
Emission enhancement and application of synthetic green fluorescent protein chromophore analogs
Hongping
Deng
and
Xinyuan
Zhu
*
School of Chemistry and Chemical Engineering, State Key Laboratory of Metal Matrix Composites, Shanghai Jiao Tong University, 800 Dongchuan Road, 200240 Shanghai, P. R. China. E-mail: xyzhu@sjtu.edu.cn; Fax: +86-21-54741297
First published on 1st November 2016
Abstract
The unique optical properties of genetically encoded green fluorescent protein (GFP) inspire the chemical synthesis, photophysical characterization and application of synthetic GFP chromophore (GFPc) analogs. In this Feature Article, we summarize the recent progress in the fluorescence enhancement and application of synthetic GFPc analogs. In view of fluorescence enhancement, various methods can be classified into physical encapsulation and chemical modification, which can inhibit the chromophores' ultrafast internal conversion. Thus, synthetic GFPc analogs hold great potential as promising candidates in the development of novel sensors or fluorescent probes.
1. Introduction
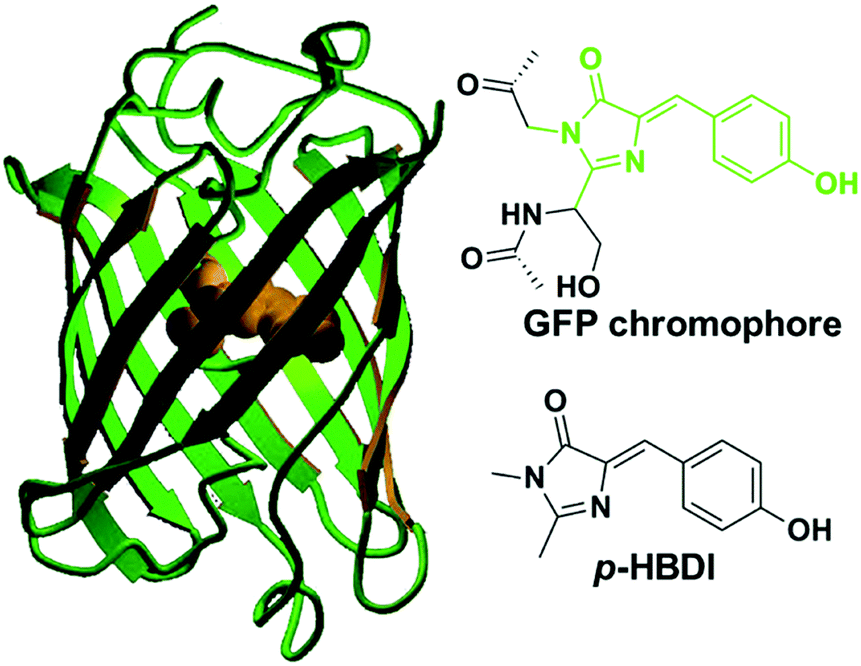
Green fluorescent protein (GFP) has been widely used as a genetically encoded fluorescent marker in pharmacology, and molecular and cell biology.1 In 1962, Shimomura and coworkers reported the separation and purification of aequorin chaperonin, GFP, from Aequorea victoria.2 Under ultraviolet (UV) irradiation, GFP can emit bright green fluorescence with a maximum emission peak at 508 nm.3 In 1992, Prasher and coworkers cloned the complete DNA sequence coding GFP by constructing the cDNA library of jellyfish, providing a universal method to express GFP artificially.4 In 1994, Chalfie et al. expressed mature GFP with bright fluorescence using Escherichia coli and Caenorhabditis elegans, confirming that the expression of GFP was not a unique function of jellyfish.1 At the same time, Tsien and coworkers put forward the bioluminescence principle of GFP, indicating that the GFP core chromophore is formed via the autocatalytic oxidation/dehydration process of a Ser65–Tyr66–Gly67 motif.5 Moreover, they also developed multicolor fluorescent proteins, providing a powerful tool for fluorescence labeling with a color palette almost across the entire visible region.6 In 2008, the Nobel Prize was awarded to Osamu Shimomura, Martin Chalfie and Roger Tsien for their discovery and development of the green fluorescent protein.6d,7Over the past few decades, fluorescent materials and dyes have drawn great attention due to their excellent properties and tremendous applications in medicine, energy and biology.8 By mimicking the bioluminescence principle of GFP, the development of fluorescent materials or dyes not only improves the understanding of the photophysical properties of GFP and its chromophore, but also offers another way towards novel fluorescent materials and dyes.9 Thus, a variety of methods have been developed for the synthesis of GFP chromophore (GFPc) analogs, benefiting from the photophysical characterization of model compounds.10 The core chromophore of GFP is a p-hydroxybenzylideneimidazolidinone (p-HBDI), which emits intense green fluorescence through excited-state proton transfer (ESPT) inside the intact protein.11 However, the fluorescence of either an isolated or synthesized core chromophore diminishes dramatically by four orders of magnitude in fluid solution at room temperature.12 At the same time, a general phenomenon of fluorescence loss for GFPc and its analogs is observed, which shows a huge gap compared to the chromophore inside the intact protein.13 To recover and enhance the emission response of these synthesized chromophores, various methods have been provided mainly through topological effects and chemical modifications.10 The fluorescence performances of these chromophores are very sensitive to the surrounding environment, which favors their application as chemical or biological fluorescent sensors.14
In this Feature Article, we summarize the basic properties of GFP and the recent progress in the synthesis, photophysics and application of GFPc analogs. Briefly, in the following sections, we will first introduce the structure, formation and photophysical properties of the core chromophore of GFP, emphasizing the bioluminescence principle of GFP. Then, we will describe the synthetic methods and the optical properties of GFPc and its analogs, paying particular attention to the emission enhancement of these chromophores in fluid solutions. Finally, we discuss the applications of synthetic GFPc analogs for fluorescence detection and imaging. This review intends to outline the exciting achievements of synthetic GFPc analogs and the inspirations in this emerging research area.
2. The green fluorescent protein chromophore
The GFP core chromophore is formed in situ without the need of any initiators or catalysts. Specifically, a Ser65–Tyr66–Gly67 tripeptide motif evolves into a p-HBDI chromophore via autocatalytic cyclization, and dehydration/oxidation processes only with the participation of an oxygen molecule.1,5,6b The p-HBDI chromophore is made up of a phenol ring and an imidazolinone heterocycle, which is covalently linked via CC single and double bonds (Fig. 1). Thus, p-HBDI can move freely along either the exocyclic C–C (ϕ) or the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C (τ) bond outside the protein matrix, which will cause an ultrafast internal conversion.12,15
C (τ) bond outside the protein matrix, which will cause an ultrafast internal conversion.12,15
 | ||
| Fig. 1 The molecular structures of green fluorescent protein and its core chromophore.1c Copyright 2002, American Chemical Society. | ||
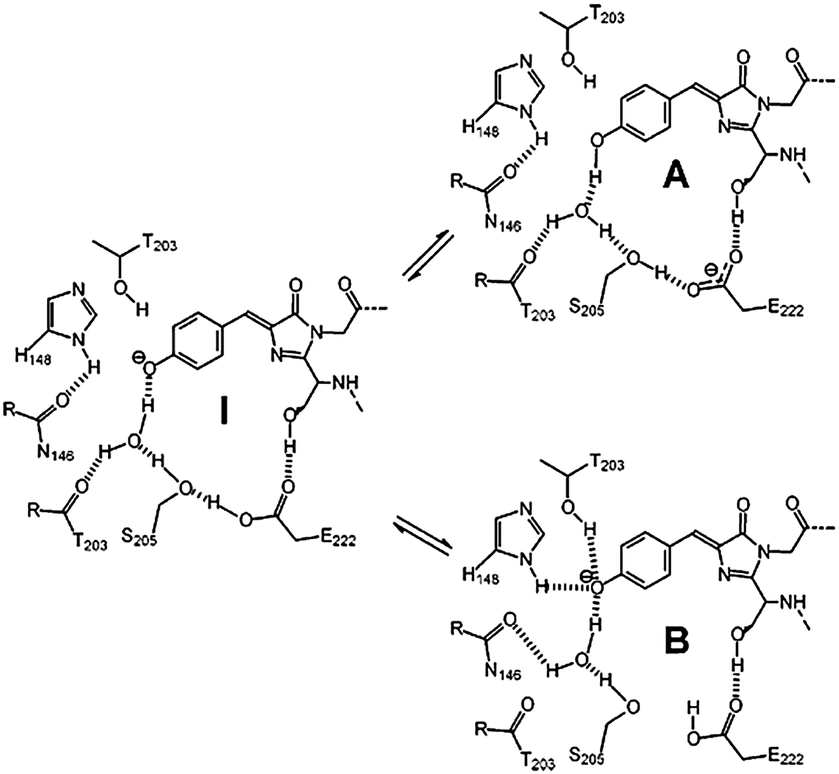
GFP shows two obvious absorption bands, defined as the A band and the B band, with the maximum at 395 nm and 480 nm, respectively.16 The main absorption band is the A band. As depicted in Fig. 2, experimental results indicate that the A band and the B band are ascribed to the different forms of the core chromophore, namely the neutral form (A-form) and the anionic form (B-form).6 In 1996, Niwa et al. prepared the core chromophore via the standard synthesis and revealed its structure, indicating that the optical properties of the chromophore were affected by its surrounding environment.17 They discovered that the absorption peaks of the neutral form of the synthetic chromophore in DMSO, EtOH and H2O were at 373 nm, 372 nm and 368 nm, respectively. These peaks are blueshifted compared to those of GFP. The solvent effect is small compared to the protein shift (25 nm), which is a significant point indicating the unique environment in the protein. With the addition of 1 M hydrochloric acid, the absorption peak of the cationic chromophore further redshifted about 10 nm in the same solvent; but the addition of 1 M sodium hydroxide greatly redshifted the absorption peaks of the anionic chromophore. Thus, the absorption spectra of p-HBDI in its different forms have also been studied.18 From its absorption spectra, only the absorption peaks at 368 nm, 391 nm and 425 nm were detected in its neutral, cationic and anionic forms, respectively, which are all blueshifted compared to those of GFP.18b Furthermore, the potential energy of the different forms of the GFP chromophore have also been probed via theoretical calculations.19 Because different charge states have a quite influence on the chromophore's optical properties, the following discussion refers to the neutral state unless otherwise stated.
 | ||
| Fig. 2 The three forms of the GFP core chromophore: the neutral form (A-form), the anionic form (B-form) and the intermediate form (I-form).6b Copyright 2005, Elsevier. | ||
Given the different forms of p-HBDI, the influence of solvent effect on the absorption properties of p-HBDI is also investigated.20 For the neutral chromophore, the solvent effect shows little influence on the absorption spectra with very weak redshifts in aprotic solvents. For the cationic chromophore, the influence of solvent effect is enhanced with relatively obvious shifts with the increase of solvent polarity, but without any regular changes. Compared with the neutral and cationic chromophore, the anionic one displays the largest solvent dependence. In aprotic solvents, the absorption peak redshifts with increasing solvent polarity, while it blueshifts in protic solvents. Through theoretical calculations, Negri and coworkers have studied the influence of solvent effect on the vibration and photodynamics of the core chromophore, indicating that fluid solvents favor the motion around CC bonds in the excited states.21
Although the absorption properties of GFPc analogs are different, the loss of fluorescence in fluid solvents is something they have in common, which is in sharp contrast to the excellent fluorescence of GFP with a quantum yield of 0.79 and a lifetime of 3.03 ns.22 Reasons for the loss of fluorescence are attributed to the ultrafast internal conversion through two torsional modes along either the exocyclic C–C or the CC bond.15 However, the confinement effect of the β-barrel can inhibit the free conformational motion of the core chromophore around the CC bonds, making fluorescence the primary form of energy release.15 Importantly, the fluorescence can be enhanced more than three orders of magnitude at 77 K.23 But, even in basic solution, the emission peak is much more blueshifted than that of GFP (about 508 nm) due to its unique excited-state proton transfer (ESPT) properties.24 The ESPT process is a complete proton transfer process, in which phenolic hydroxyl, amino acid Glu-5, water molecules and remote amino acid E222 are all involved.24b,25 Litvinenko et al. investigated the nonradiative decay process of the cationic and anionic forms of the chromophore using isoviscosity analysis.26 At room temperature, the internal conversion is favorable without any energy barriers; but it will form a small energy barrier at a lower temperature or higher solvent viscosity. Considering that the fluorescence of GFPc analogs is temperature-dependent, the related temperature refers to room temperature if not stated differently.27,28
3. Synthesis of GFP chromophore analogs
Over the past few years, a variety of methods for the synthesis of GFPc analogs have been developed.10 Among them, Erlenmeyer azlactone, Knoevenagel condensation, and 2,3-cycloaddition are most widely used.The classic method is Niwa synthesis, also called the Erlenmeyer azlactone synthesis (Scheme 1).29 Under the catalysis of sodium acetate, aromatic aldehyde reacts with N-acetylglycine to produce the corresponding azlactone, which further reacts with methylamine in the presence of K2CO3 to afford the product. Then, Tonge and coworkers modified the Niwa synthesis, which is also referred to as the Erlenmeyer azlactone synthesis.30 The only modification is that a phenolic aldehyde is used instead of the acetoxyaldehyde. Also, they replaced N-acetylglycine with N-crotonylglycine to increase the conjugation length of the chromophore, which was designed to model the chromophore in DsRed.31 Furthermore, Yampolsky et al. synthesized a model compound of the asFP595 chromoprotein from Anemonia sulcata via post-modification with selenium dioxide.32 Based on this post-modification method, a novel class of Kaede-like chromophores with additional conjugation were easily prepared.33 Chou et al. also prepared fluorescent protein red Kaede chromophores via a one-step condensation reaction catalyzed using ZnCl2.34 Moreover, they developed ortho-GFP chromophores by deprotection of a methoxy pre-chromophore.35 Furthermore, meta-GFP chromophores were synthesized directly using the corresponding meta-phenolic aldehyde.36
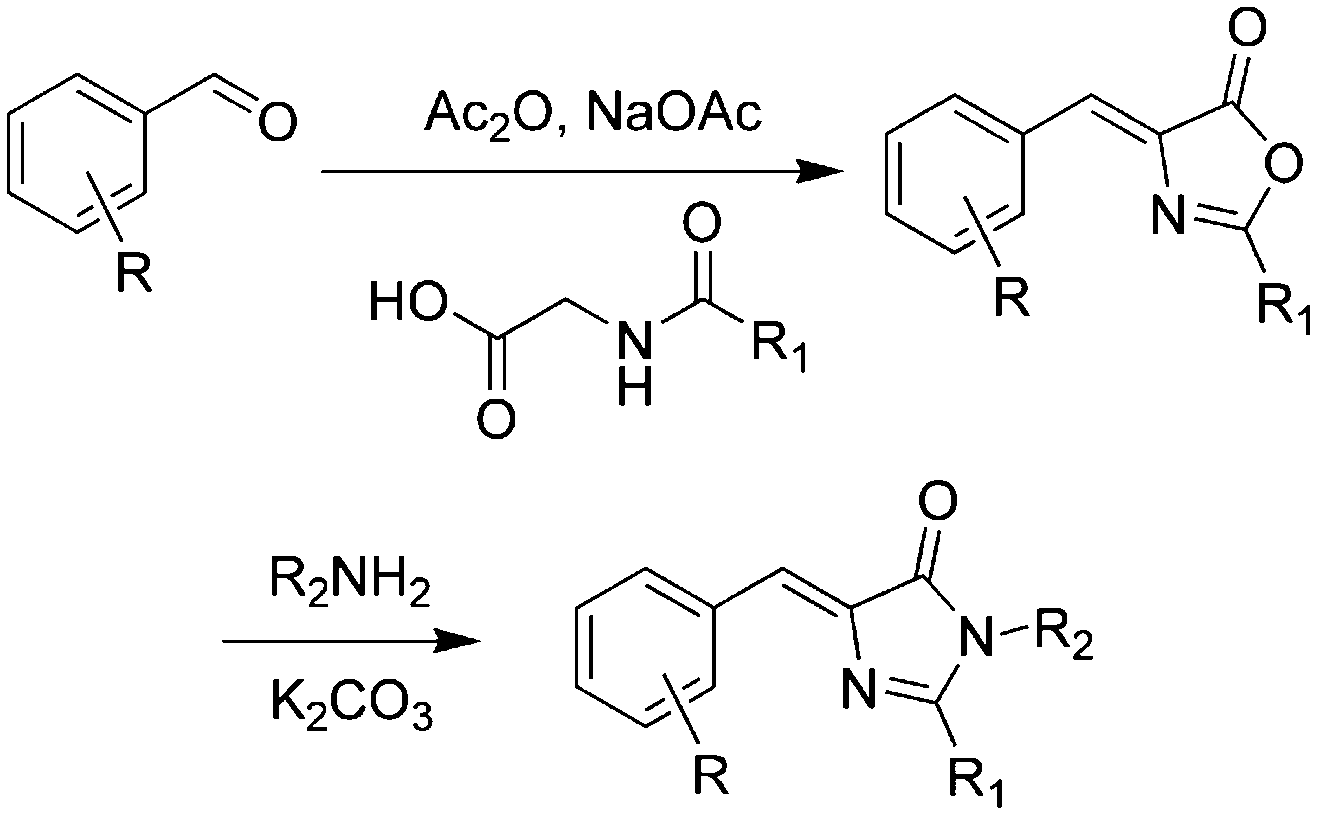 | ||
| Scheme 1 The Erlenmeyer azlactone method of GFPc analog synthesis.29 Copyright 1998, Elsevier. | ||
Meanwhile, a novel kind of Knoevenagel condensation method was developed (Scheme 2).37 In the process, imidazolidine is firstly formed with an azide-modified amino acid under the catalysis of triphenylphosphine, which further reacts with nitrogen-containing aromatic aldehyde to obtain the product. Subsequently, Tolbert and coworkers adapted this condensation reaction using ionic liquids as the catalyst for the Knoevenagel-type condensation.38 Chou et al. synthesized locked ortho- and para-core chromophores of GFP via the Knoevenagel condensation reaction, using TiCl4 as a new catalyst for the starting reactant of 5-hydroxy-1-indanone.39 This method can be applied to numerous substrates with relatively high yields, but needs multiple reactions.
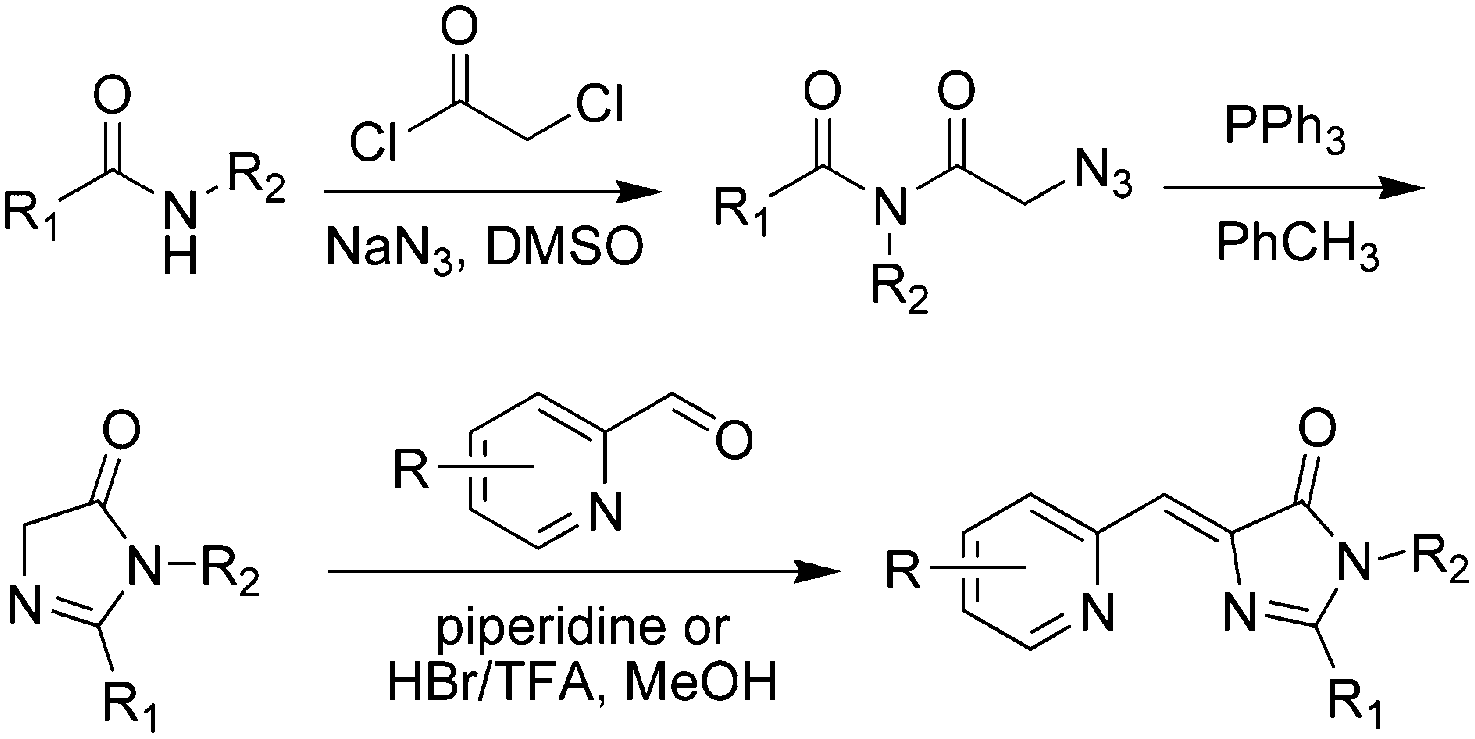 | ||
| Scheme 2 The Knoevenagel condensation method of GFPc analog synthesis.37 Copyright 2008, American Chemical Society. | ||
Another convenient synthetic method with good yields was further developed, defined as 2,3-cycloaddition (Scheme 3).40,42 Briefly, a primary amine reacts with an aromatic aldehyde to form a Schiff base, which can further react with newly prepared methyl 2-(1-ethoxyethylideneamino)acetate (MEEA) to afford the product. MEEA is often obtained through the combination of methyl glycinate hydrochloride with ethyl acetimidate hydrochloride with the catalysis of K2CO3. Since then, numerous GFPc analogs have been prepared using the 2,3-cycloaddition method.10 On the basis of this method, Clark et al. used ethyl benzimidate hydrochloride instead of ethyl acetimidate hydrochloride to prepare conjugation increased GFPc analogs with a benzene ring at the R1 position.41 2,3-cycloaddition is currently the most commonly used method for the synthesis of GFPc analogs.
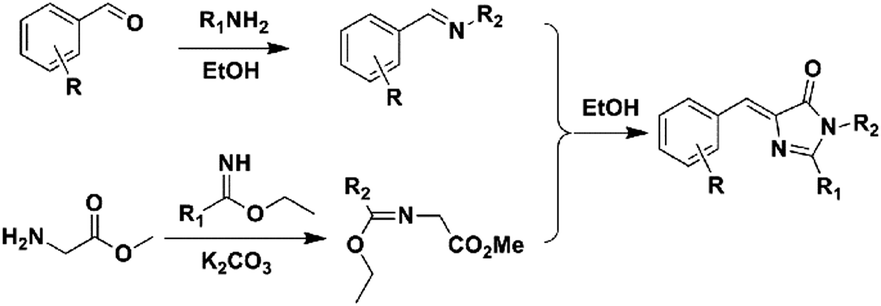 | ||
| Scheme 3 The 2,3-cycloaddition method of GFPc analog synthesis.42 Copyright 2011, American Chemical Society. | ||
Many GFPc analogs have been synthesized via the Erlenmeyer azlactone, the Knoevenagel condensation, and the 2,3-cycloaddition methods. These chromophores can be further tailored by the modification of an R1 and R2 group, but only have limited methods. For example, Lukyanov et al. modified the R1 group to an aldehyde or ketone through the oxidation of SeO2, which favors the modification at the R1 position via the Wittig reaction.33 Moreover, the formation of a double bond at the R1 position is also reported under the catalysis of ZnCl2via a one step reaction, which can increase the conjugation length of the chromophores.34 Generally, the modification of the R1 group often changes the optical properties of GFPc analogs, but shows little influence in the case of R2 group modification. To illuminate the structure–property relationship, the structures with continued numeric notations of the GFPc analogs discussed in the following figures are listed and given in Scheme 4.
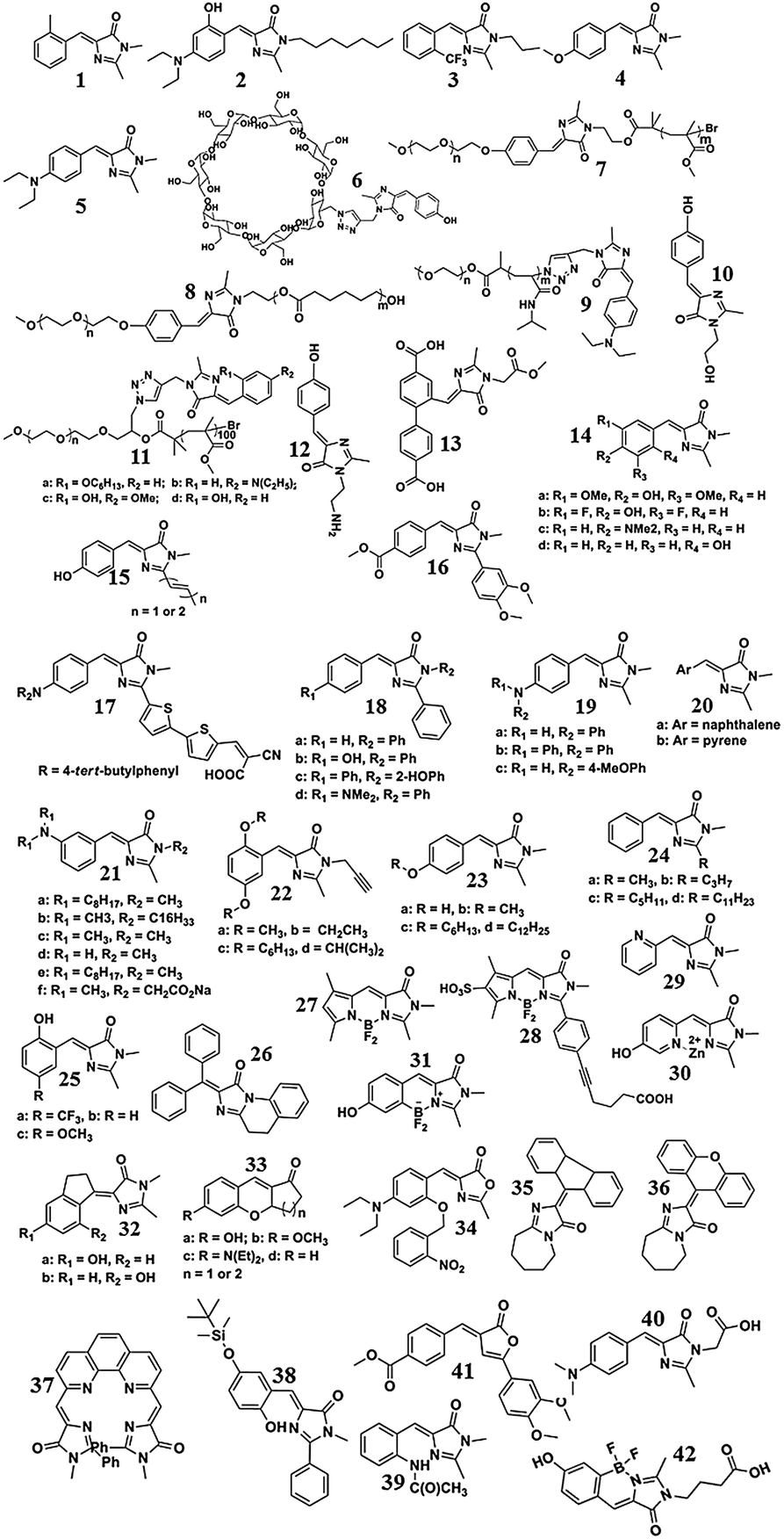 | ||
| Scheme 4 The structures of the GFPc analogs, with continued numeric notations of the GFPc analogs shown in the texts and figures. The numbers of different derivatives of a defined chromophore are labeled with continuous letters. | ||
4. Photophysics of GFP chromophore analogs
The general loss of fluorescence for GFPc analogs in fluid solvents has puzzled scientists for many years, and has drawn great attention to the research of their emission recovery and enhancement.10 Based on the way the emission recovered, physical and chemical enhancement methods are separated.4.1 Emission enhancement via physical methods
Physical encapsulation has been widely applied to enhance the performance of GFPc analogs via inhibition of the chromophores' conformational free motions. In this aspect, supramolecular hosts are firstly introduced. Baldridge et al. encapsulated GFPc analogs with octaacid (OA) to recover their fluorescence.12 Due to differences in the chromophore structure, the quantum yield increases nearly one order of magnitude only in the case of chromophore 1 after encapsulation (Fig. 3). Baldridge et al. also provided their most effective encapsulation format through theoretical calculations. Moreover, they further swapped OA with different proteins and discovered that the efficiency is better with these proteins than with OA; especially for human serum albumin (HSA), which facilitates the development of sensitive and selective “turn-on” probes for HSA using chromophore 2.42 Besides, amphiphilic sodium cholate (NaCh) has also been utilized to inhibit the chromophore's motion by forming aggregates in water.43 Among different substituents, chromophore 3 displays the best emission response. As shown in Fig. 4, the emission intensity of chromophore 3 gradually increases with the addition of NaCh. If the aggregates are broken due to light irradiation, their emission intensity decreases rapidly. Afterwards, Lee et al. screened 94 biological related analytes using 41 synthetic GFPc analogs and developed fluorescence “turn-on” sensors for pH, HSA and RNA, respectively.44 They discovered that the para-alkoxy group of the chromophore had a great influence on the selectivity of RNA with the best one (chromophore 4) showing five-fold enhancement. At the same time, they also found that chromophore 5 shows good selectivity against HSA showing a maximum blue-shift of 15 nm. Furthermore, a GFPc analog covalently modified with cyclodextrin (chromophore 6) has also prepared, which can form the cyclodextrin–chromophore complex via its hydrophobic cavity, resulting in fluorescence emission enhancement.45 But, the efficiency in enhancing the emission performance is rather low.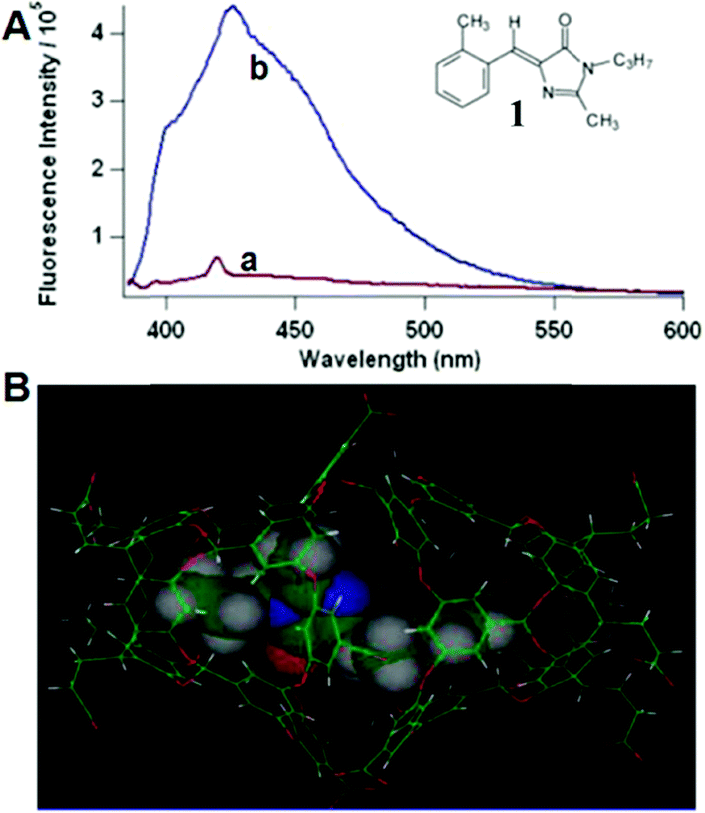 | ||
| Fig. 3 (A) Fluorescence of chromophore 1 in aerated solution (a = benzene and b = OA) together with its molecular structure; (B) chromophore 1 within the OA dimer cavity.12 Copyright 2010, American Chemical Society. | ||
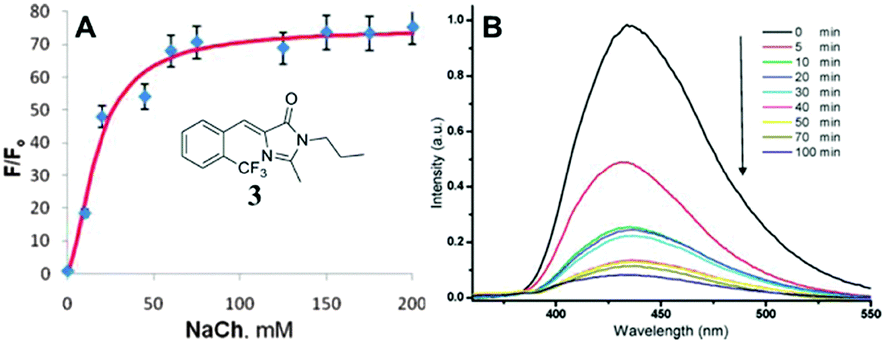 | ||
| Fig. 4 (A) Titration of chromophore 3 with NaCh in 0.2 M NaCl together with its molecular structure; (B) irradiation of chromophore 3 in 75 mM NaCh.43 Copyright 2011, American Chemical Society. | ||
Secondly, the fluorescence emission can also be enhanced via the self-assembly of amphiphilic polymers, which form fluorescent complex micelles. Specifically, we have prepared GFP–chromophore based block amphiphilic polymers (chromophore 7) via atom transfer radical polymerization, the fluorescence intensity of which shows great enhancement after assembly into complex micelles, expanding the research of the GFP chromophore and its analogs into the synthetic polymer field.46 A similar block amphiphilic polymer (chromophore 8) with a polycaprolactone chain has also been developed via ring-opening polymerization, which displays an enhanced emission once forming complex micelles.47 Meanwhile, a GFP chromophore can also be directly linked to the end of an amphiphilic polymer (chromophore 9) or be encapsulated via an amphiphilic polymer (chromophore 10), but the former format is better for fluorescence recovery due to better inhibition of the chromophore's motion.48 More importantly, multicolor GFP-mimicking fluorescent polymers (chromophore 11) were also fabricated via the combination of macromolecular self-assembly strategies with different chromophores (Fig. 5), which show an enhanced emission response and potential applications in single excitation multicolor cell imaging.49
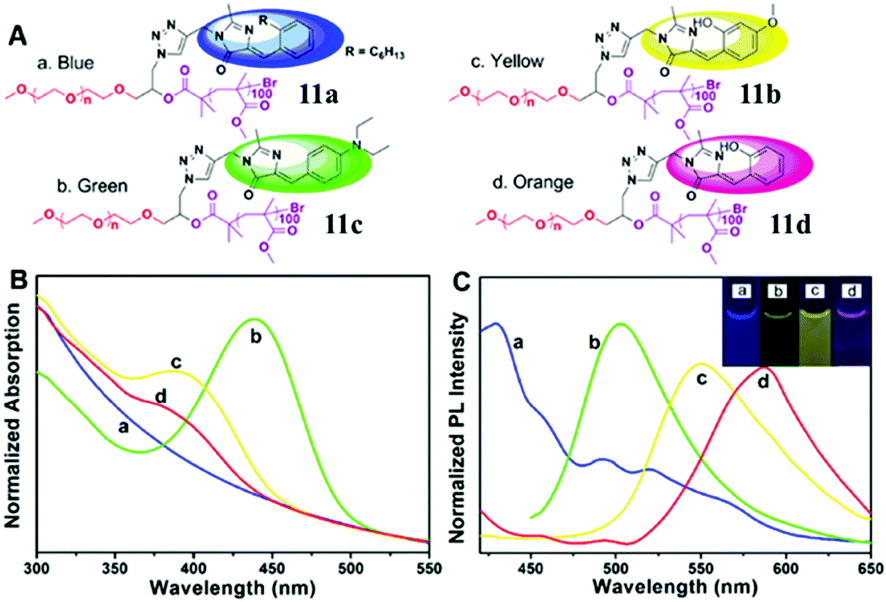 | ||
| Fig. 5 (A) Molecular structures of the multicolor fluorescent polymer, chromophore 11; normalized absorption (B) and emission (C) spectra of the multicolor fluorescent polymers in water: PEG-c1-PMMA (a, blue), PEG-c2-PMMA (b, green), PEG-c3-PMMA (c, yellow) and PEG-c4-PMMA (d, orange).49 Copyright 2015, American Chemical Society. | ||
Very recently, chromophore 12 has been used to covalently modify the tobacco mosaic virus, which shows enhanced fluorescence in several solvents via the nano-confinement effect of the virus.50 Williams et al. also synthesized a chromophore 13 metal–organic framework (MOF) to mimic the confinement effect of the β-barrel structure (Fig. 6).51 Through solid 2D NMR and theoretical calculations, they studied its low frequency vibration dynamics and demonstrated that the chromophore's molecular conformational motion is greatly inhibited. Moreover, by using the interactions between the RNA and chromophores, the fluorescence can be recovered via inhibition of free molecular motion. Thus, a series of RNA aptamers have been designed to turn-on the emission of GFPc analogs, which form RNA–chromophore complexes, termed Spinach.52 By screening the sequence of RNA, the physical interactions between RNA and the chromophore complexes can be tuned and the fluorescence is enhanced greatly (Fig. 7). Upon further modification of the RNA sequence and the structure of GFPc analogs (chromophore 14), multicolor Spinaches are developed with enhanced performance comparable to or even better than GFP, forming a color palette across the entire visible region.52 These multicolor Spinaches show enhanced fluorescence properties with good photostability, making them suitable for living cell applications.
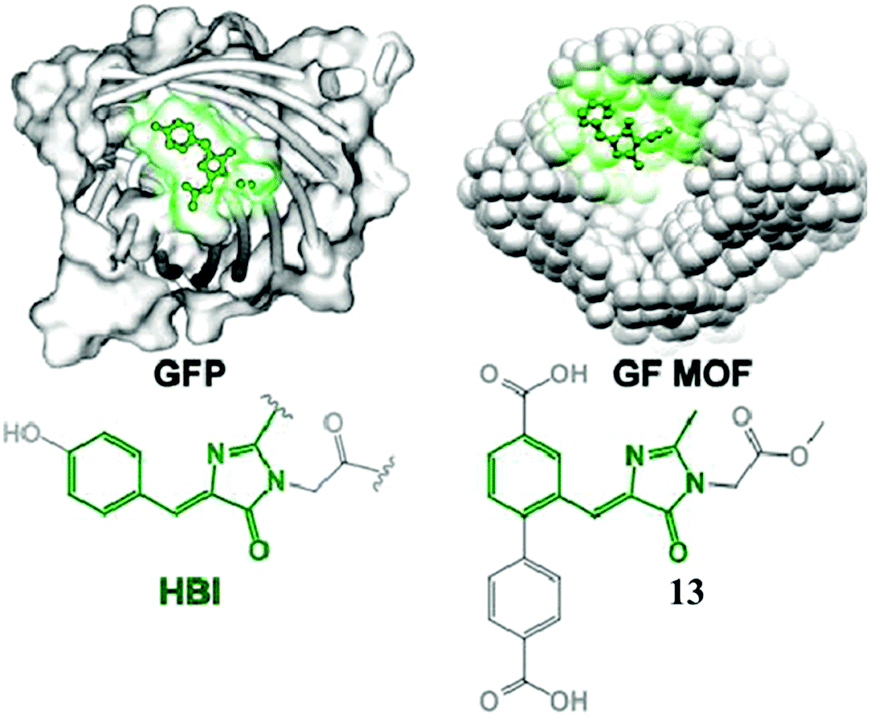 | ||
| Fig. 6 (left) GFP with an HBI chromophore structure; (right) a green fluorescent metal–organic framework constructed from chromophore 13.51 Copyright 2015, American Chemical Society. | ||
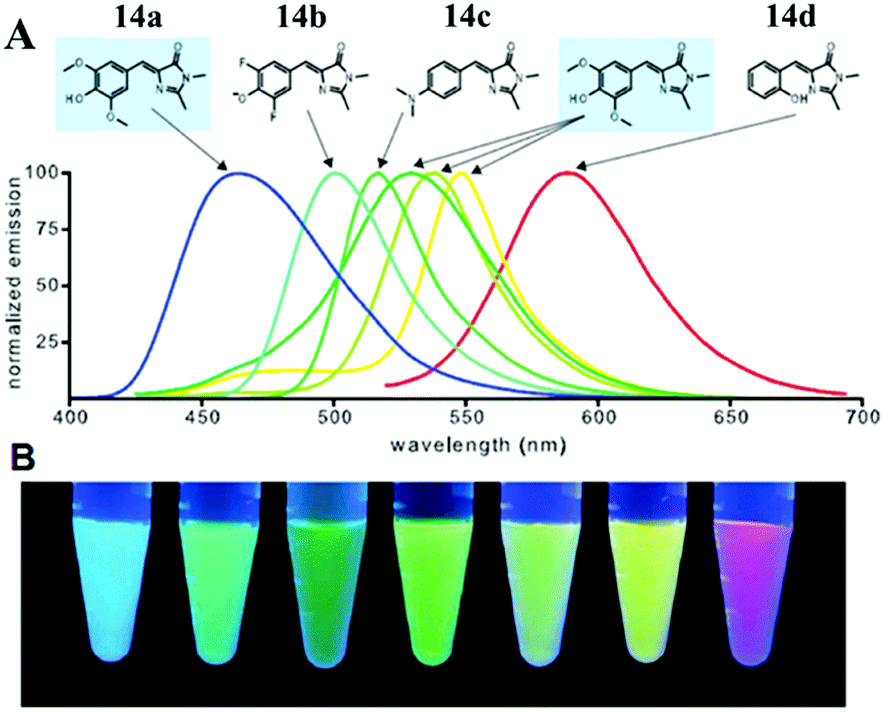 | ||
| Fig. 7 (A) Emission spectra of RNA–fluorophore complexes with the related chromophore 14; (B) RNA–fluorophore complexes were illuminated with ultraviolet light (365 nm) and photographed.52 Copyright 2011, American Association for the Advancement of Science. | ||
4.2 Emission enhancement via chemical methods
Physical encapsulation effectively inhibits the chromophores' free motion, which enhances their emission responses greatly. On the other hand, chemical modifications are also suitable for tuning the molecular structures of GFPc chromophores, which in turn will have the potential for fluorescence enhancement.The most common chemical modification is elongating the conjugation length of the chromophore, which increases the rigidity of the fluorophore and thus reduces the chromophores' motion. In 2002, Tonge et al. modified the R1 group of the GFPc analog with a double bond (chromophore 15), which enhanced the effective conjugation length.31 They found that the absorption peaks of these chromophores redshifted greatly and that the fluorescence enhanced with a maximum quantum yield (QY) of about 10% in chloroform. Then, the R1 group was further modified with various electron-donating or electron-withdrawing substituents.53 The results show that the fluorescence of chromophore 16 recovers greatly with a maximum QY of nearly 30% when the benzene ring is modified with a strong electron-withdrawing substituent and the imidazoline ring is tailored with a strong electron-donating substituent in dioxane, which could be attributed to the electronic effects of the substituents. Chou and coworkers also developed a condensation method for the formation of a double bond under the catalysis of ZnCl2, which will elongate the conjugation length of the GFPc analogs.34 By increasing the conjugation length, the absorption peak of chromophore 17 redshifts to 517 nm with enhanced emission due to increased molecular rigidity. Petkova et al. also prepared a series of GFPc analogs with an increased conjugation length via a static synthetic method.54 The absorption peaks of chromophore 18 display obvious redshifts, but their QYs are still very low with little enhancement. At the same time, a new kind of arylamine conjugated chromophore 19 with an increased conjugation length was also synthesized, resulting in a redshift of the absorption peaks.55 Its fluorescence QYs are still very low due to the motion of the chromophore's CC bonds, which is also a solvent polarity dependent result. Subsequently, Zhang et al. replaced the benzene ring of GFPc with large conjugated structures, which increased the conjugation length effectively.56 With the increase of conjugation length, the absorption peak redshifts gradually and the QY also enhances relatively. Moreover, they discovered that chromophore 20 shows solid red fluorescence, but its solid QYs are still very low.
A novel kind of meta-N,N-dimethylamino substituted chromophore 21 with an enhanced fluorescence has also been developed due to higher molecular motion barriers.57 With the increase of solvent polarity, the fluorescence peak redshifts obviously from Hex to ACN, covering across the entire visible region. Furthermore, the fluorescence of the chromophores will be quenched due to the solvent–solute hydrogen-bonding effect, which has been proved solidly.57 Moreover, the aggregation-induced emission (AIE) feature of chromophore 21 is reported, which was firstly found by Tang and coworkers.58 As shown in Fig. 8, in H2O–MeOH mixed solvents, with the increase of water content, the fluorescence intensity of chromophore 21a increases obviously, but it decreases gradually with the addition of CTAB due to the disassembly of their aggregates. More importantly, the solid powder of chromophore 21a can emit brightly with a maximum QY of about 71%.
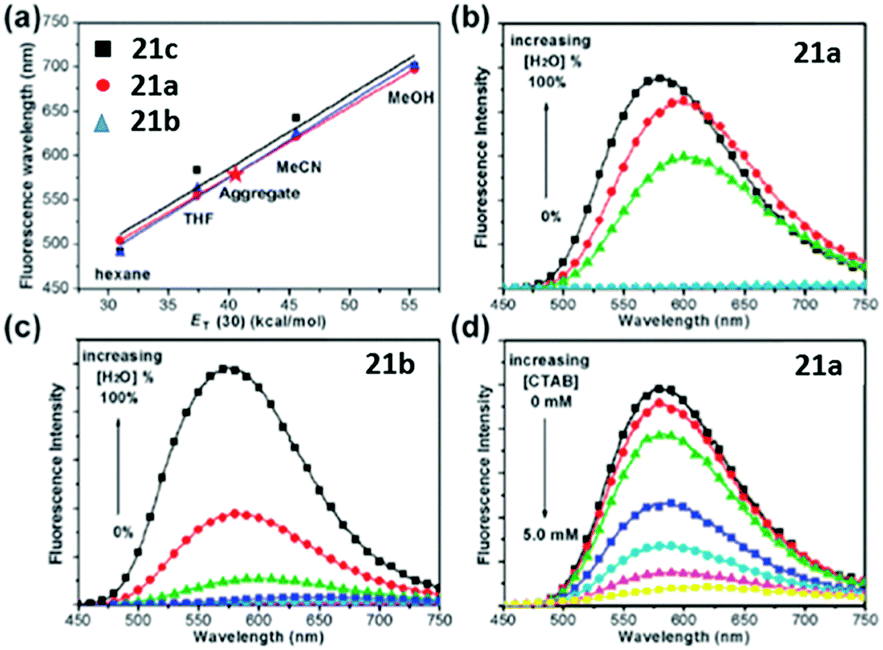 | ||
| Fig. 8 (a) Linear plots of λf against ET(30) for the chromophores; the AIE behavior of (b) chromophore 21a (0.05 mM) and (c) chromophore 21b (0.05 mM) in H2O–MeOH mixed solvents; (d) fluorescence spectra of chromophore 21a (0.05 mM) aggregates in H2O in the presence of CTAB at 0 to 5.0 mM.58a Copyright 2011, Royal Society of Chemistry. | ||
The enhancement of a 2,5-dimethoxy substituted chromophore has been previously reported.44 Very recently, our group developed a unique family of self-restricted chromophores 22 with dramatic emission enhancement and remarkable solvatofluorochromism (Fig. 9), the mechanism of which was also further revealed using fluorescence QY and lifetime measurements in combination with theoretical calculations, putting forward a universal method towards highly emissive unlocked GFPc analogs.59
 | ||
| Fig. 9 The normalized UV-Vis absorption (A, B) and fluorescence emission (C, D) spectra of chromophore 22 in aprotic (A, C) and protic (B, D) solvents; (E) a spectrum of color from blue to yellow for the fluorescence of chromophore 22 in different solvents under a 365 nm lamp.59 Copyright 2016, American Chemical Society. | ||
Although much attention has been paid to the solution performance of GFPc analogs, some studies still focuse on their solid fluorescence. For example, Dong et al. tailored p-HBDI with different carbon lengths of alkoxy at the phenolic position (chromophores 23).60 With the increase in carbon chain length, the solid fluorescence enhances gradually due to the decreased molecular interactions in the solid state prevented by the alkyl chain. Furthermore, a mild solvent exchange precipitation method was also used to prepare luminescent microcrystals with chromophore 23c, exhibiting aggregation-induced emission behavior.64 The results show that size and morphology can be affected by the experimental conditions, which further influence their solid performance. Then, the R1 position of the GFPc analog has also been modified with different carbon chain lengths.61 The solution emission of chromophore 24 increases with an increase in carbon chain length. More importantly, the solid fluorescence of 24d is enhanced greatly with a maximum QY of 37%. Chou and coworkers synthesized chromophore 25 with unique excited-state intramolecular proton transfer (ESIPT) properties.35 By changing the molecular structure, multicolor solid fluorophores were obtained with enhanced solid fluorescence.62 Then, Miyashita et al. reported a new class of multi-ring GFPc analogs using a novel condensation reaction.63 In DMSO, chromophore 26 shows little fluorescence; while in the solid or the frozen state, the molecular motion will be decreased and its fluorescence will be recovered.
Among all chemical methods, a special kind of “locking” strategy turns out to be the most effective. In 2008, Wu et al. reported locked highly emissive GFPc analogs.37 As shown in Fig. 10, a family of nitrogen heterocyclic GFPc analogs was synthesized via Knoevenagel condensation, which can be locked using BF2 to form a coplanar structure. The fluorescence of locked chromophore 27 enhances dramatically with a QY typically above 80%. After further modification, chromophore 28 can be used for fluorescence labeling. As shown in Fig. 10B, the fluorescence labeling does not affect the emission of the locked chromophore. Since then, a lot of “locked” GFPc analogs have been reported using different locking strategies, which result in wide emissive spectra from blue to red.65 Firstly, Chou et al. developed ortho-hydroxyl chromophore 25b, which formed a seven-membered ring via hydrogen bonds. Due to ESIPT, it emitted through a zwitterionic tautomer with emission peaks at about 605 nm to 580 nm from Hex to water.35 By tailoring the benzene ring, multicolor chromophores can be generated with enhanced responses, especially in the solid state (Fig. 11).62 Secondly, Baldridge et al. prepared pyridine-based chromophore 29, which could chelate with Zn2+ to lock the chromophore.38 With the addition of Zn2+ ions, the emission intensity increases gradually and the emission peak redshifts with the addition of acids. Furthermore, the reversible and irreversible locked chromophores 30 and 31 were also designed with coplanar conformation, which greatly inhibits the internal conversion of the chromophores.66 As depicted in Fig. 12, the results reveal that chromophores 30 and 31 show high photo-induced acidity; chromophore 30 was the first example of a GFPc analog with super photo-induced acidity: the pKa is 5.3 in the ground state, but it changes to −1.3 in the excited state. Besides, another kind of locking strategy toward a CC single bond is applied for the preparation of chromophore 32via Knoevenagel condensation, which will partially inhibit the chromophore's motion.39 In toluene, chromophore 32b displays enhanced fluorescence emission with a QY of 18% and also shows amplified spontaneous fluorescence (Fig. 13). In the solid, chromophore 32b forms H-aggregates and has a very narrow emission width, which facilitates its application in organic light-emitting diodes.
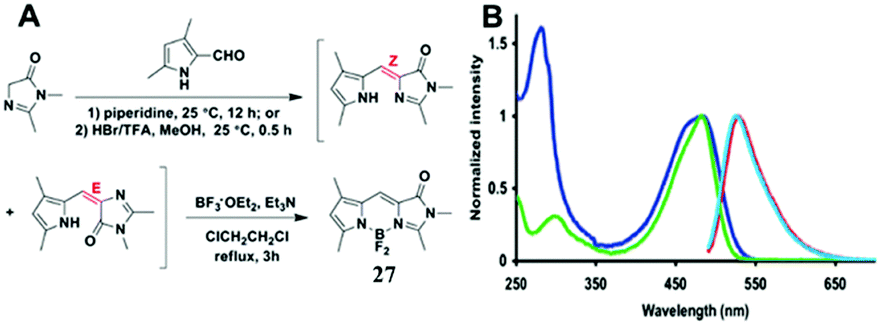 | ||
| Fig. 10 (A) The synthetic route towards the typical target chromophore 27; (B) absorbance and fluorescence of chromophore 28 (green, azure) and the labeled streptavidin with chromophore 28 (blue, red) in 0.1 M lithium phosphate buffer, pH 7.4.37 Copyright 2008, American Chemical Society. | ||
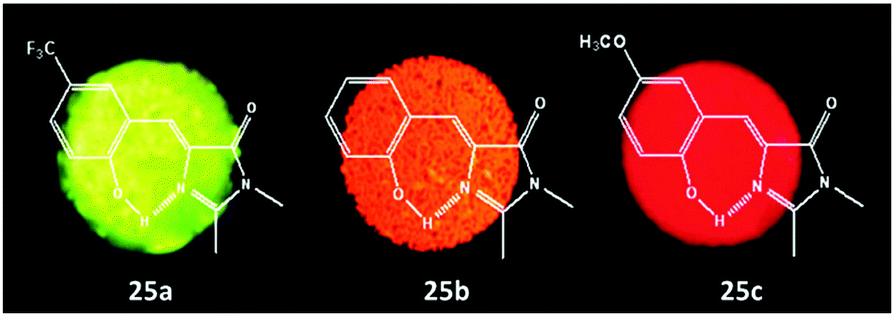 | ||
| Fig. 11 Solid-state fluorescence photographs of three typical chromophores 25a (green), 25b (orange) and 25c (red).62 Copyright 2011, American Chemical Society. | ||
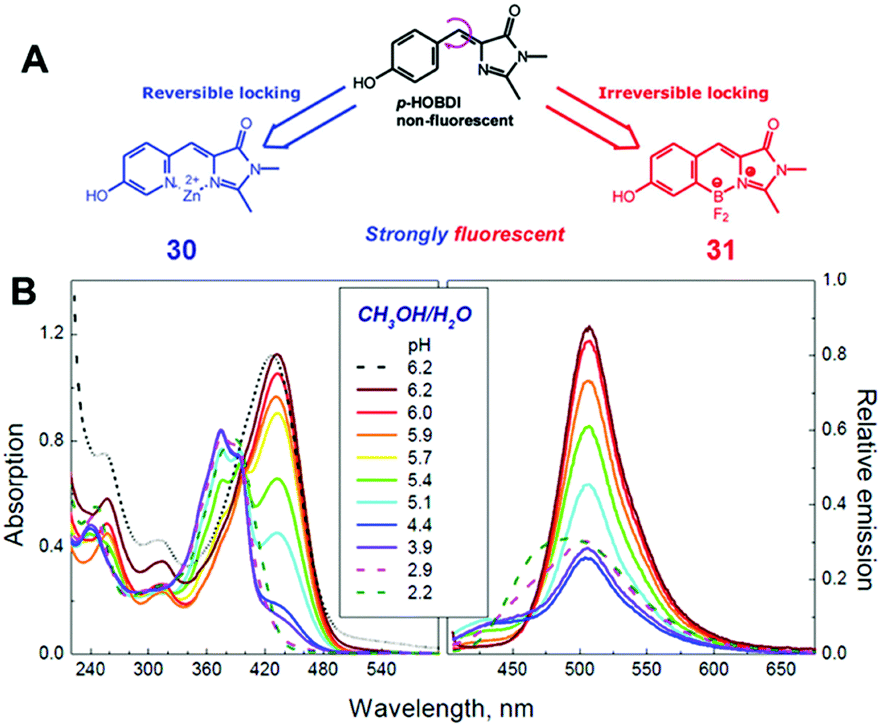 | ||
| Fig. 12 (A) The molecular structures of reversibly (chromophore 30) and irreversibly (chromophore 31) locked GFPc analogs; (B) the absorption and emission spectra of chromophore 30 in methanol/water (1/1 vol.) mixtures at various pH values.66 Copyright 2012, American Chemical Society. | ||
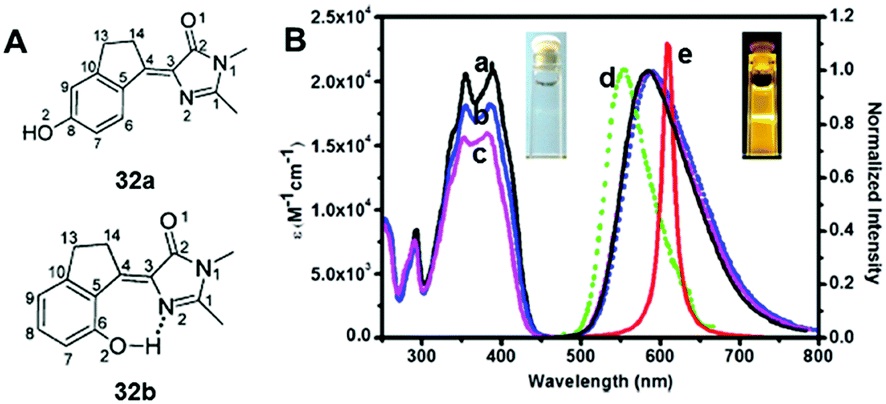 | ||
| Fig. 13 (A) The molecular structures of chromophore 32a and chromophore 32b; (B) steady-state absorption (a–c) and emission spectra of chromophore 32b in toluene (a), CH2Cl2 (b), acetonitrile (c), and solid film (d). Also shown is the amplified spontaneous emission (e) in toluene.39 Copyright 2014, American Chemical Society. | ||
Next, Yuan et al. prepared a rigid family of chromophores, 33, that have complete coplanar structures, which enhances the fluorescence greatly, especially the two-photon action cross-sections.67 In the experiments, the protection of the hydroxy group decreases its emission; once the protective group was removed via a reaction with thiols, the fluorescence could be recovered, which can be applied to imaging thiols in cells. Furthermore, Mandal et al. also converted chromophore 34 into a coumarin derivative via the nucleophilic reaction of the recovered ortho-hydroxyl group.68 Under the stimulus of light, the released hydroxyl group could attack the carbonyl of the imidazoline to rapidly form a coumarin derivative. Besides, Miyashita and coworkers also prepared the third generation of locked chromophores 35 and 36 using a locking strategy via a novel condensation reaction.69 In a good solvent, the fluorescence of these chromophores is very weak, but their emission can be enhanced at low temperatures or in the solid state.
5. Applications of GFP chromophore analogs
Enhancing the fluorescence of synthetic GFPc analogs through various physical and chemical methods benefits their application.Metal ion locking will greatly ‘turn-on’ the emission of GFPc analogs, which can be applied to fabricate metal ion sensors. For example, Baldridge et al. developed Zn2+ sensors with good selectivity and sensitivity using chromophore 29.38 To improve its binding ability and stability, Fang et al. changed the pyridine group of chromophore 29 to pyrrole.70 But, the sensitivity of this chromophore towards Zn2+ is still low. Thus, a multi-coordinative chromophore 37 was designed.71 As shown in Fig. 14, the four coordinative chromophore 37 shows a high complexing ability and stability for Zn2+ ions with excellent selectivity, which can be used for the quantitative assay of Zn2+in vitro. Then, Long and coworkers prepared the silicon-protected chromophore 38, which can react specifically with F− ions.72 Once the silicon group is removed, the optical properties of the chromophore are changed, which can be applied to the naked eye detection of F−in vitro.
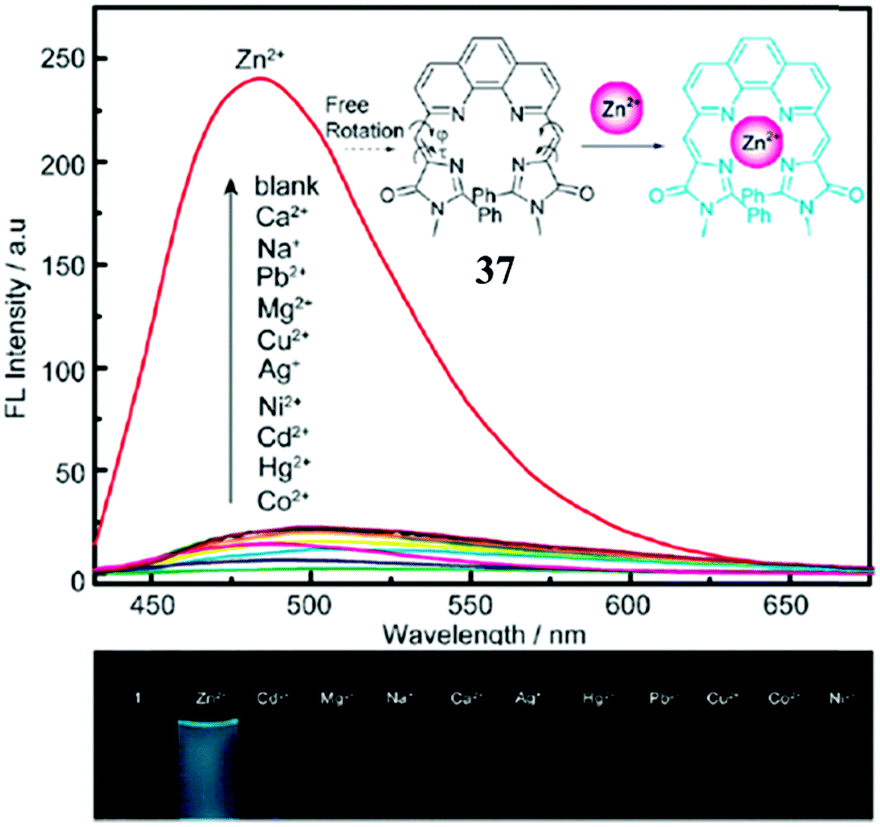 | ||
| Fig. 14 (top) Fluorescence emission of chromophore 37 (15 μM) in the presence of 1 equiv. of Co2+, Hg2+, Cd2+, Ni2+, Ag+, Cu2+, Mg2+, Pb2+, Na+, Ca2+, Zn2+ (from bottom to top) in CH3CN–HEPES; inset: proposed mechanism for the fluorescence enhancement of chromophore 37 with the Zn2+ ion; (bottom) photographs of chromophore 37 with 1 equiv. of metal ions under a 365 nm lamp.71 Copyright 2011, Royal Society of Chemistry. | ||
At the same time, Baldridge et al. also developed sensors for HSA with obvious sensitivity and selectivity via physical encapsulation.42 As shown in Fig. 15, the fluorescence intensity of chromophore 2 increases gradually with the addition of HSA; and only HSA could obviously turn-on the chromophore's emission among different proteins. Then, Lee et al. further discovered two ‘turn-on’ fluorescent sensors for pH and RNA among 41 GFPc analogs.44 Sung and coworkers measured the solvent polarity using the motion properties of chromophore 39, the mechanism of which relies on the intramolecular charge transfer and the hydrogen bonding effect of different solvents.73 Furthermore, Clark et al. reported the two-photon absorption properties of chromophore 40, which can distinguish amyloid from its aggregates via concentration dependent two-photon fluorescence.74
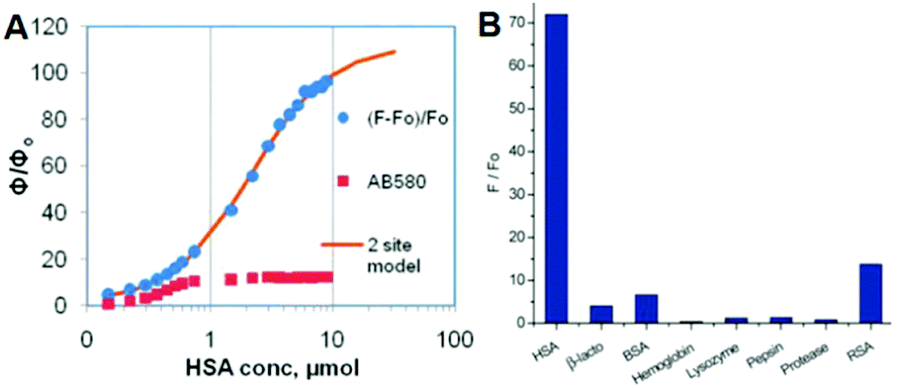 | ||
| Fig. 15 (A) Emission response of chromophore 2 and AB580 to HSA; (B) emission response of chromophore 2 with proteins and macromolecules.42 Copyright 2011, American Chemical Society. | ||
Considering the important applications of GFP in biological science, synthetic GFPc analogs are also applied for biological applications. For example, Signore and coworkers developed chromophore 41, which has a good sensitivity towards different static dielectric constants, which is used for the live cell imaging of the dielectric constant on the cell surface.75 Also, the activity of cysteine proteinase can be probed using the locked chromophore 42.76 Moreover, the cell membrane could be selectively labeled with turn-on fluorescence via the AIE feature of chromophore 21f.58 Very recently, a GFPc based amphiphilic polymer, chromophore 7, has been developed to mimic the confinement effect of the β-barrel, which can be used for cell imaging.46 And, a temperature responsive polymer, chromophore 9, covalently modified by the GFPc analog shows obvious emission enhancement with an increase in temperature, which is elegantly applied in the highly sensitive detection of Bacillus thermophilus with a 102 cfu per mL detection limit (Fig. 16).47 Finally, with a combination of macromolecular self-assembly and chemical modification strategies, single excitation multicolor cell imaging is achieved via the GFP-inspired multicolor fluorescent polymer, chromophore 11.48
 | ||
| Fig. 16 Confocal microscopy images of Bacillus thermophilus stained with chromophore 9 before (A, C) and after (B, D) heating: (up) bright field, (down) λex = 435 nm.48a Copyright 2013, Royal Society of Chemistry. | ||
RNA plays a great role in cell functions, but the real-time fluorescence labeling of RNA is very difficult to achieve.77 A novel class of RNA–chromophore complexes, termed Spinach, with enhanced emission and multicolor fluorescence have been developed.78 By tuning the sequence of RNA, the live and real-time cell imaging of different metabolites are achieved. As shown in Fig. 17, the RNA aptamer can selectively bind to specific metabolites and subsequently turn-on the fluorescence of chromophore 14b. With an increase in incubation time, the fluorescence enhances gradually due to the increase of the concentration of S-adenosylmethionine expressed by cells. Following the same principle, the selective RNA aptamers for protein were also fabricated, which can enhance the emission of chromophore 14b, making it an appropriate tool for real-time imaging and tracking of proteins expressed by Escherichia coli.79
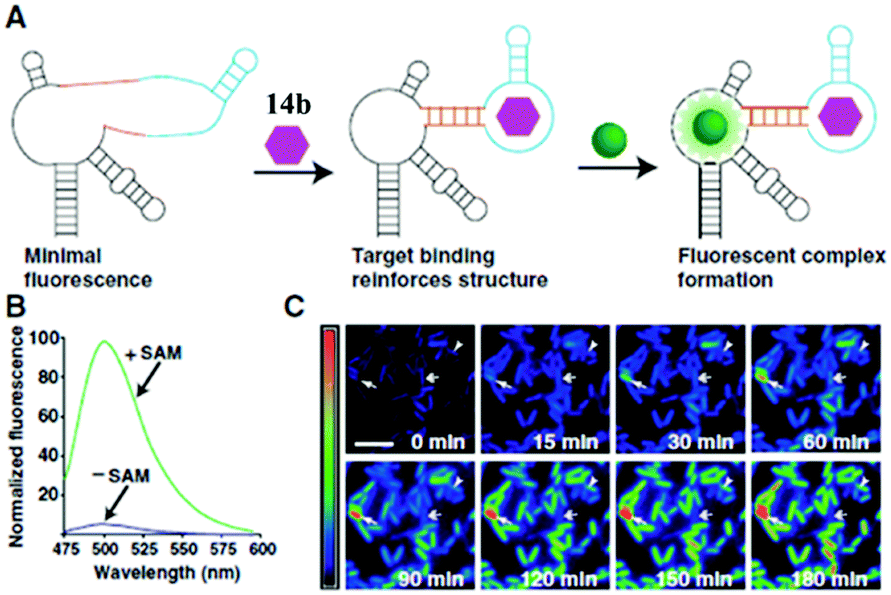 | ||
| Fig. 17 Imaging S-adenosylmethionine (SAM) in living cells with RNA; (A) the sensor RNA comprises Spinach, a transducer, a target-binding aptamer and chromophore 14b; (B) emission spectra of the SAM sensor in the presence or absence of SAM; (C) distinct patterns of SAM accumulation after adding methionine to E. coli expressing the SAM sensor RNA.78 Copyright 2012, American Association for the Advancement of Science. | ||
6. Conclusions
This Feature Article highlights the recent progress of emission enhancement and application for synthetic GFPc analogs. On the one hand, a host with a rigid/hydrophobic environment has been widely utilized to inhibit the chromophore's conformational motion via physical encapsulation, such as supramolecular hosts, protein, RNA and polymers. On the other hand, chemical modification turns out to be an effective method for the structure regulation and emission enhancement of these chromophores. Thus, their ‘turn-on’ fluorescence makes them suitable for various applications in the development of sensors or imaging probes.Future developments in the area will require further enhancement in the fluorescence performance of these chromophores, especially in highly polar solvents. Additionally, the fabrication of long wavelength chromophores with enhanced emission responses, especially in the near-infrared region, remains a challenge. Such developments will be highly attractive to chemists and biologists for their chemical and biological applications.
Acknowledgements
We acknowledge the financial support from the National Basic Research Program of China (2015CB931801) and the National Natural Science Foundation of China (51473093).Notes and references
- (a) M. Chalfie, Y. Tu, G. Euskirchen, W. W. Ward and D. C. Prasher, Science, 1994, 263, 802 CAS; (b) R. Y. Tsien, Annu. Rev. Biochem., 1998, 67, 509 CrossRef CAS PubMed; (c) M. Zimmer, Chem. Rev., 2002, 102, 759 CrossRef CAS PubMed; (d) J. Lippincott-Schwartz and G. H. Patterson, Science, 2003, 300, 87 CrossRef CAS PubMed; (e) A. Miyawaki, Annu. Rev. Biochem., 2011, 80, 357 CrossRef CAS PubMed.
- O. Shimomura, F. H. Johnson and Y. Saiga, J. Cell. Comp. Physiol., 1962, 59, 223 CrossRef CAS PubMed.
- F. H. Johnson, O. Shimomura, Y. Saiga, L. C. Gershman, G. T. Reynolds and J. R. Waters, J. Cell. Comp. Physiol., 1962, 60, 85 CrossRef CAS.
- (a) D. Prasher, R. O. McCann and M. J. Cormier, Biochem. Biophys. Res. Commun., 1985, 126, 1259 CrossRef CAS PubMed; (b) D. C. Prasher, V. K. Eckenrode, W. W. Ward, F. G. Prendergast and M. J. Cormier, Gene, 1992, 111, 229 CrossRef CAS PubMed.
- R. Heim, D. C. Prasher and R. Y. Tsien, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 12501 CrossRef CAS.
- (a) K. Brejc, T. K. Sixma, P. A. Kitts, S. R. Kain, R. Y. Tsien, M. Ormö and S. J. Remingtong, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 2306 CrossRef CAS PubMed; (b) G. Jung, J. Wiehler and A. Zumbusch, Biophys. J., 2005, 88, 1932 CrossRef CAS PubMed; (c) B. N. G. Giepmans, S. R. Adams, M. H. Ellisman and R. Y. Tsien, Science, 2006, 312, 217 CrossRef CAS PubMed; (d) R. Y. Tsien, Angew. Chem., Int. Ed., 2009, 48, 5612 CrossRef CAS PubMed.
- P. S. Weiss, ACS Nano, 2008, 2, 1977 CrossRef CAS PubMed.
- (a) J. Yao, M. Yang and Y. X. Duan, Chem. Rev., 2014, 114, 6130 CrossRef CAS PubMed; (b) C. Li, M. Y. Liu, N. G. Pschirer, M. Baumgarten and K. Müllen, Chem. Rev., 2010, 110, 6817 CrossRef CAS PubMed; (c) A. M. Breul, M. D. Hager and U. Schubert, Chem. Soc. Rev., 2013, 42, 5366 RSC; (d) H.-S. Peng and D. T. Chiu, Chem. Soc. Rev., 2015, 44, 4699 RSC; (e) T. Weil, T. Vosch, J. Hofkens, K. Peneva and K. Müllen, Angew. Chem., Int. Ed., 2010, 49, 9068 CrossRef CAS PubMed.
- A. Loudet and K. Burgess, Chem. Rev., 2007, 107, 4891 CrossRef CAS PubMed.
- L. M. Tolbert, A. Baldridge, J. Kowalik and K. M. Solntsev, Acc. Chem. Res., 2012, 45, 171 CrossRef CAS PubMed.
- J. G. Morin and J. W. Hastings, J. Cell. Physiol., 1971, 77, 313 CrossRef CAS PubMed.
- A. Baldridge, S. R. Samanta, N. Jayaraj, V. Ramamurthy and L. M. Tolbert, J. Am. Chem. Soc., 2010, 132, 1498 CrossRef CAS PubMed.
- S. Enoki, K. Saeki, K. Maki and K. Kuwajima, Biochemistry, 2004, 43, 14238 CrossRef CAS PubMed.
- C. L. Walker, K. A. Lukyanov, I. V. Yampolsky, A. S. Mishin, A. S. Bommarius, A. M. Duraj-Thatte, B. Azizi, L. M. Tolbert and K. M. Solntsev, Curr. Opin. Chem. Biol., 2015, 27, 64 CrossRef CAS PubMed.
- (a) K. B. Do and S. G. Boxer, J. Am. Chem. Soc., 2011, 133, 18078 CrossRef CAS PubMed; (b) J. N. Henderson, H.-W. Ai, R. E. Campbell and S. J. Remington, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 6672 CrossRef CAS PubMed; (c) M. Friedel, D. J. Sheeler and J.-E. Shea, J. Chem. Phys., 2003, 118, 8106 CrossRef CAS.
- M. Chattoraj, B. A. King, G. U. Bublitz and S. G. Boxer, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 8362 CrossRef CAS.
- H. Niwa, S. Inouye, T. Hirano, T. Matsuno, S. Kojima, M. Kubota, M. Ohashi and F. Tsuji, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 13617 CrossRef CAS.
- (a) X. He, A. F. Bell and P. J. Tonge, J. Phys. Chem. B, 2002, 106, 6056 CrossRef CAS; (b) G. Bublitz, B. A. King and S. G. Boxer, J. Am. Chem. Soc., 1998, 120, 9370 CrossRef CAS.
- (a) A. A. Voityuk, A. D. Kummer, M.-E. Michel-Beyerle and N. Rösch, Chem. Phys., 2001, 269, 83 CrossRef CAS; (b) I. V. Polyakov, B. L. Grigorenko, E. M. Epifanovsky, A. I. Krylov and A. V. Vemukhin, J. Chem. Theory Comput., 2010, 6, 2377 CrossRef CAS PubMed.
- J. Dong, K. M. Solntsev and L. M. Tolbert, J. Am. Chem. Soc., 2006, 128, 12038 CrossRef CAS PubMed.
- P. Altoe', F. Bernardi, M. Garavelli, G. Orlandi and F. Negri, J. Am. Chem. Soc., 2005, 127, 3952 CrossRef PubMed.
- G. Striker, V. Suvramaniam, C. A. M. Seidel and A. Volkmer, J. Phys. Chem. B, 1999, 103, 8612 CrossRef CAS.
- N. M. Webber, K. L. Litvinenko and S. R. Meech, J. Phys. Chem. B, 2001, 105, 8036 CrossRef CAS.
- (a) M. Chattoraj, B. A. King, G. U. Bublitz and S. G. Boxer, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 8362 CrossRef CAS PubMed; (b) D. Stoner-Ma, A. A. Jaye, P. Matousek, M. Towrie, S. R. Meech and P. J. Tonge, J. Am. Chem. Soc., 2005, 127, 2864 CrossRef CAS PubMed; (c) C. Fang, R. R. Frontiera, R. Tran and R. A. Mathies, Nature, 2009, 462, 200 CrossRef CAS PubMed.
- D. Stoner-Ma, E. H. Melief, J. Nappa, K. L. Ronayne, P. J. Tonge and S. R. Meech, J. Phys. Chem. B, 2006, 110, 22009 CrossRef CAS PubMed.
- K. L. Litvinenko, N. M. Webber and S. R. Meech, J. Phys. Chem. A, 2003, 107, 2616 CrossRef CAS.
- N. M. Webber, K. L. Litvinenko and S. R. Meech, J. Phys. Chem. B, 2001, 105, 8036 CrossRef CAS.
- K. L. Litvinenko, N. M. Webber and S. R. Meech, J. Phys. Chem. A, 2003, 107, 2616 CrossRef CAS.
- S. Kojima, H. Ohkawa, T. Hirano, S. Maki, H. Niwa, M. Ohashi, S. Inouye and F. I. Tsuji, Tetrahedron Lett., 1998, 39, 5239 CrossRef CAS.
- X. He, A. F. Bell and P. J. Tonge, FEBS Lett., 2003, 549, 35 CrossRef CAS PubMed.
- X. He, A. F. Bell and P. J. Tonge, Org. Lett., 2002, 1523 CrossRef CAS.
- I. V. Yampolsky, S. J. Remington, V. I. Martynov, V. K. Potapov, S. Lukyanov and K. A. Lukyanov, Biochemistry, 2005, 44, 5788 CrossRef CAS PubMed.
- I. V. Yampolsky, A. A. Kislukhin, T. T. Amatov, D. Shcherbo, V. K. Potapov, S. Lukyanov and K. A. Lukyanov, Bioorg. Chem., 2008, 36, 96 CrossRef CAS PubMed.
- W.-T. Chuang, B.-S. Chen, K.-Y. Chen, C.-C. Hsieh and P.-T. Chou, Chem. Commun., 2009, 6982 RSC.
- K.-Y. Chen, Y.-M. Cheng, C.-H. Lai, C.-C. Hsu, M.-L. Ho, G.-H. Lee and P.-T. Chou, J. Am. Chem. Soc., 2007, 129, 4534 CrossRef CAS PubMed.
- J. Dong, K. M. Solntsev, O. Poizat and L. M. Tolbert, J. Am. Chem. Soc., 2007, 129, 10084 CrossRef CAS PubMed.
- L. Wu and K. Burgess, J. Am. Chem. Soc., 2008, 130, 4089 CrossRef CAS PubMed.
- A. Baldridge, K. M. Solntsev, C. Song, T. Tanioka, J. Kowalik, K. Hardcastle and L. M. Tolbert, Chem. Commun., 2010, 46, 5686 RSC.
- Y.-H. Hsu, Y.-A. Chen, H.-W. Tseng, Z. Y. Zhang, J.-Y. Shen, W.-T. Chuang, T.-C. Lin, C.-S. Lee, W.-Y. Hung, B.-C. Hong, S.-H. Liu and P.-T. Chou, J. Am. Chem. Soc., 2014, 136, 11805 CrossRef CAS PubMed.
- A. Baldridge, J. Kowalik and L. M. Tolbert, Synthesis, 2010, 2424 CAS.
- T. B. Clark, M. E. Orr, D. C. Flynn and T. Goodson III, J. Phys. Chem. C, 2011, 115, 7331 CAS.
- A. Baldridge, S. H. Feng, Y.-T. Chang and L. M. Tolbet, ACS Comb. Sci., 2011, 13, 214 CrossRef CAS PubMed.
- A. Baldridge, A. Amador and L. M. Tolbert, Langmuir, 2011, 27, 3271 CrossRef CAS PubMed.
- J.-S. Lee, A. Baldridge, S. H. Feng, Y. SiQiang, Y. K. Kim, L. M. Tolbert and Y.-T. Chang, ACS Comb. Sci., 2011, 13, 32 CrossRef CAS PubMed.
- M. Cacciarini, M. B. Nielsen, E. M. D. Castro, L. Marinescu and M. Bols, Tetrahedron Lett., 2012, 53, 973 CrossRef CAS.
- H. P. Deng, Q. Zhu, D. L. Wang, C. L. Tu, B. S. Zhu and X. Y. Zhu, Polym. Chem., 2012, 3, 1975 RSC.
- H. P. Deng, Q. Zhu, D. L. Wang, B. S. Zhu and X. Y. Zhu, Acta Polym. Sin., 2012, 10, 1136 Search PubMed.
- (a) Y. J. Zheng, G. L. Li, H. P. Deng, Y. Su, J. H. Liu and X. Y. Zhu, Polym. Chem., 2014, 5, 2521 RSC; (b) G. J. Wang, Q. Zhu, H. P. Deng, D. L. Wang, X. Y. Zhu and D. Y. Yan, Acta Polym. Sin., 2013, 5, 660 Search PubMed.
- H. P. Deng, Y. Su, M. X. Hu, X. Jin, L. He, Y. Pang, R. J. Dong and X. Y. Zhu, Macromolecules, 2015, 48, 5969 CrossRef CAS.
- Q. Zhou, F. C. Wu, M. Wu, Y. Tian and Z. W. Niu, Chem. Commun., 2015, 51, 15122 RSC.
- D. E. Williams, E. A. Dolgopolova, P. J. Pellechia, A. Palukoshka, T. J. Wilson, R. Tan, J. M. Maier, A. B. Greytak, M. D. Smith, J. A. Krause and N. B. Shustova, J. Am. Chem. Soc., 2015, 137, 2223 CrossRef CAS PubMed.
- J. S. Paige, K. Y. Wu and S. R. Jaffrey, Science, 2011, 333, 642 CrossRef CAS PubMed.
- A. Follenius-Wund, M. Bourotte, M. Schmitt, F. Iyice, H. Lami, J.-J. Bourguignon, J. Haiech and C. Pigault, Biophys. J., 2003, 85, 1839 CrossRef CAS PubMed.
- I. Petkova, G. Dobrikov, N. Banerji, G. Duvanel, R. Perez, v. Dimitrov, P. Nikolov and E. Vauthey, J. Phys. Chem. A, 2010, 114, 10 CrossRef CAS PubMed.
- G.-J. Huang and J.-S. Yang, Chem. – Asian J., 2010, 5, 2075 CrossRef CAS PubMed.
- G. X. Huang, G. X. Zhang, Y. S. Wu, Q. Liao, H. B. Fu and D. Q. Zhang, Asian J. Org. Chem., 2012, 1, 352 CrossRef CAS.
- G.-J. Huang, J.-H. Ho, C. Prabhakar, Y.-H. Liu, S.-M. Peng and J.-S. Yang, Org. Lett., 2012, 14, 5034 CrossRef CAS PubMed.
- (a) S.-L. Tou, G.-J. Huang, P.-C. Chen, H.-T. Chang, J.-Y. Tsai and J.-S. Yang, Chem. Commun., 2014, 50, 620 RSC; (b) J. Luo, Z. Xie, J. W. Y. Lam, L. Cheng, H. Chen, C. Qiu, H. S. Kwok, X. Zhan, Y. Liu, D. Zhu and B. Z. Tang, Chem. Commun., 2001, 1740 RSC.
- H. P. Deng, C. Y. Yu, L. D. Gong and X. Y. Zhu, J. Phys. Chem. Lett., 2016, 7, 2935 CrossRef PubMed.
- J. Dong, K. M. Solntsev and L. M. Tolbert, J. Am. Chem. Soc., 2009, 131, 662 CrossRef CAS PubMed.
- S. Xiang, H. GuangXi, L. Kan, Z. GuanXin and Z. DeQing, Sci. China: Chem., 2013, 56, 1197 CrossRef.
- W.-T. Chuang, C.-C. Hsieh, C.-H. Lai, C.-H. Lai, C.-W. Shih, K.-Y. Chen, W.-Y. Hung, Y.-H. Hsu and P.-T. Chou, J. Org. Chem., 2011, 76, 8189 CrossRef CAS PubMed.
- M. Ikejiri, M. Tsuchino, Y. Chihara, T. Yamaguchi, T. Imanishi, S. Obika and K. Miyashita, Org. Lett., 2012, 14, 4406 CrossRef CAS PubMed.
- S. Fery-Forgues, S. Veesler, W. B. Fellows, L. M. Tolbert and K. M. Solntsev, Langmuir, 2013, 29, 14718 CrossRef CAS PubMed.
- (a) M. S. Baranov, K. M. Solntsev, N. S. Baleeva, A. S. Mishin, S. A. Lukyanov, K. A. Lukyanov and I. V. Yampolsky, Chem. – Eur. J., 2014, 20, 13234 CrossRef CAS PubMed; (b) N. S. Baleeva, K. A. Myannik, I. V. Yampolsky and M. S. Baranov, Eur. J. Org. Chem., 2015, 5716 CrossRef CAS.
- M. S. Baranov, K. A. Lukyanov, A. O. Borissova, J. Shamir, D. Kosenkov, L. V. Slipchenko, L. M. Tolbert, I. V. Yampolsky and K. M. Solntsev, J. Am. Chem. Soc., 2012, 134, 6025 CrossRef CAS PubMed.
- L. Yuan, W. Y. Lin, H. Chen, S. S. Zhu and L. W. He, Angew. Chem., Int. Ed., 2013, 52, 10018 CrossRef CAS PubMed.
- T. Chatterjee, D. Roy, A. Das, A. Ghosh, P. P. Bag and P. K. Mandal, RSC Adv., 2013, 3, 24021 RSC.
- M. Ikejiri, K. Matsumoto, H. Hasegawa, D. Yamaguchi, M. Tsuchino, Y. Chihara, T. Yamaguchi, K. Mori, T. Imanishi, S. Obika and K. Miyashita, Tetrahedron, 2015, 71, 4987 CrossRef CAS.
- X. X. Fang, Y. Wang, D. Wang, G. Y. Zhao, W. W. Zhang, A. M. Ren, H. Y. Wang, J. W. Xu, B.-R. Gao and W. Yang, J. Phys. Chem. Lett., 2014, 5, 92 CrossRef CAS PubMed.
- Y. Li, L. Shi, L. X. Qin, L. L. Qu, C. Jing, M. B. Lan, T. D. James and Y.-T. Long, Chem. Commun., 2011, 47, 4361 RSC.
- X.-Y. Liu, L. Shi, Z. F. Ding and Y.-T. Long, RSC Adv., 2014, 4, 53557 RSC.
- W.-J. Lo, Y.-H. Chen and K. Sung, J. Org. Chem., 2013, 78, 5925 CrossRef CAS PubMed.
- T. B. Clark, M. Ziółkowski, G. C. Schatz and T. Goodson III, J. Phys. Chem. B, 2014, 118, 2351 CrossRef CAS PubMed.
- G. Signore, G. Abbandonato, B. Storti, M. Stöckl, V. Subramaniam and R. Bizzarri, Chem. Commun., 2013, 49, 1723 RSC.
- M. Frizler, I. V. Yampolsky, M. S. Baranov, M. Stirnberg and M. Gütschow, Org. Biomol. Chem., 2013, 11, 5913 CAS.
- (a) C. C. Mello and D. Conte Jr, Nature, 2004, 431, 338 CrossRef CAS PubMed; (b) L. He and G. J. Hannon, Nat. Rev. Genet., 2004, 5, 522 CrossRef CAS PubMed.
- J. S. Paige, T. Nguyen-Duc, W. J. Song and S. R. Jaffrey, Science, 2012, 335, 1194 CrossRef CAS PubMed.
- W. J. Song, R. L. Strack and S. R. Jaffrey, Nat. Methods, 2013, 10, 873 CrossRef CAS PubMed.
| This journal is © the Partner Organisations 2017 |