organometallics, 2017, 36, 3471 CrossRef CAS; (f) L. Hu, Y. A. Liu and X. Liao, Sci. Adv., 2017, 3, e1700826 CrossRef PubMed.
- For a review of asymmetric C–H functionalization, see: C. Zheng and S.-L. You, RSC Adv., 2014, 4, 6173 RSC.
- For reviews of ruthenium catalyzed C(sp3)-H functionalization, see: (a) F. Kakiuchi and N. Chatani, Ruthenium-Catalyzed Reactions via sp C–H, sp2 C–H, sp3 C–H, and C–Halogen Bond Activations, in Ruthenium in Organic Synthesis, ed. S.-I. Murahashi, Wiley-VCH Verlag GmbH, 2005, p. 219 Search PubMed; (b) P. B. Arockiam, C. Bruneau and P. H. Dixneuf, Chem. Rev., 2012, 112, 5879 CrossRef CAS PubMed; (c) C. Bruneau, sp3 C–H Bond Functionalization with Ruthenium Catalysts, in Ruthenium in Catalysis, ed. C. Bruneau and P. H. Dixneuf, Springer, Top. Organomet. Chem., 2014, vol. 48, p. 195 Search PubMed.
- For an example of ruthenium imido intermediates, see: S.-M. Au, W.-H. Fung, M.-C. Cheng, C.-M. Che and S.-M. Peng, Chem. Commun., 1997, 1655 RSC.
- Y. Zheng, Y. Tan, K. Harms, M. Marsch, R. Riedel, L. Zhang and E. Meggers, J. Am. Chem. Soc., 2017, 139, 4322 CrossRef CAS PubMed.
- L. Zhang and E. Meggers, Acc. Chem. Res., 2017, 50, 320 CrossRef CAS PubMed.
- L. Zhang and E. Meggers, Chem.–Asian J., 2017, 12, 2335 CrossRef CAS PubMed.
- Z. Zhou, S. Chen, J. Qin, X. Nie, X. Zheng, K. Harms, R. Riedel, K. N. Houk and E. Meggers, Angew. Chem., Int. Ed., 2019, 58, 1088 CrossRef CAS PubMed.
- Several other ruthenium catalysts were tested for this C–H amination. However, none of them could catalyze this reaction to provide a Boc-protected pyrrolidine product (see Table S7†). This indicated that the ligand environment of ruthenium complexes was important for the success of C–H amination.
- Experiments in which we added fresh catalysts and optionally also additional phosphine ligands after a reaction time of 30 h did only slightly improve the yields (see Table S6,† entries 8–10).
- For a different but very convenient method to prepare non-racemic 2-aryl pyrrolidines, see: K. R. Campos, A. Klapars, J. H. Waldman, P. G. Dormer and C. Chen, J. Am. Chem. Soc., 2006, 128, 3538 CrossRef CAS PubMed.
- (a) H. Staudinger and J. Meyer, Helv. Chim. Acta, 1919, 2, 635 CrossRef CAS; (b) Y. G. Gololobov and L. F. Kasukhin, Tetrahedron, 1992, 48, 1353 CrossRef CAS.
- (a) H. F. Sleiman, S. Mercer and L. McElwee-White, J. Am. Chem. Soc., 1989, 111, 8007 CrossRef CAS; (b) K. Korn, A. Schorm and J. Sundermeyer, Z. Anorg. Allg. Chem., 1999, 625, 2125 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Characterization data and experimental procedures. See DOI: 10.1039/c9sc00054b |
| This journal is © The Royal Society of Chemistry 2019 |