
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Metal–ligand cooperative activation of nitriles by a ruthenium complex with a de-aromatized PNN pincer ligand†
Linda E.
Eijsink
a,
Sébastien C. P.
Perdriau
a,
Johannes G.
de Vries
ab and
Edwin
Otten
*a
aStratingh Institute for Chemistry, University of Groningen, Nijenborgh 4, 9747 AG Groningen, The Netherlands. E-mail: edwin.otten@rug.nl
bLeibniz-Institut für Katalyse an der Universität Rostock, Albert-Einstein-Strasse 29a, 18059 Rostock, Germany
First published on 25th August 2016
Abstract
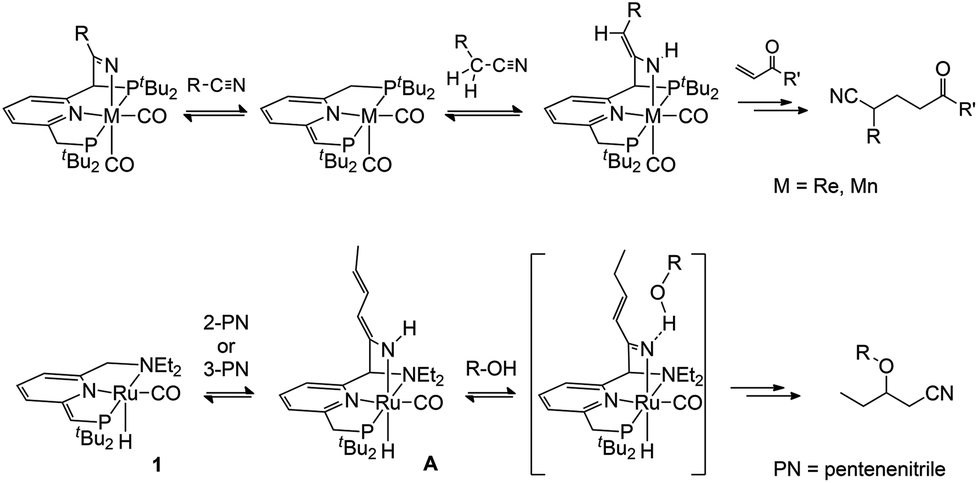
The pincer complex (PNN)RuH(CO), with a de-aromatized pyridine in the ligand backbone, is shown to react with nitriles in a metal–ligand cooperative manner. This leads to the formation of a series of complexes with new Ru–N(nitrile) and C(ligand)–C(nitrile) bonds. The initial nitrile cycloaddition products, the ketimido complexes 3, have a Brønsted basic (nitrile-derived) Ru–N fragment. This is able to deprotonate a CH2 side-arm of the pincer ligand to give ketimine complexes (4) with a de-aromatized pyridine backbone. Alternatively, the presence of a CH2 group adjacent to the nitrile functionality can lead to tautomerization to an enamido complex (5). Variable-temperature NMR studies and DFT calculations provide insight in the relative stability of these compounds and highlight the importance of their facile interconversion in the context of subsequent nitrile transformations.
Introduction
Organometallic complexes with tridentate pincer-type ligands have found widespread application in catalysis due to their tuneable nature and the robustness of the resulting transition metal complexes.1 Moreover, ligands of this type have attracted increasing attention in recent years also as a result of their ability to activate relatively strong bonds via a pathway that directly involves a (non-spectator) ligand site in bond breaking, called ‘metal–ligand cooperation’ (MLC).2 Using this strategy, a reduction in bond order within the substrate is achieved which leads to either complete bond cleavage (in case of a single bond)3 or weakening (in case of a CO or CN multiple bond). As examples for the latter, the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond in CO2
O bond in CO2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 4 or organic carbonyl compounds5 may be activated using metal complexes with de-aromatized pyridine-based pincer ligands. Similarly, metal–ligand cooperative binding/activation of nitriles was reported by Milstein,6 Pidko,7 and our group,8 and this has led to new reactivity involving organic nitriles as either Michael donors6 or acceptors8 (Chart 1). The synergistic effect of a Lewis basic fragment (the deprotonated pincer ‘arm’) and a Lewis acid (the metal centre) is reminiscent of Frustrated Lewis Pairs (FLPs),9 and nitrile activation using an Al/P-based FLP was recently reported.10
4 or organic carbonyl compounds5 may be activated using metal complexes with de-aromatized pyridine-based pincer ligands. Similarly, metal–ligand cooperative binding/activation of nitriles was reported by Milstein,6 Pidko,7 and our group,8 and this has led to new reactivity involving organic nitriles as either Michael donors6 or acceptors8 (Chart 1). The synergistic effect of a Lewis basic fragment (the deprotonated pincer ‘arm’) and a Lewis acid (the metal centre) is reminiscent of Frustrated Lewis Pairs (FLPs),9 and nitrile activation using an Al/P-based FLP was recently reported.10
 | ||
| Chart 1 Examples of metal–ligand cooperative binding of nitriles from the literature, and subsequent (catalytic) Michael addition reactivity.6,8 | ||
In many of the (catalytic) reactions involving metal–ligand cooperative bond activation, several different species are observed in solution due to competition between different potential substrates, tautomerization/rearrangement reactions of the activated substrates, or both. For example, our group reported that catalytic oxa-Michael addition of alcohols to unsaturated nitriles using 1 proceeds via metal–ligand cooperative activation of the nitrile substrate, but may be inhibited by the competing MLC activation of the alcohol.8 Milstein and co-workers showed the presence of various tautomers in equilibrium upon binding of organic nitriles to de-aromatized PNP Re and Mn pincer complexes.6 Key to the observed chemistry is that these compounds can readily interconvert and have similar energies so that the catalyst does not get trapped in a thermodynamically stable but catalytically inactive state. A better insight in the factors that govern these equilibria is important for understanding and designing improved catalysts that make use of MLC.
Results and discussion
Reaction of 1 with nitriles: identification of products
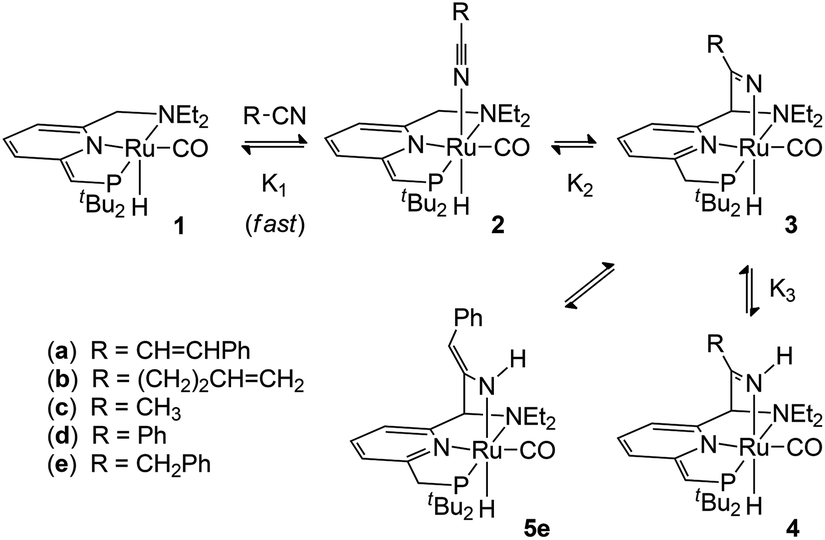
Building on our recently reported results on the activation of 2- and 3-pentenenitrile towards oxa-Michael addition reactivity with Milstein's de-aromatized PNN Ru complex 1,8 we tested the reactivity of the less activated substrate cinnamonitrile as the Michael acceptor in isopropanol with a catalytic amount of 1. While no oxa-Michael addition was observed, NMR spectroscopy indicated the catalyst 1 to be fully consumed. A stoichiometric reaction between 1 and cinnamonitrile in a teflon-sealed NMR tube in C6D6, either in the presence or absence of isopropanol, indicated clean conversion of 1 to a single ruthenium complex (4a) that contains a cinnamonitrile-derived fragment. The 1H NMR spectrum of the reaction mixture showed a broadened singlet at δ 9.68 ppm which is assigned to a NH moiety. A set of resonances at δ 6.54, 6.38 and 5.71 ppm due to the pyridine backbone of the pincer ligand indicates that it remains de-aromatized. A doublet at δ 3.77 ppm in the 1H NMR shows coupling to 31P (J = 2.8 Hz) and is correlated to a 13C resonance at δ 69.07 ppm (JP,C = 54 Hz), suggesting the presence of aCHPtBu2 moiety. The NMR data allow us to assign the structure of 4a as the NH-ketimine complex shown in Scheme 1. In analogy with the reactivity of 1 towards 2- and 3-pentenenitrile8 and the data of Milstein and co-workers for related nitrile reactivity of PNP Re and Mn complexes,6 the following pathway for the formation of 4a is proposed. Initial interaction of the nitrile with compound 1 results in the formation of the Lewis acid–base adduct 2a. Tautomerization of the reactive, unsaturated moiety in the pincer ligand from the PtBu2 to the NEt2 side-arm11 results in metal–ligand cooperative nitrile binding (via C–C and Ru–N bond formation) to give 3a. That nitrile addition takes place at the NEt2 side-arm is in agreement with previous studies in which CO/C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N addition reactions are shown to be thermodynamically more favourable at this site.4e,8 Subsequent transformation of the intermediate 3a occurs via deprotonation of the pincer CH2P sidearm by the Brønsted basic Ru–N fragment to give the de-aromatized compound 4a as the final product. Attempts to isolate the compound in pure form met with failure: after several hours in solution, decomposition is noticeable in the 1H NMR spectrum and also quick workup of the mixture by removal of the volatiles resulted in an intractable mixture.
N addition reactions are shown to be thermodynamically more favourable at this site.4e,8 Subsequent transformation of the intermediate 3a occurs via deprotonation of the pincer CH2P sidearm by the Brønsted basic Ru–N fragment to give the de-aromatized compound 4a as the final product. Attempts to isolate the compound in pure form met with failure: after several hours in solution, decomposition is noticeable in the 1H NMR spectrum and also quick workup of the mixture by removal of the volatiles resulted in an intractable mixture.
 | ||
| Scheme 1 Products obtained from the binding of nitriles to complex 1. | ||
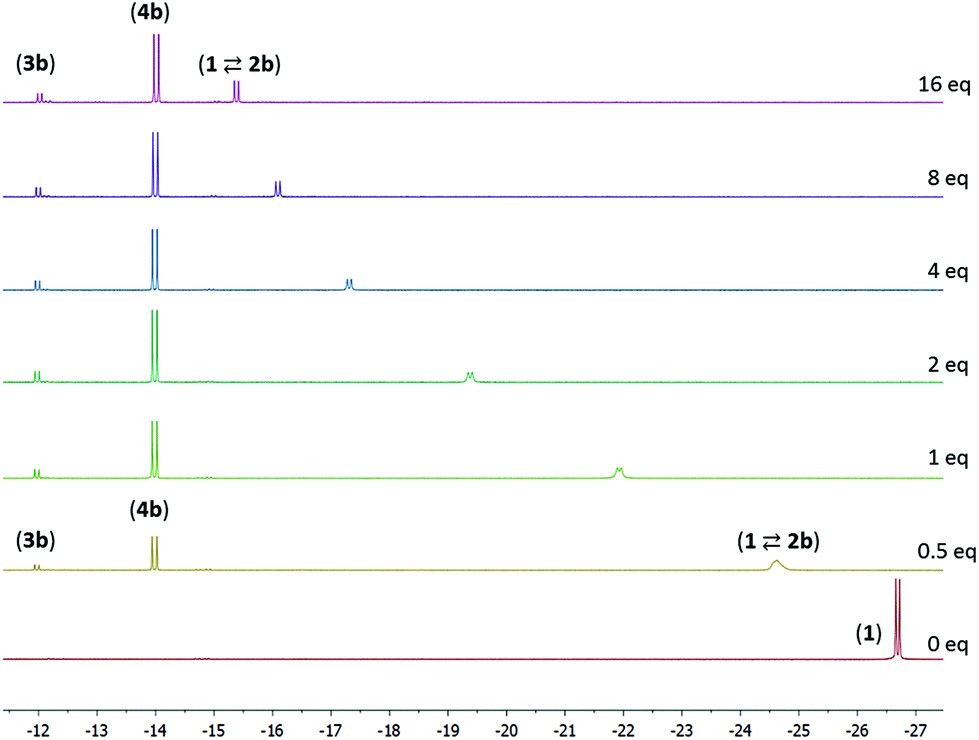
The reaction of compound 1 with 4-pentenenitrile in C6D6 was investigated. Whereas the unsaturated compounds 2- and 3-pentenenitrile (with the nitrile conjugated with the CC bond or in the allylic position, respectively) both lead to formation of compound A (Chart 1) via a series of tautomerization reactions,4e,8 the reaction with 4-pentenenitrile reproducibly leads to a different set of products. Analysis of the reaction mixture by NMR spectroscopy indicated the presence of more than one species, as evidenced for example by the appearance of two new major 31P NMR resonances at 106.0 and 96.1 and a minor species at δ 121.0 ppm, that account for ca. 49%, 43% and 8%, respectively, of the total signal intensity. The reaction is complete within 10 min as judged by NMR spectroscopy, and the composition of the mixture remains unchanged for at least 24 h. The 1H NMR spectrum contains resonances that can be attributed to two major Ru–H species at −13.85 and −21.07 ppm, and a minor component with a Ru–H resonance at −11.88 ppm with integrations that match those observed in the 31P NMR spectrum. The new Ru–H signal at −21.07 ppm is broadened, which is likely due to chemical exchange: the position of this Ru–H signal is highly dependent on the concentration of 4-pentenenitrile (Fig. 1; vide infra), suggesting that it is due to an equilibrium that involves 4-pentenenitrile. Analysis of 2D NMR data (COSY, HSQC, HMBC) allowed the assignment of the major sharp Ru–H resonance at −13.85 ppm to the 4-pentenenitrile cycloaddition product 4b, a ketimine adduct with a de-aromatized pincer backbone analogous to that observed in the reaction with cinnamonitrile (4a). For the other species that give rise to the exchange broadened Ru–H resonance at −21.07 ppm, 1H NMR resonances at 6.53, 6.42 and 5.36 ppm due to the central pyridine ring also indicate a de-aromatized ligand. This is most consistent with a rapid equilibrium (K1 in Scheme 1) between the starting materials 1 + free 4-pentenenitrile and the nitrile adduct 2b. Unfortunately, the minor species could not be further characterized by 1H NMR spectroscopy due to overlap, but its similarity in 31P and 1H (Ru–H) NMR shifts with those of compound A suggest it to be the re-aromatized ketimido product 3b. EXSY NMR spectroscopy (mixing time 0.8 s) did not provide evidence for interconversion between the Ru–H species, although it is plausible that 2b and 3b are intermediates in the formation of 4b and therefore involved in an equilibrium. The absence of exchange crosspeaks suggests that the barrier for interconversion between these compounds is sufficiently high that it is slow on the timescale of the NMR experiment. Upon irradiation of the Ru–H NMR signal assigned to 3b in a 1D-NOESY experiment results in a decrease in intensity of the Ru–H moiety of 4b but not 2b, which qualitatively suggests that the rate of 3b reacting to form 4b is faster than its conversion to 2b. Similarly, treatment of 1 with 1 equiv. of acetonitrile led to formation of a reaction mixture that consists of 1, free acetonitrile, and adduct 2c in rapid exchange (broad Ru–H: −24.02 ppm), in addition to the ketimine compound 4c (Ru–H: −13.97 ppm). In this case, signals for 3c are not observed. EXSY spectroscopy at 50 °C for the mixture in this case did show exchange crosspeaks between the Ru–H moieties of 2c and 4c, lending credence to an equilibrium between the various compounds in solution.
 | ||
| Fig. 1 Hydride region of the 1H NMR spectrum in toluene-d8 solvent for 1 with varying amounts of 4-pentenenitrile added. | ||
The reaction between 1 and benzonitrile on NMR scale was analysed 5 min after mixing, which indicated initial formation of the nitrile adduct 2d. In ca. 1 hour, this was fully converted to a mixture of two new products, which appear in a 1:4 ratio. The major species showed a Ru–H signal at −13.22 ppm (31P: 105.8 ppm), indicative of the ketimine complex 4d. The minor species was assigned as the re-aromatized ketimido 3d on the basis of a diagnostic 31P NMR resonance at 120.9 and Ru–H at −10.02 ppm.
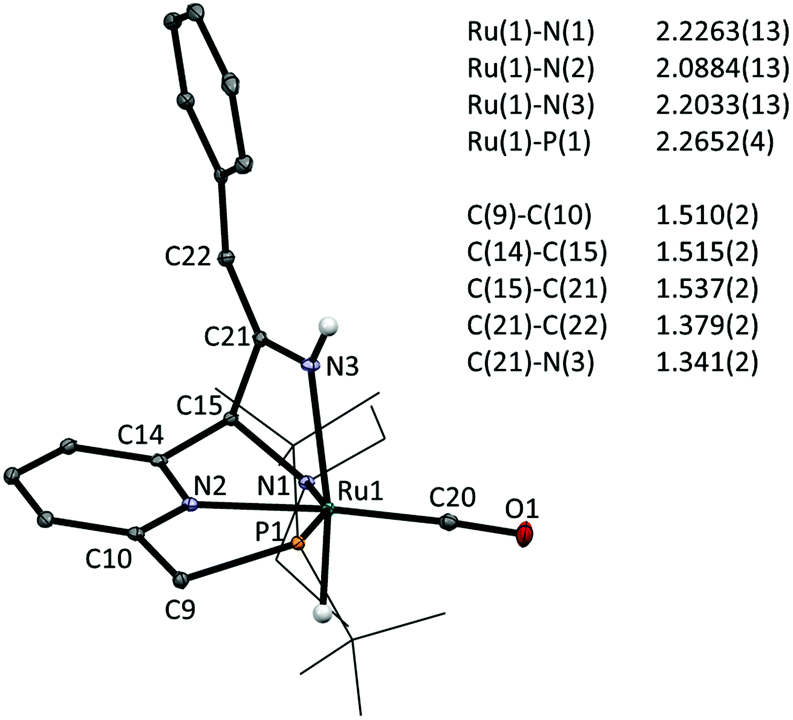
Finally, addition of benzyl cyanide to 1 on NMR scale results in clean transformation of the starting materials in ca. 1 hour to give the enamido complex 5e as the sole product (Ru–H: −12.0 ppm; 31P: 120.6 ppm). Diagnostic for the formation of the enamido group are the appearance of singlets at δ 5.35 and 5.10 ppm corresponding to the vinylic CH and the Ru–NH moieties, respectively. Formation of 5e is the result of tautomerization of the intermediate 3e, as was reported by Milstein for a Re PNP pincer complex.6a Monitoring the progress of the reaction by NMR spectroscopy allowed observation of an intermediate that is tentatively assigned as 3e on the basis of its NMR data (Ru–H: −13.71 ppm; 31P: 109.8 ppm). Its rapid disappearance, however, precluded a full NMR assignment of this intermediate. The identity of the final product 5e was confirmed by X-ray crystallography.‡ The molecular structure shows a ruthenium centre in a pseudo-octahedral environment with the tridentate PNN pincer ligand and a CO ligand in the same plane (Fig. 2). The position trans to the hydride is occupied by the nitrile-derived N atom, with a H–Ru–N(3) angle of 164.2(8)°. Metal–ligand cooperative activation of the CN is evidenced by the formation of the C(15)–C(21) and Ru(1)–N(3) bonds. The bond lengths within the central pyridine ring are indicative of re-aromatization, with C–C distances ranging between 1.387(2)–1.393(2) Å and C–N bond lengths of 1.350(2) and 1.347(2) Å. The Ru(1)–N(3) distance is relatively long (2.2033(14) Å) and from the difference Fourier map it was clear that a hydrogen atom is attached to N(3). The nitrile-derived N(3)–C(21) bond length of 1.341(2) Å and the short adjacent C(21)–C(22) bond (1.379(2) Å) are consistent with the enamido structure with a re-aromatized pyridine ring, as assigned on the basis of the NMR data for 5e.
 | ||
| Fig. 2 Molecular structure of compound 5e with selected bond lengths (Å). | ||
Study of equilibria in the 1 + 4-pentenenitrile product mixture – nitrile concentration
From the data discussed above, it is clear that the reaction of 1 with nitriles can result in a variety of products that are in equilibrium, and the final product (mixture) is highly dependent on the substituents on the nitrile. In order to gain more insight in these equilibria, we studied the system 1 + 4-pentenenitrile in more detail. Addition of increasing amounts of 4-pentenentrile to a C6D6 solution of 1 (see Fig. 1) resulted in a gradual change of the broadened Ru–H resonance from −26.69 ppm in the starting material 1 to −15.07 ppm in the presence of 32 equiv. of 4-pentenenitrile. The composition of the equilibrium mixture changes also upon addition of 4-pentenenitrile: the percentage of ketimine complex 4b increases from 31% of the total Ru–H integration when only 0.5 equiv. of substrate are present, to 64% with 32 equiv. 4-pentenenitrile. In all spectra, there is a small amount of 3b present as well (between 6 and 11%). From the data at high 4-pentenenitrile concentration it is possible to estimate the chemical shift of the nitrile adduct 2b as ca. −15.0 ppm. From the position of the exchange-averaged peak for the Ru–H the 1:2b ratio may then be determined. 1H NMR integration of the remaining Ru–H signals (from 2b and 3b) allows a complete description of the equilibria involved. Fitting of the 1H NMR data to a 1:1 binding model for the (rapidly exchanging) equilibrium 1 + 4-pentenenitrile ⇄ 2b results in a binding constant of 41 (±20%) M−1 (see ESI† for details).12 The equilibrium constants K2 and K3 (for the equilibria 2b ⇄ 3b, and 3b ⇄ 4b, respectively) are determined using the 1H NMR integrations as 0.5 and 6 M−1, respectively.
Study of equilibria in the 1 + 4-pentenenitrile product mixture – temperature dependence
NMR data were also collected at various temperatures for the product mixture obtained from 1 with 1 equiv. of 4-pentenenitrile in toluene-d8. At temperatures of −25 °C or lower, the predominant species in solution are 3b and 4b, which are observed in a ca. 1:6 ratio. Increasing the temperature results in a gradual appearance of signals due to the nitrile adduct 2b (which again is in fast exchange with 1 + 4-pentenenitrile). At +75 °C or above, <10% of the Ru–H signal intensity is due to 3b/4b, and the remaining signal approaches the chemical shift of pure 1. Keeping the sample at elevated temperature results in the gradual decrease of the total signal intensity, suggesting that the species decompose. The line broadening at low temperature and the decomposition at high temperature only allow reliable determination of concentrations in a limited temperature range (−5 to 55 °C). The temperature dependence of the 1H NMR spectra (Fig. 3) is in agreement with the components 1 + free 4-pentenenitrile being entropically favoured at high temperature, whereas binding of the nitrile in a metal–ligand cooperative manner (3b and 4b) is preferred at low temperature (exergonic) due to a favourable enthalpy contribution that results from Ru–N and C–C bond formation in these compounds. A Van 't Hoff plot afforded an estimate of the thermochemical parameters for the first equilibrium (1 + 4-pentenenitrile ⇄ 2b; K1) of ΔH = −11 kcal mol−1 and ΔS = −32 cal mol−1 K−1, and for the second equilibrium (2b ⇄ 3b; K2) it gives ΔH = −3 kcal mol−1 and ΔS = −13 cal mol−1 K−1. The negative values for ΔS are expected for the formation of the nitrile adduct (2b), and its subsequent transformation to the ketimido compound (3b). The equilibrium between 3b and 4b (K3) is hardly affected by changes in temperature, consistent with these having similar structures and thus very similar entropy.
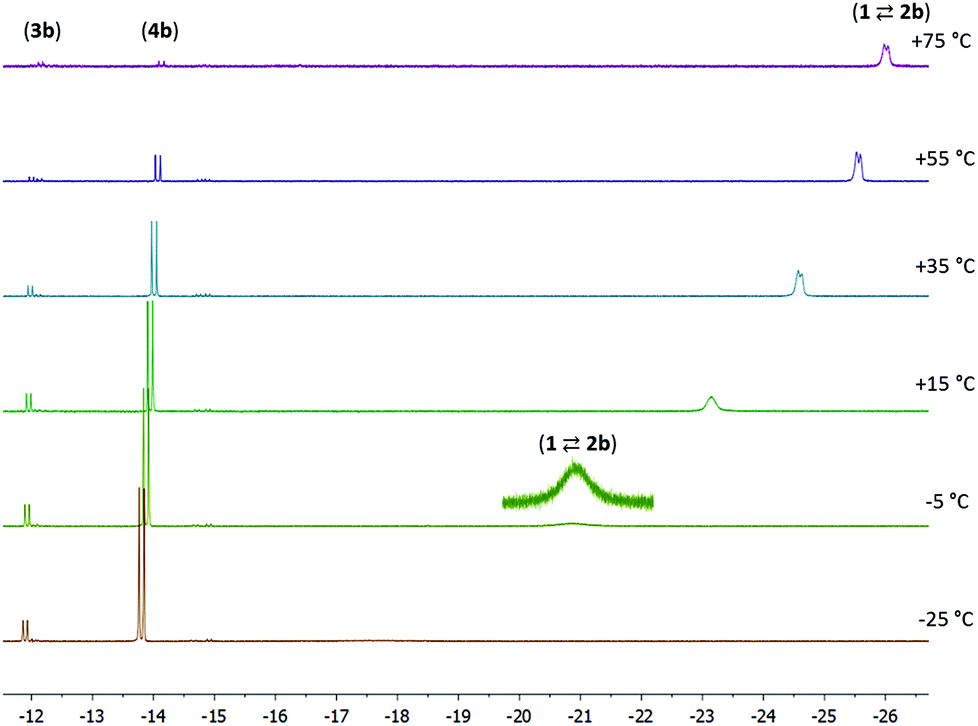 | ||
| Fig. 3 Variable temperature NMR spectra for the reaction between 1 and 4-pentenitrile (1:1, 0.04 M solution in toluene-d8). | ||
DFT calculations
A key factor that leads to formation of the various products is the Brønsted basicity of the Ru–N moiety that results from metal–ligand cooperative nitrile activation with 1. Based on the experimental data, the ketimido compounds 3 that are initially formed are either fully consumed or only present as a minor component of the reaction mixture. The geometries of the compounds 3–5 was optimized using DFT calculations at the TPSSTPSS level of theory using a LANL2DZ basis set (with effective core potential) on Ru and 6-31G(d,p) for all other atoms. Calculations were carried out in the gas phase, and the stationary points were confirmed to be local minima by subsequent frequency analyses. The energies of these geometries were further refined by carrying out single-point TPSSTPSS calculations using Alrichs’ def2-TZVP basis set13 and employing Grimme's D3 empirical dispersion correction (Table 1).14 We initially focussed on the nitriles that lead to a single product according to NMR spectroscopy, e.g., cinnamonitrile (4a) and benzyl cyanide (5e). Geometry optimizations of the possible products (3a/4a and 3e/4e/5e) from the addition of these nitriles to 1 converge on the desired structures as local minima on the potential energy surface. In case of cinnamonitrile, the difference in free energy between 3a and 4a (which is experimentally observed) is calculated to be 1.3 kcal mol−1 in favour of the latter. Evaluation of the Gibbs free energies for the benzyl cyanide addition products identifies the enamido compound 5e as the most stable product (ΔG = −6.0 and −4.6 kcal mol−1 relative to 3e and 4e, respectively), in agreement with empirical data. The stability of 5e is presumably due to conjugation with the aromatic ring. For the products of 1 with 4-pentenenitrile, the DFT calculations suggest that the energies of 3b, 4b and 5b differ by only ca. 1 kcal mol−1, with the latter being the most stable isomer computationally. This is in contrast to the experimental data, which show 4b as the major species. The relative Gibbs free energies of the reaction of 1 with the other nitriles (c/d) included in this study are shown in Table 1. In case of acetonitrile, the calculations correctly predict the de-aromatized ketimine complex 4c to be the most stable. For benzonitrile, the DFT calculations suggest that 3d is favoured by 0.3 kcal mol−1, which contrasts the experimental observation of 4d as the major isomer. Given the small computed differences in stability (and neglecting solvation in our computational model) we feel that these discrepancies are not unexpected given the complexity of the system.15 In addition, it is likely that (stabilizing) H-bonding interactions with the solvent16 exist in case of NH-containing ketimine compounds 4, which are absent in 3. We have not calculated the transition states connecting the various species, but note that recent computational studies suggest that involvement of adventitious water or other proton-shuttling agents (possibly including the nitrile substrates)17 may lead to significant lowering of the activation energies for these tautomerization steps.6b,18 In connection to this, it should be noted that during the study of the concentration- or temperature-dependence of the system 1 + 4-pentenenitrile, we observed that equilibrium is established within minutes (the time required to setup the NMR experiment).| 3a | 3b (%) | 4a | 4b (%) | 5a | 5b (%) | |
|---|---|---|---|---|---|---|
|
a Gibbs free energies in kcal mol−1 relative to the lowest energy isomer.
b Relative amount of each component as determined by 1H NMR integration of 1:1 mixture 1 + nitrile at room temperature (remainder is 1 + 2).
|
||||||
| (a) Cinnamonitrile | 1.8 | 0 | 0 | 100 | — | — |
| (b) 4-Pentenenitrile | 0.5 | 9 | 1.0 | 48 | 0 | 0 |
| (c) Acetonitrile | 0.5 | 0 | 0 | 49 | 1.0 | 0 |
| (d) Benzonitrile | 0 | 20 | 0.3 | 76 | — | — |
| (e) Benzyl cyanide | 7.9 | 0 | 5.7 | 0 | 0 | 100 |
Conclusions
We have studied how nitrile activation by a de-aromatized pincer ruthenium complex leads to a variety of different products, the stability of which depends on the substitution pattern on the nitrile. Based on our combined experimental/computational results and those reported in the literature for similar systems,6 it is clear that the highly dynamic nature of these systems is a key factor in their unusual reactivity: the barriers for interconversion between the various species is low and their Gibbs free energies are close. This allows the system to select the reaction pathway that leads to the most exergonic products, for example resulting in unusual Michael addition reactivity with 1.8 The data presented here provide additional insight in metal–ligand cooperative substrate activation pathways and subsequent reactivity (proton-transfer/tautomerization) that results from the Brønsted basicity of the bound nitrile fragment.Experimental
NMR scale reaction between 1 and cinnamonitrile to give 4a
In a J. Young NMR tube, a 1/1 mixture of 1 and cinnamonitrile was dissolved in benzene-d6. After shaking for 15 min at RT, the reaction mixture was analysed by NMR spectroscopy. Full conversion to 4a was observed. 1H NMR (500 MHz, C6D6): δ 9.68 (s, 1H, CNH), 7.12–7.01 (m, 5H, Ph), 6.81 (d, J = 16.2, 1H, PhCHCH), 6.54 (ddd, J = 8.3, 6.3, 1.8, 1H, Py-H4), 6.38 (d, J = 8.9, 1H, Py-H5), 5.93 (d, J = 16.2, 1H, PhCHCH), 5.71 (dd, J = 6.4, 1.1, 1H, Py-H3), 4.54 (s, 1H, CHN(CH2CH3)2), 3.77 (d, J = 2.8, 1H, CHPtBu2), 2.92 (dq, J = 14.4, 7.2, 1H, N(CH2CH3)2), 2.63 (m, 2H, N(CH2CH3)2), 2.31 (dq, J = 13.3, 7.1, 1H, N(CH2CH3)2), 1.65 (d, J = 13.0, 9H, PtBu2), 1.37 (d, J = 12.5, 9H, PtBu2), 0.83 (t, J = 7.0, 3H, N(CH2CH3)2), 0.75 (t, J = 7.1, 3H, N(CH2CH3)2), −12.95 (d, J = 32.2, 1H, Ru–H). 13C NMR (126 MHz, C6D6): δ 176.16 ((CHCH)(CH)CNH), 168.88 (d, J = 15.6, Py-C2), 151.43 (Py-C6), 139.47 (PhCHCH), 137.46 (Ph quaternary), 133.17 (Py-C4), 132.50 (Ph), 131.37 (Ph), 130.64 (Ph), 126.76 (PhCHCH), 114.86 (d, J = 16.4, Py-C3), 101.24 (Py-C5), 75.46 (CHN(CH2CH3)2), 69.07 (d, J = 54.3, CHPtBu2), 51.12 (s, N(CH2CH3)2), 48.55 (s, N(CH2CH3)2), 40.49 (d, J = 15.8, PC(CH3)3), 38.71 (d, J = 33.9, PC(CH3)3), 33.37 (d, J = 3.3, PC(CH3)3), 32.75 (d, J = 5.3, PC(CH3)3), 12.86 and 12.17 (N(CH2CH3)2). 31P NMR (162 MHz, C6D6): δ 105.68.
NMR scale reaction between 1 and 4-pentenenitrile
In a J. Young NMR tube, an equimolar mixture of 1 and 4-pentenenitrile was dissolved in C6D6. After shaking for 15 min at room temperature, the reaction mixture was analysed by NMR spectroscopy which showed the presence of 3 species:2b (in rapid exchange with 1 + 4-pentenenitrile):
1H-NMR (400 MHz, C6D6): δ = 6.56–6.50 (m, Py-H4), 6.42 (d, J = 8.8, Py-H3), 5.36 (d, J = 6.5, Py-H5), 3.64 (d, J = 1.6, CHP), 3.29 (d, J = 13.8, CHHN), 3.04 (d, J = 13.8, CHHN), 2.67–2.51 (m, 3H, N(CH2CH3)2), 2.35 (dq, 1H, N(CH2CH3)2, J = 13.4, 7.2), 1.42 (dd, J = 13.9, 12.9, PtBu2), 0.95 (t, J = 7.1, NEt2), 0.75 (t, J = 7.2, NEt2), −20.94 (d, J = 26.2, Ru–H). 13C{1H}-NMR (101 MHz, C6D6): δ = 207.49 (CO), 168.69 (d, J = 15.6, Py-C2), 156.46 (d, J = 2.5, Py-C6), 131.89 (d, J = 1.5, Py-C4), 119.36 (s, CN), 113.76 (d, J = 17.2, Py-C3), 96.59 (s, Py-C5), 65.02 (s, CH2NEt2), 64.94 (d, J = 54.2, CHPtBu2), 54.70 (s, N(CH2CH3)2), 50.97 (s, N(CH2CH3)2), 37.97 (d, J = 22.2, P[C(CH3)3]2), 35.70 (d, J = 29.7, P[C(CH3)3]2), 30.05 (dd, J = 2.8, 4.2, P[C(CH3)3]2), 11.28 (s, N(CH2CH3)2), 9.99 (s, N(CH2CH3)2). 31P–{1H}-NMR (162 MHz, C6D6): δ = 96.05.
4b:
1H-NMR (400 MHz, C6D6): δ = 9.66 (s, CNH), 6.56–6.50 (m, Py-H4), 6.35 (d, J = 8.8, Py-H3), 5.52 (d, J = 6.3, Py-H5), 3.73 (overlap of d, J = 1.6, CHP and CHN), 2.83 (dq, 1H, N(CH2CH3)2, J = 14.4, 7.1), 2.67–2.51 (m, 1H, N(CH2CH3)2), 2.48 (dq, 1H, N(CH2CH3)2, J = 13.3, 7.1), 2.10 (dqd, 1H, N(CH2CH3)3, J = 13.9, 6.8, 1.9), 1.61 (d, J = 13.0, PtBu2), 1.33 (d, J = 12.3, PtBu2), 0.80 (t, N(CH2CH3)2, J = 7.1), 0.68 (t, N(CH2CH3)2, J = 7.1), −13.88 (d, J = 21.6, Ru–H). 13C{1H}-NMR (101 MHz, C6D6): δ = 209.47 (d, J = 13.8, CO), 181.41 (s, CNH), 166.54 (d, J = 15.4, Py-C2), 148.99 (d, J = 2.0, Py-C6), 130.57 (d, J = 1.4, Py-C4), 112.80 (d, J = 16.4, Py-C3), 98.75 (s, Py-C5), 77.24 (s, CHNEt2), 66.85 (d, J = 54.3, CHPtBu2), 48.58 (s, N(CH2CH3)2), 46.22 (s, N(CH2CH3)2), 38.17 (d, J = 15.9, P[C(CH3)3]2), 36.34 (d, J = 34.0, P[C(CH3)3]2), 31.01 (d, J = 3.2, P[C(CH3)3]2), 30.43 (d, J = 5.5, P[C(CH3)3]2), 10.55 (s, N(CH2CH3)2), 9.59 (s, N(CH2CH3)2). 31P{1H}-NMR (162 MHz, C6D6): δ = 105.95.
Minor species (3b):
Diagnostic signals: 1H-NMR: doublet at −11.88 ppm (J = 28.5 Hz), 31P-NMR: 121.0 ppm. Further NMR assignment was unsuccessful due to overlap with 4b.
NMR scale reaction between 1 and acetonitrile
In a J. Young NMR tube, an equimolar mixture of 1 and acetonitrile was dissolved in C6D6. After shaking for 90 min at room temperature, the reaction mixture was analysed by NMR spectroscopy which showed the presence of 2 species:2c (in rapid exchange with 1 + 4-pentenenitrile):
1H-NMR (400 MHz, C6D6): δ = 6.51 (td, 1H, Py-H4, J = 6.7, 1.8), 6.41 (d, 1H, Py-H3, J = 8.9), 5.29 (d, 1H, Py-H5, J = 6.3), 3.59 (d, 1H, CHP, J = 1.9), 3.34 (d, 1H, CHHN, J = 14.0), 2.78 (d, 1H, CHHN, J = 14.0), 2.67–2.39 (m, 3H, N(CH2CH3)2), 2.20 (dqd, 1H, N(CHHCH3)2, J = 13.1, 7.3, 1.5), 1.38 (d, 9H, P(C(CH3)3)2, J = 13.4), 1.37 (d, 9H, P(C(CH3)3)2, J = 12.6), 0.89 (t, 3H, N(CH2CH3)2, J = 7.1), 0.72 (t, 3H, N(CH2CH3)2, J = 7.3), 0.60 (s, 3H, HNCCH3), −24.04 (bs, Ru–H). 13C{1H}-NMR (101 MHz, C6D6): δ = 0.27 (s, NCCH3), 10.54 (s, N(CH2CH3)2), 11.33 (s, N(CH2CH3)2), 29.60 (d, P(C(CH3)3)2, J = 4.3), 29.63 (d, P(C(CH3)3)2, J = 3.8), 35.51 (d, P(C(CH3)3)2, J = 28.3), 38.02 (d, P(C(CH3)3)2, J = 24.2), 50.84 (s, N(CH2CH3)2), 55.07 (s, N(CH2CH3)2), 64.78 (d, PyCH2NEt2, J = 1.5), 65.14 (d, PyCHPtBu2, J = 54.2), 96.58 (s, Py-C5), 114.06 (d, Py-C3, J = 17.3), 116.70 (NCCH3), 132.03 (d, Py-C4, J = 1.8), 156.71 (d, Py-C6, J = 2.6), 168.95 (d, Py-C2, 15.9), 207.12 (d, CO, J = 12.6). 31P–{1H}-NMR (162 MHz, C6D6): δ = 94.94.
4c:
1H-NMR (400 MHz, C6D6): δ = 9.24 (s, 1H, CNH), 6.53 (td, 1H, Py-H4, J = 6.7, 1.8), 6.39 (d, 1H, Py-H3, J = 8.9), 5.49 (d, 1H, Py-H5, J = 6.3), 3.73 (d, 1H, CHP, J = 2.7), 3.61 (s, 1H, CHN), 2.83 (dq, 1H, N(CHHCH3)2, J = 14.0, 7.2), 2.67–2.39 (m, 2H, NCH2CH3)2), 2.06 (dqd, 1H, N(CHHCH3)2, J = 13.2, 7.0, 2.0), 1.61 (d, 9H, P(C(CH3)3)2, J = 13.1), 1.32 (d, 9H, P(C(CH3)3)2, J = 12.5), 1.18 (d, 3H, HNCCH3, J = 1.3), 0.75 (t, 3H, N(CH2CH3)2, J = 7.2), 0.66 (t, 3H, N(CH2CH3)2, J = 7.1), −13.95 (d, 1H, Ru–H, J = 32.4). 13C{1H}-NMR (101 MHz, C6D6): δ = 9.61 (s, N(CH2CH3)2), 10.36 (s, N(CH2CH3)2), 25.48 (s, NCCH3), 30.39 (d, P(C(CH3)3)2, J = 5.5), 30.99 (d, P(C(CH3)3)2, J = 3.3), 36.37 (d, P(C(CH3)3)2, J = 33.9), 38.07 (d, P(C(CH3)3)2, J = 15.7), 46.06 (s, N(CH2CH3)2), 48.52 (s, N(CH2CH3)2), 66.79 (d, PyCHPtBu2, J = 54.2), 77.10 (s, PyCHNEt2), 98.59 (s, Py-C5), 112.88 (d, Py-C3, J = 16.5), 130.59 (d, Py-C4, J = 1.7), 148.82 (d, Py-C6, J = 2.0), 166.59 (d, Py-C2, J = 15.5), 179.06 (s, HNCCH3), 209.42 (d, CO, J = 13.8). 31P–{1H}-NMR (162 MHz, C6D6): δ = 106.00.
NMR scale reaction between 1 and benzonitrile
In a J. Young NMR tube, an equimolar mixture of 1 and benzonitrile was dissolved in C6D6. Immediately upon mixing, 1H NMR spectroscopy indicated formation of the adduct 2d. After shaking for 90 min at room temperature, the reaction mixture was analysed by NMR spectroscopy which showed the presence of 2 new species, 3d and 4d:2d (in rapid exchange with 1 + benzonitrile):
1H-NMR (400 MHz, C6D6): δ = 6.93 (d, 2H, J = 7.6, HAr, ortho), 6.80 (d, 1H, J = 7.6, HAr, para), 6.63 (t, 2H, J = 7.6, HAr,meta), 6.55 (t, 1H, J = 7.6, Py-H4), 6.45 (d, 1H, J = 8.8, Py-H3), 5.36 (d, 1H, J = 6.3, Py-H5), 3.65 (bs, 1H, PyCHPtBu2), 3.30 (d, 1H, J = 13.9, PyCHHNEt2), 3.02 (d, 1H, J = 13.9, PyCHHNEt2), 2.70–2.58 (m, 3H, N(CH2CH3)2), 2.38 (dq, 1H, J = 13.8, 6.9, N(CH2CH3)2), 1.43 (vt, 18H, J = 12.1, P(C(CH)3)2), 0.93 (t, 3H, J = 7.0, N(CH2CH3)2), 0.73 (t, 3H, J = 7.2, N(CH2CH3)2), −21.22 (s, 1H, Ru–H). 31P–{1H}-NMR (162 MHz, C6D6): δ = 95.35.
4d (major):
1H-NMR (400 MHz, C6D6): δ = 10.20 (s, 1H, CNH), 7.01–6.88 (m, Ph and undefined signals), 6.48 (ddd, 1H, J = 8.9, 6.3, 1.8, Py-H4), 6.36 (d, 1H, J = 8.9, Py-H3), 5.61 (dd, 1H, J = 6.3, 0.9, Py-H5), 4.62 (s, 1H, PyCHNEt2), 3.78 (d, 1H, J = 2.9, PyCHPtBu2), 2.94 (dq, 1H, J = 14.0, 7.1, N(CH2CH3)2), 2.70 (dqd, 1H, J = 20.9, 7.0, 1.6, N(CH2CH3)2), 2.66 (dq, 1H, J = 13.6, 7.2, N(CH2CH3)2), 2.30 (dqd, 1H, J = 13.2, 7.0, 2.0, N(CH2CH3)2), 1.65 (d, 9H, J = 12.9, P(C(CH)3)2), 1.32 (d, 9H, J = 12.5, P(C(CH)3)2), 0.77 (t, 3H, J = 7.1, N(CH2CH3)2), 0.75 (t, 3H, J = 7.1, N(CH2CH3)2), −13.23 (d, 1H, J = 32.2, Ru–H). 13C{1H}-NMR (101 MHz, C6D6): δ = 209.42 (d, J = 13.6, CO), 176.46 (s, CNH), 166.65 (d, J = 15.4, Py-C2), 148.69 (s, PyC6), 130.80 (d, J = 1.5, Py-C4), 112.86 (d, J = 16.4, Py-C3), 99.53 (s, Py-C5), 75.59 (s, CHNEt2), 67.16 (d, J = 54.3, CHPtBu2), 48.80 (s, N(CH2CH3)2), 46.39 (s, N(CH2CH3)2), 38.33 (d, J = 15.9, P(C(CH3)3)2), 36.39 (d, J = 33.7, P(C(CH3)3)2), 31.04 (d, J = 3.0, P(C(CH3)3)2), 30.29 (d, J = 5.3, P(C(CH3)3)2), 10.56 (s, N(CH2CH3)2), 9.92 (s, N(CH2CH3)2). 31P–{1H}-NMR (162 MHz, C6D6): δ = 105.97.
3d (minor):
Diagnostic signals: 1H-NMR: doublet at −10.02 ppm (J = 25.3 Hz). 31P-NMR: 120.9 ppm. Further NMR assignment was unsuccessful due to overlap with 4d.
Synthesis of 5e
Compound A was prepared in situ by stirring 25.0 mg (0.0512 mmol) of (PNN)RuCl(H)(CO) with 5.8 mg (0.0517 mmol) KtOBu in 2 mL of toluene. Subsequently, 6.0 μL (0.0522 mmol) of benzyl cyanide was added. The resulting solution was filtered and transferred to a clean vial. Slow diffusion of hexane (2 mL) into the toluene solution resulted in precipitation of the product as crystalline material. The mixture was stored at −30 °C overnight and the supernatant removed. The solid was washed with hexane (7 × 0.5 mL) and dried in vacuo to give 12.5 mg of 5e as a red crystalline solid (0.0219 mmol, 42.7%). 1H-NMR (400 MHz, C6D6): δ = 7.66 (d, 2H, HAr, ortho, J = 7.8), 7.35 (t, 2H, HAr, meta, J = 7.7), 6.95–6.79 (m, 3H, unresolved overlap HAr, para, Py-H4 and Py-H5), 6.42 (d, 1H, Py-H3, J = 7.5), 5.35 (s, 1H, Ph-CHCNH), 5.10 (br s, 1H, Ph-CHCNH), 4.13 (s, 1H, NCH-Py), 3.36 (q, 2H, N(CH2CH3), J = 7.2), 2.87 (dd, 1H, PCHH-Py, J = 16.4, 7.7), 2.79 (dd, 1H, PCHH-Py, J = 16.4, 9.9), 2.40 (dq, 1H, N(CHHCH3), J = 13.2, 7.1), 2.19 (dqd, 1H, N(CHHCH3), J = 13.3, 6.8, 3.1), 1.25 (d, 9H, PtBu2, J = 13.1), 0.94 (d, 9H, PtBu2, J = 12.7), 0.94 (t, 3H, N(CH2CH3), J = 7.2), 0.89 (t, 3H, N(CH2CH3), J = 7.1), −11.98 (d, 1H, Ru–H, J = 28.7). 13C-NMR (101 MHz, C6D6): δ = 209.43 (d, CO, J = 16.4), 160.91 (s, Py-C6), 159.71 (d, Py-C2, J = 3.9), 152.91 (s, PhCHCNH), 144.53 (s, CAr, ipso), 136.51 (s, Py-C4), 128.80 (s, CAr, meta), 123.84 (s, CAr, ortho), 119.70 (s, CAr, para), 119.09 (s, Py-C5), 118.31 (d, Py-C3, J = 8.6), 89.42 (s, PhCHCNH), 82.29 (s, NCH-Py), 49.32 (s, N(CH2CH3)2), 46.83 (s, N(CH2CH3)2), 37.28 (d, Py-CH2P, J = 10.6), 37.16 (d, P(C(CH3)3)2, J = 20.6), 34.56 (d, P(C(CH3)3)2, J = 23.3), 30.59 (d, P(C(CH3)3)2, J = 3.3), 29.16 (d, P(C(CH3)3)2, J = 4.9), 11.34 (s, N(CH2CH3)2), 9.34 (s, N(CH2CH3)2).
31P-NMR (126 MHz, C6D6): δ = 120.73. Anal. calcd for C28H42N3OPRu: C, 59.14; H, 7.39; N, 7.44. Found: C, 59.33; H, 7.64; N, 7.27.
Acknowledgements
Financial support from the Netherlands Organisation for Scientific Research (NWO) is gratefully acknowledged (VENI and VIDI grants to E. O.).Notes and references
- G. Van Koten and D. Milstein, Organometallic Pincer Chemistry, Springer, Berlin Heidelberg, 2013 Search PubMed.
- (a) C. Gunanathan and D. Milstein, Acc. Chem. Res., 2011, 44, 588–602 CrossRef CAS PubMed; (b) V. T. Annibale and D. Song, RSC Adv., 2013, 3, 11432–11449 RSC; (c) C. Gunanathan and D. Milstein, Chem. Rev., 2014, 114, 12024–12087 CrossRef CAS PubMed; (d) J. R. Khusnutdinova and D. Milstein, Angew. Chem., Int. Ed., 2015, 54, 12236–12273 CrossRef CAS PubMed.
- D. Milstein, Philos. Trans. R. Soc. London, Ser. A, 2015, 373, 20140189 CrossRef PubMed.
- (a) M. Vogt, M. Gargir, M. A. Iron, Y. Diskin-Posner, Y. Ben-David and D. Milstein, Chem. – Eur. J., 2012, 18, 9194–9197 CrossRef CAS PubMed; (b) M. Vogt, A. Nerush, Y. Diskin-Posner, Y. Ben-David and D. Milstein, Chem. Sci., 2014, 5, 2043–2051 RSC; (c) O. Rivada-Wheelaghan, A. Dauth, G. Leitus, Y. Diskin-Posner and D. Milstein, Inorg. Chem., 2015, 54, 4526–4538 CrossRef CAS PubMed; (d) M. Feller, U. Gellrich, A. Anaby, Y. Diskin-Posner and D. Milstein, J. Am. Chem. Soc., 2016, 138, 6445–6454 CrossRef CAS PubMed; (e) C. A. Huff, J. W. Kampf and M. S. Sanford, Organometallics, 2012, 31, 4643–4645 CrossRef CAS; (f) C. A. Huff and M. S. Sanford, ACS Catal., 2013, 3, 2412–2416 CrossRef CAS.
- (a) M. Montag, J. Zhang and D. Milstein, J. Am. Chem. Soc., 2012, 134, 10325–10328 CrossRef CAS PubMed; (b) C. A. Huff, J. W. Kampf and M. S. Sanford, Chem. Commun., 2013, 49, 7147–7149 RSC.
- (a) M. Vogt, A. Nerush, M. A. Iron, G. Leitus, Y. Diskin-Posner, L. J. W. Shimon, Y. Ben-David and D. Milstein, J. Am. Chem. Soc., 2013, 135, 17004–17018 CrossRef CAS PubMed; (b) A. Nerush, M. Vogt, U. Gellrich, G. Leitus, Y. Ben-David and D. Milstein, J. Am. Chem. Soc., 2016, 6985–6997 CrossRef CAS PubMed.
- G. A. Filonenko, E. Cosimi, L. Lefort, M. P. Conley, C. Copéret, M. Lutz, E. J. M. Hensen and E. A. Pidko, ACS Catal., 2014, 4, 2667–2671 CrossRef CAS.
- S. Perdriau, D. S. Zijlstra, H. J. Heeres, J. G. de Vries and E. Otten, Angew. Chem., Int. Ed., 2015, 54, 4236–4240 CrossRef CAS PubMed.
- (a) D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2015, 54, 6400–6441 CrossRef CAS PubMed; (b) D. W. Stephan, J. Am. Chem. Soc., 2015, 137, 10018–10032 CrossRef CAS PubMed.
- L. Keweloh, H. Klöcker, E.-U. Würthwein and W. Uhl, Angew. Chem., Int. Ed., 2016, 55, 3212–3215 CrossRef CAS PubMed.
- An alternative pathway, in which tautomerization to a compound with the reactive unsaturated ligand fragment at the pincer NEt2 side-arm occurs before coordination of the nitrile can not be ruled out.
- P. Thordarson, Chem. Soc. Rev., 2011, 40, 1305–1323 RSC.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- A. J. Cohen, P. Mori-Sánchez and W. Yang, Chem. Rev., 2012, 112, 289–320 CrossRef CAS PubMed.
- (a) M. Levitt and M. F. Perutz, J. Mol. Biol., 1988, 201, 751–754 CrossRef CAS PubMed; (b) M. Saggu, N. M. Levinson and S. G. Boxer, J. Am. Chem. Soc., 2012, 134, 18986–18997 CrossRef CAS PubMed; (c) M. Albertí, A. Aguilar, F. Huarte-Larrañaga, J. M. Lucas and F. Pirani, J. Phys. Chem. A, 2014, 118, 1651–1662 CrossRef PubMed.
- (a) A. Allerhand and P. von Rague Schleyer, J. Am. Chem. Soc., 1963, 85, 866–870 CrossRef CAS; (b) J. Masnovi, R. J. Baker, R. L. R. Towns and Z. Chen, J. Org. Chem., 1991, 56, 176–179 CrossRef CAS; (c) J.-Y. Le Questel, M. Berthelot and C. Laurence, J. Phys. Org. Chem., 2000, 13, 347–358 CrossRef CAS.
- (a) M. A. Iron, E. Ben-Ari, R. Cohen and D. Milstein, Dalton Trans., 2009, 9433–9439 RSC; (b) S. Qu, Y. Dang, C. Song, M. Wen, K.-W. Huang and Z.-X. Wang, J. Am. Chem. Soc., 2014, 136, 4974–4991 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1481313. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6dt02478e |
| ‡ CCDC 1481313 contains the supplementary crystallographic data for this paper. |
| This journal is © The Royal Society of Chemistry 2016 |