DOI:
10.1039/C6RA25671F
(Paper)
RSC Adv., 2017,
7, 5002-5012
Elucidating the binding interaction of andrographolide with the plasma proteins: biophysical and computational approach†
Received
22nd October 2016
, Accepted 9th December 2016
First published on 17th January 2017
Abstract
The present study focuses on the interactions of andrographolide (ANDR) with plasma proteins, human serum albumin (HSA), and α-1-acid glycoprotein (AGP), and their biological importance. To understand the pharmacological role of ANDR, its anticancer activity was studied on a breast cancer cell line (MCF-7); it showed a dose-dependent inhibition of growth, and its IC50 value was found to be 55 μM. Furthermore, to evaluate the binding mechanism of AGP and HSA with andrographolide, fluorescence emission quenching was observed as a static mechanism upon the binding of ANDR to plasma proteins. Additionally, active HSA sensor chip surfaces were prepared through an amine-coupling reaction protocol, and the equilibrium association constants for ANDR–HSA were then determined by surface plasmon resonance (SPR) analysis. The association constants of ANDR binding to HSA, obtained with fluorescence and SPR, were KA(AGD) = 1.85 ± 0.02 × 104 M−1 and 3.1 ± 0.04 × 103 M−1, respectively. Similarly, the ANDR binding affinity with AGP was analyzed through fluorescence and SPR, and the calculated binding association values were 1.5 ± 0.01 × 103 M−1 and 1.3 ± 0.04 × 103 M−1, respectively. Molecular displacement and in silico docking shows that ANDR binds to subdomain IIB. Consequently, circular dichroism analysis showed that there is partial perturbation in the structure of HSA upon an increase in the concentration of ANDR. Moreover, molecular dynamics simulation revealed that the stability of the HSA–ANDR complexes reached an equilibration state at around 3000 ps, which clearly indicates the rigidity and stability of the HSA–ANDR complexes. Thus, our results provide evidence that both plasma proteins (HSA and AGP) can act as carrier proteins for ANDR.
1. Introduction
Human serum albumin (HSA) is the most abundant carrier protein and plays an important role in the transport and disposition of many endogenous and exogenous substances, such as metabolites, drugs, and other biologically active compounds, present in the blood. It consists of three structurally homologous domains, which assemble to form a heart-shaped molecule, whereas each domain contains two subdomains. Albumin is known to bind and transport many ligands, including fatty acids, amino acids, hormones, cations and anions, and a variety of pharmaceuticals. It is suggested that the principal regions of ligand binding to HSA are located in the hydrophobic cavities in the subdomains IIA and IIIA, which are located at sites I and II, respectively.1,2 This is well characterized, as most drugs used in humans are bound to these two sites with an affinity constant of 104 to 106 M−1.3 It is widely accepted that the degree of affinity between a drug and HSA can dominate its distribution into the target tissue, affect its elimination from the body, and finally influence its therapeutic or toxic effects, biotransformation, and biodistribution.4 Besides, there is a testimony of conformational changes of the protein caused by its interaction with drugs, and these changes seem to affect the secondary and tertiary structure of protein.5 Therefore, investigating the interactions of a drug with HSA was very important, as this can provide useful information about drug actions and can be used as a model for elucidating the drug–protein complex.
Another important plasma protein, α-1-acid glycoprotein (AGP), named as orosomucoid, comprised 2.5 mg mL−1 in human blood. This glycoprotein contains 45% carbohydrate, having a molecular weight of 41–43 kDa. Since it has a carbohydrate moiety, it gives pH 2.7–3.2 in aqueous solution.6,7 This hepatic protein, made up of 183 amino acids in a single polypeptide chain, is further attached to five asparaginyl-linked glycans. During acute phase response or inflammation, its glycosylation pattern varies and gives rise to a concentration 3–4 times more than normal; thus, AGP is referred to as a positive acute-phase protein.8 As well as its immunosuppressive properties, AGP has the ability to bind and transport several drug molecules, mostly lipophilic/cationic small molecules. As it has only one binding site, it can bind one molecule at a time, which makes it different from HSA.9
Furthermore, phytochemicals are known to bind serum proteins, and this is a good model for studying pharmacokinetics. Our group has extensively studied the interaction of various phytochemicals with serum proteins, and has observed that most of the phytochemicals bind strongly to HSA,10–12 but only some of them bind to AGP.13,14 Andrographolide (ANDR) is a labdane diterpenoid, which is the main bioactive component of the medicinal plant Andrographis paniculata.15 ANDR has been reported to have a wide range of biological activities, such as anti-inflammatory,16 anti-allergic,17 anti-platelet aggregation,18 hepatoprotective,19 and anti-HIV properties. In biological systems, ANDR can interact with many inter- and intracellular constituents as a bipolar compound, thus inducing many biological responses. A recent study demonstrated that A. paniculata polysaccharides combined with ANDR can ease the recovery of diabetic nephropathy.20
Proteins are naturally-derived polymers that are advantageous in their biodegradability, low toxicity, non-antigenicity, high nutritional value, high stability, and binding capacity for various drugs, such as paclitaxel and ibuprofen.21–24 Plasma proteins are natural drug transporters, while nanocarriers are sometimes immunogenic, and their metabolite deposits may harm the body. Unlike plasma proteins, nanocarriers are artificial drug carriers, and when drug-loaded nanoparticles are injected into bodies, they cross epithelial barriers and circulate in the blood vessels before reaching the target site. There is a desire to conduct toxicology tests on the particles, since the different interactions of nanoparticles with fluids, cells, and tissues need to be considered, starting at the portal of entry and then via a range of possible pathways towards target organs.25 An alternative approach to designing nanocarriers is based on layer-by-layer (LbL) assembly technology. The assembly of LbL polymer capsules entails the sequential adsorption of interacting polymers onto sacrificial template particles, followed by template removal. The flexibility of this approach in terms of chemical nature and the properties of the polymers/template particles is limited only by the specific requirements related to therapeutic applications, such as biodegradability and biocompatibility. LbL technology confers the advantage of generating capsules with excellent structural stability,26 and allows the functionalization of nanoparticles smaller than 100 nm, while restricting aggregation. The polymer layers are expected to act as a tool to control payload retention, degradation profiles, and drug release rates.27,28 However, the thickness of the polymer wall can affect cross-transport and therefore decrease the efficacy of active compounds encapsulated inside. In capsule-mediated delivery, diffusion may be difficult due to dense layers of fibrous tissue; drug amounts cannot be readily regulated.
Many agents are bound to serum proteins, especially serum albumin, which affects their pharmacologic and pharmacokinetic properties. Studies on the interaction of ANDR with HSA/AGP will provide an insight into the chemical nature of the interaction of biomacromolecules with small molecules. This can be used as a model for elucidating ligand–protein complications. In addition, it is well known that the efficacy of many drugs in the body is based on the fact that drugs are carried by HSA/AGP; therefore, this experiment can supply some important methods to clinical research and provide a theoretical basis for designing new drugs. A variety of techniques are available for understanding the protein–ligand interaction in vitro; hence, an attempt has been made to use methods such as fluorescence quenching, surface plasmon resonance (SPR), molecular docking, and molecular dynamics simulations. Here, we emphasized particularly the mechanism and interactions of ANDR, a natural bioactive compound, with HSA/AGP, which include binding, protein–ligand stability, and conformation under physiological pH. Also, to understand the biological importance, we have used MCF-7 cell lines and studied the cell viability and cytotoxicity.
2. Experimental
2.1 Materials and methods
2.1.1 Preparation of stock solutions. Pure fat-free HSA (purchased from Sigma-Aldrich) was prepared with 1.5 mM dissolved in 0.1 M phosphate buffer at pH 7.4. Andrographolide (bioactive compound) was purchased from Natural Remedies Pvt., Ltd, Bengaluru, India, with a purity of 95%. Its stock solution (1.0 × 103 M−1) was prepared in a 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :80 ethanol:water mixture, which is equivalent to 1:4. Through our previous reports, a solution containing 20% ethanol has no effect on the secondary structure of protein.29,47 The molecular weight of andrographolide is 350.4 Da. The optimum pH for HSA was set to be 7.4, as it has the maximum absorption at this pH.29 Thus, for all the experiments, we have used 0.1 M phosphate buffer at pH 7.4 as a physiological buffer. All other chemicals are of analytical grade, purchased from Sigma-Aldrich. Research-grade CM5 sensor chips, 10× phosphate buffered saline (PBS), and an amine-coupling kit, consisting of N-ethyl-N-(3-dimethylaminopropyl)-carbodiimide (EDC), N-hydroxysuccinimide (NHS), and ethanolamine hydrochloride (pH 8.5; 1 M), were all purchased from GE Healthcare Bio-Sciences (Uppsala, Sweden).
:80 ethanol:water mixture, which is equivalent to 1:4. Through our previous reports, a solution containing 20% ethanol has no effect on the secondary structure of protein.29,47 The molecular weight of andrographolide is 350.4 Da. The optimum pH for HSA was set to be 7.4, as it has the maximum absorption at this pH.29 Thus, for all the experiments, we have used 0.1 M phosphate buffer at pH 7.4 as a physiological buffer. All other chemicals are of analytical grade, purchased from Sigma-Aldrich. Research-grade CM5 sensor chips, 10× phosphate buffered saline (PBS), and an amine-coupling kit, consisting of N-ethyl-N-(3-dimethylaminopropyl)-carbodiimide (EDC), N-hydroxysuccinimide (NHS), and ethanolamine hydrochloride (pH 8.5; 1 M), were all purchased from GE Healthcare Bio-Sciences (Uppsala, Sweden).
2.1.2 Cell response assay. To carry out cell-response anti-cancer through MTT [3-(4,5-dimethylthiazol 2-yl)-2,5-diphenyltetrazolium bromide] assay, MTT and all the cell lines, such as lung carcinoma (A549), human lung epithelial cells (BEAS-2B), prostate cancer (DU145), and breast cancer cells (MCF-7), used in the study were procured from ATCC (American Type Culture Collection). MCF-7 cells were taken in a 96-well plate, at a density of 5 × 105 cells, and sub-cultured and seeded. Then, the cells were treated with ANDR in a dose-dependent manner (10 μM to 100 μM) in a total volume of 100 μL. MTT crystals (20 μL in 5 mg mL−1 PBS) were added to the plate and mixed after 48 h with 100 μL of DMSO and incubated for 3 h. Using a μ-Quant Bio-tek Instrument, (microplate reader) the absorbance of the mixture was measured at 570 nm. According to the result, the mean ± standard error was calculated and plotted in a graph as the cell response (%) vs. concentration (μM), and IC50 values were calculated from the graph.
2.1.3 Fluorescence spectroscopy. The binding mechanisms of ANDR with macromolecules, HSA and AGP, were determined using fluorescence spectroscopy (Perkin-Elmer LS55 fluorescence spectrometer) with a 10 mm quartz cuvette. In the fluorescence emission measurement, fixed concentrations of HSA and AGP (0.001 mM), titrated with different concentrations of ANDR (0.001 mM to 0.009 mM) in 100 mM phosphate saline buffer of pH 7.4, were measured at 25 °C. The bandwidths of both excitation and emission were fixed at 5 nm, and a 1 cm quartz cuvette was used. For both HSA and AGP, the maximum excitation wavelength was kept at 285 nm. The absorbance of ANDR at higher concentrations introduces the inner filter effect (IFE), which decreases the emission intensity of HSA and AGP, hence interfering with the quenching process. To eliminate the influence of the inner filter effect, which is a common inherent problem in many fluorimetric procedures, the fluorescence intensity was corrected using the following relationship.30| | |
Fcor = Fobs × 10(Aexc + Aemi)/2
| (1) |
where Fcor is the corrected fluorescence intensity, Aexc and Aemi represent the absorbance at the fluorescence excitation (285 nm) and emission wavelengths (340 nm for AGP and 360 nm for HSA), and Fobs is the observed fluorescence.The emission spectra were collected in the wavelength range between 300 nm and 500 nm with a scan speed of 100 nm min−1. This experiment was done thrice, keeping the number of accumulations to 3 times each to reduce noise. Thus, the fluorescence intensities used in the following sections are the corrected values.
2.1.4 Displacement experiment with site I (phenylbutazone) and II (ibuprofen). Site-selective experiments were performed at room temperature using two known site-specific probes; phenylbutazone for site I (IIA) and ibuprofen for site II (IIIA), with a fixed concentration of 0.001 mM, were mixed with HSA (0.001 mM), and ANDR was added with increasing concentrations of 0.001 mM to 0.009 mM. Here, other parameters were kept the same as for intrinsic fluorescence spectroscopy. The incubation times for the known markers were kept at 5 min. The fluorescence intensity was recorded under the same experimental conditions as mentioned above. The fluorescence quenching data was analyzed using the Stern–Volmer equation. All fluorescence intensities were corrected for the absorption of excited and re-absorption of emitted light, and the IFE is corrected by using eqn (1).
2.1.5 SPR measurements. Experiments were carried out on a Biacore X100 device (GE Healthcare, Uppsala, Sweden). A carboxymethylated dextran chip (CM5-Chip, GE Healthcare, Uppsala, Sweden) was fully coupled on Fc2 with HSA or AGP (GE Healthcare), using an amine-coupling strategy (EDC/NHS) and HBS–EP (10 mM HEPES pH 7.4, 150 mM NaCl, 3 mM EDTA, and 0.005% v/v surfactant P20) as a running buffer and sodium acetate pH 4.5–5.0 as sample buffer for the ligand. At pH values above about 3.5, the carboxymethylated dextran on the sensor chip surface is negatively charged, and electrostatic attraction provides an efficient means for concentrating positively charged proteins on the surface. The pH of the protein solution should lie between 3.5 and the isoelectric point of the protein, so that the surface and the ligand carry opposite net charges. Covalent immobilization of proteins is best performed from solutions in 10 mM buffer (e.g. sodium acetate) at pH 4–5.5. These ready-to-use buffers for protein immobilization are available from GE Healthcare. The response level reached ∼14650 RU for HSA and ∼7803 RU for AGP on Fc2. Non-binding Fc1 was mock-treated. Affinities were measured using a kinetic titration series (single cycle kinetics) at 25 °C, in which five ascending concentrations of ANDR were injected consecutively for 120 s at 30 μL min−1, followed by a dissociation time of 600 s. Therefore, ANDR was diluted in the running buffer (PBS + 5% DMSO) at concentrations of 5 μM, 25 μM, 50 μM, 200 μM, and 1000 μM. The signal of the untreated flow cell was subtracted from the binding signal. Additionally, for each run, later blank injections of running buffer only were also subtracted (double referencing). Sensorgrams were analyzed after solvent correction by plotting the analyte concentration against the binding signal at the end of the injection. The resulting isotherm was fitted to obtain the KA values using the steady-state model.
2.1.5.1 Solvent refractive index correction. Since DMSO has a high refractive index, a solvent correction procedure was performed to account for the small variations in the DMSO percentage between samples and running buffer.31 PBS/DMSO mixtures ranging from 4.5% to 5.8% (v/v) (pH 7.4), were injected over the active and reference flow cells, and a calibration curve was obtained. This procedure is very important when working with small molecule–macromolecule interactions, especially when the ligand is immobilized at high density (5000 RU or more). In this experimental setup, the expected response has the same magnitude as, or a lower magnitude than, the signal arising from refractive index mismatches; therefore, including a correction protocol during the evaluation of the data becomes of great significance.
2.1.6 Circular dichroism spectroscopy. The changes in secondary elements, such as α-helix, β-sheet, and random coil content, were monitored using CD spectroscopy. For recording the spectra of free HSA and the HSA–ANDR complex, a Jasco J-815 spectropolarimeter was used at normal temperature (25 °C). Other parameters were followed as measurements ranged from 190–300 nm, including data pitch: 1 nm, bandwidth: 2.00 nm, scanning speed: 50 nm min−1, and accumulations: 3. The final concentration of HSA was fixed at 0.001 mM, and ANDR was added in increasing concentrations from 0.001 to 0.009 mM. The spectra were analyzed for secondary structure determination with CDNN 2.1 software. The experiments were repeated thrice for ANDR, and each time approximate values were obtained.
2.1.7 Molecular docking. To determine the geometrically and energetically stable conformation upon binding of ANDR to HSA and AGP, molecular docking is the best method. Here, a predominant binding model of a protein (known structure) and ligand in 3D can be predicted. AutoDock 4.2.3 was employed to generate a docked conformation of ANDR with HSA and AGP; this uses a Genetic Algorithm (GA) and a Lamarckian Genetic Algorithm to generate conformations. Using this software, the binding free energies and binding sites of ANDR on HSA were investigated. The optimized 2D/3D structure of ANDR was built using Discovery Studio 3.5 software. Using Autodock, the water molecules were removed and non-polar hydrogen atoms were added. The crystal structure of HSA (PDB ID: 1AO6) was downloaded from the Brookhaven Protein Data Bank, and its functional groups were protonated in Autodock (4.2.3), which was chosen for docking studies, where water molecules were removed and hydrogen atoms were added. To define all binding sites, a grid box was generated with a spacing of 0.586 Å and dimensions of (126 × 126 × 126) points (.gpf file). The docking parameters were inserted as the number of GA runs: 30, population size: 150, the maximum number of generations: 27000, the rate of genetic mutation: 0.02, the rate of crossover: 0.8, and GA crossover mode: 2 points. The output is selected as Lamarckian GA (.dpf file), which was implemented in Autodock (4.2.3) to conduct docking simulations. In the end, 30 possible conformers were obtained for HSA and ANDR binding. Among these, the one with the least binding free energy was considered for further analysis of binding mode and binding sites, as this was very close to experimental data. A similar protocol was followed (keeping the same parameters) to calculate the binding energy and efficiency of AGP (PDB ID: 3KQO) and ANDR interactions.
2.1.8 Molecular dynamics simulations. After the ANDR is docked into the active site of HSA, MD simulations were performed using GROMACS v4.6.3, and this package was used to perform a simulation of the previous conformation (lowest binding energy docking conformation). The topology parameters were created by using the Dundee PRODRG2.5 server (beta). The entire simulation is performed on the basis of physiological conditions, as mentioned in the experimental method. The complex structure was immersed in a box of 7.335 × 6.135 × 8.119 nm3 dimensions with extended simple point charge (SPC) water molecules, and this system was neutralized by addition of Na+ counter ions and 43626 solvent atoms. Therefore, the entire system consisted of HSA, ANDR, Na+ of the counter-ions, and solvent atoms. The energy was minimized using the steepest descent method and the conjugate gradient method. Finally, the full system was subjected to 10 ns MD at a temperature of 300 K and 1 bar pressure. The atom coordinates were recorded every 0.5 ps during the simulation. All restraints were then removed and each simulation was run for 10000 ps; simulation and analysis were performed on Linux cluster with 36 nodes (dual Xeon processor) at the Bioinformatics facility of the University of Hyderabad.32,33
3. Results and discussion
3.1 Cytotoxicity study
To reconfirm the importance of ANDR in cancer treatment, we performed a cell viability assay using MTT staining.34 Here, ANDR was used to induce apoptosis in MCF-7, a breast cancer cell line. The rate of apoptosis in the MCF-7 cell line is also dose-dependent (10, 20, 40, 60, 80, and 100 μM), as the inhibition rate increases and the cell activity decreases in a concentration-dependent manner. Furthermore, we have calculated the IC50 value of ANDR to represent the cell viability, and this was found to be 55 μM on MCF-7 cell lines (Fig. SI1†). Thus it can be concluded that ANDR has a strong anti-cancer activity, as well as other properties, such as anti-diabetic, anti-inflammatory, antiviral, antidyslipidemic, low-density lipoprotein oxidation, and antioxidant activity. Also, the biological effects of chemically synthesized ANDR and its analogs were well studied on several cancer cell lines, like lung carcinoma (A549), human lung epithelial cells (BEAS-2B), and prostate cancer (DU145), proving that ANDR is a potent drug possessing anticancer properties.35 Therefore, our study on ANDR, which is very specific to MCF-7 cells, indicates that it can be used for the treatment of cancer. We also tried to understand the molecular level of the interaction of ANDR with plasma proteins.
3.2 Binding analysis by fluorescence quenching mechanism
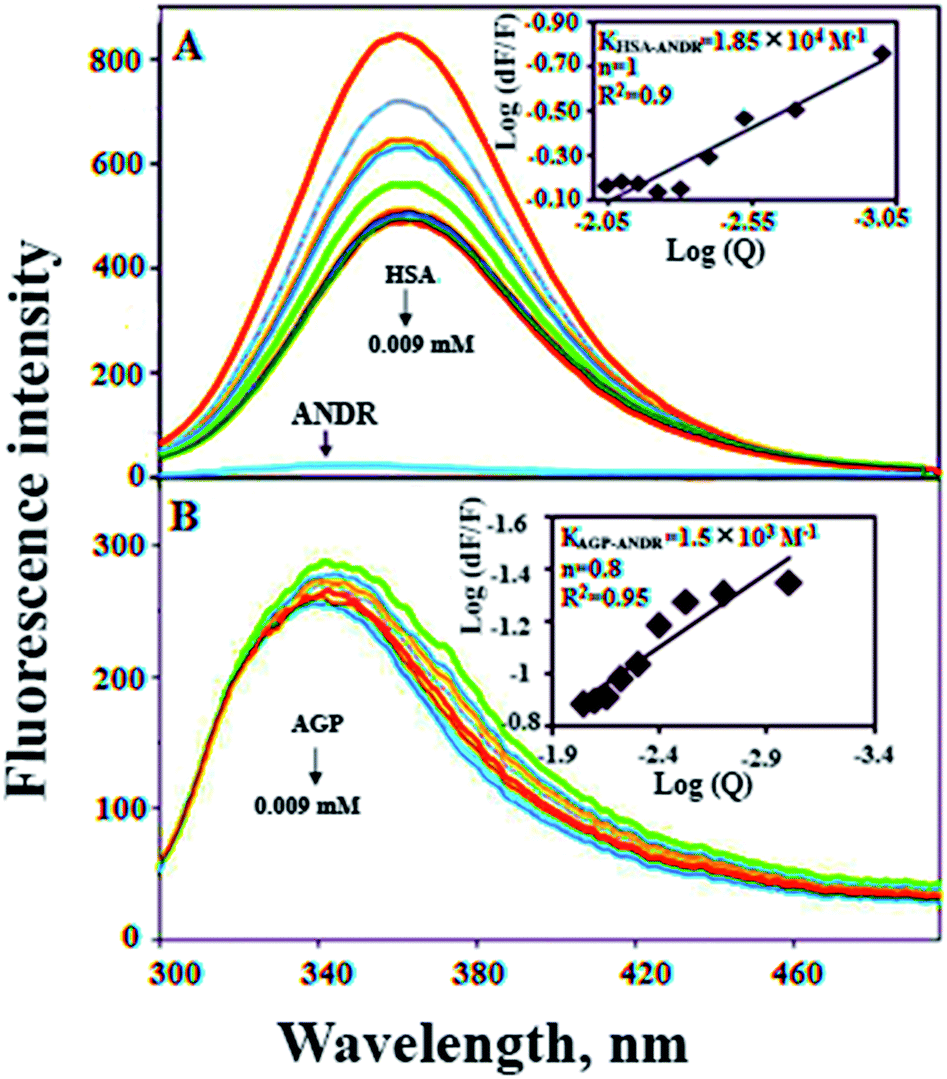
In the present study, we have used fluorescence spectroscopy to unravel the binding mechanism or binding mode of ANDR to HSA or AGP. The fluorescence quenching of AGP and HSA is due to the intrinsic tryptophan residue, which is used as a tool to understand the interaction of ANDR with these proteins (Fig. 1A and B). While titrating ANDR with HSA, the fluorescence maximum intensity at 360 nm was decreased with the addition of ANDR. The major fluorescence of HSA is due to the presence of tryptophan-214 when excited at 280 nm. This indicates that the ANDR binding site exists near to the tryptophan residue, which is located at the IIA subdomain of HSA.
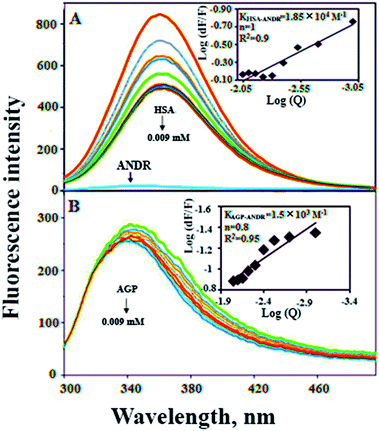 |
| | Fig. 1 Fluorescence emission spectroscopy of HSA and HSA–ANDR complex. (A) Free HSA (0.001 mM) and free HSA with different concentrations of ANDR (0.001, 0.002, 0.003, 0.004, 0.005, 0.006, 0.007, 0.008, and 0.009 mM) in 0.1 M phosphate buffer, pH 7.4, λex = 285 nm, temperature = 25 °C, inset: modified Stern–Volmer plot; plot of log(dF/F) against log[Q] λex = 285 nm, λex = 360 nm. Fluorescence emission spectra of AGP–ANDR in 0.1 M phosphate buffer with pH 7.4, λex = 285 nm, and temperature = 25 °C. (B) Free AGP (0.001 mM) and free AGP with different concentrations of ANDR at 0.001, 0.002, 0.003, 0.004, 0.005, 0.006, 0.007, 0.008, and 0.009 mM. Inset: plot of log(dF/F) against log[Q]. λex = 285 nm and λem = 340 nm. | |
In order to understand the fluorescence quenching mode (static (s) or dynamic (d)) of HSA and the AGP–ANDR complex, the quenching data were analyzed using the following equation:
| | |
F0/F = 1 + Kqt0[Q] = 1 + KD[Q]
| (2) |
where
KD is the Stern–Volmer quenching constant (
KD =
Kqt0),
Kq is the bimolecular quenching rate constant,
t0 is the lifetime of the fluorophore in the absence of quencher, [Q] is the quencher concentration, and
F0 and
F are the fluorescence intensities in the presence and absence of the quencher.
36–39 From the above equation, we have calculated the static quenching constant,
Kq(s)
(ANDR) = 5.0 ± 0.03 × 10
13 M
−1 s
−1 (Fig. SI2
†). According to the standard procedures, if
Kq is more than the maximum collisional quenching constant, then the static quenching is dominant. We found that the dynamic quenching constant is 2.06 ± 0.03 × 10
10 M
−1 s
−1.
32,40–43 Hence the binding of ANDR to HSA is ‘static’ as
Kq(s) is very much greater than the colloidal constant. This indicates the formation of the fluorophore–quencher complex. The results demonstrated that HSA fluorescence quenching by ANDR may follow a static mechanism and was initiated by HSA–ANDR complex formation, rather than dynamic collision.
When small molecules are bound independently to a set of the equivalent sites on a macromolecule, and to maintain the quenching in a linear relationship, the modified Stern–Volmer equation was used.11,44,45
| | |
log[(F0 − F)/F] = logKs + nlog[Q]
| (3) |
where
n is the number of ligands binding to the protein,
Ks is the binding constant, and [Q] is the drug concentration. The d
F is (
F0 −
F), where
F0 is the initial fluorescence of free HSA or AGP and
F is the fluorescence with different concentrations of ANDR.
We have plotted a graph of log(dF/F) against log[Q]. From the graph, using the slope and the intercept values, the binding constant and the number of binding sites were calculated. We have found that the numbers of binding sites of ANDR to HSA and AGP are 1 and 0.8 for ANDR, supporting a one-to-one interaction between HSA and ANDR. Also, the binding constant (Fig. 1A inset), KANDR = 1.85 ± 0.02 × 104 M−1, indicates strong binding of ANDR with HSA. This data is well correlated with the data found in silico i.e. 1.2 ± 0.03 × 104 M−1. The same experimental procedures were followed at 37 °C, where we found that on increasing the temperature, the fluorescence emission decreases, or in other words, the extent of lowering of fluorescence emission was higher at lower temperatures. For ANDR, the values of Kb and n were calculated at 37 °C, and the observed values are Kb = 1.2 ± 0.03 × 104 M−1 and ΔG = −5.5 kcal M−1 at 37 °C (Table 1). Thus, the data show a slight decrease in Kb on increasing the temperature as a clear indication of static quenching.46 As we know, very few drug molecules bind efficiently with AGP; therefore, we have also calculated the binding constant, which was found to be KANDR = 1.5 ± 0.03 × 103 M−1; the in silico binding affinity of AGP is 7.7 ± 0.05 × 104 M−1, and these are in the range of known FDA approved drug values (103 to 106 M−1).3 Recently, we reported that few phytochemicals bind to AGP and HSA;13,14 from this point of view, we can state that due to the acute-phase protein, AGP, binding with ANDR, in chronic inflammation/cancer conditions, the ANDR can bind to overexpressed AGP. Thus, these results indicate that ANDR binds to both HSA and AGP. This reveals that there is only one binding site for ANDR for serum proteins, suggesting that both the proteins may be used as a carrier for ANDR in different disease conditions.
Table 1 Binding parameters of ANDR interactions with HSA in 0.1 M phosphate buffer, pH 7.4, at different temperatures, obtained and calculated from fluorescence quenching results
| T (K) |
Kb (×104 M−1) |
ΔG (kcal M−1) |
| 298 |
1.85 ± 0.01 |
−5.1 |
| 310 |
1.22 ± 0.03 |
−5.5 |
3.2.1 Calculation of free energy change. As the binding of HSA to the drugs is static, there will be four types of non-covalent forces, hydrogen bonding, van der Waals, hydrophobic, and electrostatic interactions, that stabilize their binding, which can be confirmed by calculation of the thermodynamic parameters. We know that the free energy change (ΔG°) can be calculated using the following equation:| | |
ΔG° = −RTlnK
| (4) |
where ΔG° is the free energy change, R is the gas constant, T is the absolute temperature, and K is the binding constant. From the above equation, the free energy change for ANDR has been calculated as −5.79 ± 0.031 kcal mol−1 at 25 °C, when binding with HSA. Interestingly, this data also supports the in silico analysis of the HSA–ANDR complex, where 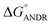 = −5.6 ± 0.031 kcal mol−1. Also, we have calculated the free energy change of binding to AGP and found this to be −4.31 ± 0.031 kcal mol−1 (
= −5.6 ± 0.031 kcal mol−1. Also, we have calculated the free energy change of binding to AGP and found this to be −4.31 ± 0.031 kcal mol−1 (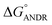 ). Similar results were found in docking with AGP and ANDR, where
). Similar results were found in docking with AGP and ANDR, where 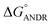 = −6.67 ± 0.21 kcal mol−1. Therefore, this data suggests that the binding between the protein and ligand can be stabilized by hydrophobic interaction and hydrogen bond formation. Similar results were recently published with other natural compounds (trans-feruloyl maslinic acid, betulinic acid, β-sitosterol, lupeol, L-dopa, 7-hydroxycoumarin, trimethoxy flavone, embelin, and corilagin).12–14,29,32,47,48 The fluorescence quenching mechanism was found to be static, which is evident from the bimolecular quenching constant for ANDR of 5.27 ± 0.02 × 1013 M−1 s−1. Quenching of the emission maximum at 340 nm of AGP was observed with ANDR. Interestingly, blue and red shifts were observed in the peaks of AGP–ANDR (Fig. 1B inset), whereas with HSA, there is no shift in the peaks of ANDR. This phenomenon might be due to the unfolding and refolding of the protein after binding. A possible explanation for the shift might be the differential exposure of tryptophan residues at various positions in the protein, which may be exposed differentially to the polar environment, leading to a shift in the emission maximum at 340 nm. The binding constants with ANDR are 10 times less than that of HSA. ANDR binds with AGP in a 1:1 ratio, indicating that only one molecule is bound with AGP, and similarly with HSA. The negative free energies with all these concentrations of ANDR are less than the values obtained with HSA. From the binding constants and free energies of HSA and AGP, it is apparent that ANDR has a higher affinity to HSA than to AGP, signifying that the HSA–ANDR complex is more stable than the AGP–ANDR complex. There are several studies showing that AGP has moderate to high affinity (103 to 106 M−1) with several drugs;49–51 some drugs showed equal binding with both HSA and AGP,52 and a few drugs, such as acetaminophen, phenobarbital, theophylline, and valproic acid, showed negligible binding to AGP.53 In our study, the ANDR compound showed less binding to AGP in comparison with HSA. These findings suggest that HSA plays a major role in binding; however, this should be verified in vivo in various pathological conditions where AGP expression is upregulated.
= −6.67 ± 0.21 kcal mol−1. Therefore, this data suggests that the binding between the protein and ligand can be stabilized by hydrophobic interaction and hydrogen bond formation. Similar results were recently published with other natural compounds (trans-feruloyl maslinic acid, betulinic acid, β-sitosterol, lupeol, L-dopa, 7-hydroxycoumarin, trimethoxy flavone, embelin, and corilagin).12–14,29,32,47,48 The fluorescence quenching mechanism was found to be static, which is evident from the bimolecular quenching constant for ANDR of 5.27 ± 0.02 × 1013 M−1 s−1. Quenching of the emission maximum at 340 nm of AGP was observed with ANDR. Interestingly, blue and red shifts were observed in the peaks of AGP–ANDR (Fig. 1B inset), whereas with HSA, there is no shift in the peaks of ANDR. This phenomenon might be due to the unfolding and refolding of the protein after binding. A possible explanation for the shift might be the differential exposure of tryptophan residues at various positions in the protein, which may be exposed differentially to the polar environment, leading to a shift in the emission maximum at 340 nm. The binding constants with ANDR are 10 times less than that of HSA. ANDR binds with AGP in a 1:1 ratio, indicating that only one molecule is bound with AGP, and similarly with HSA. The negative free energies with all these concentrations of ANDR are less than the values obtained with HSA. From the binding constants and free energies of HSA and AGP, it is apparent that ANDR has a higher affinity to HSA than to AGP, signifying that the HSA–ANDR complex is more stable than the AGP–ANDR complex. There are several studies showing that AGP has moderate to high affinity (103 to 106 M−1) with several drugs;49–51 some drugs showed equal binding with both HSA and AGP,52 and a few drugs, such as acetaminophen, phenobarbital, theophylline, and valproic acid, showed negligible binding to AGP.53 In our study, the ANDR compound showed less binding to AGP in comparison with HSA. These findings suggest that HSA plays a major role in binding; however, this should be verified in vivo in various pathological conditions where AGP expression is upregulated.
3.3 Binding analysis from SPR data
There are various techniques to understand molecular interactions; among these, fluorescence and SPR spectroscopy are widely used. Interactions between macromolecules and small molecules by fluorescence spectroscopy are based on the interaction between a quencher and a fluorophore. This allows fluorescence quenching, binding constants, and mechanisms of molecular interaction to be analyzed. SPR is an optical technique, which is based on the refractive index change near to a metal (Au) surface. The refractive index changes reflect the absorbance of analyte in solution onto the ligand HSA immobilized on the surface (Fig. 2). The advantage of SPR is that it can monitor the surface-constrained interaction in real time without the requirement for labeling. In this study, interaction between ANDR and HSA and AGP was studied by means of the above-mentioned techniques. The results showed that ANDR can be bound to HSA to form a complex. The association constants obtained from the fluorescence and SPR experiments were very close, namely, 1.85 ± 0.02 × 104 M−1 (fluorescence) and 3.1 ± 0.04 × 103 M−1 (SPR). As the interaction of ANDR with HSA is formed under flow conditions that usually cause shear stress, and takes place between an immobilized ligand and a mobile analyte, it might be slightly weaker than binding affinities derived from assays with both binding partners in solution without any shear stress forces, like fluorescence spectroscopy. Additionally, covalent coupling of HSA to the chip surface takes place randomly via primary amines in the protein. If these amines are situated next to the binding pocket for ANDR, interactions may be sterically hindered, reducing binding affinity in contrast to fluorescence spectroscopy, where no attachment is needed. However, we have also performed SPR by immobilizing AGP to investigate the interaction of ANDR with AGP under flow conditions. The binding affinities obtained from fluorescence and SPR spectroscopy were in good agreement with each other, at 1.5 ± 0.03 × 103 M−1 and 1.39 ± 0.06 × 103 M−1, respectively (Fig. SI3†). Steady-state analysis (weak and moderate interactions) data was initially extracted using Biacore X100 Control software, version 2.0.1. Hence, the binding constants obtained from the fluorescence and SPR are similar.
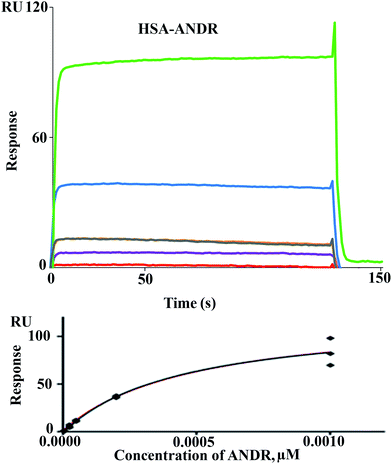 |
| | Fig. 2 Sensorgrams of binding of ANDR to HSA immobilized on CM 5 sensor chip (top) and respective Req values fitted to the steady-state model, giving the final binding isotherm (bottom). Increasing concentrations of analyte are denoted by different colors: 5 μM (orange), 25 μM (purple), 50 μM (light black and yellow), 200 μM (blue), and 1000 μM (green) for ANDR–HSA. | |
3.4 Site selectivity binding studies of ANDR with HSA
To determine the exact binding pocket of HSA upon interaction with ANDR, molecular displacement studies were carried out using site-specific markers for monitoring site I and site II of HSA. Here, we took two site-specific markers of HSA; phenylbutazone, which binds at IIA, and ibuprofen for IIIA, to determine the specific binding of ANDR with HSA through site-competitive displacement experiments. The concentration of HSA and those of site-specific markers are kept constant (0.001 mM), and a decrease in fluorescence emission intensity was observed during titration of different concentrations of ANDR into HSA–phenylbutazone. The binding constant obtained was close to that of HSA–ANDR alone. From the displacement experiment, the binding constant decreased slightly in the presence of phenylbutazone, and was found to be KANDR = 1.8 ± 0.03 × 104 M−1 (Fig. 3), which was very close to the intrinsic fluorescence data, which shows KANDR = 1.85 ± 0.02 × 104 M−1. Therefore, competition was indicated between phenylbutazone and ANDR for binding site IIA of HSA. Our experimental data are consistent with the docking studies, which show ANDR binding to the IIA subdomain of HSA. However, there are no observed changes in the binding constants of other site-specific markers for the IIIA subdomain of HSA (2.1 ± 0.05 × 104 M−1), compared with the absence of the site-specific probe; thus, ANDR displaces only phenylbutazone from subdomain IIA. Therefore, from this study, we can assume that ANDR displaces the phenylbutazone molecule and forms a stable complex due to hydrophobic interactions at the IIA domain.
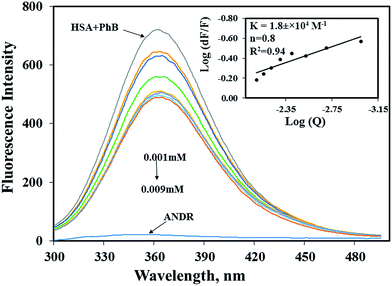 |
| | Fig. 3 Competitive binding of a ligand with HSA. Displacement of phenylbutazone from the HSA–phenylbutazone complex by ANDR. In the graph logdF/F vs. logQ (inset) was plotted to calculate the binding constant and the number of binding sites. | |
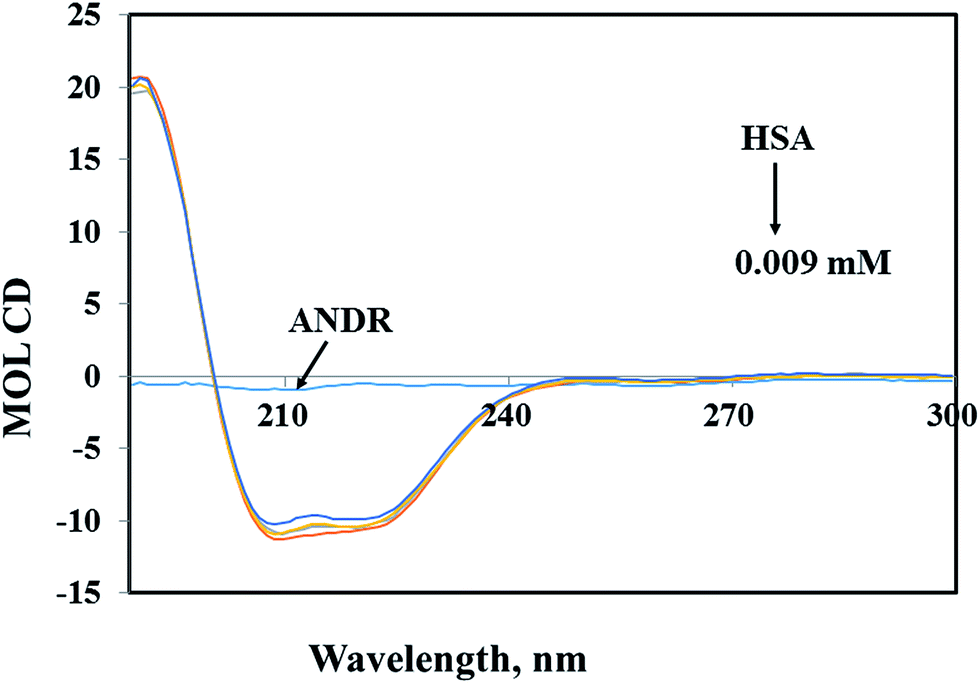
3.5 Circular dichroism analysis
Circular dichroism (CD) spectroscopy is a valuable technique for defining canonical secondary structures of proteins, based on empirically-defined spectroscopic signatures derived from proteins with known three-dimensional structures. CD (between 195 nm and 300 nm) will yield secondary structural information. The chromophore being the peptide bond, the recorded spectrum will display specific signatures that can be related to certain peptide backbone arrangements. This plays an important role in understanding the secondary structural elements (percent of α-helix, β-sheet, and random coil content) of proteins. A ligand binding to a globular protein may alter the secondary structure that results in the conformation.54 In our study, the percentages of portions of secondary structural elements of the HSA–ANDR complex were evaluated. With the addition of an increased concentration of ANDR to HSA, the CD-spectral behavior changes marginally (Fig. 4). Also, we have calculated the percentage of each of the secondary structural elements of HSA using CDNN 2.1 software. There were changes in the α-helix, β-sheet, and random coil contents, as the ANDR concentration increased. The free HSA contains 56.0 ± 2.5% α-helix, 25.6 ± 0.62% β-sheet, and 18.4 ± 0.82% random coil (see Table 2). After gradual addition of ANDR from 0.001 to 0.009 mM, the α-helical content decreased to 50.9 ± 2.25%, while the percentage of β-sheet and random coil content increased to 29.4 ± 0.74% and 19.7 ± 0.84%, respectively. Similar results were observed, with a decreased α-helical content and an increased β-sheet and random coil content, in earlier reports from our laboratory.11–13,29,55–57 It is clearly evident that there was only a marginal decrease in the negative ellipticity in the region of far-UV CD, without any significant shift in peaks. The similarity between the CD spectral shapes of free HSA and HSA–ANDR suggested that the structure of HSA was also predominantly α-helix (Table 2), indicating that the binding of the drug to HSA induced a slight decrease in the α-helical structure content of the protein, with a minor increase in the β-sheets and random coils. The changes in the α-helix of HSA indicated that the ANDR might have bound with the amino acid residues present in the HSA polypeptide chain and destroyed the hydrogen bonding networks.58 This might in turn have exerted a change in the secondary structure of the HSA. CD spectroscopy revealed conformational changes in the HSA, resulting from the change in the physiological environment of HSA after the addition of ANDR. Thus, ANDR created a variation in the local environment of HSA and changed its nature, indicating that the binding of ANDR to HSA induced a slight decrease in the α-helical structure content of the protein, with a minor increase in the β-sheets and random coils. Although the binding of ANDR is greater (1.85 × 104 M−1) to HSA, however, the protein conformation is not much observed due to orientational changes upon binding of ANDR to HSA.
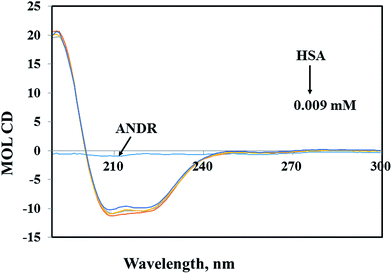 |
| | Fig. 4 Circular dichroism of the free HSA and HSA–ANDR complexes. The free HSA and HSA–ANDR complexes in aqueous solution had a protein concentration of 0.001 mM, and ANDR concentrations were 0.001, 0.005, and 0.009 mM. | |
Table 2 Percentage of the secondary structure of HSA and HSA–ANDR complex. Mean ± SE (n = 3)
| Secondary structure (%) |
HSA |
HSA-0.001 (mM) |
HSA-0.005 (mM) |
HSA-0.009 (mM) |
| Helix |
56.0 ± 2.5 |
56.4 ± 2.5 |
54.8 ± 2.3 |
50.9 ± 2.25 |
| Beta-turn |
25.6 ± 0.62 |
25 ± 0.62 |
26 ± 0.62 |
29.4 ± 0.74 |
| Random coil |
18.4 ± 0.82 |
18.6 ± 0.82 |
19.2 ± 0.83 |
19.7 ± 0.84 |
3.6 Binding studies from docking results
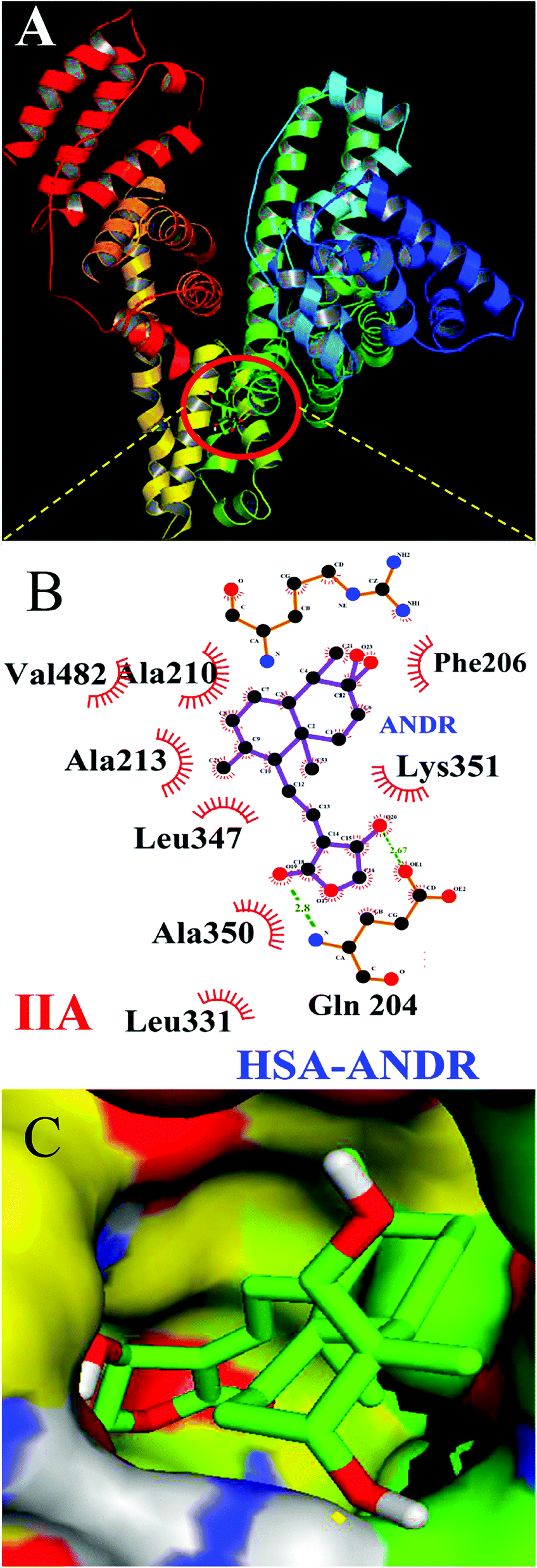
To confirm the in vitro experimental data, molecular docking studies were carried out using Autodock (4.2.3) software and generated 30 different conformations with different binding conformations. Based on the lowest binding energy of the docking results, a conformer was taken and found to be 1.2 ± 0.03 × 104 M−1, and the free energy change was −5.6 ± 0.031 kcal mol−1. Therefore, computationally calculated K and ΔG° values suggest that the binding of ANDR and HSA was similar to the results obtained with fluorescence data. These forces stabilize the docking conformation and are consistent with the binding mode obtained from the experimental results. It was also observed that ANDR binds at subdomain IIA of HSA in all of these 30 conformers. After analyzing the conformation of the complex, it was shown that ANDR binds in the hydrophobic pocket of subdomain IIA, and its binding was stabilized by the formation of 2 H-bonds between ANDR and GLN204 of HSA, with bond distances of 2.67 and 2.8 Å (Fig. 5A–C). Furthermore the microenvironment (around 4 Å region) i.e. the active site of HSA, consisting of amino acid residues, Cys, Leu, His, Thr, Leu, Gly, Phe and Lys, etc. are in the vicinity of the ligand and conjugated with ANDR. From the binding site analysis, it is clearly evident that the ANDR–HSA complex is mainly stabilized by hydrophobic interactions. This supports the quenching of fluorescence, which is evident from the location of the ANDR, which is in close proximity to the tryptophan residue of HSA (Trp 214), suggesting the existence of hydrophobic interactions. The AGP and ANDR interaction was computationally calculated, and it was found that their interaction was stabilized in the ANDR–AGP complex by formation of 3 H-bonds with Pro 131, Lys 135, and Thr 158 with distances of 2.1 Å, 3.2 Å, and 3.12 Å, respectively (Fig. 6A–C). Furthermore, the binding constant (KANDR) is 7.7 ± 0.05 × 104 M−1 and the free energy change (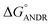 ) is −6.67 ± 0.21 kcal mol−1. These results support the experimental results (KANDR = 1.5 ± 0.03 × 103 M−1 and
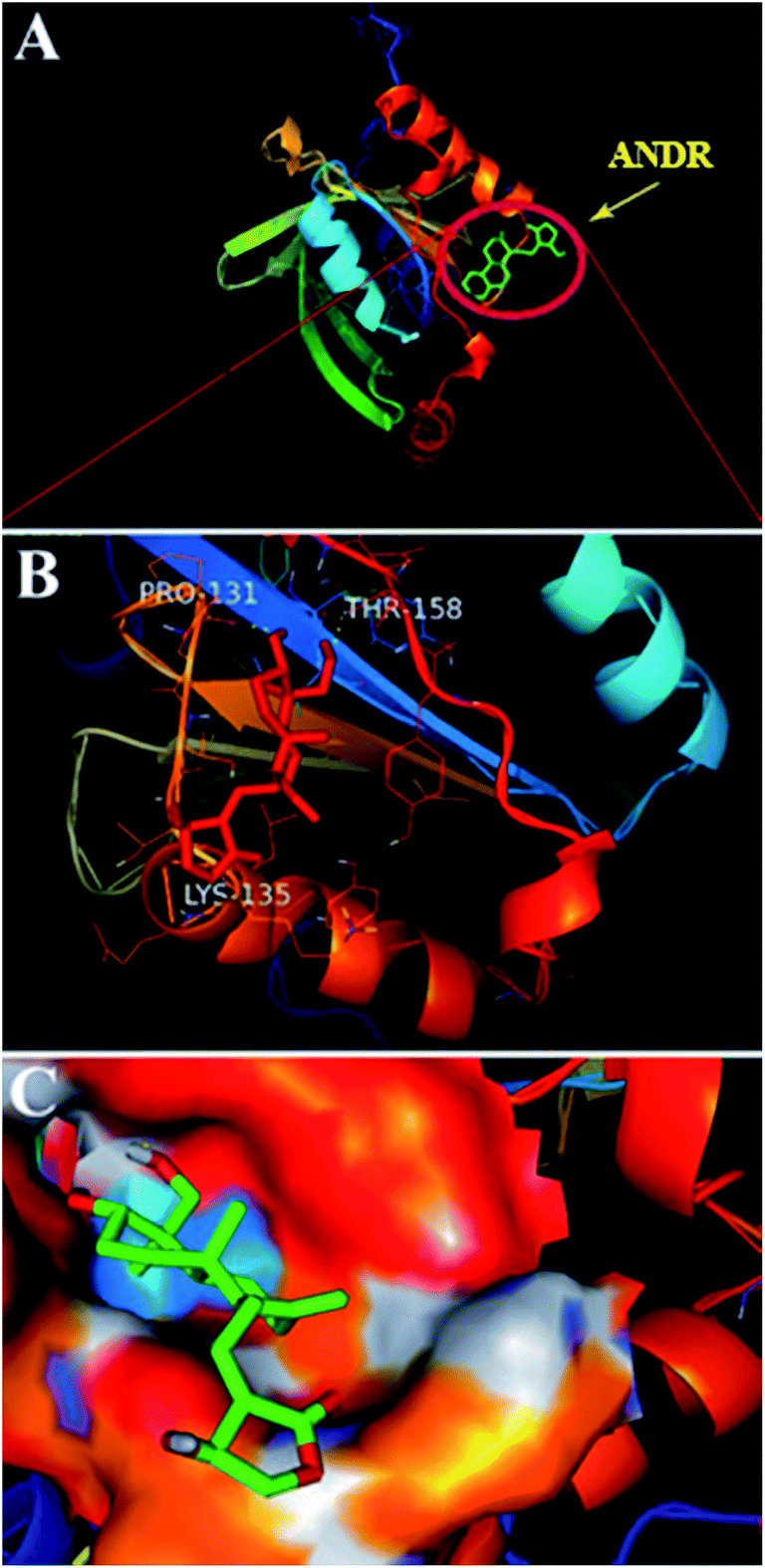
) is −6.67 ± 0.21 kcal mol−1. These results support the experimental results (KANDR = 1.5 ± 0.03 × 103 M−1 and 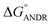 = −4.31 ± 0.031 kcal mol−1) found for the intrinsic fluorescence of the AGP and ANDR interaction. The interaction of ANDR with HSA is mainly stabilized by hydrophobic interactions, along with two hydrogen bonds, whereas with AGP, the main forces involved are three hydrogen bonds. ANDR shows robust binding to both HSA and AGP. Since AGP is an acute-phase protein expressed in pathological conditions, binding of ANDR to the AGP warrants drug-binding studies.
= −4.31 ± 0.031 kcal mol−1) found for the intrinsic fluorescence of the AGP and ANDR interaction. The interaction of ANDR with HSA is mainly stabilized by hydrophobic interactions, along with two hydrogen bonds, whereas with AGP, the main forces involved are three hydrogen bonds. ANDR shows robust binding to both HSA and AGP. Since AGP is an acute-phase protein expressed in pathological conditions, binding of ANDR to the AGP warrants drug-binding studies.
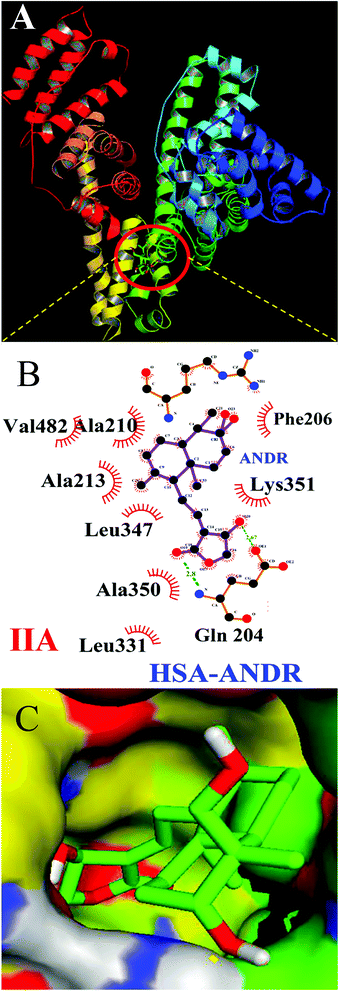 |
| | Fig. 5 Docking conformation of HSA–ANDR complex obtained from Autodock v 4.2. (A) ANDR bound to IIA domain on HSA (protein represented in ribbon model and ligand represented in stick model). (B) Ligplot of ANDR binding site, showing two hydrogen bonds (Gln 204) and hydrophobic interactions of HSA with ANDR. (C) The hydrophobic pocket of the IIA domain surrounding the ANDR. | |
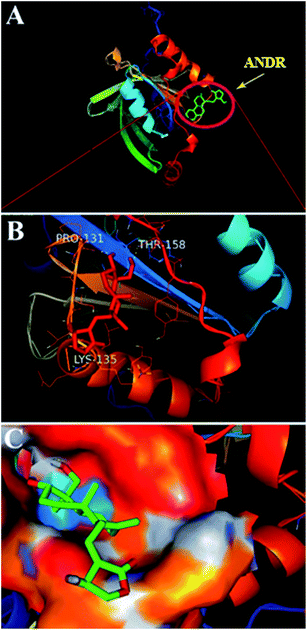 |
| | Fig. 6 Docking conformation of AGP–ANDR complex obtained from Autodock v 4.2. (A) ANDR bound to AGP (protein represented in ribbon model and ligand represented in stick model). (B) Stereo view of ANDR bound to AGP (prepared by using Pymol v 1.5) in surrounding residues is represented as lines (three hydrogen bonds are with Pro 131, Lys 135, and Thr 158). (C) The binding pocket surrounding the ANDR. | |
3.7 Rigidity of HSA–ANDR complexes observed from molecular dynamics simulation data (MD)
In the above study, to investigate the binding affinity and secondary structural changes induced by ANDR, molecular dynamics simulations of HSA and the HSA–ANDR complex were performed and compared. So, to measure the structural properties for this study, we have used root mean square deviations (RMSD), radius of gyration (Rg), and root mean square fluctuation (RMSF), and the qualitative stability, rigidity, and conformational changes of the HSA were compared during the time of simulation.
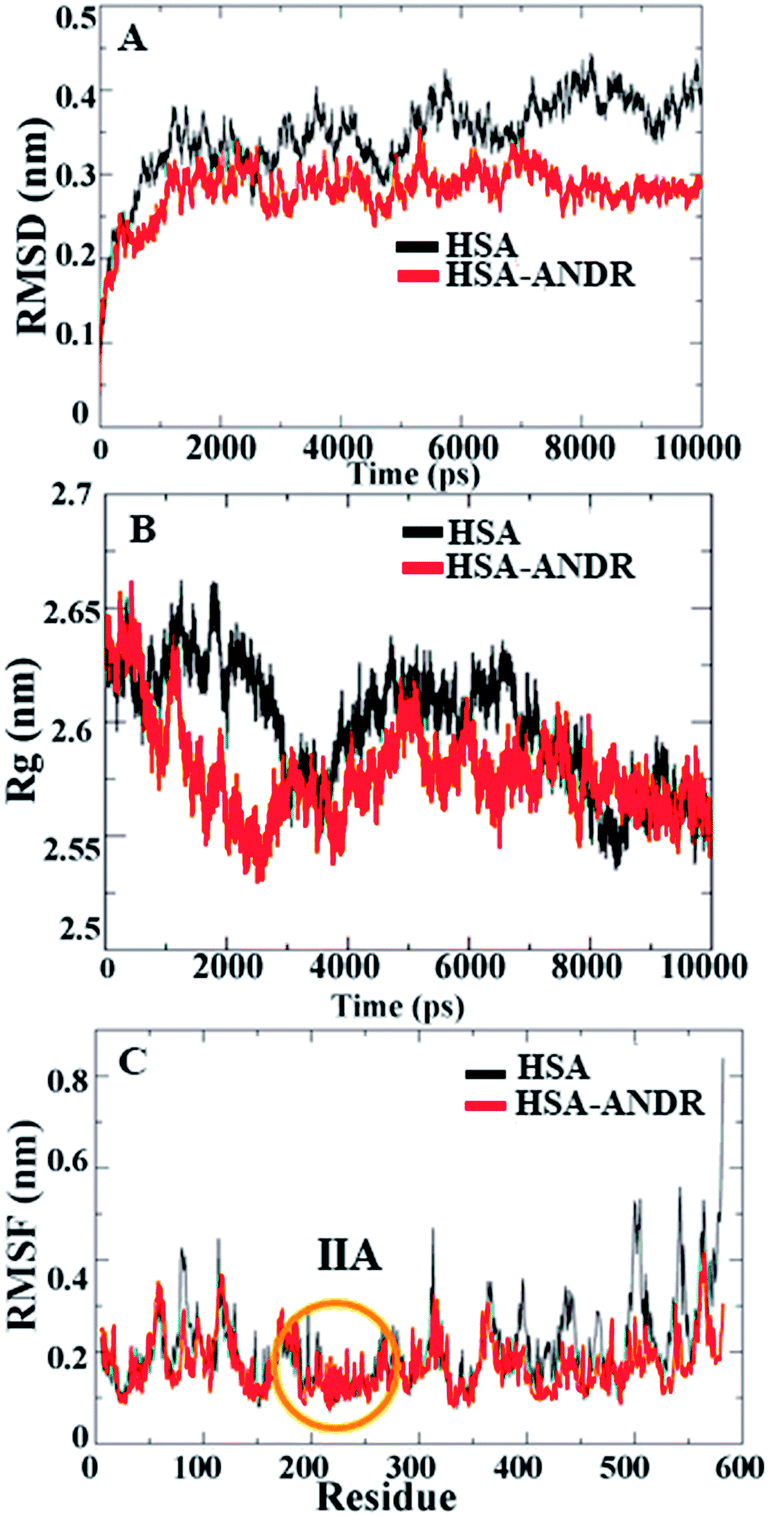
Since HSA is an important protein in blood plasma, the stability, rigidity, and conformational changes of HSA with ligand molecules play a more important role than with AGP; hence, here we focused on HSA MD simulations with ANDR. Based on the agreement of experimental and computational evidence, the lowest binding energy docked conformation of HSA–ANDR (bound to subdomain IIA) was chosen and used as a starting point for a 10 ns simulation. The RMSD values of atoms in free and ANDR-bound HSA were plotted from to 0–10 ns, as shown in Fig. 7, and the analysis indicated that the RMSD of both systems reaches equilibrium and oscillates around an average value after 3 ns simulation time. Fig. 7A shows that the data point of fluctuation was determined as follows: for HSA, 0.40 ± 0.035 nm; HSA–ANDR, 0.29 ± 0.056 nm. The rigidity could be attributed to the unfolding of the protein, which is in agreement with the CD studies that showed minor changes in the secondary structural elements. Therefore, the changes are not due to destabilization and may be due to partial unfolding of HSA. So, we conclude that during MD simulation, there is no significant structural drift from the HSA structure. Recently, similar reports were documented from our lab related to protein stability.12–14,55 We also examined the time evolution of the radius of gyration (Rg) in the course of 10 ns of MD simulations of free HSA and the HSA–ANDR complex, and the results are shown in Fig. 7. The Rg values for the complex stabilized at about 4 ns and achieved equilibrium after 6 ns (Fig. 7B). Initially, the Rg values of free HSA and the complex with the drug were 2.64 nm. The free HSA and HSA–ANDR were stabilized at 2.57 ± 0.02 nm and 2.52 ± 0.04 nm. As expected, by reducing the radius of gyration and increasing the compaction rate in the binding to ANDR, all these findings align with those for lupeol and piperine complex formation with HSA.14,55 So, based on the RMSD and Rg values obtained from our MD simulation data, it was concluded that HSA exhibits a slight structural change when combining with ANDR. Thus both experimental and computational results suggest slight conformational changes, probably at the binding region of the protein. Local protein mobility can be determined by RMSF values, which were plotted against residue numbers on a 10 ns trajectory. The RMSF plot shows the total amino acid fluctuation, which is the total polypeptide chain (Fig. 7C). The graph strongly indicates that the IIA subdomain for ANDR fluctuated less than others (IA, IB, IIB, IIIA, and IIIB). This data suggests that the drug molecule (ANDR) binds in the IIA subdomain and forms a rigid structure during MD simulation. The data clearly suggest that the ANDR binds specifically to the IIA subdomain, which supports the experimental evidence found from molecular displacement by site-specific markers. The rigidity of the binding sites from RMSF studies indicated that ANDR binds within subdomain IIA, which is in agreement with the docking studies. These results indicate that ANDR binds specifically to the respective domain, suggesting that binding sites are very specific for ANDR. Thus, during the simulation, the HSA–ANDR complexes are stable after 4 ns. Nonetheless, changes in the secondary structure were observed that many be due to partial unfolding of HSA. The unfolding of the protein structure may due to orientational changes upon binding of ANDR to HSA.
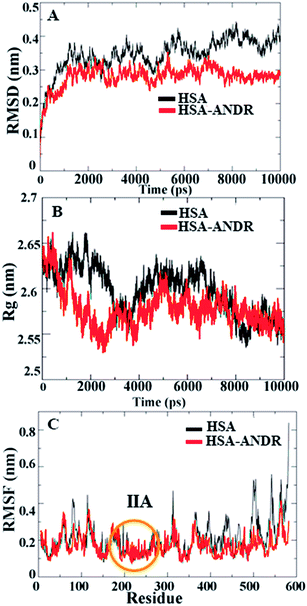 |
| | Fig. 7 Molecular dynamics simulation. (A) Time dependence of Root Mean Square Deviations (RMSD). Cα RMSD values for free HSA (black) and the HSA–ANDR (red) complex during a 10 ns MD simulation. (B) Time evolution of the radius of gyration (Rg). Radius of gyration (Rg) values during 10 ns of MD simulation of HSA (black) and HSA–ANDR (red) complex. (C) The RMSF values as a function of residue numbers. The RMSF values of free HSA (black) and HSA–ANDR (red) complex were plotted against residue numbers. | |
4. Conclusion
In the cell response study, the MCF-7 cell line of ANDR showed a concentration-dependent inhibitory effect; thus, it can be suggested that ANDR is a potent candidate against MCF-7 cell lines. Fluorescence studies of ANDR show quenching of the emission spectra of HSA and AGP proteins. Furthermore, the binding efficiency was measured by two methods, i.e. fluorescence emission and SPR, for ANDR with HSA. The binding constants from both techniques were found to be close. Furthermore, CD data reveals that the α-helical content is increased as compared with the β-sheet and random coil contents, indicating that partial changes in the secondary structural elements are shown in HSA. Additionally, site-specific marker experiments show ANDR competing with phenylbutazone, thereby replacing it for the binding site of subdomain IIA; this supports the molecular docking results, further indicating the formation of a hydrogen-bond-stabilized HSA–ANDR complex. Furthermore, MDS studies indicated that the docked conformer is stable. In a time-dependent simulation study, ANDR binds at the IIA subdomain within 4000 ps and is stabilized. Hence, our observation allowed us to understand the molecular level interaction and characterization of ANDR with plasma proteins and thus it can be further utilized as a therapeutic agent.
References
- D. Carter, X.-M. He, S. H. Munson, P. D. Twigg, K. M. Gernert, M. B. Broom and T. Y. Miller, Science, 1989, 244, 1195–1198 CAS.
- X. M. He and D. C. Carter, Nature, 1992, 358, 209–215 CrossRef CAS PubMed.
- A. Varshney, P. Sen, E. Ahmad, M. Rehan, N. Subbarao and R. H. Khan, Chirality, 2010, 22, 77–87 CrossRef CAS PubMed.
- J. Flarakos, K. L. Morand and P. Vouros, Anal. Chem., 2005, 77, 1345–1353 CrossRef CAS PubMed.
- F. Ding, W. Liu, N. Li, L. Zhang and Y. Sun, J. Mol. Struct., 2010, 975, 256–264 CrossRef CAS.
- K. Schmid, R. B. Nimberg, A. Kimura, H. Yamaguchi and J. Binette, Biochim. Biophys. Acta, 1977, 492, 291–302 CrossRef CAS.
- T. Fournier, N. Medjoubi-N and D. Porquet, Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol., 2000, 1482, 157–171 CrossRef CAS.
- J. M. Kremer, J. Wilting and L. Janssen, Pharmacol. Rev., 1988, 40, 1–47 CAS.
- Z. Huang and T. Ung, Curr. Drug Metab., 2013, 14, 226–238 CAS.
- D. P. R. Yeggoni, M. M. Darla, C. S. Reddy and R. Subramanyam, J. Biomol. Struct. Dyn., 2015, 34, 1–46 Search PubMed.
- D. P. Yeggoni and R. Subramanyam, Mol. Biosyst., 2014, 10, 3101–3110 RSC.
- D. P. Yeggoni, A. Rachamallu and R. Subramanyam, J. Photochem. Photobiol., B, 2016, 160, 248–259 CrossRef CAS PubMed.
- D. P. Yeggoni, A. Rachamallu and R. Subramanyam, RSC Adv., 2016, 6, 40225–40237 RSC.
- D. P. Yeggoni, A. Rachamallu and R. Subramanyam, J. Biomol. Struct. Dyn., 2015, 33, 1336–1351 CAS.
- W.-W. Chao and B.-F. Lin, China's Med., 2010, 5, 1 CrossRef PubMed.
- Y. C. Shen, C. F. Chen and W. F. Chiou, Br. J. Pharmacol., 2002, 135, 399–406 CrossRef CAS PubMed.
- Y.-F. Xia, B.-Q. Ye, Y.-D. Li, J.-G. Wang, X.-J. He, X. Lin, X. Yao, D. Ma, A. Slungaard and R. P. Hebbel, J. Immunol., 2004, 173, 4207–4217 CrossRef CAS.
- E. Amroyan, E. Gabrielian, A. Panossian, G. Wikman and H. Wagner, Phytomedicine, 1999, 6, 27–31 CrossRef CAS PubMed.
- N. P. Trivedi, U. M. Rawal and B. P. Patel, Integr. Cancer Ther., 2007, 6, 271–280 CrossRef CAS PubMed.
- J. Xu, Z. Li, M. Cao, H. Zhang, J. Sun, J. Zhao, Q. Zhou, Z. Wu and L. Yang, Int. J. Biol. Macromol., 2012, 51, 738–742 CrossRef CAS PubMed.
- K. Vuignier, J. Schappler, J.-L. Veuthey, P.-A. Carrupt and S. Martel, Anal. Bioanal. Chem., 2010, 398, 53–66 CrossRef CAS PubMed.
- J. Koch-Weser and E. M. Sellers, N. Engl. J. Med., 1976, 294, 526–531 CrossRef CAS PubMed.
- J. Ghuman, P. A. Zunszain, I. Petitpas, A. A. Bhattacharya, M. Otagiri and S. Curry, J. Mol.
Biol., 2005, 353, 38–52 CrossRef CAS PubMed.
- N. Desai, V. Trieu, Z. Yao, L. Louie, S. Ci, A. Yang, C. Tao, T. De, B. Beals and D. Dykes, Clin. Cancer Res., 2006, 12, 1317–1324 CrossRef CAS PubMed.
- K. Donaldson, V. Stone, C. Tran, W. Kreyling and P. J. Borm, Occup. Environ. Med., 2004, 61, 727–728 CrossRef CAS PubMed.
- H. Chen, C. Khemtong, X. Yang, X. Chang and J. Gao, Drug Discovery Today, 2011, 16, 354–360 CrossRef CAS PubMed.
- D. B. Shenoy, A. A. Antipov, G. B. Sukhorukov and H. Möhwald, Biomacromolecules, 2003, 4, 265–272 CrossRef CAS PubMed.
- F. Caruso, Adv. Mater., 2001, 13, 11–22 CrossRef CAS.
- R. Subramanyam, A. Gollapudi, P. Bonigala, M. Chinnaboina and D. G. Amooru, J. Photochem. Photobiol., B, 2009, 94, 8–12 CrossRef CAS PubMed.
- M.-B. Gholivand, A. R. Jalalvand, H. C. Goicoechea and M. Omidi, Spectrochim. Acta, Part A, 2013, 115, 516–527 CrossRef CAS PubMed.
- Å. Frostell-Karlsson, A. Remaeus, H. Roos, K. Andersson, P. Borg, M. Hämäläinen and R. Karlsson, J. Med. Chem., 2000, 43, 1986–1992 CrossRef PubMed.
- D. P. Yeggoni, M. Gokara, D. Mark Manidhar, A. Rachamallu, S. Nakka, C. S. Reddy and R. Subramanyam, Mol. Pharm., 2014, 11, 1117–1131 CrossRef CAS PubMed.
- B. Sudhamalla, M. Gokara, N. Ahalawat, D. G. Amooru and R. Subramanyam, J. Phys. Chem. B, 2010, 114, 9054–9062 CrossRef CAS PubMed.
- T. Mosmann, J. Immunol. Methods, 1983, 65, 55–63 CrossRef CAS PubMed.
- C. Aromdee, Expert Opin. Ther. Pat., 2014, 24, 1129–1138 CrossRef CAS PubMed.
- F. Zsila, Z. Bikádi and M. Simonyi, Biochem. Pharmacol., 2003, 65, 447–456 CrossRef CAS PubMed.
- N. Tayeh, T. Rungassamy and J. R. Albani, J. Pharm. Biomed. Anal., 2009, 50, 107–116 CrossRef CAS PubMed.
- E. Rahnama, M. Mahmoodian-Moghaddam, S. Khorsand-Ahmadi, M. R. Saberi and J. Chamani, J. Biomol. Struct. Dyn., 2015, 33, 513–533 CAS.
- M. Zolfagharzadeh, M. Pirouzi, A. Asoodeh, M. R. Saberi and J. Chamani, J. Biomol. Struct. Dyn., 2014, 32, 1936–1952 CAS.
- J. R. Lakowicz, Principles of fluorescence spectroscopy, Springer, 2009 Search PubMed.
- D. Agudelo, P. Bourassa, J. Bruneau, G. Berube, E. Asselin and H.-A. Tajmir-Riahi, PLoS One, 2012, 7, e43814 CAS.
- S. Tayyab, M. S. Zaroog, S. R. Feroz, S. B. Mohamad and S. N. A. Malek, Int. J. Pharm., 2015, 491, 352–358 CrossRef CAS PubMed.
- M. R. Ajmal, A. S. Abdelhameed, P. Alam and R. H. Khan, Spectrochim. Acta, Part A, 2016, 159, 199–208 CrossRef CAS PubMed.
- J. Min, X. Meng-Xia, Z. Dong, L. Yuan, L. Xiao-Yu and C. Xing, J. Mol. Struct., 2004, 692, 71–80 CrossRef CAS.
- D. Agudelo, G. Bérubé and H. Tajmir-Riahi, Int. J. Biol. Macromol., 2016, 88, 354–360 CrossRef CAS PubMed.
- S. R. Feroz, S. B. Mohamad, N. Bujang, S. N. Malek and S. Tayyab, J. Agric. Food Chem., 2012, 60, 5899–5908 CrossRef CAS PubMed.
- R. Subramanyam, M. Goud, B. Sudhamalla, E. Reddeem, A. Gollapudi, S. Nellaepalli, V. Yadavalli, M. Chinnaboina and D. G. Amooru, J. Photochem. Photobiol., B, 2009, 95, 81–88 CrossRef CAS PubMed.
- M. Gokara, B. Sudhamalla, D. G. Amooru and R. Subramanyam, PLoS One, 2010, 5, e8834 Search PubMed.
- S. Urien, E. Albengres, J.-L. Pinquier and J.-P. Tillement, Clin. Pharmacol. Ther., 1986, 39, 683–689 CrossRef CAS PubMed.
- D. Morin, N. Simon, P. Depres-Brummer, F. Levi, J.-P. Tillement and S. Urien, Pharmacology, 1997, 54, 271–275 CrossRef CAS PubMed.
- Z. Israili and P. Dayton, Drug Metab. Rev., 2001, 33, 161–235 CrossRef CAS PubMed.
- G. Kumar, U. Walle, K. Bhalla and T. Walle, Res. Commun. Chem. Pathol. Pharmacol., 1993, 80, 337–344 CAS.
- D. N. Bailey and J. R. Briggs, Ther. Drug Monit., 2004, 26, 40–43 CrossRef CAS PubMed.
- L. Whitmore and B. A. Wallace, Biopolymers, 2008, 89, 392–400 CrossRef CAS PubMed.
- M. Kallubai, A. Rachamallu, D. P. Yeggoni and R. Subramanyam, Mol. Biosyst., 2015, 11, 1172–1183 RSC.
- M. Gokara, T. Malavath, S. K. Kalangi, P. Reddana and R. Subramanyam, J. Biomol. Struct. Dyn., 2014, 32, 1290–1302 CAS.
- M. Gokara, G. B. Kimavath, A. R. Podile and R. Subramanyam, J. Biomol. Struct. Dyn., 2015, 33, 196–210 CAS.
- X.-X. Cheng, Y. Lui, B. Zhou, X.-H. Xiao and Y. Liu, Spectrochim. Acta, Part A, 2009, 72, 922–928 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra25671f |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *a
*a
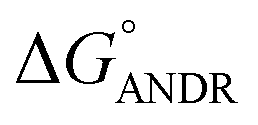 = −5.6 ± 0.031 kcal mol−1. Also, we have calculated the free energy change of binding to AGP and found this to be −4.31 ± 0.031 kcal mol−1 (
= −5.6 ± 0.031 kcal mol−1. Also, we have calculated the free energy change of binding to AGP and found this to be −4.31 ± 0.031 kcal mol−1 (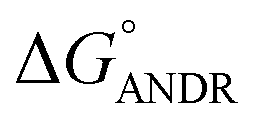 ). Similar results were found in docking with AGP and ANDR, where
). Similar results were found in docking with AGP and ANDR, where 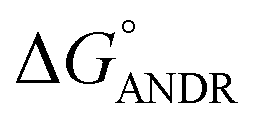 = −6.67 ± 0.21 kcal mol−1. Therefore, this data suggests that the binding between the protein and ligand can be stabilized by hydrophobic interaction and hydrogen bond formation. Similar results were recently published with other natural compounds (trans-feruloyl maslinic acid, betulinic acid, β-sitosterol, lupeol, L-dopa, 7-hydroxycoumarin, trimethoxy flavone, embelin, and corilagin).12–14,29,32,47,48 The fluorescence quenching mechanism was found to be static, which is evident from the bimolecular quenching constant for ANDR of 5.27 ± 0.02 × 1013 M−1 s−1. Quenching of the emission maximum at 340 nm of AGP was observed with ANDR. Interestingly, blue and red shifts were observed in the peaks of AGP–ANDR (Fig. 1B inset), whereas with HSA, there is no shift in the peaks of ANDR. This phenomenon might be due to the unfolding and refolding of the protein after binding. A possible explanation for the shift might be the differential exposure of tryptophan residues at various positions in the protein, which may be exposed differentially to the polar environment, leading to a shift in the emission maximum at 340 nm. The binding constants with ANDR are 10 times less than that of HSA. ANDR binds with AGP in a 1
= −6.67 ± 0.21 kcal mol−1. Therefore, this data suggests that the binding between the protein and ligand can be stabilized by hydrophobic interaction and hydrogen bond formation. Similar results were recently published with other natural compounds (trans-feruloyl maslinic acid, betulinic acid, β-sitosterol, lupeol, L-dopa, 7-hydroxycoumarin, trimethoxy flavone, embelin, and corilagin).12–14,29,32,47,48 The fluorescence quenching mechanism was found to be static, which is evident from the bimolecular quenching constant for ANDR of 5.27 ± 0.02 × 1013 M−1 s−1. Quenching of the emission maximum at 340 nm of AGP was observed with ANDR. Interestingly, blue and red shifts were observed in the peaks of AGP–ANDR (Fig. 1B inset), whereas with HSA, there is no shift in the peaks of ANDR. This phenomenon might be due to the unfolding and refolding of the protein after binding. A possible explanation for the shift might be the differential exposure of tryptophan residues at various positions in the protein, which may be exposed differentially to the polar environment, leading to a shift in the emission maximum at 340 nm. The binding constants with ANDR are 10 times less than that of HSA. ANDR binds with AGP in a 1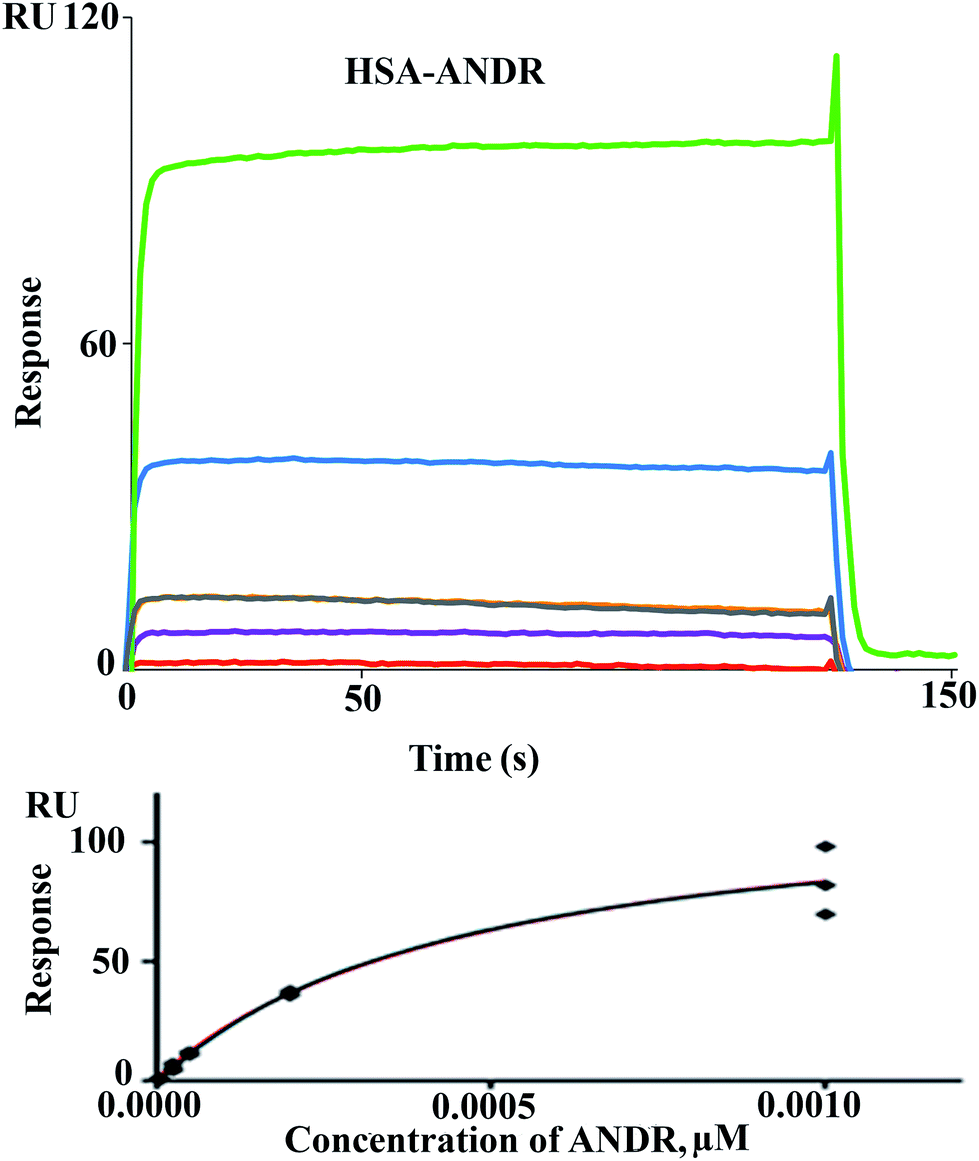

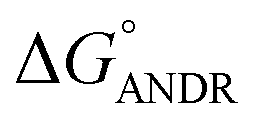 ) is −6.67 ± 0.21 kcal mol−1. These results support the experimental results (KANDR = 1.5 ± 0.03 × 103 M−1 and
) is −6.67 ± 0.21 kcal mol−1. These results support the experimental results (KANDR = 1.5 ± 0.03 × 103 M−1 and 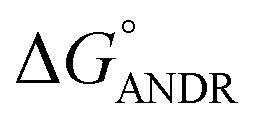 = −4.31 ± 0.031 kcal mol−1) found for the intrinsic fluorescence of the AGP and ANDR interaction. The interaction of ANDR with HSA is mainly stabilized by hydrophobic interactions, along with two hydrogen bonds, whereas with AGP, the main forces involved are three hydrogen bonds. ANDR shows robust binding to both HSA and AGP. Since AGP is an acute-phase protein expressed in pathological conditions, binding of ANDR to the AGP warrants drug-binding studies.
= −4.31 ± 0.031 kcal mol−1) found for the intrinsic fluorescence of the AGP and ANDR interaction. The interaction of ANDR with HSA is mainly stabilized by hydrophobic interactions, along with two hydrogen bonds, whereas with AGP, the main forces involved are three hydrogen bonds. ANDR shows robust binding to both HSA and AGP. Since AGP is an acute-phase protein expressed in pathological conditions, binding of ANDR to the AGP warrants drug-binding studies.