
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Direct covalent grafting of an organic radical core on gold and silver†
M. R. Ajayakumara,
I. Alcónb,
S. T. Bromley bc,
J. Veciana*a,
C. Roviraa and
M. Mas-Torrent*a
bc,
J. Veciana*a,
C. Roviraa and
M. Mas-Torrent*a
aInstitut de Ciència de Materials de Barcelona (ICMAB-CSIC), CIBER-BBN, Campus UAB, 08193 Bellaterra, Spain. E-mail: vecianaj@icmab.es; mmas@icmab.es
bDepartament de Ciència de Materials i Física Química, Institut de Química Teòrica i Computacional (IQTCUB), Universitat de Barcelona, E-08028 Barcelona, Spain
cInstitució Catalana de Recerca i Estudis Avançats (ICREA), E-08010 Barcelona, Spain
First published on 5th April 2017
Abstract
The functionalisation of surfaces with organic radicals, such as perchlorotriphenylmethyl (PTM) radicals or tris(2,4,6-trichloro-phenyl)methyl (TTM) radicals, is appealing for the development of molecular spintronic devices. Conventionally, organic radicals are chemisorbed to metal substrates by using long alkyl or aromatic spacers resulting in a weak spin–electron coupling between the radical and the substrate. To circumvent this problem, here we have employed a new design strategy for the fabrication of radical self-assembled monolayers (r-SAMs). This newly designed radical–anchor (R–A) molecule, a TTM based radical disulfide (1), can be easily synthesized and it was here characterized by electron spin resonance (ESR), cyclic voltammetry (CV) and superconducting quantum interference device magnetometry (SQUID). We have succeeded in fabricating TTM based r-SAMs by using thiolate bonds (Au–S and Ag–S) where the TTM cores are only one-atom distance from the metal surface for the first time. The resultant robust 1/Au and 1/Ag r-SAMs were well characterized, and the electrochemical and the magnetic properties were unambiguously confirmed, proving the persistence of the molecular spin.
Introduction
Metal and metal oxide surfaces decorated with organic functional molecules1–4 have widespread applications in fields such as organic electronics,5–8 sensors9,10 and biological arrays,11–14 among others. Specifically, in the field of molecular electronics, molecules grafted on surfaces are identified as one of the unique building blocks for device miniaturization since ultimately the active unit would consist of a single molecule. The question that arises then is if the molecules at the surface, which experience a very different environment compared to those in the bulk, preserve the same properties. It is noteworthy that the extent of the electronic coupling between the π-molecule and a metallic surface depends on the structure and electronic nature of the π-molecule and the Fermi level of the metal.15 Importantly, the π-conjugated molecule has to be positioned close to the metal surface to establish electronic interaction although its properties can be then altered.Amongst these materials, covalently self-assembled monolayers (SAMs)16–19 gained tremendous interest due to their ease of fabrication process and effectiveness to integrate organic molecules on inorganic (metallic) substrates. Importantly, organic radicals based SAMs20 (r-SAMs) have emerged as potential building units for spintronic materials, which could be applied for downscaling the memory devices.21,22 The general strategy for achieving r-SAMs comprises the utilization of chemisorption for anchoring rationally designed functional organic radicals, i.e. radical–linker–anchor (R–L–A) molecules, on metal or metal oxide substrates (Scheme 1). In this regard, our group has fabricated several r-SAMs23–26 based on perchlorotriphenylmethyl radical (PTM, Scheme 2), a highly persistent class of organic radicals, on ITO and Au. Although these r-SAMs are promising building blocks for bottom-up spintronics, the synthesis of appropriate PTM precursor is often challenging due to the complex multi-step synthetic protocols27,28 for integrating the PTM and the linker–anchor (L–A) functionality. In addition, the alkyl or aromatic linker can drastically reduce the spin–electron coupling between the radical entity and the metallic substrate, which is not acceptable for certain vital phenomenon such as Kondo resonance.29–32 Moreover, the integration of long linkers can cause conformational variations that may end up in several molecular self-assembly anomalies. Therefore, the utilization of a radical–anchor (R–A) type molecule (i.e., without a linker L unit) to be chemically bonded to a metal surface has fundamental importance as a model compound for the novel generation of r-SAMs (Scheme 1), even though, to the best of our knowledge, there are no examples of such kind. Additionally, the radical character persistence of a covalently grafted PTM or similar radicals at one-atom distance from the metal surface remains an open question.
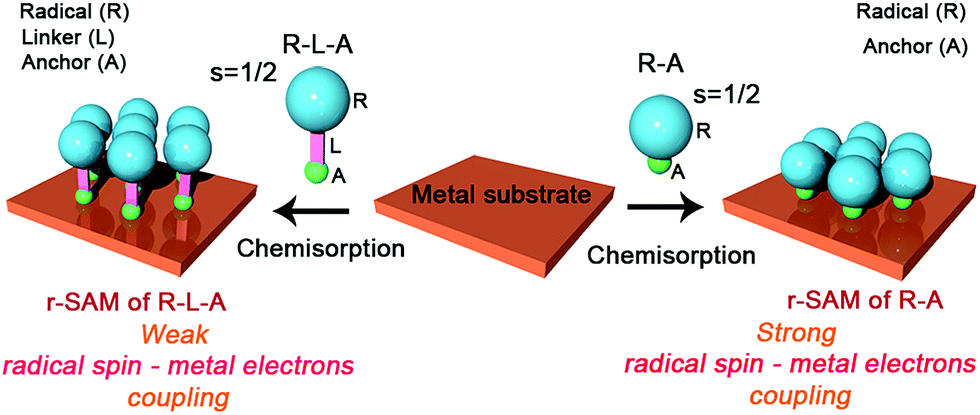 | ||
| Scheme 1 Radical–linker–anchor (R–L–A) and radical–anchor (R–A) based molecules and their chemisorption processes on a metal substrate. | ||
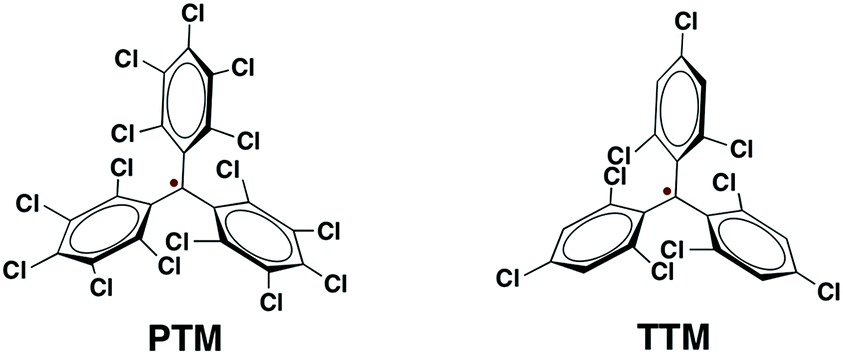 | ||
| Scheme 2 Molecular structures of PTM and TTM radicals. | ||
Herein, we describe a simple one-pot reaction strategy for the synthesis of a novel tris(2,4,6-trichloro-phenyl)methyl (TTM,33 Scheme 2) radical derivative (1), a PTM analogue, of R–A type. The diradical nature of 1 was evaluated by means of electron spin resonance (ESR), cyclic voltammetry (CV) and superconducting quantum interference device magnetometry (SQUID). The r-SAMs of 1 were generated on Au and Ag by solution based chemisorption reactions. The S atoms acted as the anchors that covalently graft the radical units to Au and Ag by forming Au–S and Ag–S bonds, respectively. The yielded robust r-SAMs were well characterized with X-ray photoelectron spectroscopy (XPS), UV photoelectron spectroscopy (UPS), CV, and ESR. Remarkably, it was clearly elucidated that the radical character of the molecules is persistent even when they are closely chemically bonded to a metal surface.
Results and discussion
Among organic radicals, the PTMs are potential candidates for the fabrication of r-SAMs due to their synthetic tailorability to attach suitable L–A functionalities.21,23,25,34 However, for this proposed R–A chemisorption pathway, we excluded PTM derivatives due to the steric hindrance of the meta-Cl atoms upon the r-SAM formation. Hence, we have chosen TTM as an ideal scaffold for establishing this strategy. We decided to utilize Au and Ag substrates since their promising applicability due to their compatibility with the well-established lithographic techniques. In addition, Au–S and Ag–S are the most widely utilized chemical bonds for SAM generation.35To pursue R–A pathway of r-SAM generation on Au and Ag, we synthesized a novel molecule 1 by replacing a Cl with S (Scheme 3). To achieve this molecule, we carried out a simple one-pot reaction employing 3,5-dichloro-4-[bis-(2,4,6-trichlorophenyl)methylen]cyclohexa-2,5-dien-1-one (2)36 as precursor. Compound 2 in dry toluene was refluxed with Lawesson's reagent for 3 h under argon and light exclusion to incorporate the S anchor to the phenyl moiety. The obtained 3 readily turned to stable diradical 1 which was isolated as a dark-brown solid by column chromatography in neutral alumina in 18% yield (see ESI†). PTM and TTM radicals are typically synthesized via the precursor analogue with the central carbon atom hydrogenated. On the contrary, in this reaction, we could avoid the usual, but difficult, deprotonation (using base such as tetrabutylammonium hydroxide) and oxidation (with oxidizing agents such as p-chloranil or AgNO3) steps that are involved in the conventional PTM/TTM radical formation from the respective hydrogenated precursor. Hence reaction–purification processes were significantly simplified.
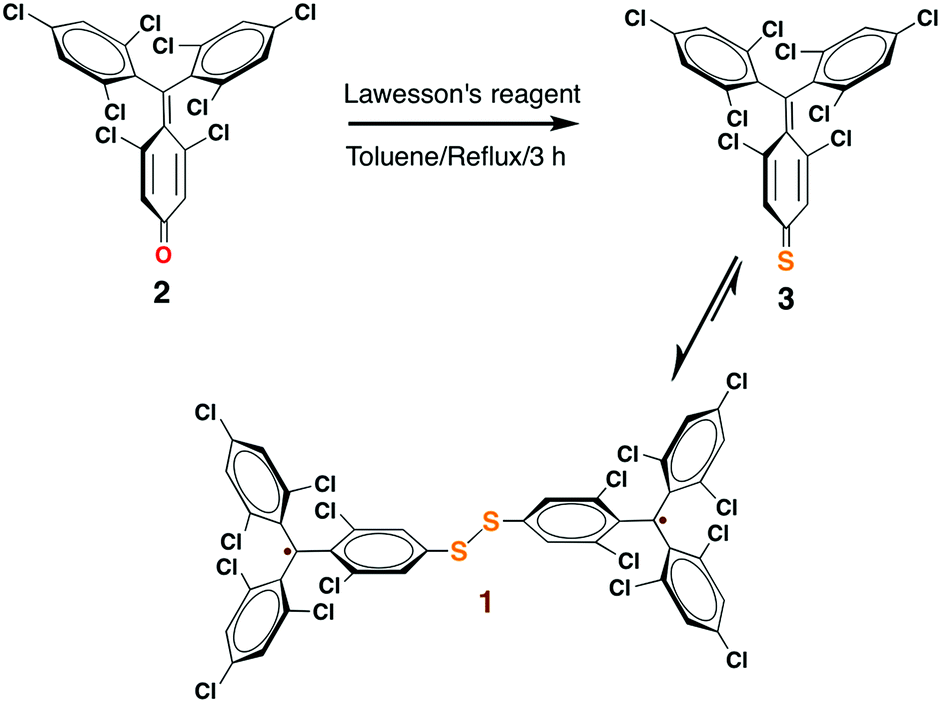 | ||
| Scheme 3 One-pot reaction strategy for the synthesis of 1. | ||
The formation of 1 was unequivocally confirmed by mass and infrared (IR) spectroscopy (Fig. S1 and S2 in the ESI†). Moreover, XPS (Fig. S3†) were employed for understanding the elemental composition of 1. UV-vis spectra of 1 (1.3 × 10−5 M) in CH2Cl2 showed the characteristic PTM/TTM radical bands at 375 (ε = 43![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 700 M−1 cm−1), 394 (29420 M−1 cm−1), 540 (2020 M−1 cm−1) and 572 (2030 M−1 cm−1) nm, which are slightly red shifted compared to the model compound TTM (Fig. S4a†). Moreover, 1 shows very strong fluorescence and the λmax (615 nm) is 49 nm bathochromically shifted compared to that of TTM radical (Fig. S4b†).
700 M−1 cm−1), 394 (29420 M−1 cm−1), 540 (2020 M−1 cm−1) and 572 (2030 M−1 cm−1) nm, which are slightly red shifted compared to the model compound TTM (Fig. S4a†). Moreover, 1 shows very strong fluorescence and the λmax (615 nm) is 49 nm bathochromically shifted compared to that of TTM radical (Fig. S4b†).
To get a detailed insight of the paramagnetic nature, the diradical 1 was examined by X-band EPR spectroscopy. The powder sample of 1 displayed EPR signal with no definite fine-structure with a g value of 2.0032 and a linewidth of 6.1 G at 300 K (Fig. S5†). Temperature dependent EPR measurements were conducted for 1 (5 × 10−4 M) in CH2Cl2/toluene (1:1 v/v). At 300 K, 1 showed a signal at g value of 2.0037 with a hyperfine splitting due to the coupling of the unpaired electron with 1H and 13C (Fig. S6†). All these spectra are consistent with that typically obtained for TTM derivatives.23,34 On cooling to 220 K, two superimposed signals are observed. A broad band and a narrower line with well-defined hyperfine structure of 7 lines corresponding to the coupling of 6 meta-1Hs together with satellite lines arising from the coupling of 13Cs are found (Fig. S6†). The simulation of this latter signal give hyperfine coupling constants values of aH = 1.2 G, and aC = 13.2 G and 11.0 G, which are typical values for monoradical derivatives.37 Thus, it is assumed that the broad band comes from the diradical species 1, but some monoradical impurity coexists in the sample. In frozen CH2Cl2/toluene (1:1 v/v) solution, the line corresponding to the monoradical impurity appears as a central broad signal, and large changes are observed in the diradical band due to strong electron–electron dipolar interactions (Fig. 1a). This band can be simulated giving a zero-field splitting parameter D of 34 G. Under these conditions also a weak peak (|ΔmS| = 2, forbidden transition) is observed at the half-field region attributed to the thermally accessible triplet state (Fig. 1a), which confirms the diradical nature of 1.38,39
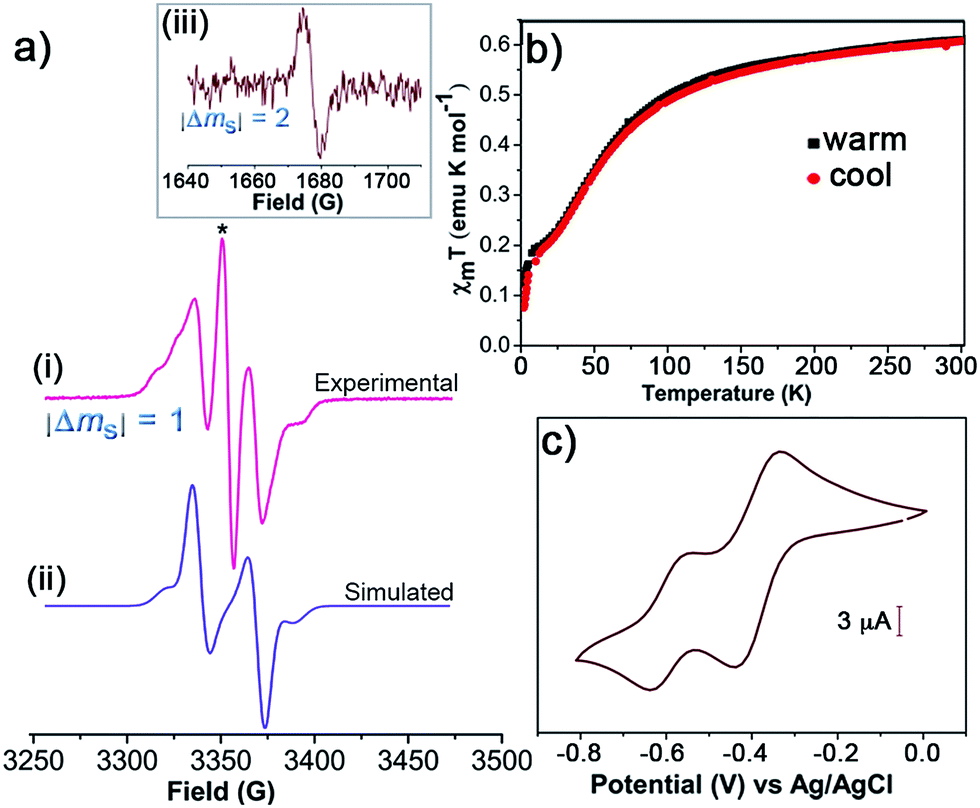 | ||
| Fig. 1 (a) The experimental (i) and simulated (ii) EPR spectra of 1 (5 × 10−4 M) in frozen CH2Cl2/toluene (1:1 v/v) at 120 K. The line marked with the symbol * corresponds to the monoradical impurity. (iii) EPR signal corresponding to the half-field transition (|ΔmS| = 2). (b) Plots of temperature-dependent χmT for 1 (solid) at 5 kOe from 2 to 300 K for the warming and cooling modes. (c) CV of 1 (5 × 10−4 M) in toluene/MeCN (1:1 v/v) with 0.1 M n-Bu4NPF6 (vs. Ag/AgCl) at 100 mV s−1 using Pt wires as working and counter electrodes. | ||
Furthermore, the magnetic character of 1 was monitored by using SQUID magnetometer at the temperature range of 2–300 K. Plots of the product of molar magnetic susceptibility (χm) and temperature (T) versus T in the warming and cooling modes at set magnetic field (H) of 5 kOe confirm the paramagnetic behaviour of 1 (Fig. 1b). χmT at 300 K gave 0.61 emu K mol−1, which is lower than the value expected for a diradical with isolated or non-interacting spins (χmT = 0.75 emu K mol−1), and hence is in agreement with the presence of some monoradical impurity. From 300 K to ∼100 K, the χmT values slowly decrease and fall off steeply on cooling down in agreement with the antiferromagnetic behaviour of 1 due to the intramolecular exchange coupling. The electrochemical properties of 1 were investigated by CV using 0.1 M of tetrabutylammonium hexafluorophosphate (n-Bu4NPF6) as supporting electrolyte in toluene/MeCN (1:1 v/v). The CV revealed two reversible redox processes with half-wave potentials (E1/2 vs. Ag/AgCl) at −0.39 and −0.60 V, which were attributed to distinct single and sequential electron transfer process of each TTM unit (Fig. 1c). This is indicative of the strong electron–electron interaction existing between the two TTM moieties.
We assume that the monoradical impurity might be the thiol derivative, which could be formed during the synthesis of the compound 1 (Fig. S7†). This would be in agreement with the CV and SQUID results. Unfortunately, the monoradical impurity was not possible to separate by the chromatographic technique due to their identical polarity to 1. However, the presence of such monoradical was not considered crucial or detrimental to proceed with the surface functionalisation since it would lead to identical SAMs.
The preparation of r-SAM on Au and Ag were conducted under similar experimental conditions. Freshly cleaned Au or template-striped Ag (AgTS)40–42 substrates were immersed in a 0.5 mM solution of 1 in dry toluene under Ar at 50 °C with the exclusion of light (Scheme 4). After 12 h, the temperature of the set-up was gradually brought to room temperature and the substrates were left in the solution for 36 h more. Afterwards, the substrates were thoroughly rinsed with fresh toluene to remove the physisorbed molecules and dried under nitrogen stream.
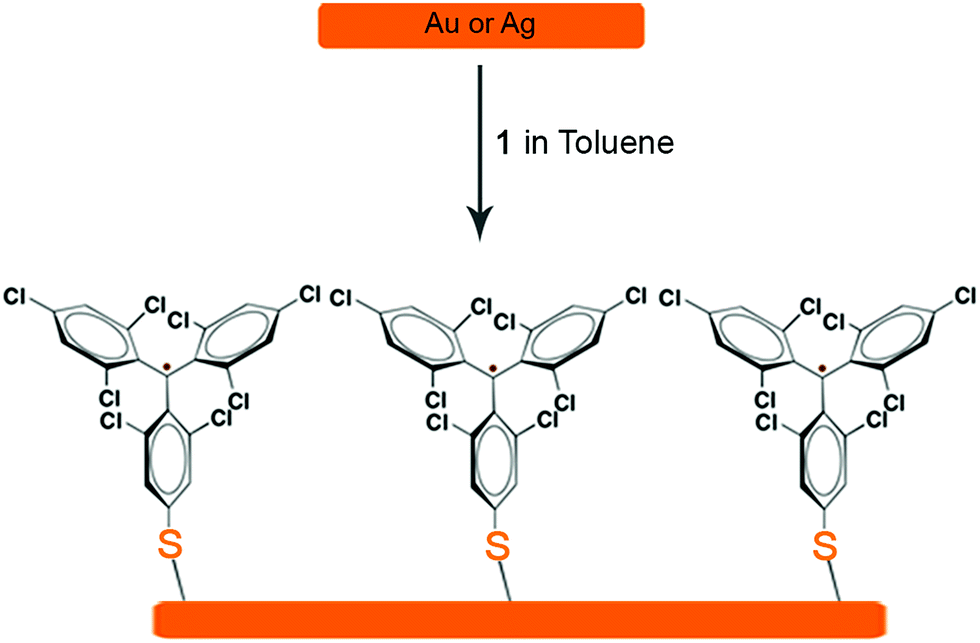 | ||
| Scheme 4 r-SAM formation of 1 on Au or Ag. | ||
The obtained r-SAMs were well characterized with the aid of XPS, UPS, EPR and CV. XPS measurements provided an in-depth chemical and structural information of r-SAMs.35 Firstly, to understand the elemental composition of surface, XPS survey scans (Fig. S8†) were collected for both 1/Au and 1/Ag and peaks corresponding to Au, Ag, S, Cl and C were identified, confirming thus the presence of TTM moiety of 1 on Ag and Au. The r-SAM formation was demonstrated by the high-resolution XPS of S 2p, Cl 2p and C 1s (Fig. 2). In detail, for S 2p data of 1/Au, the best fit could be generated with three doublets by using 2:1 peak area ratio and a 1.2 eV splitting, and hence indicated the presence of three S species (Fig. 2a). The main doublet (56%) at 162.2 and 163.5 eV was assigned to S 2p3/2 and S 2p1/2, respectively, of typical Au–S bonds.35 We also observed another component of S 2p doublet (28%) at 161.3 and 162.6 eV as the S 2p3/2 and S 2p1/2, respectively, and could be attributed to the self-assembly constraints or due to the in situ generated atomic S via C–S scission while the X-ray irradiation.43–45 Additionally, a small fraction of physisorbed 1 (16%) is also found over the substrate by observing a doublet at 163.1 and 164.4 eV. Similarly, 1/Ag also gave S 2p emission corresponding to Ag–S bonds and notably no unreacted/physisorbed molecules of 1 were observed (Fig. 2d) suggesting a more densely packed r-SAM. The Cl 2p region of both 1/Au and 1/Ag showed a doublet at 200.4 and 202.2 eV, which corresponds to Cl 2p3/2 and Cl 2p1/2, respectively, of C–Cl bonds (Fig. 2b and e). Interestingly, Ag has a tendency to interact with Cl and caused a Cl 2p doublet at 198.0 and 199.6 eV.46 The best fit of the C 1s spectra gave two peaks for both 1/Au and 1/Ag, which are consistent with the previously reported r-SAMs of PTM formed by physisorption (Fig. 2c and f).46 The peak around 284.5 eV is attributed to the C 1s of C–H and C–S bonds, whereas ∼286.0 eV peak arose from C 1s of C–Cl bonds. It is worth mentioning that the reaction of 1 on Ag is notably more favoured, compared to that on Au under similar experimental conditions, and that is reflected by the high emission counts and the excellent signal-to-noise ratio in the XPS measurements. This observation is justified by the higher surface energy of Ag which facilitates the disulfide cleavage required for the r-SAM formation.
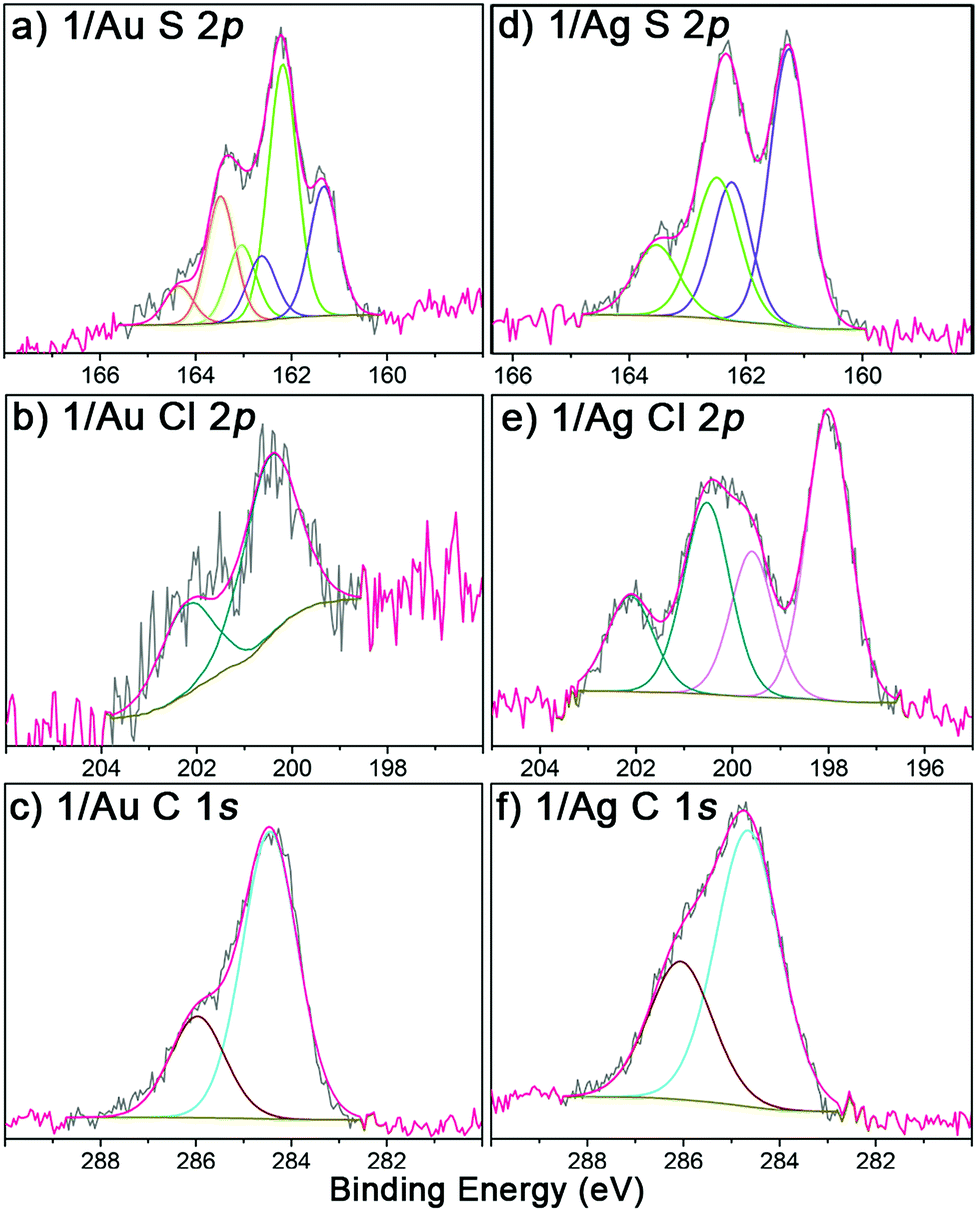 | ||
| Fig. 2 High-resolution XPS of S 2p, Cl 2p and C 1s regions (grey) of 1/Au (a–c) and 1/Ag (d–f). | ||
The paramagnetic nature of 1/Au and 1/Ag were further analyzed with UPS and EPR spectroscopy. The UPS features confirmed its open shell behaviour by observing the respective single occupied molecular orbitals (SOMO), i.e., ∼1.5 and ∼1.7 eV for 1/Au (Fig. S9†) and 1/Ag, respectively (Fig. 3a).46,47 As expected, EPR spectroscopy revealed signals at g = 2.0038 for 1/Au (Fig. S10†) with a linewidth of 4.9 G and 2.0041 for 1/Ag with a linewidth of 8.3 G under ambient conditions and, therefore, successfully confirmed that the immobilized TTM units maintain their radical nature after the chemisorptions (Fig. 3b).23,25 This result contrasts with the previous findings on physisorbed PTM radical derivatives on Ag surface where an electron charge transfer from the metal to the radical was observed.46
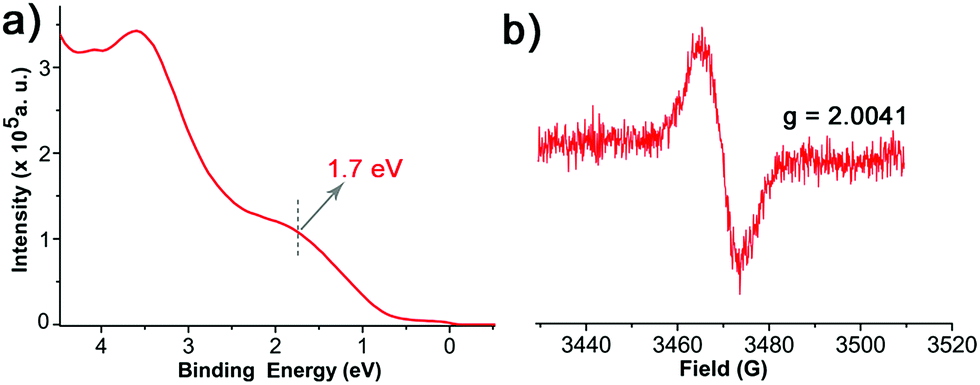 | ||
| Fig. 3 (a) UPS of 1/Ag. (b) EPR of 1/Ag at 300 K under ambient condition. | ||
Considering these results, the electrochemical characteristics of the r-SAM 1/Ag were also investigated. The CV was performed using a three-electrode cell assembly in 0.1 M n-Bu4NPF6 in degassed dry MeCN under Ar at room temperature between −0.6 and 0.1 V. A custom-built electrochemical cell was equipped with 1/Ag as the working electrode, a Ag wire as pseudo-reference electrode and a Pt wire as counter electrode. The CV displayed one quasi-reversible redox wave at −0.24 V corresponding to the one electron reduction of the TTM unit (Fig. 4a).23 Unlike the reported PTM based r-SAMs, the difference between the oxidation and reduction waves (ΔE ∼ 110 mV for scan rate of 100 mV) is significantly higher and could be rationalized by the presence of strong TTM–metal and (or) TTM–TTM interactions in 1/Ag.48 It was also observed that the anodic peak current linearly increased with scan rate and that further confirmed the surface confinement of the TTM units (Fig. 4a and b). From the integration of the anodic wave at scan rate of 100 mV s−1, we have estimated the surface coverage (Γ) of 1/Ag to be 1.5 × 10−10 mol cm−2, which is of the same order as that reported for other PTM SAMs with longer linkers. We have carried out 20 consecutive redox cycles with a scan rate of 20 mV s−1 without any noticeable current intensity decrease and thereby proved the robustness and stability of the r-SAM (Fig. 4c).
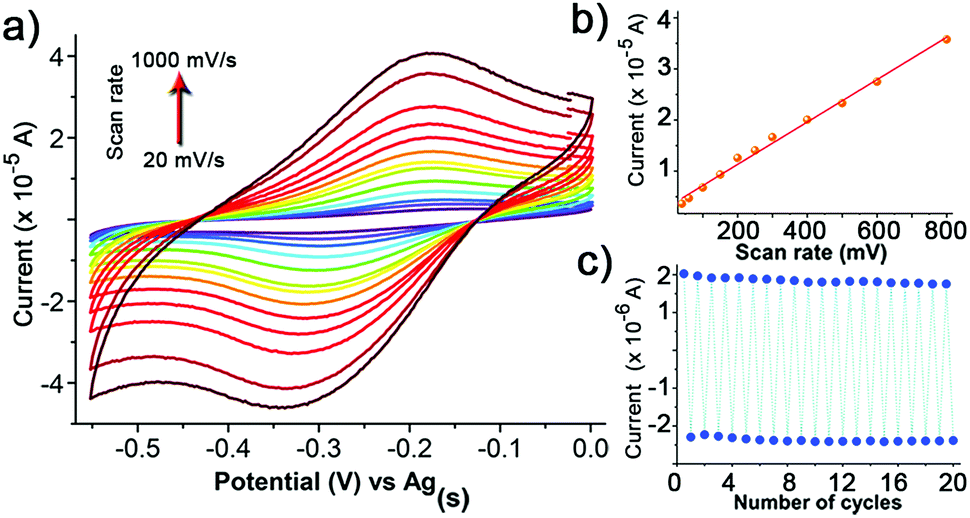 | ||
| Fig. 4 (a) CV of 1/Ag in MeCN, with 0.1 M n-Bu4NPF6 (vs. Ag(s)) at different scan rates (20–1000 mV s−1). Plots of the current intensity (b) vs. scan rate and (c) vs. the number of cycles of the redox wave of 1/Ag. | ||
Periodic density functional theory calculations, as implemented in the FHI-AIMS simulation package,49,50 were employed to study the interaction between the adsorbed 1 r-SAM and the Ag surface. All calculations were performed using the PBE functional51 and a light/tier 1 atom-centered numerical basis set, with van der Waals interactions taken into account using the Tkatchenko–Scheffler method.52 The Ag substrate was represented by a three-layered Ag(111) slab cut from a fully optimized Ag crystal. A periodic cell size of a = b = 17.7 Å; c = 40 Å; αab = 60°; αac = αbc = 90° was used, where the large c parameter was used to provide a vacuum space sufficient to avoid spurious interactions between repeated images. In the calculations the two top layers of the Ag slab were allowed to relax, with the bottom Ag layer and the cell parameters kept fixed to mimic the bulk rigidity of the substrate. Separately, a half fragment of 1 (breaking the molecule at the S–S bond, see Scheme 4) was fully optimized. This optimized molecule was placed with the S atom at a distance of 3.5 Å from the Ag surface with the S–C bond perpendicular to the plane of the Ag(111) support (Fig. 5). The substrate–molecule system was then optimized (fixing only the cell parameters and lower Ag layer). After optimization, the adsorbed molecules remain stable in a “vertical” conformation, which should facilitate the maintenance of the open-shell nature of the 1 r-SAM. More planar or inclined conformations might lead to charge transfer effects,15,46 which may destroy completely or partially the open-shell character of the monolayer. From the calculated spin densities in Fig. 5b, it can be observed that the open-shell character of the 1 r-SAM is maintained after surface chemisorption. Moreover, there appears a partial beta-spin polarization in the Ag surface atoms below the adsorbed molecular unit which is likely due to the proximity of the radical centre 1 with the metal surface.
 | ||
| Fig. 5 (a) x, y and z views of the fully optimized periodic structure of the adsorbed 1 radical SAM onto the 3-layered Ag slab. Atom colour-key: C – dark-grey, Cl – green, S – yellow, Ag – light-grey, H – white. (b) Spin density (alpha = blue, beta = red). | ||
Conclusions
In summary, we have synthesized a stable disulfide diradical by utilizing a single-pot reaction. Unlike the conventional PTM based r-SAM, these r-SAMs are fabricated without incorporating the linker unit for the first time. By using this strategy, we could avoid the conventional methodology that involves the utilization of aromatic or alkyl linkers and hence the associated synthetic difficulties. The obtained robust r-SAMs were fully characterized and, importantly, it was proved that the radical character is preserved despite the short distance with the metal and in contrast with previous works on physisorbed PTM radicals on Ag.46 Therefore, we succeed in preparing free r-SAMs with the spin-bearing cores directly and covalently grafted to a metal surface, ensuring excellent spin–spin and spin–electron interactions, as supported by ab initio density functional calculations. This design strategy and easy synthetic protocol make these r-SAMs promising building blocks for the futuristic preparation of spintronic materials and devices.Experimental
1. General procedure
NMR spectra were recorded on a Bruker Avance 400 MHz. EPR spectra were recorded in a Bruker ELEXYS E500 X-band spectrometer. The simulation of the EPR spectra was realised with software Simfonia. Electrochemical experiments were performed with a potentiostat/galvanostat Autolab/PGSTAT204 from Metrohm Autolab B.V. in a standard three-electrode cell, by using a platinum wire as working and counter electrode and Ag/AgCl as reference electrode. Tetrabutylammonium hexafluorophosphate (Fluka, 99%) was used as the supporting electrolyte. UV-vis spectra were recorded on a Varian Carey 5000 in double-beam mode. Mass spectra were recorded with a Bruker Ultraflex LDI-TOF mass spectrometer. The IR spectra were recorded with an ATR-IR Perkin Elmer Spectrum One. The synthesis and manipulation of the radicals in solution was performed under red light.2. Synthesis and characterization of 1
Compound 2 was synthesised by following previously reported method.36 2 (2.16 g, 4.05 mmol) and 2,4-bis(4-methoxyphenyl)-2,4-dithioxo-1,3,2,4-dithiadiphosphetane (Lawesson's reagent; 2.02 g, 4.99 mmol) were transferred to a 50 mL R.B. flask, and 60 mL of dry toluene was added under Ar atmosphere and under light exclusion. The reaction mixture was heated at 95 °C for 2.5 h on a magnetic stirrer with gentle stirring and cooled to RT. After drying under vacuum, the crude product was purified by column chromatography over activated neutral Al2O3 where n-hexane was used as eluent.Yield: 18%. Rf = 0.39 (SiO2 TLC, hexane as eluent). Melting point: 264 °C. MS (MALDI-TOF, positive mode): calculated for C38H12Cl16S22˙ (m/z) 1099.83, found 1100.2 [M]+, 1068.9 [M − Cl]+, 1029.9 [M − 2Cl]+, 996.1 [M − 3Cl]+, 959.2 [M − 4Cl]+, and 925.1 [M − 5Cl]+. IR (ATR, ν cm−1) = 3082, 2921, 1729, 1550, 1524, 1364, 1183, 1137, 857. UV-vis (CH2Cl2): λmax (ε) = 375 nm (43700 M−1 cm−1), 394 nm (29420 M−1 cm−1), 540 nm (2020 M−1 cm−1) and 572 nm (2030 M−1 cm−1).
3. Magnetic susceptibility measurements
Variable temperature magnetic susceptibility measurements of 1 (55.9 mg) were carried out in a Quantum Design MPMS-5S SQUID magnetometer down to 2 K. The molar magnetic susceptibility (χm) values were corrected for the temperature-independent diamagnetic contribution of the atoms/bonds by means of Pascal's tables and the magnetic contribution of the sample holder (35.9 mg).4. Preparation of the SAMs on Au and AgTS
All the glassware employed for the SAM preparation were immersed in Hellmanex II solution (2% v/v) in distilled water for overnight, thoroughly washed with MilliQ water, and dried in an oven at 80 °C. The solvents used in the surface chemistry experiments were of HPLC grade from ROMIL-SpS (Super Purity Solvent).We have used Ag wire (1.5 mm diameter) with purity of 99.99% obtained from Kurt J. Lesker Company, Spain. Si/SiOx (200 nm SiOx, sourced from Si-mat, Germany) with a thickness of 525 ± 25 μm with one side polished. This was first cleaned by blowing nitrogen and then by reactive ion etching (RIE). An O2 flux of 20 standard cubic centimeters per minute for 1 min under 100 W power and 100 mTorr pressure were used for RIE (RIE 2000 CE, South Bay Technology). For the 300 nm Ag evaporation, BOC Edwards Auto 306 electron beam evaporation systems was used at a base pressure of ∼2 × 10−6 bar. The rate of the deposition was at 0.1–0.2 Å s−1 for the first 50 nm and the rate increased gradually to 1 Å s−1 for the remaining 250 nm. Glass (2.6 × 1 cm2) or quartz (0.5 × 1.6 cm2) slides (1 mm thick) were immersed in piranha solution (concentrated H2SO4 and 30% H2O2 (aq) in 3:1 ratio. Warning: piranha solution should be handled with caution: it has been reported to detonate unexpectedly). These slides were washed thoroughly with high purity water (MilliQ, 18.2 MΩ cm at 25 °C) and isopropanol (HPLC). After drying under a nitrogen stream, the slides were further cleaned by UV ozone chamber for 25 min. Then the slides were glued onto the Ag layer with a thermally-curable epoxy adhesive (Epo-tek 353-ND) and kept at 80 °C for 15 h for curing. The glass (or quartz)/glue/Ag substrate was stripped off from the Si/SiOx wafer-template to obtain AgTS by using a sharp razor blade and immediately transferred to Ar filled reaction vessel. Au substrates (300 nm, on mica) were purchased from Georg Albert PVD-Beschichtungen. Au/mica (0.8 × 2.6 cm) were rinsed with CH2Cl2, acetone and isopropanol, respectively, and dried under nitrogen stream. These substrates were placed in UV ozone chamber for 20 minutes and thereafter immediately transferred to isopropanol. After 30 min, the substrates were rinsed with pure isopropanol, blown dry with a stream of N2 and transferred to Ar filled reaction vessel.
To the freshly prepared substrates (Au and Ag), 0.5 mM solution of 1 in dry toluene was slowly added under Ar and kept at 50 °C for 24 h. Then, the temperature was gradually decreased to RT and left the reaction vessel undisturbed for another 24 h. Then, the substrates were removed from the solution and washed with abundant toluene, followed by drying in a stream of N2.
5. X-ray photoelectron and UV photoelectron spectroscopy (XPS and UPS)
XPS and UPS measurements were performed with a Phoibos 150 analyzer (SPECS GmbH, Germany) instrument with monochromatic AlKα X-ray (1486.74 eV) and HeI UV (21.2 eV) sources under ultra-high vacuum conditions (base pressure 3 × 10−10 mbar). The X-ray radiation is at an angle of 55° to the sample with a spot size of 3.5 × 0.5 mm in ellipsoidal arrangement. A pass energy of 50 eV, a dwell time of 500 ms, and an energy step size of 1 eV were employed for the general scan. For specific element scanning, pass energy of 20 eV and step size of 0.05 eV were used. The binding energies were calibrated with respect to Au 4f7/2 at 84.0 eV for Au/SAMs and Ag 3d5/2 at 368.27 eV for Ag/SAMs. The sample for UPS was held at an angle of 55° with respect to the UV radiation and the spot size was 0.5 mm diameter. The overall resolution for XPS, as measured by the full width at half-maximum intensity (FWHM) of the Ag 3d5/2 peak for a sputtered silver foil, is 0.57 eV and for UPS as measured by FWHM of Fermi level is 0.12 eV.A 1 mM solution of 1 in CH2Cl2 was drop casted on a Au substrate and dried instantly by blowing a gentile stream of nitrogen, and the resultant 1 multilayers were examined by XPS. The spectra gave peaks corresponding to one kind of S (163.7 and 164.9 eV for 2p3/2 and 2p1/2, respectively) attributed to the disulfide group and one kind of Cl (200.3 and 201.9 eV for 2p3/2 and 2p1/2, respectively) attributed to C–Cl groups. In addition, the C 1s peaks at 284.9 and 286.0 eV indicated the existence of C–H and C–Cl bonds. However, C of methyl–C and C–S were overwhelmed by C–Cl and C–H, and could not be resolved. Along with these elements, Au from substrate and trace amount of ubiquitous O were also detected.
Acknowledgements
This work was funded by ERC StG 2012-306826 e-GAMES. The authors also thank ITN iSwitch 642196 project, the Networking Research Center on Bioengineering, Biomaterials and Nanomedicine (CIBER-BBN), DGI (Spain) BE-WELL CTQ2013-40480-R and FANCY CTQ2016-80030-R, and Generalitat de Catalunya 2014-SGR-17. The authors also acknowledge the Spanish Ministry of Economy and Competitiveness, through the “Severo Ochoa” Programme for Centres of Excellence in R&D (SEV-2015-0496). We thank Dr V. Lloveras for ESR spectroscopy characterization, Mr A. Bernabé for MALDI-TOF measurements and Dr G. Sauthier from ICN2 for XPS and UPS measurements. We also thank Dr N. Crivillers for useful discussions. S. T. B. and I. A. acknowledge support from the Spanish MINECO grant CTQ2015-64618-R grant and, in part, by Generalitat de Catalunya grants 2014SGR97 and XRQTC. IA acknowledges the Spanish Ministerio de Educación Cultura y Deporte for a FPU PhD scholarship. Access to supercomputer resources as provided through grants from the Red Española de Supercomputación is also acknowledged.Notes and references
- F. P. Netzer and M. G. Ramsey, Crit. Rev. Solid State Mater. Sci., 1992, 17, 397 CrossRef CAS.
- H. Ishii, K. Sugiyama, E. Ito and K. Seki, Adv. Mater., 1999, 11, 605 CrossRef CAS.
- S. Braun, W. R. Salaneck and M. Fahlman, Adv. Mater., 2009, 21, 1450 CrossRef CAS.
- M. Franke, F. Marchini, H. P. Steinrück, O. Lytken and F. J. Williams, J. Phys. Chem. Lett., 2015, 6, 4845 CrossRef CAS PubMed.
- C. Joachim, J. K. Gimzewski and A. Aviram, Nature, 2000, 408, 541 CrossRef CAS PubMed.
- M. Halik and A. Hirsch, Adv. Mater., 2011, 23, 2689 CrossRef CAS PubMed.
- G. Heimel, L. Romaner, E. Zojer and J. L. Bredas, Acc. Chem. Res., 2008, 41, 721 CrossRef CAS PubMed.
- A. Nitzan and M. A. Ratner, Science, 2003, 300, 1384 CrossRef CAS PubMed.
- L. Basabe-Desmonts, J. Beld, R. S. Zimmerman, J. Hernando, P. Mela, M. F. G. Parajó, N. F. van Hulst, A. van den Berg, D. N. Reinhoudt and M. A. Crego-Calama, J. Am. Chem. Soc., 2004, 126, 7293 CrossRef CAS PubMed.
- G. De Ruiter, T. Gupta and M. E. van der Boom, J. Am. Chem. Soc., 2008, 130, 2744 CrossRef CAS PubMed.
- B. A. Bejjani and L. G. Shaffer, J. Mol. Diagn., 2006, 8, 528 CrossRef CAS PubMed.
- J. Lamartine, Mater. Sci. Eng., C, 2006, 26, 354 CrossRef CAS.
- M. J. Heller, Annu. Rev. Biomed. Eng., 2002, 4, 129 CrossRef CAS PubMed.
- T. M. Herne and M. J. Tarlov, J. Am. Chem. Soc., 1997, 119, 8916 CrossRef CAS.
- G. Heimel, S. Duhm, I. Salzmann, A. Gerlach, A. Strozecka, J. Niederhausen, C. Bürker, T. Hosokai, I. Fernandez-Torrente, G. Schulze, S. Winkler, A. Wilke, R. Schlesinger, J. Frisch, B. Bröker, A. Vollmer, B. Detlefs, J. Pflaum, S. Kera, K. J. Franke, N. Ueno, J. I. Pascual, F. Schreiber and N. Koch, Nat. Chem., 2013, 5, 187 CrossRef CAS PubMed.
- J. C. Love, L. A. Estroff, J. K. Kriebel, R. G. Nuzzo and G. M. Whitesides, Chem. Rev., 2005, 105, 1103 CrossRef CAS PubMed.
- A. Ulman, Chem. Rev., 1996, 96, 1533 CrossRef CAS PubMed.
- H. Hamoudi, K. Uosaki, K. Arigaa and V. A. Esaulov, RSC Adv., 2014, 4, 39657 RSC.
- E. Marchante, M. S. Maglione, N. Crivillers, C. Rovira and M. Mas-Torrent, RSC Adv., 2017, 7, 5636 RSC.
- M. Mas-Torrent, N. Crivillers, C. Rovira and J. Veciana, Chem. Rev., 2012, 112, 2506 CrossRef CAS PubMed.
- M. Mas-Torrent, N. Crivillers, V. Mugnaini, I. Ratera, C. Rovira and J. Veciana, J. Mater. Chem., 2009, 19, 1691 RSC.
- N. Crivillers, M. Mas-Torrent, C. Rovira and J. Veciana, J. Mater. Chem., 2012, 22, 13883 RSC.
- N. Crivillers, M. Mas-Torrent, J. Vidal-Gancedo, J. Veciana and C. Rovira, J. Am. Chem. Soc., 2008, 130, 5499 CrossRef CAS PubMed.
- N. Crivillers, C. Munuera, M. Mas-Torrent, C. Simão, S. T. Bromley, C. Ocal, C. Rovira and J. Veciana, Adv. Mater., 2009, 21, 1177 CrossRef CAS.
- C. Simão, M. Mas-Torrent, N. Crivillers, V. Lloveras, J. Artés, P. Gorostiza, J. Veciana and C. Rovira, Nat. Chem., 2011, 3, 359 CrossRef PubMed.
- C. Simão, M. Mas-Torrent, J. Veciana and C. Rovira, Nano Lett., 2011, 11, 4382 CrossRef PubMed.
- N. Crivillers, M. Mas-Torrent, S. Perruchas, N. Roques, J. Vidal-Gancedo, J. Veciana, C. Rovira, L. Basabe-Desmonts, B. Ravoo, M. Crego-Calama and D. N. Reinhoudt, Angew. Chem., Int. Ed., 2007, 46, 2215 CrossRef CAS PubMed.
- O. Shekhah, N. Roques, V. Mugnaini, C. Munuera, C. Ocal, J. Veciana and C. Wöll, Langmuir, 2008, 24, 6640 CrossRef CAS PubMed.
- R. Frisenda, R. Gaudenzi, C. Franco, M. Mas-Torrent, C. Rovira, J. Veciana, I. Alcon, S. T. Bromley, E. Burzurí and H. S. J. van der Zant, Nano Lett., 2015, 15, 3109 CrossRef CAS PubMed.
- J. Liu, H. Isshiki, K. Katoh, T. Morita, B. K. Breedlove, M. Yamashita and T. Komeda, J. Am. Chem. Soc., 2013, 135, 651 CrossRef CAS PubMed.
- Y.-h. Zhang, S. Kahle, T. Herden, C. Stroh, M. Mayor, U. Schlickum, M. Ternes, P. Wahl and K. Kern, Nat. Commun., 2013, 4, 2110 Search PubMed.
- S. Müllegger, M. Rashidi, M. Fattinger and R. Koch, J. Phys. Chem. C, 2013, 117, 5718 Search PubMed.
- O. Armet, J. Veciana, C. Rovira, J. Riera, J. Castaner, E. Molins, J. Rius, C. Miravitlles, S. Olivella and J. Brichfeus, J. Phys. Chem., 1987, 91, 5608 CrossRef CAS.
- N. Crivillers, M. Paradinas, M. Mas-Torrent, S. T. Bromley, C. Rovira, C. Ocal and J. Veciana, Chem. Commun., 2011, 47, 4664 RSC.
- D. G. Castner, K. Hinds and D. W. Grainger, Langmuir, 1996, 12, 5083 CrossRef CAS.
- J. Carilla, L. Fajari, L. Julia, J. Riera and L. Viadel, Tetrahedron Lett., 1994, 35, 6529 CrossRef CAS.
- J. Veciana, C. Rovira, M. I. Crespo, O. Armet, V. M. Domingo and F. Palacio, J. Am. Chem. Soc., 1991, 113, 2552 CrossRef CAS.
- A. Rajca, K. Shiraishi, M. Vale, H. Han and S. Rajca, J. Am. Chem. Soc., 2005, 127, 9014 CrossRef CAS PubMed.
- V. Lloveras, E. Badetti, K. Wurst, V. Chechik, J. Veciana and J. Vidal-Gancedo, Chem.–Eur. J., 2016, 22, 1805 CrossRef CAS PubMed.
- E. A. Weiss, R. C. Chiechi, G. K. Kaufman, J. K. Kriebel, Z. Li, M. Duati, M. A. Rampi and G. M. Whitesides, J. Am. Chem. Soc., 2007, 129, 4336 CrossRef CAS PubMed.
- R. C. Chiechi, E. A. Weiss, M. D. Dickey and G. M. Whitesides, Angew. Chem., Int. Ed., 2008, 47, 142 CrossRef CAS PubMed.
- N. Nerngchamnong, L. Yuan, D.-C. Qi, J. Li, D. Thompson and C. A. Nijhuis, Nat. Nanotechnol., 2013, 8, 113 CrossRef CAS PubMed.
- O. Cavalleri, G. Gonella, S. Terreni, M. Vignolo, P. Pelori, L. Floreano, A. Morgante, M. Canepa and R. Rolandi, J. Phys.: Condens. Matter, 2004, 16, S2477 CrossRef CAS.
- V. Lloveras, E. Badetti, J. Veciana and J. Vidal-Gancedo, Nanoscale, 2016, 8, 5049 RSC.
- J. Jia, A. Giglia, M. Flores, O. Grizzi, L. Pasquali and V. A. Esaulov, J. Phys. Chem. C, 2014, 118, 26866 CAS.
- V. Mugnaini, A. Calzolari, R. Ovsyannikov, A. Vollmer, M. Gonidec, I. Alcon, J. Veciana and M. Pedio, J. Phys. Chem. Lett., 2015, 6, 2101 CrossRef CAS PubMed.
- F. Grillo, V. Mugnaini, M. Oliveros, S. M. Francis, D.-J. Choi, M. V. Rastei, L. Limot, C. Cepek, M. Pedio, S. T. Bromley, N. V. Richardson, J.-P. Bucher and J. Veciana, J. Phys. Chem. Lett., 2012, 3, 1559 CrossRef CAS PubMed.
- N. Nerngchamnong, D. Thompson, L. Cao, L. Yuan, L. Jiang, M. Roemer and C. A. Nijhuis, J. Phys. Chem. C, 2015, 119, 21978 CAS.
- V. Blum, R. Gehrke, F. Hanke, P. Havu, V. Havu, X. Ren, K. Reuter and M. Scheffler, Comput. Phys. Commun., 2009, 180, 2175 CrossRef CAS.
- V. Havu, V. Blum, P. Havu and M. Scheffler, J. Comput. Phys., 2009, 228, 8367 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- A. Tkatchenko and M. Scheffler, Phys. Rev. Lett., 2009, 102, 073005(1) CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra01686g |
| This journal is © The Royal Society of Chemistry 2017 |