
Non-conjugated fluorescent molecular cages of salicylaldehyde-based tri-Schiff bases: AIE, enantiomers, mechanochromism, anion hosts/probes, and cell imaging properties†
Xiaohong
Zhang‡
a,
Jun
Shi‡
b,
Guangyu
Shen
a,
Fei
Gou
a,
Jinghui
Cheng
a,
Xiangge
Zhou
 a and
Haifeng
Xiang
*a
a and
Haifeng
Xiang
*a
aCollege of Chemistry, Sichuan University, Chengdu, 610041, China. E-mail: xianghaifeng@scu.edu.cn; Fax: +86 28-8541-2291
bDepartment of Cardiovascular Surgery, West China Hospital, Sichuan University, 37 Guoxue Xiang St., Chengdu, Sichuan, China
First published on 10th April 2017
Abstract
Luminescent organic materials are commonly polycyclic aromatic molecules with planar extended π-electron conjugation. In the present work, however, we report a series of novel and simple salicylaldehyde-based tri-Schiff bases (TSBs, 15 samples), which have a non-conjugated trimethylamine bridge but emit strong blue, green, and yellow aggregation-induced emission (AIE) with large Stokes shifts (up to 167 nm) and high fluorescence quantum yields (up to 0.18). Mechanochromic fluorescence enhancement and enantiomers are also found in TSB solids. Moreover, these flexible and tripod-like molecular cages acting as ideal and universal anion hosts can be used to detect anions with turn-on fluorescence signals (fluorescence quantum yields up to 0.51). Combining their advantages of AIE and biocompatibility, TSBs have potential application in living cell imaging. The inherent relationships between their chemical structures and AIE/anion host properties are studied, which provide unequivocal insights for the design of non-conjugated fluorescent materials.
Introduction
Organic fluorescent or phosphorescent materials1 that are commonly polycyclic aromatic molecules with extended π-electron conjugation have attracted much attention in many fields including optical probes,2 organic light-emitting diodes (OLEDs),3 light-emitting electrochemical cells (LECs),4 and cell imaging.5 However, the difficult synthesis and solubility of these π-conjugated rigid materials must be considered when we design complicated organic luminescent materials. Moreover, big planar π-conjugated molecules are far more likely to encounter the problem of emission “aggregation-caused quenching” (ACQ),6 because they facilely form dimers through intermolecular π–π stacking interactions at high concentrations or in the solid state. Moreover, luminescent materials are commonly used for practical applications in the solid state (thin films for OLEDs and LECs and nanoparticles for cell imaging). Therefore, in order to solve the ACQ problem, a lot of efforts have been made recently to develop aggregation-induced emission (AIE)-active materials.7 Up to now, most AIE-active materials, such as silole8 (Fig. 1a) or tetraphenylethene9 (TPE; Fig. 1b), are propeller-type molecules that are composed of a conjugated center and multiple peripheral phenyl groups. In a free state (gas or in dilute organic solution), the dynamic intramolecular rotations (IRs) of the peripheral phenyls around the center might exhaust the excitation energy and result in the absence of luminescence. In an aggregated state (solid or precipitate), on the contrary, molecules are tightly packed, which would eliminate these IRs. Furthermore, silole or TPE molecules cannot pack through face-to-face π–π interactions due to their propeller shapes. This restriction of both IRs and π–π interactions would help to turn off the nonradiative transitions and turn on the radiative pathways.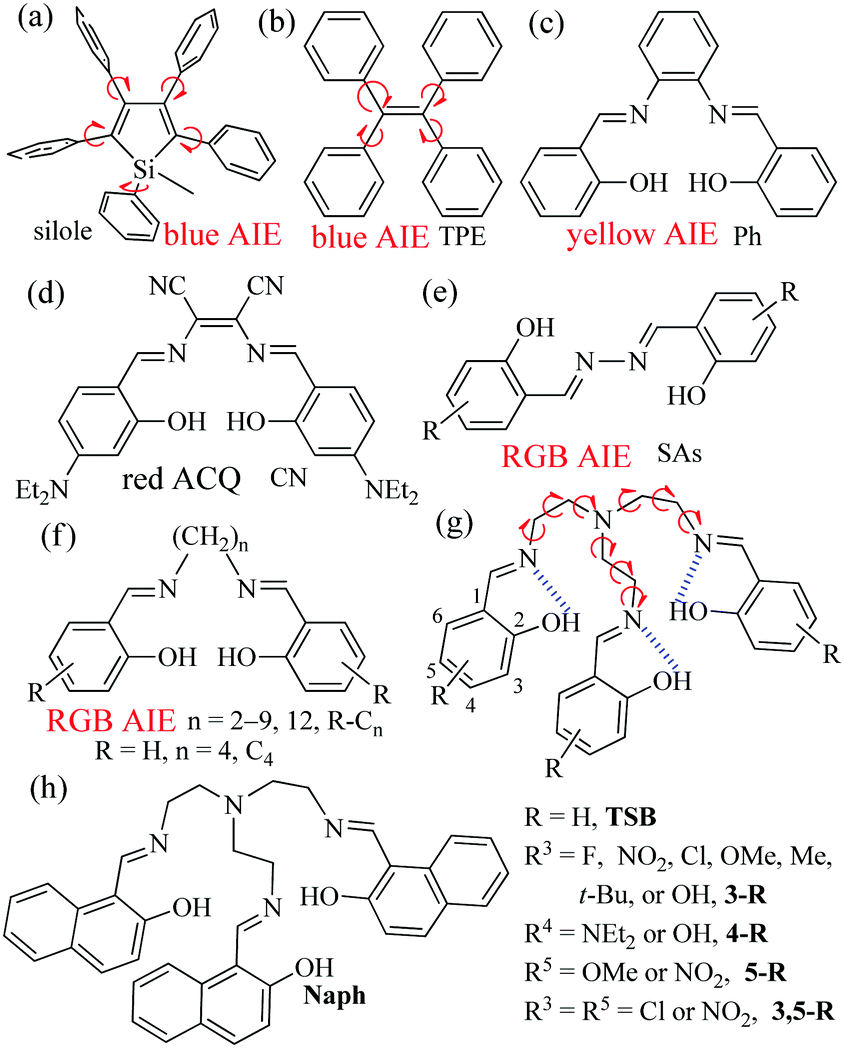 | ||
| Fig. 1 Reported AIE-active materials: silole (a), TPE (b), and salicylaldehyde-based bis-Schiff bases (c–f). TSBs (g and h) in this work. | ||
Compared with conjugated materials, non-conjugated materials have some advantages, such as better solubility, higher flexibility, lower cost, lower cytotoxicity, better biocompatibility, and naturally occurring,10 but they are usually taken for granted as non-emissive materials, due to their none or very small π-conjugated nature. Recently, several organic non-conjugated materials, including 1,1,2,2-tetraphenylethane,7b poly[(maleic anhydride)-alt-(vinyl acetate)],11 polyisobutene succinic anhydrides and imides,12 and steroidal saponin digitonin,13 have been demonstrated to emit ultraviolet (UV) and blue AIE. Our research work mainly focuses on the synthesis, optical properties, and sensing applications of salicylaldehyde-based bis-Schiff bases (Fig. 1c–f), owing to their facile preparation, good stability, biological activity, well-tuned red-green-blue (RGB) ACQ/AIE-active properties, and high emission quantum yields (Φ).6b,14 For example, π-conjugated Ph14c,15 (Fig. 1c) and SAs6b,14f,16 (Fig. 1e) show strong yellow and RGB AIE, respectively, but CN6b,14c,d (Fig. 1d) is an ACQ-active red emitter. We recently reported that step-like N,N′-bis(salicylidene)ethylenediamine (Salen) ligands (Fig. 1f) linking with different non-conjugated long alkyl bridges display strong RGB AIE.14g What would happen if we change two-arm salicylaldehyde-based bis-Schiff bases into three-arm salicylaldehyde-based tri-Schiff bases (TSBs) (Fig. 1g and h)? In the literature, however, there are only limited examples of weakly fluorescent non-conjugated TSBs17 that were used as turn-on fluorescence probes for detecting Eu3+,17a vanadate,17b and Zn2+![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 17c,d or for photochromism application.17e It is also rare that AIE-active cores have anion host/probe properties.7 Herein, we demonstrate a novel and simple class of colorful tripod-like AIE-active and anion-induced emission-active TSB cages (15 samples, Fig. 1) that contain a non-conjugated triethylamine bridge and different electron-accepting (–NO2, –F, and –Cl), electron-donating (–OH, –OMe and –NEt2), or sterically hindering (–Me and –t-butyl) substituents or π-extended system (naphthalene ring).
17c,d or for photochromism application.17e It is also rare that AIE-active cores have anion host/probe properties.7 Herein, we demonstrate a novel and simple class of colorful tripod-like AIE-active and anion-induced emission-active TSB cages (15 samples, Fig. 1) that contain a non-conjugated triethylamine bridge and different electron-accepting (–NO2, –F, and –Cl), electron-donating (–OH, –OMe and –NEt2), or sterically hindering (–Me and –t-butyl) substituents or π-extended system (naphthalene ring).
Results and discussion
Synthesis and characterization
TSBs were reasonably easy to synthesize by the condensation of primary triamine with 3 equivalents of salicylaldehyde precursor in ethanol under refluxing condition.14 Most TSBs have a poor solubility in petroleum ether, hexane, and water but a good solubility in CH3CN, CH2Cl2, DMF, and DMSO. All TSBs either in solution or in solid state are stable for several months under air. These Schiff bases have a good thermal stability (Td: 280–300 °C for TSB, 3-F, and 3,5-Cl) (Fig. S1, in ESI†). For most TSBs, good-quality single crystals can be obtained by the method of slow solvent diffusion/evaporation (CH2Cl2/hexane). For the purpose of comparison, the reference substance C414g (Fig. 1f) was prepared as well.Photophysical and AIE properties
Table S1 (in ESI†) lists the UV/visible absorption and fluorescence data of all synthesized TSBs at room temperature. For example, the absorption spectra of TSB, 3-F, 3-NO2, 3-Me, 5-OMe, 3,5-Cl, and Naph in pure organic solvent of MeCN (1.0 × 10−5 mol dm−3) are given in Fig. S2 (ESI†). To gain an insight into the nature of the excited states and transitions, gas-phase density functional theory (DFT) and time-dependent DFT (TD-DFT) were also carried out for TSB with the Gaussian 09 program package (B3LYP 6-31G(d,p)). The lower energy absorption band of TSB (λabs = 314 nm in MeCN) is reproduced well by the computation, which predicts one absorption peak at 314 nm (Fig. 2 and Fig. S3, ESI†). The lower energy absorption is contributed by the highest occupied molecular orbital (HOMO) → lowest unoccupied molecular orbital (LUMO) (S0 → S1, 323 nm, oscillator strength = 0.0084), HOMO → LUMO+1 (S0 → S2, 306 nm, oscillator strength = 0.0044), and HOMO → LUMO+2 (S0 → S3, 301 nm, oscillator strength = 0.0021). The energy level and orbital isosurface diagrams of TSB (Fig. 2) reveal that the HOMO of TSB is not composed primarily of three π-conjugated units of iminomethylphenol but of the non-conjugated triethylamine bridge, which might contribute to the electron-donating nature of trimethylamine. On the contrary, its LUMO, LUMO+1, and LUMO+2 are mainly made up of the π-functions of iminomethylphenol units rather than the triethylamine bridge. Therefore, the lower energy absorption can mainly be assigned to n → π* transition from electron-donating trimethylamine (lone pair electrons of N atom) to the π-conjugated iminomethylphenol units. For C4, however, the lower energy absorption was contributed by the π → π* transition involving molecular orbitals essentially localized on the iminomethylphenol units with little contribution from the non-conjugated alkyl bridge.14g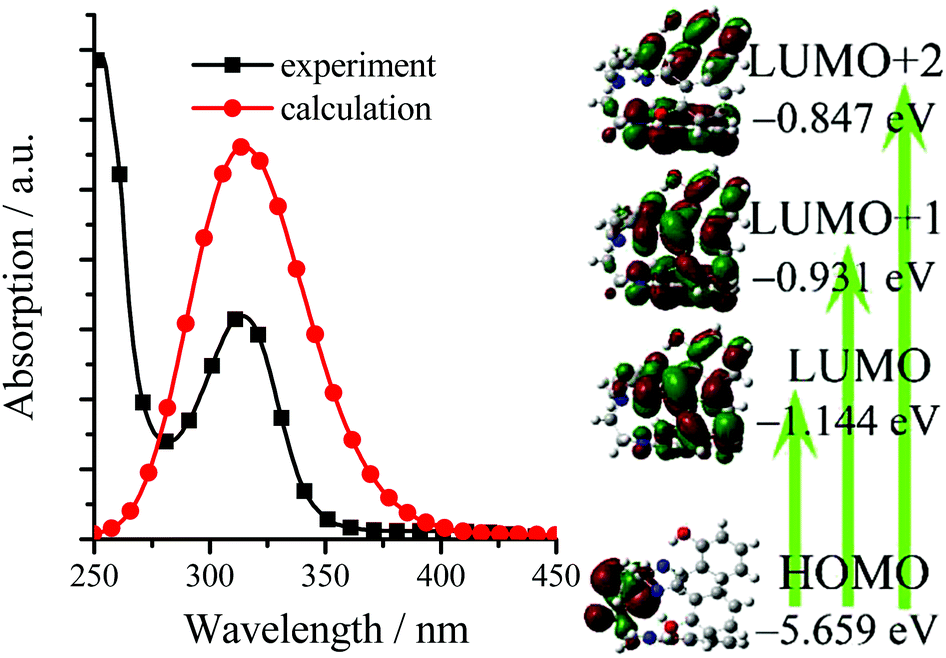 | ||
| Fig. 2 Computational (gas-phase) and experimental (in MeCN) absorption spectra, energy level diagram, and frontier molecular orbitals of TSB. | ||
The introduction of sterically hindering –Me (3-Me, λabs = 320 nm in MeCN) or –t-butyl (3-t-Bu, λabs = 320 nm) (Fig. S2 and Table S1, ESI†) substituents to the simplest TSB (λabs = 314 nm) has little effect on absorption spectra, but the presence of electron-accepting –F (3-F, λabs = 312 and 410 nm), –Cl (3-Cl, λabs = 319 and 416 nm; 3,5-Cl, λabs = 331 and 428 nm), or –NO2 (3-NO2, λabs = 357 and 409 nm; 5-NO2, λabs = 440 nm; 3,5-NO2, λabs = 381 nm) substituents, electron-donating –OMe (3-OMe, λabs = 328 and 420 nm; 5-OMe, λabs = 343 nm), –OH (3-OH, λabs = 425 nm; 4-OH, λabs = 305 and 380 nm), or –NEt2 (4-NEt2, λabs = 329 nm) substituents, or π-extended system (Naph, λabs = 402 and 420 nm) induces obvious red shifts in absorption spectra.
As we expected, all TSBs exhibit weak fluorescence in pure organic solvent of MeCN or DMSO (Table S1, ESI†). For example, 3-F shows very weak blue fluorescence (Φ = 0.0037) in dilute MeCN, because the IRs of central triethylamine bridge in 3-F molecules provide a possible way to non-radiatively annihilate its excited states and result in the absence of fluorescence. However, if water is added into MeCN, the green fluorescence of 3-F is strengthened (Fig. 3), because 3-F molecules cannot be dissolved in water, which causes 3-F molecules to precipitate and aggregate. At the same time, adding water would red shift its absorption spectrum (Fig. S4, ESI†), which further confirms this aggregation. Its green fluorescence reaches the maximum (λem = 501 nm, Φ = 0.025) with a large Stokes shift of 119 nm, when the volume fraction (f) of water is increased to 60%. Moreover, 3-F solids emit strong green-yellow fluorescence (λem = 512 nm, Φ = 0.13) under a 360 nm UV lamp (Fig. 3 and 4). All these findings reveal the AIE nature of 3-F.
 | ||
| Fig. 3 Emission spectra and photographs (under 360 nm UV light) of 3-F in MeCN–H2O with different f values (3.0 × 10−5 mol dm−3). | ||
 | ||
| Fig. 4 Photographs (top: under room light; bottom: under 360 nm UV light) of TSBs. | ||
All the TSBs have stronger AIE in solid state or water than in organic solvents (Table S1, ESI†). Most solid samples exhibit not only red-shifted but also much stronger fluorescence compared with samples in water (Fig. 3, 4 and Table S1, ESI†), and thus the later discussion will be focused on their solid-state AIE characteristics. Different solid forms of AIE-active materials that have different molecular arrangements might have different fluorescence properties. For most TSBs, however, the different solid forms, such as crystal and powder, have similar fluorescence properties, except that TSB powders show much stronger fluorescence than TSB crystals (see the later discussion).
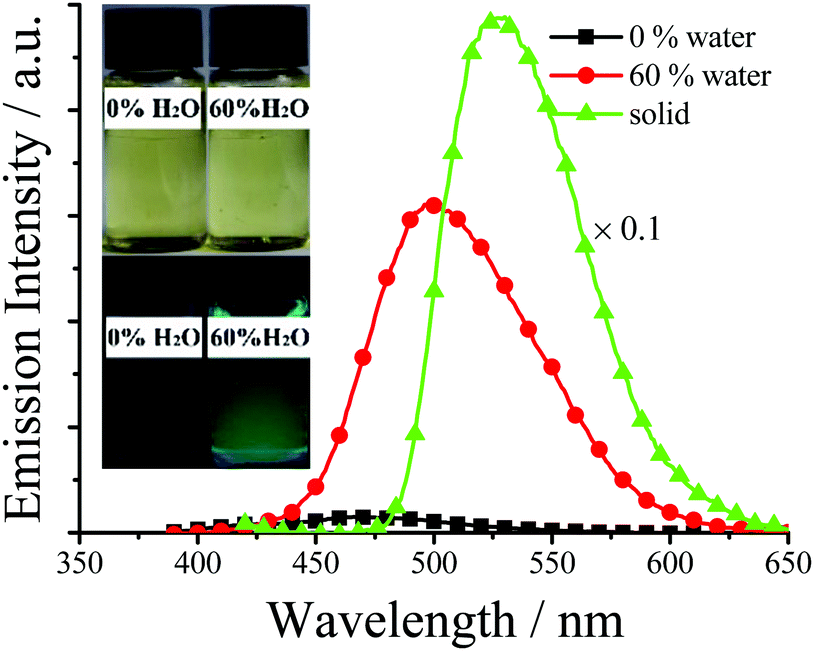
As shown in Fig. 5 and Table S1 (ESI†), for solid TSBs, different substituents have different effects on the emission peaks (λem: 470–558 nm) and colors (blue, green, and yellow). The substituents show a different effect on fluorescence spectra (Fig. 4 and 5) from absorption spectra (Fig. S2, ESI†). The presence of a π-extended system (Naph, λem = 488 nm), electron-donating –OH (4-OH, λem = 470 nm) or –NEt2 (4-NEt2, λem = 487 nm) substituents in the simplest TSB (λem = 497 nm) leads to blue shifts in fluorescence spectra. On the other hand, sterically hindering –Me (3-Me, λem = 514 nm) or –t-butyl (3-t-Bu, λem = 516 nm), electron-donating –OH (3-OH, λem = 550 nm) or –OMe (3-OMe, λem = 537 nm; 5-OMe, λem = 532 nm), or electron-accepting –F (3-F, λem = 512), –Cl (3-Cl, λem = 526 nm; 3,5-Cl, λem = 522), or –NO2 (3-NO2, λem = 556 nm; 5-NO2, λem = 548 nm; 3,5-NO2, λem = 558 nm) substituents result in red-shifted emission (Fig. 5 and Table S1, ESI†). This substituent effect is a simple and useful tool to well tune λem, which was observed in AIE-active salicylaldehyde-based SAs14f (Fig. 1e) and R–Cn14g (Fig. 1f) AIE materials as well.
 | ||
| Fig. 5 Normalized emission spectra of some solid TSBs. | ||
All fluorescence quantum yields (Table S1, ESI†) of the aggregated TSBs in water and solid state were measured by the optical dilution method of Demas and Crosby18 with a standard of quinine sulfate (Φr = 0.55, quinine in 0.05 mol dm−3 sulfuric acid) and an integrating sphere, respectively. Although the TSBs with a non-conjugated triethylamine linker have a small π-conjugated system, the fluorescence quantum yields of some TSBs are unexpectedly high (Table S1, ESI†). In general, the presence of –F (3-F, Φ = 0.13), –Cl (3-Cl, Φ = 0.12; 3,5-Cl, Φ = 0.18), or –Me (3-Me, Φ = 0.022) substituents in TSB (TSB, Φ = 0.018) would improve its fluorescence quantum yield, but the introduction of –t-butyl (3-t-Bu, Φ = 0.0058), –OH (3-OH, Φ = 0.0067; 4-OH, Φ = 0.0051) or –NEt2 (4-NEt2, Φ = 0.011) would bring adverse effects. –NO2 (3-NO2, Φ = 0.0048; 5-NO2, Φ = 0.044; 3,5-NO2, Φ = 0.0085) or –OMe (3-OMe, Φ = 0.010; 5-OMe, Φ = 0.028) substituents might influence fluorescence quantum yield positively or negatively. Therefore, chlorination or fluorination is an efficient way to improve fluorescence quantum yield.14f,g Moreover, some TSBs have different emissive colors in water and as solids. For example, 3-F displays green (λem = 501 nm) and green-yellow (λem = 512 nm) emission in water and as a solid, respectively (Fig. 3). This slight difference might be caused by the different intermolecular interactions in water and solid.
Mechanism of AIE
Since the molecular arrangements play a key role in AIE, a slight difference of the chemical structures would change the molecular arrangements and AIE properties significantly. In order to achieve a high fluorescence quantum yield, AIE-active materials should stack closely with not only a short interplanar distance (for planar molecules, such as SAs14f) or intermolecular aromatic HAr⋯π hydrogen bonds (for non-planar molecules, such as silole8 or TPE9) but also weak intermolecular face-to-face π–π interactions. The former can ensure elimination of molecular rotation; the latter would prevent the formation of excimers. For non-conjugated R–Cn, however, the restriction of C–C single bond rotations in the central alkyl chain bridges through some other non-covalent intermolecular interactions, such as N⋯H, O⋯H, C⋯H, and H⋯H, must be considered.14gIn order to investigate the effect of molecular arrangements, the X-ray single crystal structures of TSB, 3-F, 3,5-Cl, 3-t-Bu, and 3-OMe were obtained are depicted in Fig. 6–8 and Fig. S5–S13 (ESI†). Unlike plane-like SAs14f or step-like R–Cn14g molecules, these TSB molecules, except 3-OMe molecule, are tripod-like side-single-opening cages, due to the stress from lone pair electrons of N atom in the triethylamine bridge (Fig. 2).
As expected, three intramolecular N⋯H hydrogen bonds (1.8350, 1.8317, and 1.8619 Å) between H (–OH) and N (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N) atoms are found in TSB (Fig. 1g and Fig. S5, ESI†), which would eliminate free rotations of benzene rings. The dihedral angles between three panels of π-conjugated iminomethylphenol are about 60° (Fig. 6), resulting in no intramolecular face-to-face π–π interactions (overlaps in π-conjugated units) in one TSB molecule. In dilute organic solution, it is obvious that the rotatable C–N and C–C single bonds in the central triethylamine bridge afford a possible way to non-radiatively annihilate its excited states and quench the fluorescence. In the solid state, TSB molecules exhibit an edge-to-face spiral packing motif (Fig. 6a), in which the opening directions of TSB molecular cages are staggered form. No intermolecular face-to-face π–π interactions (Fig. 6a–c) are found between any neighboring TSB molecules either, which is beneficial to prevent the formation of excimers. On the contrary, many other strong intermolecular interactions including H⋯H (2.5358, 2.6370, and 2.8204 Å), N⋯H (2.8032 Å), and O⋯H (2.7643 Å) interactions are found in the two adjacent TSB molecules (Fig. 6b and c). These intramolecular and intermolecular interactions would help to restrain the IRs (Fig. 1g) and consequently lead to the presence of AIE. It is not surprising to find two enantiomers (1:1) in the single crystals of TSB (Fig. 6b–d), because the C–N and C–C single bonds cannot rotate freely in the solid state.19 However, we failed to separate two enantiomers by solution process; presumably these single bonds would rotate freely in solution.
N) atoms are found in TSB (Fig. 1g and Fig. S5, ESI†), which would eliminate free rotations of benzene rings. The dihedral angles between three panels of π-conjugated iminomethylphenol are about 60° (Fig. 6), resulting in no intramolecular face-to-face π–π interactions (overlaps in π-conjugated units) in one TSB molecule. In dilute organic solution, it is obvious that the rotatable C–N and C–C single bonds in the central triethylamine bridge afford a possible way to non-radiatively annihilate its excited states and quench the fluorescence. In the solid state, TSB molecules exhibit an edge-to-face spiral packing motif (Fig. 6a), in which the opening directions of TSB molecular cages are staggered form. No intermolecular face-to-face π–π interactions (Fig. 6a–c) are found between any neighboring TSB molecules either, which is beneficial to prevent the formation of excimers. On the contrary, many other strong intermolecular interactions including H⋯H (2.5358, 2.6370, and 2.8204 Å), N⋯H (2.8032 Å), and O⋯H (2.7643 Å) interactions are found in the two adjacent TSB molecules (Fig. 6b and c). These intramolecular and intermolecular interactions would help to restrain the IRs (Fig. 1g) and consequently lead to the presence of AIE. It is not surprising to find two enantiomers (1:1) in the single crystals of TSB (Fig. 6b–d), because the C–N and C–C single bonds cannot rotate freely in the solid state.19 However, we failed to separate two enantiomers by solution process; presumably these single bonds would rotate freely in solution.
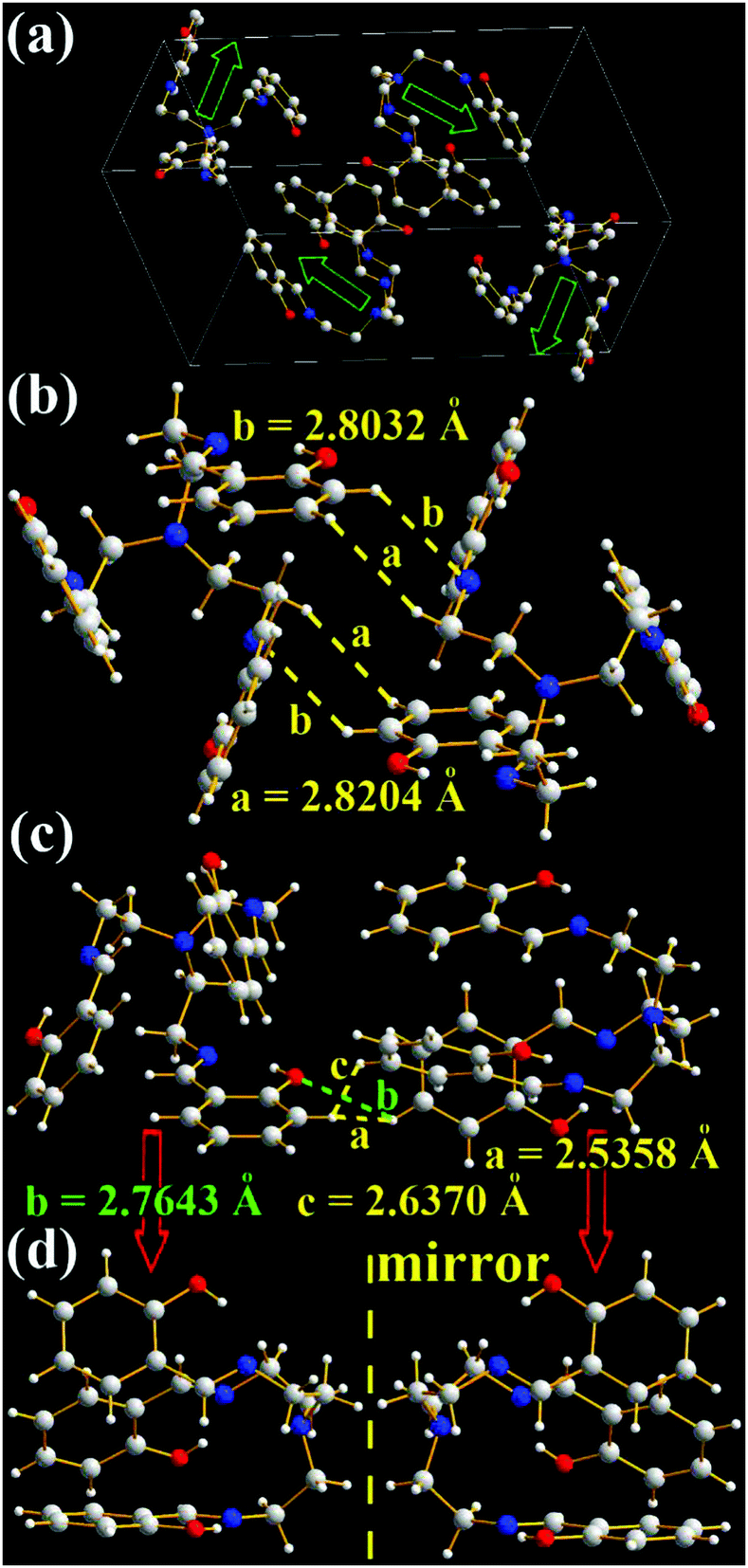 | ||
| Fig. 6 X-ray single crystal structures and packing of TSB molecules: (a) packing in a unit cell (H atoms are omitted and blue arrows indicate the opening direction of molecular cages); (b) and (c) intermolecular interactions of the two closest molecules; (d) enantiomers. | ||
Unlike TSB molecules, 3-F molecules have a much more complicated and tight hexagonal arrangement (Fig. 7a), even if their chemical structures are similar. The opening directions of 3-F molecular cages are opposite (Fig. 7b and c). No intramolecular and intermolecular face-to-face π–π interactions (Fig. 7 and Fig. S6, ESI†) are found in 3-F molecules. Like TSB molecules, many strong intermolecular interactions including H⋯H (2.6337, 2.6462, 2.6510, 2.7008, 2.7926, 2.8536, 2.8838, 2.9287, and 2.9680 Å) and O⋯H (2.4776, 2.6342, and 2.8311 Å) interactions (Fig. 7c–e and Fig. S6a, b, ESI†) and two enantiomers (1:1) (Fig. S6c, ESI†) are found in the two adjacent 3-F molecules. Moreover, many strong intermolecular F⋯H interactions (2.6780, 2.6827, 2.8139, 2.8536, 2.8732, 2.9905, and 2.9931 Å, Fig. 7d, e and Fig. S6a, b, ESI†) are observed. No such intermolecular F⋯H interactions were found in 3-F-C2 (Fig. 1f).14g It should be noted that the strongest intermolecular interaction of two neighboring 3-F molecules is 2.4776 Å (O⋯H, Fig. 7d), which is stronger than that of TSB molecules (H⋯H, 2.5358 Å). This might be one possible reason that solid 3-F has a higher fluorescence quantum yield (Φ = 0.13).
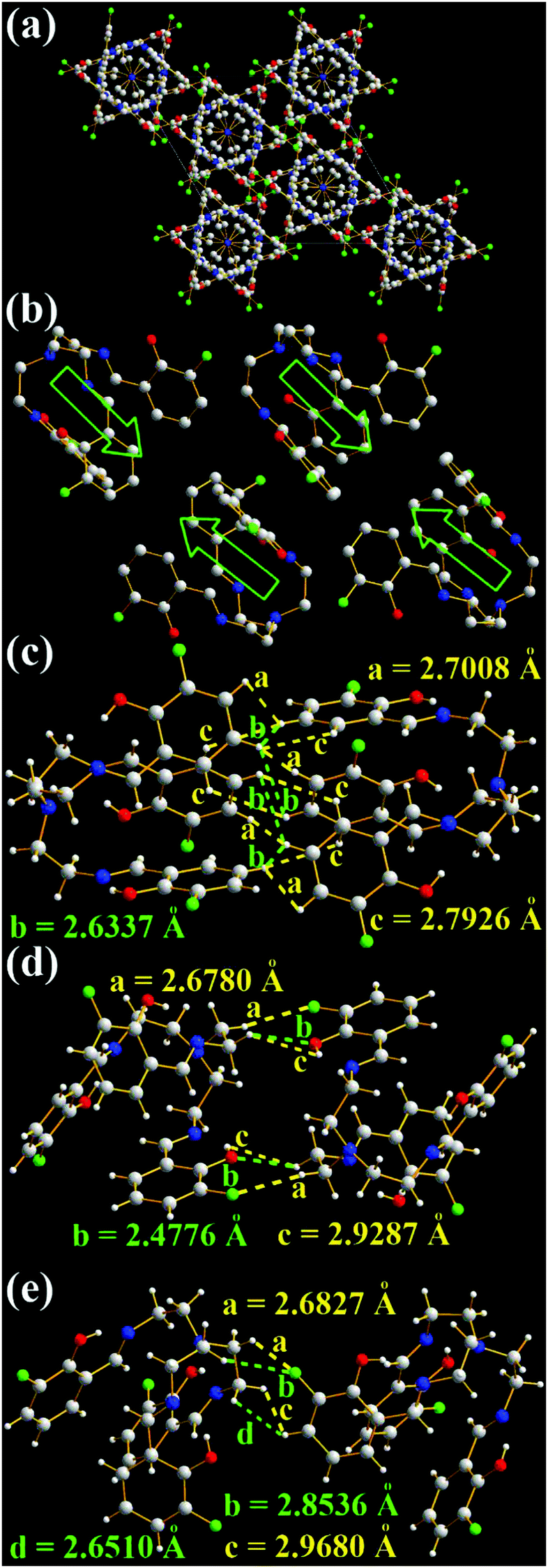 | ||
| Fig. 7 X-ray single crystal structures and packing of 3-F molecules: (a) packing in a unit cell (H atoms are omitted); (b) packing of some selected molecules (blue arrows indicate the opening direction of molecular cages); (c)–(e) intermolecular interactions of the two closest molecules. | ||
For 3,5-Cl, however, the positions of Cl atoms in one of two close 3,5-Cl molecules are not fixed (Fig. S7, ESI†), due to the disorder19,20 in its single crystals. No enantiomers are found in the 3,5-Cl single crystals (Fig. S8 and S9, ESI†). 3,5-Cl molecules tightly pack to form hexagon arrangements, similar to 3-F molecules. Besides many strong intermolecular H⋯H (2.4668, 2.7954 Å), C⋯H (2.8683 Å) (Fig. S9d, ESI†), and O⋯H (2.6269 and 2.7625 Å) (Fig. S8b, ESI†) interactions, other strong intermolecular Cl⋯H (2.8418 and 2.9448 Å) (Fig. S8a, ESI†) interactions are present as well. Moreover, strong Cl⋯Cl (3.2699 Å, Fig. S9c, ESI†) interactions are observed, because the opening directions of 3,5-Cl molecular cages (Fig. S9b, ESI†) are opposite. The F (3-F) and Cl (3,5-Cl) atoms would help to induce intermolecular F⋯H, Cl⋯H, or Cl⋯Cl interactions21 (halogen bond22) that is one possible reason why chlorination or fluorination can improve fluorescence quantum yield. The intermolecular interactions of two neighboring 3,5-Cl molecules are the strongest (2.4668 Å, H⋯H, Fig. S9d, ESI†), which is consistent with the fact that solid 3,5-Cl has the highest fluorescence quantum yield (Φ = 0.18).
Our previous work14g demonstrated that bulky –NEt2 and –t-Bu substituents might prevent the molecules from tightly packing, resulting in the absence of AIE. 3-t-Bu molecules (Fig. S10, ESI†) have a similar arrangement to TSB molecules along with two enantiomers (Fig. S11, ESI†). The density of 3-t-Bu single crystals is 1.086 g cm−3, which is much smaller than that of TSB (1.220 g cm−3), 3-F (1.389 g cm−3), and 3,5-Cl (1.480 g cm−3) single crystals, indicating that 3-t-Bu molecules have a loose packing. The intermolecular interactions of 3-t-Bu molecules are H⋯H (2.6136–2.9189 Å, Fig. S10b and c, ESI†) that mainly occur between –t-Bu substituents and are weaker than those of TSB molecules. Accordingly, 3-t-Bu has very weak AIE.
It is strange that only the 3-OMe molecule is not a molecular cage (Fig. 8 and Fig. S12, ESI†). Its three iminomethylphenol arms are separated from each other with an angle of 120°. There are three layers of 3-OMe molecules in one unit cell of X-ray single crystal structures (Fig. 8a). The panels of π-conjugated iminomethylphenol in three adjacent 3-OMe molecules are parallel with an interplanar distance (d) of 3.50 Å (Fig. 8b). This might induce intermolecular π–π interactions and result in weak AIE. Moreover, the density of 3-OMe single crystals is 1.225 g cm−3, which is similar to that of TSB (1.220 g cm−3) single crystals but smaller than that of 3-F (1.389 g cm−3) and 3,5-Cl (1.480 g cm−3) single crystals. Like 3,5-Cl, disorder (Fig. S13, ESI†) and no enantiomers are found in the 3-OMe single crystals.
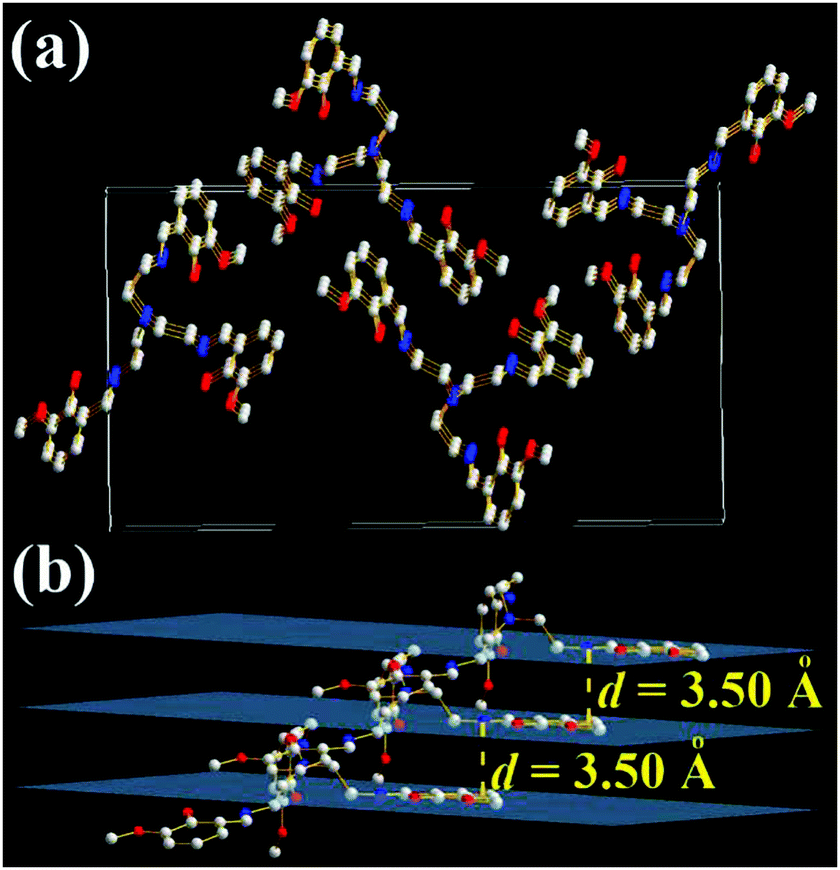 | ||
| Fig. 8 X-ray single crystal structures and packing of 3-OMe molecules: (a) packing in a unit cell; (b) intermolecular interactions of the three layers (H atoms are omitted). | ||
These TSBs have small π-conjugated systems but strong solid-state emission with large Stokes shifts, which are consistent with the characteristics of reported pure organic phosphorescent compounds.23 However, the time-resolved emission spectrum (Fig. S14, ESI†) reveals that the emission decay lifetime of 3,5-Cl powder is 1.28 ns. This indicates that the emission originates from fluorescence rather than phosphorescence. The large Stokes shifts might be contributed by the intramolecular hydrogen bonds which would lead to keto and enol tautomers through an ultrafast excited state intramolecular proton transfer (ESIPT) process.14g,24 Moreover, our previous work revealed that both the multi-Schiff base structure and –OH groups are of the greatest importance for AIE.14g
Mechanochromism properties of TSB
In general, AIE-active materials are found to have mechanochromism properties upon the transition of crystalline and amorphous states under external stimuli,7,25 because their florescence properties are highly dependent on their molecular arrangements. In most cases, applying mechanical force would lead to a red shift and intensity decrease of their florescence. It is rare to encounter mechanochromic AIE-active materials with enhanced fluorescence.14g,26 Most of the TSBs have no mechanochromism properties except TSB (Fig. 9). The pristine crystal solids of TSB are weakly emissive, but after grinding, the resulting powders emit strong green emission under irradiation with UV light. Fast evaporation of CH2Cl2 by reduced pressure distillation would bring about the same fluorescence enhancement. The same phenomenon was observed for C6 solids as well.14g | ||
| Fig. 9 Photographs of TSB solids ((a, c, e and f) crystals; (b and d) ground solids; (g) fast-evaporated solids) under sunlight or 360 nm UV light. | ||
As shown in Fig. S15 (ESI†), the powder wide-angle X-ray diffraction experiments of the pristine crystal, ground, and evaporated solids reveal that all the solid samples of TSB have intense and sharp reflection peaks, which are identical to the simulated pattern of a single crystal of TSB. This means that all the solid samples are mainly made up of microcrystalline solids. Unlike most mechanochromic AIE-active materials that show a phase transition from a crystalline to an amorphous phase,7,25TSB does not have such a phase transition. Crystal-defect-induced emission might contribute to the fluorescence enhancement of ground and evaporated TSB solids,14g,26 because a completely amorphous sample of TSB shows very weak fluorescence under UV light (Fig. S16, ESI†). Moreover, TSB and 3,5-Cl in both poly(methyl methacrylate) casting film (Fig. S17, ESI†) and frozen solvent (77 K) (Fig. S18, ESI†) also display strong fluorescence.
Anion host properties
As a major field within supramolecular chemistry, anion hosting that can be used to bind and sense anions has attracted considerable attention in recent decades due to its potential applications in pollutant sequestration, biomedical and environmental monitoring, anion exchange, anion transport and fluorescent probes.27 Since these TSBs are tripod-like side-single-opening cages with three flexible arms of iminomethylphenol, they might be used as anion hosts through hydrogen bonds or halogen bonds.The anion host/probe properties of TSB, 3-F, 3,5-Cl, 3-OMe, 3-t-Bu, and C4 are demonstrated in Fig. 10, 11, and Table S1, Fig. S19–S25 (ESI†). Various sodium or potassium salts (1.0–0.10 mol dm−3 in water) were added to dilute dye solution (1.0 × 10−5 mol dm−3 in DMSO or MeCN) by a microsyringe. As an example, upon adding many anions, including OH−, F−, Cl−, Br−, I−, NO3−, CO32−, HCO3−, SO42−, SO32−, PO43−, HPO42−, P2O74− and S2−, to a dilute 3-F DMSO solution, some of them, such as OH−, HCO3−, F−, S2−, SO42−, PO43−, P2O74−, HPO42−, CO32−, and I−, would lead to a turning on of the fluorescence of 3-F, but others have little effect on the fluorescence (Fig. 10). The fluorescence enhancement is in the order of OH− > HCO3− > F− > S2− > SO42− > PO43− > P2O74− > HPO42− > CO32− > I− > others. Alkaline OH− and neutral F− were chosen to investigate the concentration effect. The fluorescence of 3-F exhibits a linear enhancement (correlation coefficient R2 = 0.988, n = 10) upon the addition of 0–30 equivalent of OH− and then reaches maximum upon the addition of 40 equivalent of OH− (Φ = 0.50). On adding OH− further, the fluorescence would reduce and finally reach saturation (Φ = 0.42) when the amount of OH− is more than 100 equivalent (Fig. 10 and Fig. S19, ESI†). A similar fluorescence enhancement is observed upon adding F− to dilute 3-F solution (Fig. S20 and S21, ESI†).
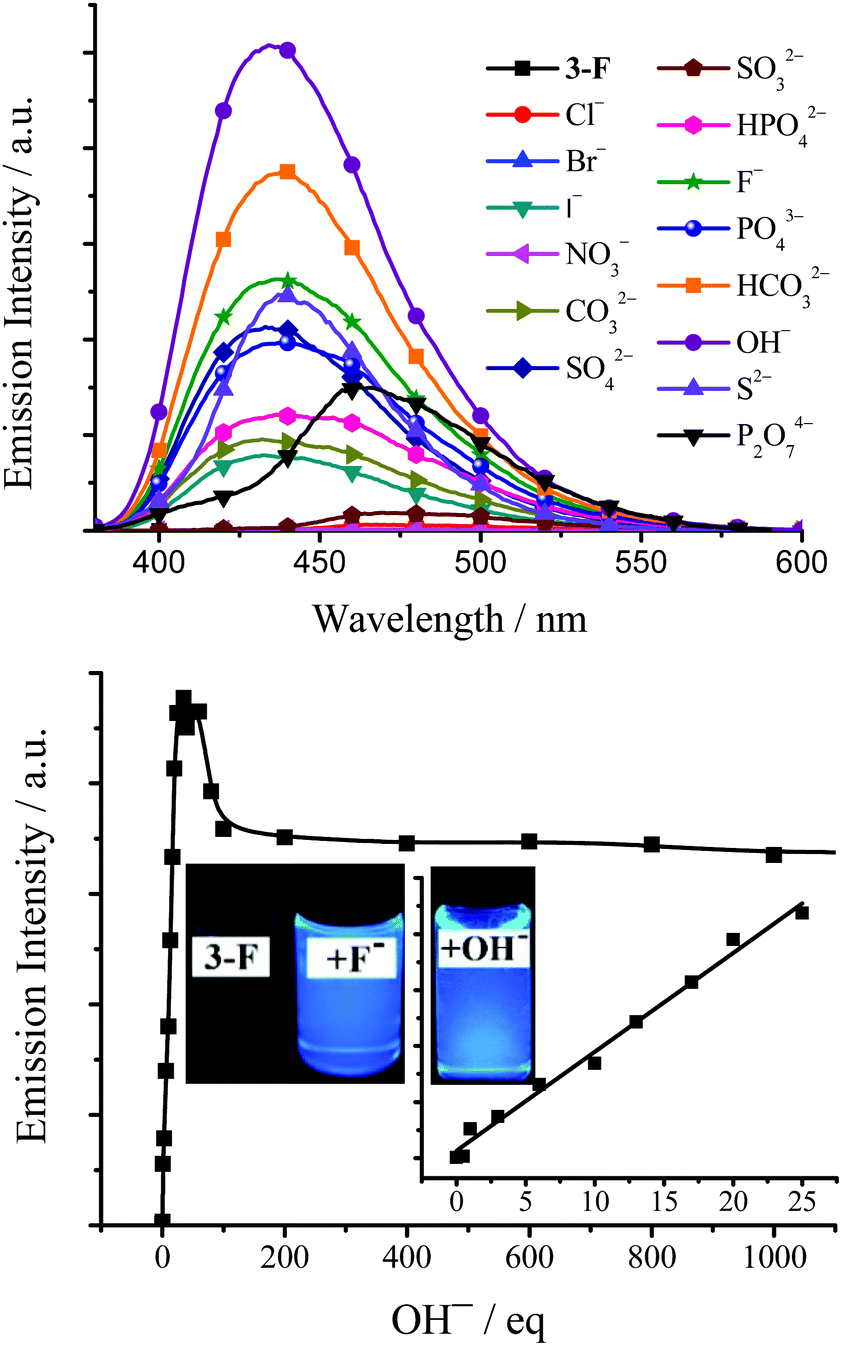 | ||
| Fig. 10 Top: Emission spectra of 3-F (1.0 × 10−5 mol dm−3 in DMSO, excited at 365 nm) upon the addition of 100 equivalent of different anions. Bottom: Plot of emission intensity of 3-F (1.0 × 10−5 mol dm−3 in DMSO) at 450 nm (excited at 365 nm) as a function of OH− concentration. Inset: Photographs of 3-F, 3-F + 100 equivalent of F− and OH− under 360 nm UV light. | ||
 | ||
| Fig. 11 Emission spectra of 3-OMe and 3-F (1.0 × 10−5 mol dm−3 in DMSO, excited at 365 nm) upon the addition of 100 equivalent of different anions and upon the addition of 100 equivalent of OH−, respectively. | ||
3,5-Cl has a similar anion effect (Fig. S22, ESI†) to 3-F with higher fluorescence quantum yields. The maximum and saturated fluorescence quantum yield is 0.51 and 0.44, upon adding 40 and 100 equivalent of OH− to dilute 3,5-Cl DMSO solution, respectively. TSB (Fig. S23, ESI†) and 3-t-Bu (Fig. S24, ESI†) have a different anion effect. The fluorescence enhancement of TSB (Φ = 0.29, adding 100 equivalent of HCO3−) and 3-t-Bu (Φ = 0.47, adding 100 equivalent of OH−) is in the order of HCO3− > CO32− > S2− > OH− > I− > SO32− > Cl− > others and OH− > PO43− > HCO3− > others, respectively. On the contrary, 3-OMe does not have an anion effect on fluorescence (Φ < 0.01 in DMSO or MeCN) (Fig. 11), which is in accordance with the fact that 3-OMe is not a molecular cage and so cannot encage anions tightly. For the same reason, it is no surprise to observe the result that step-like C4 has little anion effect on fluorescence either (Fig. S25, ESI†). Moreover, TSB and 3,5-Cl might be used as Zn2+/Mg2+ turn-on fluorescence probes (Fig. S26 and S27, ESI†).
In order to investigate the possible interaction mechanism between the host and anions, 1H nuclear magnetic resonance (1H NMR) analysis was done. As shown in Fig. 12a, the 1H NMR signal of H1 atoms in 3-F appears a small broad peak at downfield (δ = 14.2 ppm) due to the existence of intramolecular hydrogen bonds (–OH⋯N, Fig. 6).28 On adding 100 equivalent of F− (Fig. 12b) or OH− (Fig. 12c) (in D2O), this downfield peak disappears and a new small sharp peak is formed upfield (δ = 10.1 and 10.0 ppm for adding F− and OH−, respectively), which might be assigned to ArOH28,29 without intramolecular hydrogen bonds. This upfield shift indicates that the strong intramolecular hydrogen bonds are destroyed by adding F− or OH−. At the same time, the reduction (about 2/3) of peak area of the new peak reveals the formation of new intermolecular hydrogen bonds. These intermolecular hydrogen bonds would increase the molecular electron density through-bond effect and consequently lead to upfield shifts of other H atoms (H2–H7).30 Moreover, the dynamics analysis (Fig. 13) shows that the reaction between 3-F and F− or OH− is very fast. The reaction needs about one minute to reach equilibrium without shaking; however, with shaking, the reaction would finish immediately. The very fast reaction speed further confirms the fact that the reaction between 3-F and F− is not a normal chemical reaction but intermolecular hydrogen bonding. It should be noted that the 1H NMR signals of –OH in planar CN (Fig. 1d) and SAs (Fig. 1e) would disappear completely and thus their fluorescence would be quenched by adding OH−.14d,f Furthermore, the blank experiments (Fig. S28, ESI†) revealed that the 1H NMR peak at 10.1 ppm was not caused by an impurity.
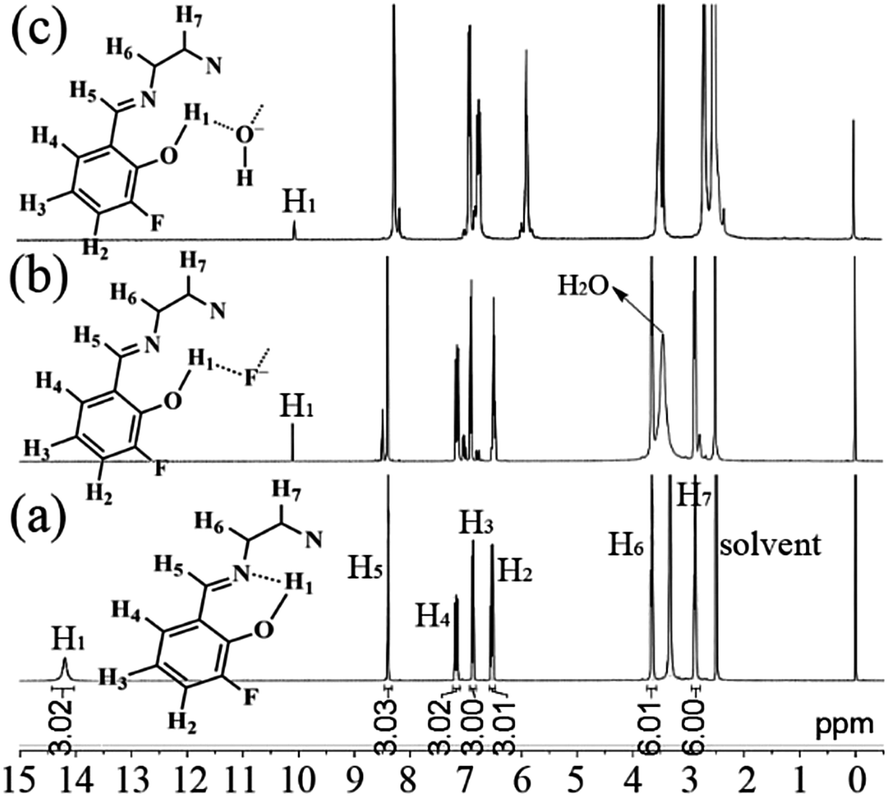 | ||
| Fig. 12 1H NMR spectra of 3-F (a), 3-F + 100 equivalent of F− (b), and 3-F + 100 equivalent of OH− (c) in DMSO-d6. | ||
 | ||
| Fig. 13 Photographs of 3-F (1.0 × 10−5 mol dm−3 in DMSO) after adding 100 equivalent of F− by a microsyringe under 360 nm UV light. Left to right: picture taken every second without shaking. Last one: shaking just after addition. | ||
The mechanisms of aggregation- and anion-induced fluorescence enhancement are totally different. For example, the green AIE of TSB originates from the aggregated state (in water or as solid) but its blue anion-induced fluorescence originates from the monomer (1.0 × 10−5 mol dm−3 in MeCN or DMSO). As mentioned above, anions, such as F− or OH−, would lead to changing –OH into O−, and thus TSB in the form of O− is used to evaluate the nature of the anion-induced excited states and transitions by TD-DFT calculation. The energy level and orbital isosurface diagram of TSB (in the form of O−, Fig. S29, ESI†) reveals that its π-functions in HOMO, LUMO, LUMO+1, and LUMO+2 are mainly made up of three π-conjugated units of iminomethylphenol rather than the triethylamine bridge. Therefore, the lower energy absorption can mainly be assigned to π → π* transition between the π-conjugated iminomethylphenol units. This also might contribute to its high fluorescence quantum yield in dilute DMSO solution.
Living cell imaging
AIE-active dyes offer higher resistance to photobleaching and thus superior photostability and higher signal reliability relative to conventional dyes.5c,d Photobleaching experiments (Fig. S30, ESI†) revealed that TSBs have preferable photostability under UV irradiation. HeLa cells were imaged by 3,5-Cl using a standard cell-staining protocol. As can be seen from Fig. 14, when incubated with 3,5-Cl, the HeLa cells grow similar as in the control experiments in the absence of the dye, which indicates that 3,5-Cl has little toxicity on the living cells. HeLa cells show negligible background fluorescence. However, intense intracellular AIE-active green fluorescence is observed if HeLa cells are incubated with 3,5-Cl. Therefore, AIE-active TSBs may have potentially applications in probing or monitoring biologically important processes in vitro as well as in vivo. | ||
| Fig. 14 Brightfield image ((a) incubated without dye) and fluorescence images ((b) incubated without dye; (c) incubated with 3,5-Cl) of HeLa cells. | ||
Conclusion
We have systematically synthesized and studied the unique AIE and anion host/probe properties, mechanofluorochromism, and cell imaging applications of a series of non-conjugated TSBs. Although they involve linking with a non-conjugated triethylamine bridge, their strong non-covalent intermolecular interactions, including N⋯H, O⋯H, H⋯H, C⋯H, and halogen bonds, ensure elimination of the IRs and achieve strong AIE consequently. Moreover, TSBs are tripod-like side-single-opening cages with three flexible arms of phenol, and thus they might be used as universal anion hosts and probes. Therefore, we believe that these simple Schiff bases provide a new paradigm in the design of non-conjugated fluorescent materials for developing advanced organic optoelectronic devices, florescent anion hosts/probes, cell imaging, and so on.Acknowledgements
This work was supported by the National Natural Science Foundation of China (no. 21372169 and 81470481). We acknowledge the comprehensive training platform of the specialized laboratory of the College of Chemistry, Sichuan University, for material analysis.Notes and references
- B. Valeur, Molecular Fluorescence: Principles and Applications, Wiley, 2002 Search PubMed.
- (a) E. M. Nolan and S. J. Lippard, Chem. Rev., 2008, 108, 3443 CrossRef CAS PubMed; (b) H. N. Kim, M. H. Lee, H. J. Kim, J. S. Kim and J. Yoon, Chem. Soc. Rev., 2008, 37, 1465 RSC; (c) Q. Zhao, F. Y. Li and C. H. Huang, Chem. Soc. Rev., 2010, 39, 3007 RSC; (d) J. F. Zhang, Y. Zhou, J. Yoon and J. S. Kim, Chem. Soc. Rev., 2011, 40, 3416 RSC; (e) Y. Feng, J. H. Cheng, L. Zhou, X. G. Zhou and H. F. Xiang, Analyst, 2012, 137, 4885 RSC; (f) Y. M. Yang, Q. Zhao, W. Feng and F. Y. Li, Chem. Rev., 2013, 113, 192 CrossRef CAS PubMed; (g) J. H. Cheng, X. G. Zhou and H. F. Xiang, Analyst, 2015, 140, 7082 RSC.
- (a) U. Mitschke and P. Bauerle, J. Mater. Chem., 2000, 10, 1471 RSC; (b) L. S. Hung and C. H. Chen, Mater. Sci. Eng., R, 2002, 39, 143 CrossRef; (c) K. Mullen and U. Scherf, Organic Light Emitting Devices–Synthesis, Properties and Applications, Wiley, 2006 Search PubMed; (d) Highly Efficient OLEDs with Phosphorescent Materials, ed. H. Yersin, Wiley, 2007 Search PubMed; (e) H. F. Xiang, J. H. Cheng, X. F. Ma, X. G. Zhou and J. Chruma, Chem. Soc. Rev., 2013, 42, 6128 RSC.
- (a) J. D. Slinker, J. Rivnay, J. S. Moskowitz, J. B. Parker, S. Bernhard, H. D. Abruoac and G. Malliaras, J. Mater. Chem., 2007, 17, 2976 RSC; (b) R. D. Costa, E. Orti and H. J. Bolink, Pure Appl. Chem., 2011, 83, 2115 CrossRef CAS.
- (a) V. Fernandez-Moreira, F. L. Thorp-Greenwood and M. P. Coogan, Chem. Commun., 2010, 46, 186 RSC; (b) Q. Zhao, C. H. Huang and F. Y. Li, Chem. Soc. Rev., 2011, 40, 2508 RSC; (c) J. Liang, B. Z. Tang and B. Liu, Chem. Soc. Rev., 2015, 44, 2798 RSC; (d) Y. F. Wang, T. b. Zhang and X. J. Liang, Small, 2016, 12, 6451 CrossRef CAS PubMed.
- (a) J. B. Birks, Photophysics of Aromatic Molecules, Wiley, New York, 1970 Search PubMed; (b) X. F. Ma, R. Sun, J. H. Cheng, J. Y. Liu, F. Gou, H. F. Xiang and X. G. Zhou, J. Chem. Educ., 2016, 93, 345 CrossRef CAS.
- (a) J. Luo, Z. Xie, J. W. Y. Lam, L. Cheng, H. Chen, C. Qiu, H. S. Kwok, X. Zhan, Y. Liu, D. Zhu and B. Z. Tang, Chem. Commun., 2001, 1740 RSC; (b) Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2011, 40, 5361 RSC; (c) J. Mei, N. L. C. Leung, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718 CrossRef CAS PubMed.
- (a) Y. Liu, Y. Tang, N. N. Barashkov, I. S. Irgibaeva, J. W. Y. Lam, R. Hu, D. Birimzhanova, Y. Yu and B. Z. Tang, J. Am. Chem. Soc., 2010, 132, 13951 CrossRef CAS PubMed; (b) Y. Yuan, R. T. K. Kwok, B. Z. Tang and B. Liu, J. Am. Chem. Soc., 2014, 136, 2546 CrossRef CAS PubMed.
- (a) J. Yang, L. Li, Y. Yu, Z. C. Ren, Q. Peng, S. H. Ye, Q. Q. Li and Z. Li, Mater. Chem. Front., 2017, 1, 91 RSC; (b) C. W. T. Leung, Y. Hong, S. Chen, E. Zhao, J. W. Y. Lam and B. Z. Tang, J. Am. Chem. Soc., 2013, 135, 62 CrossRef CAS PubMed; (c) S. Chen, Y. Hong, Y. Liu, J. Liu, C. W. T. Leung, M. Li, R. T. K. Kwok, E. Zhao, J. W. Y. Lam, Y. Yu and B. Z. Tang, J. Am. Chem. Soc., 2013, 135, 4926 CrossRef CAS PubMed; (d) L. Chen, G. W. Lin, H. R. Peng, S. Y. Ding, W. W. Luo, R. R. Hu, S. M. Chen, F. Huang, A. J. Qin, Z. J. Zhao and B. Z. Tang, Mater. Chem. Front., 2017, 1, 176 RSC; (e) J. Yang, J. Huang, Q. Q. Li and Z. Li, J. Mater. Chem. C, 2016, 4, 2663 RSC.
- B. Z. Tang, New Structural Motifs for Solid Light Emitters (Cutting-Edge Chemistry, ACS), http://https://www.acs.org/content/acs/en/pressroom/cutting-edge-chemistry/new-structural-motifs-for-solid-light-emitters.html?cid=home_promo.
- C. M. Xing, J. W. Y. Lam, A. Qin, Y. Dong, M. Haussler, W. T. Yang and B. Z. Tang, Polym. Mater.: Sci. Eng., 2007, 96, 418 CAS.
- A. Pucci, R. Rausa and F. Ciardelli, Macromol. Chem. Phys., 2008, 209, 900 CrossRef CAS.
- M. S. Mathew, K. Sreenivasan and K. Joseph, RSC Adv., 2015, 5, 100176 RSC.
- (a) L. Zhou, P. Y. Cai, Y. Feng, J. H. Cheng, H. F. Xiang, J. Liu, D. Wu and X. G. Zhou, Anal. Chim. Acta, 2012, 735, 96 CrossRef CAS PubMed; (b) L. Zhou, Y. Feng, J. H. Cheng, N. Sun, X. G. Zhou and H. F. Xiang, RSC Adv., 2012, 2, 10529 RSC; (c) J. H. Cheng, K. Y. Wei, X. F. Ma, X. G. Zhou and H. F. Xiang, J. Phys. Chem. C, 2013, 117, 16552 CrossRef CAS; (d) J. H. Cheng, Y. H. Zhang, X. F. Ma, X. G. Zhou and H. F. Xiang, Chem. Commun., 2013, 49, 11791 RSC; (e) J. H. Cheng, X. F. Ma, Y. H. Zhang, J. Y. Liu, X. G. Zhou and H. F. Xiang, Inorg. Chem., 2014, 53, 3210 CrossRef CAS PubMed; (f) X. F. Ma, J. H. Cheng, J. Y. Liu, X. G. Zhou and H. F. Xiang, New J. Chem., 2015, 39, 492 RSC; (g) J. H. Cheng, Y. X. Li, R. Sun, J. Y. Liu, F. Gou, X. G. Zhou, H. F. Xiang and J. Liu, J. Mater. Chem. C, 2015, 3, 11099 RSC.
- (a) S. Y. Li, L. M. He, F. Xiong, Y. Li and G. Q. Yang, J. Phys. Chem. B, 2004, 108, 10887 CrossRef CAS; (b) Peng Chen, R. Lu, P. C. Xue, T. H. Xu, G. J. Chen and Y. Y. Zhao, Langmuir, 2009, 25, 8395 CrossRef CAS PubMed.
- (a) W. Tang, Y. Xiang and A. Tong, J. Org. Chem., 2009, 74, 2163 CrossRef CAS PubMed; (b) R. Wei, P. Song and A. Tong, J. Phys. Chem. C, 2013, 117, 3467 CrossRef CAS; (c) X. Chen and A. Tong, J. Lumin., 2014, 145, 737 CrossRef CAS; (d) M. Gao, Q. L. Hu, G. X. Feng, B. Z. Tang and B. Liu, J. Mater. Chem. B, 2014, 2, 3438 RSC; (e) L. Peng, M. Gao, X. L. Cai, R. Y. Zhang, K. Li, G. X. Feng, A. J. Tong and B. Liu, J. Mater. Chem. B, 2015, 3, 9168 RSC; (f) L. Cui, Y. Baek, S. Lee, N. Kwon and J. Yoon, J. Mater. Chem. C, 2016, 4, 2909 RSC.
- (a) T. L. Yang and W. W. Qin, Microchim. Acta, 2007, 157, 55 CrossRef CAS; (b) Y. Zhou, J. Y. Jung, H. R. Jeon, Y. Kim, S. J. Kim and J. Yoon, Org. Lett., 2011, 13, 2742 CrossRef CAS PubMed; (c) W. H. Hsieh, C. F. Wan, D. J. Liao and A. T. Wu, Tetrahedron Lett., 2012, 53, 5848 CrossRef CAS; (d) K. B. Kim, H. Kim, E. J. Song, S. Kim, I. Noh and C. Kim, Dalton Trans., 2013, 42, 16569 RSC; (e) D. A. Safin and Y. Garcia, RSC Adv., 2013, 3, 6466 RSC.
- J. N. Demas and G. A. Crosby, J. Phys. Chem., 1971, 75, 991 CrossRef.
- F. Gou, J. H. Cheng, X. H. Zhang, G. Y. Shen, X. G. Zhou and H. F. Xiang, Eur. J. Inorg. Chem., 2016, 4862 CrossRef CAS.
- (a) P. Knuuttila, Acta Chem. Scand., Ser. A, 1982, 36, 541 CrossRef; (b) A. Adams, J. M. Guss and C. A. Collyer, Biochemistry, 1999, 38, 9221 CrossRef CAS PubMed.
- (a) W. Lu, M. C. W. Chan, N. Zhu, C. M. Che, Z. He and K. Y. Wong, Chem. – Eur. J., 2003, 9, 6155 CrossRef CAS PubMed; (b) S. C. F. Kui, S. S. Y. Chui, C. M. Che and N. Zhu, J. Am. Chem. Soc., 2006, 128, 8297 CrossRef CAS PubMed; (c) S. Kohmoto, T. Chuko, S. Hisamatsu, Y. Okuda, H. Masu, M. Takahashi and K. Kishikawa, Cryst. Growth Des., 2015, 15, 2723 CrossRef CAS.
- (a) P. Metrangolo, H. Neukirch, T. Pilati and G. Resnati, Acc. Chem. Res., 2005, 38, 386 CrossRef CAS PubMed; (b) G. Cavallo, P. Metrangolo, T. Pilati, G. Resnati, M. Sansotera and G. Terraneo, Chem. Soc. Rev., 2010, 39, 3772 RSC.
- S. Mukherjee and P. Thilagar, Chem. Commun., 2015, 51, 10988 RSC.
- V. S. Padalkar and S. Seki, Chem. Soc. Rev., 2016, 45, 169 RSC.
- (a) Y. Q. Dong, J. W. Y. Lam and B. Z. Tang, J. Phys. Chem. Lett., 2015, 6, 3429 CrossRef CAS PubMed; (b) P. C. Xue, J. P. Ding, P. P. Wang and R. Lu, J. Mater. Chem. C, 2016, 4, 6688 RSC.
- T. Han, Y. Hong, N. Xie, S. Chen, N. Zhao, E. Zhao, J. W. Y. Lam, H. H. Y. Sung, Y. Dong, B. Tong and B. Z. Tang, J. Mater. Chem. C, 2013, 1, 7314 RSC.
- (a) F. P. Schmidtchen and M. Berger, Chem. Rev., 1997, 97, 1609 CrossRef CAS PubMed; (b) C. Suksai and T. Tuntulani, Chem. Soc. Rev., 2003, 32, 192 RSC; (c) J. W. Steed, Chem. Soc. Rev., 2009, 38, 506 RSC; (d) H. B. Yu, Q. Zhao, Z. X. Jiang, J. G. Qin and Z. Li, Sens. Actuators, B, 2010, 148, 110 CrossRef CAS.
- J. H. Cheng, F. Gou, X. H. Zhang, G. Y. Shen, X. G. Zhou and H. F. Xiang, Inorg. Chem., 2016, 55, 9221 CrossRef CAS PubMed.
- J. Y. Liu, J. H. Cheng, X. F. Ma, X. G. Zhou and H. F. Xiang, Res. Chem. Intermed., 2016, 42, 5027 CrossRef CAS.
- (a) M. Boiocchi, L. Del Boca, D. E. Gomez, L. Fabbrizzi, M. Licchelli and E. Monzani, J. Am. Chem. Soc., 2004, 126, 16507 CrossRef CAS PubMed; (b) Q. Li, Y. Guo, J. Xu and S. J. Shao, Sens. Actuators, B, 2011, 158, 427 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: General, materials, computational details, crystallographic information files (CIF), crystallographic data, absorption, and fluorescence emission data. CCDC 1504826–1504830 for TSB, 3-t-Bu, 3-OMe, 3-F, and 3,5-Cl, respectively. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7qm00097a |
| ‡ These authors contributed equally to this work. |
| This journal is © the Partner Organisations 2017 |