
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Dual-initiator alkoxyamines with an N-tert-butyl-N-(1-diethylphosphono-2,2-dimethylpropyl) nitroxide moiety for preparation of block co-polymers†
Gérard Audrana,
Elena Bagryanskayabc,
Mariya Edeleva *bc,
Sylvain R. A. Marque*ab and
Toshihide Yamasakia
*bc,
Sylvain R. A. Marque*ab and
Toshihide Yamasakia
aAix Marseille Université, CNRS, ICR, UMR 7273, Case 551, Avenue Escadrille Normandie-Niemen, 13397 Marseille Cedex 20, France. E-mail: sylvain.marque@univ-amu.fr
bN. N. Vorozhtsov Novosibirsk Institute of Organic Chemistry, SB RAS, Pr. Lavrentjeva 9, Novosibirsk 630090, Russia. E-mail: edeleva@nioch.nsc.ru
cNovosibirsk State University, Pirogova Str. 2, 630090 Novosibirsk, Russia
First published on 4th January 2017
Abstract
Dual initiators for controlled radical/ionic polymerization reactions attract much attention in terms of preparation of new materials. We studied the potential of dual-initiator alkoxyamines 3 [based both on para-substituted aromatic ring for external triggering or initiation of orthogonal polymerization and on N-tert-butyl-N-(1-diethylphosphono-2,2-dimethylpropyl) nitroxide (SG1)] for nitroxide-mediated polymerization (NMP) with various monomers such as styrene, styrene sulphonate, 2-vinyl pyridine or methylmethacrylate. Alkoxyamines 3 were found to be as efficient in the NMP process as N-(2-methylpropyl)-N-(1-diethylphosphono-2,2-dimethylpropyl)-O-(2-carboxylprop-2-yl)hydroxylamine (1). Furthermore, in sharp contrast to 1, alkoxyamines 3 were easier to functionalize.
Introduction
Block co-polymers are currently used in many fields, for example, materials science,1 medicine,2 and coating3,4 for various purposes, e.g. as polymer surfactants,5 anti-bacterial agents,6 and thermoplastic elastomers.7 The use of a dual initiator gives an important advantage during preparation of a block co-polymer carrying either incompatible monomers or a monomer non-polymerisable by the same method.8 Indeed, there are two main approaches: co-polymerisation by the same method for both polymers (approach 1 in Scheme 1) and a simple initiator, or polymerisation based on a dual initiator (approach 2 in Scheme 1), which makes several routes possible because it contains two different functional groups providing orthogonal reactivity.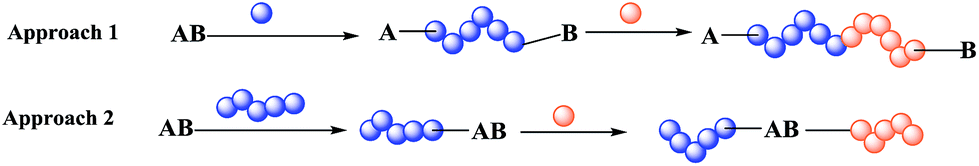 | ||
| Scheme 1 Two approaches to preparation of block co-polymers: (1) block co-polymerisation and (2) polymerisation based on a dual initiator. | ||
Indeed, preparation of a block co-polymer via approach 2 is either performed by the sequential polymerisation of two monomers (or through orthogonal polymerisation with suitable initiators and monomers) as shown in Scheme 1 or by pre- or post-functionalisation (Scheme 2). In the pre-functionalisation route, a polymer chain is grafted on the dual initiator, and then polymerisation of the second monomer is carried out. On the other hand, for the post-functionalisation, the first monomer is polymerised at the beginning using the dual initiator, and then the polymer of the second monomer is grafted onto the activated α-ω-end of the polymer. Control of the size and nature of block co-polymers plays a crucial role in the properties of such materials.1,8
 | ||
| Scheme 2 Methods for preparation of block co-polymers via pre-functionalisation (top) and post-functionalisation (bottom). | ||
In the last decades, such control was achieved using ionic polymerisation.9 Nevertheless, many of such reactions require harsh conditions, which substantially increase the cost of such polymers thus impeding their practical use.9 This problem has been partly resolved when Rizzardo and colleagues10 for the first time reported controlled radical polymerisation of several monomers using nitroxides as controlling agents (so-called nitroxide-mediated polymerisation; NMP), thus starting the era of controlled radical polymerisation along with atom transfer radical polymerisation (ATRP)11,12 and reversible radical addition fragmentation transfer radical polymerisation (RAFT),13,14 which made preparation of block co-polymers more convenient. The approach based on a dual initiator able to control both radical polymerisation and ionic polymerisation (or α-ω-end ionic reactivity) has been mainly studied for RAFT14,15 and ATRP.16,17 Application of dual alkoxyamines has also been documented in the last three decades.8,18 On the other hand, for alkoxyamine 1 (Fig. 1), which carries a carboxylic function on its alkyl moiety and is one of the most efficient initiators of NMP, both the high lability of 1 and the neopentylic carboxy function preclude easy and straightforward use as a dual initiator.19 This drawback has been circumvented by either activation of the carboxylic function for grafting of a polymer chain19,20 or by means of intermolecular radical 1,2-addition on an activated alkene.21 As far as we know, there is only one report on the esterification of 1 using the carboxylate function.22
 | ||
| Fig. 1 Alkoxyamines discussed in this article. | ||
A few years ago,23,24 we developed external triggering of C–ON homolysis with alkoxyamine 2 (Fig. 1) and demonstrated its potential for NMP.25 These results encouraged us to pursue the development of new alkoxyamines such as 3 (Fig. 1) that can be variously activated or deactivated by (de)protonation or by chemical transformation.26 In the course of this work, Gigmes et al.27 reported the use of tetramethylpiperidine-1-oxyl (TEMPO)-based carboxylic acid 4 (Fig. 1) as a dual initiator for NMP and ionic polymerisation. At the same time, Brémond et al.26 reported preparation and measurement of homolysis rate constant kd for alkoxyamines 3 at different temperatures and under various solvent conditions. Indeed, alkoxyamines showed remarkable changes in kd depending on external conditions, which can have a large impact on the polymerisation process. It should be noted that kd along with kc28 can strongly influence the outcome of NMP.
Alkoxyamines 3 possess different functionalities in the alkyl moiety, which sometimes have a striking effect on the course of NMP.29 In the present work, application of alkoxyamines 3 as a conventional initiator was studied, and alkoxyamines 3 carrying a carboxylic function were tested as a dual agent for initiation of NMP and in the grafting-onto strategy for preparation of block co-polymers. It was shown that alkoxyamines 3 afford nearly the same level of control over the NMP process as 1 does, and in sharp contrast to 1, functionalities of alkoxyamines 3 can be easily used for grafting onto polymer molecules under mild conditions. We found that alkoxyamines 3 can be used for preparation of block co-polymers based on block co-polymerisation or pre- and post-functionalisation approaches depicted in Schemes 1 and 2. Furthermore, for this type of monomeric and polymeric alkoxyamines, esterification reactions are effectively opening a convenient route for preparation of block co-polymers.
Experimental section
General
All monomers were distilled prior to use. Protonated and deuterated solvents and polyethylene glycol (Aldrich) were used as received.The synthesis and C–ON homolysis rate constants kd of alkoxyamines 3 under various conditions were reported in a recent paper.26
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. A typical polymerisation experiment was performed within 4–6 h.
:1. A typical polymerisation experiment was performed within 4–6 h.Potassium and zinc salts were prepared in situ by addition of 1.05 eq. of NaOH or ZnCl2 dissolved in methanol to alkoxyamine 3e. The solution was stirred for 1 h, then the solvent was evaporated. The resulting crude material was used in polymerisation.
For the NMR experiment, a 0.02 M solution in the mixture of D2O and methanol-d4 (2:3, v/v) was prepared, with 0.01 M tert-butyl alcohol as a standard. Potentiometry was employed to measure pH (a glass electrode [Aldrich] calibrated at pH 1.68, 7.62 and 9.05). DCl and NaOD solutions in D2O were used to adjust pH. A series of 1H NMR spectra were recorded, and evolution of the chemical shift of ortho-protons of para-substituted benzoic acid was utilised to plot the titration curve. 1H NMR spectra were acquired on a Bruker Avance 200 MHz NMR spectrometer.
The UV/Vis titration was performed on a solution of alkoxyamine 3e (20 mM) in the mixture of water and ethanol (1:1 v/v). The pH level was adjusted with solutions of HCl and NaOH in the water–ethanol mixture. UV/Vis spectra were recorded on the Agilent spectrometer. The titration curve was plotted using data on optical density at λ = 238 nm.30
For the pre-functionalisation experiment, a weighted amount of alkoxyamine 3e (5 mg in 0.5 ml of chloroform) was incubated with 30 μl of SOCl2 and 50 μl of pyridine. The reaction was conducted on an ice bath to prevent decomposition of the alkoxyamine. The reaction mixture was stirred for 30 min. Next, polyethylene glycol (11 mg) in 0.5 ml of chloroform was added, and the reaction mixture was stirred overnight. After that, the mixture was washed with a 0.01% solution of HCl, and the solvent was evaporated. The resulting product was introduced into the polymerisation reaction without further treatment. The polymerisation was conducted as described above.
Results and discussion
It should be noted that depending on the substituent in the phenyl ring, the activation energy of homolysis of 3a–j varies within a ∼8 kJ mol−1 range. The difference in activation energies after protonation (a 5 kJ mol−1 difference in the activation energy after protonation/deprotonation for 3e/3f) and the subsequent difference in kd can have significant effects on the kinetic behaviour of different monomers in NMP and even on the initiation stage of polymerisation of the same monomer. Thus, alkoxyamines 3a–j carrying various substituents in the alkyl moiety seem to hold promise as multifunctional initiators. Below, we show the potential application of alkoxyamines 3a–j to the preparation of block co-polymers based on sequential polymerisation or pre- and post-functionalisation described in Schemes 1 and 2.For pre- and post-functionalisation, different types of esterification reactions with the carboxylic group of 3e were tested to evaluate its potential as a dual initiator for preparation of block co-polymers.
NMP of styrene
Because the rate constant (kd) of C–ON bond homolysis is the key parameter of an NMP process, and because fast initiation is in general necessary to ensure control at the first moments of the polymerisation, alkoxyamines 3d, 3e and 3g were selected because they have the largest kd values: kd (3d, 110 °C) = 7 × 10−3 s−1, kd (3e, 110 °C) = 10−2 s−1 and kd (3g, 110 °C) = 1.9 × 10−2 s−1, respectively. It should be mentioned that alkoxyamines 3d, 3e and 3g differ only in the substituent at the para position of the aromatic ring of the alkyl moiety.The linear evolution of Mnversus conversion close to the theoretical line, up to 40–75% conversion, with a PDI (polydispersity index) less than 1.4, indicates strong control of the experiment as expected for SG1-based alkoxyamines (Fig. 2). The linearity of Mn at the first moments of the polymerisation means that the C–ON bond homolysis of the selected alkoxyamines is sufficiently rapid.31 Given that similar time was allocated to all experiments, i.e. 400 min for 3g, 420 min for 3d and 440 min for 3e, the clearly greater conversion (ca. 75%) for 3e than for 3d and 3g (ca. 40%) means that the conversion is faster with 3e even though the same nitroxide SG1 is involved in the control. This result is likely due to the presence of the carboxylic function in 3e, which causes degradation of SG1, resulting in a smaller excess of nitroxide and hence a higher rate of polymerisation32 at the expense of control (vide infra).
 | ||
| Fig. 2 Plots of molecular mass (filled symbols) and polydispersity index (PDI, open symbols) vs. conversion for bulk polymerisation of styrene at 110 °C initiated with 3d (squares), 3e (triangles) or 3g (circles) at the monomer/initiator ratio of 350:1. | ||
Furthermore, the efficiency of the potassium and zinc salt of carboxylate alkoxyamine 3f was also assessed. Both yielded a linear increase in Mnvs. conversion and a decrease in the PDI. These data indicate a controlled regime of polymerisation (Fig. 1 ESI†). The differences in trends were not significant.
The salts of 3f yielded a monomodal distribution of molar masses, whereas a bimodal distribution was observed when the polymerisation was initiated by 3e (Fig. 3). This is undoubtedly due to the presence of the carboxylic function in 3e, which may initiate decomposition of SG1 into new initiating species leading to a higher PDI.33
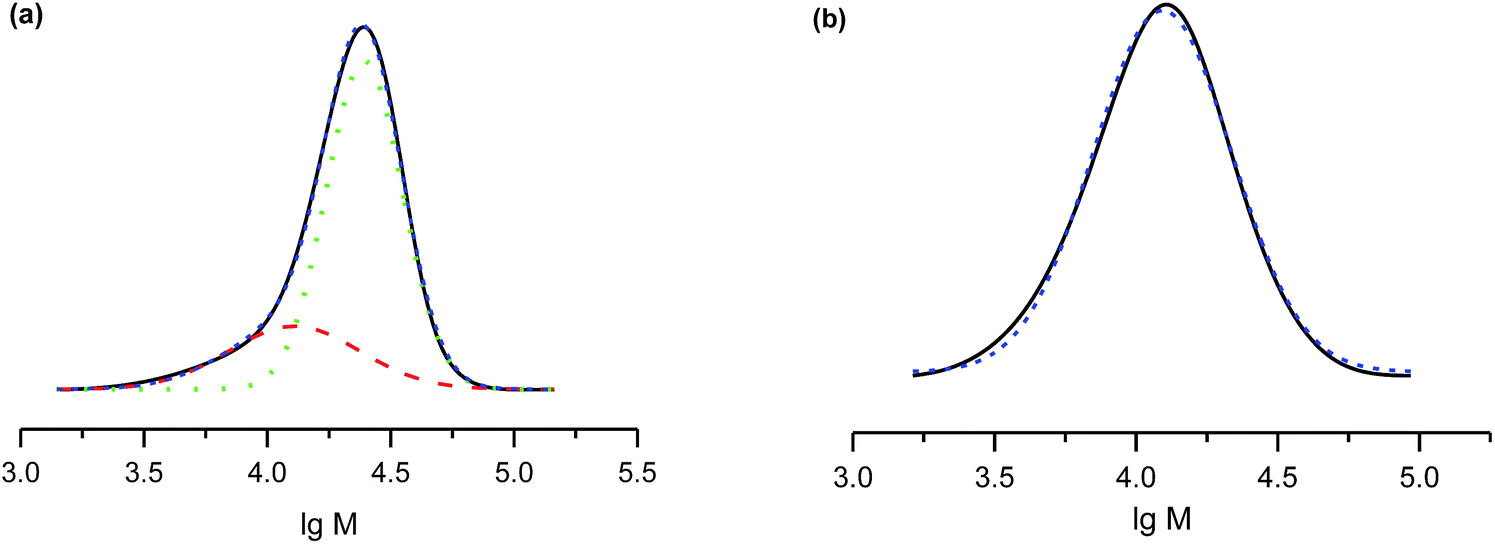 | ||
| Fig. 3 (a) A GPC trace of a polystyrene sample prepared with 3e at 51% monomer conversion (black curve). Overall molecular mass is 1.7 × 104 g mol−1, polydispersity is 1.36. Fitting to Gaussian curves shows the high-molecular-weight (dotted curve, green) and low-molecular-weight fractions (dashed line, red) at 25% and 75%, respectively. The fine dashed curves (blue) show the whole traces. (b) A GPC trace of a polystyrene sample prepared with 3f (sodium salt) at 26% monomer conversion (black curve). Overall molecular weight is 104 g mol−1, polydispersity is 1.4. Fitting to a Gaussian curve (fine dashed blue curve). | ||
Because the proportion of the low-molecular-weight fraction was less than 10% (Fig. 3), it was ignored during estimation of Mn values. Salts of 3f can be considered more efficient than 3e for the NMP of styrene because the molecular-weight distribution is monomodal and the carboxylic function is easily regenerated for further transformations.
NMP of 2-vinyl pyridine34–36
This procedure was performed in bulk at 110 °C with 3e or with the potassium salt of 3f as initiators. In both cases, linear growth of molecular weight of the polymer in the course of conversion and PDI ≈ 1.5 denote a controlled regime of polymerisation (Fig. 4). NMP with the potassium salt of 3f, in which any effect caused by the presence of the labile proton is suppressed, is faster than the polymerisation of styrene. The polymerisation rate increases as the cubic root of kd.8,14 Therefore, the increase in kd – because of an increase in polarity of the last unit with a pyridine moiety23 and because of the likely increase in the polarity with the increasing length of the polymer chain37 – accounts for the faster polymerisation of 2-vinyl pyridine in comparison with styrene when 3f and 3e serve as initiators, respectively. The fastest polymerisation observed when 3e serves as the initiator can be explained by the aforementioned effect (which may be enhanced by the presence of protonated pyridine) and by the instability of SG1 in an acidic medium (Fig. 5).33 Our results are in good agreement with those reported elsewhere.36 At the same temperature, without an accelerator within a similar period, we obtained larger molecular weight (14 kDa) though polydispersity was greater in our experiments (1.7 in our case) probably because of decomposition of nitroxide. | ||
| Fig. 4 The plots of molecular mass (filled symbols) and polydispersity indexes (open symbols) vs. conversion for bulk polymerisation of 2-vinyl pyridine at 110 °C initiated by acidic (squares) or basic forms (circles) of alkoxyamine 3e at the monomer-to-initiator ratio 400 to 1. | ||
 | ||
| Fig. 5 Comparison of bulk polymerisation kinetics of styrene (squares) and 2-vinyl pyridine (circles) at 110 °C initiated with 3e (filled symbols) or 3f (open symbols). Solid lines show a linear fit of the data. | ||
NMP involving methyl methacrylate (MMA) co-polymerisation with 2-vinyl pyridine and styrene
One of the challenges for NMP is the polymerisation of methacrylic esters.38 SG1-based alkoxyamines are effective as mediators for methacrylate only when a small percentage of styrene or acrylonitrile serves as a co-monomer,39–41 whereas other types of nitroxides can provide a controlled mode of MMA polymerisation without addition of a co-monomer.42–44Given that styrene is undesirable for some types of applications, it is worthwhile to test other types of monomers. Thus, the use of dual initiators and functionalised monomers has a potential to extend the applications of NMP to preparation of polymethacrylate materials. Therefore, the suitability of 2-vinyl pyridine as a co-monomer and of 3e as the initiator was evaluated here for the NMP of MMA.
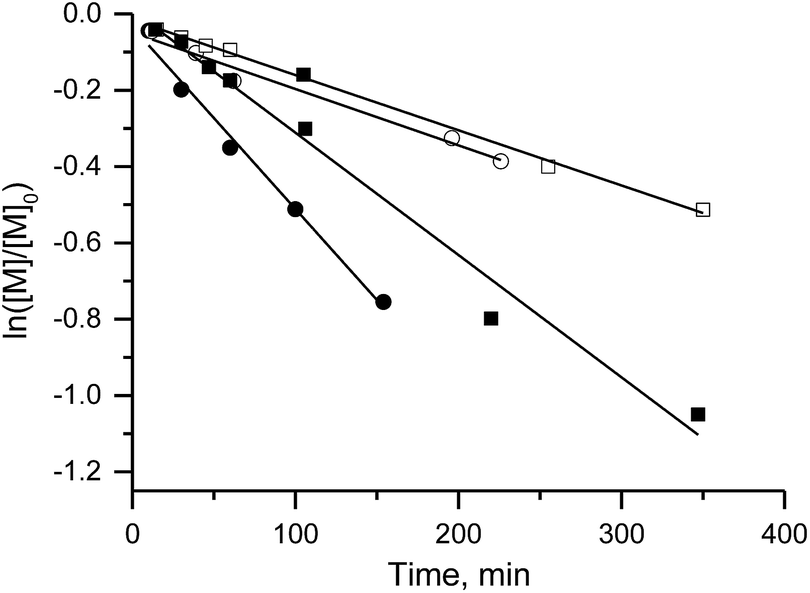
The linear evolution of Mnvs. conversion and the decrease in PDI below 1.5 with conversion denote controlled polymerisation of MMA in the presence of 2% of 2-vinylpyridine (Fig. 6) with 3e as the initiator (similar results were obtained with styrene instead of 2-vinylpyridine).
 | ||
| Fig. 6 The plot of ln([M]/[M]0) vs. time (a) and molecular mass vs. conversion (b) for co-polymerisation of methylmethacrylate with 2% of 2-vinyl pyridine in bulk initiated with 3e alkoxyamine at 80 °C and the monomer-to-initiator ratio 350 to 1. | ||
It should be noted that our results are similar to those described in the literature when styrene was used as a co-monomer and 1 as initiator. When 9% of styrene was added, the polymerization proceeded to 43.5% of conversion with polymer Mn = 14 kDa and PDI = 1.2.40
The aqueous monomer sodium styrenesulfonate45
The titration of 3e in deuterated solvents D2O:MeOH-d4 (2:3 v/v) and in the protic solvent H2O:C2H5OH (1:1 v/v) yielded pKa of 5.26 and 4.95, respectively: in the typical range for carboxylic acids (Fig. 2 ESI†).46 These data should be considered in the case of polymerisation in an aqueous solution.
Because alkoxyamines 3 show a large difference in activation energies after protonation in an aqueous medium, 3e was selected for initiation of sodium 4-styrenesulfonate in water.
The curvature in the plot of Mnvs. conversion (Fig. 7b) below 10% conversion for both pH values is indicative of an initiator with a kd value too low for efficient initiation.31 Nevertheless, the linear increase in Mn in the course of the conversion (as well as PDI < 1.5 for conversion greater than 15%) point to control of the polymerisation (Fig. 7b). The faster polymerisation at pH = 3 (0.48 M sodium 4-styrenesulfonate in water titrated with a HCl solution) than at pH = 9.5 (titration with a solution of NaOH) is expected because the nitroxide is unstable33 in an acidic environment. This situation also leads to deviation of the molecular weight from its theoretical value and to greater polydispersity in contrast to the basic conditions.
 | ||
| Fig. 7 (a) The kinetics plot of instant monomer concentration vs. time and (b) the plot of molecular mass (filled symbols, left axis) and polydispersity (open symbols, right axis) vs. conversion for polymerisation of styrenesulfonate in water at 90 °C and pH = 3 (squares) or pH = 9.5 (circles) at the ratio of sodium 4-styrenesulfonate to 3e of 192:1. | ||
Livingness and re-initiation
Livingness was studied by EPR spectroscopy47,48 for the polymerisation of styrene initiated by 3d, 3e, potassium or zinc salts of 3f, and 3g and was greater than 90%. For the polymerisation of 2-vinylpyridine initiated by 3e or the potassium salt of 3f, livingness of 75% was observed. For the polymerisation of MMA with 2-vinylpyridine and styrene initiated by 3e, livingness was 80%. For polymerisation of styrenesulfonate, livingness of 75% was obtained.Consequently, regardless of the monomer or initiator, livingness >75% is commonly observed, meaning that such alkoxyamines are suitable for preparation of block co-polymers.
To exemplify the applicability of the living polymers obtained for formation of block co-polymers, re-initiation experiments were conducted with styrene and methacrylate. In both cases, the increase in molecular weight upon heating for 0.5 or 3 h (Fig. 8) was observed, confirming the living character of polystyrene.
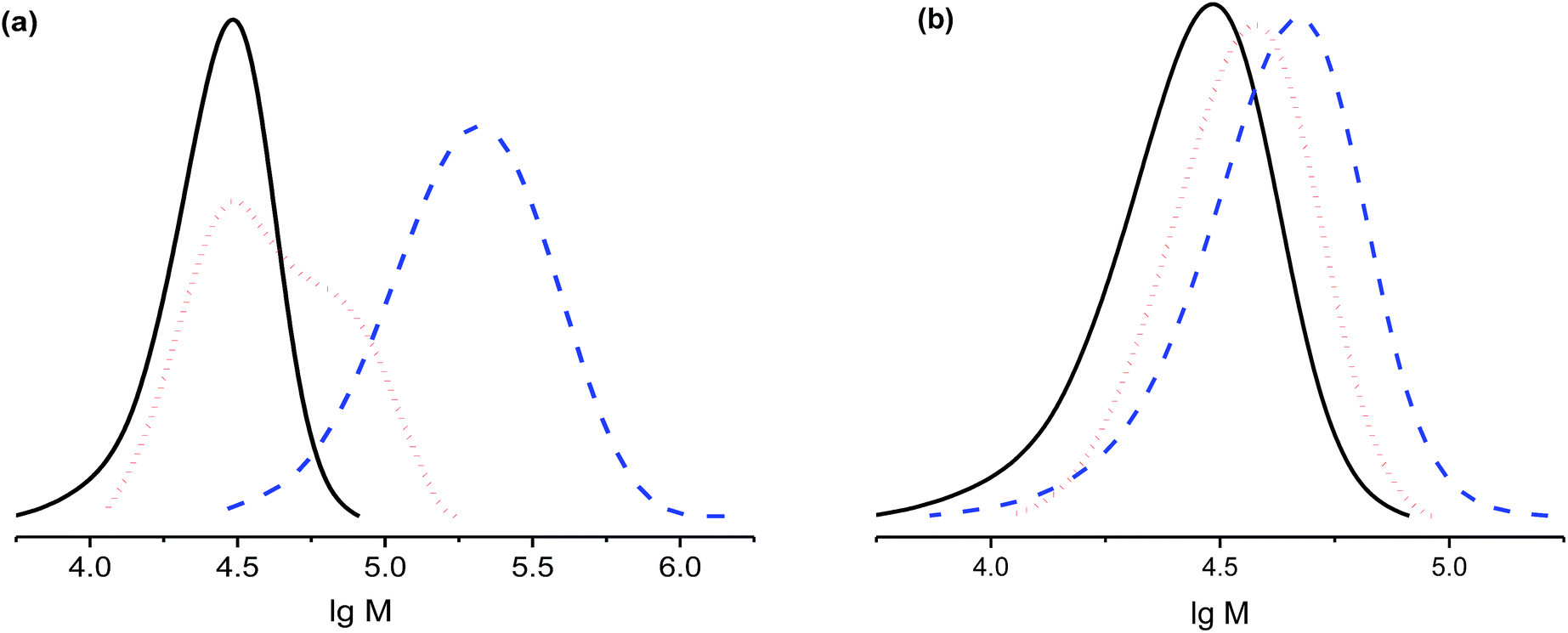 | ||
| Fig. 8 GPC traces of re-initiation experiments with polystyrene prepared with 3e as a macro-initiator (solid curve). (a) Re-initiation with styrene, at the monomer/macro-initiator ratio 0.5 g to 10 mg, T = 110 °C, sampling at 0.5 h (red dotted curve) and 3 h (blue dashed curve). (b) Re-initiation with MMA, at the monomer/macro-initiator ratio 0.5 g to 10 mg, T = 80 °C, sampling at 0.5 h (red dotted curve) and 1 h (blue dashed curve). | ||
The shoulder in the GPC trace at 30 min (Fig. 8a) means that not all macro-initiators decomposed, whereas the peak is completely shifted to greater mass after 3 h, in good agreement with the 90% livingness reported above. Re-initiation with MMA is also efficient as shown in Fig. 8b, except the growth of the polymer is likely to be not controlled, and the polymer is probably not living.49
Preparation of block co-polymers via the pre- and post-functionalisation approaches
Amongst alkoxyamines 3a–g, the greatest promise as a dual initiator was shown by 3b, 3e and 3g (or the highest potential as alkoxyamine-carrying groups to be easily applied to pre- and post-functionalisation procedures). It should be noted that for alkoxyamine 1, the typical strategy for pre- and post-functionalisation is based on the 1,2-radical addition reaction with activated olefins.21 To overcome this problem, Gigmes and co-workers27 developed 4b, which is a TEMPO-based alkoxyamine. In general, such alkoxyamines are less versatile and efficient than those containing the SG1 moiety. Consequently, 3e, which is effective at polymerisation of several monomers as mentioned above, was selected as a model to test the pre- and post-functionalisation procedures.The general strategy for pre- and post-functionalisation implemented below is shown in Schemes 3 and 4. It includes two routes: (a) post-functionalisation by means of a reaction of tosylated PEG with polystyrene obtained in polymerisation initiated by 3e (Scheme 3) and (b) the pre-functionalisation approach based on a reaction of unimolecular alkoxyamines with PEG and subsequent NMP of styrene (Scheme 4).
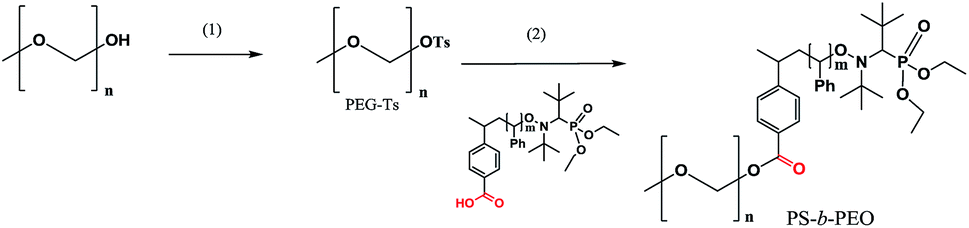 | ||
| Scheme 3 The reaction scheme for preparation of block co-polymers via the post-functionalisation approach. (1) Tosylation of PEG: a solution of PEG in pyridine and addition of TsCl at room temperature. (2) Grafting of polystyrene: addition of KOH into methanol with subsequent evaporation, addition of a solution of PEG–Ts to dimethylformamide, and heating for 80 h at 80 °C. | ||
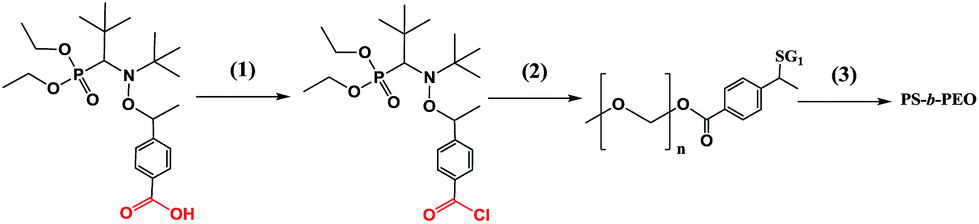 | ||
| Scheme 4 The reaction scheme for preparation of block co-polymers via the pre-functionalisation approach. (1) Formation of chloroanhydride: a solution of an alkoxyamine in CHCl3, treatment with SOCl2, pyridine, 0 °C; (2) functionalisation of chloroanhydride with PEG: a solution in CHCl3, 0 °C. (3) Nitroxide-mediated polymerisation (NMP) of bulk styrene with a macro-initiator: T = 110 °C, and monomer-to-initiator ratio 0.5 g to 10 mg. | ||
PS-b-PEG block co-polymers via the post-functionalisation procedure
Tosylate of PEG–OH (Mn = 2 kDa) was prepared at the first step, and at the second step (Scheme 3), was grafted onto polystyrene (PS-3e, Mn = 20 kDa). The initial mixture of PEG–OTs and PS-3e showed two expected curves (Fig. 9), but a unique curve was observed in crude materials after 80 h of the reaction at 80 °C, with a slight shift (2 kDa), as expected. This experiment confirmed the usefulness of 3e for post-functionalisation. | ||
| Fig. 9 A GPC trace of the reaction mixture of PEG–Ts with polystyrene (solid curve), and the final block co-polymer PS-b-PEG (dashed curve) obtained after the reaction. | ||
PS-b-PEG block co-polymers obtained by the pre-functionalisation procedure
Acyl chloride of alkoxyamine 3e was prepared and used for the coupling with PEG–OH to produce the functionalised initiator that was successfully applied to NMP of styrene (Scheme 4). The grafting of PEG onto 3e was corroborated by the initial traces, and its ability to initiate polymerisation is indicated by the rightward shift of the GPC traces with time (Fig. 10). Thus, this experiment confirmed the usefulness of 3e for pre-functionalisation. | ||
| Fig. 10 GPC traces of the PEG–SG1 macro-initiator (solid curve) and PEG–PS block co-polymers obtained by grafting after polymerisation: 20 min (dashed curve), 40 min (dotted curve), or 1 h (dash-and-dot curve). The monomer to macro-initiator ratio is 0.5 g to 10 mg, T = 110 °C. | ||
Conclusions
It is shown that carboxylic alkoxyamines 3e and 3d hold promise as a dual initiator. This potential is highlighted by the polymerisation of several monomers, either conventional such as styrene and 2-vinylpyridine or less conventional such as 4-vinylbenzenesulfonate. Moreover, in contrast to TEMPO-based alkoxyamines, 3e is suitable for the co-polymerisation of MMA by NMP with different co-monomers such as styrene and 2-vinylpyridine and thus could be used for preparation of metal-crosslinked polymers. In general, SG1-based alkoxyamines are more efficient in NMP than those based on TEMPO. In sharp contrast to 1, the carboxylic function in 3e is reactive enough for both pre- and post-functionalisation as demonstrated by the preparation of PS-b-PEG via both approaches, with preservation of the main features of 1 in NMP.Acknowledgements
We are very thankful to Dr Paul Bremond for providing alkoxyamines and for the fruitful discussion. SRAM, GA and TY thank Aix-Marseille Université, CNRS, ANR (grant NAR ANR-14-CE16-0023-01) and the A*MIDEX project (ANR-11-IDEX-0001-02) funded by the French Government program ‘Investissements d'Avenir’, managed by the French National Research Agency (ANR). SRAM and ME are grateful to the Russian Science Foundation (grant 15-13-20020) for supporting this work. ME and EB thank Russian Foundation for Basic Research (15-03-05250) for financial support of this study.Notes and references
- N. Hadjichristidis, S. Pispas and G. Floudas, Block copolymers: synthetic strategies, physical properties, and applications, John Wiley & Sons, 2003 Search PubMed.
- S. Sugihara, S. Kanaoka and S. Aoshima, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 2601 CrossRef CAS.
- R. A. Iezzi, S. Gaboury and K. Wood, Prog. Org. Coat., 2000, 40, 55 CrossRef CAS.
- T. Tüken, B. Yazıcı and M. Erbil, Prog. Org. Coat., 2004, 50, 115 CrossRef.
- M. Minoda, M. Sawamoto and T. Higashimura, Macromolecules, 1987, 20, 2045 CrossRef CAS.
- J. Pretula, K. Kaluzynski, B. Wisniewski, R. Szymanski, T. Loontjens and S. Penczek, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 830 CrossRef CAS.
- T. Hashimoto, T. Imaeda, S. Irie, M. Urushisaki and T. Sakaguchi, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 1114 CrossRef CAS.
- J. Nicolas, Y. Guillaneuf, C. Lefay, D. Bertin, D. Gigmes and B. Charleux, Prog. Polym. Sci., 2013, 38, 63 CrossRef CAS.
- G. Odian, Principles of polymerization, John Wiley & Sons, 2004 Search PubMed.
- D. H. Solomon, E. Rizzardo and P. Cacioli, EP, Appl. 135280, 1985; D. H. Solomon, E. Rizzardo and P. Cacioli, US Pat., 4, 581, 429, 1986Chem. Abstr., 1985, 102, 221335q.
- K. Matyjaszewski and J. Xia, Chem. Rev., 2001, 101, 2921 CrossRef CAS PubMed.
- M. Kamigaito, T. Ando and M. Sawamoto, Chem. Rev., 2001, 101, 3689 CrossRef CAS PubMed.
- J. Chiefari, Y. K. Chong, F. Ercole, J. Krstina, J. Jeffery, T. P. T. Le, R. T. A. Mayadunne, G. F. Meijs, C. L. Moad, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 1998, 31, 5559 CrossRef CAS.
- C. Barner-Kowollik, Handbook of RAFT polymerization, John Wiley & Sons, 2008 Search PubMed.
- A. B. Lowe and C. L. McCormick, Prog. Polym. Sci., 2007, 32, 283 CrossRef CAS.
- H. Gao and K. Matyjaszewski, Macromolecules, 2006, 39, 4960 CrossRef CAS.
- H. Gao and K. Matyjaszewski, Prog. Polym. Sci., 2009, 34, 317 CrossRef CAS.
- Y.-N. Zhou, Z.-C. Chen, C. Wei and Z.-H. Luo, Macromol. Chem. Phys., 2014, 216, 329 CrossRef.
- D. Gigmes, J. Vinas, N. Chagneux, C. Lefay, T. N. T. Phan, T. Trimaille, P.-E. Dufils, Y. Guillaneuf, G. Carrot, F. Boué and D. Bertin, ACS Symp. Ser., 2009, 1024, 245 CrossRef CAS.
- E. Guegain, V. Delplace, T. Trimaille, D. Gigmes, D. Siri, S. R. A. Marque, Y. Guillaneuf and J. Nicolas, Polym. Chem., 2015, 6, 5693 RSC.
- P.-E. Dufils, N. Chagneux, D. Gigmes, T. Trimaille, S. R. A. Marque, D. Bertin and P. Tordo, Polymer, 2007, 48(18), 5219 CrossRef CAS.
- P. Brémond, K. Kabytaev and S. R. A. Marque, Tetrahedron Lett., 2012, 53, 4543 CrossRef.
- P. Brémond and S. R. A. Marque, Chem. Commun., 2011, 47(14), 4291 RSC.
- P. Brémond, A. Koïta, S. R. A. Marque, V. Pesce, V. Roubaud and D. Siri, Org. Lett., 2012, 14(1), 358 CrossRef PubMed.
- E. Bagryanskaya, P. Brémond, M. Edeleva, S. R. A. Marque, D. Parkhomenko, V. Roubaud and D. Siri, Macromol. Rapid Commun., 2012, 33(2), 152 CrossRef CAS PubMed.
- G. Audran, P. Brémond, J.-P. Joly, S. R. A. Marque and T. Yamasaki, Org. Biomol. Chem., 2016, 14, 3574 CAS.
- D. Le, T. N. T. Phan, L. Autissier, L. Charles and D. Gigmes, Polym. Chem., 2016, 7, 1659 RSC.
- E. G. Bagryanskaya and S. R. Marque, Chem. Rev., 2014, 114, 5011 CrossRef CAS PubMed.
- E. Bagryanskaya, D. Bertin, D. Gigmes, I. Kirilyuk, S. R. A. Marque, V. Reznikov, G. Roshchupkina, I. Zhurko and D. Zubenko, Macromol. Chem. Phys., 2008, 209, 1345 CrossRef CAS.
- The dependence of chemical shift of β phenyl protons upon protonation was approximated with the following equations to obtain the value of pKa for the given conditions:
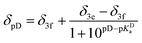 when deuterated solvent was used. When titration was performed in nondeuterated solvents by means of UV-Vis, the following dependence was used:
when deuterated solvent was used. When titration was performed in nondeuterated solvents by means of UV-Vis, the following dependence was used: 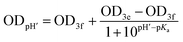 for the mixture of water and alcohol.
for the mixture of water and alcohol. - F. Chauvin, P.-E. Dufils, D. Gigmes, Y. Guillaneuf, S. R. A. Marque, P. Tordo and D. Bertin, Macromolecules, 2006, 39, 5238 CrossRef CAS.
- M. K. Georges, R. P. N. Veregin, P. M. Kazmaier, G. K. Hamer and M. Saban, Macromolecules, 1994, 27(24), 7228 CrossRef CAS.
- M. Edeleva, S. R. A. Marque, K. Kabytaev, Y. Guillaneuf, D. Gigmes and E. G. Bagryanskaya, J. Polym. Sci., Part A: Polym. Chem., 2013, 51(6), 1323 CrossRef CAS.
- A. Fischer, A. Brembilla and P. Lochon, Eur. Polym. J., 2001, 37, 33 CrossRef CAS.
- A. Fischer, A. Brembilla and P. Lochon, Macromolecules, 1999, 32, 6069 CrossRef CAS.
- C. Ioanna, S. Pispas and N. Hadjichristidis, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 2889 CrossRef.
- O. Guerret, J.-L. Couturier, F. Chauvin, H. El-Bouazzy, D. Bertin, D. Gigmes, S. Marque, H. Fischer and P. Tordo, ACS Symp. Ser., 2003, 854, 412 CrossRef CAS.
- N. Ballard, M. Aguirre, A. Simula, A. Agirre, J. R. Leiza, J. M. Asua and S. van Es, ACS Macro Lett., 2016, 5, 1019 CrossRef CAS.
- L. Couvreur, C. Lefay, J. Belleney, B. Charleux, O. Guerret and S. Magnet, Macromolecules, 2003, 36, 8260 CrossRef CAS.
- J. Nicolas, C. Dire, L. Mueller, J. Belleney, B. Charleux, S. R. A. Marque, D. Bertin, S. Magnet and L. Couvreur, Macromolecules, 2006, 39, 8274 CrossRef CAS.
- J. Nicolas, S. G. N. Brusseau and B. Charleux, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 34 CrossRef CAS.
- M. V. Edeleva, D. A. Parkhomenko, D. A. Morozov, S. A. Dobrynin, D. G. Trofimov, B. Kanagatov, I. A. Kirilyik and E. G. Bagryanskaya, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 929 CrossRef CAS.
- C. Detrembleur, C. Jérôme, J. De Winter, P. Gerbaux, J.-L. Clément, Y. Guillaneuf and D. Gigmes, Polym. Chem., 2014, 5, 335 RSC.
- D. Gigmes, A. Gaudel-Siri, S. R. A. Marque, D. Bertin, P. Tordo, P. Astolfi, L. Greci and C. Rizzoli, Helv. Chim. Acta, 2006, 89, 2312 CrossRef CAS.
- S. Brusseau, F. D'Agosto, S. Magnet, L. Couvreur, C. Chamignon and B. Charleux, Macromolecules, 2011, 44, 5590 CrossRef CAS.
- Different values of pKa are reported in ref. 26 due to different experimental conditions: a different ratio alcohol/water has been used and the methanol has been replaced by ethanol.
- D. Bertin, F. Chauvin, S. Marque and P. Tordo, Macromolecules, 2002, 35(10), 3790 CrossRef CAS.
- Error is ca. 5%.
- Y. Guillaneuf, D. Gigmes, S. R. A. Marque, P. Tordo and D. Bertin, Macromol. Chem. Phys., 2006, 207(6), 1278 CrossRef CAS.
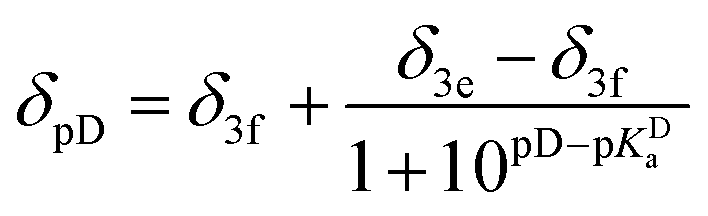

Footnote |
| † Electronic supplementary information (ESI) available: Experimental results on styrene polymerization with different forms of alkoxyamine 3e; titration curves. See DOI: 10.1039/c6ra27231b |
| This journal is © The Royal Society of Chemistry 2017 |