
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Polypyrrole salts and bases: superior conductivity of nanotubes and their stability towards the loss of conductivity by deprotonation
Jaroslav
Stejskal
*a,
Miroslava
Trchová
a,
Patrycja
Bober
a,
Zuzana
Morávková
a,
Dušan
Kopecký
b,
Martin
Vrňata
b,
Jan
Prokeš
c,
Martin
Varga
c and
Elizaveta
Watzlová
c
aInstitute of Macromolecular Chemistry, Academy of Sciences of the Czech Republic, 162 06 Prague 6, Czech Republic. E-mail: stejskal@imc.cas.cz
bFaculty of Chemical Engineering, University of Chemistry and Technology, 166 28 Prague 6, Czech Republic
cCharles University in Prague, Faculty of Mathematics and Physics, 180 00 Prague 8, Czech Republic
First published on 1st September 2016
Abstract
Polypyrrole nanotubes exhibit conductivity of tens S cm−1 which is one of the highest among the current conducting polymers. They are thus superior to the common globular form with the conductivity of units of S cm−1 or lower. The conductivity of both forms is reduced after treatment with alkalis but still remains high, units of S cm−1 and 10−2 S cm−1, respectively. The deprotonation, which is responsible for conductivity reduction, is discussed on the basis of salt–base transition in polypyrrole. It is not fully reversible, and the reprotonation with acids recovers the conductivity only in part. The role of methyl orange, which was used to support the formation of nanotubes, is proposed to be similar to that of surfactants. FTIR and Raman spectroscopies prove that methyl orange is strongly bound to polypyrrole in its acid form, and an “insertion” mechanism is proposed to explain the resistance towards the deprotonation of nanotubes. The spectra also illustrate that the molecular structure of nanotubular polypyrrole is preserved even under highly alkaline conditions at a pH close to 14, where the globular form becomes damaged. Polypyrrole, especially in its nanotubular form, is of promise in applications requiring electrical conduction even under neutral or alkaline conditions, where other conducting polymers, such as polyaniline, lose their exploitable conductivity.
Introduction
Conducting polymers, such as polypyrrole or polyaniline, find uses especially in energy storage devices and recently also in biomedical applications that exploit their intrinsic conductivity, responsivity, and electrochemical switching. They are popular because of their easy and economic preparation by the oxidation of corresponding monomers. Some of their applications, however, are limited by the pH at which conducting polymers are used. This applies especially to those operating under physiological or alkaline conditions, where the conducting polymer salts convert to non-conducting polymer bases. There are valued properties of such conducting polymers: (1) high conductivity, (2) stability towards the deprotonation and consequent loss of conductivity under alkaline conditions, (3) electrochemical activity, (4) responsivity to external stimuli, (5) catalytic and electrocatalytic properties, and (6) long-term environmental stability. The control and understanding of their underlying molecular structure and morphology is an important goal.The present study is focused on polypyrrole. The typical conductivity of polypyrrole prepared by the oxidation of pyrrole with iron(III) salts is 10−2 S cm−1.1 When the polypyrrole was prepared in the presence of surfactants, the conductivity was higher by about two orders of magnitude, and reached 3.1 S cm−1 for sodium dodecylbenzenesulfonate,1 9.9 S cm−1 for dodecylbenzenesulfonic acid,1 12.3 S cm−1 for sodium bis(2-ethylhexyl)sulfosuccinate,2 26.1 S cm−1 for sodium dodecylbenzenesulfonate3 and 42 S cm−1 in the presence of sodium n-alkylnaphthalenesulfonate.4 The surfactants thus have a beneficial influence on the conductivity of polypyrrole.
The conductivity was reduced by several orders of magnitude when polypyrrole was converted to corresponding bases in ammonia solutions, and even more in sodium hydroxide solutions.1,5–11 The certain level of conductivity is still maintained even under strongly alkaline conditions. For example, the conductivity of globular polypyrrole, 0.012 S cm−1, was reduced to 3.7 × 10−5 S cm−1 after treatment with 1 M ammonium hydroxide.12 Similarly, the conductivity of globular polypyrrole sulfate was reduced from 0.24 S cm−1 to 3.8 × 10−6 S cm−1 after being immersed in 1 M ammonium hydroxide, and to 6.4 × 10−10 S cm−1 in 1 M sodium hydroxide.1 The conductivity increased again after treatment with acid solutions but its recovery has not been complete.5,6,11 The deprotonation/reprotonation cycle in polypyrrole was exploited in ammonia sensors,13–15 silver recovery16 and sodium-ion batteries.17 Such salt–base conversion imposes restrictions to some applications, e.g., to textiles coated with conducting polymers that need to be washed.18 Both forms, a salt and a base, differ not only in conductivity but also in density and thermal conductivity,19 electron spin concentration20 or the third-order optical non-linearity,21 and other electrical and optical properties.
When polypyrrole was prepared in the presence of a dye, such as methyl orange (MO), polypyrrole nanotubes were obtained12,22–24 instead of a classical globular morphology. The conductivity of nanotubular polypyrrole was of the order of tens S cm−1,23,25i.e. considerably higher compared with the conductivity of the globular form. The preparation and application of polypyrrole nanotubes has therefore recently become a fast developing research direction,26–31 also due to the interesting nanostructured morphology. The conductivity of polypyrrole nanotubes decreased from 60 S cm−1 only to 0.012 S cm−1 after suspension in 1 M ammonium hydroxide,23i.e. nanotubes were still conducting under alkaline conditions.12,32 Similar trend was observed in the composites of polypyrrole nanotubes with silver.12,33 The high conductivity of polypyrrole nanotubes and its resistance towards the deprotonation and consequent loss of conductivity under alkaline conditions inspired the present study aimed at the discussion of salt–base transition in various forms of polypyrrole.
Experimental
Preparation of globular and nanotubular polypyrrole
Polypyrrole nanotubes were prepared by the oxidation of 0.05 M pyrrole with 0.05 M iron(III) chloride in the presence of 0.005 M of methyl orange. 3.35 g of pyrrole and 1.64 g methyl orange, sodium 4-[(4-dimethylamino)phenylazo]benzenesulfonate, were dissolved in water and the volume was adjusted to 0.5 L. 13.5 g of iron(III) chloride hexahydrate was similarly dissolved in water to 0.5 L of solution. Both solutions were mixed at room temperature to start the oxidation of pyrrole. Next day, polypyrrole precipitate was collected on filter, rinsed with 0.2 M hydrochloric acid, copious amounts of ethanol, and left to dry in air. For the purpose of present study, globular polypyrrole was prepared in the same manner, only in the absence of MO.Deprotonation and reprotonation of polypyrrole
Polypyrrole hydrochloride obtained after preparation was deprotonated by immersion in excess of 1 M ammonium hydroxide. Weighed amount of polypyrrole in sintered glass filter was placed in ammonia solution and left at rest for 24 h. The filter was then removed, rinsed with ammonia solution, and ethanol. The mass of solids was determined after drying. The ammonia solutions were evaporated and the mass of collected solids, referred to below as a filtrate, was determined.A part of the deprotonated polypyrrole was immersed again in excess of 1 M hydrochloric acid for 24 h to test the reprotonation ability. The solids were then collected, rinsed with acid solution, and ethanol. After drying, the change in mass was evaluated.
Characterization
Room temperature conductivity was determined by a four-point method in van der Pauw arrangement using a Keithley 220 Programmable Current Source, a Keithley 2010 Multimeter as a voltmeter and a Keithley 705 Scanner equipped with a Keithley 7052 Matrix Card.Energy-dispersive X-ray spectra (EDS) were recorded by Quantax 200 spectrometer with a XFlash 6|10 detector (Bruker Corp.) using 15 kV of accelerating voltage of primary electron beam in Mira LMH (Tescan) scanning electron microscope.
Fourier-transform infrared (FTIR) spectra of the powders dispersed in potassium bromide pellets were registered using a Thermo Nicolet NEXUS 870 FTIR Spectrometer with a DTGS TEC detector in the 400–4000 cm−1 wavenumber region.
Raman spectra were recorded on a Renishaw InVia Reflex Raman microspectrometer. The spectra were excited with a near infrared diode 785 nm laser. A research-grade Leica DM LM microscope with an objective magnification × 50 was used to focus the laser beam on the sample placed on an X–Y motorized sample stage. The scattered light was analyzed with a spectrograph using a holographic grating 1200 lines per mm. A Peltier-cooled CCD detector (576 × 384 pixels) registered the dispersed light.
Results and discussion
Molecular structure
The molecular structures of protonated polypyrrole (polypyrrole salt) proposed in the literature somewhat vary but there is agreement on the structure of polypyrrole base obtained after deprotonation, which is composed of both the oxidized and reduced pyrrole constitutional units (Fig. 1).7,9,13 The localization of positive charges on polymer chain, as well as the presence of unpaired spins, polarons, detected by electron paramagnetic resonance, is still open to discussion. For the purpose of present study, the rearrangement of electrons within polypyrrole chain leading to the formation of charge carriers can be proposed (Fig. 2).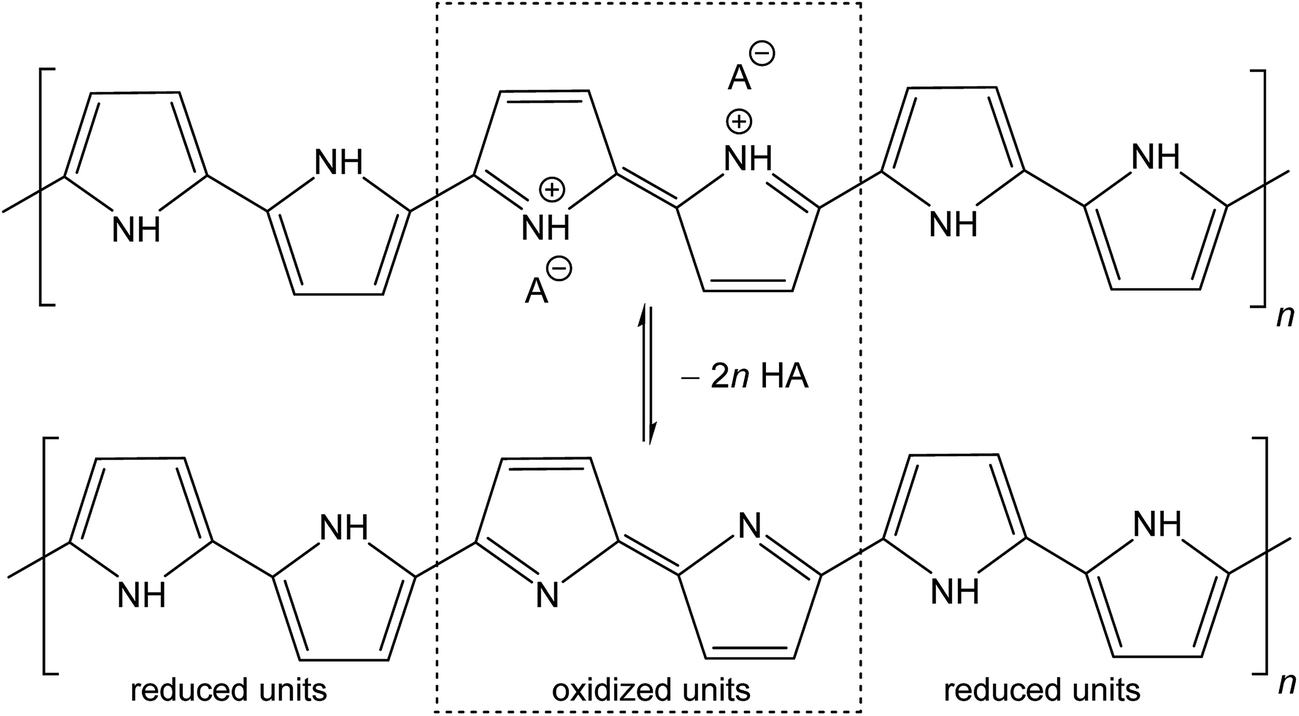 | ||
| Fig. 1 Conducting polypyrrole salt converts to non-conducting polypyrrole base under alkaline conditions. HA is an arbitrary acid, A− is a corresponding counter-ion, here chloride. | ||
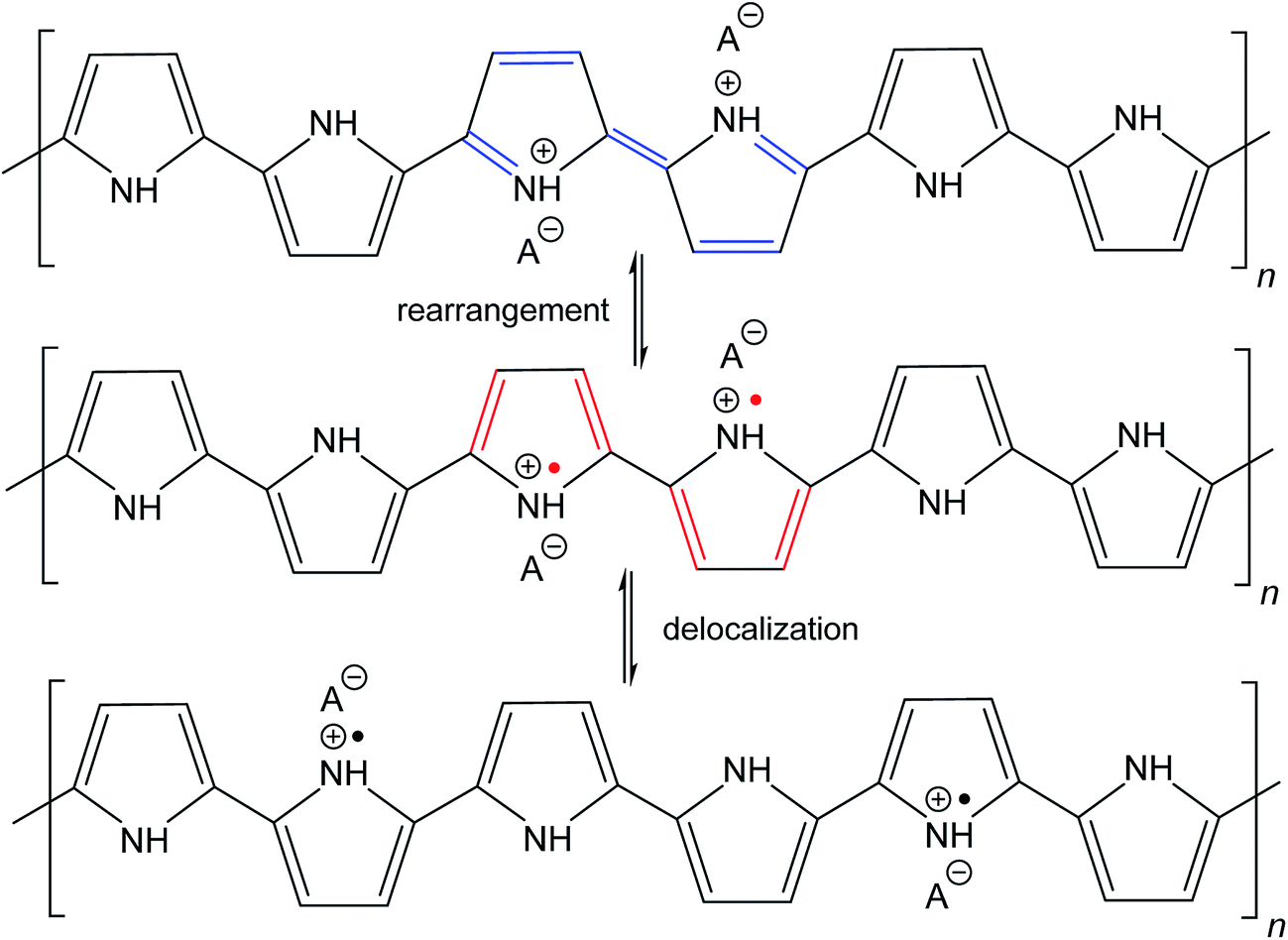 | ||
| Fig. 2 The rearrangement of electrons in polypyrrole salt may generate bipolarons and finally polarons by delocalization over the polymer chain. Polarons act as charge carriers. | ||
Conductivity
Polypyrrole exists as a conducting salt obtained after preparation and less conducting or insulating base (Fig. 1) produced by subsequent deprotonation after the treatment with alkalis.1,10,12 Both the level of conductivity and the resistance towards the deprotonation were improved when the polymerization of pyrrole was carried out in the presence of anionic surfactants, and the conductivity of polypyrrole increased by one or two orders of magnitude.1,34 The presence of anionic surfactants thus generally (1) increased the conductivity of polypyrrole and (2) improved the level of conductivity under alkaline conditions. Surfactant anions were proved to participate as counter-ions in polypyrrole (Fig. 1).1,9 The ionic bond responsible for the salt formation seems to be sterically protected by the hydrophobic surfactant part from the direct access of alkalis, hydroxyl ions, thus reducing the extent of polypyrrole deprotonation to a corresponding base (Fig. 1).The similar observation has recently been made when polypyrrole was prepared in the presence of MO dye (Fig. 3). The conductivity of polypyrrole hydrochloride was (1) considerably higher compared with the preparation in the absence of MO, and (2) the resistance to the loss of conductivity after treatment with alkalis was considerably improved (Table 1) as mentioned in the Introduction. In addition, the morphology distinctly changed from globular to nanotubular (Fig. 4). Nanotubes have also higher specific surface area,32 55 m2 g−1, than the globular form, 4.3 m2 g−1.
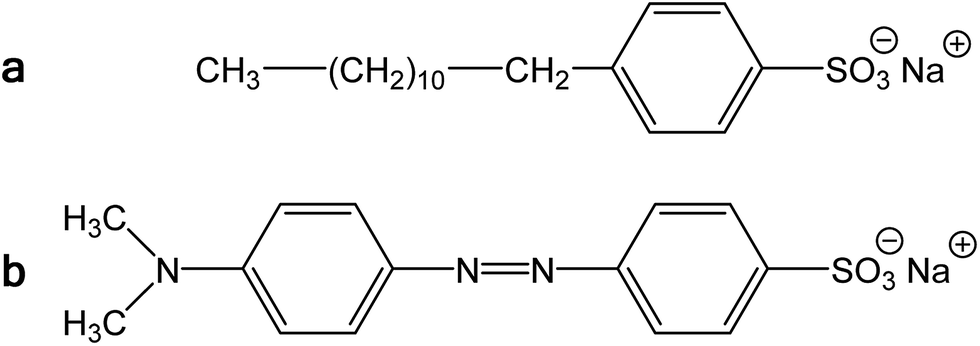 | ||
| Fig. 3 (a) Sodium dodecylbenzenesulfonate and (b) methyl orange, sodium 4-[(4-dimethylamino)phenylazo]benzenesulfonate. | ||
| Conductivity (S cm−1) | Deprotonation loss, wt% | Ammonium salt, wt% | Reprotonation gain, wt% | |||
|---|---|---|---|---|---|---|
| Original | D | R | ||||
| Globular | 1.55 | 0.012 | 0.026 | 9.35 | 17.3 | 8.06 |
| Nanotubes | 49.8 | 1.42 | 3.64 | 9.13 | 13.0 | 9.69 |
 | ||
| Fig. 4 Illustration of globular polypyrrole (left) and polypyrrole nanotubes (right). | ||
We conclude that the dye has about the similar effect on the preparation of polypyrrole as a surfactant, but its performance with respect to conductivity is even better. In aqueous media, the classical surfactant, sodium dodecylbenzenesulfonate has a tendency towards the formation of micelles with a hydrophobic core composed of dodecyl substituents. In fact, MO is similarly composed of a large hydrophobic moiety and hydrophilic sulfonate group (Fig. 3). Both types of molecules thus can sterically protect the bonding of sulfonic group to nitrogen atoms in polypyrrole chain. The marked difference, however, consists in the molecule rigidity, which is flexible in the former case and stiff in the case of MO. While dodecylbenzenesulfonate would produce amorphous micellar aggregates, MO may be better suited to form more organized objects of liquid-crystalline type. The improved organization of polypyrrole chains, enforced already to the pyrrole monomers by surfactants, then leads to the improved conductivity of this polymer.
Nanostructures where polypyrrole chains are organized are expected to have improved conductivity, and brush-like chain-ordering in electropolymerized polypyrrole films may also serve as an example. The reported conductivity was 15–36 S cm−1,9 26–28 S cm−1,7 34–52 S cm−1,35 and ≈100 S cm−1,6i.e. three orders of magnitude higher compared with globular polypyrrole. The conductivity in the first case9 was reduced to 0.3 S cm−1 after treatment with 0.5 M sodium hydroxide at 100 °C, i.e. the resistance towards the deprotonation in alkalis was also good.
Mass balance and elemental analysis
The deprotonation scheme (Fig. 1) has frequently been proposed for polypyrrole9,10,12,14,24,33,34,36 in the discussion of its properties. In the present case, the number of reduced polypyrrole units (–NH– in base) is higher than the number oxidized polypyrrole units (![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N– in base) as assumed in early reports.9,13 The protonation of oxidized units is preferred to reduced units as confirmed in experimental studies.9,17 It should be noted that some papers consider also the protonation of amine groups.37 The hypothesis that the current counter-ions are not only simply removed during deprotonation but replaced with hydroxide anions have also been proposed.20
N– in base) as assumed in early reports.9,13 The protonation of oxidized units is preferred to reduced units as confirmed in experimental studies.9,17 It should be noted that some papers consider also the protonation of amine groups.37 The hypothesis that the current counter-ions are not only simply removed during deprotonation but replaced with hydroxide anions have also been proposed.20
Generally, however, the deprotonation is associated with the reduction of mass.6 In the case of polypyrrole hydrochloride, the mass fraction of hydrochloric acid in polypyrrole salt calculated according to the formula in Fig. 1 is 15.8 wt%. The lower mass decrease by 9.4 and 9.1 wt% were found in present experiment for globular and nanotubular polypyrroles, respectively (Table 1). If all acid were neutralized with ammonium hydroxide, 1 g of polypyrrole hydrochloride should yield 232 mg of ammonium chloride. Ammonium chloride is indeed recovered as white crystals after evaporation of ammonium hydroxide solution, as identified by FTIR spectra below. Its quantity, however, is again lower, 173 and 130 mg per 1 g of globular and nanotubular polypyrrole salts. This means that either (1) a part of acid still remains attached to polypyrrole backbone and the deprotonation was not complete or (2) the amount of hydrochloric acid was smaller than that proposed. The former hypotheses seem to be supported by mass balance and elemental analysis by EDS.
The Cl/N atomic ratio expected by the proposed stoichiometry (Fig. 1) is 2/6 = 0.33. EDS yields ratio 0.21 for globular form. This suggests that globular polypyrrole is protonated less than expected. After deprotonation with 1 M ammonia solution, the Cl/N ratios is reduced from 0.21 to 0.12 but not to zero, as expected for complete deprotonation. Only in stronger alkali, in 1 M sodium hydroxide, Cl/N ratio drops to 0.02.
For polypyrrole nanotubes the Cl/N ratio was 0.30, i.e. close to theoretical expectation, and the value was reduced to 0.03 after treatment with ammonia solution, again indicating agreement with the proposed deprotonation (Fig. 1). This atomic ratio may also be affected by the presence of methyl orange. Polypyrrole nanotubes contain in addition sulfur from the sulfonic groups in MO. The atomic S/N ratio 0.10 was reduced only to 0.06 after being exposed to ammonia solutions, and changed only marginally to 0.05 in 1 M sodium hydroxide. The former hypothesis of incomplete deprotonation is also supported by conductivity measurements that confirm still a good level of conductivity, 10−2 S cm−1, and by the observation that the conductivity is further reduced after treatment with a stronger alkali, sodium hydroxide.38
The increase in mass after reprotonation of deprotonated samples with 1 M hydrochloric acid about compensates the loss after deprotonation (Table 1). The conductivity partly recovers for globular polypyrrole (Table 1). A similar deprotonation/reprotonation behavior was reported but the conductivity gradually decreased after each cycle.5,6 The literature on this process have been thoroughly reviewed.13 The recovery of conductivity in polypyrrole nanotubes has also never been complete. This is probably due to the damage of chain organization associated with mass loss and consequent volume reduction during deprotonation.
Infrared spectra during deprotonation
Infrared spectrum of the globular polypyrrole salt (spectrum G in Fig. 5a) exhibits the main bands which were previously described.1,10,32 The shape of the spectrum of polypyrrole after deprotonation with 1 M ammonium hydroxide changed32 (spectrum D). Absorption in the region above 1600 cm−1 decreased and a weak band at 1700 cm−1 appeared. It corresponds to the presence of carbonyl group previously attributed to the nucleophilic attack of water during the preparation.1 The maximum of the band observed in the spectrum of the polypyrrole salt at about 1542 cm−1 (C–C stretching vibrations in the pyrrole ring) shifted to 1553 cm−1, which corresponds to the deprotonation of the pyrrole rings (spectrum D). The maximum of the band of C–N stretching vibrations in the pyrrole rings moved from 1455 to 1478 cm−1. The broad band with a maximum at about 1305 cm−1 (C–H or C–N in-plane deformation modes) remains at the same position. The maximum of the band associated with the breathing vibrations of the pyrrole rings observed at 1167 cm−1 in the spectrum of polypyrrole salt is situated at 1180 cm−1 in the spectrum of deprotonated polypyrrole. The peak at 1092 cm−1 connected with N–H+ deformation vibrations only slightly sifted to 1096 cm−1 after deprotonation. The sharp band at 1045 cm−1 (C–H and N–H in-plane deformation vibrations) remains at the same position. The band at 905 cm−1 (the C–H out-of-plane deformation vibrations of the ring) shifted to 912 cm−1 wavelengths after deprotonation. | ||
| Fig. 5 Infrared spectra of (a) globular (G) and (b) nanotubular polypyrrole (NT) before and after deprotonation with 1 M ammonium hydroxide (D), and reprotonation with 1 M hydrochloric acid (R). Spectra of the solids collected from filtrate after deprotonation (F) and of neat methyl orange (MO) are included for comparison. | ||
The shape of the spectrum of the sample obtained after reprotonation with 1 M hydrochloric acid (spectrum R) dramatically changed. It exhibits very small absorption caused by compact stone-like structure which was difficult to disperse in potassium bromide pellets. As a consequence the aliphatic impurities (detected at 2925 and 2856 cm−1) and a relatively strong absorption bands in the region of stretching and bending vibrations of water molecules at about 3422 cm−1 and 1633 cm−1 coming from potassium bromide pellet were detected in the spectrum. Nevertheless, the main bands of protonated polypyrrole can be observed in the spectrum. The spectrum of the filtrate obtained after deprotonation (spectrum F) corresponds well to the spectrum of ammonium chloride, viz. N–H vibrations in ammonium cation, as it was expected in the Fig. 1.
Infrared spectra of the nanotubular polypyrrole (NT in Fig. 5b) and its deprotonated (D) and reprotonated (R) forms exhibit small absorption, which is a result of their compact stone-like structure and they were difficult to disperse in potassium bromide pellets. This may be connected with strong hydrogen bonding in the samples. In addition to the bands of aliphatic impurities (at 2925 and 2856 cm−1) and of humidity (at 3440 and 1633 cm−1), the band of carbonyl group at about 1722 cm−1 can be detected. The main bands of polypyrrole are present in all spectra. The band of C–N stretching vibrations in the pyrrole rings observed at 1455 cm−1 in the spectra of globular polypyrrole splits into two maxima at 1470 and 1443 cm−1, similarly as it was observed in the presence of surfactants.1 The maximum of the band connected with the breathing vibrations of the pyrrole rings observed at 1163 cm−1 in the spectrum of polypyrrole nanotubes keeps the same position after deprotonation. Some small peaks in the spectra of deprotonated and reprotonated samples marked with asterisk correspond to the presence of methyl orange (spectrum MO).
Please note that MO is, except for some traces, absent in the spectrum of the filtrate obtained after deprotonation (spectrum F). This means that MO is not simply bound to polypyrrole in its yellow salt form (Fig. 3b) as a counter-ion (Fig. 6a), but might be “inserted” into polypyrrole salt in its red acid form (Fig. 6b). Such construction would explain the absence of MO in the filtrate after treatment with ammonia, and a consequent resistance of nanotubes towards the complete deprotonation associated with the loss of polypyrrole conductivity.
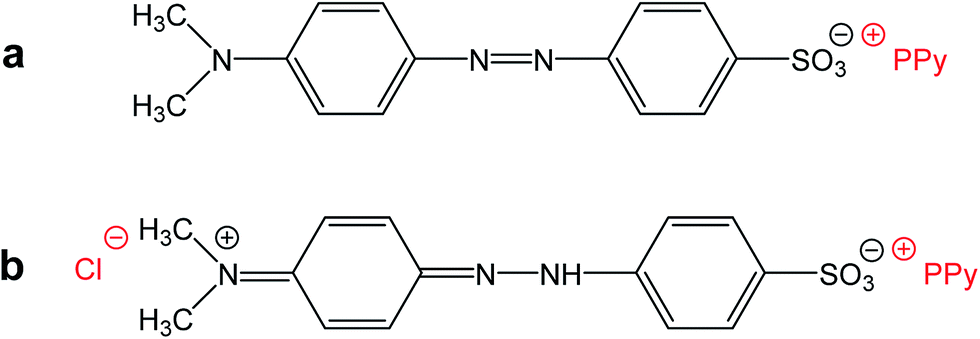 | ||
| Fig. 6 (a) Methyl orange can act as a simple counter-ion in polypyrrole (PPy) or (b) it can be “inserted” into polypyrrole hydrochloride in its acid form. | ||
Raman spectra during deprotonation
Raman spectroscopy is a convenient tool for the study of structural bonding in conjugated polymers, such as polypyrrole. A formula of polypyrrole salt includes the oxidized and reduced units in its molecular structure (Fig. 1). In reality, we have to consider the presence of localized structural defects associated with polymer backbone which are connected with the existence of polarons (radical cations) and bipolarons (dications diradicals) (Fig. 2). They can be detected in the Raman spectra of protonated polypyrrole.In the spectrum of globular polypyrrole salt (spectrum G in Fig. 7a) we observe the bands of CC in-ring of C–C inter-ring vibrations and of the stretching vibrations in cation of the polypyrrole backbone with a maximum at 1598 cm−1.39,40 The maximum of this band is shifted to 1607 cm−1 in the spectrum of polypyrrole after deprotonation with 1 M ammonium hydroxide (spectrum D), which corresponds to the CC backbone stretching vibrations in short conjugation length, a dication.39 The band of C–C and CN stretching skeletal vibrations is situated at 1492 cm−1. This band is shifted to 1495 cm−1 after deprotonation. The double peak observed in the spectrum of polypyrrole salt at about 1376 and 1333 cm−1 corresponds to ring-stretching vibrations of pyrrole. The peak situated at the higher wavenumbers is assigned to the C–C ring stretching vibrations of oxidized polypyrrole. The second maximum was assigned to the neutral C–C ring stretching vibrations of reduced units.39 This peak increased after deprotonation and it has been connected with less conducting polypyrrole.41 A shoulder at 1405 cm−1 appeared in the spectrum of deprotonated polypyrrole which corresponds to the C–N stretching vibrations in the neutral units.32 The band of C–H antisymmetric in-plane bending vibrations with a maximum at about 1243 cm−1 remained practically at the same position after deprotonation. In the double peak situated at 1088 and 1053 cm−1 in the spectrum of polypyrrole salt, the higher peak is attributed to the C–H in-plane deformation vibrations in radical cation units in protonated polypyrrole. The conductivity of polypyrrole is linked to the increase in intensity of this peak corresponding to oxidized polypyrrole units.41 The peak situated at lower wavenumber increased after deprotonation, and it is assigned to the C–H in-plane deformations in the neutral units of polypyrrole base39 (Fig. 1). The peak at 971 cm−1 in the spectrum of polypyrrole salt is attributed to the C–C ring deformation vibrations with radical cation.40 It moved to 985 cm−1 in the spectrum of deprotonated polypyrrole, and it belongs to the ring deformation in neutral units.39 The band situated at 934 cm−1 in the spectrum of polypyrrole salt moved to 922 cm−1 after deprotonation and increased in intensity. It is assigned to the C–C ring deformation vibrations within dication unit.41 After reprotonation of deprotonated samples in the solutions of acids the Raman spectrum returns to the original state (spectrum R).
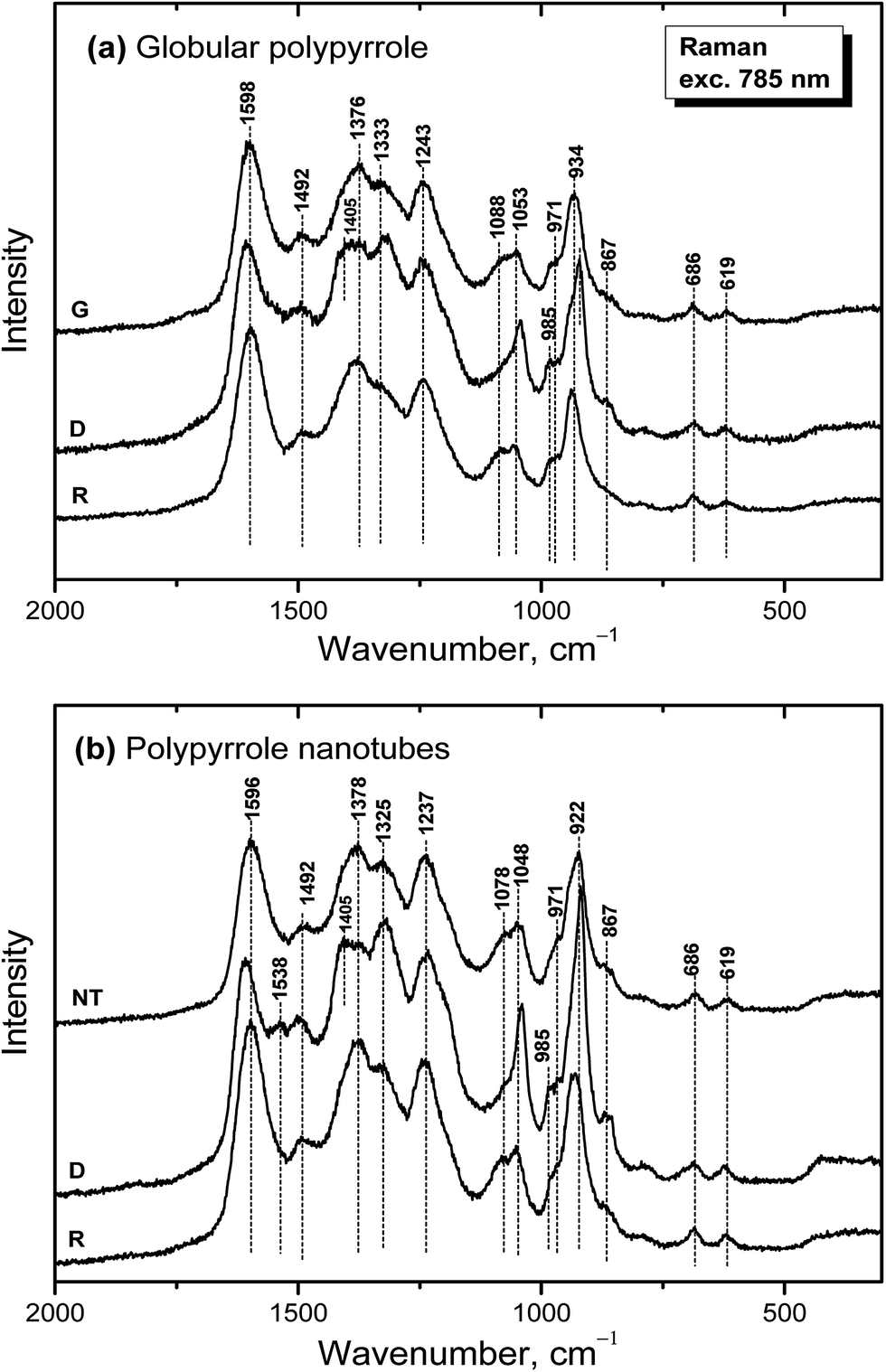 | ||
| Fig. 7 Raman spectra of (a) globular (G) and (b) nanotubular (NT) polypyrrole before and after deprotonation with 1 M ammonium hydroxide (D), and reprotonation with 1 M hydrochloric acid (R). | ||
In the Raman spectrum of nanotubular polypyrrole salt we observe the same bands practically at the same positions as in the spectrum of globular polypyrrole (spectrum NT in Fig. 7b). This means that both forms have the identical molecular structure and differ in the organization of polymer chains. After deprotonation of nanotubular polypyrrole, a new band located at 1538 cm−1 appears. It may be connected with an increase of the CN stretching skeletal vibrations in deprotonated units (Fig. 1). The spectrum exhibits a higher increase in the intensity of the band of the neutral C–C ring stretching vibrations of reduced units situated at 1325 cm−1 and also of the band of the C–C ring-deformation vibrations with dication situated at 922 cm−1 than in case of globular polypyrrole. This may be connected with interaction between polypyrrole and MO. After reprotonation of the nanotubular polypyrrole with 1 M hydrochloric acid, we recognize the bands with intensities corresponding to the protonated polypyrrole salt (spectrum R). The bands of neat MO are not observed in the Raman spectra measured with near-infrared diode 785 nm excitation laser.
The dependence of conductivity on pH
The above discussion of deprotonation of polypyrrole with 1 M ammonium hydroxide (1.7 wt% NH3) corresponded to the value of pH 11.6. The effect of the polypyrrole treatment with solutions of ammonium and sodium hydroxides of various concentrations in the pH range 10–14 on the conductivity has also been investigated (Fig. 8).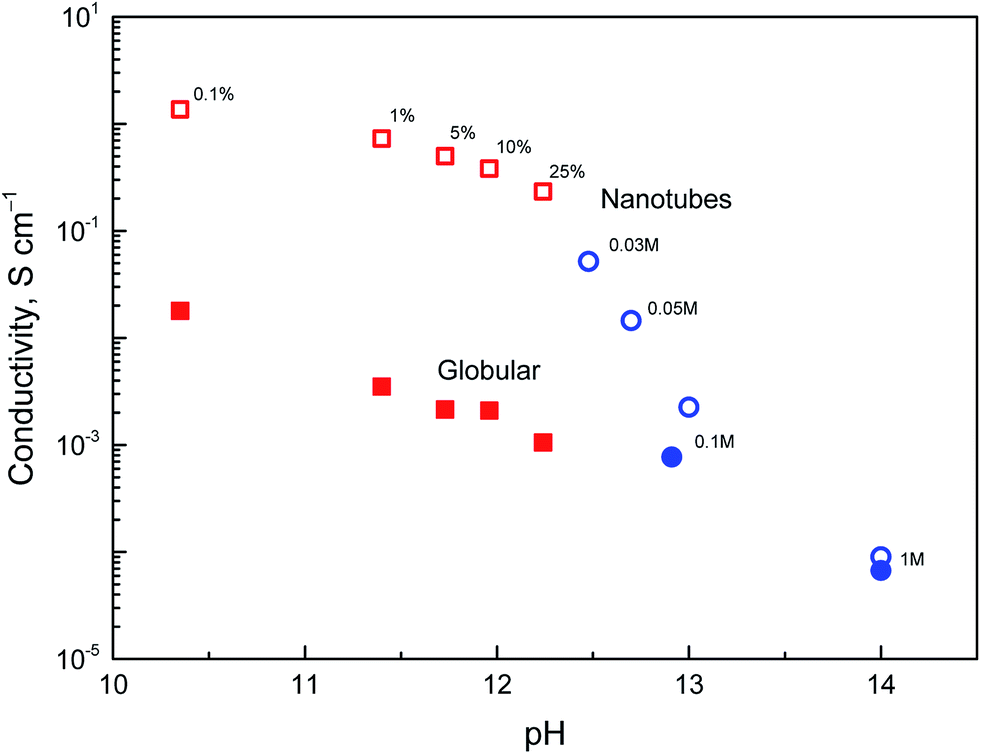 | ||
| Fig. 8 Dependence of conductivity of polypyrrole nanotubes (open symbols) and globular polypyrrole (full symbols) on pH of deprotonation medium constituted by aqueous solution of ammonium hydroxide (wt%, squares) or sodium hydroxide (mol L−1, circles). | ||
The conductivity of nanotubes treated with ammonia solution stays above 0.1 S cm−1, even at 25% ammonia concentration and only a moderate decrease is observed as the ammonia concentration increased. The same trend was found for globular polypyrrole, only the conductivity was lower by about two orders of magnitude compared with nanotubes.
In the solutions of sodium hydroxide the conductivity of nanotubes was reduced as methyl orange was liberated and hydrolytic changes in the molecular structure of polypyrrole took place, as confirmed by spectroscopic method below. At pH > 13, the conductivity of both types of polypyrrole is comparable. Some conductivity was maintained even after the exposure of polypyrrole nanotubes to 5 M and 10 M sodium hydroxide, viz. 5.1 × 10−6 and 1.2 × 10−6 S cm−1, i.e. at the level which is far from being negligible. Polypyrrole is thus suited even for the application operating at neutral or alkaline conditions where other conducting polymers, such as polyaniline, fail. It should be mentioned that the reprotonation of alkali-treated polypyrroles with acids does not recover the original conductivity, and the increase in conductivity is marginal, if any.
The dependence of infrared spectra on pH
When globular polypyrrole treated with alkali solutions in the pH range 10–14 is considered, the differences in the infrared spectra are relatively small (Fig. 9a). They correspond to the consecutive deprotonation of polypyrrole as pH increases. The maximum of the band of C–N stretching vibration in the pyrrole rings situated at 1478 cm−1 increased during deprotonation, the other maximum of this complex band at 1455 cm−1 is now well detected and a new shoulder at 852 cm−1 appeared in the spectra.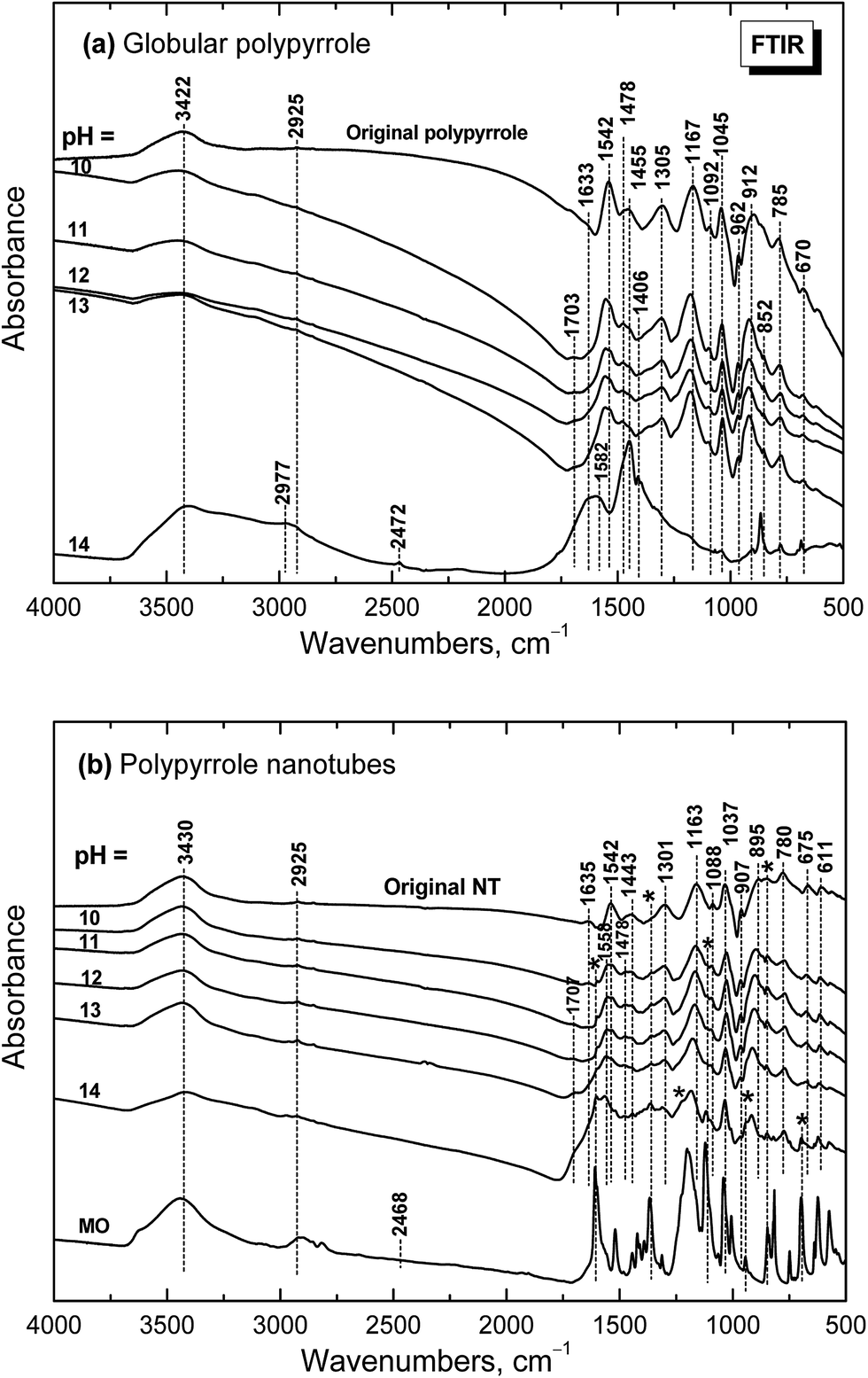 | ||
| Fig. 9 Infrared spectra of (a) globular and (b) nanotubular polypyrrole before and after exposure to the media of various pH. | ||
Under strongly alkaline medium represented by 1–10 M sodium hydroxide with pH close to 14, the spectrum of polypyrrole dramatically changed, as the chemical changes in polypyrrole chain took place. A broad band with maxima at 3422 and 2977/2925 cm−1 and a new peak at 2472 cm−1 are observed in the spectrum. A broad band with maximum at 1582 cm−1 with a shoulder at 1633 cm−1 and a strong peak at 1455 cm−1 dominate in the spectrum. A sharp maximum located at 852 cm−1 is well observed. As it was shown in electrochemical experiments provided in alkaline medium, the potential needed for overoxidation of polypyrrole is significantly reduced42 and the formation of hydroxyl and carbonyl groups by nucleophilic attack takes place,43 followed by alkaline hydrolysis to carboxylic salts and amines.42,44,45 All these products are also reflected in the present spectrum of polypyrrole exposed to strongly alkaline medium. The bands of original polypyrrole practically disappeared in the spectrum. Similar changes have previously been observed for electrochemical overoxidation of polypyrrole.45–47
Infrared spectra of the nanotubular polypyrrole (spectrum NT in Fig. 9b) are well resolved as the samples are better dispersible in potassium pellets compared with globular form. The peaks of MO marked with asterisk increased during consecutive deprotonation with increasing pH. The main bands of polypyrrole are detected in all spectra till pH 13 as it was observed in the spectra of globular polypyrrole. The situation, however, is different when pH reaches the value close to 14. Contrary to the globular polypyrrole, in the case of nanotubular polypyrrole we observe well all the bands of polypyrrole (spectrum for pH 14 in Fig. 9b) on the background corresponding to the fully oxidized globular polypyrrole (spectrum for pH 14 in Fig. 9a). Some sharp peaks of MO in its salt form are well visible in the spectrum of polypyrrole (spectrum MO). This means that the chemical stability of polypyrrole nanotubes under strongly alkaline conditions is better compared with globular form. This may be due to the fact that MO remains, at least in part, incorporated in polypyrrole as proposed in Fig. 6.
The dependence of Raman spectra on pH
Raman spectra of globular polypyrrole excited with a 785 nm laser excitation line correspond to the above described spectrum of globular polypyrrole obtained after deprotonation with 1 M ammonium hydroxide (Fig. 7a) at pH 10–13 (Fig. 10a). Changes in the spectra follow the consecutive deprotonation of polypyrrole with increasing pH. The increase in the number of the CN bonds in the neutral units is reflected in the new peaks appearing at 1548 cm−1 and 1410 cm−1. In strongly alkaline medium with pH approaching 14, the spectrum dramatically changed as in the case of FTIR spectra. We observed a broad fluorescence band corresponding to the excitation of the overoxidized units of polypyrrole.
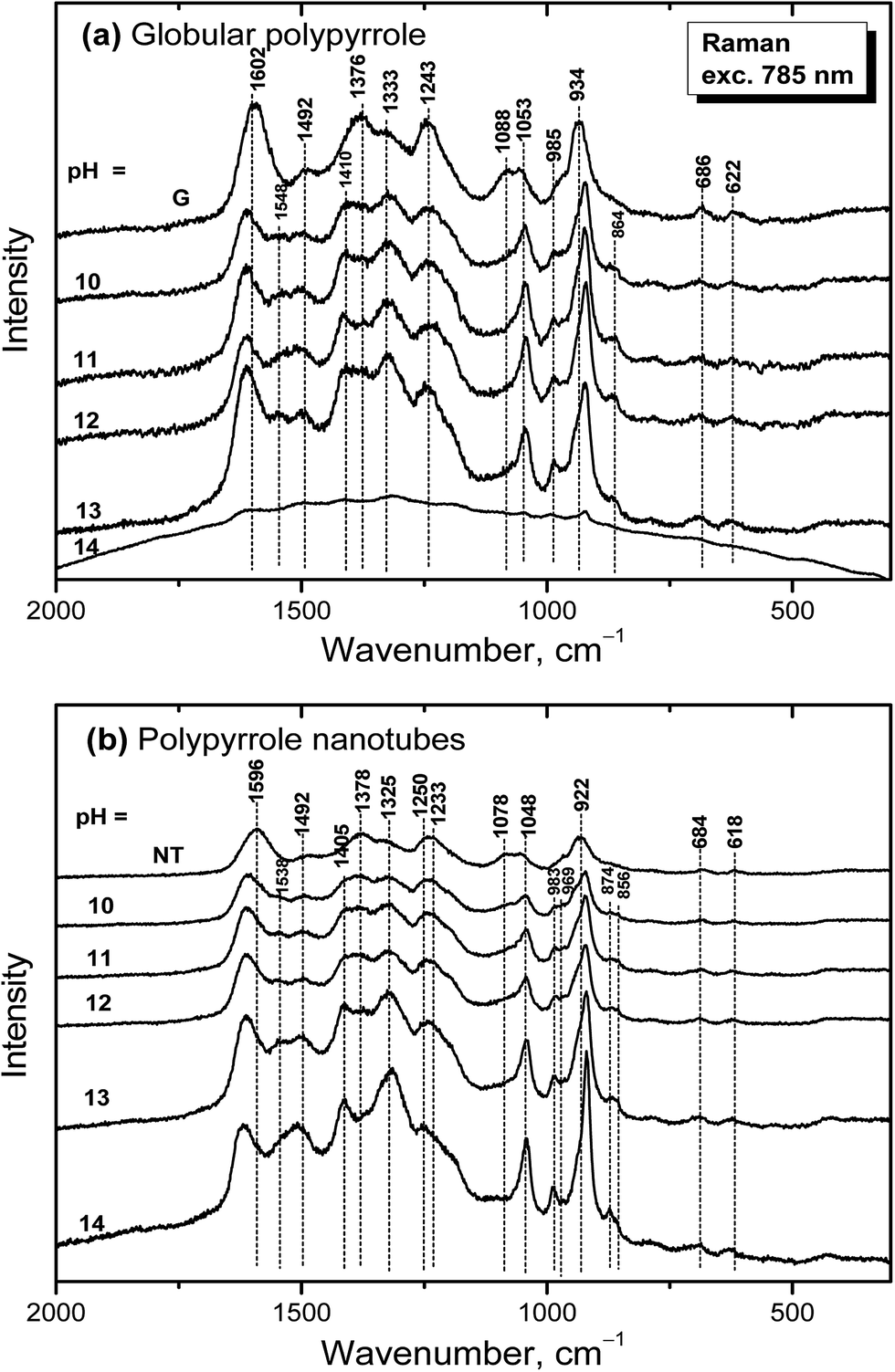 | ||
| Fig. 10 Raman spectra of original (a) globular (G) and (b) nanotubular polypyrrole (NT) and after exposure to the medium of various pH. | ||
Raman spectra of alkali-treated polypyrrole nanotubes (spectrum NT, Fig. 10b) in the medium with pH 10–13 correspond to the spectrum of nanotubular polypyrrole obtained after deprotonation with 1 M ammonium hydroxide (Fig. 7). When pH reached the value 14, the bands at about 1495, 1405, 1325, 1048, 983, 922 and 874 cm−1 increased. Contrary to the spectrum of globular polypyrrole, where no bands of polypyrrole have been detected, the spectrum of nanotubular polypyrrole preserved its features even after treatment with strongly alkaline medium. A fluorescence background corresponding to the presence of MO is present in the spectrum of polypyrrole.
Conclusions
The conductivity of globular polypyrrole salt was reduced from the units of S cm−1 to 10−2 S cm−1 after treatment with 1 M ammonia solution at pH 11.6. The conductivity of polypyrrole nanotubes, 50 S cm−1, was higher compared with globular form and after exposure to ammonia solution the conductivity dropped to the units of S cm−1. This means that polypyrrole, in particular in its nanotubular form, retains most of its conductivity under moderate alkaline conditions, up to pH 13. This may be of importance for applications operating under physiological or in alkaline region. FTIR and Raman spectra prove that the molecular structure of globular polypyrrole is irreversibly damaged at pH 14. The conductivity of both polypyrroles is further reduced under strongly alkaline conditions after treatment with 5 or 10 M sodium hydroxide solutions. The reprotonation of polypyrrole, i.e. the treatment by solutions of strong acids after exposure to alkalis, does not recover the original level of conductivity and the conductivity increase is only partial.The model molecular structure expecting the protonation of each third polypyrrole constitutional unit, i.e. Cl/N = 0.33 atomic ratio for polypyrrole hydrochloride, reasonably well reflects the experimental reality. The polypyrrole was deprotonated only in part to polypyrrole base in ammonia solution. Methyl orange, which was used as a template in the preparation of nanotubes, stayed incorporated in polypyrrole and may take a role of counter-ions resistant to the deprotonation, similarly like surfactants reported in the literature. The “insertion” of acid form of methyl orange into polypyrrole hydrochloride is proposed to account for better conductivity and stability towards the deprotonation.
Acknowledgements
The authors thank the Czech Science Foundation (13-00270S and 14-10279S) for financial support.References
- M. Omastová, M. Trchová, J. Kovářová and J. Stejskal, Synth. Met., 2003, 138, 447–455 CrossRef
.
- M. Omastová, M. Trchová, J. Pionteck, J. Prokeš and J. Stejskal, Synth. Met., 2004, 143, 153–161 CrossRef
- Y. Kudoh, Synth. Met., 1996, 79, 17–22 CrossRef CAS
- Y. Kudoh, K. Akami and Y. Matsuya, Synth. Met., 1998, 95, 191–196 CrossRef CAS
- O. Ingenäs, R. Erlandsson, C. Nylander and I. Lundström, J. Phys. Chem. Solids, 1984, 45, 427–432 CrossRef
- H. Münstedt, Polymer, 1986, 27, 899–904 CrossRef
- Q. B. Pei and R. Y. Qian, Synth. Met., 1991, 45, 35–48 CrossRef CAS
- R. Y. Qian, Q. B. Pei and Z. T. Huang, Makromol. Chem., 1991, 192, 1263–1273 CrossRef CAS
- M. Forsyth and V. T. Truong, Polymer, 1995, 36, 725–730 CrossRef CAS
- N. V. Blinova, J. Stejskal, M. Trchová, J. Prokeš and M. Omastová, Eur. Polym. J., 2007, 43, 2331–2341 CrossRef CAS
- J. Y. Ji, X. H. Yu, P. Cheng, Q. Zhang, F. P. Du, L. Li and S. M. Shang, J. Macromol. Sci., Part B: Phys., 2015, 54, 1122–1131 CrossRef CAS
- J. Škodová, D. Kopecký, M. Vrňata, M. Varga, J. Prokeš, M. Cieslar, P. Bober and J. Stejskal, Polym. Chem., 2013, 4, 3610–3616 RSC
- K. Maksymiuk, Electroanalysis, 2006, 18, 1537–1551 CrossRef CAS
- M. Joulazadeh and A. H. Navarchian, Synth. Met., 2015, 210, 404–411 CrossRef CAS
- Y. Li, H. T. Ban and M. J. Yang, Sens. Actuators, B, 2016, 224, 449–457 CrossRef CAS
- N. L. Pickup, J. S. Shapiro and D. K. Y. Wong, Anal. Chim. Acta, 1998, 364, 41–51 CrossRef CAS
- D. W. Su, J. Q. Zhang, S. X. Dou and G. X. Wang, Chem. Commun., 2015, 51, 16092–16095 RSC
- P. Bober, J. Stejskal, I. Šeděnková, M. Trchová, L. Martinková and J. Marek, Appl. Surf. Sci., 2015, 356, 737–741 CrossRef CAS
- A. Rudajevová, M. Varga, J. Prokeš, J. Kopecká and J. Stejskal, Acta Phys. Pol., A, 2015, 128, 730–736 CrossRef
- G. Zotti, G. Schiavon, S. Zecchin and G. D'Aprano, Synth. Met., 1996, 80, 35–39 CrossRef CAS
- A. J. Wang, W. Zhao and W. Yu, J. Mol. Struct., 2015, 1099, 291–296 CrossRef CAS
- J. Kopecká, D. Kopecký, M. Vrňata, P. Fitl, J. Stejskal, M. Trchová, P. Bober, Z. Morávková, J. Prokeš and I. Sapurina, RSC Adv., 2014, 4, 1551–1558 RSC
- J. Stejskal, I. Sapurina, M. Trchová, I. Šeděnková, J. Kovářová, J. Kopecká and J. Prokeš, Chem. Pap., 2015, 69, 1341–1349 CrossRef CAS
- I. Sapurina, J. Stejskal, I. Šeděnková, M. Trchová, J. Kovářová, J. Hromádková, J. Kopecká, M. Cieslar, A. Abu El-Nasr and M. M. Ayad, Synth. Met., 2016, 214, 14–22 CrossRef CAS
- M. Varga, J. Kopecká, Z. Morávková, I. Křivka, M. Trchová, J. Stejskal and J. Prokeš, J. Polym. Sci., Part B: Polym. Phys., 2015, 53, 1147–1159 CrossRef CAS
- H. Y. Mi, X. G. Zhang, X. G. Ye and S. D. Yang, J. Power Sources, 2008, 176, 403–409 CrossRef CAS
- J. Peng, L. H. Qiu, C. T. Pan, C. C. Wang, S. M. Shang and F. Yan, Electrochim. Acta, 2012, 75, 399–405 CrossRef
- D. L. Wei, X. Lin, L. Li, S. M. Shang, M. C. W. Yuen, G. P. Yan and X. H. Yu, Soft Matter, 2013, 9, 2832–2836 RSC
- S. B. Ye and J. C. Feng, ACS Appl. Mater. Interfaces, 2014, 6, 9671–9679 CAS
- M. Li, L. L. Yang and Q. Zhang, RSC Adv., 2015, 5, 1191–1197 RSC
- J. Upadhyay, A. Kumar, B. Gogoi and A. K. Buragohain, Mater. Sci. Eng., C, 2015, 54, 8–13 CrossRef CAS PubMed
- G. Ćirić-Marjanović, S. Mentus, I. Pašti, N. Gavrilov, J. Krstić, J. Travas-Sejdic, L. T. Strover, J. Kopecká, Z. Morávková, M. Trchová and J. Stejskal, J. Phys. Chem. C, 2014, 118, 14770–14784 Search PubMed
- E. Alekseeva, P. Bober, M. Trchová, I. Šeděnková, J. Prokeš and J. Stejskal, Synth. Met., 2015, 209, 105–111 CrossRef CAS
- M. Omastová, P. Bober, Z. Morávková, N. Peřinka, M. Kaplanová, T. Syrový, J. Hromádková, M. Trchová and J. Stejskal, Electrochim. Acta, 2014, 122, 296–302 CrossRef
- C. O. Yoon, M. Reghu, D. Moses and A. J. Heeger, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49, 10851–10863 CrossRef CAS
- M. Omastová, K. Mosnáčková, M. Trchová, E. N. Konyushenko, J. Stejskal, P. Fedorko and J. Prokeš, Synth. Met., 2010, 160, 701–707 CrossRef
- T. F. Otero and J. Rodríguez, J. Electroanal. Chem., 1994, 379, 513–516 CrossRef
- J. Stejskal, M. Omastová, S. Fedorova, J. Prokeš and M. Trchová, Polymer, 2003, 44, 1353–1358 CrossRef CAS
- K. Crowley and J. Cassidy, J. Electroanal. Chem., 2003, 547, 75–82 CrossRef CAS
- S. Gupta, J. Raman Spectrosc., 2008, 39, 1343–1355 CrossRef CAS
- Y.-C. Liu, J. Electroanal. Chem., 2004, 571, 255–264 CrossRef CAS
- Y. F. Li and R. Y. Quian, Electrochim. Acta, 2000, 45, 1727–1731 CrossRef CAS
- Y. F. Li and R. Y. Quian, J. Electroanal. Chem., 1993, 362, 267–272 CrossRef CAS
- I. Rodriguez, B. R. Scharifker and J. Mostany, J. Electroanal. Chem., 2000, 491, 117–125 CrossRef CAS
- H. L. Ge and G. J. Qi, Polymer, 1994, 35, 504–508 CrossRef CAS
- J. Mostany and B. R. Scharfiker, Electrochim. Acta, 1997, 42, 291–301 CrossRef CAS
- J. Mostany and B. R. Scharfiker, Synth. Met., 1997, 87, 179–183 CrossRef CAS
| This journal is © The Royal Society of Chemistry 2016 |