
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesign and synthesis of supramolecular functional monomers bearing urea and norbornene motifs†
Lulu Wangabc,
Qifeng Lind,
Yagang Zhang *abc,
Yanxia Liuabc,
Akram Yasinb and
Letao Zhangb
*abc,
Yanxia Liuabc,
Akram Yasinb and
Letao Zhangb
aDepartment of Chemical and Environmental Engineering, Xinjiang Institute of Engineering, Urumqi 830023, China. E-mail: ygzhang@ms.xjb.ac.cn; Fax: +86-991-3838957; Tel: +86-18129307169
bXinjiang Technical Institute of Physics and Chemistry, Chinese Academy of Sciences, Urumqi 830011, China
cUniversity of Chinese Academy of Sciences, Beijing 100049, China
dXinjiang Institute of Ecology and Geography, Chinese Academy of Sciences, Urumqi, Xinjiang 830011, China
First published on 27th June 2019
Abstract
Three sets of functional monomers namely urea-based, 2-ureido-4[1H]-primidone (UPy)-based and norbornene based functional monomers were designed and synthesized. These functional monomers (FM) were obtained in decent yields using amine and isocyanate/norbornene as starting materials. Methacrylate and styrene isocyanate with 1,4-diaminobutane/tris(2-aminoethyl)amine were chosen for the synthesis of symmetrical, asymmetrical and three-branched urea-functional monomers, respectively. UPy-based FMs were synthesized with isocyanate and 2-amino-4-hydroxy-6-methylpyrimidine. The synthesis of these monomers feature short reaction times, mild reaction conditions and no need for column chromatographic purification. Furthermore, the norbornene based FM was used for preparing molecularly imprinted polymers (MIPs) by Ring-Opening Metathesis Polymerization (ROMP). Results showed that these synthetic routes represent a convenient and useful approach for synthesis of novel functional monomers.
1 Introduction
The molecular imprinting technique mimics nature's enzyme and substrate interaction by choosing suitable functional monomers for specific template molecules via covalent,1 non-covalent2, electrostatic/ionic interactions to form crosslinked polymer networks called molecular imprinted polymers (MIPs). Functional monomers play a critical role in this process. The selection of the functional monomer (FM) depends upon the strength of the binding interactions between template and monomer.3 However, only a very limited number of the functional monomers are available to form complexes with the template4,5 and in most cases forming heterogeneous binding sites because the pre-polymerization step is not well defined. This resulted from the formation of complexes with different ratios of template to monomer, and the formation of nonselective binding sites.6 Methacrylic acid (MAA) was found to form hydrogen-bonded dimers and could reduce the number of background sites and enhance the specificity of molecular imprinting.7 The study indicated that the aggregation of functional monomers could help reduce the number of background sites. Specifically, the urea-based monomers were found to form linear hydrogen bonded aggregates and could significantly reduce the number of background sites in molecularly imprinted polymers.8–14The design and synthesis of molecular imprinting polymers for biomolecules and its optimization is important yet very challenging.15,16
It is practically important to design and synthesize new functional monomers capable of forming strong interactions with templates featuring high affinity and specificity. Generally, an ideal functional monomer would combine at least two types of functionality: a polymerizable vinyl group; a suitable functional group that could interact strongly with the template. What's more, appropriate adaptive cross-linkers are also needed to form pre-polymerization complex in order to form rigidly fixed polymer matrix and to improve imprinting fidelity.
Along this line, in the work reported here, three sets of functional monomers specifically urea-based, UPy(2-ureido-4[1H]-pyrimidinone)-based and multi-arm urea and norbornene based functional monomers were designed and synthesized. And the multi-arm norbornene based functional monomers were used for preparing MIPs via ring-opening metathesis polymerization.
Results showed that these synthetic routes represent a convenient and practical strategy for synthesis of functional monomers. While norbornene based multi-arm functional monomers could be used for preparing MIPs for free radical quencher type template molecules.
2 Results and discussion
2.1 Synthesis of functional monomers
The synthesis of urea-based functional monomer started out using 1,4-diaminobutane and commercially available isocyanate as a precursor to form the urea unit (Table 1). Results showed that symmetrical functional monomer was formed when CH2Cl2 was used as solvent at room temperature for 4 h (Table 1, entry 1).
|
|||||
|---|---|---|---|---|---|
| Entry | R–NCO (equiv.) | Solvent | T (°C) | Time (h) | Product (yield)a |
| a Reaction condition: 1,4-diaminobutane (1 mmol), isocyanate (1.1 mmol/2.1 mmol) in the indicated solvent (10 mL) at 0 °C (rt). | |||||
| 1 | 1a (1.1) | CH2Cl2 | rt | 4 h | 2c (43.6%) |
| 2 | 1a (1.1) | CH2Cl2 | 0 | 4 h | 2a (10.2%) |
| 2c (32.4%) | |||||
| 3 | 1a (1.1) | CH2Cl2 | 0 | 2 h | 2a (42.3%) |
| 2c (12.0%) | |||||
| 4 | 1a (1.1) | THF | 0 | 2 h | 2a (trace) |
| 5 | 1a (1.1) | DMF | 0 | 2 h | 2a (trace) |
| 6 | 1a (1.1) | PhCH3 | 0 | 2 h | 2a (trace) |
| 7 | 1a (1.1) | CH3CN | 0 | 2 h | 2a (68.4%) |
| 8 | 1a (1.1) | CH3CN | −20 | 2 h | 2a (65.1%) |
| 9 | 1a (2.1) | CH2Cl2 | rt | 2 h | 2c (99.8%) |
| 10 | 1b (1.1) | CH3CN | 0 | 2 h | 2b (78.8%) |
| 11 | 1b (2.1) | CH2Cl2 | rt | 2 h | 2d (96.7%) |
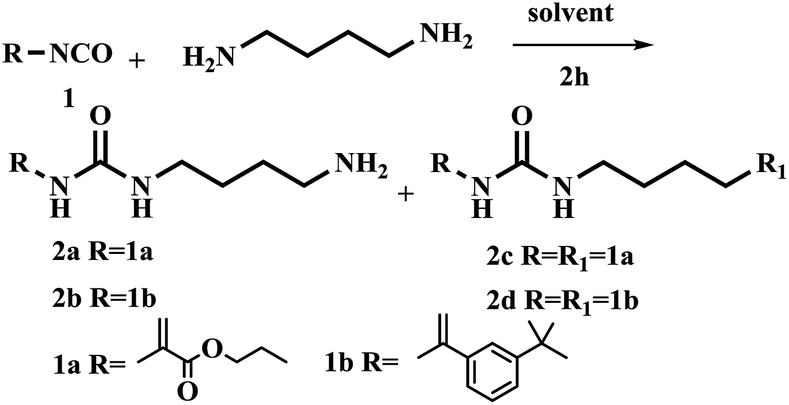
Next, reducing reaction temperature to 0 °C gave 2a in 10% and 2c in 34% yields, respectively. With reaction time 2 h at rt, the yield of 2a was improved to 42% and 2c was decreased to 12% (Table 1, entries 2 and 3). Results indicated that reaction time and temperature strongly influenced the resulting products. Based on above observations, we further explored other solvents. 1,4-Diaminobutane was slowly added dropwise into a solution of isocyanate at 0 °C using various different solvent including THF, DMF and toluene, giving only trace amount of product or no reaction (Table 1, entries 4–6). Noticeably, when using CH3CN as solvent, the yield of 2a increased to 68%. And the yield did not change significantly when the reaction was carried out at −20 °C.
With the optimized conditions in hand, the scope of the urea unit synthesis was further expanded and explored (Table 1, entries 9 and 10). It was found the reaction of urea-unit FM with 1,4-diaminobutane substrate bearing different functional groups such as styrene and methacrylic acid ester in CH2Cl2 or CH3CN gave the symmetrical urea-functional monomers in decent to excellent yield (entries 9 and 10). 2c compound was synthesized according to procedure reported by Schmitzer with 50% yield.17 In the procedure reported here, 2c was obtained in 99% yield.
The 2-ureido-4[1H]-pyrimidone (UPy) unit has been used as a building block for quadruple hydrogen bonding. Theoretically, its hydrogen-bonding ability could play a crucial role in the function and structure of polymerizable monomers.18–20 Thus, 2-amino-4-hydroxy-6-methylpyrimidine was chosen as the model substrate for the optimization of reaction conditions (Table 2). The influence of time, reaction temperature and the solvent nature were investigated. Firstly two isocyanatoethyl methacrylates and different solvents were tested. It was found that the yields of the reaction were relatively low without any solvent (Table 2, entries 1 and 2). When pyridine was used as solvent, the yield of 3a was improved to 85.4% (Table 2, entry 3). The reaction was also carried out in DMSO (Table 2, entries 5–8). Compound 3a was obtained in good yield when 1.1 or 1.3 equiv. UPy substrate was used at 170 °C in less than 5 min under optimized conditions for compound 3a (Table 2, entry 8) and for compound 3b (Table 2, entry 9).
|
|||||
|---|---|---|---|---|---|
| Entry | R–NCO (equiv.) | Solvent | T (°C) | Time (h) | Product (yield)a |
| a Reaction condition: isocyanate (1 mmol), 2-amino-4-hydroxy-6-methylpyrimidine (1.1–3 mmol) in the indicated solvent (neat/6 mL) at 100 °C (170 °C). Yield of recrystallization. | |||||
| 1 | 1a (2) | Neat | 100 | 21 h | 3a (53.1%) |
| 2 | 1a (3) | Neat | 100 | 21 h | 3a (60.7%) |
| 3 | 1a (3) | Pyridine | 100 | 16 h | 3a (85.4%) |
| 4 | 1a (3) | Pyridine | 100 | 8 h | 3a (83.9%) |
| 5 | 1a (1.1) | DMSO | 120 | 2 h | 3a (86.4%) |
| 6 | 1a (1.3) | DMSO | 120 | 2 h | 3a (87.0%) |
| 7 | 1a (1.3) | DMSO | 140 | 20 min | 3a (86.5%) |
| 8 | 1a (1.3) | DMSO | 170 | 5 min | 3a (87.6%) |
| 9 | 1b (1.3) | DMSO | 170 | 5 min | 3b (89.6%) |
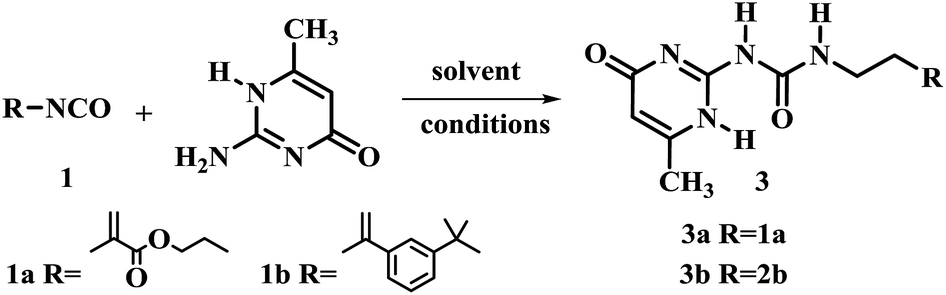
An important feature of multi-valent monomers was that they have multiple flexible “arms” which can wrap around the template molecule so to render higher affinity and selectivity than traditional MIPs based on mono-valent FMs.21 Shimizu22,23 et al. synthesized the methacrylic acid ester type three-armed functional monomer bearing urea functionality as phosphate receptor to increase the affinity and imprinting efficiency. In the work reported here, both methacrylate and styrene type precursors were used for the synthesis of FMs. The advantage of aryl groups linked to the urea motifs commonly increases the receptor's affinity toward anionic template.24 Along this line, three-branched monomer 4 was designed and obtained in 90% yield by reacting tris(2-aminoethyl)amine with 3-isopropenyl-α,α-dimethylbenzyl isocyanate (1b) in CH2Cl2 at room temperature for 6 h (Scheme 1).
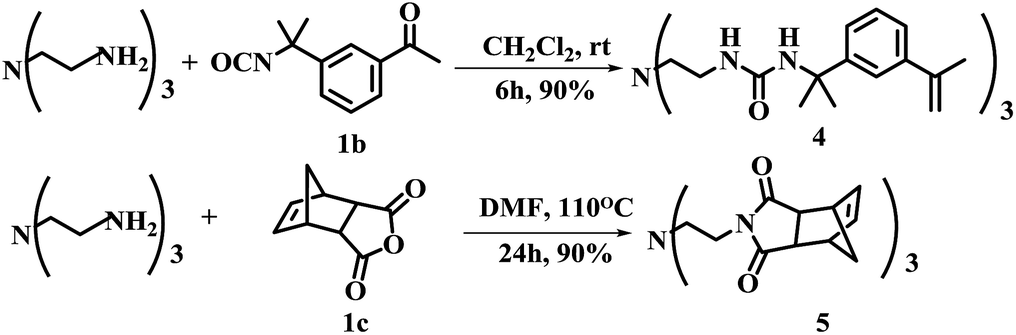 | ||
| Scheme 1 Synthesis of three-armed functional monomer 4 and monomer 5. | ||
The monomer 5 is noteworthy because it can be used as both functional monomer and cross linker. In this synthesis, cis-5-norbornene-endo-2,3-dicarboxylic anhydride was chosen as starting material. The advantages of monomer 5 include (1) basic nitrogen centered three armed structure could interact strongly with the template (2) three norbornene imide with stringed bridge double bond could participate in ring opening metathesis polymerization (ROMP) reactions and to improve the degree of crosslinking.25 More importantly, monomer 5 provides a possibility for preparing MIPs for free radical quencher type template molecules which is usually difficult to work with free radical type polymerization approach.
2.2 Synthesis of BHA/BHT-MIPs and NIPs
Generally, MIPs could be prepared with free-radical polymerization and sol–gel processes. Bulk polymerization with free-radical initiator is one of the most popular approaches which possess many attractive advantages.26,27 However, preparation of MIP for free radical quencher type template is not easy in conventional radical polymerization. For example, the amount of initiator needed was doubled for gossypol as template.28 Since the free radical quencher type template could block the radical initiation and propagation process. The ROMP using ruthenium-based Grubbs catalysts have was found highly desirable for various substituted cyclic olefins such as norbornenes.29,30 We envisioned that this approach could be useful to synthesize MIPs for free radical quencher type template molecules which is hard to be prepared with free radical type polymerization approach (Fig. 1).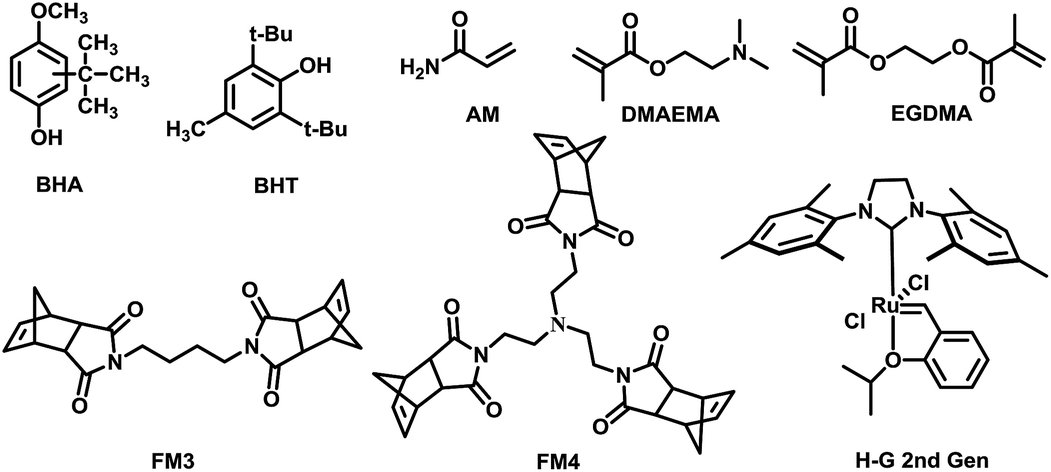 | ||
| Fig. 1 Chemical structures of template, cross-linker, functional monomers and catalyst in polymers synthesis. | ||
Butylated hydroxyanisole (BHA) and 2,6-di-tert-butyl-4-methylphenol (BHT) as classified antioxidants, has been widely used in food and pharmaceutical. However, when the total content of antioxidants are more than 0.02 wt% of the total fat/oil content of the food, it is considered carcinogenic.31,32 Therefore, recognition and detection of BHA and BHT is important for public health and food safety.
We choose two free radical quencher type molecules specifically butylated hydroxyanisole (BHA) and 2,6-di-tert-butyl-4-methylphenol (BHT) as template. Acrylamide (AM), 2-(dimethylamino) ethyl methacrylate (DMAEMA), FM3 and FM4 were used as functional monomers and ethylene glycol dimethacrylate (EGDMA) was chosen as cross-linker. The MIPs were prepared by both bulk polymerization and ring-opening metathesis polymerization. Details of synthetic procedures are summarized in the Table 3.
| Polymera,b | Template (mmol) | FM (mmol) | Cross-linker (mmol) | Initiator (catalyst) | Solvent |
|---|---|---|---|---|---|
| N1 | AM (4) | EGDMA (20) | AIBN | CH2Cl2 | |
| M1-1 | BHA (1) | AM (4) | EGDMA (20) | AIBN | CH2Cl2 |
| M1-2 | BHT (1) | AM (4) | EGDMA (20) | AIBN | CH2Cl2 |
| N2 | DMAEMA (4) | EGDMA (20) | AIBN | CH2Cl2 | |
| M2-1 | BHA (1) | DMAEMA (4) | EGDMA (20) | AIBN | CH2Cl2 |
| M2-2 | BHT (1) | DMAEMA (4) | EGDMA (20) | AIBN | CH2Cl2 |
| N3 | FM3 (2.2) | 2nd gen H–G | CH2Cl2 | ||
| M3-1 | BHA (1) | FM3 (2.2) | 2nd gen H–G | CH2Cl2 | |
| M3-2 | BHT (1) | FM3 (2.2) | 2nd gen H–G | CH2Cl2 | |
| N4 | FM4 (1.3) | 2nd gen H–G | CH2Cl2 | ||
| M4-1 | BHA (1) | FM4 (1.3) | 2nd gen H–G | CH2Cl2 | |
| M4-2 | BHT (1) | FM4 (1.3) | 2nd gen H–G | CH2Cl2 |
The preparation of MIPs and NIPs using traditional functional monomer, crosslinkers and the three-branched functional monomer bearing norbornene motif was investigated. In another words, the general procedure of free radical bulk polymerization and ring opening olefin metathesis polymerization were compared.
The free radical quencher type templates BHA and BHT were used as template for preparing MIPs with traditional free radical initiated bulk polymerization. As shown in Fig. 2A, the reaction for preparing N1 and N2 did not proceed well at RT. However, for N1 and N2, the polymerization was successful upon heating at 60 °C for 4 h. For M1-1, M2-1, M1-2 and M2-2, polymerization was not observed both at room temperature and in water bath at 60 °C for 4 h. However, in control experiments free-radical polymerization was successful in the absence of BHA and BHT in a water bath at 60 °C for 4 h (Fig. 2A1).
 | ||
| Fig. 2 Comparation of the bulk polymerization and ring-opening metathesis polymerization. N1(NIP): AM and EGDMA with AIBN; N2(NIP): DMAEMA and EGDMA with AIBN; N3(NIP): FM3 with 2nd Hoveyda–Grubbs catalyst; N4(NIP): FM4 with 2nd Hoveyda–Grubbs catalyst. M-1(MIP): BHA as template; M-2(MIP): BHT as template. (N/M 1 and N/M 2): bulk polymerization; (N/M 3 and N/M 4) ring-opening metathesis polymerization. | ||
In contrast, free radical quencher type templates BHA and BHT were used as template for preparing MIPs with functional monomer 3 and 4 via ROMP (Fig. 2B and C). Results showed that polymerization of N3 and N4 using monomer 3 and 4 were successfully via ROMP in 5 min at room temperature (The polymerization of N4 occurred faster than N3). This could possibly due to the fact that FM4 have three double bonds. Furthermore, the preparation of MIPs with the monomer 3 and monomer 4 using BHT and BHA as template were successful. This was achieved using 2nd generation Hoveyda–Grubbs catalyst via ROMP. The polymerization started after 10 min forming gel type polymer and was completed in 20 min even at room temperature. In conclusion, MIPs for BHT and BHA could be successfully prepared via ROMP with three armed norbornene based functional monomer using second generation Hoveyda–Grubbs catalyst via ROMP.
Binding capacity was measured by UV-Vis spectroscopy (see ESI Fig. S25–S28†). The adsorption peak at 290 nm (BHA) and 280 nm (BHT) were chosen for the quantitative analysis. According to binding capacity and imprinting effect, the maximum adsorption capacity of FM3-BHA-MIP, FM3-BHA-NIP, FM3-BHT-MIP, FM3-BHT-NIP, FM4-BHA-MIP, FM4-BHA-NIP, FM4-BHT-MIP, FM4-BHT-NIP were 7.1 mg g−1, 5.8 mg g−1, 5.3 mg g−1, 3.6 mg g−1, 12.2 mg g−1, 8.7 mg g−1, 11.3 mg g−1, 8.6 mg g−1, respectively. The higher binding capacity was attributed to specific binding sites in MIPs and thus higher affinity toward BHA/BHT than that of NIPs. Arco et al.32 reported BHA-imprinted polymer with the binding capacity of 6.08 mg g−1 using 4-vinylpyridine (4-VP) and 1-viny-limidazole (1-VI) as the functional monomer. Because of the anti-oxidative nature of BHA and BHT, much more initiator had to be used with typical bulk polymerization.
3 Experimental section
3.1 General method and materials
3.2 Synthetic procedure of functional monomers
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), the white solid was dried at 50 °C under reduced pressure (234.9 mg, 83.9%). 1H NMR (400 MHz, CDCl3) δ 12.97 (s, 1H), 11.92 (s, 1H), 10.48 (s, 1H), 6.18 (s, 1H), 5.78 (s, 1H), 5.55 (s, 1H), 4.27 (s, 2H), 3.59 (s, 2H), 2.20 (d, J = 26.8 Hz, 3H), 1.94 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 172.58 (s), 167.30 (s), 156.79 (s), 154.92 (s), 148.25 (s), 136.07 (s), 125.78 (s), 106.49 (s), 63.09 (s), 38.80 (s), 19.16 (s), 18.30 (s). MS-ESI (m/z) C12H16N4O4 ([M–H]−): 279.2.
:1), the white solid was dried at 50 °C under reduced pressure (234.9 mg, 83.9%). 1H NMR (400 MHz, CDCl3) δ 12.97 (s, 1H), 11.92 (s, 1H), 10.48 (s, 1H), 6.18 (s, 1H), 5.78 (s, 1H), 5.55 (s, 1H), 4.27 (s, 2H), 3.59 (s, 2H), 2.20 (d, J = 26.8 Hz, 3H), 1.94 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 172.58 (s), 167.30 (s), 156.79 (s), 154.92 (s), 148.25 (s), 136.07 (s), 125.78 (s), 106.49 (s), 63.09 (s), 38.80 (s), 19.16 (s), 18.30 (s). MS-ESI (m/z) C12H16N4O4 ([M–H]−): 279.2.3.3 Synthesis of BHA/BHT-MIPs and NIPs
3.4 Adsorption kinetic experiments
BHA/BHT-MIPs (7 samples, 10 mg each) and NIPs (7 samples, 10 mg each) samples were added to 5 mL of 50 mg L−1/100 mg L−1 BHA/BHT-CH3OH solution. The mixtures were incubated in a shaker and were sampled at time-intervals of 240 min, 360 min, 480 min, 720 min, 840 min, 960 min, and 1080 min. Each time sample was passed through a filter (0.2 μm PTFE syringe filters) to separate particles from supernatant. Residual concentrations of BHA/BHT in filtrate were quantified by measuring the UV absorbance at 290/280 nm. The adsorption capacity can be calculated according to the following equation (eqn (1)):
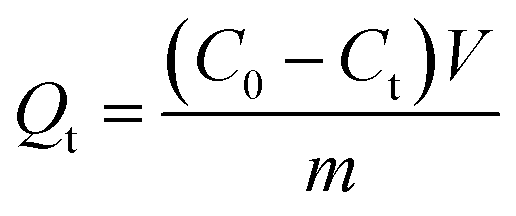 | (1) |
4 Conclusions
In summary, functional monomers bearing urea, supramolecular UPy motif and three-branched nitrogen centered norbornene motif were designed and synthesized with decent to excellent yields. The approach featured several advantages such as short reaction times, mild reaction conditions and no need for column chromatographic purification. The norbornene based FM was used for preparing molecularly imprinted polymers (MIPs) via Ring-Opening Metathesis Polymerization (ROMP). Results showed that norbornene based three-branched functional monomers could be used for preparing MIPs for free radical quencher type template molecules. Further optimization and applications of these functional monomers for advanced functional materials design and molecularly imprinted polymers are in progress.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was financially supported by National Natural Science Foundation of China (21464015, 21472235), Xinjiang Tianshan Talents Program (2018xgytsyc2-3), “One Thousand Talents” Program of China (Y32H291501), Young Elite Scientist Sponsorship Program by CAST (2017QNRC001), and the West Light Foundation of the Chinese Academy of Sciences, Grant No. 2017-XBQNXZ-B-001.Notes and references
- G. Wuff and A. Sarhan, Angew. Chem., Int. Ed., 1972, 11, 341–345 Search PubMed.
- R. Arshady and K. Mosbach, Macromol. Chem. Phys., 1981, 182, 687–692 CrossRef CAS.
- J. D. Lee, N. T. Greene, G. T. Rushton, K. D. Shimizu and J. I. Hong, Org. Lett., 2005, 7, 963–966 CrossRef CAS PubMed.
- G. Vlatakis, L. I. Andersson, R. Müller and K. Mosbach, Nature, 1993, 361, 645–647 CrossRef CAS PubMed.
- R. L. Adkins, N. Walsh, M. Edmunds and J. M. Trafford, Analyst, 1995, 120, 1433–1436 RSC.
- L. Chen, S. Xu and J. Li, Chem. Soc. Rev., 2011, 40, 2922–2942 RSC.
- Y. G. Zhang, D. Song, L. M. Lanni and K. D. Shimizu, Macromolecules, 2010, 43, 6284–6294 CrossRef CAS.
- Y. Zhang, D. Song, J. C. Brown and K. D. Shimizu, Org. Biomol. Chem., 2011, 9, 120–126 RSC.
- L. S. Shimizu, A. D. Hughes, M. D. Smith, M. J. Davis, B. P. Zhang, H. C. zur Loye and K. D. Shimizu, J. Am. Chem. Soc., 2003, 125, 14972–14973 CrossRef CAS PubMed.
- C. R. Bowers, M. Dvoyashkin, S. R. Salpage, C. Akel, H. Bhase, M. F. Geer and L. S. Shimizu, ACS Nano, 2015, 9, 6343–6353 CrossRef CAS PubMed.
- L. S. Shimizu, S. R. Salpage and A. A. Korous, Acc. Chem. Res., 2014, 47, 2116–2127 CrossRef CAS PubMed.
- J. Yang, M. B. Dewal and L. S. Shimizu, J. Am. Chem. Soc., 2006, 128, 8122–8123 CrossRef CAS PubMed.
- J. Yang, M. B. Dewal, S. Profeta, M. D. Smith, Y. Li and L. S. Shimizu, J. Am. Chem. Soc., 2008, 130, 612–621 CrossRef CAS PubMed.
- S. Dawn, M. B. Dewal, D. Sobransingh, M. C. Paderes, A. C. Wibowo, M. D. Smith, J. A. Krause, P. J. Pellechia and L. S. Shimize, J. Am. Chem. Soc., 2011, 133, 7025–7032 CrossRef CAS PubMed.
- Y. Yu, Q. Zhang, J. Buscaglia, C.-C. Chang, Y. Liu, Z. Yang, Y. Guo, Y. Wang, K. Levon and M. Rafailovich, Analyst, 2016, 141, 4424–4431 RSC.
- Y. Yu, Q. Zhang, C.-C. Chang, Y. Liu, Z. Yang, Y. Guo, Y. Wang, D. K. Galanakis, K. Levon and M. Rafailovich, Analyst, 2016, 141, 5607–5617 RSC.
- C. Gomy and A. R. Schmitzer, J. Org. Chem., 2006, 71, 3121–3125 CrossRef CAS PubMed.
- R. P. Sijbesma, F. H. Beijer, L. Brunsveld, B. J. B. Folmer, J. Hirschberg, R. F. M. Lange, J. K. L. Lowe and E. W. Meijer, Science, 1997, 278, 1601–1604 CrossRef CAS PubMed.
- F. H. Beijer, R. P. Sijbesma, H. Kooijman, A. L. Spek and E. W. Meijer, J. Am. Chem. Soc., 1998, 120, 6761–6769 CrossRef CAS.
- S. H. M. Sontjens, R. P. Sijbesma, M. H. P. van Genderen and E. W. Meijer, J. Am. Chem. Soc., 2000, 122, 7487–7493 CrossRef.
- Y. G. Zhang and K. D. Shimizu, Abstracts of Papers of the American Chemical Society, 2007, 233 Search PubMed.
- D. Song, Y. G. Zhang, M. F. Geer and K. D. Shimizu, J. Mol. Recognit., 2014, 27, 448–457 CrossRef CAS PubMed.
- X. G. Wu, K. Goswami and K. D. Shimizu, J. Mol. Recognit., 2008, 21, 410–418 CrossRef CAS PubMed.
- R. Wagner, W. Wan, M. Biyikal, E. Benito-Pena, M. C. Moreno-Bondi, I. Lazraq, K. Rurack and B. Sellergren, J. Org. Chem., 2013, 78, 1377–1389 CrossRef CAS PubMed.
- Y. G. Zhang, J. M. Lavin and K. D. Shimizu, J. Am. Chem. Soc., 2009, 131, 12062–12063 CrossRef CAS PubMed.
- L. Chen, X. Wang, W. Lu, X. Wu and J. Li, Chem. Soc. Rev., 2016, 45, 2137 RSC.
- H. Y. Yan and K. H. Row, Int. J. Mol. Sci., 2006, 7, 155–178 CrossRef CAS.
- K. Zhi, L. Wang, Y. Zhang, X. Zhang, L. Zhang, L. Liu, J. Yao and W. Xiang, J. Mol. Recognit., 2017, e2627 Search PubMed.
- S. Kanaoka and R. H. Grubbs, Macromolecules, 1995, 28, 4707–4713 CrossRef CAS.
- K. H. Yoon, K. O. Kim, C. Q. Wang, I. Park and D. Y. Yoon, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 3914–3921 CrossRef CAS.
- G. M. Williams, M. J. Iatropoulos and J. Whysner, Food Chem. Toxicol., 1999, 37, 1027–1038 CrossRef CAS PubMed.
- L. L. Vallo, R. T. Laxamana, F. U. Paredes, I. H. J. Arellano and S. D. Arco, Mater. Lett., 2015, 159, 317–320 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra01852b |
| This journal is © The Royal Society of Chemistry 2019 |