
Coumarin–chalcone hybrids: promising agents with diverse pharmacological properties
Han Wei*a,
Jinlan Ruanb and
Xiaojian Zhang*a
aDepartment of Pharmacy, The First Affiliated Hospital of Zhengzhou University, 1 Jiansedong Road, Zhengzhou 450052, Henan, China. E-mail: weihan548@126.com; firstph@163.com
bTongji School of Pharmacy, Huazhong University of Science and Technology, 13 Hangkong Road, Wuhan 430030, Hubei, China
First published on 19th January 2016
Abstract
Naturally and synthetically derived hybrid molecules are an attractive source for therapeutic agent development due to their dual or multiple modes of action and other advantages. Coumarin and chalcone, two important classes of natural products affording diverse pharmacological activities, make themselves ideal blocks for building a coumarin–chalcone hybrid scaffold as a bioactive agent. Provoked by the promising medicinal application of such hybrids, the scientific community has reported dozens of coumarin–chalcone hybrids with a wide spectrum of biological properties including anticancer, antimicrobial, antimalarial, antioxidant, antitubercular and so on, through synthetic hybridization strategy or characterization from natural sources. The present mini review provides a systematic summary on natural and synthetic agents of coumarin–chalcone hybrids on the basis of their therapeutic properties. It is expected to assist medicinal chemists in the effective and successful development of coumarin–chalcone hybrids.
Introduction
Naturally and synthetically derived hybrid molecules integrate two or more pharmacophoric units possessing different modes of action into one molecular scaffold.1 The multifunctional attributes of these hybrids offer interesting multiple biological activities, high selectivity, favorable pharmacokinetics, and/or avoid undesired drawbacks such as side effects and low oral bioavailability, which make hybrid molecules a rationally attractive source for current drug discovery.1–3 The pharmacological significance of compounds from natural sources inspires the scientific community to obtain hybrid molecules based on different types of natural product with diverse structures and versatile biological effects.4–7Coumarin and chalcone are two important classes of bioactive natural products extensively studied by medicinal chemists.8–10 Coumarin is a large group of vital lactone containing fused benzene and 2-pyrone skeletons (Fig. 1) that are widely distributed in plants. Natural and synthetic molecules based on the coumarin skeleton have been employed as medicinal agents due to their outstanding therapeutic potential such as anticancer, anticoagulant, antitubercular, antimicrobial, anti-inflammatory, anti-HIV, analgesic, anticonvulsant, antiplatelet, antifungal, antiviral, antibacterial, and antimalarial activities.11 Chalcone (1,3-diaryl-2-propen-1-ones, Fig. 1), one of bioactive secondary metabolites belonging to flavonoid family, raises enormous interests for their broad spectrum of biological properties, such as anticancer, anti-inflammatory, anti-neurodegeneration, antibacterial, antimalarial and antioxidant activities.12,13
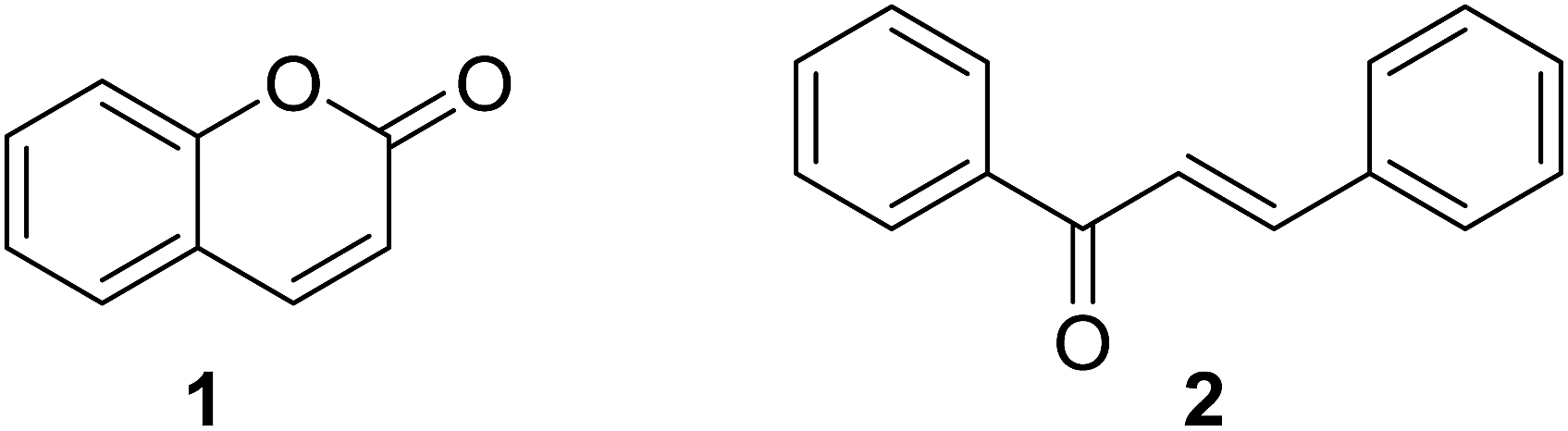 | ||
| Fig. 1 Skeletons of coumarin (1) and chalcone (2). | ||
Multifunctional features of coumarin and chalcone make them ideal blocks to create a hybrid scaffold affording interesting pharmacological properties. To this end, extensive efforts have been made on design and synthesis of coumarin–chalcone hybrids. Moreover, dozens of natural coumarin–chalcone hybrids have been reported by several groups, as listed in Table 1.
| Natural coumarin–chalcone hybrids | Sources | Natural coumarin–chalcone hybrids | Sources |
|---|---|---|---|
| a *Absolute configuration was not determined. | |||
 |
C. interruptus18 |  |
P. calomelanos20 |
| C. parasiticus19 | |||
 |
C. parasiticus21 | 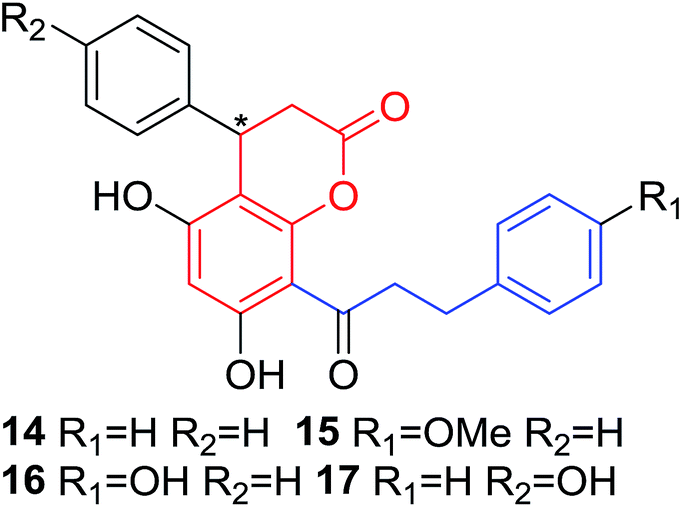 |
P. calomelanos20,22 |
 |
C. interruptus18 | 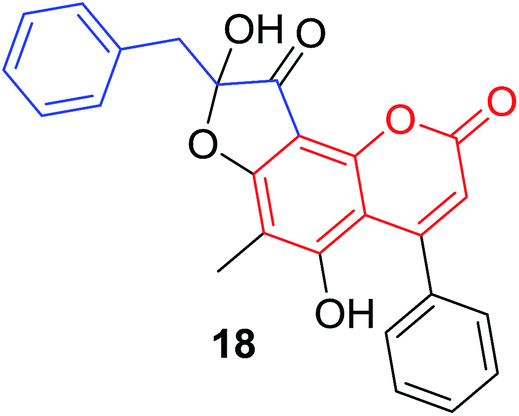 |
C. interruptus18 |
 |
C. parasiticus21 | 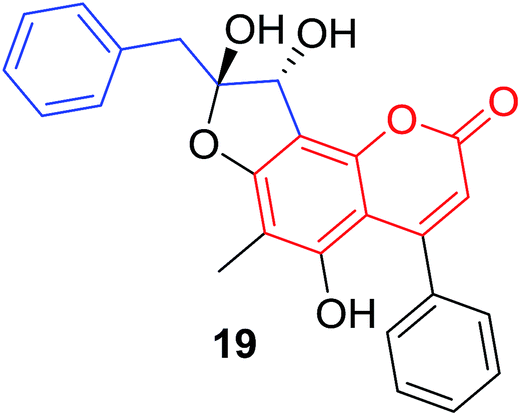 |
C. interruptus18 |
 |
P. trifoliata23 | 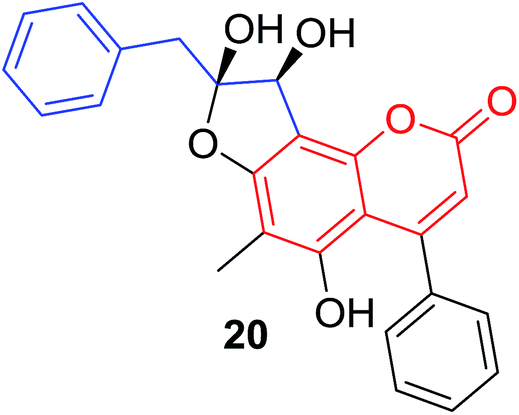 |
C. interruptus18 |
| C. parasiticus21 | |||
 |
P. trifoliata23 | 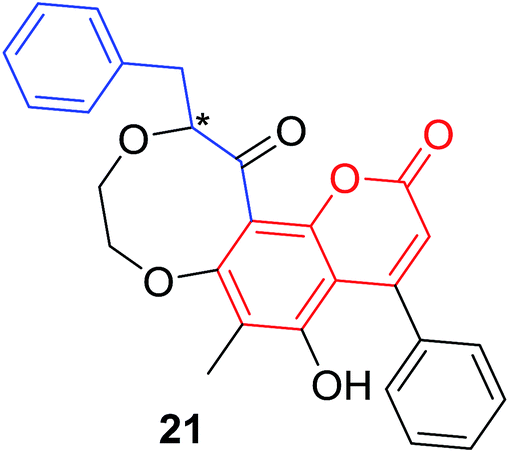 |
C. interruptus18 |
 |
P. trifoliata23 | ||
Definition of coumarin–chalcone hybrids is equivocal due to high variation in their combination form, therefore notion of coumarin–chalcone hybrid covered here just simply follows the reported literatures14–17 and mainly can be sorted into two categories: (1) full entities of coumarin and chalcone (or their derivatives, e.g. bicoumarin, dihydrocoumarin and dihydrochalcone) are fused or combined with a linker; (2) skeleton of coumarin is fused with part of chalcone moiety (e.g. benzaldehyde and styrene).
Hybrids in this class have been proven to possess diverse and impressive pharmacological activities such as anticancer, antimicrobial, antimalarial, antioxidant and antitubercular activities, indicting the development value and therapeutic potential of coumarin–chalcone hybrids.
The promising properties of coumarin–chalcone hybrid inspire us to provide a systematic summary and insight on the progress (1979–2015, patents were not covered) in this field. In this mini review, hybrids are classified on the basis of their pharmacological applications, and structural features, candidate natural sources, rational hybridization strategies, structure–activity relationship (SAR) and some mechanisms of action are discussed. This mini review is expected to assist effective study and successful development of coumarin–chalcone hybrids as promising therapeutic agents.
Anticancer activity
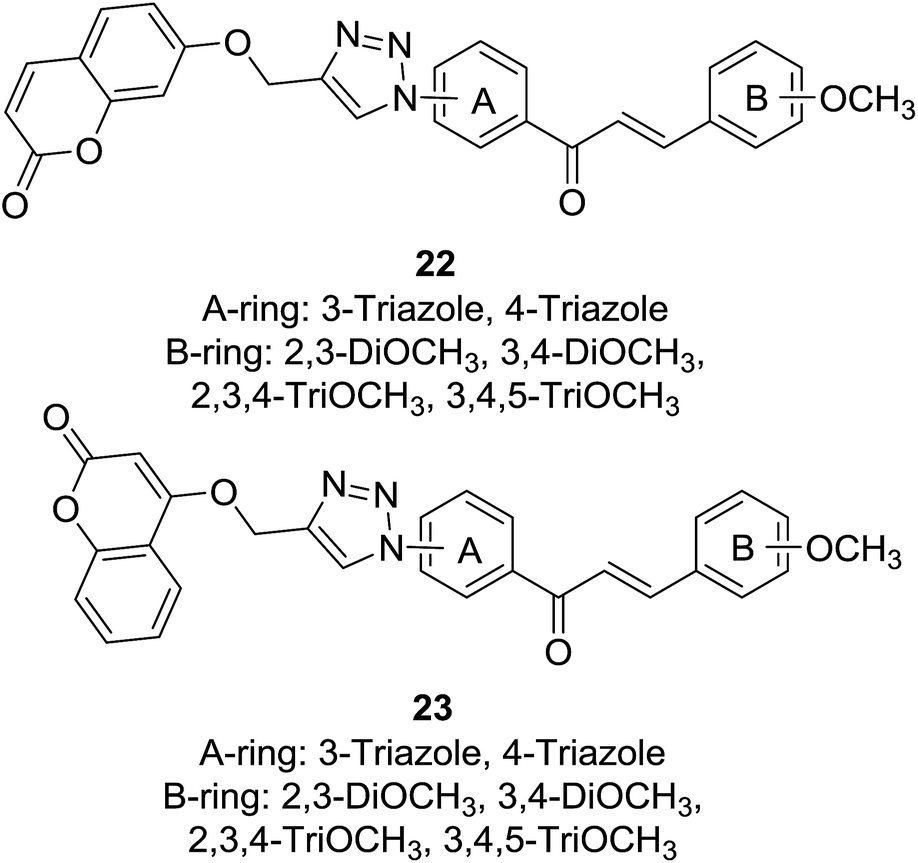
Cancer is one of the most deadly diseases threatening health of population worldwide nowadays. Although many efforts have been made to develop novel and effective anticancer agents, current clinic chemotherapy often has to face treatment failure caused by drug resistance to many anticancer drugs. The “combined chemotherapy-like” effect of anticancer hybrids due to their dual or multiple mechanisms of action is considered as an alternative solution to overcome drawbacks encountered by conventional anticancer drugs.24,25 Anticancer activities of coumarin and chalcone have been highlighted by many reviews.26–28 Accordingly, presence of coumarin–chalcone hybrids has encouraged medicinal chemists to develop novel anticancer agents bearing this hybrid scaffold.Pingaew and coworkers synthesized two sets of coumarin–chalcone derivatives (22 and 23, Fig. 2) linked by a 1,2,3-triazole ring through the azide/alkyne dipolar cycloaddition and their cytotoxicity was screened in vitro against a panel of four cancer cell lines, including HuCCA-1 (cholangiocarcinoma), HepG2 (hepatocellular carcinoma), A549 (lung carcinoma) using MTT assay and MOLT-3 (lymphoblastic leukemia) using XTT assay.15
 | ||
| Fig. 2 Coumarin–chalcone hybrids linked by 1,2,3-triazole ring. | ||
It was revealed that majority of the hybrid compounds showed cytotoxicity against MOLT-3 cells with IC50 values ranging between 0.53 and 79.49 μM, among them compounds bearing trimethoxy-substituted B-ring selectively inhibited growth of MOLT-3 cells. The most potential compound bearing 4-triazole on A-ring and 2,3-DiOCH3 on B-ring of 23 displayed significant cytotoxicity against HuCCA-1 (IC50 = 4.81 μM), followed by A549 (IC50 = 7.95 μM) and HepG2 (IC50 = 8.18 μM) with non-toxic to non-cancerous vero cell line. SAR study of these hybrids indicated that their cytotoxicity highly depended on functionalities on rings A and B as well as the coumaryl group. Further molecular docking investigation showed that promising anticancer potential of the coumarin–chalcone hybrids was possibly related to dual inhibition of α- and β-tubulins at GTP and colchicine binding sites, respectively.
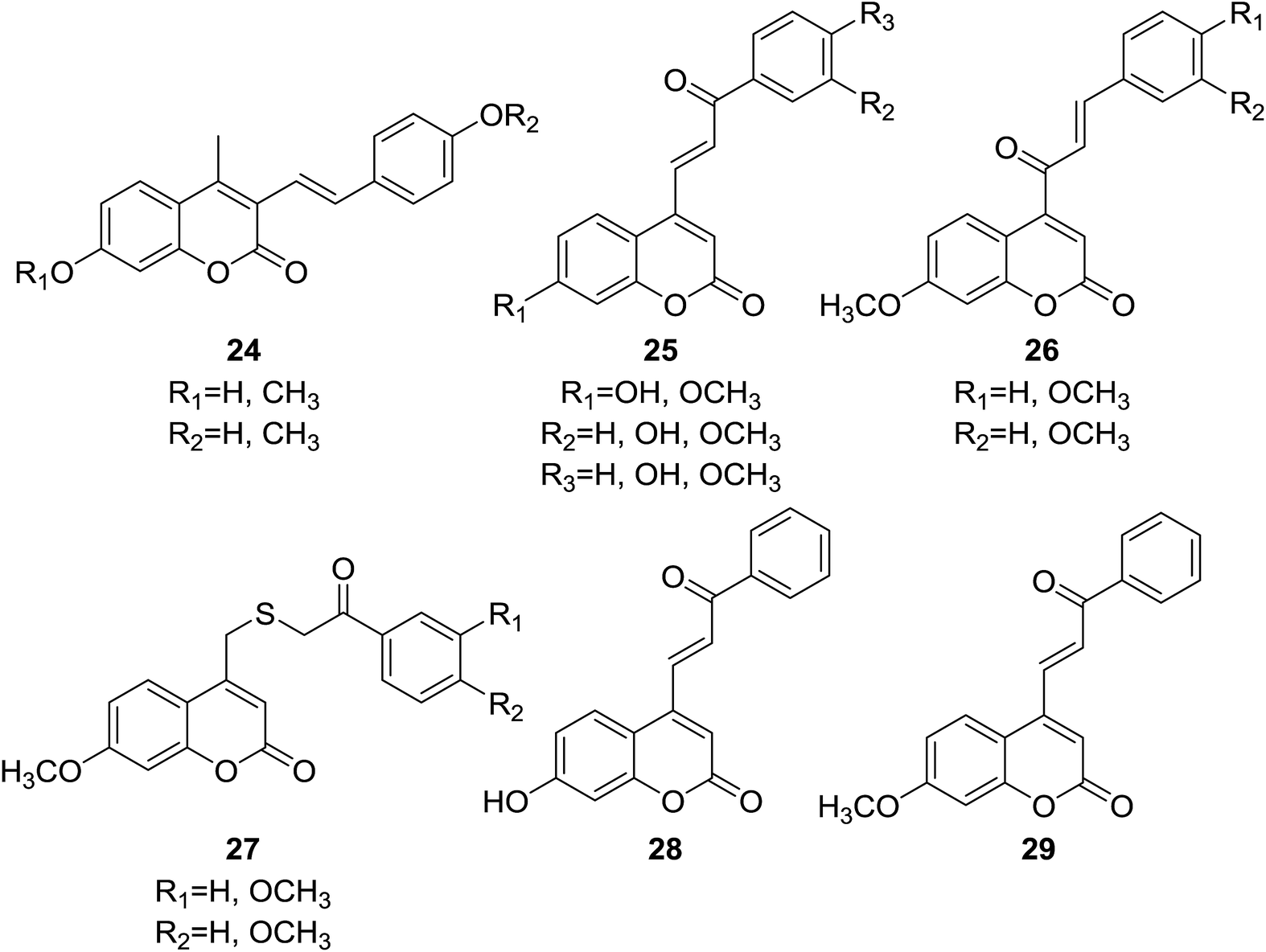
Four sets of coumarin–chalcone based inhibitors of Cdc25 (cell division cycle 25) phosphatases (24–27, Fig. 3) were designed and synthesized by Valente et al.29 The Cdc25 family of proteins are central regulators of progression through the eukaryotic cell division cycle.30 Inhibition of Cdc25 family represents a novel target for development of anticancer therapeutics.31 The inhibitory potential of hybrid compounds was evaluated on human glutathione-S-transferase (GST)-Cdc25 recombinant enzymes by a dephosphorylation assay with 3-O-methyl fluorescein phosphate.
 | ||
| Fig. 3 Coumarin–chalcone hybrid based Cdc25 phosphatases inhibitors. | ||
It was observed that coumarin–chalcone hybrids (25) bearing benzoylvinyl moiety at C-4 position of coumarin nucleus possessed most significant and selective inhibition against Cdc25A and Cdc25C, with percentage of inhibition ranging from 55.0% to 94.3% and 23.9% to 94.2%, respectively, at a concentration of 100 μM. The enzyme inhibition assay also revealed that series of hybrids with cinnamoyl coumarin scaffold (26) reduced or almost lost the inhibitory activity compared with compounds 25, suggesting that the group at C-4 position of coumarin is a crucial factor for their inhibitory activity. Among compounds 25, two most potent inhibitors 28 and 29, endowed with lowest IC50 as 27 and 28 μM against Cdc25A respectively, thus can be considered as lead compounds for further development as anticancer agents.
Inspired by the reported antiproliferative activity of some naturally occurring chalcones,26 Patel and coworkers32 synthesized a series of coumarinyl chalcone hybrids (30) bearing different substitutions at ring B of chalcone, through a two-step procedure outlined in Scheme 1. Their antiproliferative activities were evaluated against three different breast cancer cell lines (MDA-MB231, MDA-MB468 and MCF7) and one non-cancer breast epithelial cell line (184B5) by SRB-based spectrophotometry.
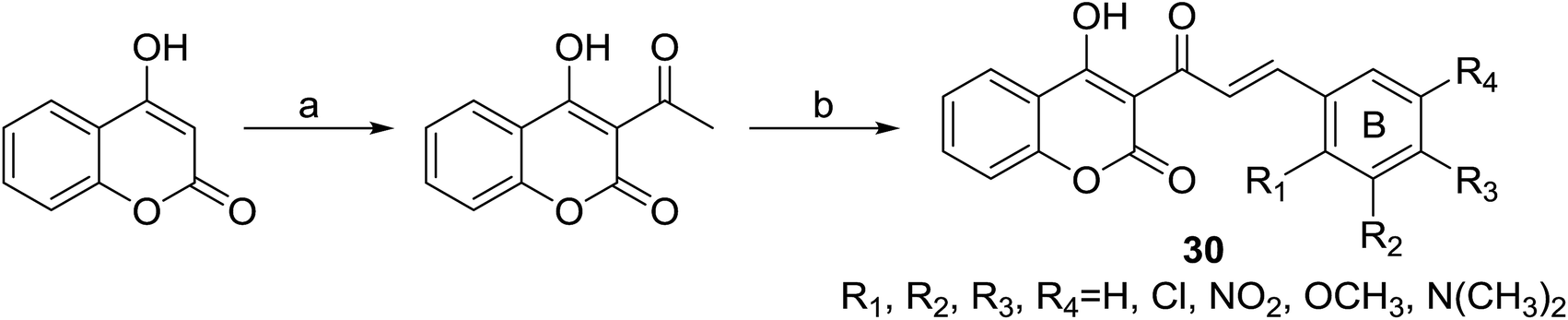 | ||
| Scheme 1 Preparation for 3-cinnamoyl-4-hydroxy-2H-chromen-2-ones (30). (a) POCl3, reflux; (b) CHCl3, piperidine (catalytic), reflux. | ||
Results of 50% growth inhibition (GI50) of hybrid compounds on three cancer cell lines revealed that all hybrids displayed antiproliferative activity at micromolar concentration. Further SAR analysis indicated that the antiproliferative activity of compounds was highly related to the position and number of methoxy substitutions on B-ring. Among this series, the most potent compound 31 (Fig. 4), with GI50 values ranging between 22.11 and 41.08 μM, displayed a comparable inhibition activity to cisplatin (GI50 ranging between 23.65 and 31.02 μM) and a better selectivity for breast cancer cell lines than non-cancer cells.
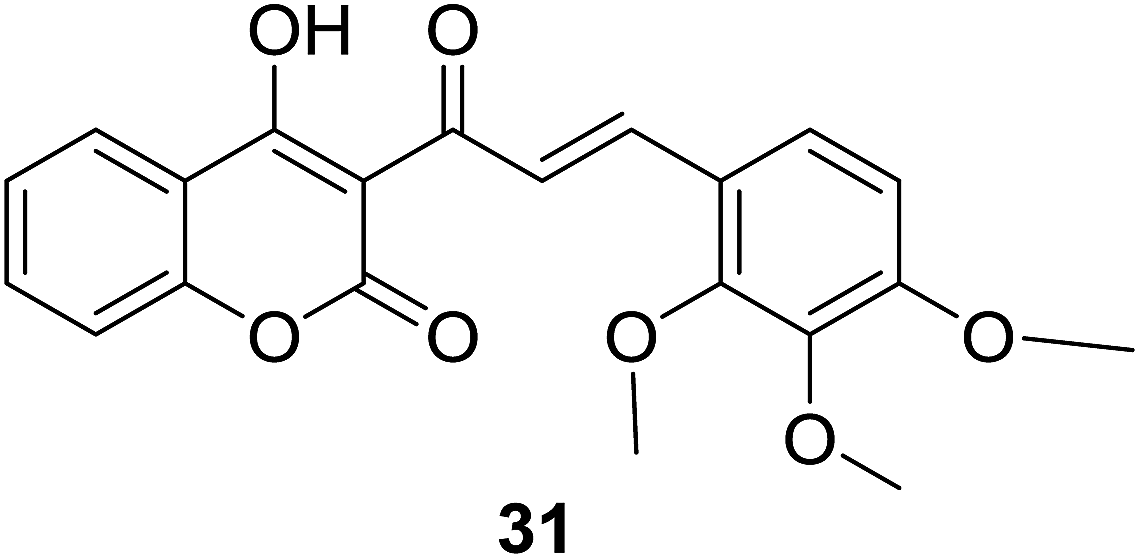 | ||
| Fig. 4 Hybrid with 2,3,4-triOCH3 groups on B-ring of chalcone moiety. | ||
Two series of novel hybrids (32 and 33, Fig. 5) containing pharmacophores of coumarin and chalcone were designed and synthesized by Sashidhara and coworkers,14 and were further screened in vitro for their growth-inhibitory effect against a panel of four human cancer cell lines, KB (oral squamous cell carcinoma), C33A (cervical carcinoma), MCF-7 (breast adenocarcinoma), A549 (lung carcinoma) and one normal cell line NIH3T3 (mouse embryo fibroblast) using sulforhodamine B assays.
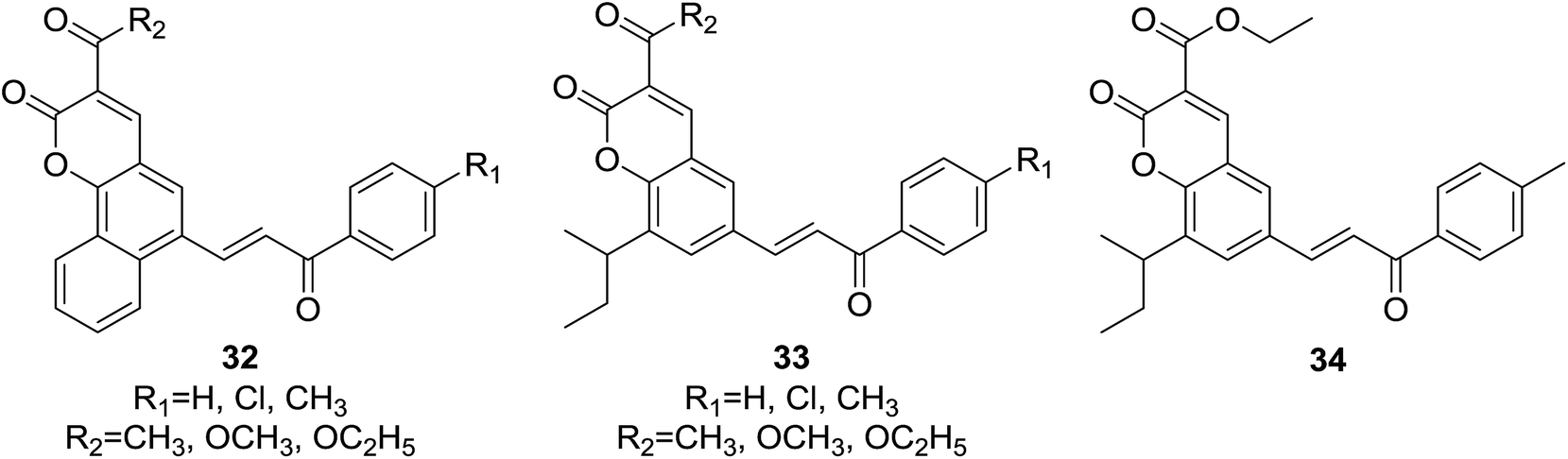 | ||
| Fig. 5 Anticancer coumarin–chalcone hybrids. | ||
It was demonstrated that, generally, series of 33 were found to be more active than 32 in cytotoxic evaluation. Moreover, compounds of 33 exhibited desirable effect against one or more cancer cell lines. Of them, several compounds showed interesting activity with IC50 < 20 μM. Detailed SAR investigation indicated that substitution at C-3 position of coumarin nucleus had a vital role in their anticancer activity, indicated by the fact that ester-containing compounds all present considerable potency, while ketone-containing one does not. SAR study also revealed that the para-chloro substituent on chalcone pharmacophore significantly decreased the selectivity of compounds 33 for cancer vs. non-cancer cells. Compound 34 (Fig. 5), containing ethyl ester at C-3 position of coumarin and para-methyl functionality at chalcone core, endowed the most significant activity against C33A cells with a lowest IC50 value of 3.59 μM, and furthermore, 34 displayed around 30 fold more selectivity towards C33A cells over normal fibroblast NIH3T3 cells.
The in vivo efficacy as well as detailed mechanism of action of compound 34 were further elucidated on tumor model induced by HeLa cell xenografts in nod SCID mice.33 Oral administration of 34 (100 mg per kg body weight) for 15 days obviously reduced the tumor volumes in xenograft mice compared to vehicle control, which was comparable with the activity of control drug, adriamycin (doxorubicin). Long-term administration of 34 did not cause any weight loss of xenograft mice, suggesting its non-toxic effect in vivo. It was demonstrated that 34 trigged apoptosis and arrested cell cycle at G2/M phase in C33A and HeLa cells. The underlying mechanism may be related to generation of ROS and regulation of Bcl-2 family proteins, leading to apoptosome mediated activation of caspase cascades. These results show that 34 is a promising candidate for anticancer agents.
Natural product is another important source for discovery of anticancer coumarin–chalcone hybrids. Until recently, a few coumarin–chalcone hybrids have been isolated from natural sources, especially fern plants of Pityrogramma and Cyclosorus species. However, coumarin–chalcone hybrids from Pityrogramm plants were reported without any biological activity study. Their chemical structures and original plants were summarized in Table 1.
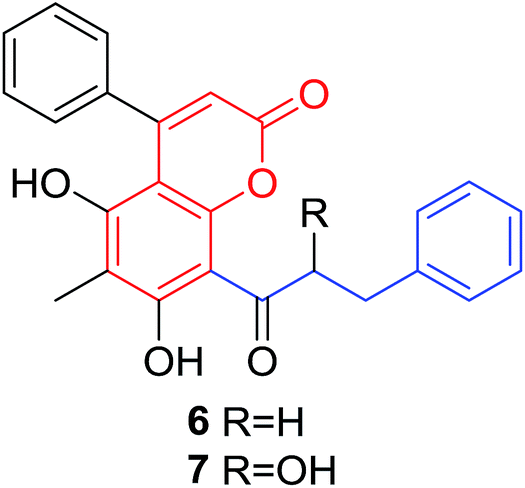
Quadri-Spinelli et al. reported a series of novel coumarin–chalcone derivatives (3, 6, 7 and 18–21, Table 1) isolated from CH2Cl2 extract of leave of fern plant Cyclosorus interruptus and cytotoxicity evaluation by inhibition of KB (human nasopharyngeal carcinoma) cell growth.18 Majority of the obtained natural coumarin–chalcone hybrids contain a dihydro-chalcone moiety; and interestingly, unusual cyclic ether or dioxocane exist between ring A and ring B of chalcone core within structures of some hybrids (18–21). Compounds 3 and 6 turned out to be active against KB cells with IC50 values of 3.8 and 5.1 ppm, respectively. SAR investigation indicated that absence of cyclic ether or dioxocane was essential for cytotoxicity for 3 and 6. However, in order to draw a comprehensive SAR conclusion, more cancer cell lines should be introduced to anticancer screening of these natural hybrids.
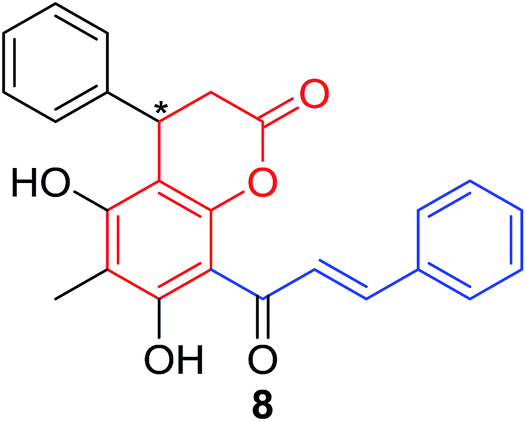
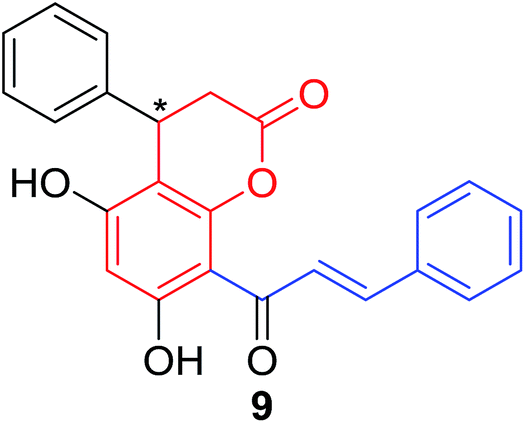
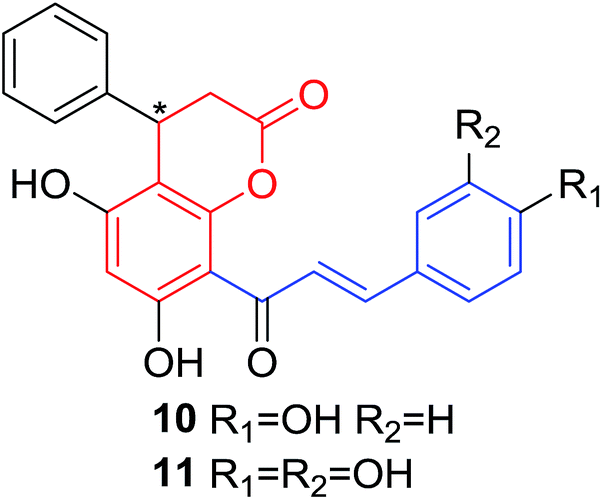
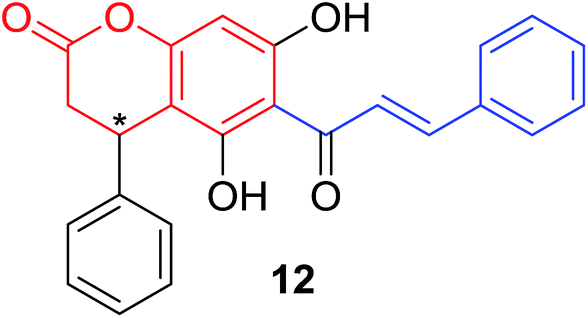
Several natural occurring coumarin–chalcone hybrids (3–5, 8 and 9, Table 1) isolated from fern Cyclosorus parasiticus and their anti-proliferative activity against a panel of six cancer cell lines, A549 (lung cancer), HepG2 (hepatocellular carcinoma), MCF-7 and MDA-MB-231 (breast cancer), ALL-SIL (human leukemia), and SW1990 (pancreatic cancer) by sulforhodamine B (SRB) assay, were reported by Han and coworkers.19,21 Some of these hybrids possess a dihydrocoumarin moiety in the scaffold, and compound 5 represents the first example of long chain fatty acid ester of compound in this class.
Anti-proliferative screen test revealed that compound 8 containing a dihydrocoumarin moiety displayed most potency against all six cell lines with IC50 ranging from 1.60 to 6.06 μM, especially towards HepG2 cells (IC50 = 1.60 μM), which was comparable with reference drug doxorubicin (IC50 = 1.19 μM). SAR investigation between compound 8 and 9 (IC50 > 9.00 μM) reveals that methyl substitution at C-6 position of dihydrocoumarin plays important role in their anti-proliferative activity. Through morphological fluorescence probe test and flow cytometry assay, it was indicated that apoptosis may contribute significantly to cytotoxicity of compound 8 against HepG2 cells. Furthermore, long-term effect of 8 was investigated by clonogenicity assay on HepG2 cells. A concentration-dependent inhibition on clonogenicity was observed in compound 8-treated cells. These results suggested that compound 8 can be employed as a promising lead for development of anticancer agents.
The research conducted by Quadri-Spinelli and Han groups suggests that ferns of Cyclosorus species are candidate plants to discovery novel and bioactive coumarin–chalcone derivatives, and these natural hybrids represent an interesting and potential source for anti-cancer agents.
Antimalarial activity
Malaria, one of the most important infectious disease problems especially in tropical countries, have caused growing mortality of two million.34 The increasing threat of malaria parasite resistance to currently used antimalarial drugs provokes scientific community to discover novel and effective agents to treat malaria.35,36 Encouraged by the antimalarial chalcone37 and coumarin compounds such as licochalcone A,38,39 quinolinyl chalcones,40,41 daphnetin42 (7,8-dihydroxycoumarin) and coumarin–trioxane hybrids43 in literatures, medicinal chemists have reported several coumarin–chalcone hybrids with antimalarial activity which are discussed as follows.Fifteen coumarinyl chalcone analogs (30, Scheme 1) were synthesized by Patel et al. through Knoevenagel condensation.44 Antimalarial activity of the synthetic hybrids was evaluated in vitro against chloroquine susceptible (3D7) and chloroquine resistant (W2) Plasmodium falciparum, a foremost strain responsible for malaria.
Majority of compounds affording the skeleton of 30 showed favorable antiplasmodial activity against both chloroquine sensitive and resistance strains at micromolar concentrations. SAR investigation revealed that para and meta substituents on the B ring of chalcone nucleus had notable impact on the antiplasmodial effect. It was also observed that the activity of para substituted compounds decreased in the order of NO2 > Cl > CH3 > OCH3 > N(CH3)2, suggesting that present of electron withdrawing substituents at para position is crucial for their antimalarial activity. Amongst the para position occupied hybrids, compound 35 (Fig. 6), possessing a –NO2 functionality, displayed most potency (IC50 < 5 μg ml−1). Therefore, the coumarinyl chalcone presented themselves as attractive lead compounds for development of efficient drugs overcoming malaria of drug-resistance.
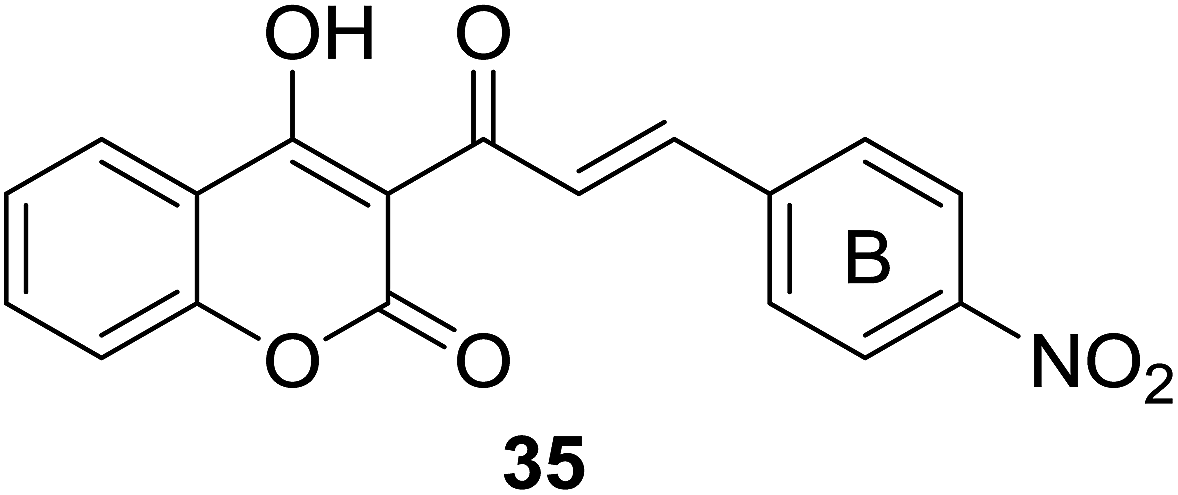 | ||
| Fig. 6 Hybrid, containing para-NO2 substituted B-ring of chalcone, exhibited outstanding antiplasmodial activity. | ||
Wanare et al. designed three series of hybrids (36–38, Fig. 7) bearing chalone and coumarin skeleton in one molecule. Synthetic hybrids were further screened for their antimalarial activity by inhibition of growth of malaria parasite Plasmodium falciparum using the microtiter plate based SYBR-Green-I in vitro assay.45
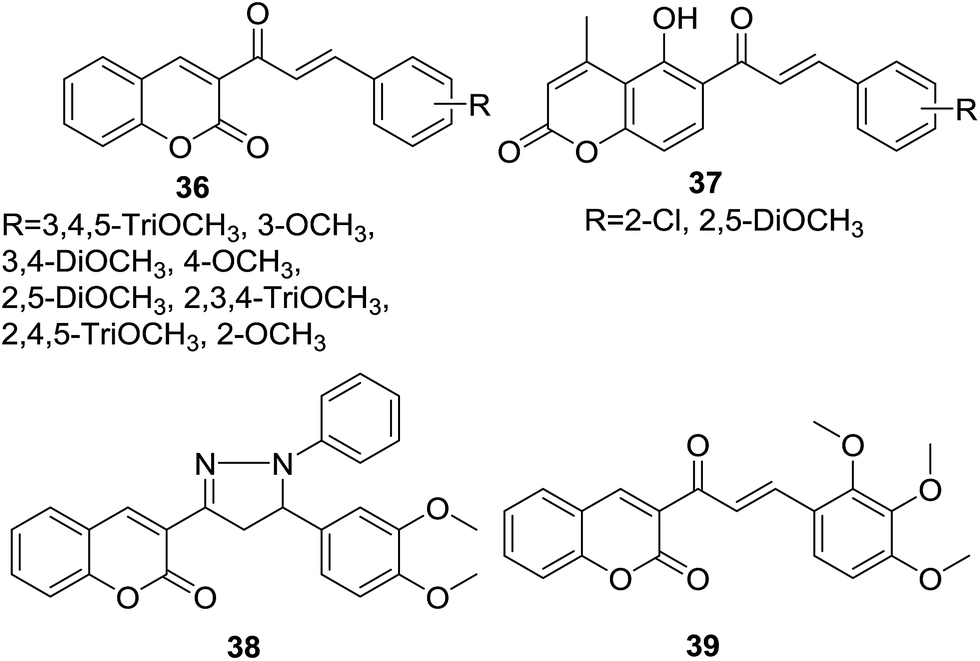 | ||
| Fig. 7 Antimalarial coumarin–chalcone hybrids. | ||
It was revealed that the most potent hybrid compound 39 (Fig. 7), bearing 2,3,4-trimethoxy groups, exhibited obvious inhibition activity against both chloroquine-sensitive (3D7) P. falciparum (IC50 = 3.1 μg ml−1) and chloroquine-resistant (RKL9) P. falciparum (IC50 = 1.1 μg ml−1). SAR study indicated that trimethoxy groups significantly polished the antimalarial activity of hybrids. Molecule docking study further inferred that 39 possessed good affinity for active site residues of falcipain enzyme. The hydrogen bonding interaction with Cys42 as predicted by the docked pose of 39 suggested that hydrogen bonding acceptor on the lactone ring of coumarin core was essential for antimalarial activity.
As discussed in last section, Pingaew et al. synthesized a group of coumarin–chalcone hybrids with anticancer effects. Moreover, they also evaluated the potency of these compounds as antimalarial agents.15 Coumarin–chalcone hybrid 40 (Fig. 8), liked by 1,2,3-triazole and substituted by trimethoxy functionality on chalcone B-ring, turned out to be the most promising antimalarial molecule affording IC50 value of 1.60 μM. Molecular docking suggested that antimalarial activity of 40 might be owing to its inhibition of falcipain-2. The coumarin moiety occupied the binding site of falcipain-2 via hydrophobic interaction with Cys42 and Trp206 residues, which was in accord with results of Wanare's research.45 This study provides novel molecules with coumarin–triazole–chalcone hybrid scaffold as potential lead for further development as antimalarial agents.
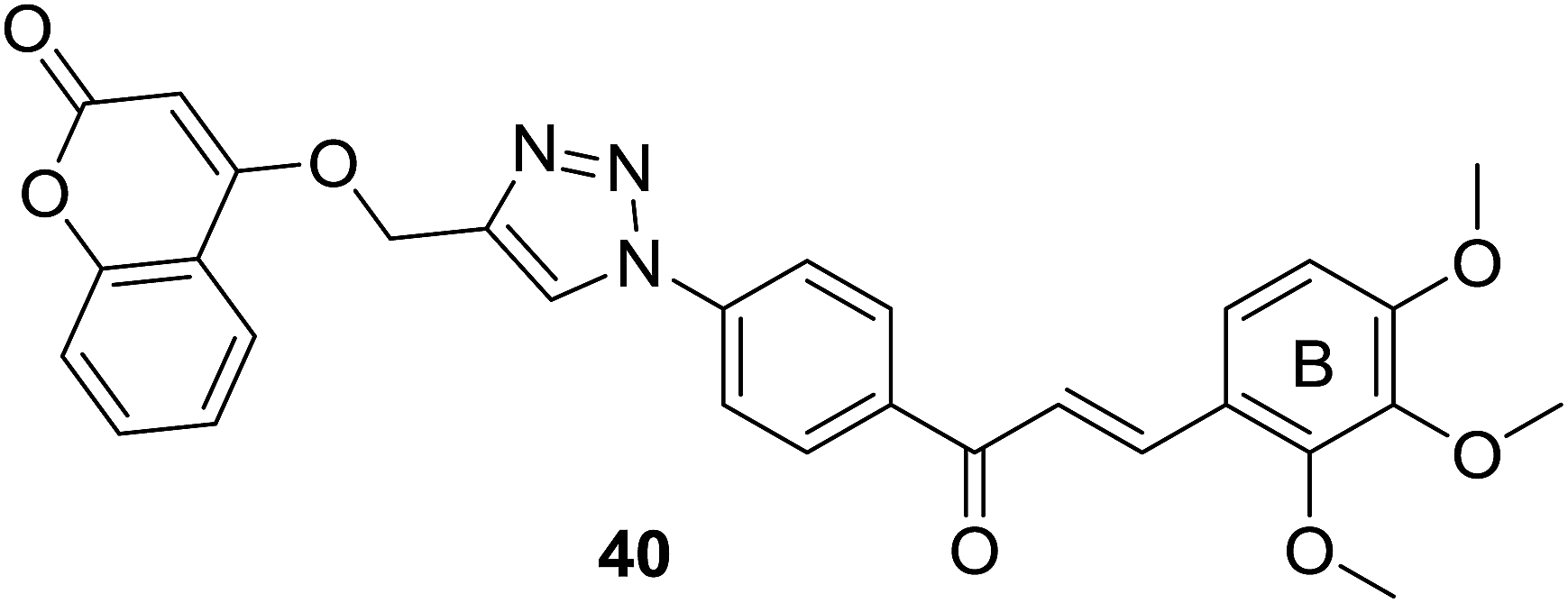 | ||
| Fig. 8 Antimalarial coumarin–chalcone hybrid linked by a 1,2,3-triazole ring. | ||
Antimicrobial activity
Infectious diseases caused by bacteria and fungi of multi-drug resistance are becoming a serious issue threatens public health worldwide.46 It prompts medicinal chemists to study more effective drugs with high potent antimicrobial activity to overcome the merging of resistant strains.47,48 Both coumarin49–51 and chalcone52,53 are known to possess favorable antimicrobial potency, which encourages medicinal chemists to develop hybrid molecules based on these two classes of natural products.Vazquez-Rodriguez et al. designed and synthesized a series of coumarin–chalcone hybrids (41) by the synthetic route outlined in Scheme 2.16 Antibacterial activity of synthetic hybrids was evaluated against several types of human bacteria strains (Escherichia coli, Staphylococcus aureus and Pseudomonas aeruginosa) and against strains of the marine pathogen including Tenacibaculum maritimum, Tenacibaculum discolor, Tenacibaculum gallaicum, Tenacibaculum soleae and Tenacibaculum ovolyticum which were responsible for tenacibaculosis in fish.
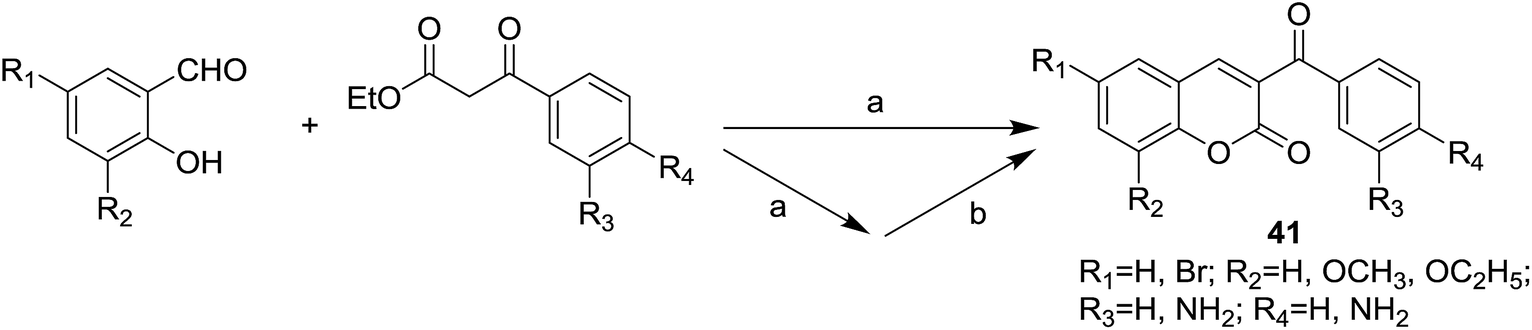 | ||
| Scheme 2 Synthesis of antibacterial coumarin–chalcone hybrids. (a) EtOH, piperidine, reflux, 2–5 h; (b) SnCl2·2H2O, EtOH, reflux, 3–7 h. | ||
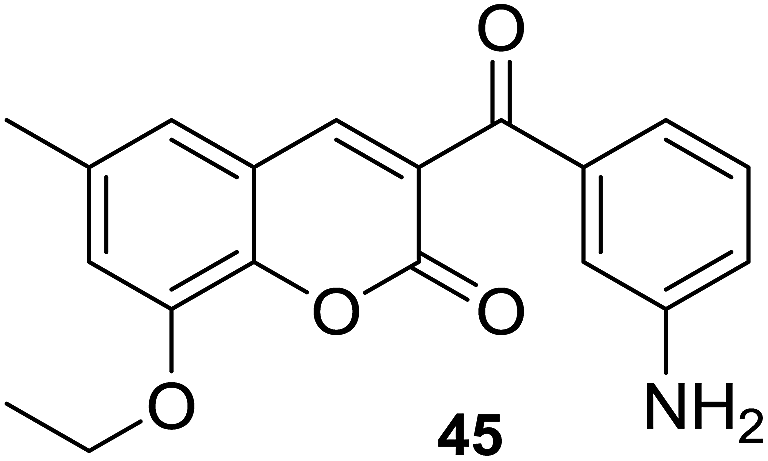
In disk diffusion test, compounds 41 displayed no significant inhibition zones neither for the human pathogenic bacteria species nor the fish pathogens T. discolor, T. gallaicum, T. soleae, and T. ovolyticum. However, several compounds (42–45, Fig. 9) bearing either methoxy or ethoxy group at C-8 position of the coumarin moiety exhibited high selectivity towards T. maritimum, with inhibition zones (IZ) ranging from 16.1 to 41.4 mm, which was comparable with that of the reference drugs enrofloxacin (25.2–35.9 mm) and ampicillin (30.3–40.4 mm). The MIC (minimum inhibitory concentration) and MBC (minimum bactericidal concentration) values of these four compounds on eight of the T. maritimum strains were further calculated. It was observed that compounds 42 and 45 (MIC/MBC in the range of 0.5–1.9 μM) possessed comparable activity as enrofloxacin (0.1–1.0 μM) towards DBa4a and 3.35 strains. The assay against T. maritimum strains LL01 8.3.8 and LL01 8.3.1 revealed that compounds 42, 43 and 45 displayed MIC values up to 20 times lower than that of enrofloxacin, ranging from 0.1 to 0.5 μM. Moreover, in the cytotoxicity test, these compounds turned out to be non-toxic against EPC (epithelioma papillosum of carp) fish cell line, suggesting these coumarin–chalcone hybrids were promising and safe candidates for aquaculture treatment.
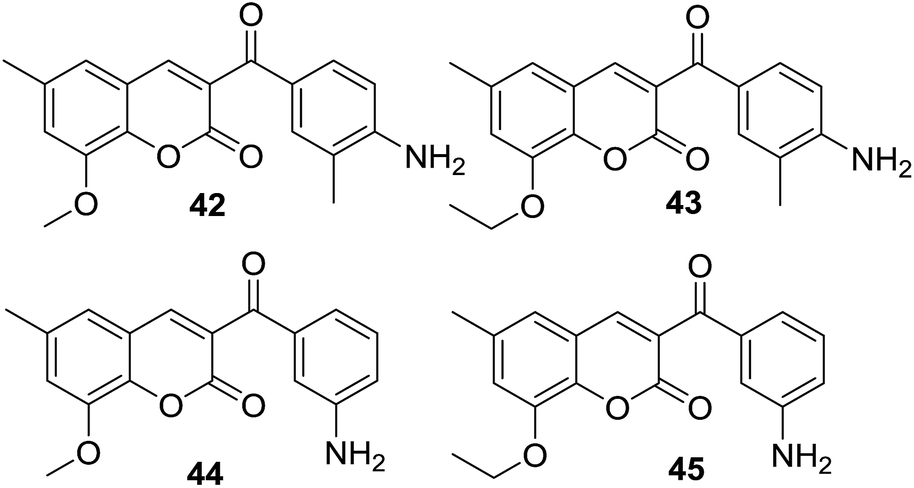 | ||
| Fig. 9 Hybrids exhibiting high selectivity towards T. maritimum. | ||
Vazquez-Rodriguez and coworkers further reported a set of coumarin–chalcone hybrids (46, Fig. 10) with varied substituted B-ring of chalcone and their trypanocidal effect was tested against the epimastigote, trypomastigote and amastigote stages of Trypanosoma cruzi parasite.54
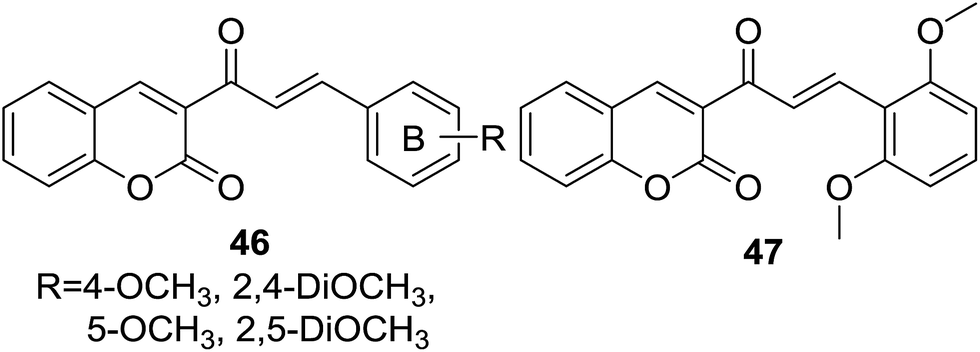 | ||
| Fig. 10 Coumarin–chalcone hybrids with varied substituted B-ring of chalcone nucleus. | ||
Epimastigote and trypomastigote viability study revealed that hybrids 46 were more active against trypomastigote stage than epimastigote stage. The most potent hybrid (47, IC50 of 2.6 μM against trypomastigote stage) possessing 2,5-dimethoxy groups on B-ring of chalcone also exhibited favorable activity against amastigote stage with a IC50 of 2.9 μM. A brief SAR investigation indicated that dimethoxy groups at C-2 and C-5 of B-ring, and no substitution at C-4 was a key feature for the trypanocidal activity of this hybrid scaffold. Although cytotoxicity assays with murin RAW 264.7 macrophages and VERO cells suggested that hybrid 47 was sensible to these two mammalian cell lines, this hybrid has considerable value for further optimization and modification as trypanocidal agents due to the promising trypanocidal activity.
Deshpande and coworkers reported a series of coumarin–chalcone hybrids (48, Fig. 11) containing an azo-linkage and their anti-bacterial activity was screened against five human pathogens including three Gram positive bacteria of Bacillus subtilis, Proteus vulgaris, and Staphylococcus aureus, and two Gram negative bacteria of Escherichia coli and Klebsiella pneumonia.55 In the agar diffusion test, compound 48 possessing a para-Cl substitution on the benzoyl ring was proven to be the most potent agent against all three Gram positive bacteria (IZ > 17 mm). Therefore this hybrid compound could be employed as a promising agent for anti-Gram positive bacteria.
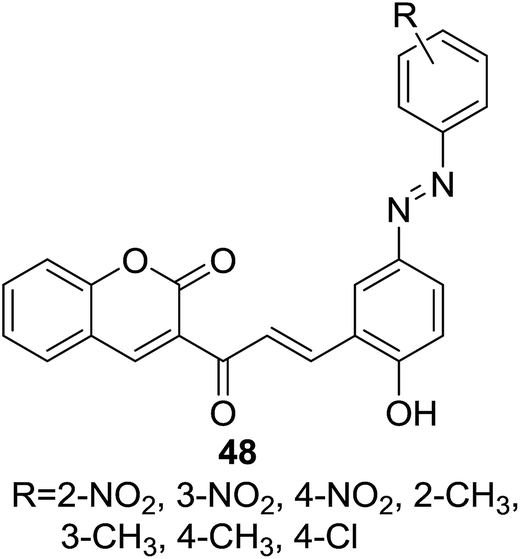 | ||
| Fig. 11 Coumarin–chalcone hybrids containing an azo-linkage. | ||
Trivedi and coworkers56 synthesized a set of 18 coumarin–chalcone derivatives (49, Fig. 12) by a rapid route of improved condition of Knoevenagel reaction, using chloroform as solvent with a mild organic base, for example, piperidine, to reduce the reaction time and facilitate the isolation of products. The synthetic hybrids were tested for their anti-HIV activity, unfortunately, no compound show any desirable effect.
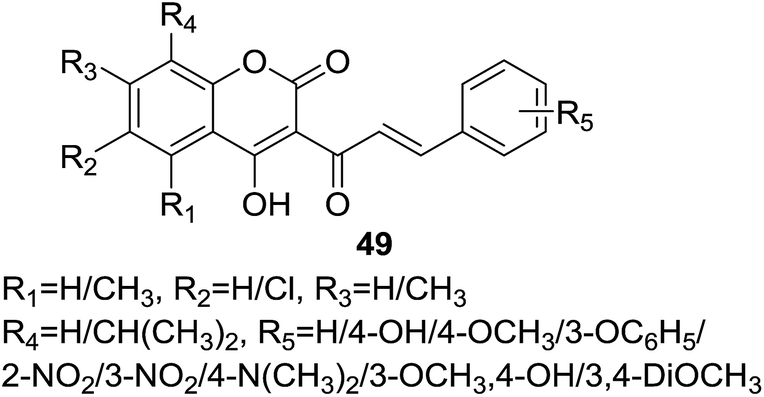 | ||
| Fig. 12 Coumarin–chalcone analogs explored for anti-HIV properties. | ||
Several coumarin–chalcone hybrids (50, Fig. 13) containing substituted B-ring with different electron donating or withdraw groups were reported by Hamdi et al.57 through the similar synthetic method reported by Trivedi.56 Obtained hybrid derivatives were screened by paper disc diffusion method for their activity against Gram-positive bacterial, Staphylococcus aureus (NCTC-7447). All tested compounds showed moderate anti-bacterial activity with diameters of IZ ranging from 8 mm to 16 mm while that of positive control gentamycin was 15–20 mm.
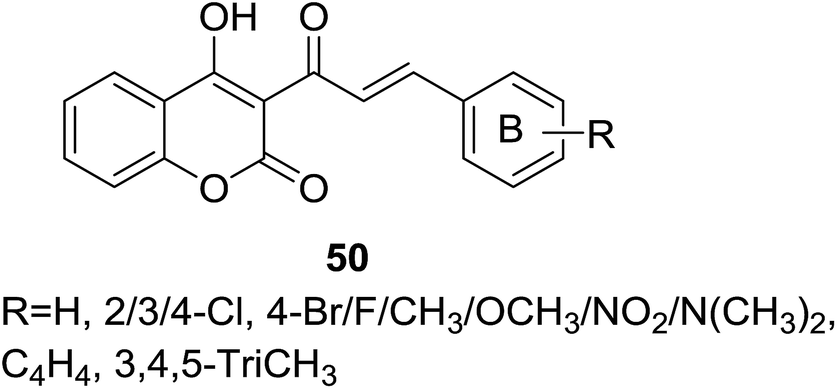 | ||
| Fig. 13 Coumarin–chalcone hybrids with varied substituted B-ring of chalcone nucleus. | ||
Špirtović-Halilović and coworkers evaluated antibacterial activities of four coumarin–chalcone analogs (51–54, Fig. 14) against two Gram-positive aerobic bacteria Bacillus subtilis (ATCC 6633) and Bacillus cereus (ATCC 11778). In silico properties of the hybrid molecules were also proposed by quantum-chemical and physicochemical calculations.58
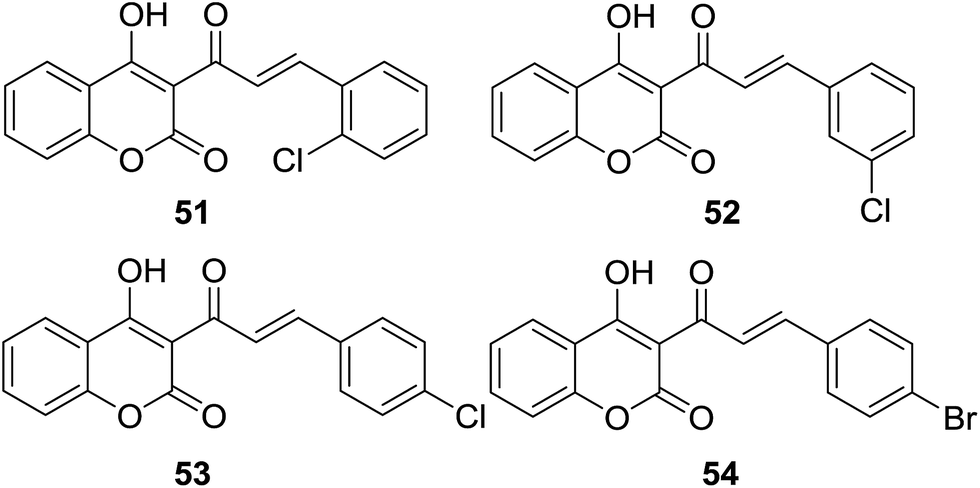 | ||
| Fig. 14 Halogen-substituted coumarin–chalcone hybrids. | ||
The diffusion method showed all hybrids had the ability to inhibit growth of two bacteria, with IZ ranging from 16.00 to 23.75 mm, of which compound 54 showed highest potency (IZ for B. subtilis and B. cereus as 22.5 mm and 23.75 mm respectively). In order to predict in silico properties such as relative stability and reactivity, the density functional theory (DFT) was employed to calculate chemical reactivity descriptors of hybrids 51–54. Amongst these hybrids, 51 is the most reactive and least potent against both bacteria whereas 54 is the most stable and least reactive, suggesting that antibacterial activity of these coumarin–chalcone analogs correlated with the calculated chemical reactivity descriptors, viz. the most chemically stable compound possessed the best antibacterial activity. This finding will be helpful for future development of effective antibacterial coumarin–chalcone agents.
A series of coumarin–chalcone hybrids (55, Fig. 15) containing different substituted B ring of chalcone scaffold were synthesized by Naruka et al. through microwave-assisted synthesis. The antimicrobial activity against Gram positive (S. aureus and P. aeruginosa), and Gram negative (E. coli and K. pneumoniae) bacteria as well as pathogenic fungi (C. albicans and A. niger) was screened.59
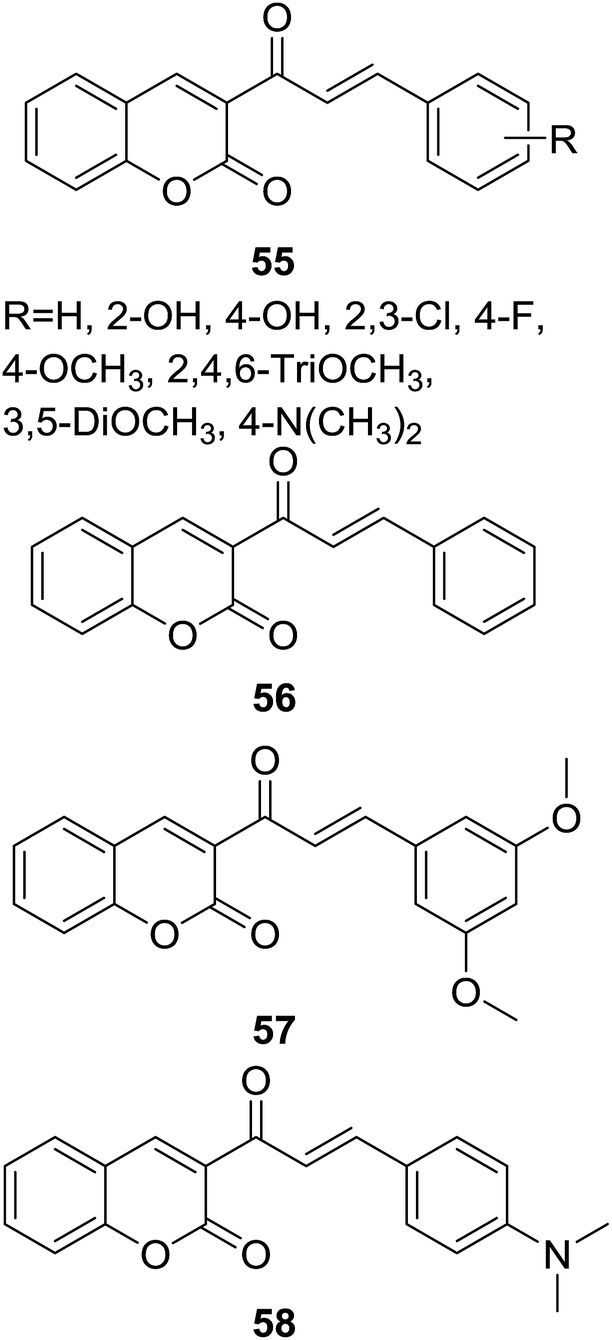 | ||
| Fig. 15 Antimicrobial coumarin–chalcone hybrids. | ||
Majority of synthetic compounds showed good antimicrobial activity against one or more strains, several hybrids (e.g. 56–58, Fig. 15) possessed even better antibacterial activity than sulfamethoxazole, the reference drug. Using simple linear regression analysis, a QSAR (quantitative structure–activity relationship) model in which parameter of WPSA (weakly polar component of the total solvent accessible area) significantly correlated to the antibacterial activity of coumarin–chalcone hybrids against E. coli was proposed. This QSAR model can be employed for the rational design of antibacterial agents based on coumarin–chalcone hybrids.
Quadri-Spinelli et al. also evaluated the anti-bacterial activity of aforementioned natural coumarin–chalcone hybrids from C. interruptus.18 Their research showed that hybrid 6 had significant anti-bacterial activity against Bacillus cereus, Staphylococcus epidermidis, and M. luteus, with values of MIC as 2, 1 and 2 ppm, respectively. The activity of 6 against three strains was comparable or even better than the reference compound chloramphenicol. Hence, compound 6 can be used as a favorable lead for antibacterial agent research.
Antioxidant activity
Oxidative stress, leaded by overproduction of free radicals and reactive oxygen species (ROS) and the consequent disturbance in the prooxidant–antioxidant balance,60 is believed to have considerable impact on the pathogenesis of many diseases, such as diabetes,61 neurodegeneration,62 cancer,63 cardiovascular diseases64 and so on. Therefore, discovery of strong antioxidants to reverse oxidative stress status is an interesting direction and has significant benefits. Hybrid molecules have been proven to be promising antioxidants against oxidative stress induced disorders.65Inspired by the antioxidant potency of natural coumarin and chalcone,66 seven coumarin–chalcone hybrids (59–65, Fig. 16) were synthesized by Xi et al. and the inhibitory effects on Cu2+/GSH-, ˙OH-, and AAPH-induced oxidation of DNA and activity on trapping ABTS+˙ [2,2′-azinobis(3-ethylbenzothiazoline-6-sulfonate) cationic radical] and DPPH (2,20-diphenyl-1-picrylhydrazyl radical) were further screened.67
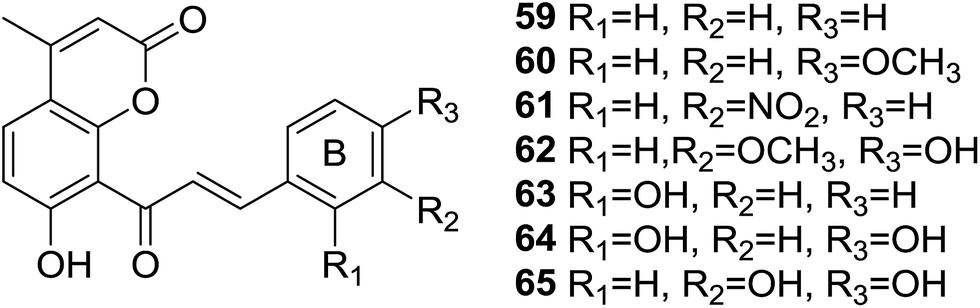 | ||
| Fig. 16 Coumarinyl chalcones with substituted B-ring. | ||
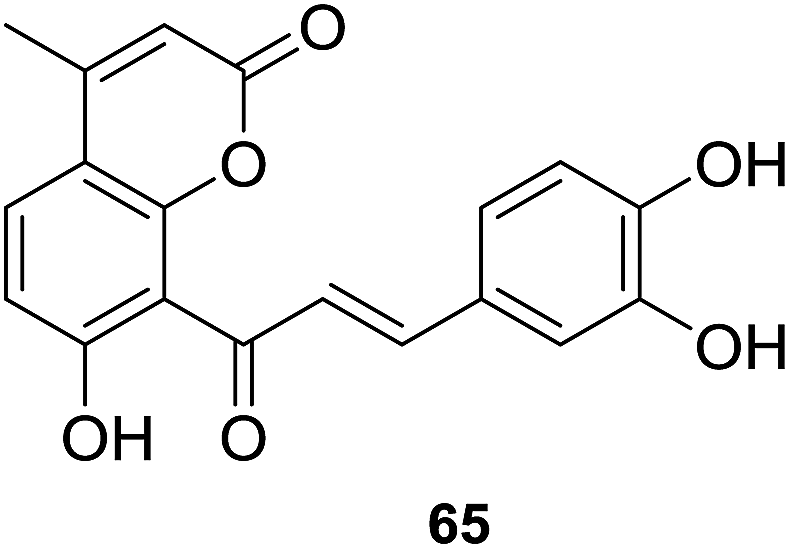
All tested coumarin–chalcone derivatives attenuated GS˙-induced destroy of DNA, of which compound 65 possessed most significant antioxidant activity on Cu2+/GSH-induced oxidation of DNA, indicating that meta-, para-dihydroxyl groups on ring B of chalcone are critical to inhibit reaction between Cu2+ and GSH to form GS˙. Hybrids 59, 61, and 63 showed good activity on ˙OH-induced oxidation of DNA, especially compound 59, which contains few functional groups and provides more positions for the addition of activity in inhibiting ˙OH, thus 59 exhibits relative high ˙OH-induced oxidation of DNA. Compound 64 showed good activity against AAPH-induced oxidation, suggesting that double hydroxyl groups at meta-position of ring B responsible for the inhibition. The most efficient scavenger against radicals, hybrid 65, trapped ABTS+˙ and DPPH with outstanding rate constants (k) being 148.0 and 10.70 mM−1 s−1, respectively. What's more, the hydroxyl group at benzene ring of 65 and an intermolecular synergistic interaction were proven to be responsible for the scavenging activity. Based on these finding, 65 can be served as an outstanding antioxidant that may be subjected to detailed pharmacological test against disorders closely related to oxidative stress.
A series of coumarin–chalcone derivatives (66, Fig. 17) were synthesized through a microwave assisted approach. In vitro antioxidant properties of these hybrids were studied by DPPH (1,1-diphenyl-2-picryl hydrazyl) method.68 Compound 67 was found to be a potential candidate for scavenging radical oxygen.
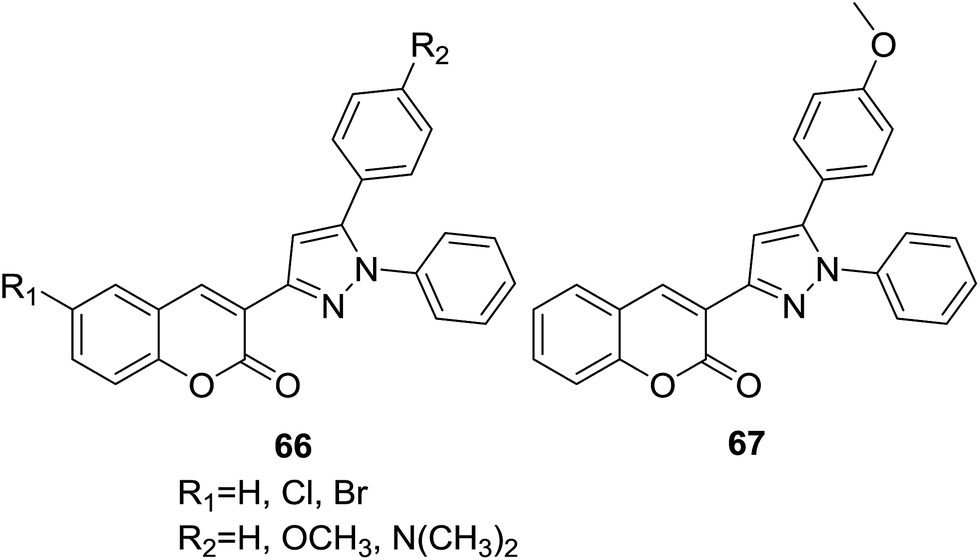 | ||
| Fig. 17 Antioxidant coumarin analogs containing a pyrazole ring. | ||
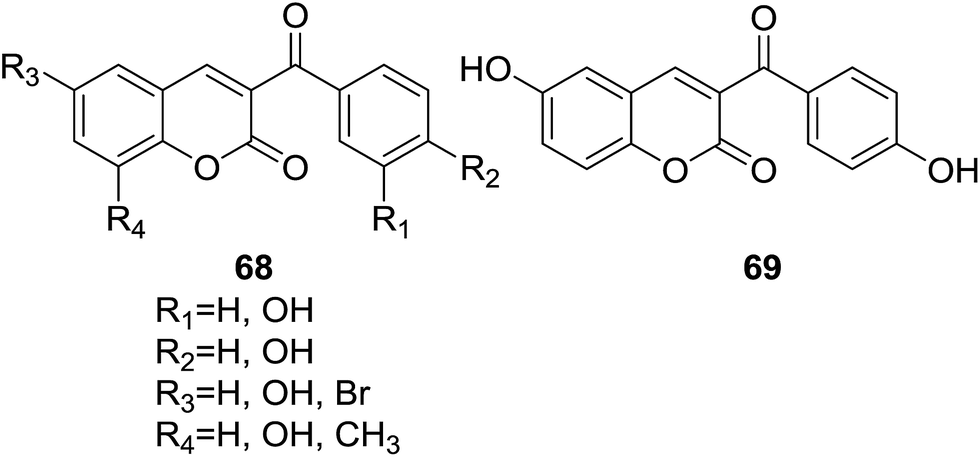
Pérez-Cruz and coworkers reported a series of coumarin–chalcone hybrids (68, Fig. 18) and their cytoprotection capacity against ROS and RNS (reactive nitrogen species) on BAEC (bovine aortic endothelial cells) by ORAC (oxygen-radical absorbance capacity) and ESR (electron spin resonance) assays.69 Moreover, the drug-like properties of studied hybrids were theoretical evaluated by calculation of log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P, TPSA (topological polar surface area) and the number of hydrogen-bond acceptors as well as hydrogen-bond donors.
P, TPSA (topological polar surface area) and the number of hydrogen-bond acceptors as well as hydrogen-bond donors.
 | ||
| Fig. 18 Coumarin–chalcone hybrids possessing a benzoyl substituent at C-3. | ||
All hybrids turned out to possess much higher ORAC values than that of the well-known antioxidants quercetin and catechin. Compound 69 (Fig. 18) had most significant ORAC value of 14.1. Relationship between the ORAC values and substituents present in these hybrids indicated that the position and number of hydroxyl groups on coumarin and benzoyl rings have key impact on their antioxidant activity. Cytotoxicity assay reveled that compound 69 showed 93% cell viability at a concentration of 50 μM, which can be considered as low cytotoxicity. In cytoprotection assay, all studied compounds exhibited observable protection against 1 mM H2O2- and 0.5 mM 3-morpholino-sydnonimine (SIN-1)-induced cytotoxicity in BAEC. Results of ADME properties calculation showed that all hybrid compounds do not break any point of the Lipinski's rule of five, suggesting that they can be promising leads for drug development.
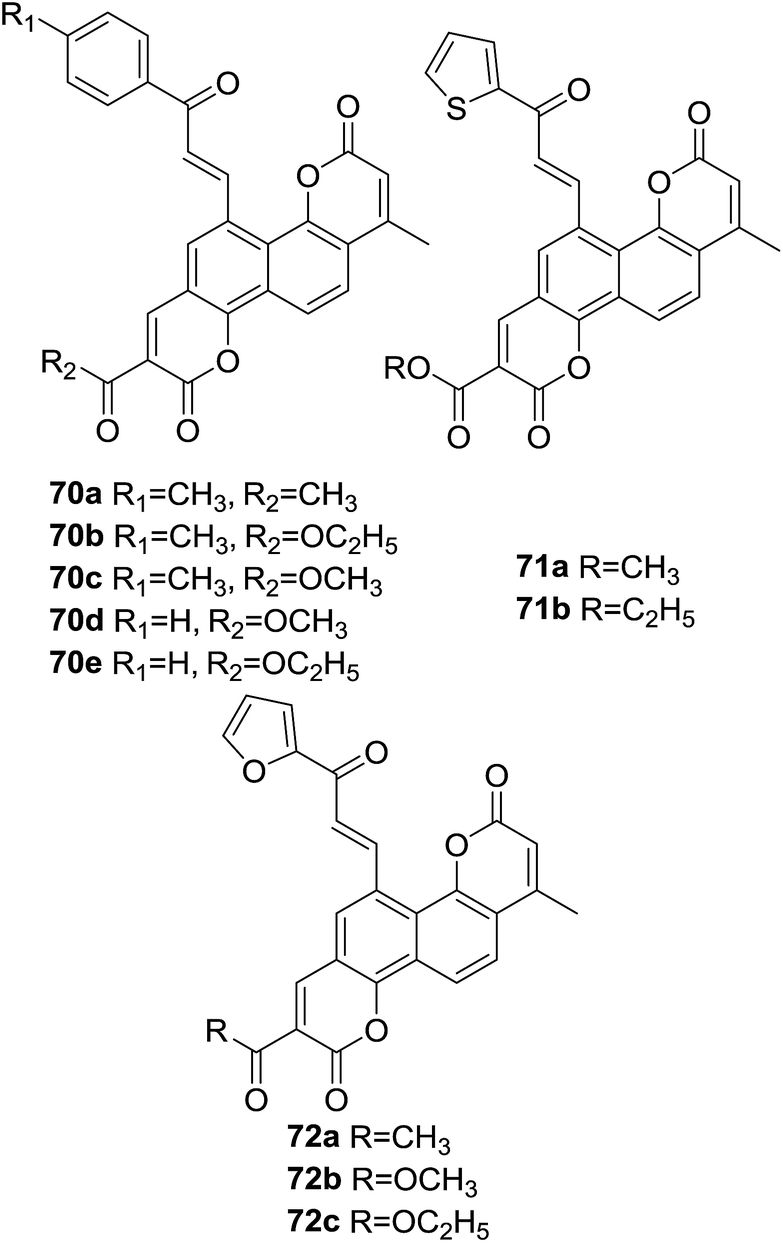
Three series of novel biscoumarin–chalcone hybrids (70–72, Fig. 19) were synthesized by Sashidhara et al.70 The scavenging potential of synthetic hybrids against formation of O2˙ and ˙OH in non-enzymic systems as well as lipid peroxidation inhibition activity in microsomes were investigated. Amongst these compounds, hybrids 70d, 71b and 72c (200 μg ml−1) showed significant activity in superoxide anions inhibition by 29%, 24%, and 30%, hydroxyl radicals inhibition by 29%, 26% and 21% and microsomal lipid peroxidation inhibition by 23%, 27% and 24%, respectively. The remaining compounds displayed modest antioxidant activity.
 | ||
| Fig. 19 Biscoumarin–chalcone hybrids. | ||
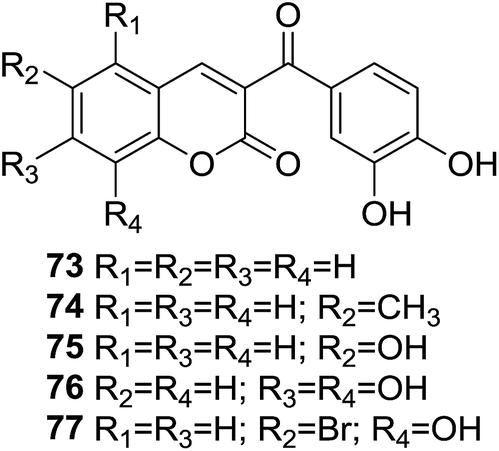
In light of potential antioxidant property of the chalcone and coumarin moieties, Vazquez-Rodriguez and coworkers synthesized a series of hybrid compounds (73–77, Fig. 20) and evaluated their antioxidant activity.71
 | ||
| Fig. 20 Hybrids bearing varied substituted coumarin nucleus. | ||
Several compounds showed desirable effects in the assay for their ORAC-FL and ORAC-PGR values as well as scavenging hydroxyl radical activity. In ORAC-FL assay, compound 77, in which one hydroxyl group is present at position C-8 and a bromide atom at position C-6 of coumarin moiety, displayed highest ORAC-FL value. In ESR assay, all compound displayed as high as 80% of radical scavenging activity, of which compound 77 exerted a highest scavenging rate of 90.9% at the concentration of 3 mM, which was correlated with the result of ORAC-FL assay. Furthermore, these coumarin–chalcone hybrids do not break the Lipinski's rule of five, making them promising candidates for drug development.
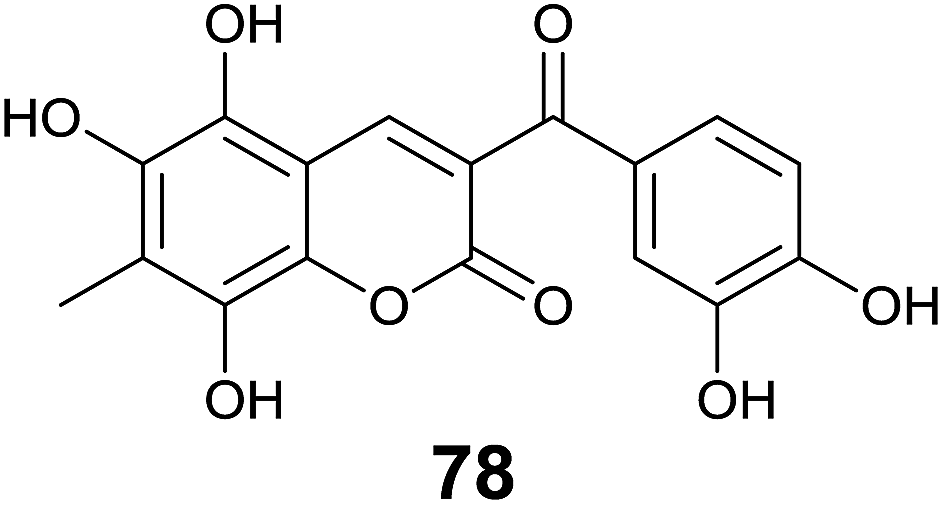
Inspired by the work of Vazquez-Rodriguez et al.,71 Mazzone and coworkers studied theoretical antioxidant properties of coumarin–chalcone hybrids based on the compounds synthesized in Vazquez-Rodriguez's experiment, through density functional theory (DFT) and time-dependent formulation of DFT (TDDFT).72 Three antioxidant mechanisms, hydrogen atom transfer (HAT), electron transfer followed by proton transfer (SET-PT), and sequential proton loss electron transfer (SPLET) were investigated on the coumarin–chalcone hybrids. HAT mechanism turned out to be the most important one for the antioxidant protection exerted by this type of compounds. Furthermore, it was indicated that poly-substitution on coumarin moiety played a key role in their antioxidant activity. The virtually designed compound, 5,6,8-trihydroxy-7-methyl-3-(3′,4′-dihydroxybenzoyl) coumarin (78, Fig. 21), represents the most promising antioxidant candidate based on the theoretical insights.
 | ||
| Fig. 21 Virtually designed potential antioxidant hybrid. | ||
Besides antibacterial activity, DPPH radical scavenging capability of coumarin–chalcone hybrids (50, Fig. 13) synthesized by Hamdi et al.57 was further evaluated. Of these hybrids with varied substituted B-ring of chalcone nucleus, compounds 79 and 80 (Fig. 22) showed the most potent antioxidant effect with EC50 values of 2.15 and 2.07 μM, respectively.
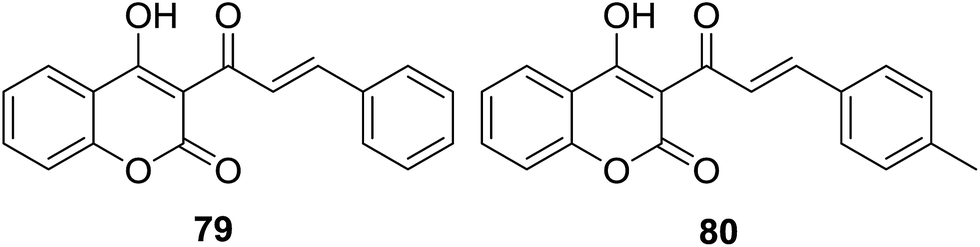 | ||
| Fig. 22 Coumarin–chalcone hybrids with promising antioxidant activity. | ||
Other activities
Six coumarin–chalcone hybrid based hA3 adenosine receptor (AR) antagonists (81–84, Fig. 23) were reported by Vazquez-Rodriguez and coworkers.73 ARs are distributed in different tissues of mammalian systems and regulate diverse physiological functions, being attractive targets for development of medicinal agents.74 The coumarin–chalcone derivatives were evaluated for their activity towards the subtypes of human AR (hA1, hA2A, and hA3) expressed in CHO (Chinese hamster ovary) cells by binding affinity assays.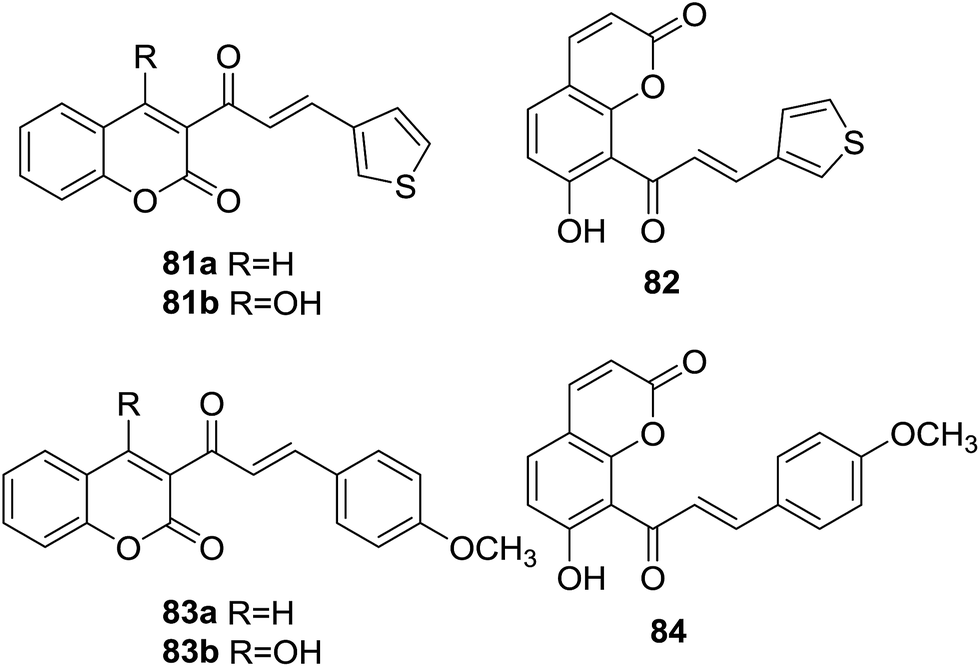 | ||
| Fig. 23 Coumarin–chalcone hybrid based hA3AR antagonists. | ||
It was demonstrated that compounds 81a and 82 bound with affinity to hA3AR in the low micromolar range [Ki values (dissociation constants) of 5160 and 5020 nM, respectively], which were comparable with the affinity of the classical naturally occurring antagonist theophylline. Detailed SAR indicated that presence of thiophenyl B-ring of chalcone scaffold was essential for their outstanding binding affinity. Absorption, distribution, metabolism and excretion (ADME) properties of these hybrids were further studied by theoretical evaluation. All six hybrids showed good logP values that were compatible with those required to cross membranes and they did not break any point of the Lipinski's rule of five, making them ideal leads for drug development as selective ligands of hA3ARs.
Provoked by the finding that lipophilicity of compounds often attributes better antitubercular activity, Ahmad et al. designed and synthesized several lipophilic hybrids based on prototype of coumarin–chalcone nucleus (85, Fig. 24). Their potency as antitubercular agents was investigated against Mycobacterium tuberculosis H37Rv strain.75
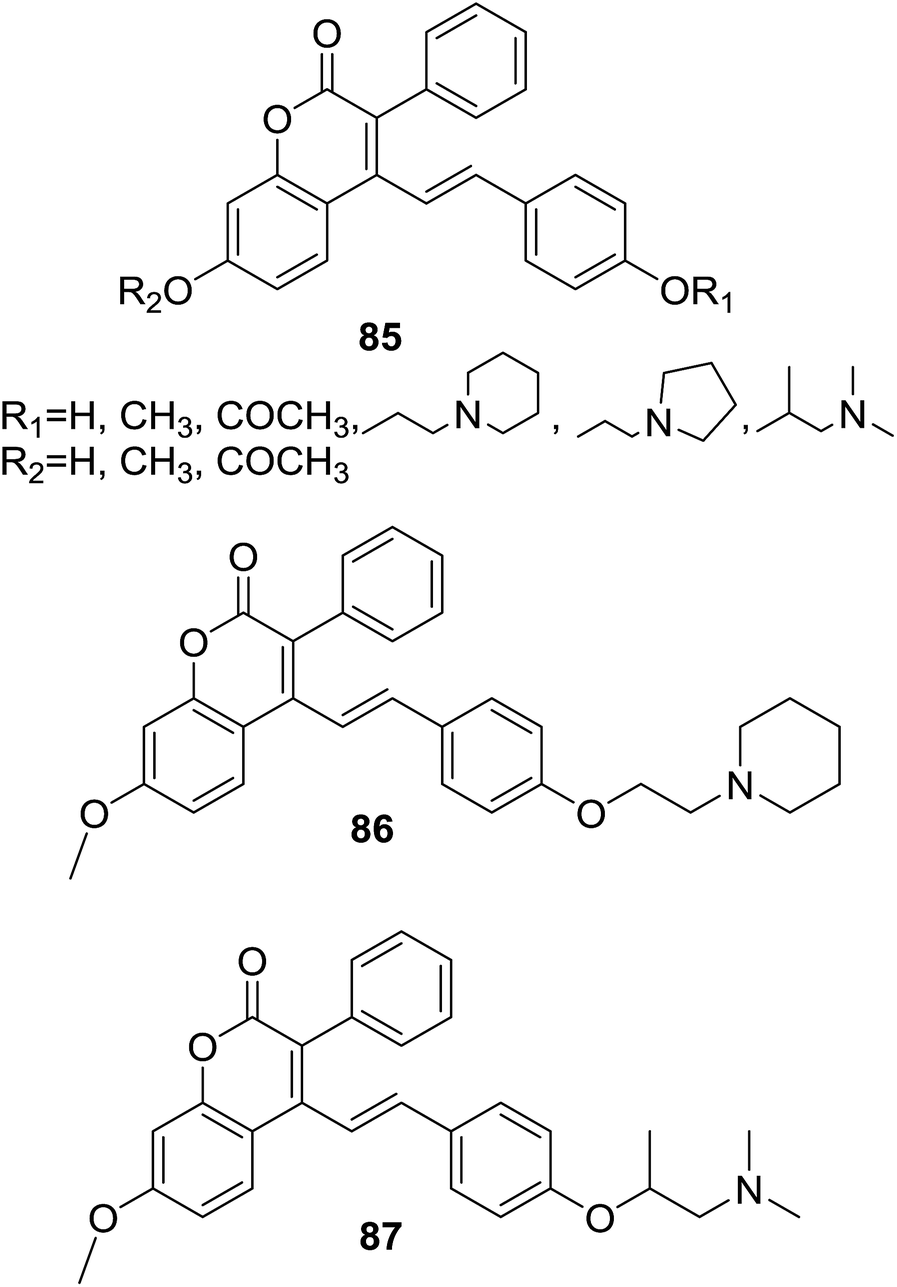 | ||
| Fig. 24 Coumarin–chalcone hybrids affording antitubercular activity. | ||
Compounds 86 and 87 (Fig. 24) exhibited promising antitubercular effect with minimum inhibitory concentrations (MIC) of 3.5 and 7.5 μg ml−1 respectively, comparable to that of antimalarial drugs rifampicin and streptomycin (MIC = 2.0 μg ml−1, respectively). Detailed investigation over these synthetic hybrids revealed that incorporation of the coumarin–chalcone hybrid with a nitrogen moiety significantly increased the antitubercular activity. Moreover, in vitro cytotoxicity assay on human epithelial kidney cell line (HEK-293) revealed that 86 was approximately 2.85 times selective towards tubercular versus healthy mammalian cells, indicating that it may be a promising molecule for development of novel antitubercular agents.
In order to assist rational design of more potent antitubercular agents, Yadav and coworker76 further conducted QSAR and docking studies on the antitubercular coumarin–chalcone hybrids (85, Fig. 24) synthesized by Ahmad et al.75 A QSAR model for screening active hybrids were developed based on multiple linear regression (MLR) statistical method by using leave-one-out (LOO) validation approach. The established QSAR model indicates a relationship between in vitro experimental activities and correlated five chemical properties, viz. heat of formation (kcal mol−1), lowest unoccupied molecular orbital energy (eV), and numbers of amine, hydroxyl, and methyl groups. Molecule docking study suggested that ENR (enoyl reductase) of M. tuberculosis (EnvM) was the potential target of active hybrids. In silico screening of ADME revealed that active coumarin–chalcone hybrids were highly correlated to the standard drug likeness parameters.
Attracted by the thought that hybrids affording dual or multiple modes of action may be able to effectively treat multifactorial diseases like metabolic syndrome, Sashidhara et al. designed a series of hybrids (88, Fig. 25) based on coumarin and chalcone scaffolds as lipid lowering agents.77 Sixteen coumarin–chalcone hybrids containing a fibrate unit were synthesized and their pharmacological activity was evaluated in triton WR-1339 induced hyperlipidemic rats.
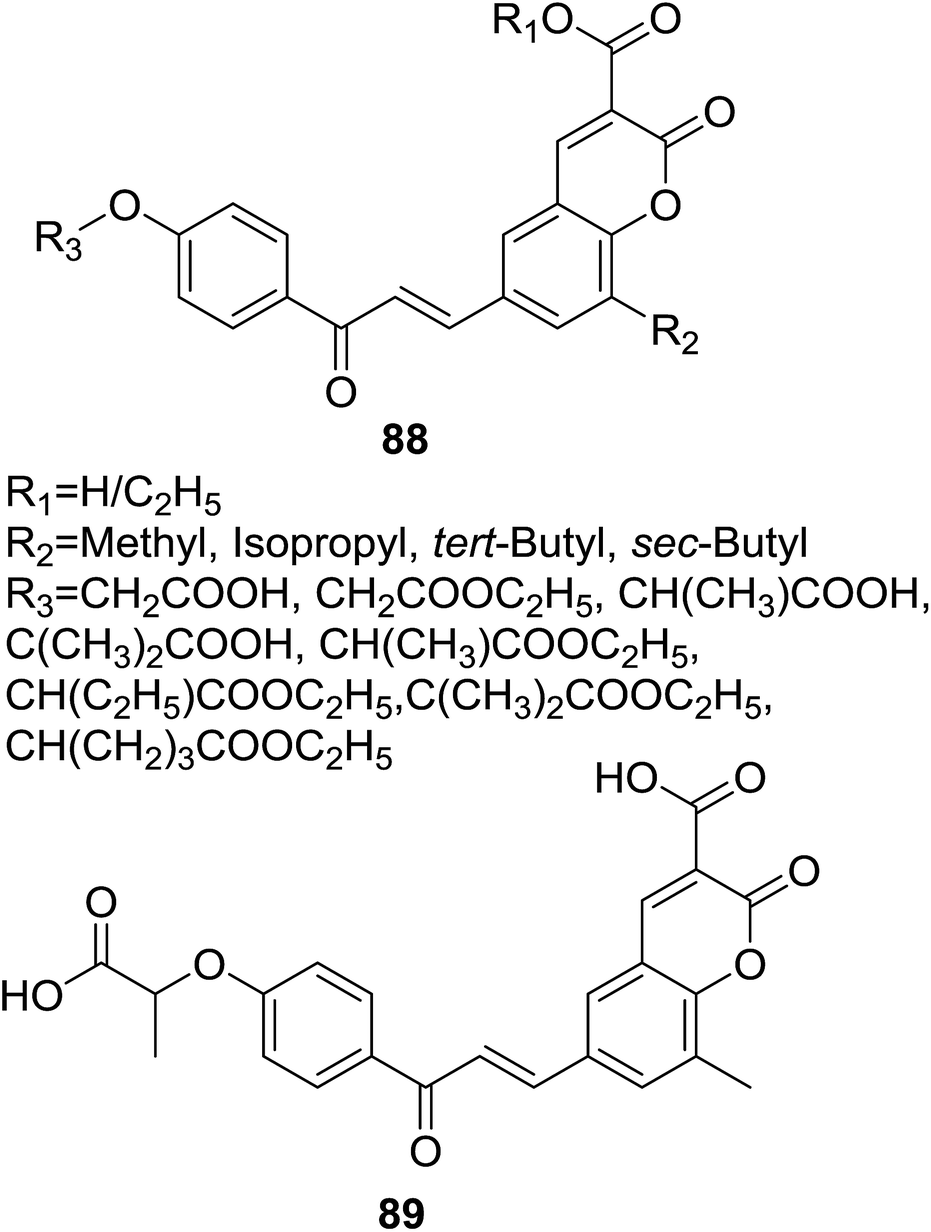 | ||
| Fig. 25 Coumarin–chalcone hybrid based fibrate like lipid lowering agents. | ||
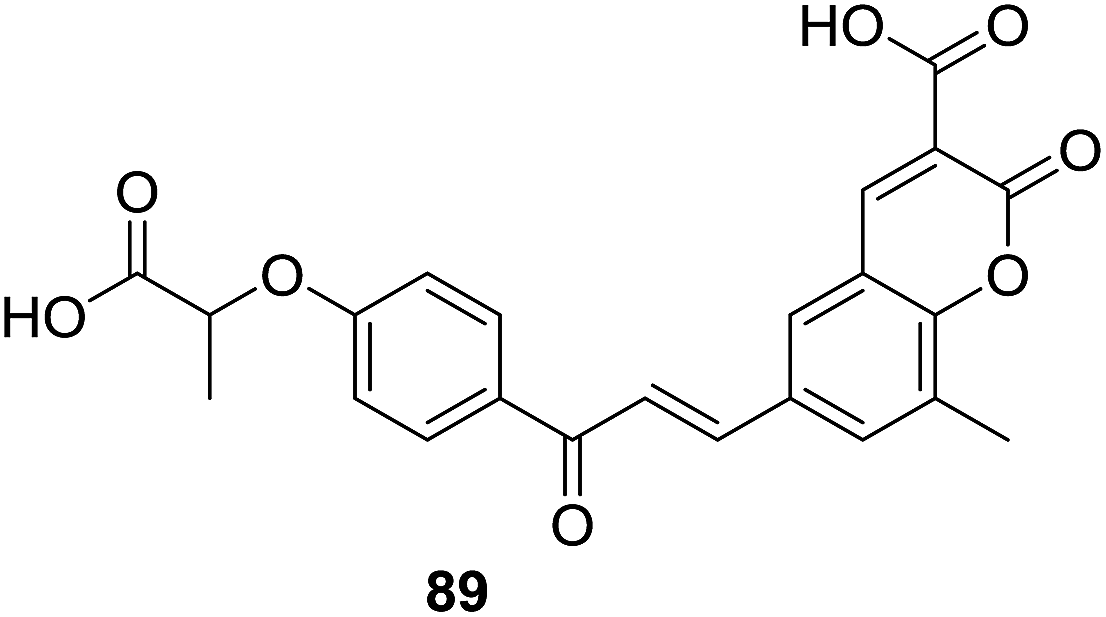
Results of lipid lowering activity screening indicated that hybrid 89 (Fig. 25) possessed most potent capability to decrease total cholesterol (TC), phospholipids (PL) and triglycerides (TG) of hyperlipidemic rats by 26%, 24%, and 25% respectively, at the dosage of 100 mg per kg body weight in triton WR-1339 induced model. Administration of 89 also reversed levels of VLDL (very low density lipoprotein), LDL (low density lipoprotein) and HDL (high density lipoprotein) as well as increased the LPL (lipoprotein lipase) activity in hyperlipidemic rats. The lipid lowering activity of 89 was comparable with the reference drug gemfibrozil which reduced levels of TC, PL and TG in plasma by 31%, 33% and 33%, respectively, at the same dose of 100 mg kg−1. SAR study of these hybrids revealed that, generally, acid derivative at C-3 position of coumarin core turned out to be more potent than their ester counterpart.
Fluorescent probes for detection and quantification of thiols and biothiols have been intensively developed due to their widely application in bioimaging analysis. García-Beltrán et al.78 reported two fluorescent probes (90, Fig. 26) affording coumarin–chalcone hybrid scaffold with a highly selective fluorescence enhancement with biothiols, such as cysteine (Cys), glutathione (GSH), homocysteine (Hcy) and cysteinylglycine (Cys-Gly) in SH-SY5Y cells. Similar reactivity toward biothiols was found for 90a and 90b, with the reactivity increases in the sequence Cys-Gly < Hcy < GSH < Cys. Test of in vivo imaging of biothiols revealed that both probes were present in the cytoplasm and exhibited a build-up detection by their fluorescence close to cell membrane. Hence, hybrids 90 are promising fluorescent probes for biothiol determination in living cells.
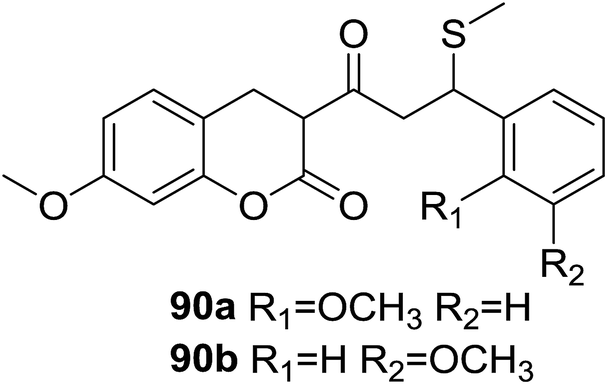 | ||
| Fig. 26 Fluorescent probes containing coumarin–chalcone hybrid scaffold. | ||
The biscoumarin–chalcone hybrids (70–72, Fig. 19) synthesized by Sashidhara and coworkers70 mentioned above were further screened for their anti-inflammatory activity of inhibiting the carrageenan-induced paw oedema in albino rats. Their study revealed that compound 91 (Fig. 27) possessed interesting anti-inflammatory property. Administration of 91 (100 mg per kg body weight) reduced 33% volume of the paw, while the reference drug, ibuprofen exhibited 59.5% protection at an equivalent dose. Moreover, potential of 91 against TNF-α by whole blood assay showed a 21% inhibition at dose of 400 μg ml−1. Thus, 91 presents an interesting molecule for development of anti-inflammatory agents.
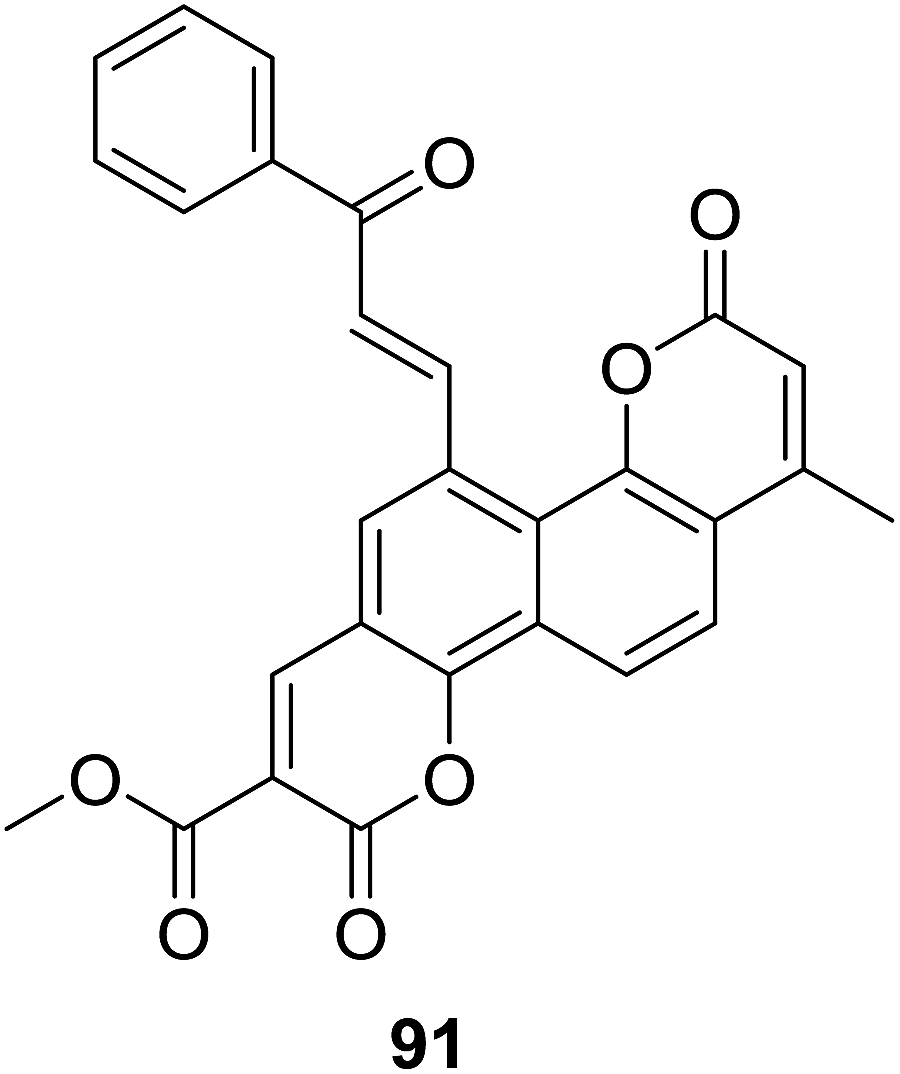 | ||
| Fig. 27 Anti-inflammatory biscoumarin–chalcone hybrid. | ||
Conclusion
Hybrid molecules are interesting templates for medicinal chemists to develop novel and effective drugs. Natural products bearing high bioactive potency are outstanding components to generate a hybrid molecule scaffold as therapeutic agents. Therefore, coumarin–chalcone hybrid that derived from two pronounced natural products with diverse bioactivities is an impressive lead that attracts many medicinal chemists' attention. Present mini review updates the knowledge of natural and synthetic coumarin–chalcone hybrids mainly as anti-cancer, anti-microbial, anti-malaria and antioxidant agents, many other effects like anti-inflammatory, antitubercular, fluorescent probe and lipid lowering activity are also covered. Some of most promising agents of coumarin–chalcone hybrid and their pharmacological properties are listed in Table 2. Moreover, the structure–activity relationships (SAR) included in this mini review may assist a researcher to choose appropriate nucleuses and functionalities to design rational coumarin–chalcone hybrids as agents against varied diseases.| Hybrid molecules | Pharmacological properties |
|---|---|
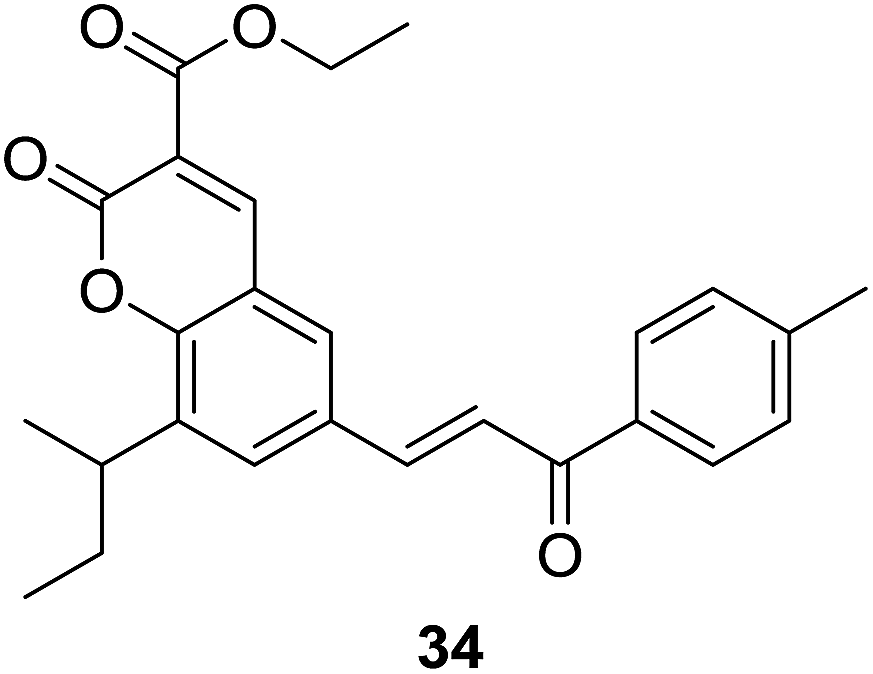 |
Anticancer |
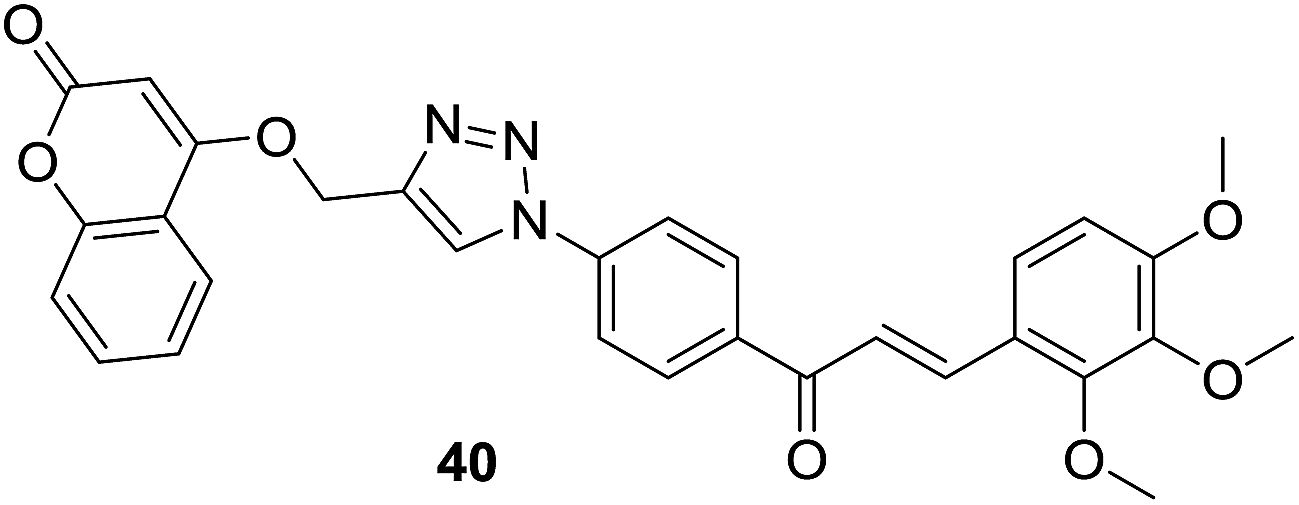 |
Antimalaria |
 |
Anti-tenacibaculosis |
 |
Antioxidant |
 |
Lipid lowering |
It is indicated that most developments of coumarin–chalcone hybrid have been achieved in anticancer, antibacterial and antioxidant fields (Fig. 28). Beside these properties, multifactorial diseases such as neurodegeneration and metabolic syndrome also may be promising fields that coumarin–chalcone hybrids could be introduced into, owing to the interesting feature of dual or multiple modes of action possessed by hybrid molecules. Rational modification or design based on the natural hybrid scaffolds listed in Table 1 is another favorable research direction in the field of coumarin–chalcone hybrids. Gather together, coumarin–chalcone hybrids possess reasonable value for development as therapeutic agents, which have been proven by the studies documented in this survey, and this mini review is expected to assist medicinal chemists for effective and successful development of coumarin–chalcone hybrids as therapeutic agents.
 | ||
| Fig. 28 Diverse pharmacological effects showed by coumarin–chalcone hybrids. | ||
Acknowledgements
Dr H. Wei is a recipient of Young Scholar Fund from The First Affiliated Hospital of Zhengzhou University.References
- C. Viegas-Junior, A. Danuello, V. da Silva Bolzani, E. J. Barreiro and C. A. Fraga, Curr. Med. Chem., 2007, 14, 1829–1852 CrossRef CAS PubMed.
- S. Sandhu, Y. Bansal, O. Silakari and G. Bansal, Bioorg. Med. Chem., 2014, 22, 3806–3814 CrossRef CAS PubMed.
- L. K. Gediya and V. C. Njar, Expert Opin. Drug Discovery, 2009, 4, 1099–1111 CrossRef CAS PubMed.
- B. Dasari, R. Jimmidi and P. Arya, Eur. J. Med. Chem., 2015, 94, 497–508 CrossRef CAS PubMed.
- X. Chen and M. Decker, Curr. Med. Chem., 2013, 20, 1673–1685 CrossRef CAS PubMed.
- M. Decker, Curr. Med. Chem., 2011, 18, 1464–1475 CrossRef CAS PubMed.
- I. A. Grigor'ev, N. I. Tkacheva and S. V. Morozov, Curr. Med. Chem., 2014, 21, 2839–2852 CrossRef.
- N. K. Sahu, S. S. Balbhadra, J. Choudhary and D. V. Kohli, Curr. Med. Chem., 2012, 19, 209–225 CrossRef CAS PubMed.
- D. I. Batovska and I. T. Todorova, Curr. Clin. Pharmacol., 2010, 5, 1–29 CrossRef CAS PubMed.
- K. N. Venugopala, V. Rashmi and B. Odhav, BioMed Res. Int., 2013, 2013, 963248 CAS.
- L. Wu, X. Wang, W. Xu, F. Farzaneh and R. Xu, Curr. Med. Chem., 2009, 16, 4236–4260 CrossRef CAS PubMed.
- N. K. Sahu, S. S. Balbhadra, J. Choudhary and D. V. Kohli, Curr. Med. Chem., 2012, 19, 209–225 CrossRef CAS PubMed.
- P. Singh, A. Anand and V. Kumar, Eur. J. Med. Chem., 2014, 85, 758–777 CrossRef CAS PubMed.
- K. V. Sashidhara, A. Kumar, M. Kumar, J. Sarkar and S. Sinha, Bioorg. Med. Chem. Lett., 2010, 20, 7205–7211 CrossRef CAS PubMed.
- R. Pingaew, A. Saekee, P. Mandi, C. Nantasenamat, S. Prachayasittikul, S. Ruchirawat and V. Prachayasittikul, Eur. J. Med. Chem., 2014, 85, 65–76 CrossRef CAS PubMed.
- S. Vazquez-Rodriguez, R. Lama Lopez, M. J. Matos, G. Armesto-Quintas, S. Serra, E. Uriarte, L. Santana, F. Borges, A. Munoz Crego and Y. Santos, Bioorg. Med. Chem., 2015, 23, 7045–7052 CrossRef CAS PubMed.
- Y. Xue, L. An, Y. Zheng, L. Zhang, X. Gong, Y. Qian and Y. Liu, Comput. Theor. Chem., 2012, 981, 90–99 CrossRef CAS.
- T. Quadri-Spinelli, J. Heilmann, T. Rali and O. Sticher, Planta Med., 2000, 66, 728–733 CrossRef CAS PubMed.
- H. Wei, G. Wu, X. Yang and J. Ruan, Chin. Tradit. Herb. Drugs, 2013, 44, 2354–2357 CAS.
- H. Wagner, O. Seligmann, M. V. Chari, E. Wollenweber, V. H. Dietz, D. M. Donnelly, M. J. Meegan and B. O'Donnell, Tetrahedron Lett., 1979, 20, 4269–4272 CrossRef.
- H. Wei, X. Zhang, G. Wu, X. Yang, S. Pan, Y. Wang and J. Ruan, Food Chem. Toxicol., 2013, 60, 147–152 CrossRef CAS PubMed.
- F. Asai, M. Iinuma, T. Tanaka and M. Mizuno, Phytochemistry, 1991, 30, 3091–3093 CrossRef CAS.
- V. H. Dietz, E. Wollenweber, J. Favre-Bonvin, P. Gómez and D. Luis, Z. Naturforsch., C: J. Biosci., 1980, 35, 36–40 Search PubMed.
- K. Nepali, S. Sharma, D. Kumar, A. Budhiraja and K. L. Dhar, Recent Pat. Anti-Cancer Drug Discovery, 2014, 9, 303–339 CrossRef CAS PubMed.
- S. Fortin and G. Berube, Expert Opin. Drug Discovery, 2013, 8, 1029–1047 CrossRef CAS PubMed.
- M. L. Go, X. Wu and X. L. Liu, Curr. Med. Chem., 2005, 12, 481–499 CrossRef CAS PubMed.
- A. J. Leon-Gonzalez, N. Acero, D. Munoz-Mingarro, I. Navarro and C. Martin-Cordero, Curr. Med. Chem., 2015, 22, 3407–3425 CrossRef CAS PubMed.
- S. Emami and S. Dadashpour, Eur. J. Med. Chem., 2015, 102, 611–630 CrossRef CAS PubMed.
- S. Valente, E. Bana, E. Viry, D. Bagrel and G. Kirsch, Bioorg. Med. Chem. Lett., 2010, 20, 5827–5830 CrossRef CAS PubMed.
- B. Aressy and B. Ducommun, Anti-Cancer Agents Med. Chem., 2008, 8, 818–824 CrossRef CAS PubMed.
- A. Lavecchia, C. Di Giovanni and E. Novellino, Expert Opin. Ther. Pat., 2010, 20, 405–425 CrossRef CAS PubMed.
- K. Patel, C. Karthikeyan, V. Raja Solomon, N. S. Hari Narayana Moorthy, H. Lee, K. Sahu, G. Singh Deora and P. Trivedi, Lett. Drug Des. Discovery, 2011, 8, 308–311 CrossRef CAS.
- N. Singh, J. Sarkar, K. V. Sashidhara, S. Ali and S. Sinha, Apoptosis, 2014, 19, 1017–1028 CrossRef CAS PubMed.
- C. J. Murray, L. C. Rosenfeld, S. S. Lim, K. G. Andrews, K. J. Foreman, D. Haring, N. Fullman, M. Naghavi, R. Lozano and A. D. Lopez, Lancet, 2012, 379, 413–431 CrossRef.
- N. J. White, J. Clin. Invest., 2004, 113, 1084–1092 CrossRef CAS PubMed.
- N. J. White, Lancet, 2014, 383, 1439–1440 CrossRef.
- N. Tadigoppula, V. Korthikunta, S. Gupta, P. Kancharla, T. Khaliq, A. Soni, R. K. Srivastava, K. Srivastava, S. K. Puri, K. S. R. Raju, P. S. Sijwali, V. Kumar and I. S. Mohammad, J. Med. Chem., 2013, 56, 31–45 CrossRef CAS PubMed.
- M. Chen, T. G. Theander, S. B. Christensen, L. Hviid, L. Zhai and A. Kharazmi, Antimicrob. Agents Chemother., 1994, 38, 1470–1475 CrossRef CAS PubMed.
- L. C. Mishra, A. Bhattacharya and V. K. Bhasin, Acta Trop., 2009, 109, 194–198 CrossRef CAS PubMed.
- J. N. Dominguez, J. E. Charris, G. Lobo, N. Gamboa de Dominguez, M. M. Moreno, F. Riggione, E. Sanchez, J. Olson and P. J. Rosenthal, Eur. J. Med. Chem., 2001, 36, 555–560 CrossRef CAS PubMed.
- M. Thillainayagam, L. Pandian, K. K. Murugan, V. Vijayaparthasarathi, S. Sundaramoorthy, A. Anbarasu and S. Ramaiah, J. Biomol. Struct. Dyn., 2015, 33, 961–977 CAS.
- Y. Z. Yang, A. Ranz, H. Z. Pan, Z. N. Zhang, X. B. Lin and S. R. Meshnick, Am. J. Trop. Med. Hyg., 1992, 46, 15–20 CAS.
- K. V. Sashidhara, A. Kumar, R. P. Dodda, N. N. Krishna, P. Agarwal, K. Srivastava and S. Puri, Bioorg. Med. Chem. Lett., 2012, 22, 3926–3930 CrossRef CAS PubMed.
- K. Patel, C. Karthikeyan, N. S. Hari Narayana Moorthy, G. S. Deora, V. R. Solomon, H. Lee and P. Trivedi, Med. Chem. Res., 2011, 21, 1780–1784 CrossRef.
- G. Wanare, R. Aher, N. Kawathekar, R. Ranjan, N. K. Kaushik and D. Sahal, Bioorg. Med. Chem. Lett., 2010, 20, 4675–4678 CrossRef CAS PubMed.
- J. Drebes, M. Kunz, C. A. Pereira, C. Betzel and C. Wrenger, Curr. Med. Chem., 2014, 21, 1809–1819 CrossRef CAS PubMed.
- M. S. Butler, M. A. Blaskovich and M. A. Cooper, J. Antibiot., 2013, 66, 571–591 CrossRef CAS PubMed.
- R. P. Rennie, in Antibiotic Resistance, Springer, Berlin, Heidelberg, 2012, pp. 45–65 Search PubMed.
- F. A. Chattha, M. A. Munawar, M. Nisa, M. Ashraf, S. Kousar and S. Arshad, Pak. J. Pharm. Sci., 2015, 28, 819–823 CAS.
- M. J. Matos, S. Vazquez-Rodriguez, L. Santana, E. Uriarte, C. Fuentes-Edfuf, Y. Santos and A. Munoz-Crego, Med. Chem., 2012, 8, 1140–1145 CAS.
- C. Canning, S. Sun, X. Ji, S. Gupta and K. Zhou, J. Ethnopharmacol., 2013, 147, 259–262 CrossRef CAS PubMed.
- D. Gupta and D. K. Jain, J. Adv. Pharm. Technol. Res., 2015, 6, 114–117 Search PubMed.
- B. Orlikova, D. Tasdemir, F. Golais, M. Dicato and M. Diederich, Genes Nutr., 2011, 6, 125–147 CrossRef CAS PubMed.
- S. Rodriguez, R. Guíñez, M. Matos, C. Azar and J. Maya, Med. Chem., 2015, 5, 173–177 Search PubMed.
- H. A. Deshpande, H. N. Chopde, C. P. Pandhurnekar and R. J. Batra, Chem. Sci. Trans., 2013, 2, 621–627 CrossRef.
- J. C. Trivedi, J. B. Bariwal, K. D. Upadhyay, Y. T. Naliapara, S. K. Joshi, C. C. Pannecouque, E. De Clercq and A. K. Shah, Tetrahedron Lett., 2007, 48, 8472–8474 CrossRef CAS.
- N. Hamdi, C. Fischmeister, M. C. Puerta and P. Valerga, Med. Chem. Res., 2010, 20, 522–530 CrossRef.
- S. Špirtović-Halilović, M. Salihovic, H. Dzudzevic-Cancar, S. Trifunovic, S. Roca, D. Softic and D. Zavrsnik, J. Serb. Chem. Soc., 2014, 79, 435–443 CrossRef.
- S. Naruka and S. Mahajan, Int. J. Res. Pharm. Chem., 2011, 1, 879–890 CAS.
- R. Mittler, Trends Plant Sci., 2002, 7, 405–410 CrossRef CAS PubMed.
- F. Giacco and M. Brownlee, Circ. Res., 2010, 107, 1058–1070 CrossRef CAS PubMed.
- K. J. Barnham, C. L. Masters and A. I. Bush, Nat. Rev. Drug Discovery, 2004, 3, 205–214 CrossRef CAS PubMed.
- M. Valko, C. Rhodes, J. Moncol, M. Izakovic and M. Mazur, Chem.-Biol. Interact., 2006, 160, 1–40 CrossRef CAS PubMed.
- H. Cai and D. G. Harrison, Circ. Res., 2000, 87, 840–844 CrossRef CAS PubMed.
- N. Tailor and M. Sharma, Mini-Rev. Med. Chem., 2013, 13, 280–297 CAS.
- G. B. Bubols, R. Vianna Dda, A. Medina-Remon, G. von Poser, R. M. Lamuela-Raventos, V. L. Eifler-Lima and S. C. Garcia, Mini-Rev. Med. Chem., 2013, 13, 318–334 CAS.
- G. L. Xi and Z. Q. Liu, Tetrahedron, 2014, 70, 8397–8404 CrossRef CAS.
- B. Jayashree, S. Arora and K. Venugopala, Asian J. Chem., 2008, 20, 1–7 CAS.
- F. Pérez-Cruz, S. Vazquez-Rodriguez, M. J. Matos, A. Herrera-Morales, F. A. Villamena, A. Das, B. Gopalakrishnan, C. Olea-Azar, L. Santana and E. Uriarte, J. Med. Chem., 2013, 56, 6136–6145 CrossRef PubMed.
- K. V. Sashidhara, M. Kumar, R. K. Modukuri, R. Sonkar, G. Bhatia, A. K. Khanna, S. Rai and R. Shukla, Bioorg. Med. Chem. Lett., 2011, 21, 4480–4484 CrossRef CAS PubMed.
- S. Vazquez-Rodriguez, R. Figueroa-Guíñez, M. J. Matos, L. Santana, E. Uriarte, M. Lapier, J. D. Maya and C. Olea-Azar, MedChemComm, 2013, 4, 993 RSC.
- G. Mazzone, N. Malaj, A. Galano, N. Russo and M. Toscano, RSC Adv., 2015, 5, 565–575 RSC.
- S. Vazquez-Rodriguez, M. J. Matos, L. Santana, E. Uriarte, F. Borges, S. Kachler and K. N. Klotz, J. Pharm. Pharmacol., 2013, 65, 697–703 CrossRef CAS PubMed.
- A. M. E. Olah and G. L. Stiles, Annu. Rev. Pharmacol. Toxicol., 1995, 35, 581–606 CrossRef PubMed.
- I. Ahmad, J. P. Thakur, D. Chanda, D. Saikia, F. Khan, S. Dixit, A. Kumar, R. Konwar, A. S. Negi and A. Gupta, Bioorg. Med. Chem. Lett., 2013, 23, 1322–1325 CrossRef CAS PubMed.
- D. K. Yadav, I. Ahmad, A. Shukla, F. Khan, A. S. Negi and A. Gupta, J. Chemom., 2014, 28, 499–507 CrossRef CAS.
- K. V. Sashidhara, G. R. Palnati, R. Sonkar, S. R. Avula, C. Awasthi and G. Bhatia, Eur. J. Med. Chem., 2013, 64, 422–431 CrossRef CAS PubMed.
- O. García-Beltrán, C. González, E. G. Pérez, B. K. Cassels, J. G. Santos, D. Millán, N. Mena, P. Pavez and M. E. Aliaga, J. Phys. Org. Chem., 2012, 25, 946–952 CrossRef.
| This journal is © The Royal Society of Chemistry 2016 |