
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Dinuclear uranium complexation and manipulation using robust tetraaryloxides†
Jordann A. L.
Wells
,
Megan L.
Seymour
,
Markéta
Suvova
and
Polly L.
Arnold
*
EaStCHEM School of Chemistry, The University of Edinburgh, The King's Buildings, Edinburgh EH9 3FJ, UK. E-mail: Polly.Arnold@ed.ac.uk; Fax: +44 (0)130 650 6453; Tel: +44 (0)130 650 5429
First published on 26th August 2016
Abstract
Two lower-oxidation state uranium cations can be readily combined in a robust, yet flexible and derivatisable, tetraaryloxide ligand framework, affording a new platform at which to use the multi-electron reductive capacity of the two actinide centres.
While single organometallic uranium centres are often capable of binding and reducing inert small molecules such as dinitrogen and carbon oxides, the most notable levels of activation and transformations are achieved almost exclusively by the combination of two uranium cations around one substrate.1,2
For example, dinitrogen overpressures as high as 80 psi are required to stabilise the terminal [(Cp*)3U(η1-N2)],3 and the first molecular uranium carbonyl [(Me3SiC5H4)3UCO] showed reversible CO binding in solution.4 However, two molecules of uranium tris(aryloxide) or tris(siloxide) UX3 (X = O-2,4,6-tBu3C6H2, OSi(2,4,6-Me3C6H2)3) in combination can effect the reduction of N2 to such an extent that the molecule [X3UIV(μ–η2:η2-N2)UIVX3] is stable in boiling toluene, and can reductively couple CO at ambient temperature and pressure to the ynediolate complex [X3UIV(OCCO)UIVX3],5,6 with further C–H and C–C bond formations possible. The conversion of aryl C–H to C–B bonds has also been possible in di-uranium(arene) complexes [X2UIV(μ–η6:η6-C6H5R)UIVX2] (R = H, alkyl, aryl).7 The recently reported reductive activation of CO2 by pairs of the uranium complexes [U(η-C8H6{SiR3}2)(η-CpR′)] (R = Me, iPr; R′ = Me4H, Me5, Me4iPr, Me4SiMe3, Me4Et) has been particularly instructive since the product (carbonate, oxo-bridged, or desirable C–C coupled oxalate) formed by trapping between the two uranium centres depends on the steric accessibility to the two U centres (rather than the redox capability).8,9
All these results suggest that a ligand pre-organised to hold two reducing U centres would be desirable if these small molecule activations are to be rendered catalytic, or better controlled. In collaboration with Love, we recently reported the use of expanded Pacman-shaped N-donor macrocycles to combine two UIII centres at a distance suitable for trapping a di-or triatomic fragment, but have been unable as yet to isolate complexes in which no X-ligand is coordinated between the two U centres.10,11 Recognising the strength of the U–aryloxide bond in a range of U oxidation states,7,12–16 we have developed a two-hour, one-pot, large-scale synthesis of three closely related analogues of a known arene-bridged tetraphenol17 in order to isolate and study the first O-donor compounds containing two discrete UIII or UIV centres in a single molecule, in geometries pre-organised for small molecule binding. The three phenols used here are H4LP and H4LM, and phenyl-substituted H4LP*, Fig. 1.
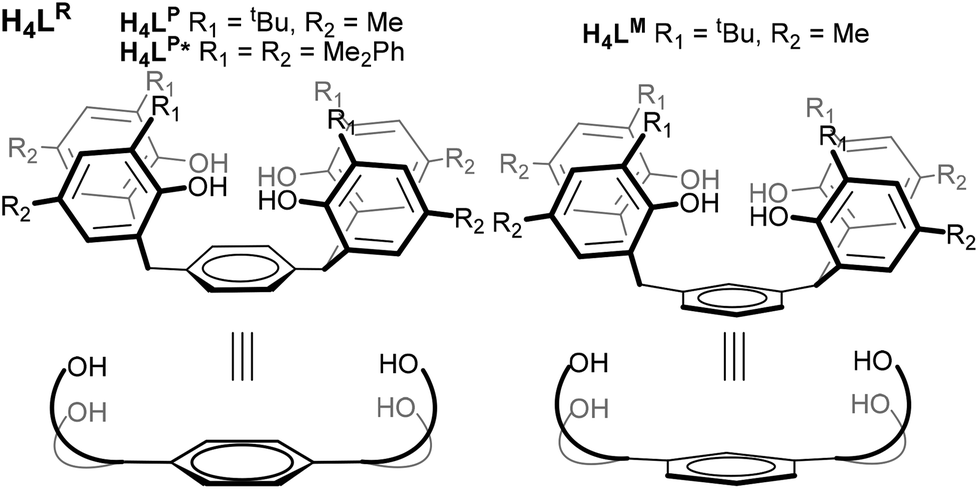 | ||
| Fig. 1 The substituted tetraphenols H4LR (with both para and meta substituted arene cores) and the new aryl-substituted H4LP*. | ||
Bimetallic salts of the phenols closely related to H4LP and H4LM in Fig. 1 (with R1 = R2 = tBu) have been demonstrated to be excellent ring opening polymerisation initiators for monomers including lactide (by H2K2LP and H2K2LM adducts),18 epoxide (by bis-AlIII adducts of LP with R1 = R2 = tBu),19 and ε-caprolactone (by bis-Nb or Ta adducts of LP with R1 = R2 = tBu).20 X-ray structural analyses in some of these complexes demonstrate a ligand flexibility that enables the metals to reside on the same or opposite sides of the central arene bridge.19,21
Both salt metathesis and protonolysis routes allow access to diuranium complexes of the tetraphenolates, as shown in Scheme 1.
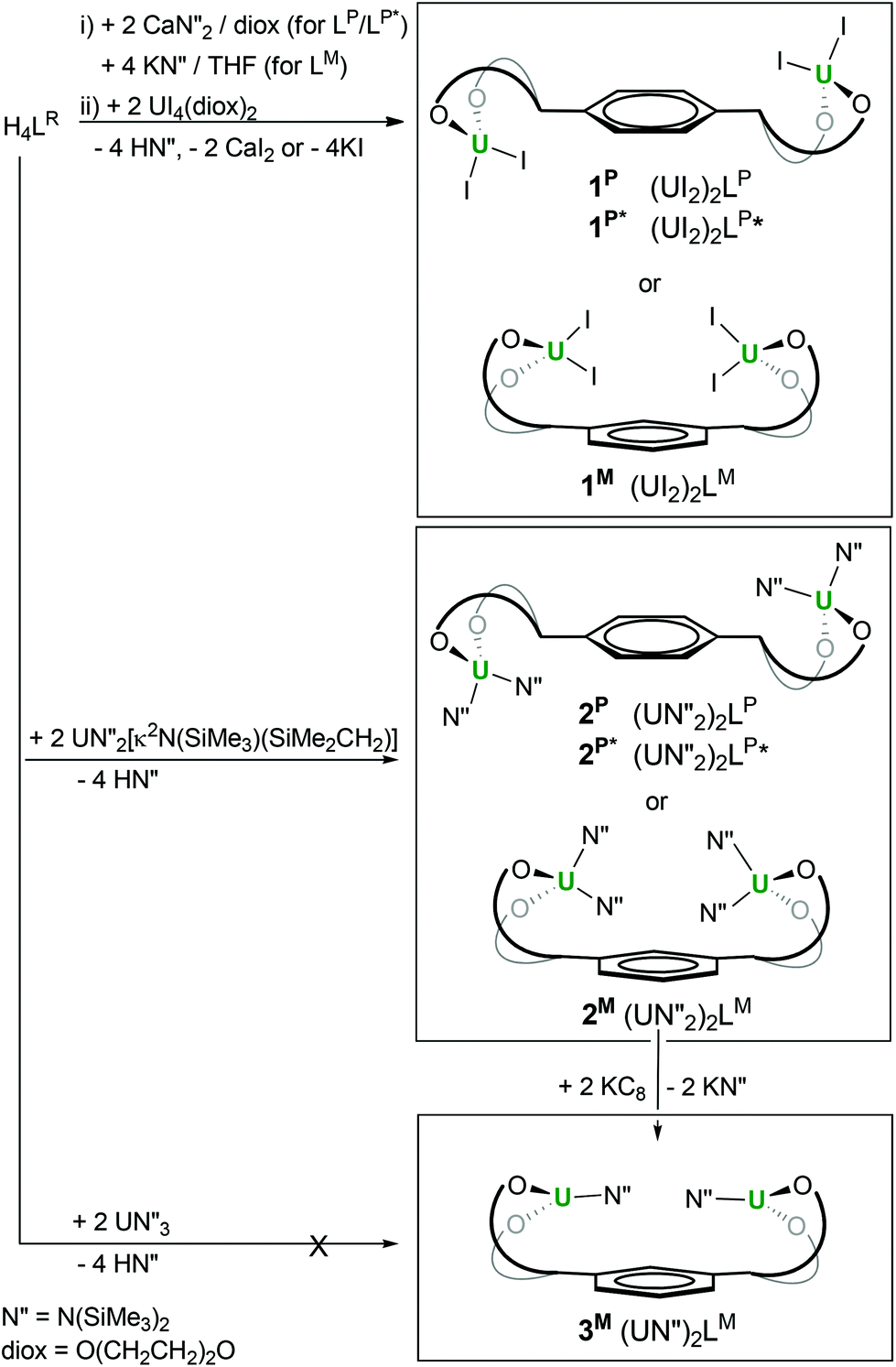 | ||
| Scheme 1 Syntheses of (UIV)2 and (UIII)2 complexes of bridged tetra-aryloxide ligands LP, LP* and LM. | ||
First, treatment of the in situ formed dicalcium salt Ca2LR (R = P, P*) or tetrapotassium K4LR (R = M) with two equivalents of UI4(diox)2 (diox = O(CH2CH2)2O, 1,4-dioxane) in THF or dioxane affords the green crystalline diuranium target complexes after work-up to remove salt by-products. The di-uranium complexes [{UI2(S)n}2LR], 1R, (R = P: S = THF, n = 3 or S = diox, n = 2; R = P*: S = THF, n = 2; R = M: S = THF, n = 2) can be isolated after work-up in excellent yields (65–80%).
Second, treatment of the proligand H4LR with two equivalents of the UIV metallacyclic silylamide UN′′2(N(SiMe3)SiMe2CH2) results in full deprotonation of all four acidic phenols to afford the unsolvated, yellow, crystalline [{UN′′2}2LR] 2R (R = P, P*, M), after work-up to eliminate the volatile, hexane soluble by-product HN′′, in essentially quantitative yields.
Complexes 1R and 2R have been fully characterised, including by single crystal X-ray diffraction. The in-situ synthesis of the calcium salts are described below as they have proven ideal metathesis precursors for some, since Group 1 bases often afford compounds that retain one or more bridging aryloxide protons.18
In our hands, direct syntheses of uranium(III) analogues of 1R and 2R from uranium(III) halide and amide starting materials were unsuccessful. We therefore investigated the electrochemical and chemical reduction of the uranium(IV) complexes. The UIV/III redox couple is known to range from −2.78 to −1.83 V versus ferrocene depending on the ligand environment.1,22,23 The cyclic voltammetry data show that the complexes 1R and 2R have one wave in the negative potential region attributable to the single electron reduction of both metal centres at the same time, and confirming the absence of UIV–UIV electronic communication through the ligand in all cases. The potentials of the complexes are collated in Table 1 and suggest that the uranium(III) complexes should be chemically accessible from a reaction with common one-electron reductants such as group 1 metals. The treatment of 2M with two equivalents of KC8 affords dark purple di-UIII [{UN′′}2LM], 3M in 63% yield after workup, which has been characterised by multinuclear NMR spectroscopy and elemental analysis, Scheme 1.
| Compound | Reduction potential at 100 mV s−1/V |
|---|---|
| 1M | −2.03 |
| 2M | −1.99 |
| 1P | −2.05 |
| 2P | −2.05 |
| 2P* | −1.53 |
Complexes 1, 2 and 3 are soluble in hot THF, dioxane, pyridine and arene solvents. The 1H NMR spectra of the iodide complexes 1 are moderately shifted by the UIV centres with resonances in the range 14 to 0 ppm whereas the amide complexes 2 are more significantly shifted with proton resonances spanning from 40 to −20 ppm.
Interestingly, following reduction of 2M to give 3M, the chemical shift range is decreased and proton resonances occur between 22 and −13 ppm. The 29Si resonance of the silylamide atom occurs at around −230 ppm for both 2P and 2M and is shifted to −100 ppm in 3M.
Single crystals of 1 and 2 were grown, details for which are in the ESI.† The molecular structures of 1P, 1P*, 1M, 2P and 2M are shown in Fig. 2 and 3; that for 2P* is in the ESI† along with the structure of a dioxane solvate of 1P, 1P(dioxane).
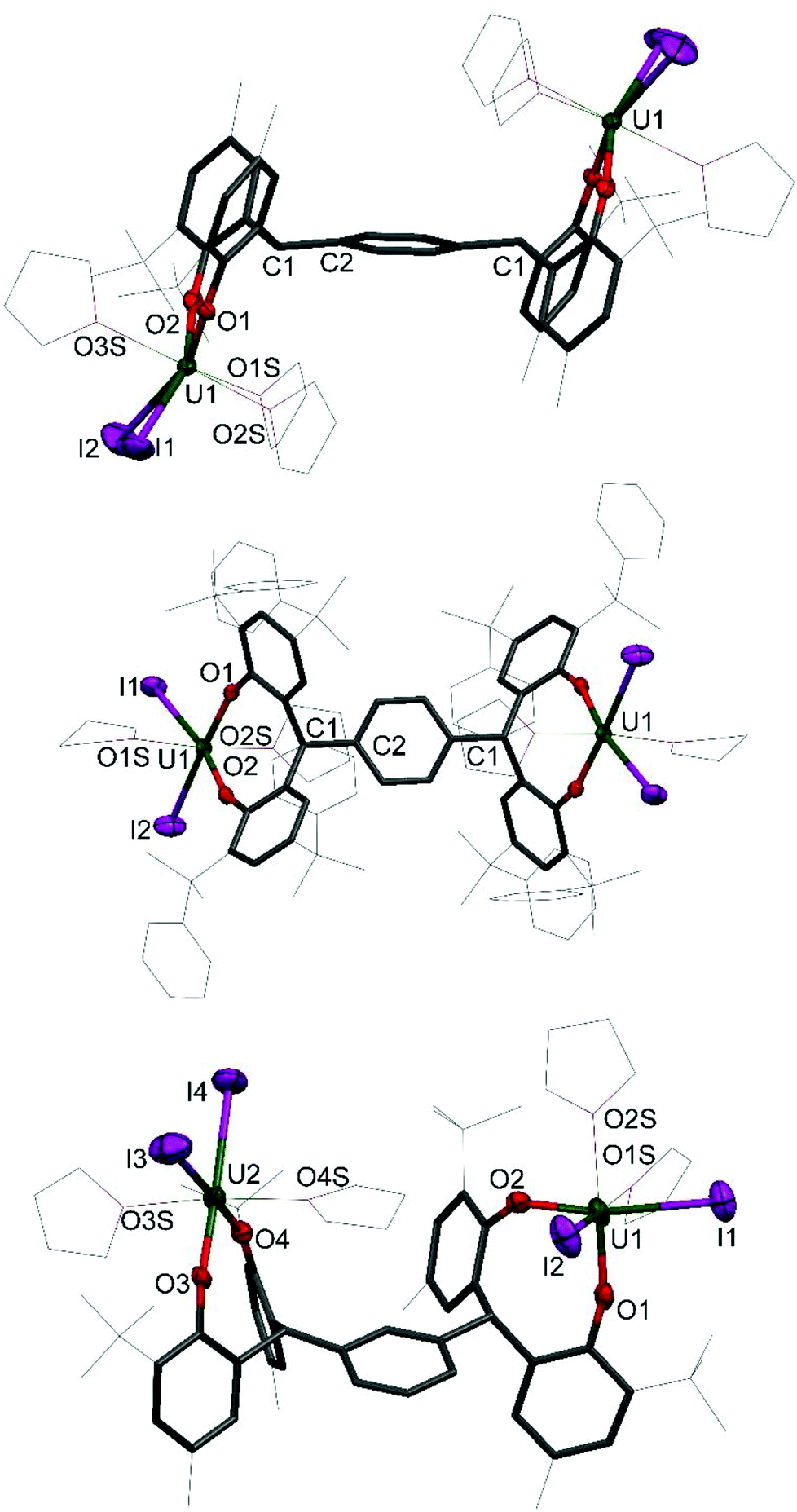 | ||
| Fig. 2 Solid-state structures of 1P (upper, side view), 1P* (middle, top view) and 1M (lower, side view). For clarity, hydrogen atoms and lattice solvent molecules are omitted (displacement ellipsoids are drawn at 50% probability, the remaining atoms and bonds shown as capped stick or wireframe). Selected bond lengths (Å) and angles (°) for 1P: U1–O1 2.108(5), U1–O2 2.117(5), U1–I1 3.0970(8), U1–I2 3.1094(8), U1–O1–C11 157.4(4), U1–O1–C21 156.8(4). For 1P*: U1–O1 2.105(4), U1–O2 2.106(4), U1–I1 3.0553(4), U1–I2 3.0472(5), U1–O1–C11 152.9(3), U1–O1–C21 159.1(3). For 1M: U1–O1 2.132(12), U1–O2 2.080(11), U2–O3 2.110(9), U2–O4 2.129(10), U1–I1 3.0643(16), U1–I2 2.9860(14), U2–I3 3.0109(18), U2–I4 3.0562(14), U1–O1–C11 138.7(9), U1–O1–C21 161.1(10), U2–O3–C31 154.1(9), U2–O4–C41 156.0(9). | ||
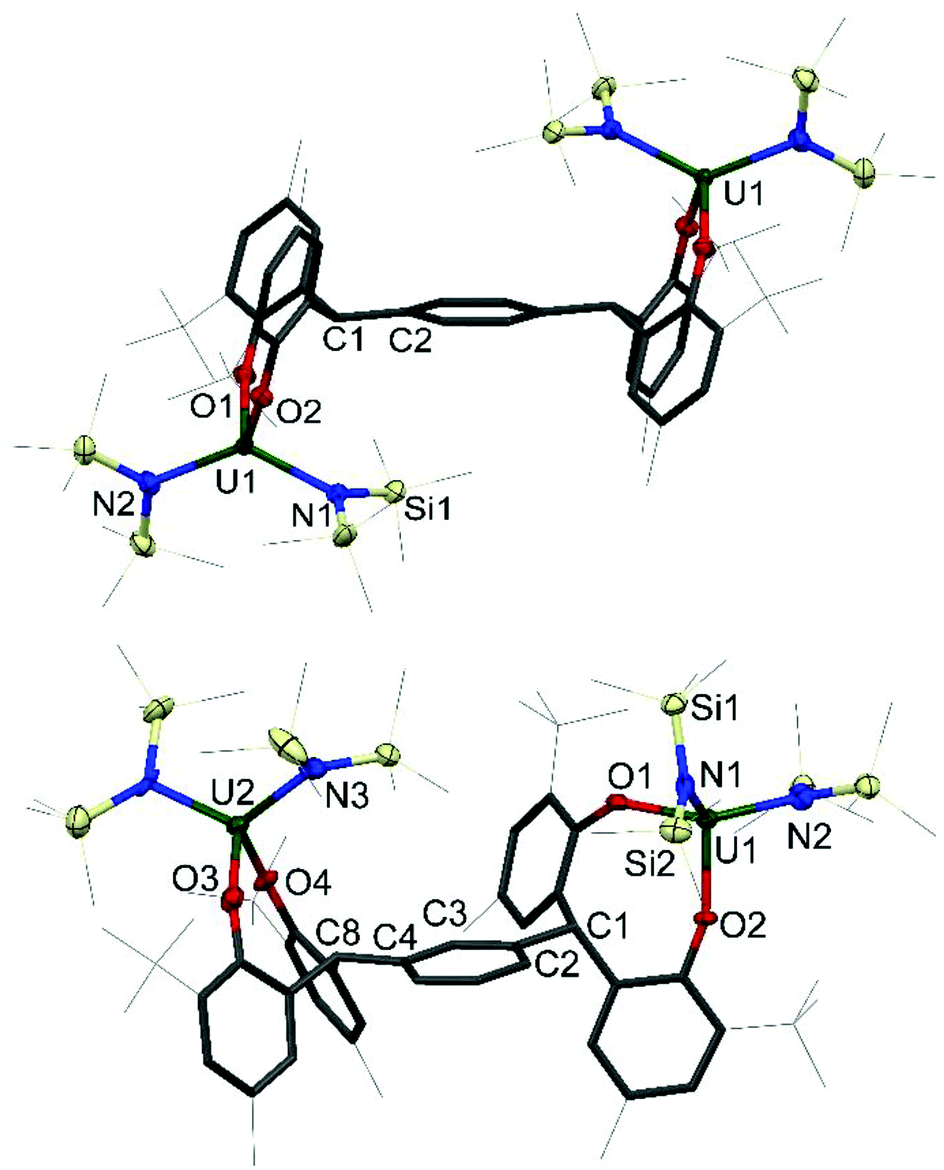 | ||
| Fig. 3 Solid-state structures of 2P (upper, side view), and 2M (lower, side view). For clarity, all methyl groups, hydrogen atoms, and lattice solvent molecules are omitted (displacement ellipsoids are drawn at 50% probability, the remaining atoms and bonds shown as capped stick). For 2P* see ESI.† Selected bond lengths (Å) and angles (°) for 2P: U1–O1 2.1033(13), U1–O2 2.1362(13), U1–N1 2.2580(16), U1–N2 2.2479(17), U1–O1–C11 152.17(12), U1–O2–C21 146.77(12); 2M: U1–O1 2.130(4), U1–O2 2.110(4), U2–O3 2.138(5), U2–O4 2.112(4), U1–N1 2.265(5), U1–N2 2.254(6), U2–N3 2.228(5), U2–N4 2.264(6), U1–O1–C11 141.5(4), U1–O2–C21 157.1(4) U2–O3–C31 138.5(4), U2–O4–C41 157.4(4). | ||
The uranium centre in 1P is seven-coordinate, adopting square face monocapped trigonal prismatic geometry, whereas the six coordinate uranium centres in 1P* and 1M adopt distorted octahedral geometry. The dioxane adduct of 1P, 1P(dioxane), also displays six coordinate uranium centres in distorted octahedral geometry. The equatorial plane is occupied by the aryloxide and iodide ligands, and the axial positions occupied by coordinated dioxane molecules. The exo-axial dioxanes act as a bridging ligand, linking the uranium centres in separate molecules to form a one-dimensional polymer in the solid state (see ESI† for further information).
The coordination environment of the two uranium centres in 1M differs. While both metal centres have a pseudo-octahedral geometry, the aryloxide and iodide ligands occupy the equatorial plane about U2 with the axial positions occupied by THF donor molecules in a trans arrangement. The THF donors about the U1 centre, however, are mutually cis occupying one equatorial and one axial position. The two iodides and one aryloxide group occupy the three remaining equatorial positions and the other aryloxide occupies the axial position. This surprising feature results in unsymmetrical bond lengths and angles in the solid state. The U1–O2 bond length is slightly shorter than the average of the three other bond lengths (2.080(11) Å and 2.124(10) Å respectively). Perhaps most notable is the distortion of the U1–O1–Cipso angle of 138.7(9)° compared to the average of the other three angles, 157.0(9)°.
The U–OAr bond distances in 1P, 1P* and 1M are very similar, with average distances of 2.112(5) Å, 2.106(4) Å and 2.120(10) Å respectively. These are comparable to previously reported uranium(IV) bis(aryloxo) bis(iodo) complexes such as I2U(ODtbp)2(thf) (ODtbp = O-2,6-tBu2C6H4) with an average U–O bond length of 2.076 Å,14 and I2U(OAr)2(thf)3 (Ar = O-4-tBuC6H4, O-2,6-Me2C6H3, C6F5) with average U–O distances of 2.068(8) Å, 2.091(8) Å and 2.120(6) Å respectively.24
The U–O–Cipso bond angles for the 1R complexes are bent, reminiscent of the homoleptic uranium(IV) complex U(ODtbp)4,25 with angles of 157.1(4)°, 156.0(3)°, 152.4(9)° for 1P, 1P* and 1M respectively, compared to 154.04(8)° for U(ODtbp)4. This is somewhat unusual, as the U–O–Cipso bond angles of other complexes of the type I2U(OAr)2 fall within the range 166.2(8)° to 176.9(8)°, and could be ascribed to the constraints imposed by the ligand frame.
The four-coordinate uranium centres in complexes 2P, 2P* and 2M all adopt a distorted tetrahedral geometry. As shown in Table 2, the complexes have comparable average U–O bond distances of 2.1198(13) Å, 2.1422(19) Å and 2.122(4) Å respectively, which is also true of the U–N bond distances of 2.2530(16) Å, 2.267(2) Å and 2.252(6) Å respectively. The slight elongation of U–O and U–N bonds in 2P* compared to 2P and 2M can be rationalised by the increased steric bulk around the metal centre in the larger tetraaryloxide framework interacting with the sterically demanding silylamide ligands. Similarly to the iodide complexes, the U–O–Cipso bond angles are closer to the homoleptic uranium(IV) aryloxide, as opposed to the I2U(OAr)2 analogues, with mean angles of 149.47(12)°, 150.72(17)° and 148.5(4)° for 2P, 2P* and 2M respectively. The U–O bond distances are comparable to that of U(ODtbp)4, as well as the tetrahedral mixed aryloxo-amido uranium(IV) complexes Et2NU(ODtbp)3 and N′′3U(ODtbp) with average U–O distances of 2.143(4) Å and 2.145(8) Å respectively.25–27 The U–N bond distances of 2R differ from the U–N distance of 2.161(5) Å exhibited by Et2NU(ODtbp)3 slightly, but agree very well with the U–N bond distances exhibited by N′′3U(ODtbp) and the uranium(V) complex N′′3U(Onapth)2 (napth = C10H7) of 2.284(10) Å and 2.222(6) Å respectively.21 This discrepancy in the U–N bond distance is presumably due to the difference in steric environment imposed by the bis(trimethylsilyl)amide ligand compared with that of the diethylamide ligand.
| Distance (Å)/angle (°) | 1p | 1* | 1m | 2p | 2* | 2m |
|---|---|---|---|---|---|---|
| U–U | — | — | 9.387 | — | — | 10.06 |
| U–O | 2.112 | 2.106 | 2.112 | 2.120 | 2.142 | 2.122 |
| U–I | 3.103 | 3.051 | 3.029 | — | — | — |
| U–N | — | — | — | 2.253 | 2.267 | 2.252 |
| OUO | 88.98 | 91.26 | 92.80 | 98.49 | 96.53 | 98.14 |
| IUI | 81.6 | 84.0 | 91.8 | — | — | — |
| NUN | — | — | — | 127.0 | 107.2 | 115.2 |
| UOC | 157.1 | 156.0 | 152.4 | 149.4 | 150.7 | 148.5 |
Inspection of the solid-state structures of these bimetallic derivatives, and of literature examples of other metal complexes suggests that the coordination of the two metals on the same side of the arene bridge is the preferred geometry in the meta-substituted LM complexes,18,20 but significantly rarer in the others, but is presumably not retained in solution for the smaller R substituted phenols. This could be due to the additional stability afforded by the generation of a dipole across the molecule.
Conclusions
Straightforward syntheses of dinuclear UIV complexes are possible using three different tetraphenolate ligands, in which a para- or meta-substituted phenyl backbone provide a strong yet flexible support to the two metal centres. The dinuclear UIII analogues are most readily accessible by chemical reduction. The meta-arene bridged ligand appears to favour the coordination of both metals on the same side of the bridge, a factor which may enhance the ability to reductively couple certain small molecules.Meta- and para-functionalised aryl imido and alkynide ligands have previously been used to demonstrate viable magnetic exchange between fn uranium centres.28,29 The properties are switched by changing the substitution patterns of the linking arene groups, and have been suggested to be of use in developing f-block magnetic materials for data storage, quantum computing or refrigeration applications. In addition to reactivity studies of these new potential multi-electron reductants, work is in progress to understand the magnetic behaviour of these new complexes.
General details
All moisture and air sensitive materials were manipulated using standard high-vacuum Schlenk-line techniques and MBraun gloveboxes and stored under an atmosphere of dried and deoxygenated dinitrogen. All gases were supplied by BOC gases UK. All glassware items, cannulae and Fisherbrand 1.2 μm retention glass microfibre filters were dried in a 160 °C oven overnight before use.Tetrahydrofuran and hexane for use with moisture and air sensitive compounds were dried using a Vac Atmospheres solvent purification system and stored over activated 4 Å molecular sieves. The solvent was cycled through a drying column containing molecular sieves for 12 hours before collection. 1,4-Dioxane for use with moisture and air sensitive compounds was refluxed over sodium for 3 days, distilled and collected into an ampoule containing 4 Å molecular sieves. All solvents were degassed and stored for 2 days prior to use. d6-Benzene and d8-tetrahydrofuran were freeze pump thaw degassed, refluxed over potassium for 24 hours and distilled by trap to trap distillation prior to use. All solvents were purchased from Sigma-Aldrich or Fisher Scientific.
Unless stated otherwise, all NMR spectroscopic analyses were recorded at 298 K using a Bruker Avance III 500.12 MHz spectrometer with 1H NMR spectra run at 500.12 MHz, and 29Si NMR spectra at 99.37 MHz. The 1H NMR spectra were referenced internally using residual solvent signals and are reported relative to external tetramethylsilane. Chemical shifts are quoted in ppm and coupling constants in Hz.
Experimental details
Potassium and sodium metal, 2-tert-butyl-4-methylphenol, 2,4-bis(dimethylbenzyl)phenol, terephthalaldehyde and isophthalaldehyde were purchased from Sigma-Aldrich and used as received. KN(SiMe3)2,30 [Ca(N(SiMe3)2]2,31 UI4·(dioxane)2,32 (((Me3Si)2)N)2U[κ2-N(SiMe3)SiMe2CH2]33 and KC8 were synthesised as previously described in literature procedures.34H4Lp
A two necked 250 cm3 round bottom flask was charged with 2-tert-butyl-4-methylphenol (41.80 g, 250 mmol, 4.4 eq.), terephthalaldehyde (7.5 g, 56 mmol, 1 eq.) and p-toluenesulfonic acid (1.06 g, 5.6 mmol, 0.1 eq.) and equipped with a stirrer bar and an oil bubbler. The flask was placed under nitrogen flow, stirred and heated to 110 °C. The solids melted to yield a yellow solution, which darkened with time. After circa 2 hours, the reaction mixture had turned to a reddish solid. The flask was allowed to cool to room temperature, at which point 50 cm3 of 20% H2O in MeCN solution was added. The resulting beige suspension was filtered to provide an off-white solid which was collected and washed with boiling ethanol. The resulting colourless solid was dried under vacuum at 65 °C overnight and stored in a glove box. Yield 27 g (64%).
1H NMR (C6D6, 500 MHz): δ 7.12 (Aryloxide H, J = 1.9 Hz, 4H), 7.06 (Aromatic H, 4H), 6.72 (Aryloxide H, J = 1.9 Hz, 4H), 5.56 (Ar3C![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) , 2H), 4.95 (ArOH, 4H), 2.06 (C3, 12H), 1.44 (tBu H, 36H).
, 2H), 4.95 (ArOH, 4H), 2.06 (C3, 12H), 1.44 (tBu H, 36H).
13C NMR (126 MHz, C6D6) δ 151.2, 140.0, 137.6, 130.1, 129.6, 128.1, (Aromatic C), 47.2 (Ar3CH), 34.6 (C(CH3)3), 29.6 (C(CH3)3), 20.7 (CH3).
Mass Spectrometry: (ESI) m/z 777.4850 [LP + Na]+.
H4Lm
A two necked 250 cm3 round bottom flask was charged with 2-tert-butyl-4-methylphenol (26.94 g, 161 mmol, 4.4 eq.), isophthalaldehyde (5.0 g, 37 mmol, 1 eq.) and p-toluenesulfonic acid (0.71 g, 3.8 mmol, 0.1 eq.) and equipped with a stirrer bar and an oil bubbler. The flask was placed under nitrogen flow, stirred and heated to 110 °C. The solids melted to yield a yellow solution, which darkened with time. After circa 2 hours, the reaction mixture had turned to a reddish solid. The flask was allowed to cool to room temperature, at which point 30 cm3 of MeCN was added. The resulting beige suspension was filtered to provide a colourless solid which was collected by filtration and washed with MeCN. The resulting colourless solid was dried under vacuum at 65 °C overnight and stored in a glove box. Yield 22.9 g (82%).
1H NMR (C6D6, 500 MHz) δ 7.09 (Aryloxide H, s, 4H), 7.06–6.96 (Aromatic H, m, 4H), 6.68 (Aryloxide H, s, 4H), 5.47 (Ar3C, s, 2H), 4.90 (ArOH, s, 4H), 2.05 (C3, s, 12H), 1.43 (tBu H, s, 36H).
13C NMR (C6D6, 126 MHz) δ 151.1, 141.8, 137.6, 130.7, 129.6, 128.4, 128.0 (Aromatic C), 47.6 (Ar3CH), 34.5 (C(CH3)3), 29.61 (C(CH3)3), 20.73 (CH3).
Elemental analysis: C 82.71%, H 8.81% calculated. C 82.83%, 8.92% found.
Mass Spectrometry: (ESI) m/z 777.4850 [LM + Na]+.
H4LP*
Analogous procedure to that used to synthesise H4LP. 82% yield.
1H NMR (C6D6, 600 MHz) δ 7.34–7.28 (Aromatic H, 12H), 7.20 (Aromatic H, 8H), 7.12 (Aromatic H, 8H), 7.07 (Aromatic H, 4H), 7.03 (Aromatic H, 4H), 6.99 (Aromatic H, 8H), 6.96–6.90 (Aromatic H, 4H), 6.89 (Aromatic H, 4H), 5.98 (Ar3C, 2H), 4.48 (ArOH, 4H), 1.65 (C3, 24H), 1.47 (C3, 24H).
13C NMR (C6D6, 151 MHz) δ 151.6, 150.2, 149.4, 142.0, 140.8, 135.4, 131.6, 129.4, 129.0, 127.4, 127.1, 126.6, 126.1, 125.8, 124.0 (Aromatic C), 44.6 (Ph3CH), 43.0 (CCH3), 42.3 (CCH3), 31.4 (CH3), 31.3 (CH3), 30.0 (CH3), 29.8 (CH3).
Mass Spectrometry: (ESI) m/z 1441.7983 [LP* + Na]+.
1P (U2I4LP)
Two 100 mL Schlenk flasks were charged respectively with [CaN′′2]2 (0.978 g, 1.36 mmol) and H4LP (1.024 g, 1.36 mmol) and equipped with stirrer bars. 30 mL of 1,4-dioxane was added to both solids to provide off-white solutions. The solutions were combined into one Schlenk with vigorous stirring and stirred for an hour at room temperature, to provide an off-white suspension. To a 250 mL Schlenk flask containing UI4(OC4H8O)2 and a stirrer bar, 100 mL of 1,4-dioxane was added to yield a slightly turbid red solution. The Ca2L suspension generated in situ was added to the UI4(OC4H8O)2 solution with vigorous stirring. Once the addition was complete, the reaction was left to stir for 48 hours to yield a light green suspension. The green brown solution was filtered and isolated from the colourless precipitate, and the solvent removed to give a yellow-brown solid (1.75 g, 65%). Crystals suitable for single crystal X-ray analysis were grown from slow evaporation of concentrated benzene, dioxane or THF solutions at room temperature.1H NMR (500 MHz, 329 K, THF-d8) δ 12.68 (Aryl, 4H), 10.39 (Aryl, 4H), 8.89 (Ar3CH, 2H), 6.63 (t-Bu, 36H), 4.25 (Me, 12H), 3.94 (Aryl, 4H).
Elemental analysis (dioxane adduct): C 38.45%, H 4.34% calculated. C 36.62%, H 4.36% found.
1P* (U2I4LP*)
Made analogously to U2I4LP. Green plates suitable for X-ray diffraction were grown from slow evaporation of a THF solution.
1H NMR (d8-THF, 329 K, 500 MHz) δ 7.13 (Aryl H, 2H), 7.08 (Aryl H, 8H), 6.98 (Aryl H, 4H), 6.94 (Aryl H, 4H), 6.84 (Aryl H, 2H), 6.69 (Aryl H, 2H), 6.57 (Aryl H, 8H), 6.47 (Aryl H, 4H), 6.42 (Aryl H, 2H), 5.65 (Aryl H, 4H), 2.31 (Ar3C, 1H), 2.21 (Ar3C, 1H), 1.45 (C3, 12H), 1.31 (Aryl H, 4H), 1.18 (C3, 12H), 1.02 (Aryl H, 4H), 0.78 (Aryl H, 4H).
Elemental analysis: C 53.62%, H 5.02% calculated. C 53.32%, H 5.16% found.
1M (U2I4LM)
A Schlenk flask was charged with H4LM (1.00 g, 1.32 mmol) and KN′′ (1.06 g, 5.30 mmol) and equipped with a stirrer bar. THF was added and the yellow solution was stirred for 1 hour at room temperature. To this solution, UI4(OC4H8O)2 (2.44 g, 2.65 mmol) in THF was added by cannula transfer from a separate Schlenk flask. The resulting dark green solution was stirred at room temperature for 48 hours, yielding a pale green suspension. The colourless precipitate was removed by filtration and the solvent was removed under reduced pressure giving a light green solid. (1.87 g, 82%). Green plate crystals suitable for single crystal X-ray analysis were grown from slow evaporation of concentrated benzene or thf solutions at room temperature.
1H NMR (d8-THF, 329 K, 500 MHz) δ 13.94 (Aryloxide H, 4H), 7.53 (Aryloxide H, 4H), 7.18 (t-Bu H, 36H), 6.92 (Aromatic H, 1H), 5.93 (C3, 12H), 5.60 (Aromatic H, 1H), 2.20 (Aromatic H, 2H), 0.90 (Ar3C, 2H).
Elemental analysis: C 40.37%, H 4.68% calculated. C 40.27%, 4.55% found.
2P (U2N′′4LP)
A Schlenk flask was charged with H4LP (100 mg, 0.133 mmol) and U(N′′)2(N{SiMe3}SiMe2CH2) (N′′ = N(SiMe3)2) (200 mg, 0.278 mmol, 2.1 eq.), a stirrer bar and hexanes (15 ml). The resulting dark brown suspension was allowed to stir at room temperature for 16 h during which time a colour change to olive green occurred. The reaction mixture was allowed to stand, and the off-white precipitate was isolated by filtration. The product was recrystallized from benzene solutions allowed to stand at room temperature to afford yellow plates of U2N′′4LP2P in 65% yield (247 mg). The yellow blocks were suitable for single crystal X-ray diffraction analysis.
1H NMR (C6D6, 500 MHz) δ 35.34 (Aryloxide H, 4H), 19.97 (Aryloxide H, 4H), 5.51 (Aromatic H, 4H), 4.40 (C3, 12H), −2.90 (Ar3C, 2H), −9.62 (t-BuH, 36H), −18.89 (SiCH3, 72H).
29Si NMR (C6D6, 99.4 MHz) δ −234.6 (Me3Si).
Elemental analysis: C 48.85%, H 7.23%, N 3.00% calculated. C 48.03%, H 7.10%, N 2.90% found.
2M (U2N′′4LM)
Made by an analogous procedure with recrystallisation by slow diffusion of hexane vapour into concentrated THF solutions to give green blocks suitable for single crystal X-ray diffraction analysis in 60% yield.
1H NMR (C6D6, 500 MHz) δ 41.19 (Aromatic H, 2H), 31.62 (Aromatic H, 1H), 27.73 (Aromatic H, 1H), 16.71 (Aryloxide H, 4H), 3.90 (Aryloxide H, 4H), 1.50 (C3, 12H), −3.03 (Ar3C, 2H), −9.76 (t-BuH, 36H), −18.51 (SiCH3, 72H) ppm; 29Si NMR (C6D6, 99.4 MHz) δ −230.8 (Me3Si).
Elemental analysis: C 48.85%, H 7.23%, N 3.00% calculated. C 48.66%, 6.91%, N 2.78% found.
2P* (U2N′′4LP*)
Made by an analogous procedure. Green plates isolated in 65% yield. Single crystals suitable for X-ray diffraction were grown by diffusion of hexanes into a THF solution of 2*.1H NMR (500 MHz, THF-d8) δ 13.12 (Aromatic H, 4H), 11.89 (Aromatic H, 8H), 9.05 (Aromatic H, 4H), 7.56 (Me H, 12H), 7.16 (Aromatic H, 8H), 7.05 (Aromatic H, 4H), 6.74 (Aromatic H, 4H), 6.65 (Aromatic H, 8H), 6.37 (Me H, 12H), 1.59 (Me H, 12H), 1.26 (Me H, 12H).
Elemental analysis: C 60.68%, H 6.92%, N 2.21% calculated. C 60.51%, H 6.93%, N 1.99% found.
3M (U2N′′2LM)
A Schlenk flask was charged with U2N′′4LM (274 mg, 0.223 mmol) and KC8 (64 mg, 0.447 mmol) and equipped with a stirrer bar. Toluene was added and the resulting dark green solution was stirred for 16 hours at room temperature, turning dark purple. The toluene was removed under reduced pressure and the product was extracted into heptane. The dark purple product was obtained as a powder following removal of volatiles under reduced pressure, (0.31 g, 74%).
1H NMR (C6D6, 500 MHz) δ 22.22 (Aromatic H, 2H), 17.50 (Aromatic H, 1H), 14.62 (Aromatic H, 1H), 11.85 (Aryloxide H, 4H), 7.50 (Aryloxide H, 4H), 2.11 (C3, 12H), 0.89 (Ar3C, 2H), −7.94 (s, 36H, tBu-H), −13.33 (s, 36H, SiCH3) ppm; 29Si NMR (C6D6, 99.4 MHz) δ −99.93 (Me3Si).
Elemental analysis: C 49.66%, H 6.38%, N 1.81% calculated. C 49.66%, H 6.38%, N 1.81% found.
Acknowledgements
We thank EaStCHEM, the University of Edinburgh and the Engineering and Physical Sciences Research Council EPSRC, grants EP/H004823/1 and EP/M010554/1. PLA also thanks the Technische Universität München – Institute for Advanced Study, funded by the German Excellence Initiative.Notes and references
- P. L. Arnold, Chem. Commun., 2011, 47, 9005 RSC.
- H. S. La Pierre and K. Meyer, in Progress in Inorganic Chemistry, John Wiley & Sons, Inc., Hoboken, New Jersey, 2014, vol. 58 Search PubMed.
- W. J. Evans, S. A. Kozimor and J. W. Ziller, J. Am. Chem. Soc., 2003, 125, 14264–14265 CrossRef CAS PubMed.
- J. G. Brennan, R. A. Andersen and J. L. Robbins, J. Am. Chem. Soc., 1986, 108, 335–336 CrossRef CAS.
- P. L. Arnold, Z. R. Turner, R. M. Bellabarba and R. P. Tooze, Chem. Sci., 2011, 2, 77–79 RSC.
- S. M. Mansell, J. H. Farnaby, A. I. Germeroth and P. L. Arnold, Organometallics, 2013, 32, 4214–4222 CrossRef CAS.
- P. L. Arnold, S. M. Mansell, L. Maron and D. McKay, Nat. Chem., 2012, 4, 668–674 CrossRef CAS PubMed.
- N. Tsoureas, O. T. Summerscales, F. G. N. Cloke and S. M. Roe, Organometallics, 2013, 32, 1353–1362 CrossRef CAS.
- N. Tsoureas, L. Castro, A. F. R. Kilpatrick, F. G. N. Cloke and L. Maron, Chem. Sci., 2014, 5, 3777 RSC.
- P. L. Arnold, C. J. Stevens, J. H. Farnaby, M. G. Gardiner, G. S. Nichol and J. B. Love, J. Am. Chem. Soc., 2014, 136, 10218–10221 CrossRef CAS PubMed.
- P. L. Arnold, J. H. Farnaby, R. C. White, N. Kaltsoyannis, M. G. Gardiner and J. B. Love, Chem. Sci., 2014, 5, 756–765 RSC.
- W. G. Van Der Sluys and A. P. Sattelberger, Chem. Rev., 1990, 90, 1027–1040 CrossRef CAS.
- W. G. Van Der Sluys, C. J. Burns, J. C. Huffman and A. P. Sattelberger, J. Am. Chem. Soc., 1988, 110, 5924–5925 CrossRef CAS.
- L. R. Avens, D. M. Barnhart, C. J. Burns, S. D. McKee and W. H. Smith, Inorg. Chem., 1994, 33, 4245–4254 CrossRef CAS.
- A. Walshe, J. Fang, L. Maron and R. J. Baker, Inorg. Chem., 2013, 52, 9077–9086 CrossRef CAS PubMed.
- R. J. Baker and A. Walshe, Chem. Commun., 2012, 48, 985–987 RSC.
- M. Janssen, L. Bini, B. Hamers, C. Müller, D. Hess, A. Christiansen, R. Franke and D. Vogt, Tetrahedron Lett., 2010, 51, 1971–1975 CrossRef CAS.
- J. Zhang, C. Jian, Y. Gao, L. Wang, N. Tang and J. Wu, Inorg. Chem., 2012, 51, 13380–13389 CrossRef CAS PubMed.
- L. Tang, E. P. Wasserman, D. R. Neithamer, R. D. Krystosek, Y. Cheng, P. C. Price, Y. He and T. J. Emge, Macromolecules, 2008, 41, 7306–7315 CrossRef CAS.
- Y. Al-Khafaji, X. Sun, T. J. Prior, M. R. J. Elsegood and C. Redshaw, Dalton Trans., 2015, 44, 12349–12356 RSC.
- A. Cottone and M. J. Scott, Organometallics, 2000, 19, 5254–5256 CrossRef CAS.
- D. E. Morris, R. E. Da Re, K. C. Jantunen, I. Castro-Rodriguez and J. L. Kiplinger, Organometallics, 2004, 23, 5142–5153 CrossRef CAS.
- A. Vallat, E. Laviron and A. Dormond, J. Chem. Soc., Dalton Trans., 1990, 921–924 RSC.
- D. D. Schnaars, G. Wu and T. W. Hayton, Dalton Trans., 2009, 3681 RSC.
- J. M. Berg, D. L. Clark, J. C. Huffman, D. E. Morris, A. P. Sattelberger, W. E. Streib, W. G. Van der Sluys and J. G. Watkin, J. Am. Chem. Soc., 1992, 114, 10811–10821 CrossRef CAS.
- P. B. Hitchcock, M. F. Lappert, A. Singh, R. G. Taylor and D. Brown, J. Chem. Soc., Chem. Commun., 1983, 561–563 RSC.
- A. J. Lewis, K. C. Mullane, E. Nakamaru-Ogiso, P. J. Carroll and E. J. Schelter, Inorg. Chem., 2014, 53, 6944–6953 CrossRef CAS PubMed.
- R. K. Rosen, R. A. Andersen and N. M. Edelstein, J. Am. Chem. Soc., 1990, 112, 4588–4590 CrossRef CAS.
- B. S. Newell, A. K. Rappé and M. P. Shores, Inorg. Chem., 2010, 49, 1595–1606 CrossRef CAS PubMed.
- S. M. Mansell, B. F. Perandones and P. L. Arnold, J. Organomet. Chem., 2010, 695, 2814–2821 CrossRef CAS.
- O. Michel, C. Meermann, K. W. Törnroos and R. Anwander, Organometallics, 2009, 28, 4783–4790 CrossRef CAS.
- M. J. Monreal, R. K. Thomson, T. Cantat, N. E. Travia, B. L. Scott and J. L. Kiplinger, Organometallics, 2011, 30, 2031–2038 CrossRef CAS.
- A. Dormond, A. El Bouadili, A. Aaliti and C. Moise, J. Organomet. Chem., 1985, 288, C1–C5 CrossRef CAS.
- L. M. Viculis, J. J. Mack, O. M. Mayer, H. T. Hahn and R. B. Kaner, J. Mater. Chem., 2005, 15, 974 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Full synthetic and characterising data. Crystallographic tables. CCDC 1478886–1478892. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6dt02630c |
| This journal is © The Royal Society of Chemistry 2016 |