
Recent progress in interfacial polymerization
Yongyang
Song
 ab,
Jun-Bing
Fan
*a and
Shutao
Wang
*ab
ab,
Jun-Bing
Fan
*a and
Shutao
Wang
*ab
aCAS Key Laboratory of Bio-inspired Materials and Interfacial Science, CAS Center for Excellence in Nanoscience, Technical Institute of Physics and Chemistry, Chinese Academy of Sciences, Beijing 100190, P. R. China. E-mail: fjb2012@mail.ipc.ac.cn; stwang@mail.ipc.ac.cn; Tel: +86-010-82543658
bUniversity of Chinese Academy of Sciences, Beijing 100049, P. R. China
First published on 30th January 2017
Abstract
Interfacial polymerization is one of the most important methods to fabricate polymer particles and membranes. As compared to bulk polymerization, interfacial polymerization is a type of step-growth polymerization that takes place at the interface of two immiscible phases, endowing the polymer materials with unique topological and chemical properties, such as anisotropic shapes, hollow structures or alternative surface chemistry. Herein, we summarize the recent progress in interfacial polymerization, generally based on the fabrication method and application field. First, two theory models of interfacial polymerization based on membrane and particles have been introduced. Second, five typical polymerization systems, i.e. liquid/monomer–solid (Lm–S), liquid/monomer–liquid (Lm–L), liquid/monomer–liquid/monomer (Lm–Lm), liquid/monomer-in-liquid (Lm-in-L), and liquid/monomer-in-liquid/monomer (Lm-in-Lm) systems, based on liquid–solid, liquid–liquid, and liquid-in-liquid emulsion interfaces, used in interfacial polymerization have been summarized. Then, three practical applications of synthetic materials, i.e. in electronic devices, separation, and cargo loading, prepared by interfacial polymerization have been shown. Finally, the current challenges and opportunities in interfacial polymerization have also been proposed and discussed.
 Yongyang Song | Yongyang Song is currently a PhD candidate at the Technical Institute of Physical Chemistry, Chinese Academy of Sciences (TIPCCAS) and University of Chinese Academy of Sciences (UCAS). He received his BE degree (2012) and ME degree (2015) in materials science from the Wuhan University of Technology. Then, he joined TIPCCAS under the supervision of Prof. Shutao Wang. His research interests include the design and synthesis of bioinspired micro/nanohybrid particles and their biological applications. |
 Jun-Bing Fan | Jun-Bing Fan is currently an Assistant Professor at TIPCCAS. He received his PhD degree (2012) from Hunan University under the supervision of Prof. Ming-Qiang Zhu and Shuming Nie. Then, he worked as a postdoctoral researcher with Prof. Lei Jiang and Shutao Wang at the Institute of Chemistry, Chinese Academy of Sciences (ICCAS) in 2012–2014. His research interests focus on the design and synthesis of polymer porous nano/microstructured particles and anisotropic particles with special properties, and their applications at the bio-nanomaterial interface. |
 Shutao Wang | Shutao Wang is currently a Professor at TIPCCAS. He received his BS degree (2000) and MS degree (2003) from the Northeast Normal University under the supervision of Prof. Enbo Wang and PhD degree (2007) from ICCAS under the supervision of Prof. Lei Jiang. He then worked in the Department of Molecular and Medical Pharmacology and California NanoSystem Institute at the University of California in Los Angeles as a postdoctoral researcher (2007–2010). He was appointed as a Professor at ICCAS (2010–2014). His research interests include the design and synthesis of bio-inspired interfacial materials with special adhesion and their applications at the bio-nanomaterial interface. |
1. Introduction
Interfacial polymerization, firstly proposed by Wittbecker and Morgan1 in 1959, initially refers to the polycondensation of diamine and diacid chloride monomers, which react to form polyamide and hydrogen chloride. When compared with bulk polymerization, interfacial polymerization exhibits great flexibility in surface topology and chemical properties of the prepared functional materials. To date, interfacial polymerization has evolved as a robust and effective approach to synthesize a wide range of functional polymer materials. It has been expanded to many polymerization mechanisms, including emulsion polymerization, atom transfer radical polymerization, and so on. Many monomers, including amide, ester, urethane, urea, alkene, aniline, pyrrole and their derivatives, are available for interfacial polymerization.2–11 Using interfacial polymerization, these monomers can be utilized to fabricate various polymer materials with unique topological and chemical properties, such as anisotropic shapes, hollow cores or alternative surface chemistry. These synthetic materials can be mainly categorized into zero-dimensional (0D) nanoparticles (NPs), one-dimensional (1D) nanofibers, two-dimensional (2D) films, and three-dimensional (3D) composite membranes. Moreover, they have been extensively applied in sensors,12,13 supercapacitors,14–19 batteries,20 separation,6,7,21–23 and cargo loading.24–28When compared with bulk polymerization, interfacial polymerization occurs at the interface of two immiscible phases.5 Therefore, the design and fabrication of polymer materials via interfacial polymerization usually refers to two main aspects: (i) construction of an interface between two immiscible phases and (ii) distribution of monomer in two immiscible phases. An interface can be generally classified as follows: liquid–solid, liquid–gas, liquid–liquid, and liquid-in-liquid emulsion interfaces. When an interface is constructed by two immiscible liquids, either one or both liquid solvents contain monomers. Due to the difference in the chemical potentials of two immiscible liquids at the interface, the monomers may diffuse to the interface and then be initiated to polymerize at interface. When the interfacial pressure of the adsorbed monomer monolayer exceeds the equilibrium spreading pressure of the polymer, the resulting polymer is anchored at the interface.5,29 As monomers diffuse and polymerization proceeds, the polymer grows at the interface. Therefore, interfacial polymerization has been considered as a popular method to prepare various polymer materials, which have been extensively used in many applications. However, to the best of our knowledge, there are no reviews that comprehensively summarize interfacial polymerization from the point of view of polymerization mechanism, fabrication methods, and applications. Therefore, it is urgently desirable to review interfacial polymerization from the controllable fabrication methods to their widespread applications.
In this review, recent progress in interfacial polymerization, generally based on the fabrication methods and application fields, has been summarized. Based on the interface classification and monomer distribution at the interface, five typical interfacial polymerization systems have been catalogued. Subsequently, recent accomplishments in the application of synthetic polymer materials, such as in electronic devices, separation, and cargo loading, prepared by interfacial polymerization have also been summarized. Finally, some of the current challenges and opportunities in interfacial polymerization are proposed.
We concluded that the interface can be classified as follows: liquid–solid interface (L–S), liquid–liquid interface (L–L), and liquid-in-liquid emulsion interface (L-in-L). In L–L and L-in-L systems, either one or both liquids may contain monomers. Therefore, Lm–S, Lm–L, Lm–Lm, Lm-in-L, and Lm-in-Lm interfacial polymerization systems have been summarized, as shown in Fig. 1 (where Lm represents the liquid phase containing the monomer). For the Lm–S system, the polymerization is initiated at the L–S interface, resulting in the formation of a polymeric film on the solid phase. For the L–L (including Lm–L and Lm–Lm) or L-in-L (including Lm-in-L and Lm-in-Lm) systems, interfacial polymerization can take place either on one side or on both sides depending on whether the monomers are present in one or both immiscible liquid phases. When the monomers only exist in a single liquid phase and the initiators are dissolved in the other liquid phase, they polymerize into homogeneous polymer materials at the interface, whereas composite polymer materials will be obtained when the two monomers separately dissolved in immiscible liquids copolymerize at the interface. However, some other interface categories, such as liquid–gas,30 solid–gas,31 and solid–solid interfaces,32 have been rarely reported in the interfacial polymerization and hence have not been discussed in this review. All these systems show their effectiveness to fabricate interfacial polymer materials. These polymer materials include particles, capsules, films, and composite membranes.
 | ||
| Fig. 1 Typical fabrication systems used in interfacial polymerization. The categories include liquid/monomer–solid (Lm–S) interfacial polymerization, liquid/monomer–liquid (Lm–L) interfacial polymerization, liquid/monomer–liquid/monomer (Lm–Lm) interfacial polymerization, liquid/monomer-in-liquid (Lm-in-L) interfacial polymerization, and liquid/monomer-in-liquid/monomer (Lm-in-Lm) interfacial polymerization, where Lm represents a liquid phase containing the monomer, L represents the liquid phase, and S represents the solid phase. | ||
2. Theory models of interfacial polymerization
Herein, two theory models to study the polymerization kinetics of interfacial polymerization have been briefly introduced. The theoretical studies of interfacial polymerization mainly focus on the polymerization kinetics, which can predict the growth rate and thickness of the polymeric materials upon controlling the polymerization time. Other parameters, such as density,33,35 porosity,33 and structure,36 have also been proposed. Ji and co-workers introduced a general theory model to study the formation of thin film composite (TFC) membranes,33 hollow fibers,37 tubular membranes,37 and capsules.34 In the Lm–S interface polymerization model, the initial interface is constructed by a porous support and monomer solution. As polymerization occurs, a thin-film is formed and polymerization proceeds at the reaction zone (Fig. 2a). The diffusion and polymerization rate are initially considered because they greatly affect the film growth rate. To simplify the calculation process, the mathematical models are generally calculated based on some assumptions; such as (i) the newly formed polymer cannot be dissolved in the liquid phase; (ii) the density of the thin film is uniform during the polymerization process; (iii) polymerization takes place at the polymerization zone having a constant thickness; and (iv) the polymerization reaction exhibits second-order kinetics with the polymerization rate being proportional to the monomer concentration, and so on. Under these assumptions, a general formula describing the relationship between polymerization time (t) and thickness (X) was obtained, as shown in eqn (1). | (1) |
 | ||
| Fig. 2 Two typical models of interfacial polymerization. (a) In the Lm–S interfacial polymerization model, the initial interface is constructed by a porous support and monomer solution. As polymerization occurs, a thin film is formed and polymerization proceeds at the reaction zone. (b) In the Lm-in-Lm interfacial polymerization model, the initial interface is constructed by an inner monomer solution and an outer monomer solution. As polymerization occurs, a capsule wall is formed and polymerization proceeds at the reaction zone. (c) The model proposed in Lm–S interfacial polymerization is well matched with the experimental data. (d) The model proposed in Lm-in-Lm interfacial polymerization matches well with the experimental data. Reproduced with permission from ref. 33 and 34. Copyright 2000, American Chemical Society. Copyright 2001, Elsevier. | ||
Moreover, they simulated the Lm-in-Lm interface model (Fig. 2b),34 in which the initial interface was constructed by an inner monomer solution and outer monomer solution. In the model, the polycondensation of immiscible monomers can only occur at the interface. As the polymerization occurs, the capsule wall is formed and polymerization proceeds at the reaction zone. Some assumptions are also proposed: (i) the monomer concentrations are uniform in their respective solution; (ii) the capsule wall has a spherical geometry; (iii) only the outer monomer is limited by diffusion, and so on. They finally obtained a general formulation between the polymerization time (t) and thickness of the capsule wall (X), as shown in eqn (2) and (3).
 | (2) |
| X = r0 − R | (3) |
These simulations succeed in simulating the thickness of the film or capsule wall formed during interfacial polymerization. Reasonably, they proposed an effective method to study the reaction kinetics and predict the size of polymer materials. Thus, the precise design and control of polymer thickness are possible. However, there are several problems to be addressed. First, the simulation results based on the two models were calculated under non-steady-state conditions, whereas they are also applicable to steady-state conditions. In the steady-state, the amount of monomer in the newly-formed polymer and that converted into a polymer are negligible when compared with amount of the monomer in the solution, which is approximately satisfied during the initial stage of polymerization. Second, the newly-formed polymer layer was assumed to be homogeneous in these simulations, whereas recent simulations demonstrate that the polymer density is different through the entire film formed via interfacial polymerization.36,39,40 These simulation results generally show that the resultant polymer films have a dense core and looser ends, leading to a heterogeneous distribution of functional groups and charge across the film.
3. The fabrication methods used for interfacial polymerization
The past few decades have witnessed great progress in fabricating polymer and hybrid materials via interfacial polymerization with the purpose of realizing various functionalities and their subsequent applications. The fabrication methods used in interfacial polymerization for the preparation of polymer materials have been discussed hereinafter, generally based on five systems, as summarized in Fig. 1.3.1. Lm–S interfacial polymerization
In Lm–S interfacial polymerization, the monomers are dissolved in the liquid solution, and then they polymerize at the L–S interface, resulting in hybrid materials. This type of interfacial polymerization can be classified into two types based on the monomers: (i) polycondensation and (ii) polyaddition. Polycondensation takes place between acyl chlorides and monomers containing hydrogen atoms, such as amines, polyhydric alcohols, and polyhydric phenols, available for the reaction. Among these, acyl chlorides and amines are the most frequently used monomers, as shown in Fig. 3. For polyaddition, alkenes are the most frequently used monomers. | ||
| Fig. 3 Lm–S interfacial polymerization. Two main aspects in the Lm–S interfacial polymerization are the monomers and solid substrates. Monomers include those for polycondensation (amines and acyl chlorides) and those for polyaddition (alkenes), whereas the solid substrates range from 0D NPs, 1D fibers, and 2D plates to 3D porous scaffolds. Reproduced with permission from ref. 41, 42, 47 and 48. Copyright 2007, Royal Society of Chemistry. Copyright 2011 and 2014, Wiley-VCH Verlag GmbH & Co. KGaA. Copyright 2016, Nature Publishing Group. | ||
For the first Lm–S interfacial polymerization type, the solid substrates are often separately wetted by two immiscible monomer solutions; after the monomers make contact at the Lm–S interface, polycondensation occurs. This polymerization system is applicable to porous substrates, such as 1D fiber scaffolds and 3D porous membranes (Fig. 3 bottom),41–45 using amines and acyl chlorides as monomers (Fig. 3 top). This type of polymerization is popular for the fabrication of composite membranes. For example, Zhang et al.43 reported a high flux TFC membrane via Lm–S interfacial polymerization. They used a porous poly(vinylidene fluoride) (PVDF) nanofibrous scaffold as a solid substrate. After PVDF was wetted using piperazine and trimesoyl chloride monomer solution, polymerization occurred at the monomer–PVDF interface. The resultant hybrid membranes exhibited hydrophilic surfaces and porous structures, which were beneficial for high-flux water permeation. For the second Lm–S interfacial polymerization system, the initiators are first immobilized on the solid substrate. Then, the monomers are initiated by the immobilized initiator to polymerize at the Lm–S interface. This type of polymerization usually employs 0D NPs and 2D plates46–48 as solid substrates (Fig. 3 bottom) and alkenes, such as styrene (St),46N-isopropylacrylamide (NiPAM),49,50 MMA,48 and butyl methacrylate,46,47 as monomers. For example, Jang et al.48 reported a CdS/poly(methyl methacrylate) (PMMA) core/shell NPs via Lm–S interfacial polymerization process. The Lm–S interface was constructed when the MMA monomer solution was wetted on Ce4+ ions-modified CdS NPs. The polymerization was triggered by Ce4+ ions with the increasing temperature. Since Ce4+ ions were immobilized on the CdS NPs, MMA was exclusively polymerized at the interface of the solution and CdS NPs to form a protective layer. This type of surface modification via interfacial polymerization can significantly improve the dispersity and stability of the NPs.
Recently, new insights on Lm–S interfacial polymerization have been demonstrated by introducing additives, which can realize alternative surface chemistry with the purpose of special applications. For example, Zhao et al.51 reported an aquaporin-doped membrane, with enhanced water permeability, via interfacial polymerization. Before polymerization, they mixed aquaporin (a water channel protein with excellent permeability and solute rejection) in the monomer solution. After the Lm–S interface polymerization, aquaporins were embedded in the polymeric layer. When compared with commercial reverse osmosis (RO) membranes, the aquaporin-doped membrane showed ∼40% higher water permeability and comparable salt rejection (∼97%), demonstrating that the introduction of additional additives in interfacial polymerization can significantly improve the properties of the materials. A composite membrane fabricated via Lm–S interfacial polymerization is an effective technique used in water treatment. Moreover, water treatment shows the challenge of surfactant-stabilized oil/water separation and their separation in highly acidic and alkaline environments.52 Therefore, the design and preparation of composite membranes via interfacial polymerization, with both resistance to acidic and alkaline solutions and ability to break emulsions, are promising in practice.
3.2. Lm–L interfacial polymerization
In Lm–L interfacial polymerization, the initiators and monomers are dissolved in separate liquid phases. When initiators and monomers diffuse to the L–L interface, polymerization is triggered (Fig. 4a). This interfacial system can produce homogeneous films or particles. As polymerization proceeds, the polymer gradually diffuses and fills one liquid phase.53 Therefore, the resulting amount of polymer material usually increases as the polymerization time increases (Fig. 4b). | ||
| Fig. 4 Lm–L interfacial polymerization. (a) A schematic description. (b) The amount of the resultant polymer materials increased as the polymerization time increased. Lm–L interfacial polymerization can realize the controllable preparation of polymer materials with various morphologies: (c) films, (d) fibers, and (e) needles. Reproduced with permission from ref. 53–55. Copyright 2004 and 2008, American Chemical Society. Copyright 2013, Royal Society of Chemistry. | ||
More importantly, Lm–L interfacial polymerization can realize the controllable preparation of polymer materials with various morphologies. Qi et al.54 synthesized free standing polypyrrole (PPy) films via Lm–L interfacial polymerization (Fig. 4c). In the initial stage, the monomers and initiators easily diffused to the interface and undergo polymerization, leading to the formation of a PPy film. Once the thin and intact PPy film was formed, the diffusion of monomers and initiators would be impeded and further polymerization would be retarded. In another case, Huang et al.53 developed an approach to synthesize polyaniline (PANI) nanofibers (Fig. 4d). Due to the linear nature of the PANI chains, PANI nanofibers were formed at the interface. However, after they migrated to the upper layer, the polymerization was terminated. Hence, the PNAI nanofibers were not subject to further polymerization when they left the interface, almost exclusively producing nanofibers. Nuraje et al.55 realized the preparation of PANI and PPy nanoneedles via Lm–L interfacial polymerization (Fig. 4e). After the monomers were oxidized using Fe3+ in the polymerization process, positively charged polymers were formed and they were bridged by the hydrophilic Cl− ions in the aqueous solution. Then, the polymerization proceeded along the normal of the interface and the polymer could be aligned between the positively charged repeated units and negatively charged Cl− ions during polymerization, forming elongated nanoneedles. In general, Lm–L interfacial polymerization is frequently used for the preparation of conducting polymers including PPy and PANI.15,56–58 This approach is applicable for the production of thin films and low-dimensional NPs, which have been widely used in electronic devices. However, the large area production of conducting polymer materials prepared via Lm–L interfacial polymerization is still difficult because the polymerization area is rather limited by the interface. Therefore, future techniques may focus on large area preparation.
3.3. Lm–Lm interfacial polymerization
In the Lm–Lm interfacial polymerization system, two different monomers are dissolved in two immiscible liquid solutions and the monomers will polymerize when they come in contact with each other at the interface.59–62 Lm–Lm interfacial polymerization is usually used to prepare Janus films. For example, Wamser et al.63 reported the fabrication of Janus porphyrin–polyamide composite films via interfacial polymerization. In a typical procedure, tetrakis(4-aminophenyl)porphyrin (TAPP) or diethylenetriamine (DETA) and tetrakis[4-(chlorocarbonyl)phenyl]porphyrin (TCCPP) were separately dissolved in two immiscible liquids. Interfacial polymerization occurred via dechlorohydrogenation between two monomers, resulting in a porphyrin–polyamide film, as described in Fig. 5a. The typical microscopy images of the film showed the easy deformation properties, as presented in Fig. 5b. Upon determination of the contact angle, the N side (TAPP/TCCPP film) was more hydrophobic than the C side, as shown in Fig. 5c, which demonstrated the Janus properties of the film. | ||
| Fig. 5 Lm–Lm interfacial polymerization. (a) A schematic describing the polymerization system. (b) SEM images showing the ease of deformation of the film. (c) The N side (TAPP/TCCPP film) was more hydrophobic than the C side. Reproduced with permission from ref. 63. Copyright 1989, American Chemical Society. | ||
These Janus films fabricated via Lm–Lm interfacial polymerization demonstrate flexible and anisotropic deformation properties, indicating their great potential in artificial muscles and soft robotics. Moreover, various properties, such as wettability, optical, and swelling properties, of these Janus films can be thoroughly studied.64
3.4. Lm-in-L interfacial polymerization
A Lm-in-L interface is fabricated when the monomers are dissolved in the inner liquid phase of a surfactant-stabilized emulsion, whereas the initiators are dissolved in the outer liquid phase. After the initiator radicals are generated via the decomposition of the initiators, they can diffuse into the L-in-L interface and trigger the polymerization, producing homogeneous capsules. This type of interfacial polymerization demonstrates the effectiveness in controlling the capsule shell thickness. Recently, Kang et al.65 developed a controllable up-conversion (UC) system composed of uniform capsules fabricated via Lm-in-L interfacial polymerization combined with a microfluidic approach. As shown in Fig. 6a, the chromophore-dispersed monomer solution was injected as an inner liquid phase, whereas an aqueous solution containing a photoinitiator and surfactant was introduced as an outer liquid phase. After the oil-in-water capsule was irradiated with UV light, free radicals were produced in the aqueous phase and they diffused into the interface. Subsequently, the monomers diffused towards the interface and polymerization occurred at the Lm-in-L interface. The resultant capsules comprised a fluidic active core and an elastomeric shell (Fig. 6b). The thickness of the shell layer increased as the polymerization time increased, by which the thickness of the up-converted and down-converted layers could be controlled (Fig. 6c). | ||
| Fig. 6 Lm-in-L interfacial polymerization. (a) Construction of the oil-in-water emulsion droplet interface. (b) Lm-in-L interfacial polymerization. When the radicals from water phase and monomers from oil core diffuse towards the Lm-in-L interface, the polymerization occurs, finally resulting in the formation of UC capsules. (c) The shell thickness increase as the polymerization time increase. Reproduced with permission from ref. 65. Copyright 2012, Wiley-VCH Verlag GmbH & Co. KGaA. | ||
The controllable preparation of capsules with uniform and tunable size is still a challenge in Lm–L interfacial polymerization. Although the microfluidic approach is highly effective in producing uniform capsules, their size is usually in the range of micrometers and is difficult to tune.66 Moreover, nanocapsules are favorable in drug delivery. Therefore, the preparation of uniform and size-tunable capsules is desirable in the future.
3.5. Lm-in-Lm interfacial polymerization
In the Lm-in-Lm interface polymerization system, two types of monomers are simultaneously present in the outer and inner liquid phases of the emulsion, whereas the initiators are dissolved in the outer liquid phase. After the initiator radicals are generated and they diffuse into the Lm-in-L interface, monomers in the two immiscible liquids will only copolymerize at the interface. This type of interfacial polymerization is usually used to prepare composite polymer capsules for cargo loading. Scott et al.67 fabricated nanoscale composite capsules via interfacial polymerization (Fig. 7a). A miniemulsion was first prepared by emulsifying an oil phase containing dibutyl maleate as the hydrophobic monomer, an aqueous phase containing poly(ethylene glycol) divinyl ether as the hydrophilic monomer and dodecyltrimethylammonium bromide as the surfactant. Then, the temperature was increased to 60 °C and the initiator radicals generated from the amphiphilic initiators were anchored on the Lm-in-Lm interface. As a result, the ester and ether can only copolymerize at the Lm-in-Lm interface, producing the composite polymer capsules (Fig. 7b). These capsules are effective for the encapsulation of drugs, dyes, and enzymes, endowing them with great potential in drug delivery and other applications. | ||
| Fig. 7 Lm-in-Lm interfacial polymerization. Schematic describing (a) the interfacial polymerization process and (b) interfacial polymerization mechanism. Reproduced with permission from ref. 67. Copyright 2005, American Chemical Society. | ||
Due to the limitation of interfacial tension, particles prepared from emulsion polymerization are usually spherical. However, interfacial polymerization at the Lm-in-Lm interface proposes a new approach to fabricate anisotropic Janus particles. Wang et al.68 fabricated an oil-in-water emulsion, which contained hydrophobic monomers, hydrophilic monomers, and oil-soluble species. When the hydrophobic monomers polymerized in the inner liquid phase, phase separation occurred. The newly-formed cross-linked polymer spheres migrated towards the interface and subsequently, the monomers in the two phases copolymerized along the interface, producing asymmetric Janus particles (Fig. 8). As anisotropic Janus particles have been extensively used as optical probes, solid surfactants, and self-propulsion motors,69–72 it will be significant to simultaneously synthesize chemically and topologically anisotropic Janus particles via Lm-in-Lm interfacial polymerization.
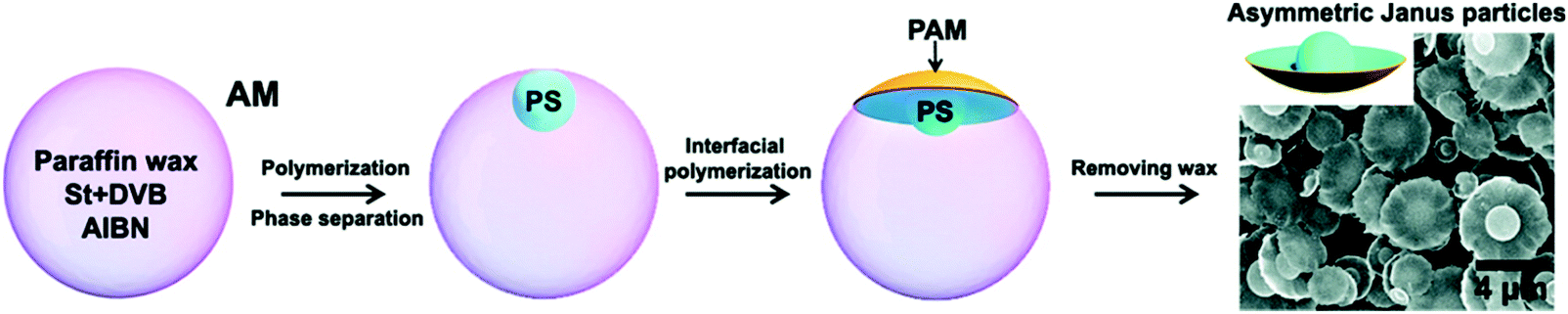 | ||
| Fig. 8 An asymmetric Janus particle produced via Lm-in-Lm interfacial polymerization. Reproduced with permission from ref. 68. Copyright 2011, American Chemical Society. | ||
4. Applications of polymer materials via interfacial polymerization
4.1. Conducting polymers for electronic devices
In recent years, interfacial polymerization is very popular for the preparation of conducting polymer materials such as, PANI, PPy, poly(3,4-ethylenedioxythiophene) (PEDOT), and polythiophene (PTh). These conducting polymer materials are superb conductors due to their fast electron transportation properties and have been widely used in electronic devices such as sensors, switches, supercapacitors, and cells.73–83 | ||
| Fig. 9 Conductor materials fabricated via interfacial polymerization for electrical applications: (a) sensor, (b) switch, and (c) supercapacitor. Reproduced with permission from ref. 13, 14, 55 and 84. Copyright 2004, Wiley-VCH Verlag GmbH & Co. KGaA. Copyright 2004 and 2008, American Chemical Society. Copyright 2015, Royal Society of Chemistry. | ||
Nanoscale conducting polymer materials used in electronic devices can be readily synthesized using interfacial polymerization. As the miniaturization of electrical devices is the trend of future industry,86 nano-sized conducting polymer particles are promising in the next-generation microelectronics.
4.2. Membranes used for separation
With an urgent demand for water pollution treatment and seawater desalination, scientists have devoted great efforts to fabricate highly efficient separation materials.52,87–103 Separation of oil, ions, and biomolecules from water is the main focus in water treatment, which requires both high water flux and separation selectivity. Membranes, especially RO membranes, are a popular approach for foul water treatment. Generally, Lm–S interfacial polymerization is one of the most commonly used techniques for the fabrication of RO membranes. Recently, Solomon et al.42 reported a cross-linked and rigid nanofilm by manipulating the molecular structure via Lm–S interfacial polymerization. These films showed enhanced microporosity and higher interconnectivity in the intermolecular network voids. Moreover, these pores endowed smaller molecules to freely pass through, whereas reject larger molecules (Fig. 10a). Due to this, they exhibited outstanding performance in dye separation from water (Fig. 10c). In addition, these porous membranes are able to separate larger gas molecules from smaller gas molecules, such as the separation of CO2 and H2 (Fig. 10b and d). | ||
| Fig. 10 Membranes fabricated via interfacial polymerization used for separation. The separation of (a and c) solvent and solute and (b and d) bi-component gas. Reproduced with permission from ref. 42. Copyright 2016, Nature Publishing Group. | ||
As one of the most popular separation materials, membranes will continuously attract interests in environmental, energy, and some other industrial applications. To date, liquid separation and gas separation have been realized by membranes fabricated via interfacial polymerization. Moreover, the separation of various ions, macromolecules, particles, and biological units (such as cell, bacterium, and virus) is also very important.104–116 Interfacial polymerization is promising in various applications because membranes engineered via interfacial polymerization possess advantages such as tunable porosity, alternative surface chemistry, and easy doping.
4.3. Capsules used for cargo loading
Capsules have attracted significant attention in pharmaceuticals, microreactors, catalysis, diagnostics, and other biotechnological application fields.25,26,28,117–122 Polymer capsules are generally composed of a polymeric shell and hollow core, in which various cargo can be loaded. Many synthetic methods, including self-assembly, layer-by-layer assembly, single-step polymer adsorption, and surface/interfacial polymerization have been developed for capsule preparation.27,123 In comparison, L-in-L interfacial polymerization is a controllable, facile, and flexible route to prepare hollow polymer capsules with diverse functionalities and properties.Cui et al.124 developed a Lm-in-L interfacial polymerization for the synthesis of monodispersed polymer capsules with tunable size (from 400 nm to 2.4 μm) and shell thickness (from ∼10 nm to 140 nm). Moreover, this method was well suited for the encapsulation of a series of hydrophobic species including drugs (thiocoraline), CdSe/CdS quantum dots (QDs), and Fe3O4 NPs (Fig. 11). In the polymerization process, the functionalized cargo is initially dispersed in the emulsion droplets. After the monomer polymerization at the liquid–liquid interface and removal of internal liquid, the functionalized particles or molecules remain encapsulated in the polymer capsules. The capsules are generally well-dispersed, providing a confined space for cargo loading and transportation with good dispersity. This method will be promising in opening up new avenues for the preparation of polymer capsules that are used in biomedical applications.
 | ||
| Fig. 11 Capsules fabricated via interfacial polymerization for cargo loading. (a) A schematic. (b) The loading of drugs (thiocoraline), (c) The loading of CdSe/CdS QDs. Insets is high resolution transmission microscopy (HRTEM) images, indicating the QDs are successfully loaded. (d) The loading of Fe3O4 NPs. Insets is HRTEM images, indicating the Fe3O4 NPs are successfully loaded. Reproduced with permission from ref. 124. Copyright 2010, Wiley-VCH Verlag GmbH & Co. KGaA. | ||
In conclusion, interfacial polymerization offers a one-step method for the preparation of cargo/vehicle hybrids. When applied in practical disease therapy, the microenvironments in vivo require the capsules that are equipped with multiple properties. These include switches from “close” to “open”, robust enough to resist decomposition on untargeted sites, the controlled release of drugs, good biocompatibility and biodegradation, and so on. These properties are generally determined by the chemical and topological properties of polymer materials fabricated via interfacial polymerization.
5. Conclusion and perspective
In this review, we have summarized the recent progress of interfacial polymerization as an effective approach to fabricate polymer and hybrid materials, and their widespread applications. Various categories of interfacial polymerization system demonstrate their good control over the topology and chemistry and generality for monomers and interface categories. A series of polymer materials, including particles, capsules, and membranes, have been prepared via interfacial polymerization and are widely used in electronic devices, separation, and cargo loading.However, there are still several challenges that need to be addressed in interfacial polymerization: (i) large area production of materials is difficult because the interface surface is limited; (ii) due to the limitation of interfacial tension, the preparation of anisotropic Janus particles during interfacial polymerization is still a challenge; and (iii) the preparation of uniform and size-tunable capsules via interfacial polymerization is still difficult. Although there are still challenges, new opportunities from new materials will shed light on interfacial polymerization in the future; for example, asymmetric Janus materials have served as platforms for advanced applications, such as separation, auto propulsion, artificial muscle, and soft robotics. The preparation of Janus materials with both topologically and chemically anisotropic properties via interfacial polymerization will be important and significant.
Acknowledgements
This research is supported by the National Natural Science Foundation of China (21504098, 21425314, 21501184, 21434009 and 21421061), the National Research Fund for Fundamental Key Projects (2012CB933800), the Key Research Program of the Chinese Academy of Sciences (KJZD-EW-M01), the National High Technology Research and Development Program of China (863 Program) (2013AA032203), MOST (2013YQ190467), the Top-Notch Young Talents Program of China, and the Beijing Municipal Science & Technology Commission (Z161100000116037).Notes and references
- E. L. Wittbecker and P. W. Morgan, J. Polym. Sci., 1959, 40, 289–297 CrossRef CAS.
- K. H. Shen, D. B. Qi, Y. Li, X. L. Zhao and Y. Li, Chem. Commun., 2015, 51, 10166–10169 RSC.
- S. Karan, Z. W. Jiang and A. G. Livingston, Science, 2015, 348, 1347–1351 CrossRef CAS PubMed.
- N. C. Mougin, P. van Rijn, H. Park, A. H. E. Muller and A. Boker, Adv. Funct. Mater., 2011, 21, 2470–2476 CrossRef CAS.
- V. V. Yashin and A. C. Balazs, J. Chem. Phys., 2004, 121, 11440–11454 CrossRef CAS PubMed.
- W. J. Lau, A. F. Ismail, N. Misdan and M. A. Kassim, Desalination, 2012, 287, 190–199 CrossRef CAS.
- G. D. Kang and Y. M. Cao, Water Res., 2012, 46, 584–600 CrossRef CAS PubMed.
- B. X. Yang, K. P. Pramoda, G. Q. Xu and S. H. Goh, Adv. Funct. Mater., 2007, 17, 2062–2069 CrossRef CAS.
- K. Y. Shin, S. Cho and J. Jang, Small, 2013, 9, 3792–3798 CrossRef CAS PubMed.
- W. Choi, J. E. Gu, S. H. Park, S. Kim, J. Bang, K. Y. Baek, B. Park, J. S. Lee, E. P. Chan and J. H. Lee, ACS Nano, 2015, 9, 345–355 CrossRef CAS PubMed.
- X. G. Li, Y. Z. Liao, M. R. Huang and R. B. Kaner, Chem. Sci., 2015, 6, 2087–2101 RSC.
- J. Jang, M. Chang and H. Yoon, Adv. Mater., 2005, 17, 1616–1620 CrossRef CAS.
- J. Huang, S. Virji, B. H. Weiller and R. B. Kaner, Chem. – Eur. J., 2004, 10, 1314–1319 CrossRef CAS PubMed.
- D. J. Li, Y. Li, Y. Y. Feng, W. P. Hu and W. Feng, J. Mater. Chem. A, 2015, 3, 2135–2143 CAS.
- H. Guan, L. Z. Fan, H. C. Zhang and X. H. Qu, Electrochim. Acta, 2010, 56, 964–968 CrossRef CAS.
- U. Male, B. S. Singu and P. Srinivasan, J. Appl. Polym. Sci., 2015, 132, 42013 Search PubMed.
- G. A. Snook, P. Kao and A. S. Best, J. Power Sources, 2011, 196, 1–12 CrossRef CAS.
- K. Wang, H. P. Wu, Y. N. Meng and Z. X. Wei, Small, 2014, 10, 14–31 CrossRef CAS PubMed.
- B. Anothumakkool, R. Soni, S. N. Bhange and S. Kurungot, Energy Environ. Sci., 2015, 8, 1339–1347 CAS.
- Y. S. Choi, S. H. Joo, S. A. Lee, D. J. You, H. Kim, C. Pak, H. Chang and D. Seung, Macromolecules, 2006, 39, 3275–3282 CrossRef CAS.
- Y. Z. Zhu, W. Xie, S. J. Gao, F. Zhang, W. B. Zhang, Z. Y. Liu and J. Jin, Small, 2016, 12, 5034–5041 CrossRef CAS PubMed.
- S. C. Li, Z. Wang, X. W. Yu, J. X. Wang and S. C. Wang, Adv. Mater., 2012, 24, 3196–3200 CrossRef CAS PubMed.
- H. Q. Zhang and J. D. Liu, Chin. Chem. Lett., 2004, 15, 609–610 CAS.
- S. Kango, S. Kalia, A. Celli, J. Njuguna, Y. Habibi and R. Kumar, Prog. Polym. Sci., 2013, 38, 1232–1261 CrossRef CAS.
- M. M. Conn and J. Rebek, Chem. Rev., 1997, 97, 1647–1668 CrossRef CAS PubMed.
- S. De Koker, R. Hoogenboom and B. G. De Geest, Chem. Soc. Rev., 2012, 41, 2867–2884 RSC.
- J. F. Shi, Y. J. Jiang, X. L. Wang, H. Wu, D. Yang, F. S. Pan, Y. L. Su and Z. Y. Jiang, Chem. Soc. Rev., 2014, 43, 5192–5210 RSC.
- S. F. M. van Dongen, H. P. M. de Hoog, R. Peters, M. Nallani, R. J. M. Nolte and J. C. M. van Hest, Chem. Rev., 2009, 109, 6212–6274 CrossRef CAS PubMed.
- F. Macritch, Trans. Faraday Soc., 1969, 65, 2503–2507 RSC.
- J. E. Biegajski, D. A. Cadenhead and P. N. Prasad, Macromolecules, 1991, 24, 298–303 CrossRef CAS.
- A. M. Coclite, R. M. Howden, D. C. Borrelli, C. D. Petruczok, R. Yang, J. L. Yague, A. Ugur, N. Chen, S. Lee, W. J. Jo, A. D. Liu, X. X. Wang and K. K. Gleason, Adv. Mater., 2013, 25, 5392–5422 CrossRef CAS PubMed.
- J. Stejskal, I. Sapurina and M. Trchova, Prog. Polym. Sci., 2010, 35, 1420–1481 CrossRef CAS.
- J. Ji, J. M. Dickson, R. F. Childs and B. E. McCarry, Macromolecules, 2000, 33, 624–633 CrossRef CAS.
- J. Ji, R. F. Childs and M. Mehta, J. Membr. Sci., 2001, 192, 55–70 CrossRef CAS.
- V. Freger and S. Srebnik, J. Appl. Polym. Sci., 2003, 88, 1162–1169 CrossRef CAS.
- A. V. Berezkin and Y. V. Kudryavtsev, Langmuir, 2015, 31, 12279–12290 CrossRef CAS PubMed.
- J. Ji and M. Mehta, J. Membr. Sci., 2001, 192, 41–54 CrossRef CAS.
- L. J. J. M. Janssen, A. Boersma and K. Tenijenhuis, J. Membr. Sci., 1993, 79, 11–26 CrossRef CAS.
- R. Nadler and S. Srebnik, J. Membr. Sci., 2008, 315, 100–105 CrossRef CAS.
- R. Oizerovich-Honig, V. Raim and S. Srebnik, Langmuir, 2010, 26, 299–306 CrossRef CAS PubMed.
- J. C. Chen, J. A. Wu, K. H. Lin, P. J. Lin and J. H. Chen, Polym. Eng. Sci., 2014, 54, 430–437 CAS.
- M. F. Jimenez-Solomon, Q. Song, K. E. Jelfs, M. Munoz-Ibanez and A. G. Livingston, Nat. Mater., 2016, 15, 760–767 CrossRef CAS PubMed.
- H. Y. Zhang, J. F. Zheng, Z. G. Zhao and C. C. Han, J. Membr. Sci., 2013, 442, 124–130 CrossRef CAS.
- J. J. Zhao, Y. Zhang, Y. L. Su, J. Z. Liu, X. T. Zhao, J. M. Peng and Z. Y. Jiang, J. Membr. Sci., 2013, 445, 1–7 CrossRef CAS.
- C. M. Zhou, Y. L. Shi, C. S. Sun, S. C. Yu, M. H. Liu and C. J. Gao, J. Membr. Sci., 2014, 471, 381–391 CrossRef CAS.
- P. A. Levkin, F. Svec and J. M. Frechet, Adv. Funct. Mater., 2009, 19, 1993–1998 CrossRef CAS PubMed.
- D. Zahner, J. Abagat, F. Svec, J. M. J. Frechet and P. A. Levkin, Adv. Mater., 2011, 23, 3030–3034 CrossRef CAS PubMed.
- J. S. Jang, S. Kim and K. J. Lee, Chem. Commun., 2007, 2689–2691 RSC.
- M. Kaholek, W. K. Lee, S. J. Ahn, H. W. Ma, K. C. Caster, B. LaMattina and S. Zauscher, Chem. Mater., 2004, 16, 3688–3696 CrossRef CAS.
- H. Liu, X. Liu, J. Meng, P. Zhang, G. Yang, B. Su, K. Sun, L. Chen, D. Han, S. Wang and L. Jiang, Adv. Mater., 2013, 25, 922–927 CrossRef CAS PubMed.
- Y. Zhao, C. Q. Qiu, X. S. Li, A. Vararattanavech, W. M. Shen, J. Torres, C. Helix-Nielsen, R. Wang, X. Hu, A. G. Fane and C. Y. Tang, J. Membr. Sci., 2012, 423, 422–428 CrossRef.
- J.-B. Fan, Y. Song, S. Wang, J. Meng, G. Yang, X. Guo, L. Feng and L. Jiang, Adv. Funct. Mater., 2015, 25, 5368–5375 CrossRef CAS.
- J. X. Huang and R. B. Kaner, J. Am. Chem. Soc., 2004, 126, 851–855 CrossRef CAS PubMed.
- G. J. Qi, Z. L. Wu and H. L. Wang, J. Mater. Chem. C, 2013, 1, 7102–7110 RSC.
- N. Nuraje, K. Su, N. L. Yang and H. Matsui, ACS Nano, 2008, 2, 502–506 CrossRef CAS PubMed.
- C. Bora and S. K. Dolui, Polymer, 2012, 53, 923–932 CrossRef CAS.
- G. J. Qi, L. Y. Huang and H. L. Wang, Chem. Commun., 2012, 48, 8246–8248 RSC.
- J. Song, H. Liu, M. X. Wan, Y. Zhu and L. Jiang, J. Mater. Chem. A, 2013, 1, 1740–1744 CAS.
- S. Liu, H. Zhang, R. A. Remy, F. Deng, M. E. Mackay, J. M. Fox and X. Q. Jia, Adv. Mater., 2015, 27, 2783–2790 CrossRef CAS PubMed.
- G. Y. Chai and W. B. Krantz, J. Membr. Sci., 1994, 93, 175–192 CrossRef CAS.
- H. Yamaguch and Y. Minoura, J. Polym. Sci., Polym. Chem. Ed., 1973, 11, 763–778 CrossRef.
- A. Provatas, J. Matisons, S. Clarke, D. Graiver and W. Lomas, Macromolecules, 2000, 33, 9156–9159 CrossRef CAS.
- C. C. Wamser, R. R. Bard, V. Senthilathipan, V. C. Anderson, J. A. Yates, H. K. Lonsdale, G. W. Rayfield, D. T. Friesen, D. A. Lorenz, G. C. Stangle, P. Vaneikeren, D. R. Baer, R. A. Ransdell, J. H. Golbeck, W. C. Babcock, J. J. Sandberg and S. E. Clarke, J. Am. Chem. Soc., 1989, 111, 8485–8491 CrossRef CAS.
- S. Wang, K. Liu, X. Yao and L. Jiang, Chem. Rev., 2015, 115, 8230–8293 CrossRef CAS PubMed.
- J. H. Kang and E. Reichmanis, Angew. Chem., Int. Ed., 2012, 51, 11841–11844 CrossRef CAS PubMed.
- J.-B. Fan, C. Huang, L. Jiang and S. Wang, J. Mater. Chem. B, 2013, 1, 2222–2235 RSC.
- C. Scott, D. Wu, C. C. Ho and C. C. Co, J. Am. Chem. Soc., 2005, 127, 4160–4161 CrossRef CAS PubMed.
- Y. H. Wang, C. L. Zhang, C. Tang, J. Li, K. Shen, J. G. Liu, X. Z. Qu, J. L. Li, Q. Wang and Z. Z. Yang, Macromolecules, 2011, 44, 3787–3794 CrossRef CAS.
- A. Walther and A. H. E. Mueller, Chem. Rev., 2013, 113, 5194–5261 CrossRef CAS PubMed.
- F. Liang, K. Shen, X. Qu, C. Zhang, Q. Wang, J. Li, J. Liu and Z. Yang, Angew. Chem., Int. Ed., 2011, 50, 2379–2382 CrossRef CAS PubMed.
- F. Mou, C. Chen, H. Ma, Y. Yin, Q. Wu and J. Guan, Angew. Chem., Int. Ed., 2013, 52, 7208–7212 CrossRef CAS PubMed.
- J.-B. Fan, Y. Song, H. Li, J.-P. Jia, X. Guo and L. Jiang, J. Mater. Chem. B, 2014, 2, 3911–3914 RSC.
- J. E. Frommer, Acc. Chem. Res., 1986, 19, 2–9 CrossRef CAS.
- N. Gospodinova and L. Terlemezyan, Prog. Polym. Sci., 1998, 23, 1443–1484 CrossRef CAS.
- J. D. Stenger-Smith, Prog. Polym. Sci., 1998, 23, 57–79 CrossRef CAS.
- H. Shirakawa, Angew. Chem., Int. Ed., 2001, 40, 2575–2580 CrossRef CAS.
- H. Bai and G. Q. Shi, Sensors, 2007, 7, 267–307 CrossRef CAS.
- C. Li, H. Bai and G. Q. Shi, Chem. Soc. Rev., 2009, 38, 2397–2409 RSC.
- H. D. Tran, D. Li and R. B. Kaner, Adv. Mater., 2009, 21, 1487–1499 CrossRef CAS.
- Y. Z. Long, M. M. Li, C. Z. Gu, M. X. Wan, J. L. Duvail, Z. W. Liu and Z. Y. Fan, Prog. Polym. Sci., 2011, 36, 1415–1442 CrossRef CAS.
- X. F. Lu, W. J. Zhang, C. Wang, T. C. Wen and Y. Wei, Prog. Polym. Sci., 2011, 36, 671–712 CrossRef CAS.
- Z. G. Yin and Q. D. Zheng, Adv. Energy Mater., 2012, 2, 179–218 CrossRef CAS.
- R. Green and M. R. Abidian, Adv. Mater., 2015, 27, 7620–7637 CrossRef CAS PubMed.
- S. Virji, J. X. Huang, R. B. Kaner and B. H. Weiller, Nano Lett., 2004, 4, 491–496 CrossRef CAS.
- K. Su, N. Nuraje, L. Z. Zhang, I. W. Chu, R. M. Peetz, H. Matsui and N. L. Yang, Adv. Mater., 2007, 19, 669–672 CrossRef CAS.
- P. S. Peercy, Nature, 2000, 406, 1023–1026 CrossRef CAS PubMed.
- X. Zhang, Z. Li, K. Liu and L. Jiang, Adv. Funct. Mater., 2013, 23, 2881–2886 CrossRef CAS.
- L. P. Xu, B. Dai, J. B. Fan, Y. Q. Wen, X. J. Zhang and S. T. Wang, Nanoscale, 2015, 7, 13164–13167 RSC.
- X. Gao, L.-P. Xu, Z. Xue, L. Feng, J. Peng, Y. Wen, S. Wang and X. Zhang, Adv. Mater., 2014, 26, 1771–1775 CrossRef CAS PubMed.
- Z. Xue, S. Wang, L. Lin, L. Chen, M. Liu, L. Feng and L. Jiang, Adv. Mater., 2011, 23, 4270–4273 CrossRef CAS PubMed.
- L. Feng, Z. Y. Zhang, Z. H. Mai, Y. M. Ma, B. Q. Liu, L. Jiang and D. B. Zhu, Angew. Chem., Int. Ed., 2004, 43, 2012–2014 CrossRef CAS PubMed.
- L. Hu, S. J. Gao, X. G. Ding, D. Wang, J. Jiang, J. Jin and L. Jiang, ACS Nano, 2015, 9, 4835–4842 CrossRef CAS PubMed.
- L. Hu, S. J. Gao, Y. Z. Zhu, F. Zhang, L. Jiang and J. Jin, J. Mater. Chem. A, 2015, 3, 23477–23482 CAS.
- J. Liu, L. Wang, F. Y. Guo, L. L. Hou, Y. E. Chen, J. C. Liu, N. Wang, Y. Zhao and L. Jiang, J. Mater. Chem. A, 2016, 4, 4365–4370 CAS.
- Z. Shi, W. B. Zhang, F. Zhang, X. Liu, D. Wang, J. Jin and L. Jiang, Adv. Mater., 2013, 25, 2422–2427 CrossRef CAS PubMed.
- M. M. Tao, L. X. Xue, F. Liu and L. Jiang, Adv. Mater., 2014, 26, 2943–2948 CrossRef CAS PubMed.
- D. L. Tian, X. F. Zhang, Y. Tian, Y. Wu, X. Wang, J. Zhai and L. Jiang, J. Mater. Chem., 2012, 22, 19652–19657 RSC.
- D. L. Tian, X. F. Zhang, X. Wang, J. Zhai and L. Jiang, Phys. Chem. Chem. Phys., 2011, 13, 14606–14610 RSC.
- Q. Wen, J. C. Di, L. Jiang, J. H. Yu and R. R. Xu, Chem. Sci., 2013, 4, 591–595 RSC.
- Z. X. Xue, Y. Z. Cao, N. Liu, L. Feng and L. Jiang, J. Mater. Chem. A, 2014, 2, 2445–2460 CAS.
- F. Zhang, W. B. Zhang, Z. Shi, D. Wang, J. Jin and L. Jiang, Adv. Mater., 2013, 25, 4192–4198 CrossRef CAS PubMed.
- W. B. Zhang, Z. Shi, F. Zhang, X. Liu, J. Jin and L. Jiang, Adv. Mater., 2013, 25, 2071–2076 CrossRef CAS PubMed.
- Y. Z. Zhu, D. Wang, L. Jiang and J. Jin, NPG Asia Mater., 2014, 6, e101 CrossRef CAS.
- E. Gouaux and R. MacKinnon, Science, 2005, 310, 1461–1465 CrossRef CAS PubMed.
- J. Kim, Y. Piao, N. Lee, Y. I. Park, I.-H. Lee, J.-H. Lee, S. R. Paik and T. Hyeon, Adv. Mater., 2010, 22, 57 CrossRef CAS PubMed.
- H. Yu, X. Qiu, S. P. Nunes and K.-V. Peinemann, Nat. Commun., 2014, 5, 4110 CAS.
- J. Han and H. G. Craighead, Science, 2000, 288, 1026–1029 CrossRef CAS PubMed.
- P. S. Doyle, J. Bibette, A. Bancaud and J. L. Viovy, Science, 2002, 295, 2237 CrossRef CAS PubMed.
- K. Xiong, W. Wei, Y. Jin, S. Wang, D. Zhao, S. Wang, X. Gao, C. Qiao, H. Yue, G. Ma and H. Y. Xie, Adv. Mater., 2016, 28, 7929–7935 CrossRef CAS PubMed.
- S. T. Wang, H. Wang, J. Jiao, K. J. Chen, G. E. Owens, K. I. Kamei, J. Sun, D. J. Sherman, C. P. Behrenbruch, H. Wu and H. R. Tseng, Angew. Chem., Int. Ed., 2009, 48, 8970–8973 CrossRef CAS PubMed.
- P. Zhang and S. Wang, ChemPhysChem, 2014, 15, 1550–1561 CrossRef CAS PubMed.
- C. Huang, G. Yang, Q. Ha, J. Meng and S. Wang, Adv. Mater., 2015, 27, 310–313 CrossRef CAS PubMed.
- Y. Y. Li, Q. H. Lu, H. L. Liu, J. F. Wang, P. C. Zhang, H. G. Liang, L. Jiang and S. T. Wang, Adv. Mater., 2015, 27, 6848–6854 CrossRef CAS PubMed.
- Y.-T. Yeh, Y. Tang, A. Sebastian, A. Dasgupta, N. Perea-Lopez, I. Albert, H. Lu, M. Terrones and S.-Y. Zheng, Sci. Adv., 2016, 2, e1601026 Search PubMed.
- K. A. Stevens and L. A. Jaykus, Crit. Rev. Microbiol., 2004, 30, 7–24 CrossRef PubMed.
- X. Liu and S. Wang, Chem. Soc. Rev., 2014, 43, 2385–2401 RSC.
- L. Adriaenssens and P. Ballester, Chem. Soc. Rev., 2013, 42, 3261–3277 RSC.
- L. J. De Cock, S. De Koker, B. G. De Geest, J. Grooten, C. Vervaet, J. P. Remon, G. B. Sukhorukov and M. N. Antipina, Angew. Chem., Int. Ed., 2010, 49, 6954–6973 CrossRef CAS PubMed.
- B. G. De Geest, N. N. Sanders, G. B. Sukhorukov, J. Demeester and S. C. De Smedt, Chem. Soc. Rev., 2007, 36, 636–649 RSC.
- S. De Koker, L. J. De Cock, P. Rivera-Gil, W. J. Parak, R. A. Velty, C. Vervaet, J. P. Remon, J. Grooten and B. G. De Geest, Adv. Drug Delivery Rev., 2011, 63, 748–761 CrossRef CAS PubMed.
- P. Degen, Curr. Opin. Colloid Interface Sci., 2014, 19, 611–619 CrossRef CAS.
- A. P. R. Johnston, C. Cortez, A. S. Angelatos and F. Caruso, Curr. Opin. Colloid Interface Sci., 2006, 11, 203–209 CrossRef CAS.
- J. W. Cui, M. P. van Koeverden, M. Mullner, K. Kempe and F. Caruso, Adv. Colloid Interface Sci., 2014, 207, 14–31 CrossRef CAS PubMed.
- J. W. Cui, Y. J. Wang, A. Postma, J. C. Hao, L. Hosta-Rigau and F. Caruso, Adv. Funct. Mater., 2010, 20, 1625–1631 CrossRef CAS.
| This journal is © the Partner Organisations 2017 |