
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEfficient route for the construction of polycyclic systems from bioderived HMF†
F. A.
Kucherov
,
K. I.
Galkin
,
E. G.
Gordeev
and
V. P.
Ananikov
 *
*
Zelinsky Institute of Organic Chemistry, Russian Academy of Sciences, Leninsky pr. 47, Moscow 119991, Russia. E-mail: val@ioc.ac.ru; Web: http://AnanikovLab.ru
First published on 30th August 2017
Abstract
The first synthesis of tricyclic compounds from biobased 5-hydroxymethylfurfural (HMF) is described. The Diels–Alder reaction was used to implement the transition from HMF to a non-planar framework, which possessed structural cores of naturally occurring biologically active compounds and building blocks of advanced materials. A one-pot, three-step sustainable synthesis in water was developed starting directly from HMF. The reduction of HMF led to 2,5-bis(hydroxymethyl)furan (BHMF), which could be readily involved in the Diels–Alder cycloaddition reaction with HMF-derived maleimide, followed by hydrogenation of the double bond. The described transformation was diastereoselective and proceeded with a good overall yield. The applicability of the chosen approach for the synthesis of analogous structures containing amine functionality on the side chain was demonstrated. To produce the target compounds, only platform chemicals were used with carbohydrate biomass as the single carbon source.
Introduction
In recent decades, the development of renewable resource processing for organic synthesis has obtained considerable attention.1–8 Currently, one of the most important challenges in the global chemical industry deals with the implementation of sustainable processes with the full integration of plant bioresources.9 5-(Hydroxymethyl)furfural (HMF) is a versatile bioderived platform chemical with several applications.1–10 An important advantage of such a useful compound is the possibility of its synthesis directly from biomass without using other (non-renewable) precursors.1–12 Accordingly, the preparation of practically important compounds using only platform chemicals as the single carbon source represents a highly efficient and sustainable approach.The outstanding platform potential of HMF for the production of liquid biofuels, bioplastics and chemicals has been discussed.1–15 The well-established strategies of HMF chemistry involve transformations of the side groups or reduction/hydrolysis of the furanic core, which lead to two-dimensional molecular structures. Although HMF chemistry is very well developed currently, the access to only a limited carbon skeleton diversity imposes severe limitations. Particularly, the preparation of compounds with non-planar 3D molecular frameworks is required to obtain bioactive products, as it provides more opportunities to “fine tune” their chemical properties.16–21 Indeed, the vast majority of natural bioactive compounds possess evolved skeletal diversity.22,23 Smart and stimuli responsive materials represent additional fascinating applications for chemical structures with diverse molecular skeleton frameworks.
The construction of three-dimensional polycyclic frameworks from HMF is a challenging task. The Diels–Alder reaction is a powerful synthetic tool for the creation of non-planar heterocyclic systems.24,25 It is known that furans readily undergo the Diels–Alder reaction to form oxabicyclic systems.26–31 However, HMF is an exception and does not directly participate in the cycloaddition reaction in contrast to 2,5-dimethylfuran.32 As a result, the synthesis of oxabicyclic derivatives from HMF is not possible (Scheme 1).33
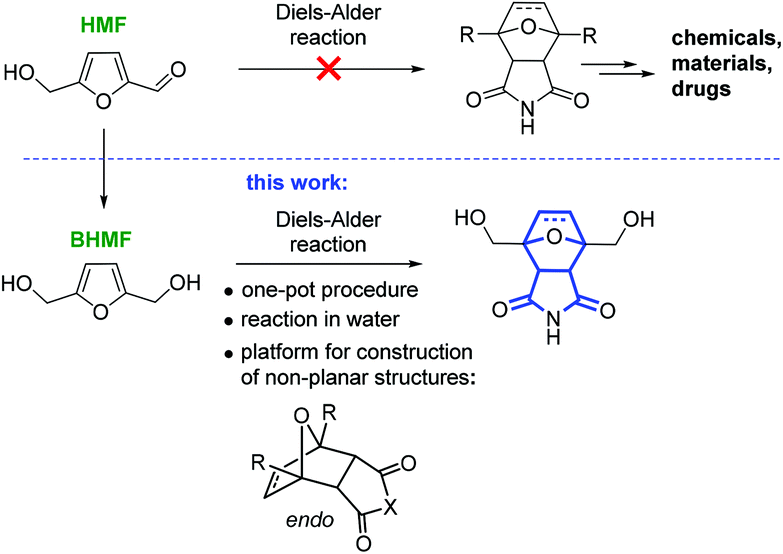 | ||
| Scheme 1 Sustainable production of tricyclic non-planar compounds from HMF via the Diels–Alder reaction (norcantharimide core is shown in blue). | ||
From an application point of view, the product of the cycloaddition to HMF with a 7-oxabicyclo[2.2.1]heptane core is a structural component of the naturally occurring anticancer drugs cantharidin and norcantharimide, and such compounds possess considerable practical potential.34–38 Materials science is another area of application for this functionalized tricyclic product, which can be used as a monomer for controlled polymerization either involving two OH groups or via a ring-opening process.
In this work, we report the selective synthesis of non-planar tricyclic molecular structures starting from HMF by a one-pot Diels–Alder reaction in water (Scheme 1). The reagents were chosen based on the concept of a sustainable process: only platform chemicals (HMF and bioethanol) were used as the carbon source.39–41 HMF can serve as the starting material for both the diene (BHMF) and the dienophile (maleimide). An additional approach to functionalized amines, ethers and esters by the Diels–Alder reaction was also explored in the present study.
To the best of our knowledge, there have not been any published examples of the Diels–Alder reaction of unsubstituted BHMF with alkenes. Only a few examples of cycloaddition reactions of substituted polyesters and peptide macrocycles have been reported.42–46
Results and discussion
The high reactivity of furan and dimethylfuran in the Diels–Alder reaction has been shown experimentally and theoretically.32 However, the reactivity of HMF and the possibility to tune the bioderived core for Diels–Alder chemistry remain unknown. To evaluate the possibility of performing the Diels–Alder reaction with HMF and BHMF, we carried out theoretical calculations at the DFT level (Scheme 2).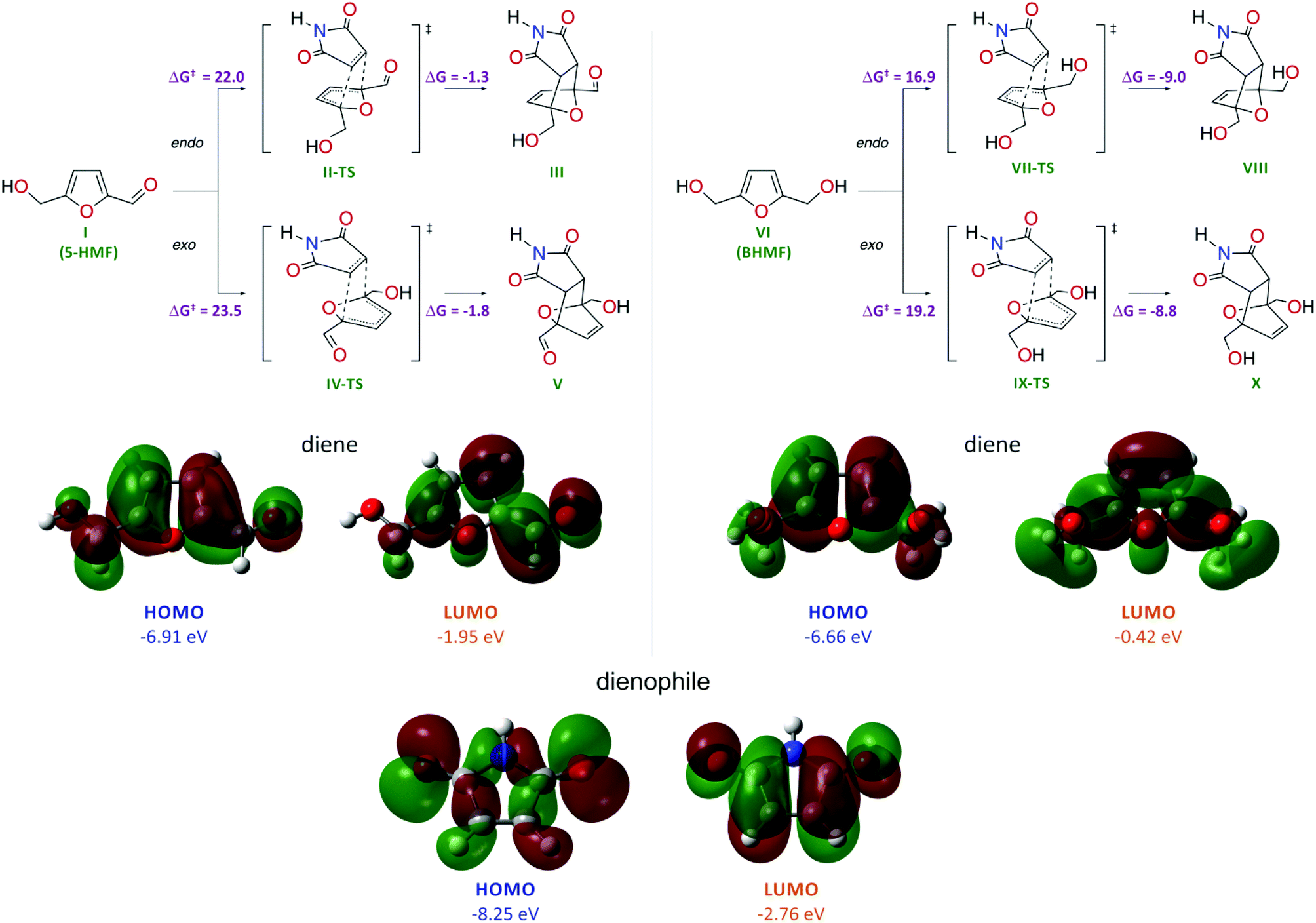 | ||
| Scheme 2 Schemes of the cycloaddition reactions for HMF and BHMF and representations of the frontier molecular orbitals for the reagents. Gibbs activation energies (ΔG‡, kcal mol−1) and Gibbs reaction energies (ΔG, kcal mol−1) are shown; theory level: PBE1PBE/6-311++G(d,p), GD3BJ and SCRF (SMD, water). | ||
Quantum chemical calculations were performed at the PBE1PBE/6-311++G(d,p) level with dispersion interaction corrections by Grimme D3 with Becke–Johnson damping and with the consideration of solvent effects at the SCRF(SMD, water) level. A comparative computational study showed that, for BHMF, the reaction with maleimide is energetically much more favorable compared with that for HMF (Scheme 2). The low reactivity of HMF is reflected by the considerably higher activation energies for this molecule in the Diels–Alder reaction: ΔG‡I→II-TS = 22.0 kcal mol−1 (endo) and ΔG‡I→IV-TS = 23.5 kcal mol−1 (exo) compared to BHMF (ΔG‡VI→VII-TS = 16.9 kcal mol−1 (endo) and ΔG‡VI→IX-TS = 19.2 kcal mol−1 (exo)). In the case of HMF, the calculated energy change shows a nearly thermoneutral transformation with ΔGI→III = −1.3 kcal mol−1 (endo) and ΔGI→V = −1.8 kcal mol−1 (exo).
In the case of BHMF, the process is much more exothermic with ΔGVI→VIII = −9.0 kcal mol−1 (endo) and ΔGVI→X = −8.8 kcal mol−1 (exo). Thus, in the case of BHMF, both kinetic and thermodynamic factors are more preferable for the Diels–Alder reaction. Regarding the product geometry, the energetic factors are more favorable for the endo form of the product.
The different reactivities of the HMF and BHMF molecules in the reaction with maleimide acting as a dienophile were also confirmed by analyzing the electronic structures of the reacting molecules (Scheme 2). The HOMO energy of the BHMF molecule is higher than the HOMO energy of the HMF molecule by 0.25 eV, which reflects the greater favorability of BHMF to interact with the dienophile in the cycloaddition reaction (interaction of the diene HOMO with the dienophile LUMO). The maleimide molecule is characterized by the lowest HOMO energy (−8.25 eV) and the lowest LUMO energy (−2.76 eV). Consequently, the maleimide molecule will act as an electron acceptor from the sides of the dienes (−6.91 eV HOMO and −1.95 eV LUMO for HMF; −6.66 eV HOMO and −0.42 eV LUMO for BHMF). Thus, the calculations clearly show that BHMF should be more reactive in the Diels–Alder cycloaddition process than HMF.
To verify the computational modeling, the following sequence of steps was performed: HMF 1 was reduced to BHMF 2 in an aqueous solution (Scheme 3). BHMF was subjected to the cycloaddition reaction with maleimide, and the corresponding tricyclic product 3 was successfully formed.
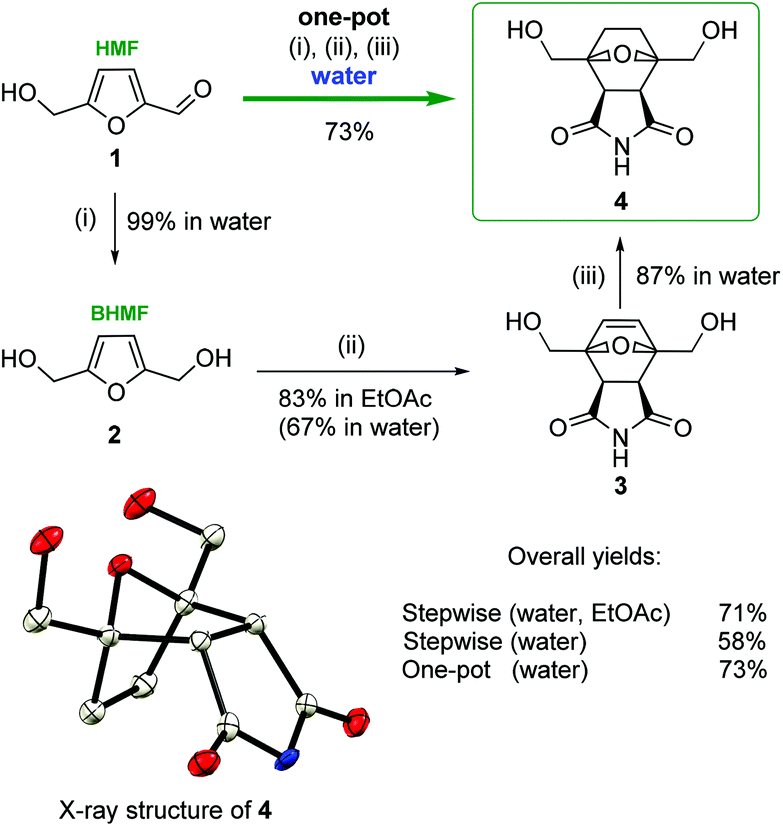 | ||
| Scheme 3 Synthesis of bis(hydroxymethyl)norcantharimide 4 in water. (i) NaBH4, water; (ii) maleimide, water; and (iii) H2, 1 atm, 10% Pd/C or Ni/Ra, water. Single-crystal X-ray structure of bis(hydroxymethyl)norcantharimide 4. | ||
It is known that Diels–Alder coupling products are capable of undergoing a reverse transformation. Conducting the reaction in ethyl acetate allows for the isolation of cycloadduct 3 in a pure form, as the Diels–Alder product was removed from the reaction media as a precipitate (Scheme 3). In ethyl acetate compound 3 was obtained in a high yield (83%). To exclude the possibility of the reverse process, unsaturated product 3 was reduced to bis(hydroxymethyl)norcantharimide 4 under an atmosphere of gaseous hydrogen in the presence of a metal catalyst (10% Pd/C or Ni/Ra).
There are two possible routes to attack the dienophile, which lead to the corresponding exo- and endo-cycloaddition products. The key stage involves BHMF interacting with maleimide diastereoselectively to form the endo product with de = 96%, in full accordance with the theoretical calculations. An even higher purity of final compound 4 was achieved by a simple recrystallization from ethanol. The structure of the obtained bis(hydroxymethyl)norcantharimide 4 was unambiguously determined by single-crystal X-ray diffraction (Scheme 3).
Carrying out the reaction in water and avoiding organic solvents would be an important improvement. Preliminary experiments showed that the step-by-step synthesis of 4 in aqueous media is possible, but it is less efficient. The reductive transformations of HMF → BHMF and 3 → 4 proceeded selectively with high yields (99% and 87%, respectively). However, the second stage, BHMF → 3, in water was quite problematic. In this case, the isolation of 3 was complicated due to the high solubility of the target compound in water. Moreover, the reaction sequence of retro-DA makes it difficult to obtain the substance in its pure form. Thus, derivative 3 was prepared from BHMF in water with a 67% yield, thus resulting in a much lower overall yield: 58% vs. 71% (Scheme 3). This problem was solved by the introduction of a one-pot protocol in water. The total yield of bis(hydroxymethyl)norcantharimide 4 in the one-pot, three-step synthesis in water proved to be more efficient with a yield of 73%, while for the three consecutive steps in water, the yield was considerably lower at 58% (Scheme 3).
To assess the scope of the developed synthesis approach, several substituted derivatives were tested in the cycloaddition/reduction sequence. BHMF was transformed into 2,5-bis(ethoxymethyl)furan 6 in ethanol in the presence of a catalytic amount of an acid (TsOH) with an 81% yield (Scheme 4). The resulting compound 6 was subjected to the one-pot cycloaddition–reduction sequence, which led to a 62% yield of tricycle 8.
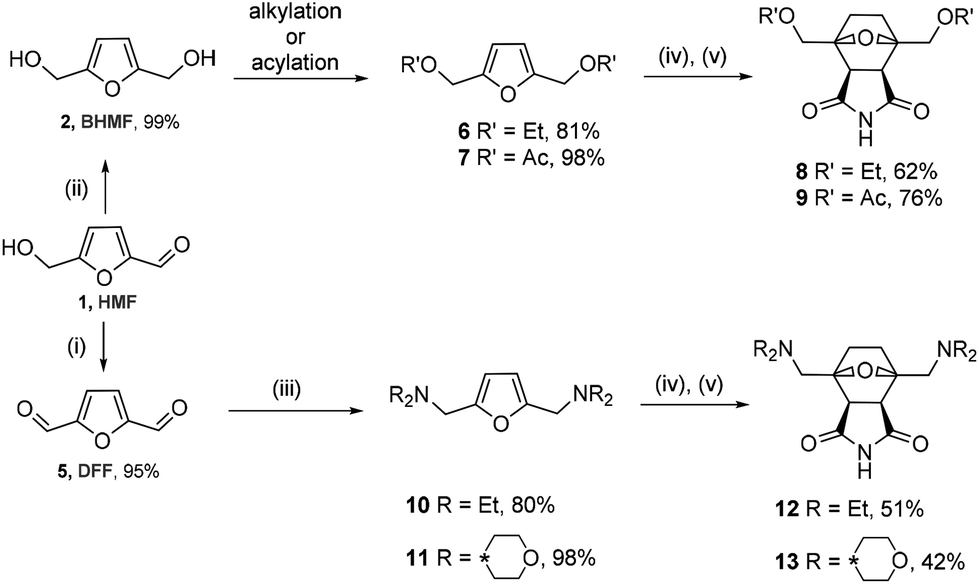 | ||
| Scheme 4 Synthesis of the norcantharimide derivatives. (i) [Pip*(O)][BF4], [BMIm][Cl]; (ii) NaBH4, water; (iii) HNEt2 or morpholine, STAB; (iv) maleimide; and (v) H2, 1 atm, 10% Pd/C. | ||
2,5-Bis(acetoxymethyl)furan 7 was obtained through the acylation of BHMF with acetyl chloride in pyridine in a 98% yield. The one-pot reaction of compound 7 with maleimide followed by hydrogenation by molecular hydrogen led to product 9 in a 76% yield. These transformations also proceeded diastereoselectively with de > 97%, leading to the corresponding endo products.
The developed synthesis strategy can be applied to not only alcohols but also amines. A dialkylaminomethyl moiety can be introduced into the molecule through reductive amination. For this purpose, HMF was oxidized to 2,5-diformylfuran (DFF, 5) by a recyclable system containing 4-acetamido-2,2,6,6-tetramethyl-1-oxopiperidinium tetrafluoroborate ([Pip*(O)][BF4]) in ionic liquid media in a 95% yield.47 The reaction of dialdehyde 5 with two equivalents of diethylamine or morpholine, followed by the treatment with sodium triacetoxyborohydride as a reducing agent, led to the corresponding tertiary amine 10 or 11, respectively, in a high yield (80% or 98%). Derivatives 10 and 11 interacted in a similar way with maleimide, followed by the hydrogenation of the cycloadducts. This reaction led to compounds 12 and 13 containing two dialkylaminomethyl groups at the head of the 1,4-epoxycyclohexane bridge. Somewhat lower yields of 51% and 42% are associated with more bulky substituents on the side chain.
To assess the synthesis potential of the obtained heterocyclic system, it was important to probe the reactivity of its functional groups. For this, compound 4 was subjected to acylation and alkylation reactions (Scheme 5).
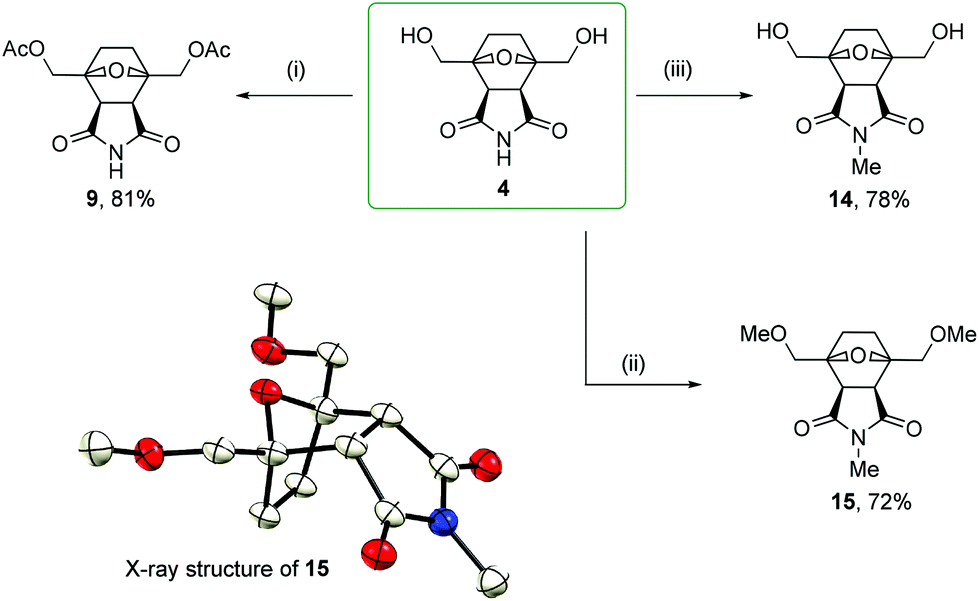 | ||
| Scheme 5 Possible modifications by alkylation and acylation: (i) AcCl, Py; (ii) MeI, NaH, DMF; and (iii) MeI, K2CO3, DMF. Single-crystal X-ray structure of N-methyl-bis(methoxymethyl)norcantharimide 15. | ||
The imide moiety was readily methylated by methyl iodide with potassium carbonate in DMF. In the presence of a weak base, none of hydroxyl groups were affected, but one alkyl group was introduced into the molecule with the selective formation of the N-Me derivative 14 in a 78% yield. When sodium hydride was used in the reaction of 4 with methyl iodide in dry DMF, complete alkylation took place with the formation of derivative 15 in a 72% yield. The structure of permethylated compound 15 was determined by single-crystal X-ray diffraction (Scheme 5). Thus, it can be concluded that the endo configuration of starting compound 4 was not affected during alkylation even under strongly basic conditions. A comparison of the chemical shifts of the MeO–CH2 and EtO–CH2 characteristic protons in the 1H NMR spectra of homologous compounds 15 and 8 revealed a difference of only 0.01 ppm (3.73 and 3.74 ppm, respectively). Considering the endo configuration of 15 and based on the similarity of the chemical shifts of 15 and 8, most likely, the studied 2,5-bis(ethoxymethyl)furan 6 reacted with maleimide in an endo-selective manner.
Bis(hydroxymethyl)norcantharimide 4 reacted with acetyl chloride in pyridine to form di-O-acetyl derivative 9 in an 81% yield. Considering the proven endo structure of starting compound 4 (Scheme 3), an endo configuration can also be assigned to reaction product 9. Diacetyl derivative 9 was also obtained through an alternative synthesis route by Diels–Alder cycloaddition (Scheme 4). A comparison of the chemical shifts in the 1H NMR spectra showed the full equivalence of both products obtained by the counter syntheses 7 → 9 and 4 → 9. This confirms the endo-selective nature of the Diels–Alder reaction of substituted derivative 7 with maleimide. The independent confirmation of the structure assignment was also performed by carrying out nuclear Overhauser effect spectroscopy (NOESY) and analysis of the obtained NOE data (see the ESI†).
Such reactivity clearly shows the possibility to vary the order of modification of the functional groups and gain access to a wide range of compounds.
Conclusions
Computational modeling showed that unsubstituted 2,5-bis(hydroxymethyl)furan (BHMF) is a more reactive compound in the Diels–Alder reaction with maleimide compared to 5-(hydroxymethyl)furfural (HMF). Following the theoretical predictions, the reduction of HMF to BHMF was used to raise the reactivity of the platform-chemical-derived molecule. An efficient three-step, one-pot procedure in water was implemented, and the first example of the Diels–Alder reaction of unsubstituted BHMF with maleimide was carried out in water with high diastereoselectivity. The developed synthesis approach was extended to functionalized amines, ethers and esters with a furanic core.The present study developed an approach to solve the synthesis problem in preparing tricyclic non-planar derivatives from HMF. The implementation of this protocol opens new opportunities for the synthesis of demanded functional derivatives.
Experimental section
General
NMR spectra were recorded on a Bruker DRX500 NMR spectrometer using residual solvent peaks as internal standards. The NMR spectra were processed using ACDLabs SpecManager 6.0. Mass spectra were measured on a high-resolution time-of-flight Bruker maXis instrument using electrospray ionization (ESI-MS). Measurements were performed in the positive ion mode, with an interface capillary voltage of 4.5 kV, at an effective scan range of m/z 100–1200, with external calibration (0.016 M sodium formate in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of MeCN–water or ESI-L Low Concentration Tuning Mix, Agilent Technologies), with a direct syringe injection at a flow rate of 3 μL min−1, with nitrogen as a dry gas (4 L min−1), and at an interface temperature of 180 °C. The spectra were processed using the Bruker Data Analysis 4.0 software package. Chromatographic separations were performed on silica gel (Merck Kieselgel 230–400 mesh) with analytical grade solvents. Analytical TLC was performed on Merck silica gel plates with a QF-254 indicator. Visualization was accomplished with a UV light and/or p-anisaldehyde–MeOH–H2SO4 and/or an acidic solution of KMnO4.
:1 mixture of MeCN–water or ESI-L Low Concentration Tuning Mix, Agilent Technologies), with a direct syringe injection at a flow rate of 3 μL min−1, with nitrogen as a dry gas (4 L min−1), and at an interface temperature of 180 °C. The spectra were processed using the Bruker Data Analysis 4.0 software package. Chromatographic separations were performed on silica gel (Merck Kieselgel 230–400 mesh) with analytical grade solvents. Analytical TLC was performed on Merck silica gel plates with a QF-254 indicator. Visualization was accomplished with a UV light and/or p-anisaldehyde–MeOH–H2SO4 and/or an acidic solution of KMnO4.
One-pot synthesis of endo-4,7-bis(hydroxymethyl) hexahydro-1H-4,7-epoxyisoindole-1,3(2H)-dione, 4
5-(Hydroxymethyl)furfural111 (0.15 g, 1.2 mmol) was dissolved in water (2.5 mL), and a solution of sodium borohydride (45 mg, 1.2 mmol) in water (0.7 mL) was added dropwise. The reaction mixture was stirred at 24 °C for 2 h and at 60 °C for 1 h. Then, the solution was cooled to 24 °C, and maleimide (0.23 g, 2.4 mmol, 2 eq.) was added. The solution was stirred for 16 h. 10% Pd/C (20 mg) or RANEY® nickel (30 mg) was added, and the reaction mixture was placed under a hydrogen atmosphere for 12 h at 24 °C. The catalyst was filtered off and washed thoroughly with water (3 × 1 mL), and the filtrate was evaporated under reduced pressure. The residue was washed with diethyl ether (3 × 2 mL) to remove succinimide, and then washed with acetone (3 × 4 mL) to separate the target compound. The acetone filtrate was evaporated under reduced pressure, and the residue was recrystallized from ethanol. Target compound 4 was obtained as a white solid, yield 73% (0.2 g).General procedure for the amination of 2,5-diformylfuran
A solution of 2,5-diformylfuran475 (0.5 g, 4 mmol) and the corresponding amine (8.4 mmol) in chloroform (15 mL) was stirred at 24 °C for 20 min, and then, sodium triacetoxyborohydride (3.4 g, 16 mmol) was added portionwise. The reaction mixture was stirred overnight, then poured into a saturated sodium bicarbonate solution and extracted with chloroform (3 × 7 mL). The combined organic phases were washed with water, dried with sodium sulfate, evaporated and dried under high vacuum. The target compounds were obtained as slightly yellow oils.2,5-Bis(N,N-diethylaminomethyl)furan10, yield 80%.
2,5-Bis(morpholinomethyl)furan11, yield 98%.
2,5-Bis(hydroxymethyl)furan (BHMF), 2
HMF 1 (5 g, 40 mmol) was dissolved in distilled water (70 mL), and a solution of sodium borohydride (1.5 g, 40 mmol) in water (15 mL) was added dropwise while stirring vigorously. The reaction mixture was stirred for 1 h at 24 °C, and then extracted with ethyl acetate (4 × 20 mL). The combined organic phases were dried with sodium sulfate, evaporated and dried under high vacuum. 2,5-Bis(hydroxymethyl)furan 2 was obtained as a white solid, yield 99% (5 g).2,5-Bis(ethoxymethyl)furan, 6
A solution of BHMF 2 (1 g, 7.8 mmol) and TsOH (20 mg) in absolute ethanol (30 mL) was heated at 60 °C for 3 h and then evaporated at reduced pressure. The residue was purified by flash chromatography (ethyl acetate/petroleum ether, 1:8). 2,5-Bis(ethoxymethyl)furan 6 was obtained as a yellow oil, yield 81% (1.16 g).
2,5-Bis(acetoxymethyl)furan, 7
To a solution of BHMF 2 (1 g, 7.8 mmol) in dry pyridine (15 mL), acetic anhydride (1.85 mL, 19.6 mmol, 2.5 eq.) was added dropwise. The reaction mixture was stirred at 24 °C for 6 h, the volatiles were evaporated under reduced pressure, 1 M HCl (15 mL) was added to the residue, and the product was extracted with chloroform (3 × 8 mL). The combined organic phases were washed with water, dried with sodium sulfate, evaporated and dried under high vacuum. 2,5-Bis(acetoxymethyl)furan 7 was obtained as a slightly yellow solid, yield 98% (1.62 g).endo-4,7-Bis(hydroxymethyl)-3a,4,7,7a-tetrahydro-1H-4,7-epoxyisoindole-1,3(2H)-dione, 3
BHMF 2 (0.2 g, 1.56 mmol) was dissolved in ethyl acetate (1.5 mL), maleimide (0.3 g, 3.1 mmol) was added, and the reaction mixture was stirred at 24 °C for 16 h. The precipitate was filtered off, washed with ethyl acetate (2 × 1 mL), and dried on a filter. Target compound 3 was obtained as a white solid, yield 83% (0.29 g).endo-1,3-(Dioxohexahydro-1H-4,7-epoxyisoindole-4,7-diyl)bis(methylene) diacetate, 9 (method A)
Diacetate 7 (0.2 g, 0.94 mmol) was dissolved in ethyl acetate, and maleimide (0.18 g, 1.9 mmol, 2 eq.) was added. The reaction mixture was stirred for 24 h at 24 °C; 10% Pd/C was added (20 mg), and the reaction mixture was placed under a hydrogen atmosphere for 8 h at 24 °C. The catalyst was filtered off and washed thoroughly with hot ethyl acetate (3 × 4 mL). The filtrate was evaporated under reduced pressure. The residue was washed with diethyl ether (3 × 3 mL) to remove succinimide. Target compound 9 was obtained as a white solid, yield 76% (0.22 mg).endo-1,3-(Dioxohexahydro-1H-4,7-epoxyisoindole-4,7-diyl)bis(methylene) diacetate, 9 (method B)
Compound 4 (64 mg, 0.28 mmol) was dissolved in dry pyridine (2 mL), acetyl chloride (120 μL, 1.68 mmol) was added dropwise, and the reaction mixture was stirred for 24 h at 24 °C. The volatiles were evaporated under reduced pressure, and the residue was poured into 1 M HCl and extracted with ethyl acetate (4 × 2 mL). The combined organic phases were dried with sodium sulfate, evaporated and dried under high vacuum. Target compound 9 was obtained as a white solid, yield 81% (71 mg).endo-4,7-Bis(ethoxymethyl)hexahydro-1H-4,7-epoxyisoindole-1,3(2H)-dione, 8
2,5-Bis(ethoxymethyl)furan 6 (0.17 g, 0.92 mmol) was dissolved in ethyl acetate (2 mL), and maleimide (0.18 g, 1.9 mmol, 2 eq.) was added. The reaction mixture was stirred for 24 h at 24 °C; 10% Pd/C was added (20 mg), and the reaction mixture was placed under a hydrogen atmosphere for 8 h at 24 °C. The catalyst was filtered off and washed thoroughly with hot ethyl acetate (3 × 4 mL). The filtrate was evaporated under reduced pressure. The residue was purified by flash chromatography (hexane–ethyl acetate, 4:1). Target compound 8 was obtained as a white solid, yield 62% (0.16 g).
endo-4,7-Bis((diethylamino)methyl)hexahydro-1H-4,7-epoxyisoindole-1,3(2H)-dione, 12
2,5-Bis(N,N-diethylaminomethyl)furan 10 (0.14 g, 0.59 mmol) was dissolved in ethyl acetate (2 mL), and maleimide (0.12 g, 1.2 mmol, 2 eq.) was added. The reaction mixture was stirred for 24 h at 24 °C; 10% Pd/C was added (20 mg), and the reaction mixture was placed under a hydrogen atmosphere for 8 h at 24 °C. The catalyst was filtered off and washed thoroughly with hot ethyl acetate (3 × 4 mL). The filtrate was evaporated under reduced pressure. The residue was purified by flash chromatography (ethyl acetate). Target compound 12 was obtained as a slightly yellow oil, yield 51% (0.11 g).endo-4,7-Bis(morpholinomethyl)hexahydro-1H-4,7-epoxy-isoindole-1,3(2H)-dione, 13
2,5-Bis(morpholinomethyl)furan 11 (0.17 g, 0.64 mmol) was dissolved in ethyl acetate (2 mL), and maleimide (0.12 g, 1.3 mmol, 2 eq.) was added. The reaction mixture was stirred for 24 h at 24 °C; 10% Pd/C was added (20 mg), and the reaction mixture was placed under a hydrogen atmosphere for 8 h at 24 °C. The catalyst was filtered off and washed thoroughly with hot ethyl acetate (3 × 4 mL). The filtrate was evaporated under reduced pressure. The residue was purified by flash chromatography (ethyl acetate). Target compound 13 was obtained as a slightly yellow oil, yield 42% (98 mg).endo-4,7-Bis(hydroxymethyl)-2-methylhexahydro-1H-4,7-epoxyisoindole-1,3(2H)-dione, 14
Compound 4 (28 mg, 0.12 mmol) was dissolved in dry DMF (1 mL), and methyl iodide (10 μL, 0.16 mmol) and potassium carbonate (34 mg, 0.24 mmol) were added successively. The reaction mixture was stirred for 16 h at 24 °C, and the volatiles were evaporated under reduced pressure. The residue was washed with ethyl acetate (3 × 1 mL). The filtrate was evaporated and dried under high vacuum. Target compound 14 was obtained as a white solid, yield 78% (23 mg).endo-4,7-Bis(methoxymethyl)-2-methylhexahydro-1H-4,7-epoxyisoindole-1,3(2H)-dione, 15
Compound 4 (55 mg, 0.24 mmol) was dissolved in dry DMF (2 mL), sodium hydride (60% dispersion in mineral oil, 32 mg, 0.79 mmol) was added and the mixture was stirred for 1 h at 24 °C. The solution of methyl iodide (91 μL, 1.44 mmol) in DMF (1 mL) was added dropwise, and the reaction mixture was stirred for 24 h at 24 °C. The volatiles were evaporated under reduced pressure, and the residue was poured into 1 M HCl and extracted with ethyl acetate (4 × 2 mL). Combined organic phases were dried with sodium sulfate, evaporated and dried under high vacuum. Target compound 15 was obtained as a white solid, yield 72% (51 mg).Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This study was supported by the Russian Science Foundation (Grant RSF 17-13-01176). The authors are grateful to Dr. Victor Khrustalev for the help with X-Ray structure analysis.Notes and references
- M. Besson, P. Gallezot and C. Pinel, Chem. Rev., 2014, 114, 1827–1870 CrossRef CAS PubMed.
- D. Li, X. Li and J. Gong, Chem. Rev., 2016, 116, 11529–11653 CrossRef CAS PubMed.
- H. Y. Luo, J. D. Lewis and Y. Roman-Leshkov, Annu. Rev. Chem. Biomol. Eng., 2016, 7, 663–692 CrossRef CAS PubMed.
- K. A. Rogers and Y. Zheng, ChemSusChem, 2016, 9, 1750–1772 CrossRef CAS PubMed.
- V. S. Sikarwar, M. Zhao, P. Clough, J. Yao, X. Zhong, M. Z. Memon, N. Shah, E. J. Anthony and P. S. Fennell, Energy Environ. Sci., 2016, 9, 2939–2977 CAS.
- X. Zhang, K. Wilson and A. F. Lee, Chem. Rev., 2016, 116, 12328–12368 CrossRef CAS PubMed.
- Z. Zhang, J. Song and B. Han, Chem. Rev., 2017, 117, 6834–6880 CrossRef CAS PubMed.
- H. Zhu, W. Luo, P. N. Ciesielski, Z. Fang, J. Y. Zhu, G. Henriksson, M. E. Himmel and L. Hu, Chem. Rev., 2016, 116, 9305–9374 CrossRef CAS PubMed.
- C. K. Williams, A. J. Ragauskas, B. H. Davison, G. Britovsek, J. Cairney, C. A. Eckert, W. J. Frederick, J. P. Hallett, D. J. Leak and C. L. Liotta, et al. , Science, 2006, 311, 484–489 CrossRef PubMed.
- R.-J. van Putten, J. C. van der Waal, E. de Jong, C. B. Rasrendra, H. J. Heeres and J. G. de Vries, Chem. Rev., 2013, 113, 1499–1597 CrossRef CAS PubMed.
- K. I. Galkin, E. A. Krivodaeva, L. V. Romashov, S. S. Zalesskiy, V. V. Kachala, J. V. Burykina and V. P. Ananikov, Angew. Chem., Int. Ed., 2016, 55, 8338–8342 CrossRef CAS PubMed.
- A. S. Kashin, K. I. Galkin, E. A. Khokhlova and V. P. Ananikov, Angew. Chem., Int. Ed., 2016, 55, 2161–2166 CrossRef CAS PubMed.
- I. Delidovich, P. J. C. Hausoul, L. Deng, R. Pfützenreuter, M. Rose and R. Palkovits, Chem. Rev., 2016, 116, 1540–1599 CrossRef CAS PubMed.
- L. Hu, L. Lin, Z. Wu, S. Zhou and S. Liu, Renewable Sustainable Energy Rev., 2017, 74, 230–257 CrossRef CAS.
- L. V. Romashov and V. P. Ananikov, Org. Biomol. Chem., 2016, 14, 10593–10598 CAS.
- W. R. Galloway, A. Isidro-Llobet and D. R. Spring, Nat. Commun., 2010, 1, 80 Search PubMed.
- A. J. Cruz Cabeza, G. M. Day, W. D. S. Motherwell and W. Jones, Cryst. Growth Des., 2007, 7, 100–107 CAS.
- S. R. Vasudevan, J. B. Moore, Y. Schymura and G. C. Churchill, J. Med. Chem., 2012, 55, 7054–7060 CrossRef CAS PubMed.
- A. Kumar and K. Y. J. Zhang, Methods, 2015, 71, 26–37 CrossRef CAS PubMed.
- A. Stimac, M. Sekutor, K. Mlinaric-Majerski, L. Frkanec and R. Frkanec, Molecules, 2017, 22, 297 CrossRef PubMed.
- D. S. Tan, Nat. Chem. Biol., 2005, 1, 74–84 CrossRef CAS PubMed.
- J. Clardy and C. Walsh, Nature, 2004, 432, 829–837 CrossRef CAS PubMed.
- S. Kortagere, M. D. Krasowski and S. Ekins, Trends Pharmacol. Sci., 2009, 30, 138–147 CrossRef CAS PubMed.
- Z. Wang, in Comprehensive Organic Name Reactions and Reagents, John Wiley & Sons, Inc., 2010, DOI:10.1002/9780470638859.conrr190.
- K. C. Nicolaou, S. A. Snyder, T. Montagnon and G. Vassilikogiannakis, Angew. Chem., Int. Ed., 2002, 41, 1668–1698 CrossRef CAS PubMed.
- X. Ding, S. T. Nguyen, J. D. Williams and N. P. Peet, Tetrahedron Lett., 2014, 55, 7002–7006 CrossRef CAS PubMed.
- F. I. Zubkov, V. P. Zaytsev, E. S. Puzikova, E. V. Nikitina, V. N. Khrustalev, R. A. Novikov and A. V. Varlamov, Chem. Heterocycl. Compd., 2012, 48, 514–524 CrossRef CAS.
- G. Papeo, H. Posteri, D. Borghi, A. A. Busel, F. Caprera, E. Casale, M. Ciomei, A. Cirla, E. Corti, M. D'Anello, M. Fasolini, B. Forte, A. Galvani, A. Isacchi, A. Khvat, M. Y. Krasavin, R. Lupi, P. Orsini, R. Perego, E. Pesenti, D. Pezzetta, S. Rainoldi, F. Riccardi-Sirtori, A. Scolaro, F. Sola, F. Zuccotto, E. R. Felder, D. Donati and A. Montagnoli, J. Med. Chem., 2015, 58, 6875–6898 CrossRef CAS PubMed.
- G. Caillot, S. Hegde and E. Gras, New J. Chem., 2013, 37, 1195–1200 RSC.
- M. E. Jung and L. J. Street, J. Am. Chem. Soc., 1984, 106, 8327–8329 CrossRef CAS.
- S. Higson, F. Subrizi, T. D. Sheppard and H. C. Hailes, Green Chem., 2016, 18, 1855–1858 RSC.
- A. E. Settle, L. Berstis, N. A. Rorrer, Y. Roman-Leshkov, G. T. Beckham, R. M. Richards and D. R. Vardon, Green Chem., 2017, 19, 3468–3492 RSC.
- T. J. Connolly, J. L. Considine, Z. Ding, B. Forsatz, M. N. Jennings, M. F. MacEwan, K. M. McCoy, D. W. Place, A. Sharma and K. Sutherland, Org. Process Res. Dev., 2010, 14, 459–465 CrossRef CAS.
- G. Hirai and M. Sodeoka, Acc. Chem. Res., 2015, 48, 1464–1473 CrossRef CAS PubMed.
- Y. Baba, N. Hirukawa, N. Tanohira and M. Sodeoka, J. Am. Chem. Soc., 2003, 125, 9740–9749 CrossRef CAS PubMed.
- M. Shen, M. Y. Wu, L. P. Chen, Q. Zhi, F. R. Gong, K. Chen, D. M. Li, Y. Wu, M. Tao and W. Li, Sci. Rep., 2015, 5, 11836 CrossRef PubMed.
- J.-Y. Wu, C.-D. Kuo, C.-Y. Chu, M.-S. Chen, J.-H. Lin, Y.-J. Chen and H.-F. Liao, Molecules, 2014, 19, 6911 CrossRef PubMed.
- C. E. Puerto Galvis, L. Y. Vargas Méndez and V. V. Kouznetsov, Chem. Biol. Drug Des., 2013, 82, 477–499 CAS.
- E. Mahmoud, D. A. Watson and R. F. Lobo, Green Chem., 2014, 16, 167–175 RSC.
- H. Zabed, J. N. Sahu, A. Suely, A. N. Boyce and G. Faruq, Renewable Sustainable Energy Rev., 2017, 71, 475–501 CrossRef CAS.
- J. A. Posada, A. D. Patel, A. Roes, K. Blok, A. P. C. Faaij and M. K. Patel, Bioresour. Technol., 2013, 135, 490–499 CrossRef CAS PubMed.
- C. Zeng, H. Seino, J. Ren, K. Hatanaka and N. Yoshie, Polymer, 2013, 54, 5351–5357 CrossRef CAS.
- T. Ikezaki, R. Matsuoka, K. Hatanaka and N. Yoshie, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 216–222 CrossRef CAS.
- Y. S. Ryu, K. W. Oh and S. H. Kim, Macromol. Res., 2016, 24, 874–880 CrossRef CAS.
- C. Zeng, H. Seino, J. Ren, K. Hatanaka and N. Yoshie, Macromolecules, 2013, 46, 1794–1802 CrossRef CAS.
- S. Verlinden, S. Ballet and G. Verniest, Eur. J. Org. Chem., 2016, 5807–5812 CrossRef CAS.
- V. P. Kashparova, E. A. Khokhlova, K. I. Galkin, V. M. Chernyshev and V. P. Ananikov, Russ. Chem. Bull. Int. Ed., 2015, 64, 1069–1073 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1557878 and 1557879. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7gc02211e |
| This journal is © The Royal Society of Chemistry 2017 |