
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnraveling the impact of the Pd nanoparticle@BiVO4/S-CN heterostructure on the photo-physical & opto-electronic properties for enhanced catalytic activity in water splitting and one-pot three-step tandem reaction†
Subhajyoti
Samanta
 a,
Biswarup
Satpati
b and
Rajendra
Srivastava
*a
a,
Biswarup
Satpati
b and
Rajendra
Srivastava
*a
aDepartment of Chemistry, Indian Institute of Technology Ropar, Rupnagar, Punjab-140001, India. E-mail: rajendra@iitrpr.ac.in; Fax: +91-1881-223395; Tel: +91-1881-242175
bSurface Physics and Material Science Division, Saha Institute of Nuclear Physics, 1/AF, Bidhannagar, Kolkata-700 064, India
First published on 8th January 2019
Abstract
Herein, a Pd nanoparticle-embedded SBVCN-37 heterostructure photocatalyst was synthesized and employed in the water-splitting reaction and for the synthesis of imines via a one-pot tandem reaction involving the photocatalytic reduction of nitrobenzene and oxidation of benzyl alcohol, followed by their condensation reaction. The embedded Pd nanoparticles (mean diameter ∼ 5–7 nm) act as an electron mediator and enhance the catalytic activity of SBVCN-37 during the oxidation and reduction reactions. The experimental results confirm that the light-induced holes owing to the favourable redox potential of the catalyst oxidize N2H4 to N2 and liberate H+ ions, which subsequently react with photogenerated electrons to facilitate the reduction of nitrobenzene. The obtained quantum yields for benzyl alcohol oxidation and nitrobenzene reduction were calculated to be 2.08% and 6.53% at λ = 420 nm light illumination, respectively. Furthermore, the obtained apparent quantum yields for the OER and HER were calculated to be 10.22% and 12.72% at 420 nm, respectively, indicating the excellent potential of the investigated photocatalyst for solar fuel production. Photoelectrochemical (PEC) and time-resolved and steady-state photoluminescence measurements reveal that the optimum amount of Pd nanoparticles over SBVCN-37 is the crucial factor for achieving the highest photocurrent response, lowest charge transfer resistance, and efficient carrier mobility, leading to prominent catalytic activity. Furthermore, the Mott–Schottky (M–S) analysis confirmed that the deposition of Pd nanoparticles effectively reduced the over-potential and fine-tuned the band edge potential required for the HER and OER reactions, respectively.
1. Introduction
The strategic design and development of multi-functional heterogeneous catalysts are of significant importance to carry out one-pot multi-component reactions1 and one-pot multi-step tandem reactions2 for fine chemical synthesis, energy production, etc.3 In general, conventional thermal catalytic pathways are adopted to carry out tandem reactions. However, very recently, visible light has been utilized to trigger these catalytic reactions as an strong alternative to the conventional thermal-induced reactions owing to its several environmental advantages.3 Besides solar fuel production, the applications of photocatalysis also include the activation and utilization of renewable reactants such as O2, H2O, and CO2.4 Moreover, heterogeneous photocatalysis has become a hot topic for carrying out several important reactions, such as water splitting for the production of H2/O2 gas and CO2 reduction for the production of methanol/methane/CO.5–8 Similarly, the photocatalytic activation of O2 produces various reactive oxygen species (ROS) such as O2−˙ and HO˙ (through electrons and holes), which oxidize aromatic and aliphatic compounds, leading to the production of various commercially important fine chemicals.7 For this purpose, a wide range of semiconductor photocatalysts have been synthesized and further modified via composition tuning, band gap engineering, morphology modifications, alteration of textural properties, noble metal deposition, etc. to achieve the desired activity.9–14 Recently, metal-free and inexpensive graphitic C3N4-based materials have been developed for visible light-assisted catalytic reactions.15 The objective of this study is to develop a multi-functional catalyst based on g-C3N4 for the synthesis of imines via the tandem reaction of nitroaromatics and benzyl alcohols and water splitting reactions via suspension photocatalysis for the production of H2 and O2 gases.Several sustainable routes are known for the synthesis of aromatic imines.16 Researchers have developed noble metal-based catalysts for the dehydrogenation of secondary amines in the presence of molecular O2.17 Aromatic imines can also be synthesized via the condensation of aromatic aldehydes and anilines.18 However, it should be noted that these reactants are susceptible to oxidation by O2. Therefore, it is important to design synthetic strategies using less reactive substrates such as nitroaromatics and benzyl alcohols.19–24 The synthesis of aromatic imines from nitroaromatics and benzyl alcohols can be realized via one-pot tandem synthesis protocols involving three consecutive steps: (1) selective oxidation of benzyl alcohols to benzaldehydes, (2) selective reduction of nitroaromatics to anilines, and (3) successive condensation reaction between aldehydes and anilines. Noble metal- and non-noble metal-mediated and C3N4-based photocatalysts have been developed for the selective oxidation of benzyl alcohol to benzaldehyde.25–27 Further, catalytic hydrogenation of nitroaromatics using Pt-, Au-, Pd-, and Ir-based systems has been developed using H2 as a reducing agent.28–30 It is also possible to use the catalytic transfer hydrogenation strategy to reduce nitroaromatics in the presence of hydrogen donors, such as hydrazine, formic acid, and NaBH4.31 This strategy avoids the utilization of high pressure reaction conditions, which are generally required when H2 is used as a reducing agent. These hydrogen donors make the reaction set-up simpler, especially when the aim is to reduce nitroaromatics under photocatalytic conditions.
Photoelectrochemical (PEC) water splitting emerges as the most efficient process in terms of solar to fuel conversion efficiency.5,32,33 However, the controlled synthetic strategy for the fabrication of photoanodes/photocathodes, complicated reaction setup, and requirement of external applied potential bias restrict its wider application.5 Therefore, liquid-phase suspension photocatalysis has become a strong alternative owing to its simple operational procedure for solar to fuel production. Photocatalytic H2 production using g-C3N4 under visible light illumination is one of the initial pioneering contributions of Wang et al.34 However, bare g-C3N4 exhibits low activity for this reaction due to its small surface area (<10 m2 g−1), low absorption of visible light, and fast recombination of charge carriers compared to metals and metal oxides reported for this reaction.35 Thus, the photocatalytic research community has devoted tremendous efforts towards improving the H2 evolution efficiency of g-C3N4 by adopting several promising strategies such as increasing its surface area,36 doping with heteroatoms (such as S, N, O, P, and C),36 and introduction of mesoporosity (for higher surface area).36 It is worth mentioning that all the aforementioned modifications require Pt as a co-catalyst to catalyze the H2-evolution reaction.36 Therefore, it is of utmost importance to fabricate g-C3N4-based nanocomposite photocatalysts that do not require the additional photo-deposition of Pt as a co-catalyst during the H2-evolution reaction. Among the O2 evolution photocatalysts reported to date, BiVO4 emerges as one of the most promising materials for this application owing to its suitable band edge potential required for the OER with the maximum efficiency of 90% of its theoretical value under 1.5 AM solar simulated light.37 However, its sluggish reaction kinetics, low surface area, and fast charge carrier recombination limit the application of this photocatalyst.38–40 Thus, to overcome these limitations and improve its quantum efficiency for OER application, various concepts have implemented.41–45 Among them, the formation of heterostructures seems to be one of the most promising strategies. Using this strategy, the separation of charge carriers is maximized at the interface of two materials through carrier mobility alteration.41 Therefore, the development of highly efficient BiVO4-based heterostructure photocatalysts for the OER is another important milestone to achieve.
Herein, a Pd nanoparticle-decorated BiVO4/S-CN (hereafter represented as SBVCN, where S-CN is the sulphur-doped carbon nitride) heterostructure photocatalyst is developed for the first time to overcome the above mentioned problems associated with the HER, OER, and imine synthesis. This catalyst exhibits excellent activity in the HER, OER and one-pot tandem reaction of nitroaromatics and benzyl alcohols for the synthesis of imines under visible light illumination under mild conditions. The detailed experimental investigation with the help of analytical and spectroscopic measurements reveals that the embedded Pd nanoparticles (hereafter represented as Pd NPs) in the SBVCN-37 photocatalyst (having optimum amounts of BiVO4 and S-doped g-C3N4) play a crucial role in obtaining high yield and selectivity of imines. Additionally, they are important for achieving excellent activity for water splitting in the HER and OER reactions in the absence of a Pt co-catalyst (for HER reaction) with an apparent quantum yield (AQY) of 12.72% and 10.22%, respectively. Thus, this meaningful approach demonstrates a promising pathway for the development of g-C3N4-based Pt co-catalyst-free multi-functional photocatalysts.
2. Experimental
2.1 Materials, methods and photocatalyst characterization
Details of the materials, catalyst synthesis, catalyst characterization, procedures for the photocatalytic HER/OER, details for the catalytic reactions for benzyl alcohol oxidation/nitrobenzene reduction, tandem reaction, apparent quantum yield (%) calculation, photoelectrochemical measurements (PEC), and photographs of the photochemical reaction set-up (Fig. S1†) are given in the ESI.†3. Result and discussion
3.1 Physico-chemical properties
The phase purity, crystalline nature, and composition of the prepared materials were analyzed via powder XRD (Fig. 1a and Fig. S2, ESI†). The XRD pattern of graphitic C3N4 exhibits reflections at 2θ = 13.1° and 27.4°, which correspond to the 100 and 002 planes, respectively, and are attributed to the in-plane structural packing of the aromatic units (Fig. S2, ESI†).46,47 The 002 plane in S-CN appears at a slightly higher 2θ value than 27.4°, which may be due to the H2SO4 treatment of the parent g-C3N4 (Fig. 1a and Fig. S2, ESI†).47,48 The d-spacings of the sulphated g-C3N4(S-CN) are found to be 0.675 nm and 0.319 nm, which are similar to that of the standard g-C3N4.46 S-CN also exhibits some very low intense unknown reflections, which can be ascribed to the presence of different oligomeric units of g-C3N4 generated after the H2SO4 treatment.46,49 The XRD pattern of BiVO4 exhibits reflections at 2θ values of 14.79°, 19.01°, 27.31°, 28.97°, 30.75°, 34.62°, 35.40°, 39.95°, 42.56°, 46.04°, 46.97°, 47.59°, 50.25°, 53.46°, 58.44°, and 59.78°, which correspond to the 020, 110, , 121, 040, 200, 002, 211,
, 121, 040, 200, 002, 211,  , 150, 240, 042,
, 150, 240, 042,  , 202,
, 202, 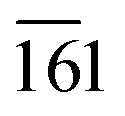 , and 161 planes, respectively, for monoclinic BiVO4 (JCPDS No. 014-0688) (Fig. 1a). The XRD pattern of the composite photocatalyst SBVCN-37 exhibits reflections corresponding to both S-CN and BiVO4, with very high intensity reflections from the BiVO4 phase (Fig. 1a). Further, the incorporation of Pd NPs in SBVCN-37 is confirmed by the appearance of two new less prominent reflections at 40.1° and 47.2°, corresponding to the 111 and 200 planes of Pd NPs, respectively (Fig. 1a).50 The textural properties of all the samples were determined by N2-sorption experiments. Fig. 1b shows the isotherms of S-CN, SBVCN-37, and (Pd3%)/SBVCN-37. The detailed explanation and determined values are provided in the ESI.†
, and 161 planes, respectively, for monoclinic BiVO4 (JCPDS No. 014-0688) (Fig. 1a). The XRD pattern of the composite photocatalyst SBVCN-37 exhibits reflections corresponding to both S-CN and BiVO4, with very high intensity reflections from the BiVO4 phase (Fig. 1a). Further, the incorporation of Pd NPs in SBVCN-37 is confirmed by the appearance of two new less prominent reflections at 40.1° and 47.2°, corresponding to the 111 and 200 planes of Pd NPs, respectively (Fig. 1a).50 The textural properties of all the samples were determined by N2-sorption experiments. Fig. 1b shows the isotherms of S-CN, SBVCN-37, and (Pd3%)/SBVCN-37. The detailed explanation and determined values are provided in the ESI.†
 | ||
| Fig. 1 (a) Powder XRD patterns of all the materials synthesized in this study, (b) N2 adsorption–desorption isotherms of S-CN, SBVCN-37, and (Pd3%)/SBVCN-37, (inset shows BJH pore size distribution) and FESEM images of (c) BiVO4, (d) S-CN, (e) SBVCN-37, and (f) (Pd3%)/SBVCN-37 photocatalyst. | ||
The microstructures of BiVO4 exhibit a dendrite-like structure (Fig. 1c); whereas, S-CN exhibits a spherical morphology (Fig. 1d) in the FESEM images. In the synthesis of BiVO4, sodium dodecyl sulphate (SDS) was used as a surfactant, which is responsible for this dendrite-like morphology.48 The nanocomposite (Pd3%)/SBVCN-37 exhibits a somewhat distorted morphology (Fig. 1e and f) compared to that of pristine BiVO4 and S-CN, which may be due to the ultrasonication, annealing, and reducing environment during its synthesis. Furthermore, this also indicates that aggregated BiVO4 crystals are supported over the S-CN microstructures (Fig. 1e and f). The Pd NPs are not visible in the FESEM image of (Pd3%)/SBVCN-37; however, their presence was confirmed from the elemental mapping (Fig. 2). Further, the elemental mapping and EDAX spectrum confirm the presence of all the elements in the (Pd3%)/SBVCN-37 heterostructure (Fig. 2).
 | ||
| Fig. 2 (a) FESEM image of (Pd3%)/SBVCN-37, (b) covered area for elemental mapping and EDAX, (c) overall mapping, single element mapping of (d) C, (e) N, (f) O, (g) S, (h) Bi, (i) Pd, (j) V, and (k) EDAX spectrum. | ||
The TEM images indicate that BiVO4 exhibits large particle sizes in the range of 300–500 nm (Fig. S3a and b, ESI†). The bright rectangular spots in its SAED pattern indicates that BiVO4 is highly crystalline in nature and the obtained pattern well matches with the standard 110, 200, and 220 planes of BiVO4 (Fig. S3c, ESI†).48 High-angle annular dark-field (HAADF), STEM and EDAX confirmed the presence of individual elements in BiVO4 (Fig. S3d–g, ESI†). The TEM image of the SBVCN-37 nanocomposite shows that it contains a nanomesh morphology corresponding to S-CN and large crystal-like morphology corresponding to BiVO4 (Fig. 3a). In the nanocomposite (Pd3%)/SBVCN-37, BiVO4, S-CN, and Pd NPs are clearly visible (Fig. 3b and c). Moreover, in the case of (Pd3%)/SBVCN-37, Pd NPs with an average particle size of 5–7 nm were found to be embedded successfully over the surface of the SVBCN-37 heterostructure (inset in Fig. 3c, and Fig. 3d). The TEM images also confirmed the uniform dispersion of Pd NPs on the surface of the SBVCN-37 heterostructure, which is one of the desired targets for the fabrication of highly active heterogeneous photocatalysts (Fig. 3b and c). An overview of the drift-corrected TEM, HAADF, and EDAX results is presented in Fig. 4. STEM and EDAX confirmed the presence of all the elements in the (Pd3%)/SBVCN-37 photocatalyst (Fig. 4).
 | ||
| Fig. 3 TEM images of (a) SBVCN-37 and (b and c) (Pd3%)/SBVCN-37, with the inset of “c” showing the average particle size distribution of the Pd NPs. (d) HRTEM image of the Pd NPs present in (Pd3%)/SBVCN-37. | ||
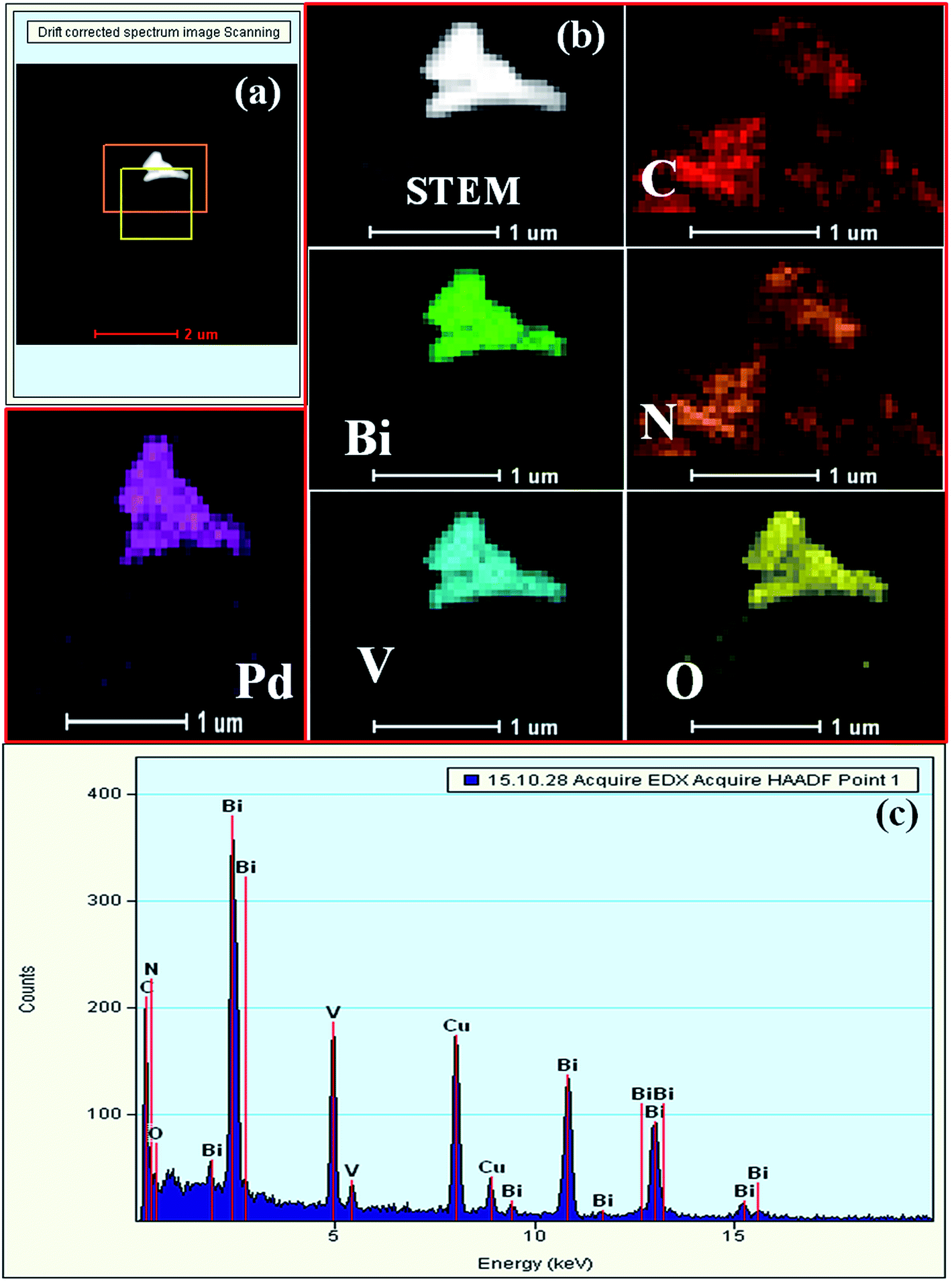 | ||
| Fig. 4 (a) HAADF and (b) STEM-HAADF images of individual elements, and (c) EDAX spectrum of (Pd3%)/SBVCN-37. | ||
Our previous study showed that acidity plays an important role in the final condensation step of tandem reactiond.48,51,52 Therefore, the acidity of the materials was analyzed via NH3-TPD (Fig. S4, ESI†). The detailed explanation and determined values are provided in the ESI.† The presence of various functional groups in the material was confirmed by FT-IR spectroscopy (Fig. 5a) and a detailed explanation is provided in the ESI.†
 | ||
| Fig. 5 (a) FTIR spectra, (b) DRUV-visible spectra, (c) Steady-state photoluminescence spectra, and (d) time-resolved spectra of the materials investigated in this study (inset shows a photograph of the synthesized powder photocatalysts). | ||
The light absorption and related optoelectronic properties of the photocatalysts were investigated via diffuse reflectance ultraviolet-visible spectroscopy (DRUV-vis) (Fig. 5b). The absorbance band edge positions for S-CN and pristine BiVO4 are 460 nm and 530 nm, respectively (Fig. 5b). The absorption spectra indicate that both materials are visible light-active materials. SBVCN-37 exhibits an absorption spectrum similar to BiVO4 and it has the highest absorbance co-efficient among all the investigated photocatalysts. The Pd-incorporated SBVCN-37 samples exhibited similar visible light spectra to that of the parent SBVCN-37 with some additional features (Fig. 5b). Interestingly, the peak in the visible region at 455 nm for SBVCN-37 was red-shifted to 460 nm when different contents of Pd nanoparticles were embedded in it (inset, Fig. 5b). This peak arises due to the surface plasmon resonance (SPR) phenomenon of Pd NPs.53 Generally, Pd NPs exhibit SPR in the UV region’ however, in this case the ‘d’ electrons of Bi and V are disrupted by the valance electrons of the Pd NPs, leading to the red shift of this peak, which is consistent with a recent literature report.53 The average diameter of the Pd NPs is in the range of 5–7 nm; therefore, the synthesized catalyst exhibited the SPR phenomenon.53,54 This observation further confirms the successful incorporation of Pd NPs in the SBVCN-37 heterostructure (Fig. 5b). The calculated band gap energies for S-CN, BiVO4, SBVCN-37, (Pd1%)/SBVCN-37, (Pd3%)/SBVCN-37, and (Pd5%)/SBVCN-37 are 2.77 eV, 2.45 eV, 2.48 eV, 2.42 eV, 2.25 eV, and 2.40 eV, respectively (Fig. S5, ESI†). The conduction band and valance band edge potentials were found to be 0.35 and 2.80 eV for BiVO4 (χBiVO4 = 6.04 eV) and −1.20 eV and 1.57 eV for S-CN, respectively (χC3N4 = 4.66 eV) using the Mulliken electronegativity theory (Fig. S5, ESI†).46,47
The steady-state photoluminescence spectroscopic analysis revealed that among the photocatalysts, S-CN exhibits the highest intensity PL spectrum, which indicates its inferior charge carrier separation efficiency (Fig. 5c).47,55–57 However, the spectrum of BiVO4 indicates that it has better charge separation ability than S-CN (Fig. 5c). Interestingly, SBVCN-37 exhibited a relatively lower intensity peak in its steady-state PL spectrum (Fig. 5c) than S-CN, which signifies that the formation of the heterostructure is beneficial for efficient charge carrier separation. Further, the Pd-incorporated SBVCN-37 materials exhibited further quenching in their PL spectrum in comparison to SBVCN-37. This investigation confirmed that the Pd NPs further assisted in the charge carrier migration and separation in the SBVCN-37 photocatalyst through carrier mobility modulation. Notably, (Pd3%)/SBVCN-37 exhibited the lowest intensity PL spectrum among the Pd NP-decorated SBVCN-37 photocatalysts. A higher loading of Pd NPs covered the active surface and caused interference for light absorption and migration and separation of charge carriers, which is responsible for the higher intensity PL spectrum of (Pd5%)/SBVCN-37 than (Pd3%)/SBVCN-37. The combination of DRUV-visible and steady-state PL investigations demonstrated that among the synthesized photocatalysts, (Pd3%)/SBVCN-37 exhibited the best photo-physical and optoelectronic properties, which may be beneficial for obtaining high photocatalytic activity. Further, the time-resolved PL spectroscopic analysis revealed detailed information related to the dynamics of the charge transfer together with the carrier decay time and lifetime of the charge carriers in the excited state (Fig. 5d). The average emission lifetimes and their associated amplitudes for all the catalysts are provided in Table S2 (ESI†). The average emission lifetime for (Pd3%)/SBVCN-37 and SBVCN-37 were 5.18 ns and 6.01 ns, respectively (details about the charge carrier lifetime and kinetics calculation from TRPL are provided in the ESI†). Therefore, it can be concluded that the incorporation of Pd NPs on the surface of SBVCN-37 lowered the average lifetime, which is mainly associated with promoted charge carrier separation with immediate migration over the interface of the heterostructure photocatalyst. The kinetics of the charge carriers was found to be the highest for (Pd3%)/SBVCN-37 (0.19 (ns−1)), which corroborates that an optimum Pd NP loading is crucial for achieving high photocatalytic activity. Based on this study, the obtained decay time and lifetime of all the charge carriers of the photocatalysts are presented in Table S2 (ESI†). The combination of steady-state and time-resolved photoluminescence demonstrated that the separation, migration, and transportation of electrons/holes to their respective band positions were the most effective for (Pd3%)/SBVCN-37 and facilitated only when an optimum amount of Pd NPs (3 wt%) is present at the interface. Hence, the Pd NPs act as mediators in the charge carrier separation at the heterostructure interface, and (Pd3%)/SBVCN-37 is proven to be the best heterostructure photocatalyst among the materials prepared in this study.
Fig. 6a presents the surface survey XPS spectrum of (Pd3%)/SBVCN-37, which confirms the presence of all the elements in the photocatalyst. Fig. 6b–h present the individual high resolution XPS spectra of Pd 3d, S 2p, Bi 4f, V 2p, C 1s, N 1s, and O 1s of (Pd3%)/SBVCN-37. The two peaks at the binding energies of 340.2 eV and 335.1 eV correspond to 3d3/2 and 3d5/2, respectively, which confirm the incorporation of Pd0 NPs (Fig. 6b).50 The two peaks at the binding energies of 164.1 eV and 165.2 eV can be assigned to the S 2p3/2 and S 2p1/2 states, respectively.46 The appearance of an intense peak at 169.3 eV is due to the presence of sulphur in the form of –SO3H/HSO4− groups in the g-C3N4 framework (Fig. 6c).46 This confirms the existence of ‘S’ in the +6 oxidation state in the resulting (Pd3%)/SBVCN-37 photocatalyst (Fig. 6c). The high-resolution spectrum of Bi 4f exhibits two sharp peaks at 164.1 eV and 159.2 eV, which can be assigned to Bi 4f1/2 and Bi 4f3/2, respectively, and confirm the existence of Bi in the +4 oxidation state (Fig. 6d).48,49 The V 2p spectrum also exhibits two peaks at binding energies of 515 eV and 524.2 eV, corresponding to V 2p3/2 and V 2p1/2, respectively (Fig. 6e).52 Further, the high-resolution XPS spectrum of C 1s displays two peaks at the binding energies of 284.6 eV and 288.5 eV, which can be assigned to the sp2-hybridized carbon atom of the melam framework and tri-coordinated carbon atom attached to the nitrogen atom of S-CN (Fig. 6f).46,48 Three, different types of N atoms can be seen from the N 1s high-resolution XPS spectrum. The sharp peak at 397.4 eV is due to the two carbon atoms coordinated to nitrogen, while the broad peak at 399.1 eV can be assigned to the tertiary nitrogen atom (–C–N3–) (Fig. 6g).46,47 The satellite peak at 400.2 eV corresponds to the hydrogen atom attached to the nitrogen atom in SBVCN-37. Further, three peaks at the binding energies of 528 eV, 530.2 eV, and 532.4 eV are observed in the O 1s high-resolution XPS spectrum, which indicate the existence of three different oxygen species in the (Pd3%)/SBVCN-37 nanocomposite (Fig. 6h).46,49
 | ||
| Fig. 6 (a) Full survey XPS spectrum of (Pd3%)/SBVCN-37. High-resolution XPS spectra of (b) Pd 3d, (c) S 2p, (d) Bi 4f, (e) V 2p, (f) C 1s, (g) N 1s, and (h) O 1s. | ||
The photocatalysts exhibited an impressive photocurrent response upon the illumination of light on the surface of all the photocatalyst-coated FTO, as evident from their LSV curves (Fig. 7a and b and Fig. S6, ESI†). The Pd NP-decorated nanocomposite (Pd3%)/SBVCN-37 produced a photocurrent three times higher than that SVBCN-37, which clearly reflects that the incorporation of an optimum amount of Pd NPs accelerates the electronic relay over the photocatalyst surface, resulting in a higher photocurrent response (Fig. 7a and b). Among the investigated photocatalysts, (Pd3%)/SBVCN-37 displayed the highest current (∼0.45 mA cm−2) response (Fig. 7b and Fig. S6, ESI†). The transient photocurrent analysis confirmed the high photo-reversibility of the photocatalyst in the presence and absence of light irradiation (Fig. 7c and d).48,50,51 This observation clearly proves that light illumination produces excessive electrons at the electrode–electrolyte junction, which are responsible for the generation of a photocurrent. The obtained current density of SBVCN-37 was found to be two times and three times higher than that of BiVO4 and S-CN, respectively (Fig. 7c). The magnitude of the photocurrent response was almost 23 times and 36 times higher for (Pd1%)/SBVCN-37 (∼0.07 mA cm−2) and (Pd3%)/SBVCN-37 (∼0.11 mA cm−2), respectively, than that of SBVCN-37 (∼0.003 mA cm−2), indicating that the incorporation of Pd NPs is highly beneficial for the enhancement of electron density (Fig. 7c and d). However, beyond 3 wt% Pd, the photocurrent density decreased for (Pd5%)/SBVCN-37 (∼0.08 mA cm−2) (Fig. 7d). This observation suggests that an excessive Pd content blocks the active surface of SBVCN-37 for light absorption, resulting in a lower photocurrent response. Moreover, EIS was also employed to gain insight into the charge carrier transfer in the vicinity of electrode–electrolyte with the help of the Nyquist plot, which consists of a semicircle at the low frequency followed by straight line at the high frequency region (Fig. 7e). Further, the results are better represented in the form of an equivalent circuit, which is composed of solution resistance (Rs), charge transfer resistance (Rct), and constant phase elements (CPE) (inset, Fig. 7e). A smaller Rct value reflects the efficient charge transfer capability of the corresponding photocatalyst and vice versa. Here, the trend of Rct follows the order of S-CN > BiVO4 > SBVCN-37 > (Pd1%)/SBVCN-37 > (Pd5%)/SBVCN-37 > (Pd3%)/SBVCN-37 (Table S3, ESI†). Therefore the incorporation of an optimum content (3 wt%) of Pd NPs over the surface of SBVCN-37 fine-tuned the charge transfer properties, leading to the lowest Rct value. This can be attributed to the efficient generation and separation of charge carriers (e−–h+) at the electrode electrolyte interface under light illumination, resulting in enhanced photo-electrochemical activity. Due to the smallest Rct value of the (Pd3%)/SBVCN-37 photocatalyst, its transient and LSV responses under light illumination were also the highest (Fig. 7b–d). Additionally, the charge carrier density and the flat-band potentials (Efb) of BiVO4, S-CN, and (Pd3%)/SBVCN-37 were calculated using Mott–Schottky analysis (Fig. 7f) at the constant frequency of 1 kHz and applied potential of 1 V according to the equation 1/C2 = (2/eεεoA2Nd) × [E−Efb−kbT/e],51 where, C is the capacitance of the space charge layers, ε is the dielectric constant of the photocatalyst, e is the electronic charge (1.602 × 10−19 C), εo is the permittivity of the vacuum (8.854 × 10−14 F cm−1), k is the Boltzmann constant, A refers to the geometric surface area of the electrodes, T is the temperature, E is the applied potential, Efb is the flat-band potential, and Nd is the donor charge carrier density. The x-intercept of 1/C2vs. applied potential corresponds to the flat-band potential (Efb) of the photocatalyst. A shift in the negative side of the applied potential refers to the efficient charge carrier separation and migration at the photocatalyst surface.58 All the photocatalysts exhibited a positive slope in their M–S plot, indicating that they are all are n-type semiconductors (Fig. 7f).51
 | ||
| Fig. 7 LSV curves of (a) SBVCN-37 and (b) (Pd3%)/SBVCN-37 in the dark and under light illumination. (c and d) Transient photocurrent (i–t) of all the photocatalysts, (e) EIS profiles (inset shows enlarged profiles at the low frequency region and simulated equivalent circuit), and (f) Mott–Schottky plots of BiVO4, S-CN, and (Pd3%)/SBVCN-37 at the constant frequency of 1000 Hz. | ||
Further, the obtained flat-band potentials (Efb) were used to calculate the conduction band edge potential (ECB) of the tested photocatalysts. For an n-type semiconductor, its flat band potential (Efb) should be lowered by 0.1 V.58 The calculated flat-band potentials are presented in Table S3.† The flat-band potential (Efb) of (Pd3%)/SBVCN-37 was found to be −1.67 eV vs. Ag/AgCl, which is −1.09 vs. NHE using the equation ENHE = EAg/AgCl + 0.059 pH + EAg/AgCl0.51 Therefore, its conduction band edge potential was calculated to be −0.99 eV vs. NHE by considering the reduction in its magnitude by 0.1 V as an n-type semiconductor. The band gap (Eg) energy of (Pd3%)/SBVCN-37 was found to be 2.25 eV, as evident from its DRUV-visible spectrum. Hence, its valance band edge potential was calculated to be +1.26 eV vs. NHE. Therefore, both the valance band and conduction band edge potentials of (Pd3%)/SBVCN-37 fulfil the thermodynamic requirements for hydrogen and oxygen gas production from the water splitting reaction. The detailed photoelectrochemical measurements confirmed that Pd (3 wt%) NPs incorporated in SBVCN-37 modulate the charge carriers separation process, minimize the charge carrier recombination probability, and fulfil the thermodynamic potential requirements for H2/O2 production during the water splitting reaction.
3.2 Catalytic activity evaluation
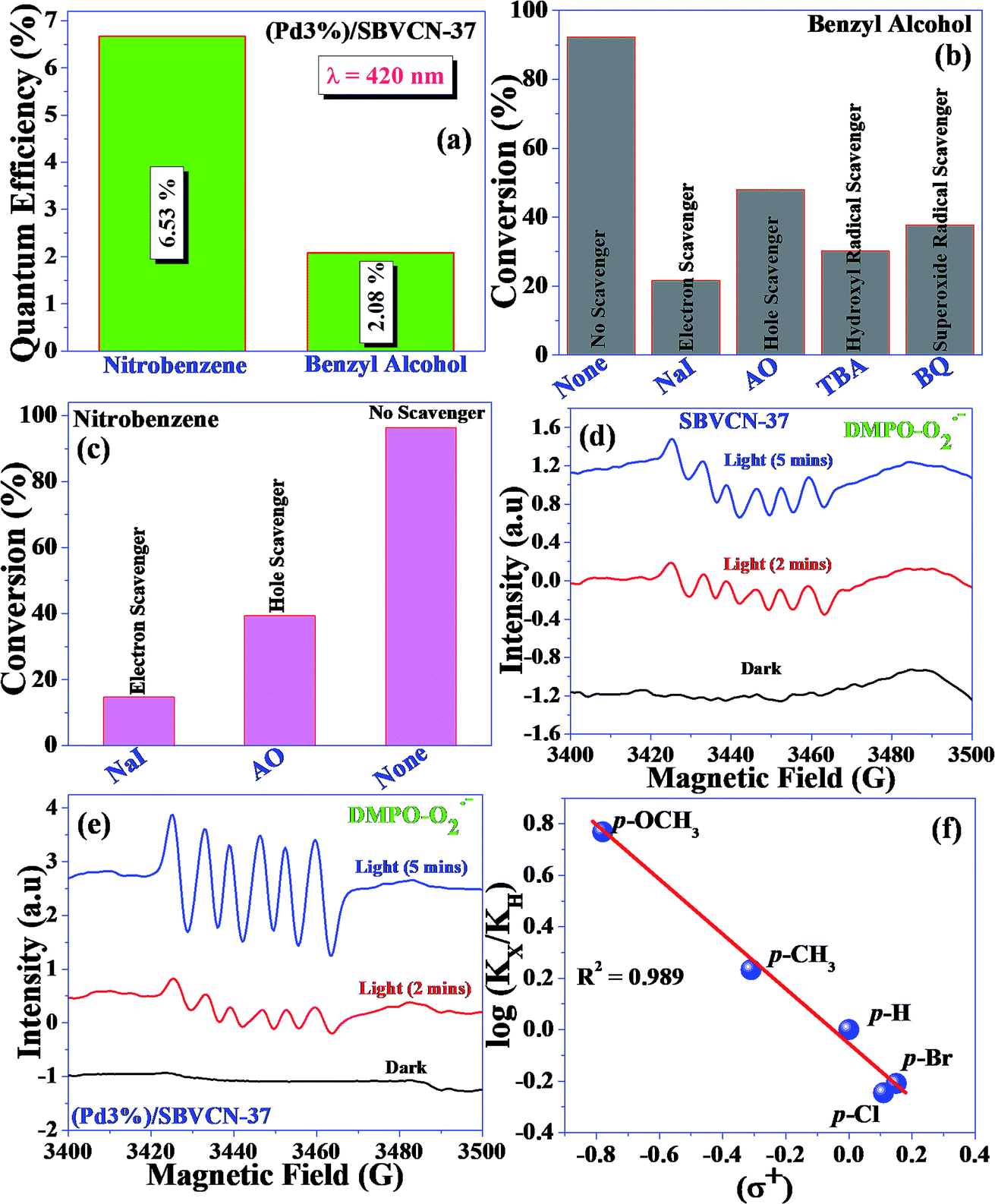 | ||
| Fig. 8 (a) Calculated quantum efficiency of nitrobenzene reduction and benzyl alcohol oxidation at 420 nm for 8 h with (Pd3%)/SBVCN-37 photocatalyst. Influence of scavengers in the (b) benzyl alcohol oxidation and (c) nitrobenzene reduction reactions (NaI = sodium iodide, AO = ammonium oxalate, TBA = tertiarybutyl alcohol, and BQ = benzoquinone). EPR spectra recorded using (d) SBVCN-37 and (e) (Pd3%)/SBVCN-37 for DMPO-O2−˙ in the presence of benzyl alcohol and (f) Hammett plot for benzyl alcohol oxidation. | ||
After the optimization of the two independent steps (oxidation and reduction), the one-pot tandem reaction was performed (Table 1). In this reaction, initially nitrobenzene was reacted with hydrazine hydrate for 8 h. Subsequently, benzyl alcohol was added and the reaction mixture was flushed with O2 and the reaction was performed in an O2 atmosphere (balloon) for 8 h. In the one-pot tandem reaction, after 16 h in the absence of catalyst, no product was formed. In contrast, in the one-pot tandem reaction after 16 h of reaction using SBVCN-37, the product distribution showed the formation of (43.7%) N-benzylideneaniline. However, when the same reaction was performed using (Pd1%)/SBVCN-37 and (Pd3%)/SBVCN-37, the yields of N-benzylideneaniline of 69.3% and 92.1%, respectively, were obtained. Furthermore, in these one-pot tandem reactions, in addition to the desired product N-benzylideneaniline, no side-product was observed. Then, the same reaction was performed in the presence of the (Pd3%)/SBVCN-37 catalyst, but by reversing the sequence of the reaction, i.e. first benzyl alcohol was reacted under an O2 atmosphere followed by the addition of nitrobenzene, and the reaction was performed using hydrazine hydrate. After completion of the reaction, the product distribution showed a somewhat lower yield of N-benzylideneaniline (81.1%) compared to the reaction performed using the same catalyst but in the opposite sequential addition of reactants, as stated earlier. However, in this case, in addition to N-benzylideneaniline, a small amount of phenylhydroxylamine was also observed in the product distribution together with the reactants. It was surprising to note that when the same reaction was performed by reacting nitrobenzene, benzyl alcohol, hydrazine hydrate together in one-pot in the presence of O2, a very low amount of imine was formed. This reaction was not favoured and a large amount of reactants remained after the course of the reaction. Based on these reactions, it can be concluded that the correct sequence of independent steps is required to achieve a high yield of N-benzylideneaniline in the one-pot tandem reaction, mediated by photocatalyst. Furthermore, to investigate the origin of phenylhydroxylamine, benzyl alcohol was reacted with aniline under an O2 atmosphere and the reaction was monitored at regular intervals up to 8 h (Table S5, ESI†). In this case, N-benzylideneaniline was obtained as the selective product (Table S5†). Further, when nitrobenzene was reacted with benzaldehyde in the presence of hydrazine hydrate, imine and phenylhydroxylamine were obtained (Table S6, ESI†). With the progress of the reaction, the phenylhydroxylamine selectivity diminished and the imine selectivity increased. Based on these controlled reactions, it can be concluded that the reduction of nitrobenzene proceeded through the direct phenylhydroxylamine pathway. No azo compound was detected during the reaction, which confirms that the indirect pathway via azo-hydrazo intermediates can be ruled out in this photocatalytic experiment. After 16 h of reaction, the catalyst was separated, washed with acetonitrile and dried in an oven and then reused in the next cycle. No appreciable change in the photocatalytic activity was observed after five cycles, which confirms that (Pd3%)/SBVCN-37 is highly stable for the photocatalytic tandem reaction (Fig. S9a, ESI†).
|
|
||||
|---|---|---|---|---|
| Entry | Catalyst | Sequence of substrate addition | Time | Product distribution |
| a Reaction conditions: substrate 1 (nitrobenzene, 50 μM) + substrate 2 (benzyl alcohol, 50 μM), N2H4 (0.1 mL 0.48 mmol−1), catalyst (25 mg), acetonitrile (12 mL), O2 in balloon (for 2nd step), and Visible light λ > 420 nm. | ||||
| 1 | — | I, II | 8 + 8 | — |
| 2 | SBVCN-37 | I, II | 8 + 8 | N-benzylideneaniline (43.7%), benzyl alcohol (20.3%), nitrobenzene (16.5%), benzyl benzoate (10.1%), and aniline (9.4%) |
| 3 | (Pd1%)/SBVCN-37 | I, II | 8 + 8 | N-benzylideneaniline (69.3%), benzyl alcohol (12.8%), nitrobenzene (11.5%), aniline (3.4%), benzyl benzoate (3.0%) |
| 4 | (Pd3%)/SBVCN-37 | I, II | 8 + 8 | N-benzylideneaniline (92.1%), benzyl alcohol (3.1%), nitro benzene (2.9%), and aniline (1.9%) |
| 5 | (Pd3%)/SBVCN-37 | II, I | 8 + 8 | N-benzylideneaniline (81.1%), benzyl alcohol (8.3%), nitrobenzene (4.2%), aniline (1.7%), and phenylhydroxylamine (4.7%) |
| 6 | (Pd3%)/SBVCN-37 | All together I + II + NH2NH2 | 8 + 8 | N-benzylideneaniline (30.7%), benzyl benzoate (6.5%), benzyl alcohol (32.2%), and nitrobenzene (30.6%) |

To understand the mechanism of the reduction, control experiments were performed as follows. First, the reaction was carried out in the presence of H2 gas (H2 filled balloon) without hydrazine hydrate. In this case, a somewhat low yield of aniline (46.2%) was obtained. In another set of experiments, the reaction medium was flushed with Ar gas and the reaction was performed under an Ar atmosphere using hydrazine hydrate as a reducing agent. In this case, a very low yield of aniline (6.9%) was obtained. These control reactions confirm that during the reduction reaction, hydrazine hydrate produces H2 and N2 in the reaction medium via diimide in the presence of dissolved oxygen. The liberated dissolved H2 is easily accessible to nitrobenzene to form the reduced product aniline. To further confirm the liberation of H2via diimide, one more control experiment was performed. The reaction was performed in the absence of nitrobenzene using excess hydrazine hydrate (0.1 M, 2 mL). After 8 h reaction, 1 mL gas mixture was withdrawn using a Gas Tight Syringe and analyzed using GC coupled with a TCD. H2 gas (7.4 μM) was measured during the photochemical treatment, confirming the liberation of H2 in the presence of catalyst under visible light irradiation. Additionally, the influence of various radicals (electrons and holes) during the nitrobenzene reduction was investigated (Fig. 8c). Both scavengers influenced the aniline yield. The scavenging study suggests that electrons dominate the reduction reaction. However, holes are also involved in the reduction reaction via the decomposition of hydrazine hydrate and production of N2 and H+ ions. The liberated H+ ions are reduced by the electrons and produce H2, which is responsible for the reduction of nitrobenzene.
Based on the independent steps, control reactions, and scavenging studies the following reaction mechanism is proposed. The overall reaction proceeds through the reduction of nitrobenzene, oxidation of benzyl alcohol, followed by the condensation between benzaldehyde and aniline, leading to the formation of N-benzylideneaniline as the desired product. Considering the band alignment of the photocatalyst, the conduction band edge potential (ECB = −0.99 eV vs. NHE) is more negative than the standard reduction potential of O2/O2−˙ (−0.33 eV vs. NHE) (Scheme 1), which results in the spontaneous formation of O2−˙.60,61 Moreover, h+ and HO˙ are also responsible for the oxidation of benzyl alcohol. The generated OH− can be transformed to HO˙ through H2O2 assisted by h+ (H2O2 + e− → HO˙ + OH−; h+ + OH− → HO˙). Again, the (Pd3%)/SBVCN-37 photocatalyst facilitates the selective production of benzaldehyde. The incorporated Pd helps in the electron transfer process and maximizes the charge carrier separation, and thereby helps in the benzyl alcohol oxidation reaction. The reasonable linear relationship between the log(kx/kh) values and the Brown-Okamoto constant (σ+) parameters for the oxidation of benzyl alcohol is consistent with the suggested mechanism (Fig. 8f).62 Similarly, the reduction of nitrobenzene also takes place in the presence of photogenerated electrons and holes. The reduction potential of nitrobenzene to nitroso benzene and aniline is lower than −0.75 V, which confirms the thermodynamic feasibility of this reduction process.63 The reduction of nitrobenzene involves six electrons and six protons.64 Also, the reduction of nitrobenzene is mediated by the oxidation of hydrazine, which involves the release of four electrons and four protons. This is the reason why excess hydrazine was used in the reaction since it can provide more electrons and protons required for the reduction of nitrobenzene. The light-induced holes on the valance band have a potential of +1.26 eV vs. NHE, which is higher than the standard oxidation potential of hydrazine (EN2H4/N20 = 1.16 eV) (Scheme 1).65 Thus, the holes easily oxidize the hydrazine to di-nitrogen together with the liberation of H+ ions. These H+ ions are reduced to H2 by the light-induced electrons on the surface of (Pd3%)/SBVCN-37 (Scheme 1). The in situ-generated H2 assists in the reduction of nitrobenzene to aniline. Further, the incorporated Pd nanoparticles facilitate the reduction process. At the Pd sites, protons are reduced by the generated electrons and chemisorbed H–Pd sites are generated, which selectively reduce nitrobenzene to aniline. The reduction of nitrobenzene to aniline in the presence of molecular hydrogen, as discussed above, further confirms this phenomenon. Finally, the acidic sites present in the catalyst facilitate the condensation reaction between benzaldehyde and aniline to form the desired N-benzylideneaniline.
 | ||
| Scheme 1 Proposed mechanism for the photocatalytic one-pot tandem reaction for the synthesis of imine via the reduction of nitrobenzene, oxidation of benzyl alcohol, followed by a condensation reaction and photocatalytic hydrogen evolution reaction (HER) and oxygen evolution reaction (OER) at the surface of the (Pd3%)/SBVCN-37 heterostructure photocatalyst. | ||
 | ||
| Fig. 9 (a) Photocatalytic HER and (b) OER reactions over the various photocatalysts. Action spectra of (c) HER and (d) OER (inset shows the apparent quantum yields). (e) Time-dependent and (f) weight-dependent HER and OER experiments. (Errors bars represent n = 3 and ±0.1). | ||
| S no. | Catalyst | Co-catalyst | Light source | Reactant sol. | Efficiency | Ref. |
|---|---|---|---|---|---|---|
| 1 | g-C3N4/Ag/BiVO4 | — | 300 W Xe lamp | 20 V% methanol solution | A.Q.Y for HER = 1.23% at 420 nm | 67 |
| 2 | g-C3N4@Ag/BiVO4(040) | — | 350 W Xe lamp | 0.2 M NaIO3 aqueous solution | A.Q.Y: not provided for OER; (O2 evolution rate = 14.68 μmol h−1) | 68 |
| 3 | 5 mol% Cu doped-BiVO4 | 5 wt% CoOx | 300 W Xe lamp (>420 nm) | H2O (0.02 M Na2S2O8 + 0.1 M NaOH) | A.Q.Y for OER = 2.63% at 420 nm | 69 |
| 4 | SiC/QD-BiVO4 | — | 300 W Xe lamp (>420 nm) | 0.04 mol l−1 FeCl3 solution | A.Q.Y for OER = 11.4% at 420 nm | 70 |
| 5 | BiVO4 | — | 300 W Xe lamp (>420 nm) | 0.05 mol L−1 AgNO3 aqueous solution | A.Q.Y for OER = 0.5% at 450 nm | 71 |
| 6 | Ce-doped BiVO4 | 300 W Xe lamp (>420 nm) | 0.015 M AgNO3 aqueous solution | A.Q.Y: not provided for OER; (O2 evolution rate = 90 μmol h−1) | 72 | |
| 7 | Cr-doped BiVO4 | 300 W Xe lamp (>400 nm) | 0.02 mol l−1 AgNO3 with 0.1 g of La2O3 | A.Q.Y: for OER not provided; (O2 evolution rate = 3.5 μmol h−1) | 73 | |
| 8 | g-C3N4 | 2 wt% Pt | 500 W Hg lamp (>400 nm) | 10 V% triethanol amine solutions | A.Q.Y for HER = 4.19% at 420 nm | 74 |
| 9 | ZNS/g-C3N4 | — | 300 W Xe lamp (>420 nm) | N2SO3/Na2S | A.Q.Y: for HER not provided; (H2 evolution rate = 713.6 μmol g−1 h−1) | 75 |
| 10 | g-C3N4 | 1-3 wt% Pt | 300 W Xe lamp (>420 nm) | 10 V% methanol solution | A.Q.Y for HER = 6.8% at 420 nm | 76 |
| 11 | Phosphorene/g-C3N4 | 1-3 wt% Pt | 420 nm, 3 W LED | 88 V% lactic acid | A.Q.Y for HER = 1.2% at 420 nm | 66 |
| 12 | Co(OH)2/g-C3N4 | — | 300 W Xe lamp (>420 nm) | 0.01 mol l−1 AgNO3 with 0.1 g of La2O3 | A.Q.Y for OER not provided; (O2 evolution rate 27.4 μmol h−1) | 77 |
| 13 | S-C3N4/BiVO4 | — | 300 W Xe lamp (>420 nm) | 0.05 M AgNO3 solution | A.Q.Y for OER not provided; (O2 evolution rate = 328 μmol g−1 h −1). | 78 |
| 14 | (Pd3%)/SBVCN-37 | — | 300 W Xe lamp | 10 V% triethanol amine Sol./0.1 M AgNO3 | A.Q.Y for OER (10.22%) & HER (12.72%) at 420 nm | This work |
3.6 Origin and mechanism of enhanced HER and OER activity
When a heterostructure is formed between BiVO4 and S-CN in the weight ratio of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :7, the excited electrons are accumulated on the conduction band of S-CN, leaving behind bare holes at the valance band of the photocatalyst, resulting in efficient charge carrier separation. Further, the conduction band potential of S-CN (−1.20 eV vs. NHE) is more negative than the conduction band potentials of BiVO4 (−0.33 eV vs. NHE).41–43,48 Similarly, the valance band potential of BiVO4 (+2.85 eV vs. NHE) is more positive than the valance band potential of S-CN (+1.52 eV vs. NHE).41,48,78 Therefore, according to the principle of electronic charge carrier transportation, the conduction band electrons will flow toward the CB of BiVO4 and in the case of holes, the reverse sequence occurs.
:7, the excited electrons are accumulated on the conduction band of S-CN, leaving behind bare holes at the valance band of the photocatalyst, resulting in efficient charge carrier separation. Further, the conduction band potential of S-CN (−1.20 eV vs. NHE) is more negative than the conduction band potentials of BiVO4 (−0.33 eV vs. NHE).41–43,48 Similarly, the valance band potential of BiVO4 (+2.85 eV vs. NHE) is more positive than the valance band potential of S-CN (+1.52 eV vs. NHE).41,48,78 Therefore, according to the principle of electronic charge carrier transportation, the conduction band electrons will flow toward the CB of BiVO4 and in the case of holes, the reverse sequence occurs.
The ECB and EVB potentials of SBVCN-37 are high enough to exhibit HER and OER reactions, respectively. Therefore, due to the proper band alignments, the synthesized heterostructure SBVCN-37 exhibited high photocatalytic activity for the HER and OER, separately. When visible light falls on the surface of the semiconductor, the conduction band electrons and valance band holes are immediately separated, resulting in the formation of a Mott–Schottky junction at the interface between the Pd NPs and SBVCN-37. The Fermi level of the Pd NPs prevails between the CBmin and VBmax of S-BVCN-37, resulting in the formation of an efficient photocatalytic system. Furthermore, the Mott–Schottky analysis from the PEC measurements revealed that the conduction band edge potential of (Pd3%)/SBVCN-37 is −0.99 eV vs. NHE, while its valance band edge potential is +1.26 eV vs. NHE (Scheme 1). In the next step, the conduction band electrons of SBVCN-37 shift to the Fermi level of the Pd NPs, lying just −0.1 eV lower than that of the conduction band of the (Pd3%)/SBVCN-37 photocatalyst (Scheme 1), leading to an increment in the electron density.58 This type of electronic relay efficiently separates the holes from the electrons, and therefore noticeable H2 gas production activity was observed with the (Pd3%)/SBVCN-37 photocatalyst. The accumulated electrons on the active sites of (Pd3%)/SBVCN-37 assisted in the reduction of the adsorbed H+ ions to produce H2 gas. However, when the amount of Pd NPs increased to 5 wt%, a decrease in the H2 gas production activity was observed. The large content of Pd NPs reduced the visible light absorption and SPR phenomenon, which are dependent on the inter-particle interaction and local environment.53,54 Therefore, the excessive amount of Pd NPs interfered in the electronic relay to the Fermi level of the Pd NPs, resulting in a decrease in HER activity. In the case of O2 production, the Pd NPs have no direct influence, but they help to harvest a larger number of photons and also lower the band gap. Further, upon Pd NP loading, the valance band edge becomes closer to the standard potential (EVB = 1.26 eV vs. NHE) for the water oxidation reaction (EH2O/O2 = 1.23 eV vs. NHE) (Scheme 1).41 Therefore, the kinetics of the water oxidation is smoother over the (Pd3%)/SBVCN-37 photocatalyst. Since, the electrons are gathered at the Fermi level of the Pd NPs, the prominent separation of holes from the electrons takes place on the valance band maximum of the (Pd3%)/SBVCN-37 photocatalyst. This phenomenon is also consistent with the TRPL measurements. Thus, the Pd NPs indirectly modulate the separation of holes on the catalyst junction, leading to higher water oxidation to produce O2 gas. In the absence of Pd NPs, this charge separation is less prominent, which is responsible for the inferior OER activity by the SVBCN-37 photocatalyst. Again, the photoelectrochemical (PEC) measurements reveal that among the investigated photocatalysts, (Pd3%)/SBVCN-37 produces the highest transient photocurrent (0.12 mA cm−2), the lowest charge transfer resistance (27.4 Ω) from the EIS measurements, and the highest photo-reversibility, as evident from the transient photocurrent measurements. Further, steady state PL spectroscopy results indicates that it has the most efficient charge carrier separation and migration among the investigated photocatalysts. Moreover, the TRPL spectrum furnishes evidence that it has the highest charge carrier dynamics (k = 0.19 (ns)−1) and the lowest charge carrier average lifetime (τ = 5.18 ns). Thus, the TRPL analysis further confirms that effective separation and promotion of electrons–holes take place at the plasmonic (Pd3%)/SBVCN-37 reactive surface. Notably, the Pd NPs deposited over the surface of the SBVCN-37 photocatalyst act as a mediator through their SPR effect, which promptly helps in the charge carrier separation and results in excellent water splitting activity. Thus, considering all these photo-physical parameters, it can be concluded that the optimum content of Pd in SBVCN-37, lowering of the band gap, and the highest charge carrier separation are some of the prime reasons for the impressive HER and OER activity exhibited by the (Pd3%)/SBVCN-37 heterostructure photocatalyst.
4. Conclusions
In summary, a Pd nanoparticle-decorated SBVCN-37 heterostructure photocatalyst was synthesized. Pristine g-C3N4 was doped with –SO3H groups through H2SO4 treatment and coupled with BiVO4 in a 7:3 weight ratio, resulting in the formation of the SBVCN-37 heterostructure photocatalyst. The developed photocatalyst was used for a one-pot, three-step tandem reaction for the synthesis of N-benzylideneaniline. The photocatalytic reaction revealed that the one-pot, three-step tandem reaction involving nitrobenzene reduction, benzyl alcohol oxidation, followed by condensation sequence steps was suitable to obtain the maximum yield of the desired N-benzylideneaniline. The EPR and scavenging experiments revealed that the light-induced electrons from the conduction band (ECB = −0.99 eV vs. NHE) easily reduced O2 to O2−˙ (−0.33 eV vs. NHE) due to its favourable redox potential, which was found to be the main influencing reactive oxygen species (ROS) in this photocatalytic tandem reaction. Inevitably, due to the proper band potential alignment on (Pd3%)/SBVCN-37, the separated holes on its valance band oxidized N2H4 and produced H+ ions, which served as the reducing agent for the reduction of nitrobenzene to aniline. Moreover, the developed photocatalyst with an optimum content of Pd nanoparticles, (Pd3%)/SBVCN-37, exhibited excellent activity in the independent HER and OER reactions. In this photocatalytic system, the Pd nanoparticles served three important purposes. First, they avoided the requirement of an additional Pt co-catalyst, which is necessary for H2 evolution reaction over g-C3N4. Second, they lowered the valance band edge potential (EVB = 1.26 eV vs. NHE) close to the standard water oxidation potential (EH2O/O20 = 1.23 eV vs. NHE), and thereby the sluggish water oxidation kinetics of pristine BiVO4 was overcome by this (Pd3%)/SBVCN-37 photocatalyst, as evident from the Mott–Schottky analysis. Third, the photocatalytic activity for independent HER and OER was enhanced through efficient charge carrier separation due to the SPR effect of the Pd nanoparticles on the surface of SBVCN-37. The obtained catalytic activity was consistent with the photoelectrochemical measurements and time-resolved photoluminescence studies, which confirmed that the efficient charge carrier separation with low average lifetime in the excited state, suitable alignment of the band edge potential, and the lowest charge transfer resistance were the prime reasons behind the excellent photocatalytic activity of (Pd3%)/SBVCN-37. This constructive and conceptual investigation will surely promote some new directions in the development of multifunctional photocatalysts.
Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
The financial support provided by Nano Mission, DST, New Delhi through the grant (SR/NM-NS-1054/2015) is gratefully acknowledged. SS sincerely thanks MHRD, New Delhi, for awarding him with a Senior Research Fellowship. We are grateful to DST-FIST funded XPS facility at Department of Physics, IIT Kharagpur for XPS analysis. TRPL analysis facility availed from CIF, IIT Guwahati is also gratefully acknowledged. Authors are also thankful to Dr Anil Kumar Vardaman, IICT-Hyderabad and Dr Murali Banavoth, School of Chemistry, University of Hyderabad for helping in the EPR analysis. Authors also acknowledged the FESEM facility from CRF, IIT Kharagpur. Credits are due to Mr Abhinav Kumar, Department of Chemistry, IIT Ropar for helping in BET and TPD analysis.Notes and references
- M. J. Climent, A. Corma and S. Iborra, RSC Adv., 2012, 2, 16–58 RSC.
- N. R. Shiju, A. H. Alberts, S. Khalid, D. R. Brown and G. Rothenberg, Angew. Chem., Int. Ed., 2011, 50, 9615–9619 CrossRef CAS PubMed.
- X. Lang, X. Chen and J. Zhao, Chem. Soc. Rev., 2014, 43, 473–486 RSC.
- H. Wang, L. Zhang, Z. Chen, J. Hu, S. Li, Z. Wang, J. Liu and X. Wang, Chem. Soc. Rev., 2014, 43, 5234–5244 RSC.
- S. Chen, T. Takata and K. Domen, Nat. Rev. Mater., 2017, 2, 17050 CrossRef CAS.
- K. Li, B. Peng and T. Peng, ACS Catal., 2016, 6, 7485–7527 CrossRef CAS.
- Y. Nosaka and A. Y. Nosaka, Chem. Rev., 2017, 117, 11302–11336 CrossRef CAS PubMed.
- M. Oelgemöller, Chem. Rev., 2016, 116, 9664–9682 CrossRef PubMed.
- N. Zhang, Y. Zhang, M.-Q. Yang, Z.-R. Tang and Y.-J. Xu, J. Catal., 2013, 299, 210–221 CrossRef CAS.
- M.-Q. Yang, C. Han, N. Zhang and Y.-J. Xu, Nanoscale, 2015, 7, 18062–18070 RSC.
- C. Han, M.-Q. Yang, N. Zhang and Y.-J. Xu, J. Mater. Chem. A, 2014, 2, 19156–19166 RSC.
- N. Zhang, Y. Zhang, X. Pan, M.-Q. Yang and Y.-J. Xu, J. Phys. Chem. C, 2012, 116, 18023–18031 CrossRef CAS.
- N. Zhang, M.-Q. Yang, Z.-R. Tang and Y.-J. Xu, ACS Nano, 2014, 8, 623–633 CrossRef CAS PubMed.
- X. Li, N. Zhang and Y.-J. Xu, ChemCatChem, 2015, 7, 2047–2054 CrossRef CAS.
- W.-J. Ong, L.-L. Tan, Y. H. Ng, S.-T. Yong and S.-P. Chai, Chem. Rev., 2016, 116, 7159–7329 CrossRef CAS PubMed.
- R. W. Layer, Chem. Rev., 1963, 63, 489–510 CrossRef CAS.
- M. T. Schümperli, C. Hammond and I. Hermans, ACS Catal., 2012, 2, 1108–1117 CrossRef.
- M. M. Sprung, Chem. Rev., 1940, 26, 297–338 CrossRef CAS.
- Y. Wu, X. Ye, S. Zhang, S. Meng, X. Fu, X. Wang, X. Zhang and S. Chen, J. Catal., 2018, 359, 151–160 CrossRef CAS.
- Z.-R. Tang, Q. Yu and Y.-J. Xu, RSC Adv., 2014, 4, 58448–58452 RSC.
- N. Zhang, M.-Q. Yang, S. Liu, Y. Sun and Y.-J. Xu, Chem. Rev., 2015, 115, 10307–10377 CrossRef CAS PubMed.
- C. Han, N. Zhang and Y.-J. Xu, Nano Today, 2016, 11, 351–372 CrossRef CAS.
- N. Zhang, M.-Q. Yang, Z.-R. Tang and Y.-J. Xu, ACS Nano, 2014, 8, 623–633 CrossRef CAS PubMed.
- L. Yuan, B. Weng, J. C. Colmenares, Y. Sun and Y.-J. Xu, Small, 2017, 13, 1702253 CrossRef PubMed.
- M. J. Pavan, H. Fridman, G. Segalovich, A. I. Shames, N. G. Lemcoff and T. Mokari, ChemCatChem, 2018, 10, 2541–2545 CrossRef CAS.
- T. Jiang, C. Jia, L. Zhang, S. He, Y. Sang, H. Li, Y. Li, X. Xu and H. Liu, Nanoscale, 2015, 7, 209–217 RSC.
- F. Su, S. C. Mathew, G. Lipner, X. Fu, M. Antonietti, S. Blechert and X. Wang, J. Am. Chem. Soc., 2010, 132, 16299–16301 CrossRef CAS PubMed.
- R. Negishi, S. Naya and H. Tada, J. Phys. Chem. C, 2015, 119, 11771–11776 CrossRef CAS.
- S. Higashimoto, N. Suetsugu, M. Azuma, H. Ohue and Y. Sakata, J. Catal., 2010, 274, 76–83 CrossRef CAS.
- T. Aditya, A. Pal and T. Pal, Chem. Commun., 2015, 51, 9410–9431 RSC.
- R. A. W. Johnstone and A. H. Wilby, Chem. Rev., 1985, 85, 129–170 CrossRef CAS.
- R. Zhang, L. Yang, X. Huang, T. Chen, F. Qu, Z. Liu, G. Du, A. M. Asiri and X. Sun, J. Mater. Chem. A, 2017, 5, 12086–12090 RSC.
- R. Zhang, Y. Fang, T. Chen, F. Qu, Z. Liu, G. Du, A. M. Asiri, T. Gao and X. Sun, ACS Sustainable Chem. Eng., 2017, 5, 7502–7506 CrossRef CAS.
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76–80 CrossRef CAS PubMed.
- S. Zhang, J. H. Sun, K. Maeda, K. Domen, P. Liu, M. Antonietti, X. J. Fu and X. Wang, Energy Environ. Sci., 2011, 4, 675–678 RSC.
- D. Masih, Y. Ma and S. Rohani, Appl. Catal., B, 2017, 206, 556–588 CrossRef CAS.
- T. W. Kim and K.-S. Choi, Science, 2014, 343, 990–994 CrossRef CAS PubMed.
- H. L. Tan, R. Amal and Y. H. Ng, J. Mater. Chem. A, 2017, 5, 16498–16521 RSC.
- B. Lamm, B. Trzesniewski, H. Döscher, W. A. Smith and M. Stefik, ACS Energy Lett., 2018, 3, 112–124 CrossRef CAS.
- Z.-F. Huang, L. Pan, J.-J. Zou, X. Zhang and L. Wang, Nanoscale, 2014, 6, 14044–14063 RSC.
- B. Zhang, G. Dong, L. Wang, Y. Zhang, Y. Ding and Y. Bi, Catal. Sci. Technol., 2017, 7, 4971–4976 RSC.
- S. Kim, Y. Yu, S. Y. Jeong, M. G. Lee, H. W. Jeong, Y. M. Kwon, J. M. Baik, H. Park, H. W. Jang and S. Lee, Catal. Sci. Technol., 2018, 8, 3759–3766 RSC.
- S. Obregón and G. Colón, Catal. Sci. Technol., 2014, 4, 2042–2050 RSC.
- S. Gu, W. Li, F. Wang, H. Li and H. Zhou, Catal. Sci. Technol., 2016, 6, 1870–1881 RSC.
- J. Song, M. J. Seo, T. H. Lee, Y.-R. Jo, J. Lee, T. V. Kim, S. Y. Kim, S.-M. Kim, S. Y. Jeong, H. An, S. Kim, B. H. Lee, D. Lee, H. W. Jang, B. J. Kim and S. Lee, ACS Catal., 2018, 8, 5952–5962 CrossRef CAS.
- S. Samanta and R. Srivastava, Sustainable Energy Fuels, 2017, 1, 1390–1404 RSC.
- S. Samanta, S. Martha and K. Parida, ChemCatChem, 2014, 6, 1453–1462 CAS.
- S. Samanta, S. Khilari, D. Pradhan and R. Srivastava, ACS Sustainable Chem. Eng., 2017, 5, 2562–2577 CrossRef CAS.
- S. Patnaik, S. Martha, G. Madras and K. Parida, Phys. Chem. Chem. Phys., 2016, 18, 28502–28514 RSC.
- M. N. Patil, M. A. Bhosale and B. M. Bhanage, RSC Adv., 2015, 5, 86529–86535 RSC.
- S. Samanta, S. Khilari and R. Srivastava, ACS Appl. Nano Mater., 2018, 1, 423–436 Search PubMed.
- S. Samanta and R. Srivastava, Appl. Catal., B, 2017, 218, 621–636 CrossRef CAS.
- S. Cao, H. Li, Y. Li, B. Zhu and J. Yu, ACS Sustainable Chem. Eng., 2018, 6, 6478–6487 CrossRef CAS.
- Y. Xiong, J. Chen, B. Wiley, Y. Xia, Y. Yin and Z.-Y. Li, Nano Lett., 2005, 5, 1237–1242 CrossRef CAS PubMed.
- S. Cao, Q. Huang, B. Zhu and J. Yu, J. Power Sources, 2017, 351, 151–159 CrossRef CAS.
- M. S. Ansari, A. Banik and M. Quershi, Carbon, 2017, 121, 90–105 CrossRef CAS.
- Y. Zeng, X. Liu, C. Liu, L. Wang, Y. Xia, S. Zhang, S. Luo and Y. Pei, Appl. Catal., B, 2018, 224, 1–9 CrossRef CAS.
- A. Verma, A. Srivastav, S. A. Khan, V. R. Satsangi, R. Shrivastav, D. Avasthi and D. Dass, Phys. Chem. Chem. Phys., 2017, 19, 15039–15049 RSC.
- X. Xiao, J. Jiang and L. Zhang, Appl. Catal., B, 2013, 142–143, 487–493 CrossRef CAS.
- M. J. Islam, A. D. Reddy, J. Choi and T. K. Kim, RSC Adv., 2016, 6, 19341–19350 RSC.
- F. Dong, X. Xiao, G. Jiang, Y. Zhang, W. Cui and J. Ma, Phys. Chem. Chem. Phys., 2015, 17, 16058–16066 RSC.
- S. Yurdakal, G. Palmisano, V. Loddo, O. Alagoz, V. Augugliaro and L. Palmisano, Green Chem., 2009, 11, 510–516 RSC.
- S. Zhang, W. Huang, X. Fu, X. Zheng, X. Meng, X. Ye and S. Chen, Appl. Catal., B, 2018, 233, 1–10 CrossRef CAS.
- Y. P. Bhoi and B. G. Mishra, Chem. Eng. J., 2017, 316, 70–81 CrossRef CAS.
- Q. Yi and W. Yu, J. Electroanal. Chem., 2009, 633, 159–164 CrossRef CAS.
- J. Ran, W. Guo, H. Wang, B. Zhu, J. Yu and S.-Z. Qiao, Adv. Mater., 2018, 30, 1800128 CrossRef PubMed.
- M. Song, Y. Wu, X. Wang, M. Liu and Y. Su, J. Colloid Interface Sci., 2018, 529, 375–384 CrossRef CAS PubMed.
- M. Ou, S. Wan, Q. Zhong, S. Zhang, Y. Song, L. Guo, W. Cai and Y. Xu, Appl. Catal., B, 2018, 221, 97–107 CrossRef CAS.
- B. He, Z. Li, D. Zhao, H. Liu, Y. Zhong, J. Ning, Z. Zhang, Y. Wang and Y. Hu, ACS Appl. Nano Mater., 2018, 1, 2589–2599 CrossRef CAS.
- D. Wang, L. Huang, Z. Guo, S. Jin, C. Liu, W. Wang and W. Yuan, ACS Appl. Nano Mater., 2018, 1, 4594–4601 CrossRef CAS.
- A. Kudo, K. Ueda, H. Kato and I. Mikami, Catal. Lett., 1998, 53, 229–230 CrossRef CAS.
- Z. Jiang, Y. Liu, T. Jing, B. Huang, X. Zhang, X. Qin, Y. Dai and M. H. Whangbo, J. Phys. Chem. C, 2016, 120, 2058–2063 CrossRef CAS.
- K. Okuno, H. Kato, J. Jhon, M. Vequizo, A. Yamakata, H. Kobayashi, M. Kobayashi and M. Kakihana, RSC Adv., 2018, 8, 38140–38145 RSC.
- Y. Zhang, J. Gao and J. Chen, J. Colloid Interface Sci., 2019, 535, 331–340 CrossRef CAS PubMed.
- G. Zhang, S. Zang and X. Wang, ACS Catal., 2015, 5, 941–947 CrossRef CAS.
- X. Hao, J. Zhou, Z. Cui, Y. Wang, Y. Wang and Z. Zou, Appl. Catal., B, 2018, 221, 97–107 CrossRef.
- L. Lina, W. Rena, C. Wang, A. M. Asiric, J. Zhang and X. Wang, Appl. Catal., B, 2018, 231, 234–241 CrossRef.
- H. J. Kong, D. Y. Won, J. Kim and S. I. Woo, Chem. Mater., 2016, 28, 1318–1324 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Details of the materials, catalyst preparation, materials characterization, electrode fabrication, details for the calculation of band gap, charge carrier lifetime, FTIR, NH3-TPD analysis, and optimization of the catalytic reactions. Tables S1 to S4 provide elemental analysis data, resistance behaviour, comparison of the electrocatalytic activities and rate constant determined in this study, respectively. Fig. S1–S11 include photographs of the photocatalytic reaction set-up, XRD patterns, FE-SEM images, HRTEM, EDAX analysis, HAADF, STEM, NH3-TPD, Tau plots, LSV profiles, wavelength-dependent HER/OER, recyclability data and catalyst stability characterization (XRD, FESEM, and EIS) data. See DOI: 10.1039/c8na00372f |
| This journal is © The Royal Society of Chemistry 2019 |